

Abstract
Diabetes predisposes affected individuals to a significant spectrum of cardiovascular complications, one of the most debilitating in terms of prognosis is heart failure. Indeed, the increasing global prevalence of diabetes and an aging population has given rise to an epidemic of diabetes-induced heart failure. Despite the significant research attention this phenomenon, termed diabetic cardiomyopathy, has received over several decades, understanding of the full spectrum of potential contributing mechanisms, and their relative contribution to this heart failure phenotype in the specific context of diabetes, has not yet been fully resolved. Key recent preclinical discoveries that comprise the current state-of-the-art understanding of the basic mechanisms of the complex phenotype that is the diabetic heart, form the basis of this review. Abnormalities in each of cardiac metabolism, physiological and pathophysiological signaling and the mitochondrial compartment, in addition to oxidative stress, inflammation, myocardial cell death pathways and neurohumoral mechanisms are addressed. Further, the interactions between each of these contributing mechanisms and how they align to the functional, morphological and structural impairments that characterize the diabetic heart, are considered in light of the clinical context: from the disease burden, its current management in the clinic, and where the knowledge gaps remain. The need for continued interrogation of these mechanisms (both known and those yet to be identified) is essential to not only decipher the how and why of diabetes-induced heart failure, but also to facilitate improved inroads into the clinical management of this pervasive clinical challenge.
Keywords: Diabetes mellitus; heart failure; cardiac remodeling; diastolic dysfunction; Diabetes, Type 2; Heart Failure; Obesity
INTRODUCTION
The global prevalence of diabetes (particularly type 2 diabetes, T2D) has been progressively increasing over many years in a troubling manner1,2. The most recent estimates from the International Diabetes Federation Diabetes Atlas considered in excess of 450 million adults with diabetes across the globe, a number that was projected to reach 693 million by 20452. This diabetes pandemic imposes a significant burden on society, both in terms of the substantial healthcare costs and the poor health outcomes for affected patients2. Almost five decades ago, the Framingham Heart study established the epidemiological links between diabetes and increased risk of heart failure (HF)3. Indeed, diabetes has long been recognized to not only escalate the risk of heart failure, but also to increase its incidence approximately 2.5-fold (and double this in females), independent of age or concomitant comorbidities such as coronary artery disease and dyslipidemia4–8. Diabetic patients account for up to one third of patients in clinical HF trials, with diabetes persisting as an independent predictor of poor outcome9–11 (and as recently reviewed12–15). Together with an aging population, this has now given rise to a worldwide epidemic of heart failure5. Indeed, the existence of ‘diabetic cardiomyopathy” as a distinct entity became increasingly apparent over the almost five decades since its first description in a small cohort of 4 patients in 197216.
The phenomenon defined as diabetic cardiomyopathy comprises impairments in cardiac structure and function independent of the macrovascular complications of diabetes (including hypertension, coronary artery disease and atherosclerosis)17,18, described in a plethora of clinical and experimental studies (as reviewed19–21). Despite these statistics, there has been a dearth of effective treatments for diabetes-induced cardiac dysfunction, (and even traditional means of intensive blood glucose control often have failed to improve cardiac function or reduce risk of heart failure)22–24. Conventional therapy for established heart failure is the same, whether or not the patient has diabetes. Mechanism-specific therapy for the diabetic-associated heart failure is currently not available (although the new era of sodium glucose cotransporter-2 inhibition [SGLT2i] offers some promise in this direction). Thus, therapies that target diabetes-induced cardiac dysfunction and subsequent heart failure are urgently needed. Further, patients with concomitant diabetes and heart failure in the absence of other comorbidities such as obesity, hypertension and atherosclerosis are likely the exception rather than the rule in clinical medicine. It is thus now timely in this compendium article to both reconsider the nomenclature and definition of the diabetic heart, and to update the contributing mechanisms to the complex phenotype, identified from preclinical discoveries reported in the last five years.
FUNCTIONAL IMPAIRMENTS THAT CHARACTERIZE THE DIABETIC HEART
Cardiac dysfunction is often clinically silent in diabetes and frequently is not detected until later stages of disease. Even in asymptomatic, normotensive patients with well-controlled diabetes, ~50% are considered to exhibit some degree of cardiac dysfunction12,25–27. It is widely accepted that one of the hallmarks of the diabetic heart is left ventricular (LV) diastolic dysfunction, which is one of the first signs of diabetic cardiomyopathy, often detectable earlier than clinically-significant LV systolic dysfunction17,19–21,28. Systolic and diastolic dysfunction have been attributed to differences in gross cardiac morphology29. Notably, the existence of isolated diastolic dysfunction as an indicator of diabetic cardiomyopathy was until recently challenged at times, as patients in the early stages of diabetes were not routinely subject to careful assessment of diastolic function. The cardiac complications of diabetes were usually only investigated after overt heart failure symptoms became evident. The era of more sensitive imaging techniques, and the recognition that heart failure is becomingly increasingly common in people affected by diabetes, may overcome this. Regardless of whether this is assessed in the clinical or preclinical/laboratory context, cardiac magnetic resonance imaging (CMRI) is largely considered the gold standard approach for assessment of cardiac function4,15. However, although diabetes is associated with impaired CMRI-derived parameters of LV diastolic function in the clinic28,30, CMRI is not always readily available (with limited access in regional, remote and/or low socioeconomic circumstances). Likewise, synchrotron-based cardiac imaging in experimental laboratory animals enables considerable high-resolution imaging information31 but does not lend itself to high-throughput, serial assessment. Advancement in ultrasound-based echocardiography imaging approaches in recent years, in both clinical and experimental contexts, now permits relatively high-resolution, detailed, non-invasive, serial assessment of cardiac function without the time-consuming or availability/access restraints often posed by CMRI and synchrotron-based imaging and as such is the imaging approach of choice for the majority of clinical and preclinical cardiac routine and interventional research studies32,33.
Traditionally, the initial impairments in LV diastolic function that are considered characteristic of diabetes-induced cardiomyopathy include prolonged and delayed LV filling and LV relaxation32, often in the absence of concomitant impairments in LV systolic function15,27. As detailed by the most recent guidelines published by the American Society of Echocardiography and the European Association of Cardiovascular Imaging, a range of Doppler-based signals can be used for the assessment of diastolic function, with several parameters (usually at least 3-4) of diastolic function assessed. These parameters include those determined at the end of diastole (which correlate with LV end-diastolic pressure, EDP). These are obtained by measuring blood flow velocity across the mitral valve (via Doppler flow echocardiography), or the analogous velocity of motion of the adjacent myocardial tissue (via tissue Doppler imaging). These approaches yield two major phases of movement, an early phase (denoted as the E wave on Doppler flow, and E’ on tissue Doppler) and a later, atrial phase (denoted as the A wave on Doppler flow, and A’ on tissue Doppler)19. Mitral peak A wave velocity and tissue Doppler–derived mitral annular a′ wave velocity, as well as those that are measured earlier in diastole, such as mitral E/A ratio, E wave deceleration time and E/e′ ratio, are common parameters derived from these approaches. E/e’ is one of the most reproducible and reliable of these examples32. Isovolumetric ventricular relaxation time (IVRT) has also been widely used to assess diastolic function, but is highly-dependent on heart rate and afterload32. Interested readers are referred to the guidelines for a comprehensive viewpoint on the strengths and potential limitations in the presence and absence of comorbidities such as age, hypertension, rhythm disturbances, etc12,32. There is considerable evidence supporting the presence of impairments in LV diastolic function in the diabetic population, often manifest in a significant proportion of otherwise healthy, complication-free diabetic individuals34,35. Prolonged deceleration time, reduced e’ and e’/a’, and increased E/e’ are reported in diabetic patients in the absence of clinically-significant impairments in LV systolic function30,33,35–38, in addition to impairments in other (less reliable) measures of diastolic function, such as reduced E/A and prolonged IVRT30,35,38. Diastolic dysfunction can also be reliably determined in the diabetic population by other echocardiography-derived parameters, including LV global longitudinal strain, speckle-tracking echocardiography and strain rate imaging18,39,40. Again, these approaches confirm that LV systolic dysfunction is only evident later than diastolic dysfunction in the diabetic human myocardium, and/or is milder, and often in the context of heart failure with preserved ejection fraction (HFpEF, discussed later)15,32,41,42. These more sensitive means of detection can reveal mild (subclinical) impairments in LV systolic function in diabetic patients, even when LV ejection fraction (EF) is preserved15,28,37,43,44. Regardless of whether concomitant LV systolic function is impaired, evidence from epidemiological studies suggests that LV diastolic dysfunction imparts significant prognostic implications, with a four-fold risk of death relative to healthy individuals45. These functional impairments in the diabetic heart are likely independent of concomitant atherosclerosis in both the human and preclinical contexts. Indeed, they are clearly manifest in diabetic rodents which are resistant to coronary atherosclerosis20.
AVAILABLE PRE-CLINICAL MODELS OF DIABETES
A selection of animal models of diabetes have been described over the years, predominantly in rodents, for the study of diabetes and its complications. As has been comprehensively reviewed14,19,46, these each mimic different aspects of disease pathology, and the choice of model should reflect the clinical aspects relevant to the study goals. For example, streptozotocin (STZ)-induced mouse models of diabetes are amongst the most common models, and background mouse strain can affect the cardiac phenotype observed. In male FVB/N mice, LV diastolic dysfunction (evident from 4 weeks) precedes LV systolic dysfunction by at least several weeks47,48, and in some studies, the timeframe may not have been sufficiently long for this later systolic dysfunction to become manifest (in both C57/Bl6 and FVB/N strains)49–51. STZ-induced diabetes superimposed on the FVB/N strain is often associated with negligible differences in body weight at study end compared to non-diabetic sham animals, in contrast to STZ-induced diabetes in C57/Bl6 mice, where a smaller bodyweight at endpoint may be evident in diabetic mice52,53. Increased levels of reactive oxygen species (ROS), inflammation, fibrosis and apoptosis are routinely seen in the myocardium following STZ-induced diabetes in mice. A clear strength of the STZ-induced mouse model of diabetes is the functional phenotype of LV diastolic dysfunction, evident earlier (and perhaps even in the absence of) LV systolic dysfunction, which thus mimics the common functional phenotype of the diabetic human heart. The weakness of this model is the need to use a toxin to induce diabetes and severe untreated hyperglycemia. In contrast to the STZ mouse model, in male STZ rats, both systolic and diastolic dysfunction are often present, mimicking the later functional phenotype of the diabetic human heart. In addition, the STZ rat retains the cardiac phenotype of increased ROS, inflammation, fibrosis and apoptosis, but weaknesses of this model in rats are the significantly retarded weight gain and bradycardia that are also evident54–56, which can confound interpretation of results obtained. Other models of type 1 diabetes (T1D) have also been used to study the cardiac complications of the disease, including the Akita (on the C57/Bl6 background) and OVE26 mouse models (on the FVB/N background). The strength of both of these models is that they avoid use of the toxin STZ to limit insulin availability and hence induce T1D57–62. Both of these genetic models of T1D however do not always reproduce all aspects of the human disease. For example, Akita mice also exhibit retarded weight gain, the cardiac dysfunction is not as reproducible as the STZ mouse and the animals often lack cardiac oxidative stress and cardiac remodeling that are considered common features of the diabetic human heart (as detailed below)58,59. The OVE26 mouse model of T1D reproduces a cardiac functional phenotype similar to that seen in the STZ rat, whereby both systolic and diastolic dysfunction are evident60–62. Weaknesses of the OVE26 model is the very early T1D onset (during first week of postnatal development), it is not the most readily-available mouse line, and the disease results from overexpression of pancreatic β-islet cell calmodulin, not a common cause of T1D in humans60–62. For these reasons, the STZ-induced mouse model of T1D remains a preferred model for many preclinical scientists.
A widely-used model of T2D is the spontaneously-diabetic db/db mouse63–66. The strength of this model is that it reliably reproduces LV diastolic dysfunction characteristic of the diabetic human heart, together with cardiac oxidative stress and inflammation. A weakness of the db/db mouse is however the morbid obesity and a systemic metabolic phenotype including hyperleptinemia and severe insulin resistance that is quite distinct to the human condition. Moreover, homozygotes are sterile. In contrast, rodent models that combine a milder STZ regime than that used to induce T1D combined with a high fat diet may be a more appropriate choice in some instances, where a milder systemic phenotype combined with a robust cardiomyopathy phenotype of cardiac remodeling and dysfunction is observed67. Strengths of these diet-based models is the extent to which they can be widely-available, are not limited by breeding constraints (particularly useful when crossing with another mouse line) and that their systemic phenotype more closely parallels what is observed in human diabetes. Their weakness is the longer amount of time required to produce a cardiac phenotype (perhaps six months), and the milder cardiac dysfunction that may result, compared to the db/db mouse. Rat models of T2D include spontaneously diabetic models such as the Zucker diabetic fatty rat68,69 and Goto Kakizaki rat (difficult to source outside of Japan)70 as well as dietary-based models (high fructose, sucrose or fat)71–73 and combined STZ-high fat diet models71,74. Of these models, the spontaneously-diabetic db/db mouse model of T2D, followed by a diet-based mouse model (such as STZ-high fat) have been the preferred model by many preclinical scientists. For a more detailed analysis of the preclinical models of diabetes available, and their strengths and weaknesses, readers are referred to several comprehensive reviews devoted to this topic14,19,46.
In general, rodent models of diabetic cardiomyopathy do not recapitulate all of the aspects of diabetic cardiomyopathy and although they have provided important mechanistic insights, a strong imperative exists for the development of additional models in other species. Human-derived cells such as inducible pluripotent stem (iPS) stem cells and/or three-dimensional human cardiac organoids to complement animal studies are also being developed, including some with a more mature phenotype75–80.
MORPHOLOGICAL AND STRUCTURAL PHENOTYPES THAT CHARACTERIZE THE DIABETIC HEART
There has been considerable assessment of the structural abnormalities that are characteristic of the diabetic heart, in clinical and experimental settings, ranging from indices of cardiac remodeling to impairment in coronary microvascular perfusion19,20,81. These morphological defects likely contribute to the functional impairments evident in the diabetic human heart, but evaluation of whether these alterations precede deficits in cardiac function, relies predominantly from observations in animal models of the disease, where they have been attributed to the pathophysiological mechanisms that lead to diabetic heart disease, as discussed in the next section.
• Cardiac fibrosis:
There is robust evidence of cardiac fibrosis in the diabetic human heart. Early evidence of diabetes-associated cardiac fibrosis in the human heart was largely derived from post-mortem or biopsy analyses82,83. Increased deposition of interstitial collagen types I and III (detected using immunohistochemistry) is evident in diabetic compared to non-diabetic human myocardial biopsies82, confirming earlier reports of interstitial and perivascular fibrosis detected by standard histological and electron microscopic ultrastructural analyses in post-mortem samples 83,84. However, recent approaches using either CMRI-derived T1 mapping and late gadolinium enhancement (LGE) imaging, or echocardiography-derived integrated backscatter, have proved useful in the non-invasive detection of cardiac fibrosis in humans85–88. These approaches reveal that diabetes is associated with increased cardiac fibrosis, even in the absence of prior ischemic injury15,30,84–86,89,90, but some observations suggest fibrosis may emerge later in disease progression (after deficits in cardiac function have manifest)12,16. It has been suggested that a component of the diabetes-induced increase in cardiac fibrosis may be replacement fibrosis83. Further, diabetic patients with LGE-derived evidence of cardiac fibrosis, exhibit poor prognosis across major adverse cardiovascular events (including survival), highlighting a potential role for these non-invasive cardiac imaging approaches in clinical management or risk stratification of the diabetic patient85. This clinical evidence of cardiac fibrosis in the diabetic heart is supported by a plethora of preclinical studies in experimental rodent models of diabetes reporting diabetes-associated cardiac fibrosis47,49,56,63,65,81,91–96.
• Cardiac hypertrophy:
An association between diabetes and increased LV mass is well-substantiated97–101 and is regarded as a structural hallmark of the disease in humans19,20. CMRI revealed increased LV mass in the human heart in patients with T2D, even in relatively younger patients (e.g. at 40 years of age)4,102. Echocardiography-derived increases in LV mass are also evident18,37,38,103,104, and may even be exaggerated in diabetic females relative to males15,36,99,101. It is however pertinent to note that these increases in LV mass in humans affected by diabetes reflects the net effects at the level of cardiac fibrosis (outlined above), cardiomyocyte hypertrophy and cardiomyocyte loss via myocardial cell death pathways (discussed later). Thus, increased LV mass in diabetic patients might not exclusively reflect the impact of cardiomyocyte hypertrophy per se in the diabetic human heart. Observations from diverse animal models, including STZ-induced T1D mice and rats51,105, 6, high-fat-fed mice107,108, Goto-Kakizaki T2D rats109 and spontaneously-T2D db/db mice63,65 suggest that diabetes is commonly associated with cardiomyocyte hypertrophy. However cardiomyocyte hypertrophy has not been observed in other animal models such as in some studies utilizing the Akita and STZ-induced mouse models of T1D15,110. Although the reason for this controversy regarding whether diabetes increases cardiomyocyte size remains to be resolved, concomitant obesity and/or insulin resistance are potential contributing mechanisms to diabetes-induced cardiomyocyte hypertrophy111–113, that may exacerbate mechanisms by which high glucose alone may promote cardiomyocyte hypertrophy, at least in experimental settings106,114.
• Impaired coronary microvascular perfusion:
There are several reports in the literature in which impairments in each, of coronary flow reserve, myocardial blood volume and myocardial blood flow, with increased coronary resistance, are observed in humans affected by diabetes. Myocardial specimens studied at autopsy in diabetic patients reveal reduced numbers of capillaries and arterioles with increased thickness of arteriolar walls115. Diabetic patients also exhibit reduced sensitivity to dipyridamole-induced increases in coronary blood flow, with increased coronary resistance and reduced coronary reserve during cardiac catheterization in vivo116. Similar findings of increased coronary resistance and reduced coronary reserve have been obtained using whole-body positron emission tomography (PET)117. Evidence of microvascular disease, including reduced myocardial blood flow reserve, has also been obtained in the diabetic human heart using contrast echocardiography36,103. Whilst much of the clinical evidence has been obtained in T2D patients, similar observations have been reported in human subjects with T1D in vivo. For example, reduced sensitivity to cold pressure test and adenosine-induced increases in myocardial blood flow, with increased coronary resistance and reduced coronary reserve (via PET)117, as well as reduced myocardial blood volume and blood flow (via contrast echocardiography)118, have been reported in T1D patients. These diabetes-induced microvascular impairments would reduce the delivery of oxygen and other essential nutrients to the myocardium. Indeed, coronary rarefaction in the human heart has been identified as a predictor of cardiac fibrosis119. This evidence of microcirculation impairment in humans with diabetes is recapitulated in experimental models of diabetes120–122. Diabetic rodents exhibit significantly decreased myocardial capillary density; this impairment precedes diabetes-induced cardiac fibrosis and in turn, reduced density of cardiomyocytes (at least in STZ-induced diabetic rats)94,123. Similarly, T2D rats exhibit reduced coronary flow reserve and increased coronary vascular resistance109,120. Indirect evidence for impaired capillary density in the diabetic myocardium also comes from reduced cardiac expression of pro-angiogenic vascular endothelial growth factor (VEGF), and both of its receptors, VEGF-R1 and VEGF-R2, in both diabetic rats and humans94,123,124. These microvascular impairments are likely further exacerbated by impaired coronary endothelial function and increased microvascular stiffness as a result of sustained diabetes19,117,123,125–129.
MECHANISMS CONTRIBUTING TO DIABETIC HEART DISEASE
With each passing decade, the list of potential contributing mechanisms to diabetes-induced cardiomyopathy continues to expand. As illustrated in Figure 1, well-established mediators identified for a number of years have included oxidative stress, inflammation and impaired Ca2+ handling, as well as alterations in substrate metabolism/utilization, insulin signaling, gene regulation, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, neurohumoral activation and cardiac cell death19,20. An overview of each of these contributing mechanisms is provided below, with consideration of recent insights to have now emerged. Figure 2 illustrates these contributing mechanisms, their complexity and sites of likely overlap in more comprehensive detail.
Figure 1.

Oxidative stress, inflammation, alterations in metabolic pathways (including abnormalities in substrate utilization, mitochondrial function, advanced glycation end-product [AGE] formation and O-GlcNAcylation), as well as changes at the level of insulin signaling, gene regulation, endoplasmic reticulum (ER) stress, neurohumoral activation and cardiac cell death, have all been widely accepted as mediators of diabetes-induced myocardial remodeling and dysfunction (see text for references).
Figure 2.
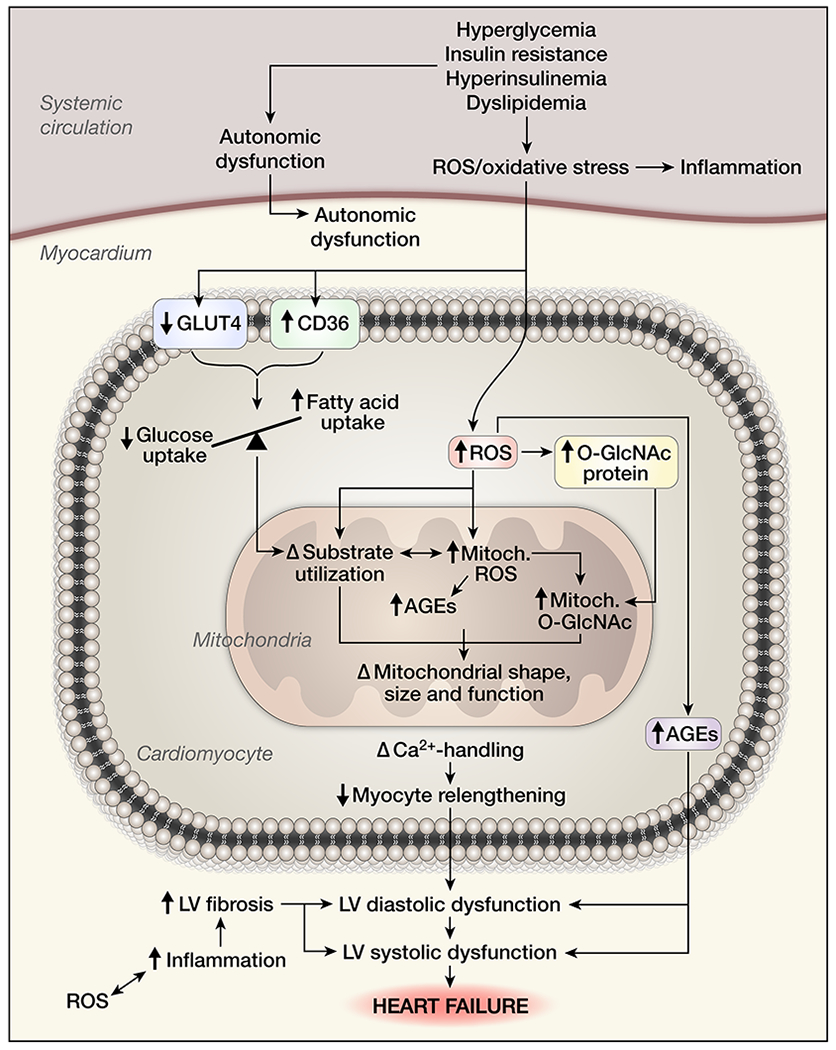
The contributing mechanisms to the cardiac phenotype of diabetes-induced cardiomyopathy are complex and multifactorial, as are their consequences. Indeed, there is significant intersection between many of these, driving the diabetic myocardium towards failure (see text for references). Illustration Credit: Ben Smith.
• Oxidative stress:
Oxidative stress is defined as an imbalance between inappropriate (i.e. excessive) ROS generation and the capacity for these to be degraded. Major sources of ROS such as superoxide in the heart include NADPH oxidase, mitochondrial respiration (normally a minor byproduct but increased in diabetes) and uncoupled nitric oxide synthases; the latter two becoming particularly problematic in pathological contexts such as diabetes19,81,130–132. In addition to increased capacity for ROS generation in settings of diabetes (as a result of induction of all three major cardiac sources of ROS), endogenous antioxidant mechanisms are often also impaired in diabetes (as reviewed19,133,134). Intervention with appropriate antioxidants such as superoxide dismutase (SOD) mimetics or coenzyme Q10, in animal models of diabetes, either early as a preventative measure to limit progression to diabetic cardiomyopathy, or later in the disease process (once cardiac impairment is evident) have established the causal contributing role of oxidative stress to diabetic heart disease (in both T1D and T2D)65,113,135–141. Similar evidence for a causal role in clinical settings of diabetic cardiomyopathy is less abundant, largely hampered by use of relatively poor antioxidant approaches (e.g., vitamin E)19,132,142,143, although increased ROS levels are apparent in human diabetic myocardium and vasculature144–146. In addition to the direct oxidative damage (to proteins, lipids, DNA) as a result of inappropriate cardiac ROS levels, ROS is also a key trigger of inflammasome activation (discussed below) 18,142,147. ROS also activate multiple other pathological signaling cascades, such as protein kinase C (PKC), apoptosis signal-regulating kinase-1 (Ask1), p38 mitogen-activated protein kinase (p38-MAPK), NH2-terminal Jun kinases (JNK) and JAK-STAT pathways19,20,105,130,148–155, which themselves have been implicated in diabetes-induced cardiac complications156–158. Moreover, several of these (including PKC, p38-MAPK and JAK-STAT)130,152,157,159 can also induce ROS generation, creating a detrimental feedforward loop. Two of the most promising approaches for limiting diabetes-induced oxidative damage in the heart in recent years is through pharmacological activation of Nrf2 (regarded as the master antioxidant regulator)147,160,161 and inhibition of NADPH oxidase with the joint NOX1/NOX4 inhibitor GKT137831, as demonstrated in several preclinical models of diabetes129,162–165. Their efficacy with respect to limiting diabetic cardiomyopathy (particularly in humans) remains to be confirmed.
• Inflammation:
Diabetes is an inflammatory disorder, with increased levels of ROS playing a key contributing role in this context19,131,136,147,166. Indeed, a detrimental feedforward loop in which increased ROS induces inflammatory cytokines and vice versa is thought to provide the momentum for this highly pro-inflammatory environment21,147,166,167. Systemic inflammation is clearly present in both T1D and T2D patients, and is considered a contributing mechanism to peripheral disease progression in several organ systems (e.g. liver, pancreas, kidney and vasculature). Increased circulating cytokines, chemokines, immune cells and other inflammatory biomarkers are clearly evident168–170. Moreover, diabetes promotes tissue infiltration of macrophages and their polarization towards an M1-like ‘pro-inflammatory’ phenotype in affected patients, with upregulated leukocyte inflammatory cytokine signaling171. Mouse and rat models of T1D and T2D also display systemic inflammation relatively early in disease progression, as indicated by increased circulating monocytes and neutrophils170, as well as increased tumor necrosis factor-α (TNFα), interleukins (IL-1β, IL-6), transforming growth factor-β (TGF-β), interferon-γ (IFNγ), the inflammatory transcriptional regulator NFκB, and various chemokines and vascular adhesion molecules155,172–177. A pathophysiological role for inflammatory signaling is thus clearly implicated in diabetic complications. The influx of infiltrating immune cells has emerged as a significant contributor to the development and progression of myocardial injury and LV dysfunction in a range of cardiac pathologies, including myocardial ischemia, HF with reduced ejection fraction (HFrEF) and HFpEF178–185. Inflammatory processes are also activated in human diabetic myocardium, as evidenced by increased cytokines, chemokines, and various leukocyte populations. This implicates cardiac inflammation in the pathogenesis of cardiomyopathy in diabetic patients169. Induction of pro-inflammatory genes and proteins (TNFα, IL-1β, IL-6, TGF-β, IFNγ and NFκB) have also been reported (at timepoints later than systemic inflammatory markers) in the LV of T1D and T2D rodent models of diabetes54,131,136, 141,153–155,158,166,167,186–188. Damage-associated molecular pattern molecules (DAMPs, e.g. S100A8/S100A9), infiltration of other inflammatory cells (e.g. T cells, B cells, neutrophils, dendritic cells and mast cells) and/or TH1/TH17 immune responses could also contribute to cardiomyopathy in this context170,172,182,187. Much of this cardiac inflammation in the diabetic heart may be triggered or amplified by increased ROS levels, as revealed by the ability of ROS-lowering strategies to ameliorate the pro-inflammatory state in T1D and T2D rodent models of diabetes136,167,189. This pro-inflammatory state is thought to be kickstarted to a large extent by ROS-triggered activation of the NLRP3 inflammasome in the diabetic heart190, as has been observed in multiple preclinical models141,161,189,191. NLRP3 activation triggers a cascade of events, recruiting pro-caspase-1 to facilitate caspase-1 activation, and cleavage of IL-1β and IL-18 precursors to generate their active products21,147. As a result, a plethora of the above pro-inflammatory mechanisms are enhanced189, favoring an altered macrophage polarization towards an M1-like phenotype in diabetes21,192. The net effect of infiltrating inflammatory cells such as macrophages can be implicated in diabetes-induced LV fibrosis and dysfunction.
In addition to the pro-inflammatory state evident in diabetes, how the body attempts to recover from this continuous insult may also be impaired. Infiltrating macrophages exhibit defective phagocytosis of apoptotic neutrophils at the site of vascular inflammatory injury (efferocytosis)182. It is thus possible that defective resolution of inflammation may hence represent an additional mechanism of diabetes-induced cardiac damage secondary to macrophage infiltration, as is evident in other tissues192–194. In support of this, approaches including targeting of the chemokine receptor CCR2, or enhancement of heme-oxygenase-1 (downstream of Nrf2), which favor macrophage polarization towards an M2-like state, suppress diabetes-induced oxidative stress, remodeling and dysfunction in the myocardium in both T1D and T2D settings155,186. There is thus considerable interest in the development of novel pro-resolving therapies to target a range of inflammatory diseases195,196 and diabetes and the associated cardiomyopathy could be one such indication for such an approach.
• Changes at the level of insulin sensitivity and signaling:
Impaired systemic insulin sensitivity is evident in, and characteristic of, people with diabetes197–199. In the heart, sensitivity to insulin as a result of diabetes is impaired at multiple levels. This includes reduced insulin-stimulated glucose uptake into cardiomyocytes and other cardiac cell types, with decreases in both LV GLUT4 expression as well as reduced translocation of GLUT4 from the cytoplasm to the cell membrane19. Responsiveness to insulin is also evident at the level of myocardial contractility and regulation of vascular tone, in which phosphoinositide 3-kinase[p110α], PI3Kα, and its mediator Akt play contributing roles200. Moreover, the relationship between altered insulin signaling and diabetic cardiomyopathy is complex, reflecting consequences of increased or imbalanced activation of certain insulin signaling pathways that could promote cardiac remodeling on the one hand versus inhibition of cardioprotective pathways on the other201. Activation of pathways that may promote LV remodeling include signaling pathways downstream of insulin receptor substrate 1 (IRS1) or crosstalk between insulin signaling and beta adrenergic signaling via the G-coupled receptor kinase (GRK2)202,203. Altered effectiveness of endogenous physiological mechanisms in the heart, including insulin sensitivity and downstream signaling, through both insulin and insulin-like growth factor-1 (IGF1) receptors (via activation of PI3Kα, and its mediator Akt), have also been observed in the diabetic myocardium70,204,205. Indeed, defects in PI3Kα/Akt signaling exaggerate diabetic cardiomyopathy105,135 and indeed may be sufficient to impair insulin responsiveness, at least in the liver and adipose206–208. On the other hand, increased cardiac activity of PI3Kα in the context of diabetic cardiomyopathy is cardioprotective; these actions are closely associated with suppression of diabetes-induced cardiac ROS and inflammation47,105,209. This evidence of impairments in insulin sensitivity and signaling in the diabetic myocardium (and their likely mechanistic role in the diabetic heart) is largely reliant on observations in diabetic rodent models47,65,70,109,210–212, as corresponding observations in myocardial biopsies are logistically challenging in humans, although a small number of studies have provided evidence to support imbalanced insulin signaling201,213. Therapeutic targeting of these changes thus represents innovative means of limiting the cardiac consequences of diabetes, particularly if selective for the myocardium47,49,105 (a feat not yet met by pharmacological means).
• Altered cardiac metabolic pathways
Diabetes alters myocardial substrate utilization20, 21, such that there is reduced glucose oxidation, increased fatty acid oxidation and reduced glycolysis. In addition, ROS-induced mitochondrial uncoupling in the diabetic myocardium markedly reduces cardiac efficiency64,204. Formation of advanced glycation end-products (AGEs) and a maladaptive hexosamine biosynthesis pathway (HBP) also contribute to glucotoxicity19, 20.
Advanced glycation end-products (AGEs):
As described by Brownlee and others, AGEs are long-lasting molecules that result from advanced, non-enzymatic glycosylation of excess sugars which can induce an irreversible form of posttranslational protein modification20,214,215. AGE-modification of extracellular matrix (ECM) and other structural proteins are thought to provide a physical impediment to myocardial compliance. Preclinical studies targeting AGEs and their receptors, RAGE, reveal that hyperglycemia-induced increases in the AGE-RAGE axis may contribute to diabetic heart disease20,216. Moreover, a detrimental feed-forward cycle has been proposed, whereby ROS trigger increased AGE formation and RAGE activation20,217, and the AGE-RAGE axis also drives ROS formation and concomitant inflammation (particularly in the context of diabetes and its complications)218,219. The crosslink breaker Alagebrium (ALT-711, which disrupts crosslinks between proteins and AGEs) attenuates the morphological characteristics of both the T1D rat heart95 and the high fat fed mouse heart219. These cardioprotective effects of Alagebrium mirror those seen in mice lacking RAGE219,220. Indeed, enhancing levels of AGEs even in the absence of diabetes is sufficient to drive endothelial inflammation and atherogenesis221. More recently, the mitochondrial-targeted methylglyoxyl scavenger, mitoGamide, has been shown to also protect LV diastolic function in Akita T1D mice57. AGEs thus likely contribute to diabetes-induced cardiac remodeling and dysfunction. AGE modifications of cardiac proteins is not restricted to ECM proteins but occurs more broadly in the diabetic myocardium, as observed for example with the Ca2+-handling protein, sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA2a), and myofilament proteins including troponin, actin and α-myosin heavy chain220,222–224. Although Alagebrium has been taken into clinical trials in humans with heart failure in the absence and presence of diabetes215, these have however largely failed to be completed due to financial reasons. As such, therapies specifically targeting AGEs have not crossed over into routine clinical care.
Hexosamine biosynthesis pathway (HBP):
Under normal physiological conditions, the majority of glucose metabolized in the heart is attributed to glycolysis, the pentose phosphate pathway and glycogen synthesis. Hexosamine biosynthesis represents a relatively minor consequence of glucose metabolism (crudely estimated as <5% of all glucose metabolism)225–227, with UDP-N-acetylglucosamine (UDP-GlcNAc) its ultimate product. UDP-GlcNAc provides O-GlcNAc (an effector of glucose) for post-translational modification of more than 3000 nuclear, mitochondrial, and cytosolic proteins228. Analogous to phosphorylation, O-GlcNAcylation occurs specifically at Ser/Thr residues, changing activity of affected proteins. In contrast to phosphorylation, only two functionally-opposing enzymes control all protein modification by O-GlcNAcylation. The O-GlcNAc-transferase (OGT) and O-GlcNAcase (OGA) are responsible for the addition and removal of O-GlcNAc from susceptible proteins, respectively225–228. Whilst it has been known for some time that glutamine fructose-6-phosphate aminotransferase (GFAT) is the rate-limiting enzyme of the HBP225–227, recent work now reveals that whereas HBP flux increases in direct proportion with glucosamine levels, glucose metabolism via the HBP only represents ~0.006% of that which is consumed by glycolysis229. This is much lower than previous estimates. The O-GlcNAc modification (‘O-GlcNAcylation’) is distinct from other forms of glycosylation that occur in the ER and Golgi apparatus (N-linked, mucin-like, O-mannosylation), as well as advanced glycation227. In diabetes, protein O-GlcNAcylation becomes maladaptive, and the sustained induction of O-GlcNAcylation contributes to cardiac glucotoxicity198,225–227,230. Both sustained hyperglycemia and increased cardiac ROS generation increase the flux of glucose metabolism through hexosamine biosynthesis, enhancing UDP-GlcNAc levels and subsequent O-GlcNAcylation225–227,231. In contrast, an acute increase in glucose load (over minutes rather than hours) has minimal impact on cardiac UDP-GlcNAc levels229. In addition to the known ability of ROS to drive protein O-GlcNAcylation, hyper O-GlcNAcylation in return drives NADPH oxidase, illustrating the intimate relationship between O-GlcNAcylation and ROS/oxidative stress81,232–234. Further, both O-GlcNAcylation and ROS are regulators of inflammation, autophagy and ER stress81,235.
Activity of the rate-limiting enzyme of the HBP, GFAT and of its downstream product, UDP-GlcNAc, are increased in human T2D at the level of skeletal muscle198,230 and the heart233,234. Gene expression and activity of GFAT were also reported to be increased in human T2D myocardium, in conjunction with increased total protein O-GlcNAcylation236. Similar correlations have been described in leucocytes237,238. This might be exacerbated by reduced OGA expression in human T2D (both systemically and in the myocardium), impairing removal of O-GlcNAc from affected proteins and impairing O-GlcNAc balance233,239. Interestingly, genetic mutations resulting in a truncated, less-effective form of OGA carries a 3-fold increased risk of developing T2D238,240. O-GlcNAcylation has also emerged as a novel regulator of inflammation, of both pro- and anti-inflammatory pathways, as recently reviewed241. For example, O-GlcNAc-modified TGF‑β-activated kinase-1 triggers pro-inflammatory signals such as NFκB, which enhances levels of cytokines and promotes macrophage polarization towards an M1-like phenotype241,242. O-GlcNAcylation has also been implicated in the activation of both T-cells and B-cells243. There are still many unanswered questions as to functional correlates of the many (>3000) proteins susceptible to this posttranslational modification. O-GlcNAc-modified Ca2+/calmodulin-dependent protein kinase II (CaMKII) under hyperglycemic conditions is directly linked to enhanced CaMKII activity and cardiac Ca2+ sparks236. The cardioprotective signals PI3Kα and Akt are O-GlcNAc-modified in diabetes, an effect that is ablated by cardiac gene delivery of O-GlcNAc-ase in parallel with blunting of the structural and functional characteristics of the diabetic mouse heart233,234. Further studies interrogating the functional consequences of O-GlcNAc-modifications of specific proteins is thus warranted. On the other hand, O-GlcNAc modification can favor protective anti-inflammatory actions, such as in the atherosclerotic vasculature, particularly in an acute context244.245, but this is unlikely to apply in the context of diabetes, and the diabetic myocardium, where sustained O-GlcNAcylation is evident, and detrimental233,234. Together, these observations demonstrate clinical relevance of maladaptive O-GlcNAcylation in human T2D myocardium.
Other metabolic alterations in the diabetic heart:
In addition to these glucose-driven abnormalities in cardiac metabolism, other impairments in substrate utilization, particularly of fatty acids and the resultant lipotoxicity, are also evident. Circulating levels of free fatty acids, and other lipids (triglycerides, etc.) are elevated in diabetes, particularly T2D63,64,246,247, likely as a result of impaired insulin action in adipose tissue and liver201,248. Importantly, diabetes-associated cardiac lipotoxicity is not unique to the T2D heart, but is also observed in T1D247,248. As recently reviewed21,248,249, the diabetic heart exhibits altered substrate utilization for energy (i.e. ATP) generation, with an enhanced reliance on fatty acids (at the expense of glucose utilization)64,250. This altered utilization is facilitated in part by the increased expression of sarcolemmal transport proteins on cardiomyocytes that mediate free fatty acid uptake by the heart (particularly CD36), whilst expression and sarcolemmal localization of glucose transport proteins on the heart (i.e. GLUT4) is reduced20,54,205,247,249–251. Myocardial lipid content increases as a consequence, as observed in both humans and animal models of diabetes64,247,252. Increased cardiac lipid accumulation has been observed as a relatively early change in the diabetic heart (preceding onset of impaired LV function), and hence represents another potential contributor to diabetic heart disease20,246–248. Concomitant with this switch in substrate preference, abnormalities at the level of the mitochondria (across oxidative phosphorylation, dynamics, morphology and ROS generation) in the diabetic myocardium are not only manifest but have been convincingly implicated as contributing causal mechanisms of diabetes-induced impairments in mitochondrial function21,249, as detailed below. ER stress is also evident in the diabetic myocardium, likely resulting from multiple abnormalities, including increased ROS/oxidative stress, altered substrate utilization (and subsequent lipotoxicity), activation of cell death pathways (apoptosis, autophagy, etc.), amongst other contributing mechanisms20,253.
• Diabetes-induced impairments in the mitochondrial structure and function:
Mitochondria are essential integrators of redox signals and metabolic flux14,254. In T2D, in both patients and preclinical models, mitochondrial dynamics are impaired and mitochondrial ROS generation exceeds the endogenous scavenging capacity, leading to cardiac oxidative stress and inflammation19,144,249,255,256. Mitochondrial number may actually be increased, but mitochondria are smaller in size, and often fragmented256,257, suggestive of an imbalance in fission/fusion quality control, as supported by cardiac mitochondrial fragmentation and reduced expression of the mitochondrial fusion protein, mitofusin-1, in the diabetic human myocardium255. This mitochondrial dysfunction plays an essential part in the progression of diabetic cardiomyopathy19,144,255,256. Multiple mechanisms impair mitochondrial function in diabetic hearts, much of which directly results from the altered myocardial substrate utilization outlined above. Impaired insulin signaling itself also has a profound effect on mitochondrial morphology and function258,259. Further, disruption of mitochondrial physiology following impaired glycemic control is a major contributing mechanism to diabetic heart disease, with many of the above cytoplasmic perturbations reciprocated within the mitochondrial environment.
Mitochondrial ROS:
Metabolic perturbations such as hyperglycemia and increased cardiac lipid load increase mitochondrial ROS generation131,260,261. This augmented mitochondrial ROS generation has been observed in human myocardium144. Evidence from preclinical models suggests this is sufficient to impair mitochondrial dynamics, and trigger mitochondrial fragmentation and dysfunction, with dysregulation of mitochondrial fission/fusion proteins such as DRP1 and OPA1 implicated in this process253,260. The net effect of these diabetes-induced impairments in mitochondrial function correlate with worsening diabetes144.
Mitochondrial O-GlcNAcylation:
A functional O-GlcNAc cycle, including OGT and OGA, is housed within mitochondria231,262,263, and the pyrimidine nucleotide carrier can transport UDP-GlcNAc into mitochondria from the cytoplasm262. Overexpression of cytosolic OGT is sufficient to increase O‑GlcNAcylation of mitochondrial proteins264, but whether his is also true for mitochondrial OGT is yet to be determined. Hyperglycemia increases O-GlcNAcylation of several proteins key to mitochondrial respiration (e.g. Complexes I, III, IV and their subunits, e.g. NDUFA9). Evidence from short-term rodent models of T1D (over 2–4 weeks, prior to impairment in cardiac function) suggests that O-GlcNAcylation of numerous proteins key to mitochondrial dynamics is elevated226,262,263,265. Affected proteins include DRP1, OPA1, pyruvate dehydrogenase, mitochondrial transcription factor A (TFAM) and mitofusin-2. Whether O-GlcNAcylation of these mitochondrial components is increased in the heart over the longer-term (when onset of cardiac dysfunction is evident) and/or in the more prevalent T2D, is currently not known. How sustained O-GlcNAc-modifications of specific mitochondrial proteins affects both their activity and net cardiac function also remains unresolved, but we predict that maladaptive O-GlcNAcylation of key mitochondrial proteins may provide another mechanistic link between T2D and cardiac dysfunction.
• Myocardial cell death pathways:
As outlined above, altered homeostatic pathways such as oxidative and ER stress have been implicated as drivers of cell death pathways in metabolic disease, with input from impaired mitochondrial dynamics likely contributing to this. Key forms of cell death evident in the diabetic heart, in both T1D and T2D models, include apoptosis, autophagy and necrosis14,19,20,47,48,53,94,211,266–271. Under physiological conditions, a low-level of apoptosis and autophagy are essential for tissue homeostasis, permitting timely removal of unwanted cells and organelles/proteins respectively whose time has come20,272. Whilst the increased apoptosis evident in the diabetic heart (reflected to a considerable extent as cardiomyocyte death) and subsequent replacement fibrosis are conventionally accepted as a detrimental phenomenon19,20 whether this also applies to autophagy remains less clear14. Insulin signaling is an important regulator of myocardial autophagy201,273, with reduced insulin action being associated with increased myocardial autophagy, whereas excessive insulin signaling suppressing autophagy. Thus the relationship between insulin action and autophagy in diabetes, might depend on the relationship between activation or repression of insulin signaling pathways. Additionally, lipotoxicity has also been implicated in autophagy dysfunction in the context of diabetes274,275. Increased autophagy observed in high-fructose-fed insulin-resistant mice for example is associated with modest increases in both cardiac ROS and collagen levels268, but the impact on LV function was not determined. In contrast, blunted cardiac autophagic flux has been observed in the obese ob/ob mouse heart276, and the T1D heart, in at least 2 different mouse models277,278, hence the impact of diabetes on cardiac autophagy, and its consequences, still remain to be resolved14. However, Mst1 has been implicated as a possible master switch regulating whether the cell progresses towards apoptotic or autophagic mechanisms in the diabetic heart279. In addition to activation of cell death pathways in diabetic hearts, it is also apparent that a number of endogenous protective mechanisms that would otherwise defend against cell death are also negatively affected by diabetes205. Taken together, these observations suggest that the diabetic myocardium appears destined to suffer increased cardiac cell death. Indeed subclinical myocardial injury has long been described in epidemiological studies as evidenced by increased circulating concentrations of troponin (detected by highly sensitive assays) in individuals with T2D and prediabetes280.
• Impaired calcium handling:
A slower decay of the Ca2+ transient has repeatedly been reported in studies of isolated diabetic cardiomyocytes, even before any evidence of LV systolic dysfunction becomes manifest15,281–286. Putative mechanisms of these impairments in decay of the Ca2+ transient include reduced gene and protein expression, and/or reduced activity, of SERCA2a15. Post-translational modifications of this Ca2+-handling protein as a result of advanced glycation end-products and/or O-GlcNAcylation have also been implicated in this deficit, as well as oxidative modifications15,19,20,224,233,234,287–290. Multiple other aspects of Ca2+-handling, as a result of impaired Ca2+ influx and efflux to/from the cytoplasm from the extracellular space, altered Ca2+levels in intracellular organelles (including both the SR and mitochondria), altered expression and/or activity of other Ca2+-handling proteins (e.g. phospholamban, ryanodine receptors, etc.), as well as myofibrillar responsiveness to Ca2+, are also defective in the diabetic heart19–21,220,282,286,288,289,291–295. Indeed Ca2+-handling defects were one of the earliest defective mechanisms identified in the diabetic heart291,292,296,297. Moreover, decreased PI3Kα signaling has been associated with blunted inward Ca2+ currents (and thus contractility), and defects in both PI3Kα and inward Ca2+ currents have been reported in the T1D Akita mouse298–300. This spectrum of diabetes-induced defects in Ca2+-handling and responsiveness to Ca2+ underlie delays in cardiomyocyte relengthening in the diabetic heart, and thus have been attributed to account for a significant component of diabetes-induced LV diastolic dysfunction in vivo.
• Neurohumoral mechanisms:
It has been appreciated for several decades now that the renin-angiotensin-aldosterone-system (RAAS)19,21,301–306 and the endothelin-1 system19,307 are activated in diabetes. This is evident both systemically and within the heart, and across both clinical and preclinical contexts. As the RAAS is a critical regulator of blood pressure308,309, both the resultant increased afterload secondary to diabetes-induced systemic RAAS upregulation, as well as the direct actions of the cardiac RAAS on the myocardium, contribute to diabetes-induced remodeling. Indeed pharmacological and gene knockout approaches to targeting these systems is effective at limiting diabetic cardiomyopathy in animal models65,136,302,303,310, implicating them as contributing mechanisms to diabetic heart disease. For this reason, in heart failure patients with concomitant diabetes the use of ACE inhibitors or angiotensin receptor antagonists81,143,311 has been advocated in patients with adequate renal function. Diabetes is also associated with autonomic dysfunction, including at the level of the heart (such as abnormalities in heart rhythm regulation), observed in patients and animals with diabetes312–315. Diabetes-induced autonomic dysfunction likely further exacerbates inflammation in this context, with a resulting increase in levels of immune cells, cytokines and DAMPs316. Demonstrating a potential role for cardiac autonomic dysfunction in diabetic heart disease however is precluded by lack of available approaches with which to manage this dysfunction. More recently, fibroblast growth factor 21 (FGF21) has emerged as a novel skeletal hormone that is important in the regulation of glucose and lipid metabolism in diabetes317. Interestingly, its expression is induced in this context, with increased levels detected in the circulation in T2D humans318,319. Given that FGF21 has been shown to both enhance fatty acid oxidation and glucose utilization, whilst concomitantly blunting lipogenesis320,321, it likely attempts to serve as a compensatory brake against further metabolic disturbances in diabetes, as recently reviewed322. A direct role of FGF21 in ameliorating cardiac dysfunction in diabetes remains to be determined.
• Changes at the level of cardiac gene regulation, such as epigenetic and microRNA-mediated mechanisms, have also been reported as regulators of the phenotype evident in preclinical models of the diabetic heart, as previously reviewed19–21,81,323,324. Increased cardiac expression and/or circulating levels of several small, non-coding single-stranded miRNAs have been described in both experimental settings of diabetes, including miR-199a, miR-2119,81,325–327, as well as in the diabetic human myocardium (including miR-199b, miR-210, miR-223, miR-34b, miR-34c and miR-650 amongst multiple others)19,328. Other miRNAs are downregulated in the diabetic heart, including miR-1, miR-133a, miR-30c, miR-181a and miR-20319,324,325,327,329,330. Whether each of these alterations are a consequence versus a causal contributing mechanism to diabetic cardiomyopathy is yet to be resolved for many of these323. miR-1, miR-133a and MiR-203 may normally serve protective roles against glucose-driven impairments in cardiomyocyte death and/or size, and induction of fibrosis20,327,331, whilst rescue of myocardial miR-92a expression in the T2D mouse heart preserves LV systolic and diastolic function332. An imbalance of detrimental miRs increased in the myocardium (and/or loss of protective miRs) as a result of sustained diabetes may thus contribute to the net cardiac phenotype in this context. Other non-coding RNA molecules including long non-coding RNAs (lncRNAs) and circRNAs have been shown to be regulated by high glucose in cardiac fibroblasts and cardiomyocytes in in vitro studies323. Likewise, small non-coding RNAs (snoRNAs), including the family U32a, U33 and U35a, have been shown to negatively regulate both lipotoxicity and systemic glucose metabolism, blunting oxidative stress333,334. These non-coding RNA molecules may potentially also emerge as contributing to components of the diabetic myocardial phenotype. Lastly, DNA methylation and histone modifications (acetylation and deacetylation) are altered as a result of diabetes in multiple organs, and have been suggested as potential co-conspirators in the progression of diabetic cardiomyopathy, at least an animal models of the disease81. Consensus on their causal role in this process with respect to the diabetic heart is however yet to be reached. Indeed, a small number of recent studies have been exploring the area of epigenetic changes in diabetic cardiomyopathy271,335. For example, diabetes is associated with abnormalities in DNA methylation in both human (cardiac mesenchymal cells and cardiac biopsies) and rodent myocardium (mouse and rat models of T1D and T2D)271,335. We anticipate that these might yield additional insights in the future into the causal factors underlying diabetes-associated heart failure over time.
As the significant plethora of above studies indicates, both the consequences of, and contributing mechanisms to, the cardiac phenotype is complex and multifactorial. As illustrated in Figure 2, many of the drivers of this phenomenon interact, creating a web of multiple, detrimental feed-forward pathways that, without an effective intervention, push the diabetic myocardium towards a fate of poor prognosis. The status of current understanding of the time of onset of each of the major players in the phenotype of the diabetic myocardium is stylized in Figure 3; this timing is considered in the context of common key comorbidities.
Figure 3.

A stylized overview of current understanding of the time-course of diabetes progression, from the initial onset of hyperglycemia, through the mid and late stages of the disease, and their consequences for the phenotype of the diabetic myocardium (see text for references).
THE DIABETIC HEART AND HFpEF
HFpEF describes a diagnosis of heart failure in symptomatic patients whose LV EF is >40–50% (and in whom non-cardiac causes of symptoms have been excluded)119,336–338. This HF phenotype is now slightly more common than HFrEF in hospital admissions for HF336,339, an increased prevalence secondary to the increased global prevalence of diabetes, obesity and hypertension in an aging population337,340. HFpEF likely represents a spectrum of several etiologies depending on which comorbidities and risk factors are also present. These can include aging, obesity, and hypertension amongst others. Both female subjects (particularly elderly females) and diabetic subjects are over-represented in the HFpEF population (compared to HFrEF)336,340–342. Hypertension is a common feature of HFpEF. patients338. Considering HFpEF as a single phenotype is however a misconception, as the disorder is particular heterogeneous, with at least three distinct clusters described338,340. This includes the obesity-cardiometabolic phenotype that has been classically associated with HFpEF (patients with a high body mass index, marked diastolic dysfunction, and commonly accompanied by diabetes and/or hypertension). Other subgroups include the natriuretic peptide deficiency syndrome (obese patients with low BNP who are often hypertensive as a consequence), as well as the high cardiorenal phenotype with right ventricular (RV) failure340,343,344. Of these three clusters, event-free survival is highest for those in the natriuretic peptide deficiency cluster and poorest in the cardiorenal-RV failure cluster340. Intersection between these phenotype clusters is also evident337.
The increasing emergence and awareness of HFpEF in the HF research field might tempt some to (falsely) consider diabetic cardiomyopathy simply the manifestation of HFpEF coexistent with diabetes, given that both share a number of common characteristics. Indeed, diabetic patients may be affected by either HFpEF or HFrEF341,345,346. If these diabetic patients also exhibit microvascular complications, then a HFpEF phenotype (rather than HFrEF) is however more prevalent, as is a poorer prognosis341,345. Post-mortem characteristics of the human HFpEF myocardium include clear evidence of increases in both cardiac mass and fibrosis, with reduced microvascular density119. These observations are also evident via echocardiography336,347. It is likely that a large component of the increased cardiac mass is attributable to fibrosis rather than hypertrophy per se, as clinically-significant hypertrophy may only be evident in approximately 50% of HFpEF patients119. In addition to cardiac fibrosis and microvascular dysfunction, inflammation and cardiac stiffness are also evident338. In contrast to HFpEF cohorts (where females tend to be over-represented)336, diabetic cardiomyopathy does not appear to “favor” one gender over the other348. Further, diabetes-associated HF also includes patients with HFrEF and those with HFpEF346. Thus although diabetic cardiomyopathy is more complex than simply the manifestation of HFpEF coexistent with diabetes, it is clear that the HFpEF phenotype is enriched in diabetes. Future study is required to further understand the mechanistic contribution of diabetes to HFpEF.
BRIEF COMMENTARY ON CURRENT THERAPY FOR THE DIABETIC HEART
Poor glycemic control contributes to increased risk of HF in diabetes, with risk of HF correlated to HbA1c349. Prior to the current era, intense glucose-lowering therapies have largely failed to reduce the risk of incident HF, and may even exacerbate HF risk (particularly thiazolidinediones, sulfonylureas, certain dipeptidyl peptidase-4 (DPP-4) inhibitors and potentially insulin and glucagon-like peptide [GLP-1] agonists)350–355. Although the GLP-1 agonist was shown to be safe and tolerable in T2D patients in the REWIND study, with evidence of sustained glucose-lowering (reduction in HbA1c of 0.61 percentage units over 5 months) together with modest but significant reductions in bodyweight (by 1.5kg) and systolic blood pressure (BP, by 1.7mmHg) compared to placebo over 5 years, no differences in cumulative incidence of cardiovascular outcomes was seen356. The arrival of sodium-glucose co-transporter-2 inhibitors (SGLT2i) has brought a welcome change355,357,358. The EMPA-REG OUTCOME study provided the first observation that SGLT2i represented robust cardioprotective actions for an effective glucose-lowering therapy. Risk of HF hospitalization and cardiovascular mortality were reduced by 35–50% with empagliflozin compared to placebo in T2D patients358. Similar findings with respect to benefits at the level of both reduced HF hospitalization and cardiovascular mortality have subsequently been observed in clinical studies in T2D with both canagliflozin (CANVAS, CREDENCE, CVD-REAL)359–361 and dapagliflozin (DECLARE-TIMI 58)362,363. The reduction in HF hospitalizations has thus now emerged as a class effect364,365. Moreover, SGLT2i appears equally beneficial in T2D patients affected by either HFpEF or HFrEF346,355.
Contributing mechanisms for this SGLT2i cardioprotection particularly with respect to beneficial impact on HF in the context of diabetes remains to be resolved, with a plethora of putative mechanisms reported in the literature366–369. The benefit with respect to HF outcomes is surprisingly early, at least at the level of hospitalizations for HF (with the event curves separating within weeks of treatment commencement). Whether beneficial effects at the level of cardiac remodeling and/or dysfunction are evident at this point in the diabetic human heart remains to be determined. Whilst it has been suggested that the benefits of SGLT2i simply reflect mechanisms such as enhanced glycosuria, natriuresis and reduced BP365, consensus on the mechanism of SGLT2i mechanism of cardioprotection is yet to be reached. SGLT2 is not detected in heart370,371, hence the mechanism of cardioprotection is likely extracardiac in nature; moreover, more than one mechanism may contribute. Although enhanced glycosuria and natriuresis are likely to aid renoprotection, they are not likely to be sufficient to completely account for the cardiac benefit, particularly with respect to HF. Glucose-lowering therapies historically have largely been ineffective with respect to cardiovascular endpoints (prior to the current era), potentially arguing against glucosuria as the sole, standalone mechanism of benefit of SGLT2i. Diuretics do not match the efficacy seen with SGLT2i to date, hence volume unloading on its own is similarly insufficient to fully account for cardioprotection in this context. BP-lowering SGLT2i actions have also come under the spotlight as a contributing mechanism of benefit359,372,373, but the antihypertensive effects of SLGT2i are generally more modest than seen for example with ACE inhibitors367, although of note patients in the EMPA-REG study were well-matched for BP control358,365. Preclinical studies have implicated the impact of empagliflozin on sodium/hydrogen exchanger-1 in the myocardium366, and effects on sympathetic tone (which in turn regulates BP). Most recently, it was revealed that increased hematocrit and enhanced renal erythropoietin synthesis, boosting the systemic oxygen-carrying capacity of erythrocytes, could contribute a significant component of SGLT2i cardioprotection374. Whether these latter putative mechanisms are unique to empagliflozin or a class effect also remains to be resolved. A multifactorial mechanism of SGLT2i benefit, combining glucose-lowering with other, diuretic, natriuretic, antihypertensive and metabolic benefits (analogous to a poly-pill) may indeed explain the cardioprotective consequences of SGLT2i therapy. Despite this lack of consensus regarding the mechanism responsible for SGLT2i benefit, this benefit has been accepted with certainty and excitement. Indeed clinical trials are currently underway for SGLT2i in HF even in the absence of diabetes, and evidence has already emerged that SGLT2i are efficacious in the context of non-diabetic HF375–378. For example, the DAPA-HF phase 3 randomized clinical trial of dapagliflozin or placebo (on top of standard care) in >4700 HF patients with an LVEF <40% revealed a markedly reduced risk of worsening HF or cardiovascular death, in both the diabetic and non-diabetic cohorts378. The smaller DEFINE-HF trial in HFrEF patients reported a greater proportion of patients with improved health status in the dapagliflozin treatment arm than placebo, an effect that was comparable between the diabetic and non-diabetic HFrEF patients379. Readers are referred to the position statement from the European Society of Cardiology/Heart Failure Association for a comprehensive review355. Even with the promising signs that SGLT2i may offer new hope for the debilitating combination of diabetes and HF, the problem of the diabetic heart (and its effective clinical management) is not yet solved. It is likely that more than one component of the complex and multifaceted pathophysiology of diabetic heart disease (and hence more than one treatment/target) are required to optimally manage this growing clinical burden.
CONCLUDING REMARKS:
Given that other comorbidities (atherosclerosis, hypertension, coronary artery disease, etc.) commonly co-manifest with HF in patients with diabetes (Figure 3), it is thus now timely in 2020 to reconsider the definition of diabetic cardiomyopathy. Originally defined as impairments in cardiac structure and function independent of the macrovascular complications of diabetes, perhaps the phenomenon commonly referred to as “diabetic cardiomyopathy” should instead be known as “diabetic heart disease” or “diabetes-associated HF”. While the knowledge base around the mechanisms of diabetic heart disease has continued to expand, there remains several unanswered questions, by no means the least of which can lead us towards development of better therapeutic approaches for tackling this major health burden (and how these might be distinct from conventional approaches for systolic HF in the absence of diabetes). We in the diabetic heart disease research community await with excitement the elucidation of remaining unknown pathways, novel contributors and their interactions that will inform the basic mechanisms of diabetic disease, and new treatments likely to emerge in the coming decade that will arise from the intensive ongoing research activity.
Acknowledgments
Sources of Funding
RHR is supported by the National Health and Medical Research Council (NHMRC) of Australia (ID1059960, ID1158013). EDA is supported by the American Heart Association (16SFRN31810000, 20SFRN35120123) and the National Institutes of Health (HL127764; HL112413, HL141783, HL108379).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- AGEs
advanced glycation end-products
- Ask1
apoptosis signal-regulating kinase-1
- BP
blood pressure
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CMRI
cardiac magnetic resonance imaging
- DAMPs
Damage-associated molecular pattern molecules
- DPP-4
dipeptidyl peptidase-4
- GLP-1
glucagon-like peptide
- EDP
end-diastolic pressure
- ECM
extracellular matrix
- EF
ejection fraction
- ER
endoplasmic reticulum
- FGF21
fibroblast growth factor 21
- GFAT
glutamine fructose-6-phosphate aminotransferase
- GRK2
G-coupled receptor kinase
- HBP
hexosamine biosynthesis pathway
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- IFNγ
interferon-γ
- IGF1
insulin-like growth factor-1
- IL
interleukin
- IRS1
insulin receptor substrate 1
- IVRT
isovolumetric ventricular relaxation time
- JNK
NH2-terminal Jun kinase
- LGE
late gadolinium enhancement
- lncRNAs
long non-coding RNAs
- LV
left ventricular
- miR
microRNA
- OGA
O-GlcNAcase
- OGT
O-GlcNAc-transferase
- p38-MAPK
p38 mitogen-activated protein kinase
- PET
positron emission tomography
- PI3Kα
phosphoinositide 3-kinase[p110α]
- PKC
protein kinase C
- RAAS
renin-angiotensin-aldosterone-system
- RAGE
AGE receptors
- ROS
reactive oxygen species
- SERCA2a
sarco(endo)plasmic reticulum Ca2+-ATPase
- SGLT2i
sodium glucose cotransporter-2 inhibition
- snoRNAs
small, non-coding RNAs
- STZ
streptozotocin
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TFAM
mitochondrial transcription factor A
- TGF-β
transforming growth factor-β
- TNFα
tumor necrosis factor-α
- UDP-GlcNAc
UDP-N-acetylglucosamine
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures
RHR has no conflicts of interest to disclose. EDA has no conflicts of interest to disclose.
References
- 1.Guariguata L, Whiting DRR, Hambleton I, Beagley J, Linnenkamp U, Shaw JEE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Research and Clinical Practice. 2014;103:137–149. [DOI] [PubMed] [Google Scholar]
- 2.Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, Malanda B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Research and Clinical Practice. 2018;138:271–281. [DOI] [PubMed] [Google Scholar]
- 3.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. American Journal of Cardiology. 1974;34:29–34. [DOI] [PubMed] [Google Scholar]
- 4.Gulsin GS, Swarbrick DJ, Hunt WH, Levelt E, Graham-Brown MPM, Parke KS, Wormleighton JV, Lai FY, Yates T, Wilmot EG, Webb DR, Davies MJ, McCann GP. Relation of Aortic Stiffness to Left Ventricular Remodeling in Younger Adults With Type 2 Diabetes. Diabetes. 2018;67:1395–1400. [DOI] [PubMed] [Google Scholar]
- 5.Nichols GA, Gullion CM, Koro CE, Ephross SA, Brown JB. The Incidence of Congestive Heart Failure in Type 2 Diabetes: An update. Diabetes Care. 2004;27:1879–1884. [DOI] [PubMed] [Google Scholar]
- 6.Thrainsdottir IS, Aspelund T, Thorgeirsson G, Gudnason V, Hardarson T, Malmberg K, Sigurdsson G, Rydén L. The association between glucose abnormalities and heart failure in the population-based Reykjavik study. Diabetes Care. 2005;28:612–6. [DOI] [PubMed] [Google Scholar]
- 7.Aksnes TA, Kjeldsen SE, Rostrup M, Omvik P, Hua TA, Julius S. Impact of new-onset diabetes mellitus on cardiac outcomes in the Valsartan Antihypertensive Long-term Use Evaluation (VALUE) trial population. Hypertension. 2007;50:467–73. [DOI] [PubMed] [Google Scholar]
- 8.Ohkuma T, Komorita Y, Peters SAE, Woodward M. Diabetes as a risk factor for heart failure in women and men: a systematic review and meta-analysis of 47 cohorts including 12 million individuals. Diabetologia. 2019;62:1550–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsue Y, Suzuki M, Nakamura R, Abe M, Ono M, Yoshida S, Seya M, Iwatsuka R, Mizukami A, Setoguchi M, Nagahori W, Ohno M, Matsumura A, Hashimoto Y. Prevalence and prognostic implications of pre-diabetic state in patients with heart failure. Circ J. 2011;75:2833–9. [DOI] [PubMed] [Google Scholar]
- 10.Gustafsson I, Brendorp B, Seibaek M, Burchardt H, Hildebrandt P, Kober L, Torp-Pedersen C. Influence of diabetes and diabetes-gender interaction on the risk of death in patients hospitalized with congestive heart failure. J Am Coll Cardiol. 2004;43:771–7. [DOI] [PubMed] [Google Scholar]
- 11.Echouffo-Tcheugui JB, Masoudi FA, Bao H, Spatz ES, Fonarow GC. Diabetes Mellitus and Outcomes of Cardiac Resynchronization With Implantable Cardioverter-Defibrillator Therapy in Older Patients With Heart Failure. Circ Arrhythm Electrophysiol. 2016;9. [DOI] [PubMed] [Google Scholar]
- 12.Marwick TH, Ritchie RH, Shaw JE, Kaye D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J Am Coll Cardiol. 2018;71:339–351. [DOI] [PubMed] [Google Scholar]
- 13.Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, Ramu V, Bhogal S, Kumar G, Shanmugasundaram M, Paul TK. Diabetic cardiomyopathy - A comprehensive updated review. Progress in Cardiovascular Diseases. 2019;62:315–326. [DOI] [PubMed] [Google Scholar]
- 14.Riehle C, Bauersachs J. Of mice and men: models and mechanisms of diabetic cardiomyopathy. Basic Research in Cardiology. 2019;114:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Failure Reviews. 2013;18:149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. American Journal of Cardiology. 1972;30:595–602. [DOI] [PubMed] [Google Scholar]
- 17.de Simone G, Devereux RB, Chinali M, Lee ET, Galloway JM, Barac A, Panza JA, Howard BV. Diabetes and incident heart failure in hypertensive and normotensive participants of the Strong Heart Study. J Hypertens. 2010;28:353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holscher ME, Bode C, Bugger H. Diabetic Cardiomyopathy: Does the Type of Diabetes Matter? Int J Mol Sci. 2016;17:2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacology & Therapeutics. 2014;142:375–415. [DOI] [PubMed] [Google Scholar]
- 20.Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57:660–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia G, Hill MA, Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circulation Research. 2018;122:624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oe H, Nakamura K, Kihara H, Shimada K, Fukuda S, Takagi T, Miyoshi T, Hirata K, Yoshikawa J, Ito H. Comparison of effects of sitagliptin and voglibose on left ventricular diastolic dysfunction in patients with type 2 diabetes: results of the 3D trial. Cardiovascular Diabetology. 2015;14:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Accord Study Group. Nine-Year Effects of 3.7 Years of Intensive Glycemic Control on Cardiovascular Outcomes. Diabetes Care. 2016;39:701–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert RE, Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet. 2015;385:2107–2117. [DOI] [PubMed] [Google Scholar]
- 25.Boyer JK, Thanigaraj S, Schechtman KB, Pérez JE. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. American Journal of Cardiology. 2004;93:870–875. [DOI] [PubMed] [Google Scholar]
- 26.Diamant M, Lamb HJ, Groeneveld Y, Endert EL, Smit JW, Bax JJ, Romijn JA, de Roos A, Radder JK. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J Am Coll Cardiol. 2003;42:328–35. [DOI] [PubMed] [Google Scholar]
- 27.Zabalgoitia M, Ismaeil MF, Anderson L, Maklady FA. Prevalence of diastolic dysfunction in normotensive, asymptomatic patients with well-controlled type 2 diabetes mellitus. Am J Cardiol. 2001;87:320–3. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Yang ZG, Gao Y, Xie LJ, Jiang L, Hu BY, Diao KY, Shi K, Xu HY, Shen MT, Ren Y, Guo YK. Left ventricular subclinical myocardial dysfunction in uncomplicated type 2 diabetes mellitus is associated with impaired myocardial perfusion: a contrast-enhanced cardiovascular magnetic resonance study. Cardiovasc Diabetol. 2018;17:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood P, Piran S, Liu PP. Diastolic heart failure: progress, treatment challenges, and prevention. Can J Cardiol. 2011;27:302–10. [DOI] [PubMed] [Google Scholar]
- 30.Paiman EHM, van Eyk HJ, Bizino MB, Dekkers IA, de Heer P, Smit JWA, Jazet IM, Lamb HJ. Phenotyping diabetic cardiomyopathy in Europeans and South Asians. Cardiovasc Diabetol. 2019;18:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenkins MJ, Pearson JT, Schwenke DO, Edgley AJ, Sonobe T, Fujii Y, Ishibashi-Ueda H, Kelly DJ, Yagi N, Shirai M. Myosin heads are displaced from actin filaments in the in situ beating rat heart in early diabetes. Biophys J. 2013;104:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagueh SF, Smiseth OA, Appleton CP, Byrd BF, Dokainish H, Edvardsen T, Flachskampf FA, Gillebert TC, Klein AL, Lancellotti P, Marino P, Oh JK, Alexandru Popescu B. Waggoner AD. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. European Heart Journal – Cardiovascular Imaging. 2016;17:1321–1360. [DOI] [PubMed] [Google Scholar]
- 33.Grigorescu E-D, Lacatusu C-M, Floria M, Mihai B-M, Cretu I, Sorodoc L. Left Ventricular Diastolic Dysfunction in Type 2 Diabetes-Progress and Perspectives. Diagnostics. 2019;9:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarich SW, Arbuckle BE, Cohen LR, Roberts M, Nesto RW. Diastolic abnormalities in young asymptomatic diabetic patients assessed by pulsed Doppler echocardiography. J Am Coll Cardiol. 1988;12:114–20. [DOI] [PubMed] [Google Scholar]
- 35.Vukomanovic V, Suzic-Lazic J, Celic V, Cuspidi C, Petrovic T, Grassi G, Tadic M. The relationship between functional capacity and left ventricular strain in patients with uncomplicated type 2 diabetes. Journal of Hypertension. 2019;37:1871–1876. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno R, Fujimoto S, Saito Y, Nakamura S. Exercise-induced delayed onset of left ventricular early relaxation in association with coronary microcirculatory dysfunction in patients with diabetes mellitus. J Card Fail. 2010;16:211–7. [DOI] [PubMed] [Google Scholar]
- 37.Fang ZY, Yuda S, Anderson V, Short L, Case C, Marwick TH. Echocardiographic detection of early diabetic myocardial disease. J Am Coll Cardiol. 2003;41:611–7. [DOI] [PubMed] [Google Scholar]
- 38.Loncarevic B, Trifunovic D, Soldatovic I, Vujisic-Tesic B. Silent diabetic cardiomyopathy in everyday practice: a clinical and echocardiographic study. BMC Cardiovascular Disorders. 2016;16:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ernande L, Rietzschel ER, Bergerot C, De Buyzere ML, Schnell F, Groisne L, Ovize M, Croisille P, Moulin P, Gillebert TC, Derumeaux GG. Impaired myocardial radial function in asymptomatic patients with type 2 diabetes mellitus: A speckle-tracking imaging study. Journal of the American Society of Echocardiography. 2010;23:1266–1272. [DOI] [PubMed] [Google Scholar]
- 40.Enomoto M, Ishizu T, Seo Y, Kameda Y, Suzuki H, Shimano H, Kawakami Y, Aonuma K. Myocardial dysfunction identified by three-dimensional speckle tracking echocardiography in type 2 diabetes patients relates to complications of microangiopathy. J Cardiol. 2016;68:282–7. [DOI] [PubMed] [Google Scholar]
- 41.Ng ACT, Bertini M, Ewe SH, van der Velde ET, Leung DY, Delgado V, Bax JJ. Defining Subclinical Myocardial Dysfunction and Implications for Patients With Diabetes Mellitus and Preserved Ejection Fraction. American Journal of Cardiology. 2019;124:892–898. [DOI] [PubMed] [Google Scholar]
- 42.Lassen MCH, Jensen MT, Biering-Sorensen T, Mogelvang R, Fritz-Hansen T, Vilsboll T, Rossing P, Jorgensen PG. Prognostic value of ratio of transmitral early filling velocity to early diastolic strain rate in patients with Type 2 diabetes. European Heart Journal - Cardiovascular Imaging. 2019;20:1171–1178. [DOI] [PubMed] [Google Scholar]
- 43.Muranaka A, Yuda S, Tsuchihashi K, Hashimoto A, Nakata T, Miura T, Tsuzuki M, Wakabayashi C, Watanabe N, Shimamoto K. Quantitative assessment of left ventricular and left atrial functions by strain rate imaging in diabetic patients with and without hypertension. Echocardiography. 2009;26:262–71. [DOI] [PubMed] [Google Scholar]
- 44.Jensen MT, Sogaard P, Gustafsson I, Bech J, Hansen TF, Almdal T, Theilade S, Biering-Sorensen T, Jorgensen PG, Galatius S, Andersen HU, Rossing P. Echocardiography improves prediction of major adverse cardiovascular events in a population with type 1 diabetes and without known heart disease: the Thousand & 1 Study. Diabetologia. 2019;62:2354–2364. [DOI] [PubMed] [Google Scholar]
- 45.Galderisi M Diastolic dysfunction and diastolic heart failure: diagnostic, prognostic and therapeutic aspects. Cardiovascular Ultrasound. 2005;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Disease Models & Mechanisms. 2009;2:454–466. [DOI] [PubMed] [Google Scholar]
- 47.Prakoso D, De Blasio MJ, Qin C, Rosli S, Kiriazis H, Qian H, Du X- J, Weeks KL, Gregorevic P, McMullen JR, Ritchie RH. Phosphoinositide 3-kinase (p110α) gene delivery limits diabetes-induced cardiac NADPH oxidase and cardiomyopathy in a mouse model with established diastolic dysfunction. Clinical Science. 2017;131:1345–1360. [DOI] [PubMed] [Google Scholar]
- 48.De Blasio MJ, Huynh N, Deo M, Dubrana LE, Walsh J, Willis A, Prakoso D, Kiriazis H, Donner DG, Chatham JC, Ritchie RH. Defining the progression of diabetic cardiomyopathy in a mouse model of type 1 diabetes. Frontiers in Physiology. 2020;11:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huynh K, McMullen JR, Julius TL, Tan JW, Love JE, Cemerlang N, Kiriazis H, Du X-J, Ritchie RH. Cardiac-specific IGF-1 receptor transgenic expression protects against cardiac fibrosis and diastolic dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes. 2010;59:1512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandramouli C, Reichelt ME, Curl CL, Varma U, Bienvenu LA, Koutsifeli P, Raaijmakers AJA, De Blasio MJ, Qin CX, Jenkins AJ, Ritchie RH, Mellor KM, Delbridge LMD. Diastolic dysfunction is more apparent in STZ-induced diabetic female mice, despite less pronounced hyperglycemia. Sci Rep. 2018;8:2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao N, Wong YG, Rosli S, Kiriazis H, Huynh K, Qin C, Du X-J, Kemp-Harper BK, Ritchie RH. Chronic administration of the nitroxyl donor 1-nitrosocyclo hexyl acetate limits left ventricular diastolic dysfunction in a mouse model of diabetes mellitus in vivo. Circulation Heart Failure. 2015;8:572–81. [DOI] [PubMed] [Google Scholar]
- 52.Xiao Q, Yang Y, Zhao X-Y, He L-S, Qin Y, He Y-H, Zhang G-P, Luo J-D. Oxidative stress contributes to the impaired sonic hedgehog pathway in type 1 diabetic mice with myocardial infarction. Experimental and Therapeutic Medicine. 2015;10:1750–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang D, Zhong P, Hu J, Lin F, Qian Y, Xu Z, Wang J, Zeng C, Li X, Liang G. EGFR inhibition protects cardiac damage and remodeling through attenuating oxidative stress in STZ-induced diabetic mouse model. Journal of Molecular and Cellular Cardiology. 2015;82:63–74. [DOI] [PubMed] [Google Scholar]
- 54.Tate M, Deo M, Cao AH, Hood SG, Huynh K, Kiriazis H, Du XJ, Julius TL, Figtree GA, Dusting GJ, Kaye DM, Ritchie RH. Insulin replacement limits progression of diabetic cardiomyopathy in the low-dose streptozotocin-induced diabetic rat. Diab Vasc Dis Res. 2017;14:423–433. [DOI] [PubMed] [Google Scholar]
- 55.Joffe II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, Douglas PS. Abnormal cardiac function in the streptozotocin-induced non-insulin-dependent diabetic rat: noninvasive assessment with doppler echocardiography and contribution of the nitric oxide pathway. Journal of the American College of Cardiology. 1999;34:2111–9. [DOI] [PubMed] [Google Scholar]
- 56.Linthout S, Seeland U, Riad A, Eckhardt O, Hohl M, Dhayat N, Richter U, Fischer JW, Böhm M, Pauschinger M, Schultheiss H- P, Tschöpe C. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Research in Cardiology. 2008;103:319–327. [DOI] [PubMed] [Google Scholar]
- 57.Tate M, Higgins GC, De Blasio MJ, Lindblom R, Prakoso D, Deo M, Kiriazis H, Park M, Baeza-Garza CD, Caldwell ST, Hartley RC, Krieg T, Murphy MP, Coughlan MT, Ritchie RH. The Mitochondria-Targeted Methylglyoxal Sequestering Compound, MitoGamide, Is Cardioprotective in the Diabetic Heart. Cardiovasc Drugs Ther. 2020;34:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. Tissue-Specific Remodeling of the Mitochondrial Proteome in Type 1 Diabetic Akita Mice. Diabetes. 2009;58:1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, Kassiri Z. Type 1 diabetic cardiomyopathy in the Akita ( Ins2 WT/C96Y ) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. American Journal of Physiology-Heart and Circulatory Physiology. 2009;297:H2096–H2108. [DOI] [PubMed] [Google Scholar]
- 60.Epstein PN, Overbeek PA, Means AR. Calmodulin-induced early-onset diabetes in transgenic mice. Cell. 1989;58:1067–73. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y, Sun W, Du B, Miao X, Bai Y, Xin Y, Tan Y, Cui W, Liu B, Cui T, Epstein PN, Fu Y, Cai L. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-κB. American Journal of Physiology-Heart and Circulatory Physiology. 2013;304:H567–H578. [DOI] [PubMed] [Google Scholar]
- 62.Li Y, Ma J, Zhu H, Singh M, Hill D, Greer PA, Arnold JM, Abel ED, Peng T. Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes. 2011;60:2985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bowden MA, Tesch GH, Julius TL, Rosli S, Love JE, Ritchie RH. Earlier onset of diabesity-Induced adverse cardiac remodeling in female compared to male mice. Obesity. 2015;23:1166–1177. [DOI] [PubMed] [Google Scholar]
- 64.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–9. [DOI] [PubMed] [Google Scholar]
- 65.Huynh K, Kiriazis H, Du X-J, Love JE, Jandeleit-Dahm KA, Forbes JM, McMullen JR, Ritchie RH. Coenzyme Q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes. Diabetologia. 2012;55:1544–53. [DOI] [PubMed] [Google Scholar]
- 66.Mori J, Patel VB, Abo Alrob O, Basu R, Altamimi T, Desaulniers J, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY. Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circulation Heart Failure. 2014;7:327–39. [DOI] [PubMed] [Google Scholar]
- 67.Tate M, Prakoso D, Willis AM, et al. Characterising an Alternative Murine Model of Diabetic Cardiomyopathy. Front Physiol. 2019;10:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sultan A, Singh J, Howarth FC. Mechanisms underlying electro-mechanical dysfunction in the Zucker diabetic fatty rat heart: a model of obesity and type 2 diabetes. Heart Failure Reviews. 2019; 10.1007/s10741-019-09872-4. [DOI] [PubMed] [Google Scholar]
- 69.Aragon-Herrera A, Feijoo-Bandin S, Otero Santiago M, et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochemical Pharmacology. 2019;170:113677–113677. [DOI] [PubMed] [Google Scholar]
- 70.Desrois M Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovascular Research. 2004;61:288–296. [DOI] [PubMed] [Google Scholar]
- 71.Ti Y, Xie G-l, Wang Z-h, Bi X-l, Ding W-y, Wang J, Jiang G-H, Bu P-L, Zhang Y, Zhong M, Zhang W. TRB3 gene silencing alleviates diabetic cardiomyopathy in a type 2 diabetic rat model. Diabetes. 2011;60:2963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Axelsen LN, Lademann JB, Petersen JS, Holstein-Rathlou N- H, Ploug T, Prats C, Pedersen HD, Kjølbye AL. Cardiac and metabolic changes in long-term high fructose-fat fed rats with severe obesity and extensive intramyocardial lipid accumulation. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2010;298:R1560–R1570. [DOI] [PubMed] [Google Scholar]
- 73.Vasanji Z, Cantor EJF, Juric D, Moyen M, Netticadan T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. American Journal of Physiology-Cell Physiology. 2006;291:C772–C780. [DOI] [PubMed] [Google Scholar]
- 74.Fricovsky ES, Suarez J, Ihm S- H, Scott BT, Suarez-Ramirez, Banerjee I, Torres-Gonzalez M, Wang H, Ellrott I, Maya-Ramos L, Villarreal, Dillmann WH. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2012;303:R689–R699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Figtree GA, Bubb KJ, Tang O, Kizana E, Gentile C. Vascularized Cardiac Spheroids as Novel 3D in vitro Models to Study Cardiac Fibrosis. Cells Tissues Organs. 2017;204:191–198. [DOI] [PubMed] [Google Scholar]
- 76.Hernandez D, Millard R, Sivakumaran P, Wong RCB, Crombie DE, Hewitt AW, Liang H, Hung SSC, Pebay A, Shepherd RK, Dusting GJ, Lim SY. Electrical Stimulation Promotes Cardiac Differentiation of Human Induced Pluripotent Stem Cells. Stem Cells International. 2016;2016:1718041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim SY, Sivakumaran P, Crombie DE, Dusting GJ, Pebay A, Dilley RJ. Enhancing Human Cardiomyocyte Differentiation from Induced Pluripotent Stem Cells with Trichostatin A. Methods in Molecular Biology. 2016;1357:415–21. [DOI] [PubMed] [Google Scholar]
- 78.Mills RJ, Parker BL, Quaife-Ryan GA, et al. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell. 2019;24:895–907. [DOI] [PubMed] [Google Scholar]
- 79.Mills RJ, Titmarsh DM, Koenig X, et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E8372–E8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Polonchuk L, Chabria M, Badi L, Hoflack J-C, Figtree G, Davies MJ, Gentile C. Cardiac spheroids as promising in vitro models to study the human heart microenvironment. Scientific Reports. 2017;7:7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tate M, Grieve DJ, Ritchie RH. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clinical Science. 2017;131:897–915. [DOI] [PubMed] [Google Scholar]
- 82.Shimizu M, Umeda K, Sugihara N, Yoshio H, Ino H, Takeda R, Okada Y, Nakanishi I. Collagen remodelling in myocardia of patients with diabetes. Journal of Clinical Pathology. 1993;46:32–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, Haider B. Evidence for cardiomyopathy in familial diabetes mellitus. Journal of Clinical Investigation. 1977;60:885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Hoeven KH, Factor SM. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation. 1990;82:848–55. [DOI] [PubMed] [Google Scholar]
- 85.Kwong RY, Sattar H, Wu H, Vorobiof G, Gandla V, Steel K, Siu S, Brown KA. Incidence and prognostic implication of unrecognized myocardial scar characterized by cardiac magnetic resonance in diabetic patients without clinical evidence of myocardial infarction. Circulation. 2008;118:1011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turkbey EB, Backlund JY, Genuth S, Jain A, Miao C, Cleary PA, Lachin JM, Nathan DM, van der Geest RJ, Soliman EZ, Liu CY, Lima JA, Bluemke DA. Myocardial structure, function, and scar in patients with type 1 diabetes mellitus. Circulation. 2011;124:1737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lorenzo-Almorós A, Tuñón J, Orejas M, Cortés M, Egido J, Lorenzo. Diagnostic approaches for diabetic cardiomyopathy. Cardiovascular Diabetology. 2017;16:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Negishi K Echocardiographic feature of diabetic cardiomyopathy: where are we now? Cardiovasc Diagn Ther. 2018;8:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Di Bello V, Talarico L, Picano E, Di Muro C, Landini L, Paterni M, Matteucci E, Giusti C, Giampietro O. Increased echodensity of myocardial wall in the diabetic heart: an ultrasound tissue characterization study. J Am Coll Cardiol. 1995;25:1408–15. [DOI] [PubMed] [Google Scholar]
- 90.Fischer VW, Barner HB, Larose LS. Pathomorphologic aspects of muscular tissue in diabetes mellitus. Hum Pathol. 1984;15:1127–36. [DOI] [PubMed] [Google Scholar]
- 91.Westermann D, Rutschow S, Jager S, Linderer A, Anker S, Riad A, Unger T, Schultheiss HP, Pauschinger M, Tschope C. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes. 2007;56:641–6. [DOI] [PubMed] [Google Scholar]
- 92.Mizushige K, Yao L, Noma T, Kiyomoto H, Yu Y, Hosomi N, Ohmori K, Matsuo H. Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model. Circulation. 2000;101:899–907. [DOI] [PubMed] [Google Scholar]
- 93.Singh VP, Baker KM, Kumar R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production. American Journal of Physiology-Heart and Circulatory Physiology. 2008;294:H1675–H1684. [DOI] [PubMed] [Google Scholar]
- 94.Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, Hanley A, Isner JM, Losordo DW. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111:2073–85. [DOI] [PubMed] [Google Scholar]
- 95.Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, Burrell LM. A Breaker of Advanced Glycation End Products Attenuates Diabetes-Induced Myocardial Structural Changes. Circulation Research. 2003;92:785–792. [DOI] [PubMed] [Google Scholar]
- 96.Connelly KA, Kelly DJ, Zhang Y, Prior DL, Martin J, Cox AJ, Thai K, Feneley MP, Tsoporis J, White KE, Krum H, Gilbert RE. Functional, structural and molecular aspects of diastolic heart failure in the diabetic (mRen-2)27 rat. Cardiovasc Res. 2007;76:280–91. [DOI] [PubMed] [Google Scholar]
- 97.Devereux RB, Roman MJ, Paranicas M, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. 2000;101:2271–6. [DOI] [PubMed] [Google Scholar]
- 98.Lee M, Gardin JM, Lynch JC, Smith VE, Tracy RP, Savage PJ, Szklo M, Ward BJ. Diabetes mellitus and echocardiographic left ventricular function in free-living elderly men and women: The Cardiovascular Health Study. Am Heart J. 1997;133:36–43. [DOI] [PubMed] [Google Scholar]
- 99.Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241:2035–8. [DOI] [PubMed] [Google Scholar]
- 100.Dawson A, Morris AD, Struthers AD. The epidemiology of left ventricular hypertrophy in type 2 diabetes mellitus. Diabetologia. 2005;48:1971–9. [DOI] [PubMed] [Google Scholar]
- 101.Galderisi M, Anderson KM, Wilson PW, Levy D. Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (the Framingham Heart Study). Am J Cardiol. 1991;68:85–9. [DOI] [PubMed] [Google Scholar]
- 102.Lam B, Stromp TA, Hui Z, Vandsburger M. Myocardial native-T1 times are elevated as a function of hypertrophy, HbA1c, and heart rate in diabetic adults without diffuse fibrosis. Magnetic Resonance Imaging. 2019;61:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moir S, Hanekom L, Fang ZY, Haluska B, Wong C, Burgess M, Marwick TH. Relationship between myocardial perfusion and dysfunction in diabetic cardiomyopathy: a study of quantitative contrast echocardiography and strain rate imaging. Heart. 2006;92:1414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Eguchi K, Boden-Albala B, Jin Z, Rundek T, Sacco RL, Homma S, Di Tullio MR. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. American Journal of Cardiology. 2008;101:1787–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ritchie RH, Love JE, Huynh K, Bernardo BC, Henstridge DC, Kiriazis H, Tham YK, Sapra G, Qin C, Cemerlang N, Boey EJH, Jandeleit-Dahm K, Du X- J, McMullen JR. Enhanced phosphoinositide 3-kinase(p110α) activity prevents diabetes-induced cardiomyopathy and superoxide generation in a mouse model of diabetes. Diabetologia. 2012;55:3369–81. [DOI] [PubMed] [Google Scholar]
- 106.Feng B, Chen S, Chiu J, George B, Chakrabarti S. Regulation of cardiomyocyte hypertrophy in diabetes at the transcriptional level. American Journal of Physiology-Endocrinology and Metabolism. 2008;294:E1119–E1126. [DOI] [PubMed] [Google Scholar]
- 107.Ceylan-Isik AF, Kandadi MR, Xu X, Hua Y, Chicco AJ, Ren J, Nair S. Apelin administration ameliorates high fat diet-induced cardiac hypertrophy and contractile dysfunction. Journal of Molecular and Cellular Cardiology. 2013;63:4–13. [DOI] [PubMed] [Google Scholar]
- 108.Hua Y, Zhang Y, Dolence J, Shi G-P, Ren J, Nair S. Cathepsin K knockout mitigates high-fat diet-induced cardiac hypertrophy and contractile dysfunction. Diabetes. 2013;62:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Desrois M, Sidell RJ, Gauguier D, Davey CL, Radda GK, Clarke K. Gender differences in hypertrophy, insulin resistance and ischemic injury in the aging type 2 diabetic rat heart. Journal of Molecular and Cellular Cardiology. 2004;37:547–55. [DOI] [PubMed] [Google Scholar]
- 110.Tate M, Robinson E, Green BD, McDermott BJ, Grieve DJ. Exendin-4 attenuates adverse cardiac remodelling in streptozocin-induced diabetes via specific actions on infiltrating macrophages. Basic Research in Cardiology. 2016;111:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiological Reviews. 2008;88:389–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bell DS. Hypertension and diabetes: a toxic combination. Endocr Pract. 2008;14:1031–9. [DOI] [PubMed] [Google Scholar]
- 113.Ritchie RH, Quinn JM, Cao AH, Drummond GR, Kaye DM, Favaloro JM, Proietto J, Delbridge LMD. The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo. Journal of Molecular and Cellular Cardiology. 2007;42:1119–1128. [DOI] [PubMed] [Google Scholar]
- 114.Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, Singh S, Prasad R, Khullar M. miR-30c and miR-181a synergistically modulate p53–p21 pathway in diabetes induced cardiac hypertrophy. Molecular and Cellular Biochemistry. 2016;417:191–203. [DOI] [PubMed] [Google Scholar]
- 115.Gherasim L, Tasca C, Havriliuc C, Vasilescu C. A morphological quantitative study of small vessels in diabetic cardiomyopathy. Morphol Embryol. 1985;31:191–5. [PubMed] [Google Scholar]
- 116.Strauer BE, Motz W, Vogt M, Schwartzkopff B. Impaired coronary flow reserve in NIDDM: a possible role for diabetic cardiopathy in humans. Diabetes. 1997;46 Suppl 2:S119–24. [DOI] [PubMed] [Google Scholar]
- 117.Di Carli MF, Janisse J, Grunberger G, Ager J. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol. 2003;41:1387–93. [DOI] [PubMed] [Google Scholar]
- 118.Hansen A, Johansson BL, Wahren J, von Bibra H. C-peptide exerts beneficial effects on myocardial blood flow and function in patients with type 1 diabetes. Diabetes. 2002;51:3077–82. [DOI] [PubMed] [Google Scholar]
- 119.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu Y, Ohmori K, Kondo I, Yao L, Noma T, Tsuji T, Mizushige K, Kohno M. Correlation of functional and structural alterations of the coronary arterioles during development of type II diabetes mellitus in rats. Cardiovasc Res. 2002;56:303–11. [DOI] [PubMed] [Google Scholar]
- 121.Hayashi T, Sohmiya K, Ukimura A, Endoh S, Mori T, Shimomura H, Okabe M, Terasaki F, Kitaura Y. Angiotensin II receptor blockade prevents microangiopathy and preserves diastolic function in the diabetic rat heart. Heart. 2003;89:1236–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Factor SM, Minase T, Cho S, Fein F, Capasso JM, Sonnenblick EH. Coronary microvascular abnormalities in the hypertensive-diabetic rat. A primary cause of cardiomyopathy? Am J Pathol. 1984;116:9–20. [PMC free article] [PubMed] [Google Scholar]
- 123.Joshi MS, Berger PJ, Kaye DM, Pearson JT, Bauer JA, Ritchie RH. Functional relevance of genetic variations of endothelial nitric oxide synthase and vascular endothelial growth factor in diabetic coronary microvessel dysfunction. Clin Exp Pharmacol Physiol. 2013;40:253–61. [DOI] [PubMed] [Google Scholar]
- 124.Chou E, Suzuma I, Way KJ, Opland D, Clermont AC, Naruse K, Suzuma K, Bowling NL, Vlahos CJ, Aiello LP, King GL. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation. 2002;105:373–9. [DOI] [PubMed] [Google Scholar]
- 125.Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2006;291:H1780–7. [DOI] [PubMed] [Google Scholar]
- 126.Kahlberg N, Qin CX, Anthonisz J, Jap E, Ng HH, Jelinic M, Parry LJ, Kemp-Harper BK, Ritchie RH, Leo CH. Adverse vascular remodelling is more sensitive than endothelial dysfunction to hyperglycaemia in diabetic rat mesenteric arteries. Pharmacol Res. 2016;111:325–335. [DOI] [PubMed] [Google Scholar]
- 127.Ng HH, Leo CH, Prakoso D, Qin C, Ritchie RH, Parry LJ. Serelaxin treatment reverses vascular dysfunction and left ventricular hypertrophy in a mouse model of Type 1 diabetes. Sci Rep. 2017;7:39604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tare M, Kalidindi RS, Bubb KJ, Parkington HC, Boon WM, Li X, Sobey CG, Drummond GR, Ritchie RH, Kemp-Harper BK. Vasoactive actions of nitroxyl (HNO) are preserved in resistance arteries in diabetes. Naunyn Schmiedebergs Arch Pharmacol. 2017;390:397–408. [DOI] [PubMed] [Google Scholar]
- 129.Sharma A, Rizky L, Stefanovic N, Tate M, Ritchie RH, Ward KW, de Haan JB. The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activator dh404 protects against diabetes-induced endothelial dysfunction. Cardiovascular Diabetology. 2017;16:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Faria A, Persaud SJ. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacology & Therapeutics. 2017;172:50–62. [DOI] [PubMed] [Google Scholar]
- 131.Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. 2010;59:2033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Elbatreek MH, Pachado MP, Cuadrado A, Jandeleit-Dahm K, Schmidt HHHW. Reactive Oxygen Comes of Age: Mechanism-Based Therapy of Diabetic End-Organ Damage. Trends in Endocrinology and Metabolism. 2019;30:312–327. [DOI] [PubMed] [Google Scholar]
- 133.Hansen SS, Aasum E, Hafstad AD. The role of NADPH oxidases in diabetic cardiomyopathy. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2017;1864:1908–1913. [DOI] [PubMed] [Google Scholar]
- 134.Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, Suzuki H, Toyama K, Spin JM, Tsao PS. Diabetic cardiovascular disease induced by oxidative stress. International Journal of Molecular Sciences. 2015;16:25234–25263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.De Blasio MJ, Huynh K, Qin C, Rosli S, Kiriazis H, Ayer A, Cemerlang N, Stocker R, Du X- J, McMullen JR, Ritchie RH. Therapeutic targeting of oxidative stress with coenzyme Q10 counteracts exaggerated diabetic cardiomyopathy in a mouse model of diabetes with diminished PI3K(p110α) signaling. Free Radical Biology and Medicine. 2015;87:137–147. [DOI] [PubMed] [Google Scholar]
- 136.Huynh K, Kiriazis H, Du X-J, Love JE, Gray SP, Jandeleit-Dahm KA, McMullen JR, Ritchie RH. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radical Biology and Medicine. 2013;60:307–317. [DOI] [PubMed] [Google Scholar]
- 137.Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–43. [DOI] [PubMed] [Google Scholar]
- 138.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. [DOI] [PubMed] [Google Scholar]
- 139.Rajesh M, Mukhopadhyay P, Batkai S, Mukhopadhyay B, Patel V, Hasko G, Szabo C, Mabley JG, Liaudet L, Pacher P. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J Cell Mol Med. 2009;13:2330–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fiordaliso F, Bianchi R, Staszewsky L, Cuccovillo I, Doni M, Laragione T, Salio M, Savino C, Melucci S, Santangelo F. Antioxidant treatment attenuates hyperglycemia-induced cardiomyocyte death in rats. Journal of Molecular and Cellular Cardiology. 2004;37:959–968. [DOI] [PubMed] [Google Scholar]
- 141.Li X, Li Z, Li B, Zhu X, Lai X. Klotho improves diabetic cardiomyopathy by suppressing the NLRP3 inflammasome pathway. Life Sciences. 2019;234. [DOI] [PubMed] [Google Scholar]
- 142.Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. 2017;104:293–299. [DOI] [PubMed] [Google Scholar]
- 143.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. N Engl J Med. 2000;342:154–60. [DOI] [PubMed] [Google Scholar]
- 144.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–62. [DOI] [PubMed] [Google Scholar]
- 146.Shah MS, Brownlee M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ Res. 2016;118:1808–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, de Haan JB. Oxidative Stress and NLRP3-Inflammasome Activity as Significant Drivers of Diabetic Cardiovascular Complications: Therapeutic Implications. Front Physiol. 2018;9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu Q, Wang S, Cai L. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. Journal of Diabetes Investigation. 2014;5:623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution. Journal of the American College of Cardiology. 2017;70:230–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. [DOI] [PubMed] [Google Scholar]
- 151.Yang Y-C, Tsai C-Y, Chen C-L, Kuo C-H, Hou C-W, Cheng S-Y, Aneja R, Huang C-Y, Kuo W-W. PKC delta Activation is Involved in ROS-Mediated Mitochondrial Dysfunction and Apoptosis in Cardiomyocytes Exposed to Advanced Glycation End Products. Aging and Disease. 2018;9:647–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Eid RA, Alkhateeb MA, El-Kott AF, Eleawa SM, Zaki MSA, Alaboodi SA, Al-Shudiefat AAIRS, Aldera H, Namar NMA, Alassiri M, Khalil MA. A high-fat diet rich in corn oil induces cardiac fibrosis in rats by activating JAK2/STAT3 and subsequent activation of ANG II/TGF-1 beta/Smad3 pathway: The role of ROS and IL-6 trans-signaling. Journal of Food Biochemistry. 2019;43:e12952. [DOI] [PubMed] [Google Scholar]
- 153.Luo W, Jin Y, Wu G, Zhu W, Qian Y, Zhang Y, Li J, Zhu A, Liang G. Blockage of ROS and MAPKs-mediated inflammation via restoring SIRT1 by a new compound LF10 prevents type 1 diabetic cardiomyopathy. Toxicology and Applied Pharmacology. 2019;370:24–35. [DOI] [PubMed] [Google Scholar]
- 154.Ge Q, Zhao L, Ren X-M, Ye P, Hu Z-Y. Feature article: LCZ696, an angiotensin receptor-neprilysin inhibitor, ameliorates diabetic cardiomyopathy by inhibiting inflammation, oxidative stress and apoptosis. Experimental Biology and Medicine. 2019;244:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jadhav A, Tiwari S, Lee P, Ndisang JF. The Heme Oxygenase System Selectively Enhances the Anti-Inflammatory Macrophage-M2 Phenotype, Reduces Pericardial Adiposity, and Ameliorated Cardiac Injury in Diabetic Cardiomyopathy in Zucker Diabetic Fatty Rats. Journal of Pharmacology and Experimental Therapeutics. 2013;345:239–249. [DOI] [PubMed] [Google Scholar]
- 156.Ruiz M, Coderre L, Lachance D, Houde V, Martel C, Legault JT, Gillis M- A, Bouchard B, Daneault C, Carpentier AC, Gaestel M, Allen BG, Rosiers CD. MK2 Deletion in Mice Prevents Diabetes-Induced Perturbations in Lipid Metabolism and Cardiac Dysfunction. Diabetes. 2016;65:381–392. [DOI] [PubMed] [Google Scholar]
- 157.Wang S, Ding L, Ji H, Xu Z, Liu Q, Zheng Y. The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy. International Journal of Molecular Sciences. 2016;17:1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Westermann D, Rutschow S, Van Linthout S, Linderer A, Bucker-Gartner C, Sobirey M, Riad A, Pauschinger M, Schultheiss HP, Tschope C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia. 2006;49:2507–13. [DOI] [PubMed] [Google Scholar]
- 159.Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, Hirano M, Morrow JP. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. Plos One. 2016;11:e0145750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ge Z-D, Lian Q, Mao X, Xia Z. Current Status and Challenges of NRF2 as a Potential Therapeutic Target for Diabetic Cardiomyopathy. International Heart Journal. 2019;60:512–520. [DOI] [PubMed] [Google Scholar]
- 161.Zhang H, Chen X, Zong B, Yuan H, Wang Z, Wei Y, Wang X, Liu G, Zhang J, Li S, Cheng G, Wang Y, Ma Y. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. Journal of Cellular and Molecular Medicine. 2018;22:4437–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang J, Wang S, Wang W, Chen J, Zhang Z, Zheng Q, Liu Q, Cai L. Protection against diabetic cardiomyopathy is achieved using a combination of sulforaphane and zinc in type 1 diabetic OVE26 mice. Journal of Cellular and Molecular Medicine. 2019;23:6319–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Guan Y, Zhou L, Zhang Y, Tian H, Li A, Han X. Effects of PP2A/Nrf2 on experimental diabetes mellitus-related cardiomyopathy by regulation of autophagy and apoptosis through ROS dependent pathway. Cellular Signalling. 2019;62:109339. [DOI] [PubMed] [Google Scholar]
- 164.Gu J, Cheng Y, Wu H, Kong L, Wang S, Xu Z, Zhang Z, Tan Y, Keller BB, Zhou H, Wang Y, Xu Z, Cai L. Metallothionein Is Downstream of Nrf2 and Partially Mediates Sulforaphane Prevention of Diabetic Cardiomyopathy. Diabetes. 2017;66:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gray SP, Jha JC, Kennedy K, van Bommel E, Chew P, Szyndralewiez C, Touyz RM, Schmidt H, Cooper ME, Jandeleit-Dahm KAM. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia. 2017;60:927–937. [DOI] [PubMed] [Google Scholar]
- 166.Rendra E, Riabov V, Mossel DM, Sevastyanova T, Harmsen MC, Kzhyshkowska J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology. 2019;224:242–253. [DOI] [PubMed] [Google Scholar]
- 167.Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J. NF-κB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovascular Research. 2010;85:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. [DOI] [PubMed] [Google Scholar]
- 169.Dinh W, Futh R, Nickl W, Krahn T, Ellinghaus P, Scheffold T, Bansemir L, Bufe A, Barroso MC, Lankisch M. Elevated plasma levels of TNF-alpha and interleukin-6 in patients with diastolic dysfunction and glucose metabolism disorders. Cardiovasc Diabetol. 2009;8:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nagareddy PR, Murphy AJ, Stirzaker RA, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ururahy MA, Loureiro MB, Freire-Neto FP, de Souza KS, Zuhl I, Brandao-Neto J, Hirata RD, Doi SQ, Arrais RF, Hirata MH, Almeida M, de Rezende AA. Increased TLR2 expression in patients with type 1 diabetes: evidenced risk of microalbuminuria. Pediatr Diabetes. 2012;13:147–54. [DOI] [PubMed] [Google Scholar]
- 172.Park JH, Jung JH, Yang JY, Kim HS. Olive leaf down-regulates the oxidative stress and immune dysregulation in streptozotocin-induced diabetic mice. Nutr Res. 2013;33:942–51. [DOI] [PubMed] [Google Scholar]
- 173.Koga K, Kenessey A, Ojamaa K. Macrophage migration inhibitory factor antagonizes pressure overload-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013;304:H282–93. [DOI] [PubMed] [Google Scholar]
- 174.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palu G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–25. [DOI] [PubMed] [Google Scholar]
- 175.Kassan M, Choi SK, Galan M, Bishop A, Umezawa K, Trebak M, Belmadani S, Matrougui K. Enhanced NF-kappaB activity impairs vascular function through PARP-1-, SP-1-, and COX-2-dependent mechanisms in type 2 diabetes. Diabetes. 2013;62:2078–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sato-Horiguchi C, Ogawa D, Wada J, Tachibana H, Kodera R, Eguchi J, Nakatsuka A, Terami N, Shikata K, Makino H. Telmisartan attenuates diabetic nephropathy by suppressing oxidative stress in db/db mice. Nephron Exp Nephrol. 2012;121:e97–e108. [DOI] [PubMed] [Google Scholar]
- 177.Toyama K, Nakamura T, Kataoka K, Yasuda O, Fukuda M, Tokutomi Y, Dong YF, Ogawa H, Kim-Mitsuyama S. Telmisartan protects against diabetic vascular complications in a mouse model of obesity and type 2 diabetes, partially through peroxisome proliferator activated receptor-gamma-dependent activity. Biochem Biophys Res Commun. 2011;410:508–13. [DOI] [PubMed] [Google Scholar]
- 178.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–71. [DOI] [PubMed] [Google Scholar]
- 179.Anker SD, von Haehling S. Inflammatory mediators in chronic heart failure: an overview. Heart. 2004;90:464–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, Reichelt ME, Delbridge LM, Young MJ. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension. 2012;60:1443–50. [DOI] [PubMed] [Google Scholar]
- 181.Qin CX, May LT, Li R, et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nature Communications. 2017;8:14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Gao XM, Liu Y, White D, et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: a predominant role of anti-inflammation. J Mol Cell Cardiol. 2011;50:991–9. [DOI] [PubMed] [Google Scholar]
- 184.Qin CX, Finlayson SB, Al-Sharea A, et al. Endogenous Annexin-A1 Regulates Haematopoietic Stem Cell Mobilisation and Inflammatory Response Post Myocardial Infarction in Mice In Vivo. Scientific Reports. 2017;7:16615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chrysohoou C, Pitsavos C, Barbetseas J, Kotroyiannis I, Brili S, Vasiliadou K, Papadimitriou L, Stefanadis C. Chronic systemic inflammation accompanies impaired ventricular diastolic function, detected by Doppler imaging, in patients with newly diagnosed systolic heart failure (Hellenic Heart Failure Study). Heart Vessels. 2009;24:22–6. [DOI] [PubMed] [Google Scholar]
- 186.Tan X, Hu L, Shu Z, Chen L, Li X, Du M, Sun D, Mao X, Deng S, Huang K, Zhang F. Role of CCR2 in the Development of Streptozotocin-treated Diabetic Cardiomyopathy. Diabetes. 2019;68:2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang ZG, Jin Q, Fan M, Cong XL, Han SF, Gao H, Shan Y. Myocardial remodeling in diabetic cardiomyopathy associated with cardiac mast cell activation. PLoS One. 2013;8:e60827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 188.Botta A, Laher I, Beam J, Decoffe D, Brown K, Halder S, Devlin A, Gibson DL, Ghosh S. Short term exercise induces PGC-1alpha, ameliorates inflammation and increases mitochondrial membrane proteins but fails to increase respiratory enzymes in aging diabetic hearts. PLoS One. 2013;8:e70248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pal PB, Sonowal H, Shukla K, Srivastava SK, Ramana KV. Aldose Reductase Mediates NLRP3 Inflammasome-Initiated Innate Immune Response in Hyperglycemia-Induced Thp1 Monocytes and Male Mice. Endocrinology. 2017;158:3661–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Luo B, Huang F, Liu Y, Liang Y, Wei Z, Ke H, Zeng Z, Huang W, He Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Frontiers in Physiology. 2017;8:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, Zhang M, Zhang Y, An F. NLRP3 Gene Silencing Ameliorates Diabetic Cardiomyopathy in a Type 2 Diabetes Rat Model. PLoS ONE. 2014;9:e104771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jia G, Habibi J, DeMarco VG, Martinez-Lemus LA, Ma L, Whaley-Connell AT, Aroor AR, Domeier TL, Zhu Y, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension. 2015;66:1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Brennan EP, Mohan M, McClelland A, et al. Lipoxins Regulate the Early Growth Response-1 Network and Reverse Diabetic Kidney Disease. Journal of the American Society of Nephrology. 2018;29:1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.de Gaetano M, McEvoy C, Andrews D, Cacace A, Hunter J, Brennan E, Godson C. Specialized Pro-resolving Lipid Mediators: Modulation of Diabetes-Associated Cardio-, Reno-, and Retino-Vascular Complications. Frontiers in Pharmacology. 2018;9:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128:2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fu T, Mohan M, Brennan EP, Woodman OL, Godson C, Kantharidis P, Ritchie RH, Qin CX. Therapeutic Potential of Lipoxin A4 in Chronic Inflammation: Focus on Cardiometabolic Disease. ACS Pharmacology & Translational Science. 2020;3:43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yki-Jarvinen H, Helve E, Koivisto VA. Hyperglycemia decreases glucose uptake in type I diabetes. Diabetes. 1987;36:892–6. [DOI] [PubMed] [Google Scholar]
- 198.Pouwels MJ, Span PN, Tack CJ, Olthaar AJ, Sweep CG, van Engelen BG, de Jong JG, Lutterman JA, Hermus AR. Muscle uridine diphosphate-hexosamines do not decrease despite correction of hyperglycemia-induced insulin resistance in type 2 diabetes. J Clin Endocrinol Metab. 2002;87:5179–84. [DOI] [PubMed] [Google Scholar]
- 199.Fröjdö S, Vidal H, Pirola L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2009;1792:83–92. [DOI] [PubMed] [Google Scholar]
- 200.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocrine Reviews. 2007;28:463–491. [DOI] [PubMed] [Google Scholar]
- 201.Riehle C, Abel ED. Insulin Signaling and Heart Failure. Circulation Research. 2016;118:1151–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED, Xiang YK. Inhibiting insulin-mediated β 2-adrenergic receptor activation prevents diabetes-associated cardiac dysfunction. Circulation. 2017;135:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fu Q, Xu B, Liu Y, et al. Insulin Inhibits Cardiac Contractility by Inducing a Gi-Biased beta(2)-Adrenergic Signaling in Hearts. Diabetes. 2014;63:2676–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. [DOI] [PubMed] [Google Scholar]
- 205.Hou N, Mai Y, Qiu X, Yuan W, Li Y, Luo C, Liu Y, Zhang G, Zhao G, Luo J-d. Carvacrol Attenuates Diabetic Cardiomyopathy by Modulating the PI3K/AKT/GLUT4 Pathway in Diabetic Mice. Frontiers in Pharmacology. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Miyake K, Ogawa W, Matsumoto M, Nakamura T, Sakaue H, Kasuga M. Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3-kinase signaling in the liver. Journal of Clinical Investigation. 2002;110:1483–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nelson VLB, Jiang Y- P, Dickman KG, Ballou LM, Lin RZ. Adipose tissue insulin resistance due to loss of PI3K p110α leads to decreased energy expenditure and obesity. American Journal of Physiology-Endocrinology and Metabolism. 2014;306:E1205–E1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Solheim MH, Winnay JN, Batista TM, Molven A, Njølstad PR, Kahn CR. Mice carrying a dominant-negative human PI3K mutation are protected from obesity and hepatic steatosis but not diabetes. Diabetes. 2018;67:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Prakoso D, De Blasio MJ, Tate M, Kiriazis H, Donner DG, Qian H, Nash D, Deo M, Weeks KL, Parry LJ, Gregorevic P, McMullen JR, Ritchie RH. Gene therapy targeting cardiac phosphoinositide 3-kinase (p110alpha) attenuates cardiac remodeling in type 2 diabetes. American Journal of Physiology Heart and Circulatory Physiology. 2020;318:H840–H852. [DOI] [PubMed] [Google Scholar]
- 210.Fang CX, Dong F, Thomas DP, Ma H, He L, Ren J. Hypertrophic cardiomyopathy in high-fat diet-induced obesity: role of suppression of forkhead transcription factor and atrophy gene transcription. American Journal of Physiology-Heart and Circulatory Physiology. 2008;295:H1206–H1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12:144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia. 2005;48:1229–37. [DOI] [PubMed] [Google Scholar]
- 213.Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, Strickland N, Matsui T, Das S, Rosenzweig A, Punjabi P, Camici PG. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. European Heart Journal. 2010;31:100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–21. [DOI] [PubMed] [Google Scholar]
- 215.Borg DJ, Forbes JM. Targeting advanced glycation with pharmaceutical agents: where are we now? Glycoconj J. 2016;33:653–70. [DOI] [PubMed] [Google Scholar]
- 216.Jandeleit-Dahm K, Cooper ME. The role of AGEs in cardiovascular disease. Current Pharmaceutical Design. 2008;14:979–86. [DOI] [PubMed] [Google Scholar]
- 217.Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, Boccuzzi G. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology. 2006;147:5967–74. [DOI] [PubMed] [Google Scholar]
- 218.Brownlee M The pathobiology of diabetic complications: A unifying mechanism. Diabetes. 2005;54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 219.Tikellis C, Thomas MC, Harcourt BE, Coughlan MT, Pete J, Bialkowski K, Tan A, Bierhaus A, Cooper ME, Forbes JM. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am J Physiol Endocrinol Metab. 2008;295:E323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kranstuber AL, del Rio C, Biesiadecki BJ, Hamlin RL, Ottobre J, Gyorke S, Lacombe VA. Advanced glycation end product cross-link breaker attenuates diabetes-induced cardiac dysfunction by improving sarcoplasmic reticulum calcium handling. Frontiers in Physiology. 2012;3:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tikellis C, Pickering RJ, Tsorotes D, Huet O, Cooper ME, Jandeleit-Dahm K, Thomas MC. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes. 2014;63:3915–25. [DOI] [PubMed] [Google Scholar]
- 222.Janssens JV, Ma B, Brimble MA, Van Eyk JE, Delbridge LMD, Mellor KM. Cardiac troponins may be irreversibly modified by glycation: novel potential mechanisms of cardiac performance modulation. Sci Rep. 2018;8:16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, Besch HR Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. 2004;53:463–73. [DOI] [PubMed] [Google Scholar]
- 224.Papadaki M, Holewinski RJ, Previs SB, Martin TG, Stachowski MJ, Li A, Blair CA, Moravec CS, Van Eyk JE, Campbell KS, Warshaw DM, Kirk JA. Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight. 2018;3:e121264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Chatham JC, Marchase RB. Protein O-GlcNAcylation: A Critical Regulator of the Cellular Response to Stress. Current Signal Transduction Therapy. 2010;5:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wright JN, Collins HE, Wende AR, Chatham JC. O-GlcNAcylation and cardiovascular disease. Biochemical Society Transactions. 2017;45:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Qin CX, Sleaby R, Davidoff AJ, Bell JR, De Blasio MJ, Delbridge LM, Chatham JC, Ritchie RH. Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications. Pharmacological Research. 2017;116:45–56. [DOI] [PubMed] [Google Scholar]
- 228.Groves JA, Lee A, Yildirir G, Zachara NE. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress & Chaperones. 2013;18:535–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Olson AK, Bouchard B, Zhu WZ, Chatham JC, Des Rosiers C. First characterization of glucose flux through the hexosamine biosynthesis pathway (HBP) in ex vivo mouse heart. J Biol Chem. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yki-Jarvinen H, Daniels MC, Virkamaki A, Makimattila S, DeFronzo RA, McClain D. Increased glutamine:fructose-6-phosphate amidotransferase activity in skeletal muscle of patients with NIDDM. Diabetes. 1996;45:302–7. [DOI] [PubMed] [Google Scholar]
- 231.Wende AR, Young ME, Chatham J, Zhang J, Rajasekaran NS, Darley-Usmar VM. Redox biology and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radical Biology and Medicine. 2016;100:94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Darley-Usmar VM, Ball LE, Chatham JC. Protein O-linked beta-N-acetylglucosamine: A novel effector of cardiomyocyte metabolism and function. Journal of Molecular and Cellular Cardiology. 2012;52:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Prakoso D, Lim S, Erickson J, et al R. Fine-tuning the cardiac O-GlcNAcylation regulatory enzymes governs the functional and structural phenotype of the diabetic heart. European Heart Journal. 2020;(under review, submitted 20th April 2020). [DOI] [PubMed] [Google Scholar]
- 234.Prakoso D, Kiriazis H, Tate M, Qian H, Deo M, Parry L, Gregorevic P, Du X, Chatham J, De Blasio M, Ritchie RH. Manipulation of cardiac O-GlcNAc modification alters cardiac function and remodelling in the setting of diabetic cardiomyopathy (abstract). European Heart Journal. 2018;39.29020337 [Google Scholar]
- 235.De Blasio MJ, Ramalingam A, Cao AH, Prakoso D, Ye JM, Pickering R, Watson AMD, de Haan JB, Kaye DM, Ritchie RH. The superoxide dismutase mimetic tempol blunts diabetes-induced upregulation of NADPH oxidase and endoplasmic reticulum stress in a rat model of diabetic nephropathy. European Journal of Pharmacology. 2017;807:12–20. [DOI] [PubMed] [Google Scholar]
- 236.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Srinivasan V, Sandhya N, Sampathkumar R, Farooq S, Mohan V, Balasubramanyam M. Glutamine fructose-6-phosphate amidotransferase (GFAT) gene expression and activity in patients with type 2 diabetes: inter-relationships with hyperglycaemia and oxidative stress. Clin Biochem. 2007;40:952–7. [DOI] [PubMed] [Google Scholar]
- 238.Lehman DM, Fu D- J, Freeman AB, et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-Acetyl- -D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes. 2005;54:1214–1221. [DOI] [PubMed] [Google Scholar]
- 239.Coomer M, Essop MF. Differential hexosamine biosynthetic pathway gene expression with type 2 diabetes. Molecular Genetics and Metabolism Reports. 2014;1:158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Duggirala R, Blangero J, Almasy L, Dyer TD, Williams KL, Leach RJ, O’Connell P, Stern MP. Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet. 1999;64:1127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li Y, Xie M, Men L, Du J. O-GlcNAcylation in immunity and inflammation: An intricate system. International Journal of Molecular Medicine. 2019;44:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pathak S, Borodkin VS, Albarbarawi O, Campbell DG, Ibrahim A, van Aalten DMF. O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO Journal. 2012;31:1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Golks A, Tran T- TT, Goetschy JF, Guerini D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. EMBO Journal. 2007;26:4368–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hilgers RHP, Xing D, Gong K, Chen Y- F, Chatham JC, Oparil S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. American Journal of Physiology-Heart and Circulatory Physiology. 2012;303:H513–H522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Xing D, Feng W, Noet LG, Miller AP, Zhang Y, Chen Y- F, Majid-Hassan E, Chatham JC, Oparil S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluminal arterial injury. American Journal of Physiology-Heart and Circulatory Physiology. 2008;295:H335–H342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus - A H-1-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. [DOI] [PubMed] [Google Scholar]
- 247.Schulze PC, Drosatos K, Goldberg IJ. Lipid Use and Misuse by the Heart. Circulation Research. 2016;118:1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ritchie RH, Zerenturk EJ, Prakoso D, Calkin AC. Lipid metabolism and its implications for type 1 diabetes-associated cardiomyopathy. Journal of Molecular Endocrinology. 2017;58:R225–R240. [DOI] [PubMed] [Google Scholar]
- 249.Eid S, Sas KM, Abcouwer SF, Feldman EL, Gardner TW, Pennathur S, Fort PE. New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia. 2019;62:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57:2924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. American Journal of Physiology-Endocrinology and Metabolism. 2000;279:E1104–E1113. [DOI] [PubMed] [Google Scholar]
- 252.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB Journal. 2004;18:1692–700. [DOI] [PubMed] [Google Scholar]
- 253.Pereira RO, Tadinada SM, Zasadny FM, et al. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J. 2017;36:2126–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341–52. [DOI] [PubMed] [Google Scholar]
- 255.Montaigne D, Marechal X, Coisne A, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130:554–64. [DOI] [PubMed] [Google Scholar]
- 256.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–66. [DOI] [PubMed] [Google Scholar]
- 257.Liang Q, Kobayashi S. Mitochondrial quality control in the diabetic heart. Journal of Molecular and Cellular Cardiology. 2016;95:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Boudina S, Bugger H, Sena S, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bugger H, Riehle C, Jaishy B, et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J Mol Cell Cardiol. 2012;52:1019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tsushima K, Bugger H, Wende AR, et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circulation Research. 2018;122:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes - Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. [DOI] [PubMed] [Google Scholar]
- 262.Banerjee PS, Ma J, Hart GW. Diabetes-Associated Dysregulation of O -GlcNAcylation in Rat Cardiac Mitochondria. Proceedings of the National Academy of Sciences. 2015;112:6050–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Wende AR. Post-translational modifications of the cardiac proteome in diabetes and heart failure. Proteomics Clin Appl. 2016;10:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Trapannone R, Mariappa D, Ferenbach AT, van Aalten DMF. Nucleocytoplasmic human O-GlcNAc transferase is sufficient for O-GlcNAcylation of mitochondrial proteins. Biochemical Journal. 2016;473:1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ma J, Banerjee P, Whelan SA, Liu T, Wei AC, Ramirez-Correa G, McComb ME, Costello CE, O’Rourke B, Murphy A, Hart GW. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. Journal of Proteome Research. 2016;15:2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circulation Research. 2000;87:1123–1132. [DOI] [PubMed] [Google Scholar]
- 267.Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, Kypson AP. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. American Journal of Physiology-Heart and Circulatory Physiology. 2011;300:H118–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LMD. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. Journal of Molecular and Cellular Cardiology. 2011;50:1035–1043. [DOI] [PubMed] [Google Scholar]
- 269.Park JH, Ku HJ, Kim JK, Park JW, Lee JH. Amelioration of high fructose-induced cardiac hypertrophy by naringin. Scientific Reports. 2018;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chowdhry MF, Vohra HA, Galinanes M. Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: Role of caspases and poly-adenosine diphosphate-ribose polymerase. Journal of Thoracic and Cardiovascular Surgery. 2007;134:124–U25. [DOI] [PubMed] [Google Scholar]
- 271.Spallotta F, Cencioni C, Atlante S, et al. Stable Oxidative Cytosine Modifications Accumulate in Cardiac Mesenchymal Cells From Type2 Diabetes Patients: Rescue by alpha-Ketoglutarate and TET-TDG Functional Reactivation. Circulation Research. 2018;122:31–46. [DOI] [PubMed] [Google Scholar]
- 272.Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pires KM, Torres NS, Buffolo M, Gunville R, Schaaf C, Davis K, Selzman CH, Gottlieb RA, Boudina S. Suppression of Cardiac Autophagy by Hyperinsulinemia in Insulin Receptor-Deficient Hearts Is Mediated by Insulin-Like Growth Factor Receptor Signaling. Antioxidants & Redox Signaling. 2019;31:444–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. Journal of Lipid Research. 2015;56:546–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Jaishy B, Abel ED. Lipotoxicity: Many Roads to Cell Dysfunction and Cell Death Lipids, lysosomes, and autophagy. Journal of Lipid Research. 2016;57:1619–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Pires KM, Buffolo M, Schaaf C, Symons JD, Cox J, Abel ED, Selzman CH, Boudina S. Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. Journal of Molecular and Cellular Cardiology. 2017;113:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou M-H. Improvement of Cardiac Functions by Chronic Metformin Treatment Is Associated With Enhanced Cardiac Autophagy in Diabetic OVE26 Mice. Diabetes. 2011;60:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zhao Y, Zhang L, Qiao Y, et al. Heme Oxygenase-1 Prevents Cardiac Dysfunction in Streptozotocin-Diabetic Mice by Reducing Inflammation, Oxidative Stress, Apoptosis and Enhancing Autophagy. Plos One. 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Zhang M, Zhang L, Hu J, Lin J, Wang T, Duan Y, Man W, Feng J, Sun L, Jia H, Li C, Zhang R, Wang H, Sun D. MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia. 2016:2435–2447. [DOI] [PubMed] [Google Scholar]
- 280.Selvin E, Lazo M, Chen Y, Shen L, Rubin J, McEvoy JW, Hoogeveen RC, Sharrett AR, Ballantyne CM, Coresh J. Diabetes mellitus, prediabetes, and incidence of subclinical myocardial damage. Circulation. 2014;130:1374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Lagadic-Gossmann D, Buckler KJ, LePrigent K, Feuvray D. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. American Journal of Physiology-Heart and Circulatory Physiology. 1996;270:H1529–H1537. [DOI] [PubMed] [Google Scholar]
- 282.Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. 2004;53:3201–3208. [DOI] [PubMed] [Google Scholar]
- 283.Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. American Journal of Physiology-Heart and Circulatory Physiology. 1997;272:H148–H158. [DOI] [PubMed] [Google Scholar]
- 284.Dutta R, Podolin DA, Davidson MB, Davidoff AJ. Cardiomyocyte dysfunction in sucrose-fed rats is associated with insulin resistance. Diabetes. 2001;50:1186–1192. [DOI] [PubMed] [Google Scholar]
- 285.Davidoff AJ, Davidson MB, Carmody MW, Davis ME, Ren J. Diabetic cardiomyocyte dysfunction and myocyte insulin resistance: Role of glucose-induced PKC activity. Molecular and Cellular Biochemistry. 2004;262:155–163. [DOI] [PubMed] [Google Scholar]
- 286.Lacombe VA, Viatchenko-Karpinski S, Terentyev D, Sridhar A, Emani S, Bonagura JD, Feldman DS, Gyorke S, Carnes CA. Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2007;293:R1787–R1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circulation Research. 2005;96:1006–1013. [DOI] [PubMed] [Google Scholar]
- 288.Bidasee KR, Zhang YN, Shao CH, Wang M, Patel KP, Dincer UD, Besch HR. Diabetes increases formation of advanced glycation end products on sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. 2004;53:463–473. [DOI] [PubMed] [Google Scholar]
- 289.Li SY, Yang X, Ceylan-Isik AF, Du M, Sreejayan N,d Ren J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006;49:1434–1446. [DOI] [PubMed] [Google Scholar]
- 290.Shao CH, Capek HL, Patel KP, Wang M, Tang K, DeSouza C, Nagai R, Mayhan W, Periasamy M, Bidasee KR. Carbonylation Contributes to SERCA2a Activity Loss and Diastolic Dysfunction in a Rat Model of Type 1 Diabetes. Diabetes. 2011;60:947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Penpargkul S, Fein F, Sonnenblick EH, Scheuer J. Depressed cardiac sarcoplasmic reticular function from diabetic rats. Journal of Molecular and Cellular Cardiology. 1981;13:303–309. [DOI] [PubMed] [Google Scholar]
- 292.Pierce GN, Dhalla NS. Cardiac myofibrillar ATPase activity in diabetic rats. Journal of Molecular and Cellular Cardiology. 1981;13:1063–1069. [DOI] [PubMed] [Google Scholar]
- 293.Zarainherzberg A, Yano K, Elimban V, Dhalla NS. Cardiac sarcoplasmic-reticulum Ca2+-ATPase expression in streptozotocin-induced diabetic rat-heart. Biochemical and Biophysical Research Communications. 1994;203:113–120. [DOI] [PubMed] [Google Scholar]
- 294.Fauconnier J, Lanner JT, Zhang SJ, Tavi P, Bruton JD, Katz A, Westerblad H. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca(2+) transients and reveal mitochondrial Ca(2+) handling defects in cardiomyocytes of ob/ob mice. Diabetes. 2005;54:2375–2381. [DOI] [PubMed] [Google Scholar]
- 295.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, Gomez AM. Mechanisms of Ca2+ (i) transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–615. [DOI] [PubMed] [Google Scholar]
- 296.Lopaschuk GD, Tahiliani AG, Vadlamudi R, Katz S, McNeill JH. Cardiac sarcoplasmic-reticulum function in insulin-treated or carnitine-treated diabetic rats. American Journal of Physiology. 1983;245:H969–H976. [DOI] [PubMed] [Google Scholar]
- 297.Dillmann WH. Diabetes-mellitus induces changes in cardiac myosin of the rat. Diabetes. 1980;29:579–582. [DOI] [PubMed] [Google Scholar]
- 298.Lu Z, Jiang YP, Wang W, Xu XH, Mathias RT, Entcheva E, Ballou LM, Cohen IS, Lin RZ. Loss of cardiac phosphoinositide 3-kinase p110 alpha results in contractile dysfunction. Circulation. 2009;120:318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Lu Z, Jiang YP, Xu XH, Ballou LM, Cohen IS, Lin RZ. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes. 2007;56:2780–9. [DOI] [PubMed] [Google Scholar]
- 300.Ghigo A, Laffargue M, Li M, Hirsch E. PI3K and Calcium Signaling in Cardiovascular Disease. Circulation Research. 2017;121:282–293. [DOI] [PubMed] [Google Scholar]
- 301.Wang Y, Fan P- S, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis & Rheumatism. 2006;54:2271–2279. [DOI] [PubMed] [Google Scholar]
- 302.Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Jia G, Habibi J, Bostick BP, Ma L, DeMarco VG, Aroor AR, Hayden MR, Whaley-Connell AT, Sowers JR. Uric acid promotes left ventricular diastolic dysfunction in mice fed a Western diet. Hypertension. 2015;65:531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Cohen-Solal A, Beauvais F, Logeart D. Heart failure and diabetes mellitus: Epidemiology and management of an alarming association. Journal of Cardiac Failure. 2008;14:615–625. [DOI] [PubMed] [Google Scholar]
- 305.Miller JA, Floras JS, Zinman B, Skorecki KL, Logan AG. Effect of hyperglycaemia on arterial pressure, plasma renin activity and renal function in early diabetes. Clinical Science. 1996;90:189–195. [DOI] [PubMed] [Google Scholar]
- 306.Xu Y-Z, Zhang X, Wang L, Zhang F, Qiu Q, Liu M-L, Zhang G-R, Wu X-L. An Increased Circulating Angiotensin II Concentration is Associated with Hypoadiponectinemia and Postprandial Hyperglycemia in Men with Nonalcoholic Fatty Liver Disease. Internal Medicine. 2013;52:855–861. [DOI] [PubMed] [Google Scholar]
- 307.Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M, Hirata K- I. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–18. [DOI] [PubMed] [Google Scholar]
- 308.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiological Reviews. 2013;93:137–188. [DOI] [PubMed] [Google Scholar]
- 309.McFarlane SI, Kumar A, Sowers JR. Mechanisms by which angiotensin-converting enzyme inhibitors prevent diabetes and cardiovascular disease. American Journal of Cardiology. 2003;91:30H–37H. [DOI] [PubMed] [Google Scholar]
- 310.Verma S, Arikawa E, McNeill JH. Long-term endothelin receptor blockade improves cardiovascular function in diabetes. American Journal of Hypertension. 2001;14:679–687. [DOI] [PubMed] [Google Scholar]
- 311.McFarlane SI. Role of angiotensin receptor blockers in diabetes: implications of recent clinical trials. Expert Review of Cardiovascular Therapy. 2009;7:1363–71. [DOI] [PubMed] [Google Scholar]
- 312.Pappachan JM, Sebastian J, Bino BC, Jayaprakash K, Vijayakumar K, Sujathan P, Adinegara LA. Cardiac autonomic neuropathy in diabetes mellitus: prevalence, risk factors and utility of corrected QT interval in the ECG for its diagnosis. Postgrad Med J. 2008;84:205–10. [DOI] [PubMed] [Google Scholar]
- 313.Stables CL, Glasser RL, Feldman EL. Diabetic cardiac autonomic neuropathy: Insights from animal models. Autonomic Neuroscience-Basic & Clinical. 2013;177:74–80. [DOI] [PubMed] [Google Scholar]
- 314.Voulgari C, Pagoni S, Vinik A, Poirier P. Exercise improves cardiac autonomic function in obesity and diabetes. Metabolism-Clinical and Experimental. 2013;62:609–621. [DOI] [PubMed] [Google Scholar]
- 315.Spallone V, Ziegler D, Freeman R, Bernardi L, Frontoni S, Pop-Busui R, Stevens M, Kempler P, Hilsted J, Tesfaye S, Low P, Valensi P, Toronto Consensus Panel D. Cardiovascular autonomic neuropathy in diabetes: clinical impact, assessment, diagnosis, and management. Diabetes-Metabolism Research and Reviews. 2011;27:639–653. [DOI] [PubMed] [Google Scholar]
- 316.Vinik AI, Erbas T, Casellini CM. Diabetic cardiac autonomic neuropathy, inflammation and cardiovascular disease. Journal of Diabetes Investigation. 2013;4:4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Tezze C, Romanello V, Sandri M. FGF21 as Modulator of Metabolism in Health and Disease. Front Physiol. 2019;10:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Mraz M, Bartlova M, Lacinova Z, Michalsky D, Kasalicky M, Haluzikova D, Matoulek M, Dostalova I, Humenanska V, Haluzik M. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin Endocrinol. 2009;71:369–75. [DOI] [PubMed] [Google Scholar]
- 319.Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care. 2009;32:1542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, Moller DE, Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–27. [DOI] [PubMed] [Google Scholar]
- 321.Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Zhang X, Yang L, Xu X, Tang F, Yi P, Qiu B, Hao Y. A review of fibroblast growth factor 21 in diabetic cardiomyopathy. Heart Failure Reviews. 2019;24:1005–1017. [DOI] [PubMed] [Google Scholar]
- 323.Zhang W, Xu W, Feng Y, Zhou X. Non-coding RNA involvement in the pathogenesis of diabetic cardiomyopathy. Journal of Cellular and Molecular Medicine. 2019;23:5859–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Leon LE, Rani S, Fernandez M, Larico M, Calligaris SD. Subclinical Detection of Diabetic Cardiomyopathy with MicroRNAs: Challenges and Perspectives. Journal of Diabetes Research. 2016;2016:6143129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Diao X, Shen E, Wang X, Hu B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Molecular Medicine Reports. 2011;4:633–640. [DOI] [PubMed] [Google Scholar]
- 326.Raut S, Kumar A, Khullar M. Epigenetic role of micrornas in diabetic cardiomyopathy. Journal of the Practice of Cardiovascular Sciences. 2016;2:79–85. [Google Scholar]
- 327.Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes-Metabolism Research and Reviews. 2010;26:40–49. [DOI] [PubMed] [Google Scholar]
- 328.Greco S, Fasanaro P, Castelvecchio S, D’Alessandra Y, Arcelli D, Di Donato M, Malavazos A, Capogrossi MC, Menicanti L, Martelli F. MicroRNA Dysregulation in Diabetic Ischemic Heart Failure Patients. Diabetes. 2012;61:1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, Singh S, Prasad R, Khullar M. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Molecular and Cellular Biochemistry. 2016;417:191–203. [DOI] [PubMed] [Google Scholar]
- 330.Yang X, Li X, Lin Q, Xu Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene. 2019;715:143995. [DOI] [PubMed] [Google Scholar]
- 331.Katare R, Caporali A, Zentilin L, Avolio E, Sala-Newby G, Oikawa A, Cesselli D, Beltrami AP, Giacca M, Emanueli C, Madeddu P. Intravenous Gene Therapy With PIM-1 Via a Cardiotropic Viral Vector Halts the Progression of Diabetic Cardiomyopathy Through Promotion of Prosurvival Signaling. Circulation Research. 2011;108:1238–1251. [DOI] [PubMed] [Google Scholar]
- 332.Li H, Dai B, Fan J, Chen C, Nie X, Yin Z, Zhao Y, Zhang X, Wang DW. The Different Roles of miRNA-92a-2-5p and let-7b-5p in Mitochondrial Translation in db/db Mice. Molecular Therapy Nucleic Acids. 2019;17:424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Lee J, Harris AN, Holley CL, et al. Rpl13a small nucleolar RNAs regulate systemic glucose metabolism. Journal of Clinical Investigation. 2016;126:4616–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Michel CI, Holley CL, Scruggs BS, Sidhu R, Brookheart RT, Listenberger LL, Behlke MA, Ory DS, Schaffer JE. Small Nucleolar RNAs U32a, U33, and U35a Are Critical Mediators of Metabolic Stress. Cell Metabolism. 2011;14:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Tao H, Tao J-Y, Song Z-Y, Shi P, Wang Q, Deng Z-Y, Ding X-S. MeCP2 triggers diabetic cardiomyopathy and cardiac fibroblast proliferation by inhibiting RASSF1A. Cellular Signalling. 2019;63:109387–109387. [DOI] [PubMed] [Google Scholar]
- 336.Pieske B, Tschoepe C, de Boer RA, et al. How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). European Heart Journal. 2019;40:3297–3317. [DOI] [PubMed] [Google Scholar]
- 337.Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nature Reviews Cardiology. 2017;14:591–602. [DOI] [PubMed] [Google Scholar]
- 338.Redfield MM. Heart Failure with Preserved Ejection Fraction. New England Journal of Medicine. 2016;375:1868–1877. [DOI] [PubMed] [Google Scholar]
- 339.van Riet EES, Hoes A, Wagenaar KP, Limburg A, Landman MAJ, Rutten FH. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. European Journal of Heart Failure. 2016;18:242–252. [DOI] [PubMed] [Google Scholar]
- 340.Shah SJ. Precision Medicine for Heart Failure with Preserved Ejection Fraction: An Overview. Journal of Cardiovascular Translational Research. 2017;10:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Seferovic PM, Petrie MC, Filippatos GS, et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. European Journal of Heart Failure. 2018;20:853–872. [DOI] [PubMed] [Google Scholar]
- 342.Beale AL, Nanayakkara S, Segan L, Mariani JA, Maeder MT, van Empel V, Vizi D, Evans S, Lam CSP, Kaye DM. Sex Differences in Heart Failure With Preserved Ejection Fraction Pathophysiology A Detailed Invasive Hemodynamic and Echocardiographic Analysis. JACC-Heart Failure. 2019;7:239–249. [DOI] [PubMed] [Google Scholar]
- 343.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation. 2017;136:6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Unger ED, Dubin RF, Deo R, Daruwalla V, Friedman JL, Medina C, Beussink L, Freed BH, Shah SJ. Association of chronic kidney disease with abnormal cardiac mechanics and adverse outcomes in patients with heart failure and preserved ejection fraction. European Journal of Heart Failure. 2016;18:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Tromp J, Lim SL, Tay WT, et al. Microvascular Disease in Patients With Diabetes With Heart Failure and Reduced Ejection Versus Preserved Ejection Fraction. Diabetes Care. 2019;42:1792–1799. [DOI] [PubMed] [Google Scholar]
- 346.Figtree GA, Radholm K, Barrett TD, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Matthews DR, Shaw W, Neal B. Effects of Canagliflozin on Heart Failure Outcomes Associated With Preserved and Reduced Ejection Fraction in Type 2 Diabetes Mellitus Results From the CANVAS Program. Circulation. 2019;139:2591–2593. [DOI] [PubMed] [Google Scholar]
- 347.Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM. Longitudinal Changes in Ejection Fraction in Heart Failure Patients With Preserved and Reduced Ejection Fraction. Circulation-Heart Failure. 2012;5:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Durgan DJ, Pat BM, Laczy B, et al. O -GlcNAcylation, Novel Post-Translational Modification Linking Myocardial Metabolism and Cardiomyocyte Circadian Clock. Journal of Biological Chemistry. 2011;286:44606–44619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lind M, Bounias I, Olsson M, Gudbjörnsdottir S, Svensson A- M, Rosengren A. Glycaemic control and incidence of heart failure in 20,985 patients with type 1 diabetes: an observational study. Lancet. 2011;378:140–6. [DOI] [PubMed] [Google Scholar]
- 350.Margulies KB, Hernandez AF, Redfield MM, et al. Effects of Liraglutide on Clinical Stability Among Patients With Advanced Heart Failure and Reduced Ejection Fraction A Randomized Clinical Trial. JAMA-Journal of the American Medical Association. 2016;316:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Weir DL, McAlister FA, Senthilselvan A, Minhas-Sandhu JK, Eurich DT. Sitagliptin Use in Patients With Diabetes and Heart Failure A Population-Based Retrospective Cohort Study. JACC-Heart Failure. 2014;2:573–582. [DOI] [PubMed] [Google Scholar]
- 352.Lincoff AM, Tardif JC, Schwartz GG, et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA. 2014;311:1515–25. [DOI] [PubMed] [Google Scholar]
- 353.Li L, Li S, Deng K, et al. Dipeptidyl peptidase-4 inhibitors and risk of heart failure in type 2 diabetes: systematic review and meta-analysis of randomised and observational studies. British Medical Journal. 2016;352:i610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Hippisley-Cox J, Coupland C. Diabetes treatments and risk of heart failure, cardiovascular disease, and all cause mortality: cohort study in primary care. British Medical Journal. 2016;354:i3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Seferovic PM, Coats AJS, Ponikowski P, et al. European Society of Cardiology/Heart Failure Association position paper on the role and safety of new glucose-lowering drugs in patients with heart failure. European Journal of Heart Failure. 2020;22:196–213. [DOI] [PubMed] [Google Scholar]
- 356.Gerstein HC, Colhoun HM, Dagenais GR, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. 2019;394:121–130. [DOI] [PubMed] [Google Scholar]
- 357.Cefalu WT, Kaul S, Gerstein HC, Holman RR, Zinman B, Skyler JS, Green JB, Buse JB, Inzucchi SE, Leiter LA, Raz I, Rosenstock J, Riddle MC. Cardiovascular Outcomes Trials in Type 2 Diabetes: Where Do We Go From Here? Reflections From a Diabetes Care Editors’ Expert Forum. Diabetes Care. 2018;41:14–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. New England Journal of Medicine. 2015;373:2117–2128. [DOI] [PubMed] [Google Scholar]
- 359.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M,Matthews DR. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. New England Journal of Medicine. 2017;377:644–657. [DOI] [PubMed] [Google Scholar]
- 360.Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. New England Journal of Medicine. 2019;380:2295–2306. [DOI] [PubMed] [Google Scholar]
- 361.Kosiborod M, Cavender MA, Fu AZ, et al. Lower Risk of Heart Failure and Death in Patients Initiated on SGLT-2 Inhibitors Versus Other Glucose-Lowering Drugs: The CVD-REAL Study. Circulation. 2017;136:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. New England Journal of Medicine. 2019;380:347–357. [DOI] [PubMed] [Google Scholar]
- 363.Cahn A, Mosenzon O, Wiviott SD, et al. Efficacy and Safety of Dapagliflozin in the Elderly: Analysis From the DECLARE-TIMI 58 Study. Diabetes Care. 2020;43:468–475. [DOI] [PubMed] [Google Scholar]
- 364.Kluger AY, Tecson KM, Lee AY, Lerma EV, Rangaswami J, Lepor NE, Cobble ME, McCullough PA. Class effects of SGLT2 inhibitors on cardiorenal outcomes. Cardiovascular Diabetology. 2019;18:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Kramer CK, Zinman B. Sodium-Glucose Cotransporter-2 (SGLT-2) Inhibitors and the Treatment of Type 2 Diabetes. Annual Review of Medicine , Vol 70; 2019(70): 323–334. [DOI] [PubMed] [Google Scholar]
- 366.Scheen AJ. Cardiovascular Effects of New Oral Glucose-Lowering Agents DPP-4 and SGLT-2 Inhibitors. Circulation Research. 2018;122:1439–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Thomas MC, Cherney DZI. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia. 2018;61:2098–2107. [DOI] [PubMed] [Google Scholar]
- 368.Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia. 2017;60:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review. Diabetologia. 2018;61:2108–2117. [DOI] [PubMed] [Google Scholar]
- 370.Van Steenbergen A, Balteau M, Ginion A, et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Scientific Reports. 2017;7:41166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Vrhovac I, Eror DB, Klessen D, Burger C, Breljak D, Kraus O, Radovic N, Jadrijevic S, Aleksic I, Walles T, Sauvant C, Sabolic I, Koepsell H. Localizations of Na+-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Archiv-European Journal of Physiology. 2015;467:1881–1898. [DOI] [PubMed] [Google Scholar]
- 372.Taylor SI, Leslie BR. Cardiovascular outcome trials of diabetes drugs: lessons learned. J Clin Invest. 2018;128:893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Wanner C, Lachin JM, Inzucchi SE, Fitchett D, Mattheus M, George JT, Woerle H- J, Broedl UC, von Eynatten M, Zinman B. Empagliflozin and Clinical Outcomes in Patients with Type 2 Diabetes, Established Cardiovascular Disease and Chronic Kidney Disease. Circulation. 2018;137:119–129. [DOI] [PubMed] [Google Scholar]
- 374.Mazer CD, Hare GMT, Connelly PW, et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores and Red Blood Cell Morphology in Patients with Type 2 Diabetes and Coronary Artery Disease. Circulation. 2020;141:704–707. [DOI] [PubMed] [Google Scholar]
- 375.Kosiborod MN, Jhund PS, Docherty KF, et al. Effects of Dapagliflozin on Symptoms, Function, and Quality of Life in Patients With Heart Failure and Reduced Ejection Fraction Results From the DAPA-HF Trial. Circulation. 2020;141:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Martinez FA, Serenelli M, Nicolau JC, et al. Efficacy and Safety of Dapagliflozin in Heart Failure With Reduced Ejection Fraction According to Age Insights From DAPA-HF. Circulation. 2020;141:100–111. [DOI] [PubMed] [Google Scholar]
- 377.McMurray JJV, Solomon SD, Docherty KF, Jhund PS. The Dapagliflozin And Prevention of Adverse outcomes in Heart Failure trial (DAPA-HF) in context. European Heart Journal. 2020:ehz916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med. 2019;381:1995–2008. [DOI] [PubMed] [Google Scholar]
- 379.Nassif ME, Windsor S, Tang F, et al. Dapagliflozin Effects on Biomarkers, Symptoms, and Functional Status in Patients With Heart Failure With Reduced Ejection Fraction: The DEFINE-HF Trial. Circulation. 2019;140:1463–1476. [DOI] [PubMed] [Google Scholar]