

Abstract
We recently described GPX4 as a promising target for killing therapy-resistant cancer cells via ferroptosis. Remarkably, the induction of therapy resistance by multiple types of treatments results in a stable cell state marked by high levels of polyunsaturated lipids and an acquired dependency on GPX4. Unfortunately, all existing inhibitors of GPX4 act covalently via a reactive alkyl chloride moiety that confers poor selectivity and pharmacokinetic properties. Here, we report our discovery that masked nitrile-oxide electrophiles, which have not been explored previously as covalent cellular probes, undergo remarkable chemical transformations in cells and provide an effective strategy for selective targeting of GPX4. The novel GPX4-inhibiting compounds we describe exhibit unexpected proteome-wide selectivity and, in some instances, vastly improved physiochemical and pharmacokinetic properties compared to existing chloroacetamide-based GPX4 inhibitors. These features make them superior tool compounds for biological interrogation of ferroptosis and constitute starting points for development of improved inhibitors of GPX4.
Graphical Abstract
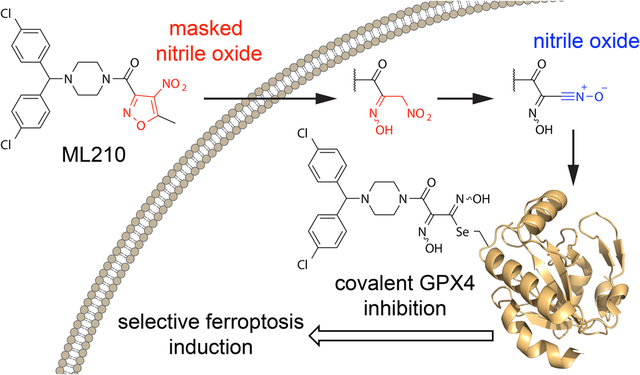
We have reported previously that cancer cells in a therapy-induced, drug-resistant state have an enhanced dependence on phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4) to prevent undergoing lipid hydroperoxide-promoted ferroptotic cell death1,2. In vitro and in vivo evidence support the possibility that rational combination of GPX4 inhibitors and current standard of care agents can achieve durable responses in a range of cancer types1–3.
GPX4 is a selenoprotein that plays a critical role in protecting cells from lipid peroxidation and ferroptosis by reducing lipid hydroperoxides to their corresponding lipid alcohols4. GPX4 is unique in its ability to reduce complex lipid hydroperoxides and is the sole mammalian protein known thus far to be capable of performing this crucial function5,6. The broad substrate scope of GPX4 likely derives from its monomeric structure and the relatively flat surface adjacent to the catalytic selenocysteine residue in the active site7,8.
Unfortunately, these same structural features also make GPX4 a challenging target for the development of small-molecule inhibitors. The lack of a drug-like binding pocket and reliance on a nucleophilic selenocysteine residue for enzymatic activity suggest that covalent inhibitors may be necessary for inhibition of cellular GPX4. Indeed, all known direct and cell-active GPX4 inhibitors are alkylating agents that covalently bind the selenocysteine residue via an activated alkyl chloride (Fig. 1a and Supplementary Fig. 1)4,9,10. Such inhibitors exhibit low selectivity and poor pharmacokinetic properties. This limits their utility as tool compounds in vitro, hinders their use in vivo, and makes them undesirable starting points for the development of drug-like GPX4 inhibitors. It is further notable that cellular4,11 and biochemical12 screening efforts have not identified other common electrophiles, such as acrylamides, that can serve as selective GPX4-targeting warheads. Likewise, replacing the chloroacetamide warheads with less reactive electrophiles produces analogs that no longer inhibit GPX44,13.
Figure 1. ML210 is a selective covalent inhibitor of cellular GPX4.
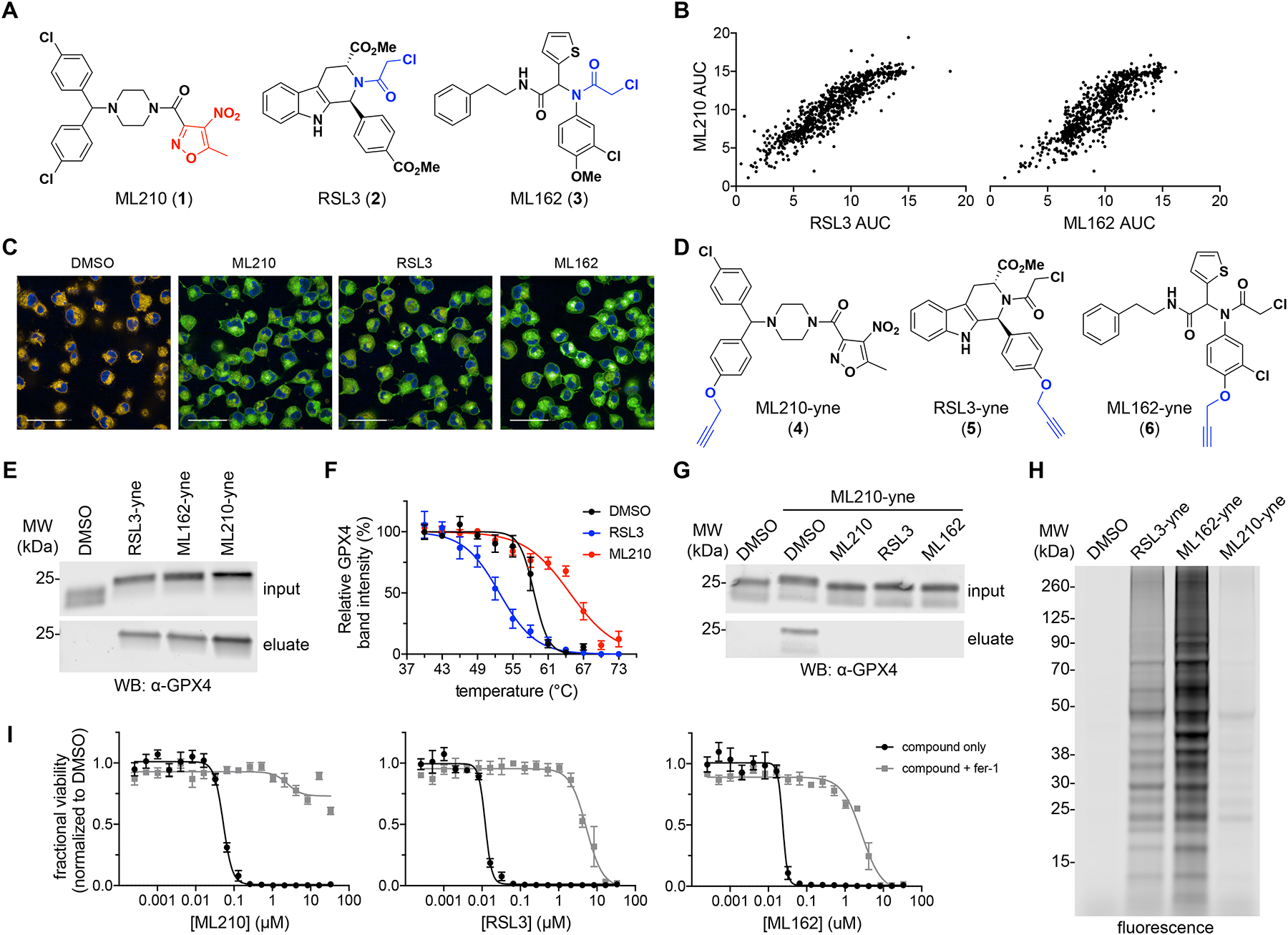
(A) Chemical structures of RSL3, ML162, and ML210 (1–3). Chloroacetamide groups are shown in blue and the nitroisoxazole group is colored red. (B) ML210 exhibits cell-killing activity similar to RSL3 and ML162 across a panel of 821 cancer cell lines. Each dot represents a single cancer cell line. Data plotted reflect an area-under-the-curve (AUC) metric of cell-line sensitivity to small molecules. (C) Treatment of cells with RSL3, ML162, or ML210 (10 μM, 90 min) leads to accumulation of lipid hydroperoxides in LOX-IMVI cells as assessed by fluorescence imaging with C11-BODIPY 581/591. The C11-BODIPY dye emission shifts from orange to green upon oxidation. Scale bars, 50 μM. See also Supplementary Fig. 3a,b. (D) Chemical structures of GPX4-inhibitor affinity probes RSL3-yne, ML162-yne, and ML210-yne (4–6) with alkyne groups shown in blue. (E) RSL3-yne, ML162-yne, and ML210-yne (10 μM, 1 h) pull down GPX4 from LOX-IMVI cells. Full-length western blots are shown in Supplementary Fig. 4. (F) GPX4 CETSA profiles for intact HCC4006 cells treated with DMSO (black), RSL3 (blue; destabilizing), or ML210 (red; stabilizing). Cells were treated with 10 μM compound for 1 h. Data are plotted as mean ± s.e.m., n ≥ 4 biologically independent samples. Representative western blots are shown in Supplementary Fig. 4. (G) Competitive affinity enrichment between ML210-yne probe (10 μM, 30 min) and chloroacetamide GPX4 inhibitors (10 μM, 30 min pretreatment in LOX-IMVI cells). Full-length western blots are shown in Supplementary Fig. 4. (H) Fluorescent labeling of proteins modified by RSL3-yne, ML162-yne, and ML210-yne probes (10 μM, 1 h) in LOX-IMVI cells. See also Supplementary Fig. 4a. (I) Co-treatment with fer-1 (1.5 μM) rescues the cell-killing effects of RSL3, ML162, and ML210 in LOX-IMVI cells. Data are plotted as mean ± s.e.m., n = 4 technical replicates.
Thus, the development of improved GPX4 inhibitors with drug-like potential would benefit from broadening the scope of electrophilic chemotypes and mechanisms of action. We hypothesized that the study of small molecules that are not overtly reactive, yet selectively kill GPX4-dependent cancer cells, may reveal novel strategies for the development of improved inhibitors. One such molecule is the nitroisoxazole-containing compound ML210 (1). ML210 has emerged as a popular ferroptosis-inducing tool compound and has been presumed, without substantiation, to be a direct GPX4 inhibitor. However, ML210 lacks an apparent covalent reactive group through which it might engage GPX4 or other nucleophiles and previous efforts have failed to demonstrate any binding interaction between ML210 and GPX49.
Here, we report on our finding that ML210 is a prodrug that is converted in cells into a nitrile-oxide electrophile that covalently inhibits GPX4 with remarkable proteome-wide selectivity. This discovery not only establishes the molecular mechanism of action of ML210, but also illuminates nitrile oxides as novel, cell-active warheads uniquely suited for engaging the GPX4 active-site selenocysteine. While the intrinsic reactivity of nitrile oxides would normally preclude their use as cellular tool compounds, we demonstrate that masking elements, such as those exemplified by ML210, afford these compounds with superior physiochemical and pharmacokinetic properties compared to chloroacetamide GPX4 inhibitors.
Insights from our mechanistic studies have led us to rationally design and validate several structurally-distinct masked nitrile-oxide chemotypes that also selectively target GPX4. Importantly, we demonstrate that masked nitrile-oxide GPX4 inhibitors constitute the most selective ferroptosis-inducing tool compounds known to date and we provide evidence of the importance this selectivity holds for biologists investigating ferroptosis using small molecules.
RESULTS
ML210 is a covalent inhibitor of cellular GPX4
To identify small-molecule GPX4 inhibitors that do not contain a reactive alkyl chloride warhead, we focused our attention on the nitroisoxazole-containing compound ML210 (Fig. 1a)14. ML210 exhibits a pattern of cell killing across 821 cancer cell lines (Cancer Therapeutics Response Portal at http://portals.broadinstitute.org/ctrp.v2.1/)15–17 that is strikingly similar to that of chloroacetamide-containing GPX4 inhibitors (1S,3R)-RSL3 (RSL3, 2)4,9,11 and ML162 (3)4,9,14 (Fig. 1a,b), yet previous efforts have failed to uncover evidence of direct interaction between ML210 and GPX49. While ML210 does suppress GPX4 enzymatic activity in a lysate-based assay1,4, prompting some investigators to refer to ML210 as a GPX4 inhibitor, this assay is not diagnostic of direct GPX4 inhibition. An illustration of this is found in two recently described ferroptosis-inducing compounds, FIN5610 and FINO218, which eliminate GPX4 activity in a lysate-based enzymatic activity assay yet are not direct GPX4 binders. Moreover, ML210 lacks an obvious protein-reactive group through which it could act as a covalent GPX4 inhibitor and it does not react with small-molecule thiols (Supplementary Fig. 2).
To investigate the possibility that ML210 may yet be a direct GPX4 inhibitor through some novel mechanism, we first confirmed its ferroptosis-inducing effects. As for RSL3 and ML162, treatment of cells with ML210 results in the accumulation of cellular lipid hydroperoxides whose lethal effects can be prevented by co-treatment with the radical-trapping antioxidant ferrostatin-1 (fer-1)19–21 (Fig. 1c and Supplementary Fig. 3a,b). GPX4 enzymatic activity is eliminated in cells treated with both ML210 and chloroacetamide GPX4 inhibitors (Supplementary Fig. 3c), leaving open the possibility that ML210 acts as a direct GPX4 inhibitor.
To probe direct interactions between ML210 and GPX4, we synthesized alkyne-containing analogs (denoted with “-yne” suffix; 4–6) of ML210, as well as RSL3 and ML162 (Fig. 1d and Supplementary Fig. 3d), that would enable affinity enrichment of target proteins following click chemistry conjugation to a tag. ML210-yne, like the RSL3 and ML162 probes, pulls down GPX4 from cells (Fig. 1e), indicating that ML210 or an ML210-derived metabolite interacts covalently with GPX4. To validate this result using an orthogonal method, we used a label-free cellular thermal shift assay (CETSA)22. CETSA revealed that both ML210 and chloroacetamides affect the thermal stability of GPX4 (Fig. 1f and Supplementary Fig. 3e), consistent with cellular target engagement. However, these CETSA results also revealed an intriguing distinction between ML210 and chloroacetamide GPX4 inhibitors: while chloroacetamide inhibitors result in GPX4 thermal destabilization, ML210 stabilizes GPX4 (Fig. 1f and Supplementary Fig. 3e). This difference may reflect underlying differences in the way these two classes of molecules interact with GPX4.
To characterize further the details of covalent interaction between GPX4 and ML210, we investigated the GPX4 residue that ML210 covalently modifies using competitive pulldown experiments (Fig. 1g and Supplementary Fig. 3f). These experiments revealed that ML210 binding is mutually exclusive with that of chloroacetamide inhibitors, which are known to bind the catalytic selenocysteine 46 residue of GPX49,23. These results therefore signify that ML210 also likely binds the GPX4 selenocysteine residue. To confirm this, we performed intact-cell CETSA experiments with GPX4 mutants containing cysteine or alanine in place of the selenocysteine residue. We found that ML210 stabilizes the selenocysteine-to-cysteine (U46C) mutant form of GPX4 (Supplementary Fig. 3g) but has no effect on the selenocysteine-to-alanine (U46A) mutant protein (Supplementary Fig. 3h). Together, these data establish the importance of a nucleophilic (seleno)cysteine residue within the active site of GPX4 for covalent interaction with inhibitors. Moreover, these findings indicate that despite differences between ML210 and chloroacetamide GPX4 inhibitors, the mechanisms of action of both classes converge on covalent modification of the GPX4 selenocysteine residue.
ML210 exhibits selectivity for GPX4
A major drawback of chloroacetamide GPX4 inhibitors is their high chemical reactivity, which imparts them with relatively low proteome-wide selectivity. To test whether ML210 may act in a more selective manner, we performed cellular proteome reactivity profiling experiments using the ML210, RSL3, and ML162 alkyne probes described above (4–6). These experiments revealed that ML210-yne exhibits markedly lower proteome reactivity compared to RSL3-yne and ML162-yne, which each engage in a large number of covalent interactions with cellular proteins (Fig. 1h and Supplementary Fig. 6a). Competitive labeling experiments in cells indicate that ML210 and chloroacetamide GPX4 inhibitors do not share many protein targets in common (Supplementary Fig. 6b,c), and mass spectrometry-based proteomics reveal that the off-targets of ML210 are highly abundant proteins (e.g. tubulins) (Supplementary Fig. 6d). The cellular selectivity of ML210 is further demonstrated by the ability of ferroptosis inhibitors, including fer-1, liproxstatin-1 (lip-1)24, and deferoxamine (DFO)19, to rescue completely the cell-killing activity of ML210 but not that of either RSL3 or ML162 (Fig. 1i and Supplementary Fig. 6e). The impressive selectivity of ML210 promises to overcome a key limitation of GPX4-inhibiting tool compounds and makes it a favorable starting point for further development. This recognition motivated us to further investigate the mechanism of action of ML210 further and gain insights into the basis of its selectivity.
ML210 requires the intact cell context to bind GPX4
To understand how ML210 covalently modifies GPX4 without a chloroacetamide warhead, we developed an intact protein mass spectrometry-based workflow to detect covalent GPX4-inhibitor complexes generated in cells (Fig. 2a and Supplementary Fig. 7). Subjecting ML210 to this approach revealed the formation of a +434 Da ML210-GPX4 adduct (Fig. 2a), which corresponds to an unusual 41 Da loss from ML210. This cannot be explained by the displacement of an obvious leaving group and suggests a novel covalent mechanism of action for ML210.
Figure 2. ML210 requires the intact cell context to bind GPX4.
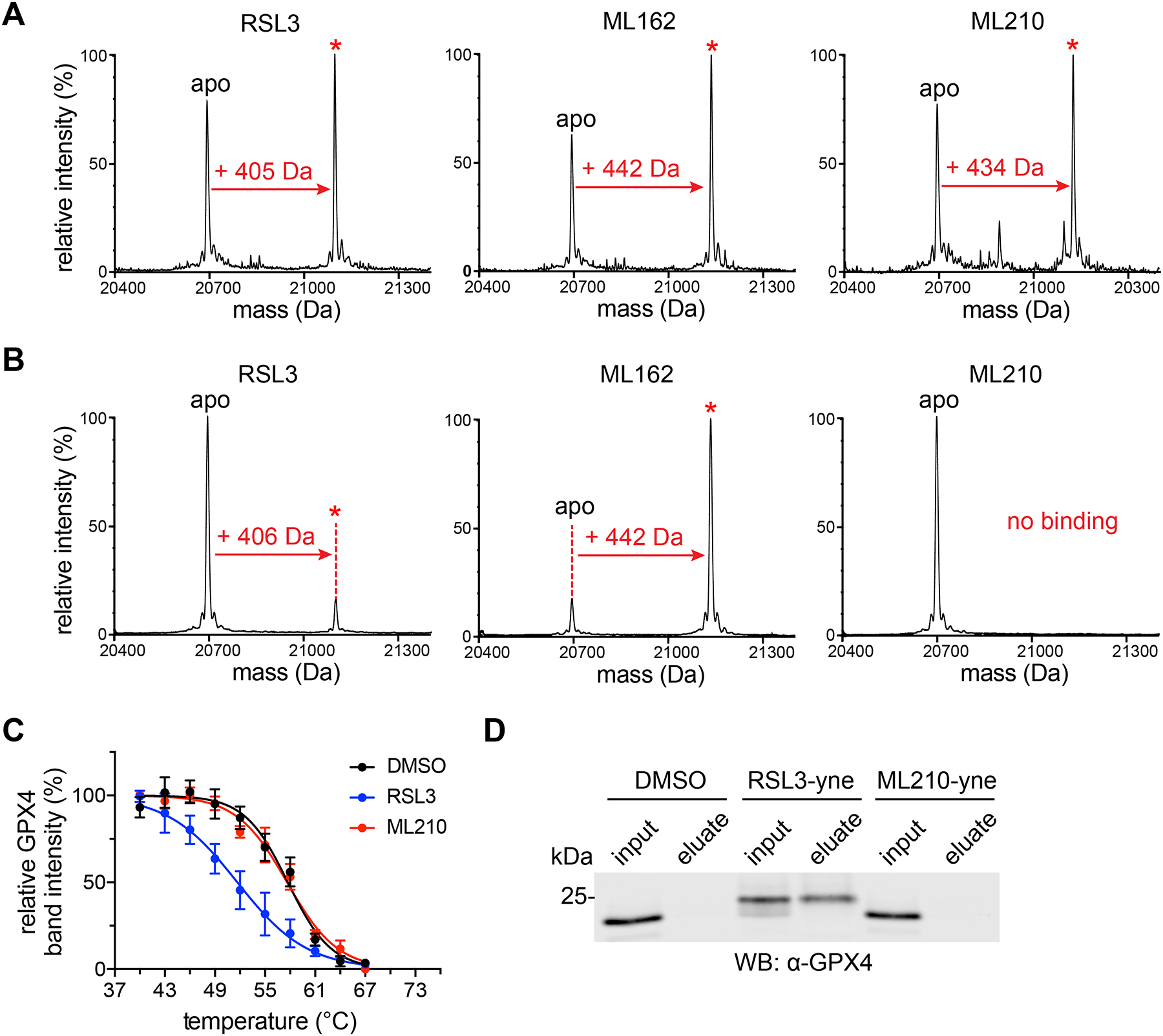
(A) RSL3, ML162, and ML210 (10 μM, 24 h) show evidence of covalent interaction with FLAG-GPX4WT in HEK293–6E cells by intact protein mass spectrometry. Covalent adduct peaks are marked with a red asterisk (*). See also Supplementary Fig. 7b. (B) RSL3 and ML162, but not ML210, (100 μM, 2 h) covalently bind purified FLAG-GPX4WT (10 μM) as assessed by intact protein mass spectrometry. Adduct peaks are marked with a red asterisk (*). (C) GPX4 CETSA profiles for cell lysates (HCC4006) treated with DMSO (black), RSL3 (blue), or ML210 (red). Cells were treated with 10 μM compound for 1 h. Data are plotted as mean ± s.e.m., n = 3 biologically independent samples. Representative western blots are shown in Supplementary Fig. 8a (D) RSL3-yne, but not ML210-yne, pulls down GPX4 from cell lysate. Lysates were treated for 1 h with the indicated alkyne probe (10 μM). Full gel image is shown in Supplementary Fig. 8b.
We probed this mechanism further by investigating binding of ML210 with purified GPX4 protein. However, these experiments revealed that the ML210-GPX4 adduct forms only when ML210 is added to intact cells and not when ML210 is incubated with purified wild-type GPX4 (Fig. 2b). This context-dependent binding contrasts with chloroacetamide inhibitors, which form adducts with GPX4 both when added to cells and incubated with purified protein (Fig. 2b). We confirmed this unique intact-cell dependence of ML210 binding using a lysate-based variant of CETSA (Fig. 2c) and by performing pulldown experiments from lysates (Fig. 2d). In both assays, ML210 shows no evidence of covalent interaction with GPX4 outside of intact cells while RSL3 and ML162 show consistent target engagement regardless of context.
The intact-cell dependence of ML210 binding, together with its lack of an obvious protein-reactive moiety compared to chloroacetamide inhibitors, led us to hypothesize that ML210 may be a prodrug that undergoes chemical transformation in cells to covalently modify GPX4. Such a mechanism could also explain the unusual mass of the ML210-GPX4 adduct, which may be a result of mass loss during cellular activation. Finally, the prodrug nature of ML210 and unique reactivity of its unleashed active electrophilic form may also underlie the cellular selectivity of ML210 (Fig. 1h,i).
ML210 requires cellular conversion to JKE-1674
To investigate how ML210 may give rise in cells to a GPX4-targeting metabolite, we focused our attention on the nitroisoxazole group, which contains structural features susceptible to diverse chemical transformations in cells25–27. Structure–activity relationship (SAR) studies with compounds 7–36 demonstrated that both the isoxazole and nitro groups of the nitroisoxazole are unable to be replaced without complete loss of activity (Fig. 3a and Supplementary Fig. 9). In contrast to this steep SAR of the nitroisoxazole itself, modifications of substituents at the 3- and 5-positions of the isoxazole ring (26–35) are well tolerated with little effect on potency relative to ML210. Most surprisingly, SAR of the nitroisoxazole 5-position (26–33) revealed that cellular activity is unaffected by the steric bulk or polarity of this substituent. On the basis of the SAR trends and unusual mass loss associated with GPX4 binding (Fig. 2a), we hypothesized that cellular activation of ML210 involves elimination of the nitroisoxazole 5-position. Consistent with this hypothesis, treatment of cells with both isopropyl-ML210 (27) (Fig. 3b) and ML210 produces identical GPX4 adducts (Fig. 3c).
Figure 3. ML210 requires conversion in cells to the α-nitroketoxime JKE-1674.
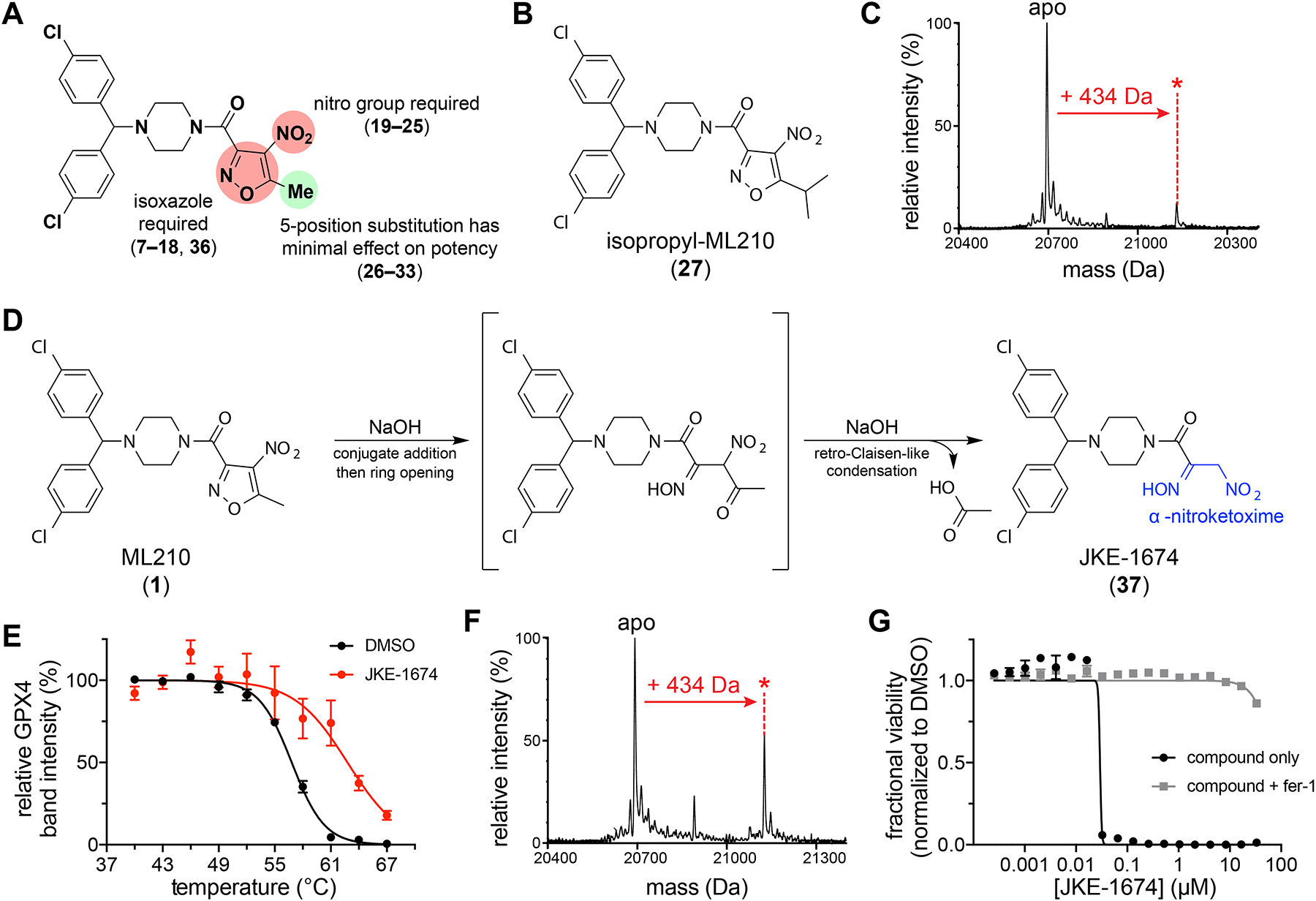
(A) Summary of ML210 nitroisoxazole group SAR studies. Chemical structures of compounds 7–36 are shown in Supplementary Fig. 9. (B) Chemical structure of isopropyl-ML210 (27). (C) Treatment of HEK293–6E cells with isopropyl-ML210 (10 μM, 24 h) produces the same covalent GPX4 adduct mass increase (+434 Da) as ML210. (D) Scheme showing proposed ML210 hydrolysis and structure of JKE-1674 with α-nitroketoxime group highlighted in blue. (E) GPX4 CETSA of intact cells (LOX-IMVI) treated with JKE-1674 (red, 10 μM, 1 h) reveals thermal stabilization of GPX4 compared to treatment with DMSO (black). Data are plotted as mean ± s.e.m., n ≥ 4 biologically independent samples. Representative western blots are shown in Supplementary Fig. 12. (F) Treatment of cells with JKE-1674 (10 μM, 1 h) produces the same covalent GPX4 adduct (+434 Da) as ML210. (G) Co-treatment with fer-1 rescues the LOX-IMVI cell-killing effects of JKE-1674 and ML210 to a similar extent. Data are plotted as mean ± s.e.m., n = 4 technical replicates. See also Supplementary Fig. 10f.
Nitroisoxazoles have been reported in the literature to undergo base-promoted hydrolysis to yield an α-nitroketoxime and carboxylic acid28. By investigating the relevance of this reaction to ML210 activation, we identified conditions that rapidly convert ML210 to its corresponding α-nitroketoxime, JKE-1674 (37) (Fig. 3d and Supplementary Fig. 10a). JKE-1674 exhibits activity indistinguishable from that of ML210 in cellular target engagement assays (Fig. 3e,f), including yielding the same +434 Da GPX4 adduct in cells (Fig. 3f). An alkyne analog of JKE-1674, JKE-1674-yne (38) (Supplementary Fig. 10b,c), also pulls down GPX4 from cells (Supplementary Fig. 10d) and proteome-wide reactivity studies performed with this compound reveal a degree of selectivity similar to ML210 (Supplementary Fig. 10e). Finally, JKE-1674 kills cells in a manner that is equipotent to ML210 and is completely rescued by ferroptosis inhibitors (Fig. 3g and Supplementary Fig. 10f). We further confirmed the mechanistic link between JKE-1674 and ML210 by observing the time-dependent formation of JKE-1674 in cells treated with ML210 (Supplementary Fig. 11a–g). Finally, ML210 and JKE-1674 form identical adducts with cysteine and glutathione in cells (Supplementary Fig. 11d,e,g), consistent with their reactivity via a common intermediate.
Together, these data indicate that JKE-1674 is a key metabolite in the mechanism of action of ML210. However, like ML210, JKE-1674 is unable to engage either purified recombinant GPX4 or GPX4 in cell lysate (Supplementary Fig. 11h,i), indicating that additional cellular activation of JKE-1674 must be required to yield a protein-reactive electrophile.
JKE-1674 forms a nitrile-oxide electrophile in cells
The +434 Da mass of the ML210-derived GPX4 adduct suggests that the final protein-reactive metabolite of ML210 is related to JKE-1674 through the loss of water. In cells, ML210 and JKE-1674 react with cysteine and glutathione to form +434 Da adducts, presumably the corresponding thiohydroximates (Supplementary Fig. 11e). Thiohydroximate adducts have been reported as products of the nitroalkane-containing isocitrate lyase inhibitor, 3-nitropropionate, which acts as a masked electrophile via its nitronic acid tautomer29. However, direct reactivity between nitroalkane and nucleophile (Supplementary Fig. 13a) does not explain the requirement of intact cells for JKE-1674 to bind GPX4. We therefore speculated that JKE-1674 must first be dehydrated in order to generate a GPX4-reactive species (Supplementary Fig. 13b).
To test this dehydration hypothesis, we attempted experimentally to eliminate water from JKE-1674. Unfortunately, we found that published conditions for the dehydration of α-nitroketoximes30,31 do not promote this transformation with JKE-1674. However, we developed a two-step reaction sequence via bis-oxime 39 (Supplementary Fig. 14a) that enables the isolation of a dehydrated analog of JKE-1674, JKE-1777 (40) (Fig. 4a). JKE-1777 is unstable in solution, precluding detailed characterization, but it exhibits infrared absorption characteristic of a nitrile oxide group32–34 and forms dimeric side products consistent with nitrile oxide reactivity32 (41) (Supplementary Fig. 14b).
Figure 4. Dehydration of JKE-1674 yields a nitrile-oxide electrophile that binds GPX4.
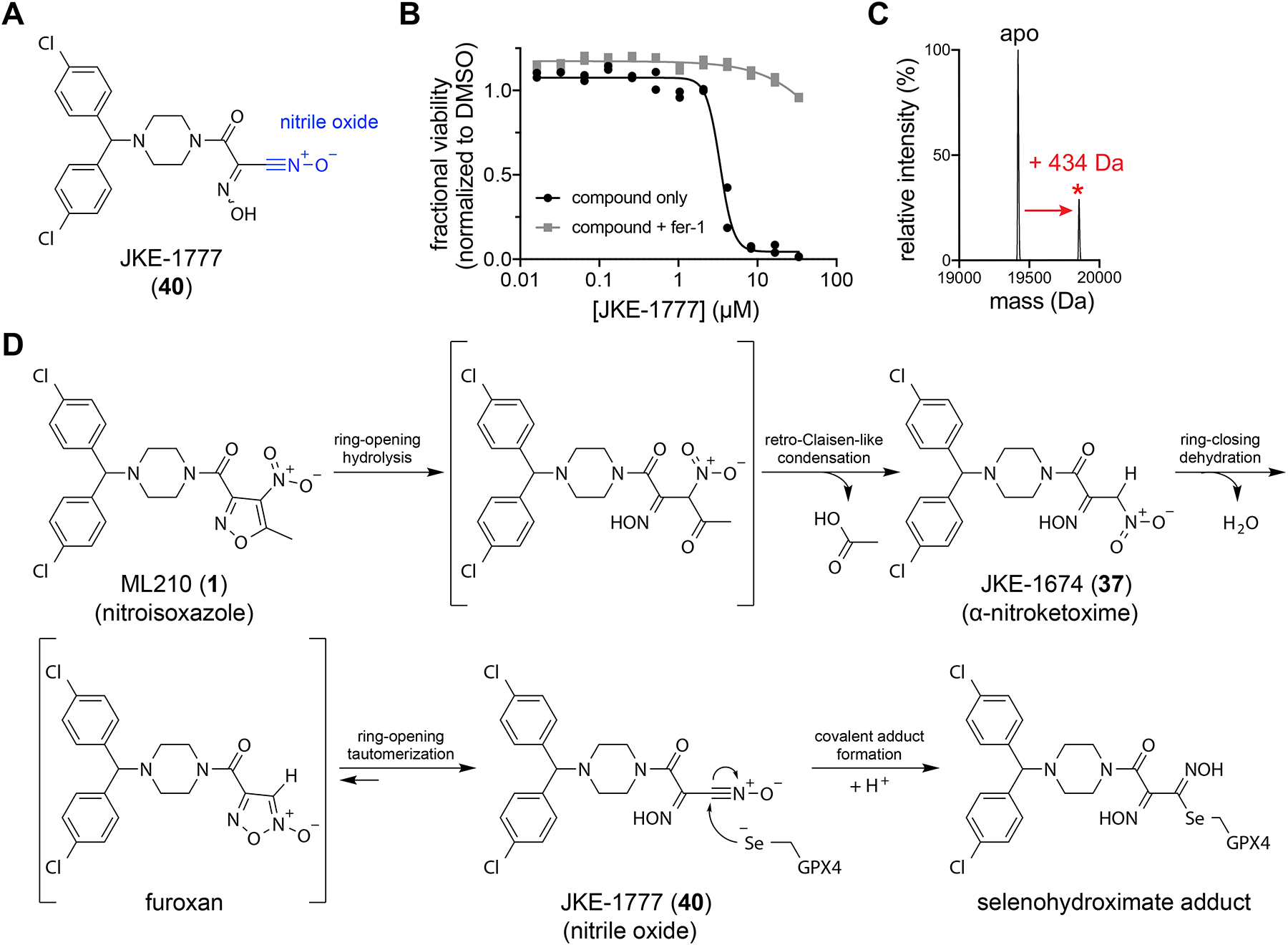
(A) Proposed structure of JKE-1777 (40) with nitrile-oxide group shown in blue. (B) Co-treatment with fer-1 rescues the cell-killing effects of JKE-1777 in LOX-IMVI cells. Data are plotted as two individual technical replicates. (C) JKE-1777 (50 μM, 1 h) is able to form a +434 Da covalent adduct with purified GPX4U46C allCys(−) (5 μM). Adduct peak is marked with a red asterisk (*). (D) Proposed cellular transformation of masked nitrile-oxide GPX4 inhibitors ML210 and JKE-1674 into JKE-1777.
We found that JKE-1777 exhibits cell-killing activity that is rescuable by fer-1 (Fig. 4b), albeit with diminished potency compared to ML210 and JKE-1674 that is likely due to the chemical instability of the nitrile oxide group and inactivity of side products derived from JKE-1777 (41 and 42) (Supplementary Fig. 14b–e). Based on the observation that JKE-1777 reacts with purified GPX4 to yield the same +434 Da adduct that other ML210-derived analogs generate only in cells (Fig. 4c), we hypothesized that the nitrile oxide JKE-1777 is the ultimate ML210-derived electrophile that targets GPX4 in cells. With JKE-1777 in hand, we demonstrated that reaction with a small-molecule thiol generates an adduct containing a thiohydroximate group (43) (Supplementary Fig. 14f) that is consistent with the thiohydroximate adducts of cysteine and glutathione that form in cells treated with ML210 or JKE-1674 (Supplementary Fig. 11e). We therefore conclude that ML210-derived inhibitors likely bind GPX4 to form the analogous selenohydroximate adduct (Fig. 4d).
We next investigated JKE-1777 precursor 39 to determine if the formation of JKE-1777 from JKE-1674 in cells occurs via a two-step reduction-oxidation process. However, the inactivity of 39 (Supplementary Fig. 15a) suggests that the formation of JKE-1777 likely occurs through a distinct process.
To explore alternative cellular activation mechanisms, we performed SAR studies focused on the α-nitroketoxime group of JKE-1674 (Supplementary Fig. 15). We found that both the oxime and nitro groups play crucial roles in enabling GPX4 binding as indicated by compounds 39 and 44–54 (Supplementary Fig. 11). A possible explanation for these results is that dehydration of the α-nitroketoxime initially yields an unstable monosubstituted furoxan heterocycle (Fig. 4d), the formation of which is dependent on the presence of the oxime group. This furoxan then readily undergoes a ring-opening isomerization reaction to yield a nitrile oxide32,33 (Fig. 4d).
Diverse masked nitrile oxides target GPX4
A key finding of our studies is the surprising cellular selectivity of masked nitrile-oxide electrophiles. To explore further the scope of masked nitrile-oxide electrophiles as GPX4 targeting moieties, we investigated JKE-1674 analogs that incorporate structurally distinct nitrile-oxide precursors. We were unable to prepare analogs containing a hydroximoyl chloride group, which is commonly used for in situ preparation of nitrile oxides35. Instead, we found that JKE-1674 can be converted directly into its nitrolic acid analog JKE-1708 (55) (Fig. 5a and Supplementary Fig. 16a,b), which also acts as a nitrile-oxide precursor (Supplementary Fig. 16c)36–38.
Figure 5. Diverse masked nitrile oxides target GPX4.
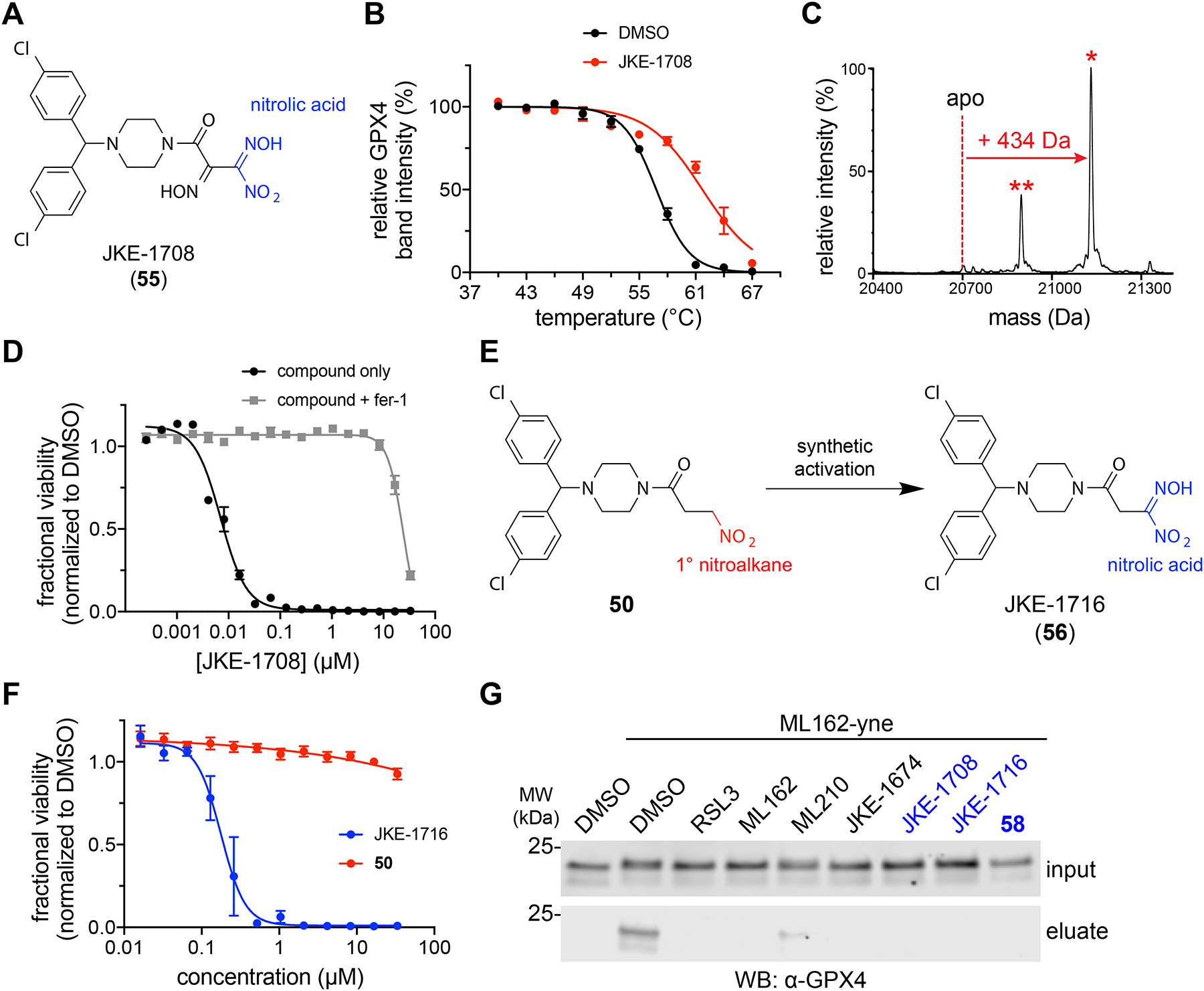
(A) Structure of JKE-1708 (55) with nitrolic acid group shown in blue. (B) GPX4 CETSA with intact cells (LOX-IMVI) treated with JKE-1708 (red, 10 μM, 1 h) reveals thermal stabilization of GPX4 compared to treatment with DMSO (black). Data are plotted as mean ± s.e.m., n ≥ 3 biologically independent samples. DMSO data is reproduced from Fig. 3e. Representative western blots are shown in Supplementary Fig. 18a. (C) JKE-1708 forms covalent adduct when incubated with purified GPX4 protein. Adduct peak is marked with a red asterisk (*). A second peak (denoted with red **) indicates covalent protein adduct after benzhydryl group fragmentation. See also Supplementary Fig. 11c. (D) Co-treatment with fer-1 (1.5 μM) rescues the cell-killing effects of JKE-1708 in LOX-IMVI cells. Data are plotted as mean ± s.e.m., n = 4 technical replicates. See also Supplementary Fig. 16f,g. (E) Inactive nitroalkane 50 can be synthetically transformed into the GPX4-inhibiting nitrolic acid JKE-1716 (56). (F) Viability measurements of cells treated with inactive nitroalkane 50 (red) and active nitrolic acid JKE-1716 (blue). Data are plotted as mean ± s.e.m., n = 4 technical replicates. See also Supplementary Fig. 17a. (G) Pretreatment of LOX-IMVI cells with nitrolic acids (10 μM, 30 min) prevents GPX4 pulldown by ML162-yne. Nitrolic acid-containing compounds are denoted with blue labels. Full-length western blots are shown in Supplementary Fig. 18b.
Assessment of JKE-1708 by CETSA revealed thermal stabilization of GPX4 identical to that induced by ML210 and JKE-1674 (Fig. 5b). Like nitrile oxide JKE-1777, nitrolic acid JKE-1708 does not require cellular activation and reacts spontaneously with purified GPX4 to form the expected +434 Da GPX4 adduct (Fig. 5c). JKE-1708 also reacts with small-molecule thiols to yield thiohydroximate adducts identical to those formed with JKE-1777 directly (Supplementary Fig. 16d) or with ML210 and JKE-1674 in cells (Supplementary Fig. 16e). Most compellingly, JKE-1708 exhibits cell-killing effects that are rescued almost completely by ferroptosis inhibitors, indicating ferroptosis-inducing selectivity similar to ML210 and JKE-1674 (Fig. 5d and Supplementary Fig. 16f,g).
The critical role that the oxime group of JKE-1674 plays in nitrile oxide formation led us to hypothesize that incorporation of a nitrolic acid group into inactive analogs would yield cell-active GPX4 inhibitors. Indeed, several inactive primary nitroalkane analogs (50, 53, and 54) could be transformed into potent nitrolic acid-containing GPX4 inhibitors (56–58) (Fig. 5e–g and Supplementary Fig. 17a–d). This ability of nitrolic acids to selectively induce ferroptosis is unexpected given the reactivity of this group, which spontaneously generates nitrile oxides in aqueous solutions36. Indeed, these results suggest that nitrile-oxide electrophiles may have intrinsic selectivity for GPX4.
Our discovery of GPX4-targeting nitrolic acids reinforces insights from our SAR studies of JKE-1674 (Supplementary Fig. 15), which indicate that the oxime group is critical for the cellular conversion of the α-nitroketoxime in JKE-1674 into an active electrophile. While either the nitrile oxide of JKE-1777 or its proposed furoxan isomer (Fig. 4d and Supplementary Fig. 13b) could plausibly act as an electrophile, other nitrile-oxide precursors that are incapable of forming monosubstituted furoxans (Fig. 5e and Supplementary Fig. 17b) are also able to induce ferroptosis selectively (Fig. 5f and Supplementary Fig. 17c,d) through covalent GPX4 inhibition (Fig. 5g). Together, our findings are consistent with a nitrile oxide as the most likely ML210-derived electrophile and support the crucial role of the oxime moiety of JKE-1674 in the formation of this electrophilic species.
Our findings also suggest possible approaches to modulate the reactivity of nitrile-oxide precursors. For example, a thiohydroximate analog (43) derived from the reaction between thiophenol with either JKE-1777 or JKE-1708 (Supplementary Fig. 14f and Supplementary Fig. 16d) exhibits cell-killing activity rescuable by fer-1 co-treatment (Supplementary Fig. 17e). Thiohydroximate 43 plausibly reacts via loss of thiophenol in a manner similar to nitrolic acids, which lose HNO2 to generate a reactive nitrile-oxide electrophile (Supplementary Fig. 16c)36–38. These observations suggest that it may be possible to tune the reactivity and off-target effects of these compounds through careful selection of the leaving group.
Masked nitrile oxides are useful tool compounds
Our efforts to elucidate the mechanism of action of ML210 have identified a family of previously uncharacterized masked nitrile oxides that act as covalent GPX4 inhibitors. This class of compounds is structurally distinct from previously reported GPX4 inhibitors and represents a novel electrophilic chemotype capable of selectively targeting GPX4 in cells. These structurally diverse masked nitrile oxides exhibit a range of chemical reactivities and physiochemical properties, providing a new wealth of cellular and biochemical tool compounds for studying GPX4 inhibition and ferroptosis induction.
To encourage the experimental adoption of key masked nitrile-oxide GPX4 inhibitors (i.e. nitroisoxazoles, α-nitroketoximes, and nitrolic acids) as cellular probes of GPX4, we now detail their use in cellular assays relevant to ferroptosis. We confirmed that these compounds share a pattern of cell killing with chloroacetamide GPX4 inhibitors and genetic loss of GPX439 across a range of cancer cell lines (Fig. 6a). However, a key distinction of masked nitrile-oxide GPX4 inhibitors compared to chloroacetamide inhibitors is the far greater degree to which ferroptosis inhibitors (fer-1, zileuton40,41) rescue their lethal effects (Fig. 6b). As tools to study ferroptosis biology, masked nitrile-oxide GPX4 inhibitors enable the isolation of the cellular circuitry of ferroptosis from that of other cell-death modes. This point is illustrated by the results of genome-scale CRISPR suppressor screens1, performed side-by-side with RSL3 and ML210, to identify genes necessary for cell death by ferroptosis. While selection of CRISPR library-infected cells with RSL3 recovered three of the hits identified by selection with ML210, three others uncovered by ML210 failed to score in the RSL3 treatment condition, likely overwhelmed by off-target, non-ferroptotic effects of RSL3 (Fig. 6c and Supplementary Tables 1,2).
Figure 6. Profiling of structurally diverse GPX4 inhibitors in cellular and pharmacokinetic assays.
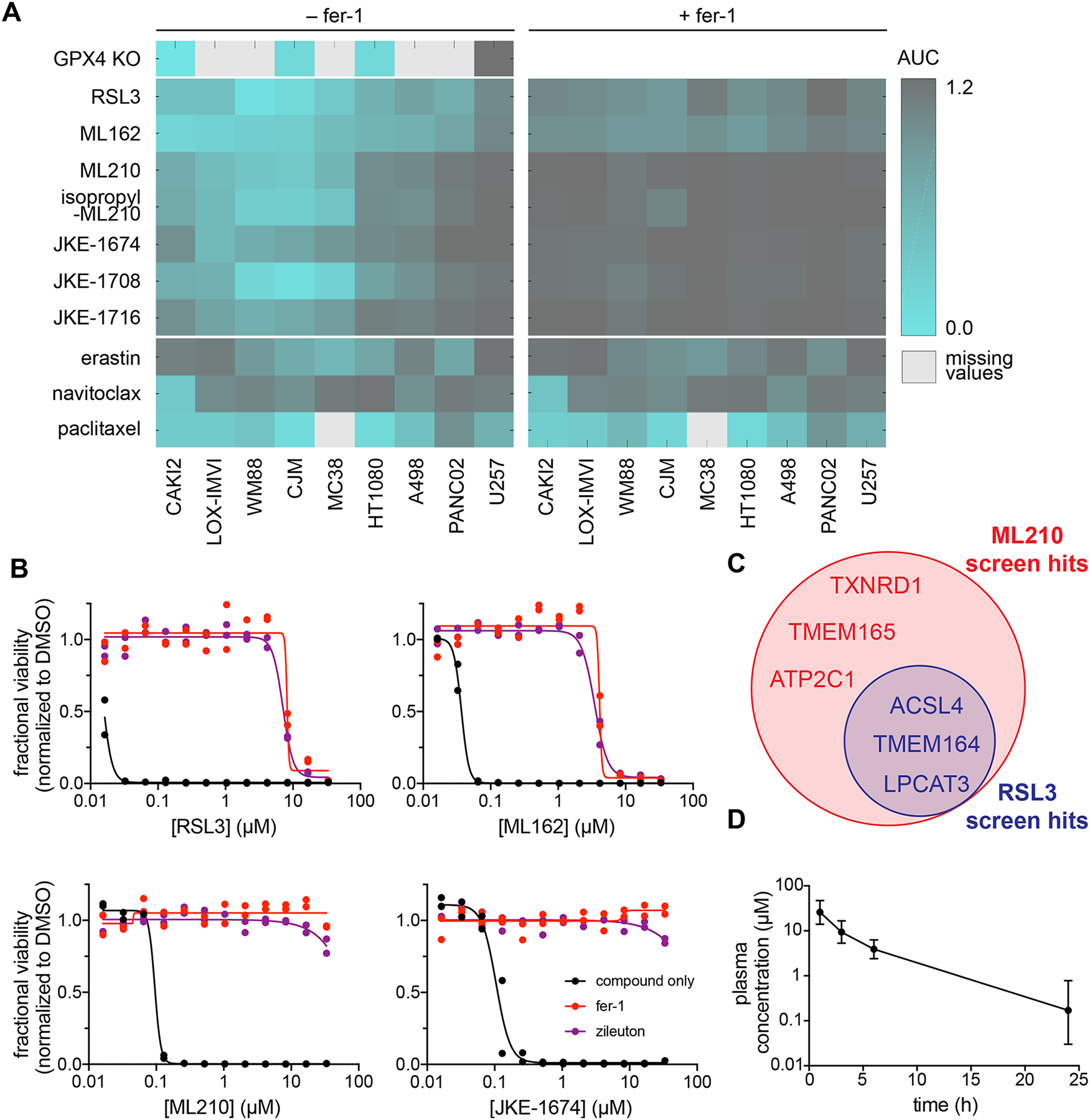
(A) GPX4 inhibitors bearing chloroacetamide, nitroisoxazole, and α-nitroketoxime warheads exhibit a similar pattern of cell killing across a range of cancer cell lines relative to control lethal agents. Nitrile-oxide precursors show enhanced rescuability by fer-1 (1.5 μM) relative to chloroacetamide GPX4 inhibitors. WM88, LOX-IMVI, CJM and U257 are human melanoma cell lines. CAKI2 and A498 are human renal cell carcinoma cell lines. HT1080 is a human fibrosarcoma cell line. MC38 is a mouse colon cancer cell line and PANC02 is a mouse pancreatic cancer cell line. (B) Compared to GPX4-targeting chloroacetamides, ML210 and JKE-1674 exhibit fewer off-target effects in LOX-IMVI cells that cannot be rescued by ferroptosis inhibitors. Data are plotted as two individual technical replicates. (C) Summary of hits identified in ML210 and RSL3 genome-wide CRISPR suppressor screens. See also Supplementary Tables 1,2. (D) In vivo PK assessment of JKE-1674 in SCID mice. Plasma concentration of JKE-1674 was determined by LCMS after oral administration of JKE-1674 (50 mg/kg) aver a 24-hour period. Data are plotted as mean ± s.d., n = 4 biologically independent samples.
The masked reactivity of nitrile-oxide precursors also has the potential to overcome key pharmacokinetic liabilities of highly-reactive alkyl chloride GPX4 inhibitors4,9, which we found do indeed exhibit poor physiochemical and pharmacokinetic properties (Supplementary Table 3). Attempts to improve these properties by using alternate warheads with attenuated electrophilic groups, such as clinically-relevant acrylamides, produced inactive analogs (59–64) (Supplementary Fig. 19). In contrast, both ML210 and JKE-1674 exhibit far greater stability than chloroacetamide inhibitors (Supplementary Table 3). Additionally, the α-nitroketoxime group of JKE-1674 improves upon the solubility of ML210 and chloroacetamide inhibitors, crossing the threshold necessary for formulation and dosing in animals. Indeed, even without medicinal chemistry optimization, JKE-1674 can be detected in the serum of mice dosed orally with the compound (Fig. 6d). It is possible that systematic optimization of JKE-1674 will yield further-improved tool compounds for understanding the basic biology of GPX4 and therapeutic potential of ferroptosis induction.
Alongside a discussion of the advantages of masked nitrile-oxide GPX4 inhibitors in biological experiments, we also note several caveats. The first relates to diminished potency of this class of inhibitors in certain contexts relative to chloroacetamide GPX4 inhibitors (Fig. 6a). The second concerns potential discrepancies in the activity of masked nitrile-oxide versus chloroacetamide GPX4 inhibitors in one of the two mouse cancer cell lines that we have examined (Fig. 6a), an observation that warrants further investigation. This and other yet-to-be-discovered nuances between different GPX4-inhibiting chemotypes argues for the inclusion of several chemotypes in all biological experiments probing ferroptosis. A final caution relates to the need for careful storage and handling of JKE-1674, which can decompose if stored for prolonged periods at room temperature as a solution in DMSO.
DISCUSSION
Drugs that form covalent bonds with their targets constitute important medicines in pharmacopeia (e.g. penicillin, aspirin, omeprazole) but historically have raised concerns about potential lack of selectivity. Interest in covalent binders has recently undergone a resurgence encouraged by identification of selective and tunable covalent reactive groups (e.g. acrylamides) and development of FDA-approved drugs (e.g. afatinib, ibrutinib, neratinib).
Chloroacetamide groups feature prominently in covalent chemical probes and small-molecule tools to assess proteome ligandability. In particular, recent studies have shown that a significant proportion of the ‘undruggable’ proteome contains nucleophilic residues that are chemically accessible via chloroacetamide groups42–44. Unfortunately, chloroacetamide-containing compounds are poor starting points for further development given their promiscuity, instability, and low bioavailability45. Therefore, therapeutic translation of the many tantalizing leads generated by ligandability studies42–44 requires substitution of the chloroacetamide warhead with a more drug-like electrophile. While acrylamides or other attenuated electrophiles may be appropriate alternatives in certain cases45,46, in other contexts they may be insufficient substitutes for chloroacetamide groups, as we have found to be the case for GPX4 inhibitors.
In this manuscript, we describe nitrile-oxide electrophiles as effective substitutes for chloroacetamide groups within GPX4 inhibitors. Masked nitrile oxides exhibit remarkable selectivity for GPX4 and diminished off-target effects compared to chloroacetamide-based GPX4 inhibitors. This selectivity is unexpected given that nitrile oxides are regarded as one of the most reactive organic functional groups. While the requirement for cellular activation may play a role in this cellular selectivity, we speculate that the more important contribution derives from the intrinsic reactivity properties of the nitrile oxides themselves. By virtue of their transient nature, we expect that these reactive moieties do not persist in cells for prolonged periods of time and thereby avoid extensive covalent interactions with the proteome. Similar kinetic selectivity arising from metabolically labile covalent inhibitors has been reported for cysteine-targeting fumarate esters47 and amine- and carboxylate-targeting isoxazolium48 probes.
The specific mechanisms underlying cellular activation of ML210 remain unclear. While ML210 seemingly requires intact cells to target GPX4, we have not found direct evidence to support the role of any specific enzyme(s) in this process. Moreover, the lack of discrepancy between ML210 cell killing and that of chloroacetamide GPX4 inhibitors across a large panel of cell lines (Fig. 1b) suggests that even if specific enzymes play a role in ML210 activation, they are ubiquitous across cancer cell lines. Attempts to uncover candidate enzymes from ML210 suppressor screens using genome-scale CRISPR perturbations did not yield any genes that are unique to ML210 and are not general ferroptosis regulators. It should be noted that this same result could be expected if the putative enzymes underlying ML210 activation are either essential to cell viability or comprise a class of enzymes with multiple members having redundant and overlapping functions. While CYP enzymes have been reported previously to promote the formation of nitrile oxides in the context of natural product biosynthesis49 and drug metabolism50, in principle, the chemical transformations required to activate ML210 do not necessarily require enzymes.
We hope these results, which highlight the sometimes-surprising differences between in vitro reactivity and cellular selectivity of electrophilic compounds, will spur a re-examination of conventional wisdom and lead to an expansion of the roster of covalent warheads available for tool compounds and therapeutics aiming to target currently undruggable proteins. Nevertheless, the biological utility of nitrile-oxide electrophiles is predicated on the unique electrophile-masking elements present in the ML210 nitroisoxazole chemical pathway. These prodrug features are necessary for the isolation, storage, and delivery of nitrile-oxide electrophiles to cells, and in certain cases even bestow GPX4 inhibitors with pharmacokinetic properties and bioavailability that far surpass those of unmasked electrophiles such as chloroacetamides. The unique GPX4-targeting ability of masked nitrile oxides is further supported by our discovery that diacylfuroxans, structurally unrelated to ML210, inhibit GPX4 via nitrile-oxide electrophiles generated through a distinct unmasking mechanism34.
The toolkit of GPX4 inhibitors uncovered by our mechanistic studies represents an unprecedented wealth of diverse GPX4-targeting chemotypes (nitroisoxazoles, α-nitroketoximes, and nitrolic acids). These compounds promise to overcome a major shortcoming of the GPX4-biology field, namely, reliance on a single non-specific chemotype (chloroacetamides) to perform and interpret biological experiments. We therefore encourage scientists probing GPX4 and ferroptosis biology to incorporate into their experimental designs the chemically diverse GPX4 inhibitors we have illuminated. This approach promises to mitigate chemotype-specific off-target effects and enable biological discoveries that require highly selective ferroptosis induction such as that achieved by masked nitrile-oxide GPX4 inhibitors.
Finally, we expect that the numerous tool compounds, mechanistic insights, cellular target engagement assays, and biochemical data we make available will be catalytic for the development of improved GPX4-inhibiting small molecules by the academic and pharmaceutical communities.
ONLINE METHODS
Mammalian cell lines.
LOX-IMVI (melanoma), CJM (melanoma), WM88 (melanoma), KP4 (pancreatic cancer), HCC4006 (lung adenocarcinoma), and HT1080 (fibrosarcoma) cell lines were procured from the Broad Institute Biological Samples Platform. A498 (renal cell carcinoma), CAKI2 (renal cell carcinoma), and U2OS (osteosarcoma) cells were procured from ATCC. PANC02 (pancreatic cancer) and MC38 (colon cancer) cell lines were a gift from the Dougan Lab (DFCI). HEK293–6E cells used for protein expression were licensed and received directly from Prof. Yves Durocher (National Research Council, Canada) and maintained in FreeStyle F17 Expression Medium (ThermoFisher). Caco-2 cells were purchased from DSMZ. All cell lines were maintained at 37 °C under a humidified 5% CO2 atmosphere according to supplier recommendations.
Chemical synthesis.
Synthesis and analytical characterization of all compounds are detailed in the Supplementary Note.
Cell viability assays.
Cell viability experiments were performed by seeding 1000 cells/well (30 μL volume) in opaque white 384-well plates (Corning). Cells were allowed to adhere for 24 h after which they were exposed to compounds for 72 h. DMSO stock solutions of compounds were added to cells using an CyBio Well Vario liquid dispenser (Analytik Jena AG). Cellular ATP levels were measured using CellTiter-Glo (Promega) as a surrogate for viability. Rescue experiments were performed using ferrostatin-1 (fer-1; 1.5 μM), liproxstatin-1 (lip-1; 1 μM), deferoxamine (DFO; 50 μM), or zileuton (10 μM) added to cells at the time of seeding. All ferroptosis inhibitors were purchased from Sigma-Aldrich in ≥ 92.5% purity.
Western blotting.
Cell lysates were diluted with SDS sample buffer (Boston BioProducts, reducing, 6x) and heated to 95 °C for 5 minutes. Samples (typically 30 μg total protein) were separated by SDS/PAGE (using buffers, gels, and equipment from Invitrogen) and transferred to a nitrocellulose membrane using an iBlot 2 Gel Transfer Device (ThermoFisher). Antibodies for GPX4 (ab41787, Abcam) and ACTB (sc-47778, SCBT) were used at a 1:1000 dilution. IRDye secondary antibodies (680RD and 800CW, LI-COR Biosciences) were used at a 1:10000 dilution. Blots were imaged using an Odyssey imaging system (LI-COR Biosciences).
Lipid peroxidation assay by imaging.
LOX-IMVI cells were seeded at 5,000 cells per well in a CellCarrier Ultra 96-well plate (Perkin-Elmer) in 150 μL of RPMI medium supplemented with 10% FBS, 1% pen-strep, 5 μg/mL plasmocin, and 1 μM fer-1 (where indicated). Cells were incubated for 24 h at 37 °C and then treated with the indicated compounds or DMSO (90 min, 37 °C). During the last 30 min of incubation, 60 nM DRAQ7 (Abcam), 1 μg/mL Hoechst 33342 (ThermoFisher), and 1 μM BODIPY 581/591 C11 (ThermoFisher) dyes were added. Cells were imaged using an Opera Phenix High-Content Screening System (Perkin-Elmer) equipped with 405, 488, 560, and 647 nm lasers. Image analysis was performed with Harmony High-Content Imaging and Analysis software (Perkin-Elmer).
Lipid peroxidation assay by flow cytometry.
U2OS cells were seeded at 15,000 cells per well in 96-well plates. After 48 h, culture media was replaced with 200 μL media containing either DMSO or the indicated GPX4 inhibitor (10 μM) and 1 μM fer-1 (where indicated). Cultures were incubated at 37°C for 2 h. Thirty minutes before the end of the incubation period, 10 μM BODIPY 581/591 C11 (Molecular Probes #C10445) was added to cells. Cells were harvested in 200 μL PBS + 0.1% BSA and subjected to flow cytometry analysis (BD FACSCanto II).
Preparation of phosphatidylcholine hydroperoxide (PCOOH).
Preparation of PCOOH was accomplished by adapting literature procedures describing lipoxygenase-mediated peroxidation of phosphatidylcholine (PC)4,51,52. A solution of 16:0/18:2 PC (100 mg; Avanti Polar Lipids) in methanol (1 mL) was dissolved in 50 mM borate buffer (170 mL, containing 2.2 w/v% sodium deoxycholate, pH 9.0). Soybean lipoxygenase (2.5 million units; Cayman Chemical) was added and the mixture was stirred at room temperature for 30 minutes. The reaction was acidified with 10 N HCl and extracted with dichloromethane. The combined fractions were concentrated and filtered through a silica plug using chloroform/methanol/acetic acid/water (100:75:7:4, v/v) solvent. Fractions containing PCOOH were identified by TLC with N,N,N’,N’-tetramethyl-p-phenylenediamine (TPD; Sigma-Aldrich) stain according to a reported protocol53. Combined fractions were dried, dissolved in methanol, and filtered through a 0.2 μm filter. The methanolic PCOOH solution was stored at −20 °C until use.
GPX4 activity assay.
A mass spectrometry-based GPX4 enzymatic activity assay was adapted from a previously described procedure4. LOX-IMVI cells were treated with indicated compounds (10 μM) or DMSO for 1 h at 37 °C. Cells were washed with PBS and lysed by freeze-thaw method (x3) in GPX4 reaction buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1 mM EDTA, 0.1 mM DFO; pH 7.4). Lysates were cleared by centrifugation (10 min, 20000 × g, 4 °C) and total protein concentration was adjusted to 1.67 mg/mL. Typical enzymatic activity assay mixtures were prepared as follows: 200 μL lysate (1.67 mg/mL in GPX4 reaction buffer), 2 μL of PCOOH in MeOH, and 20 μL GSH solution (100 mM; ~5 mM final concentration). Reactions were vortexed briefly and incubated at 37 °C for 15 min. Reaction mixtures were then extracted using 250 μL of a 2:1 chloroform/methanol (v/v) solution. Extracts were dried under a stream of nitrogen and reconstituted in methanol before analysis.
LC-MS analysis was performed with Acquity RP UPLC system coupled to a Xevo G2XS QToF mass spectrometer (Waters). Reconstituted extract was separated on a Waters Acquity RP UPLC BEH-C18 column (2.1 × 50 mm; 1.7 μm particle size; 45 °C). Mobile phase consisted of 10 mM aqueous ammonium acetate (solvent A) and 95:5 acetonitrile/10 mM ammonium acetate (solvent B). The total run time was 8 minutes. UPLC eluate was introduced into the mass spectrometer by positive mode electrospray ionization. Source settings were 120 °C, 50 V cone voltage, 1 kV capillary voltage, 500 °C desolvation temperature, and 1100 L/h desolvation gas flow. Mass spectrometry experiments were performed in sensitivity mode with a resolution of 20,000 and a mass accuracy of <1 ppm. The lockmass (Leu-Enk, m/z 556.2771) was infused continuously at 5 μL/minute and sampled every 15 seconds. MassLynx and TargetLynx software (Waters) were used for analysis of mass spectra, PCOOH identification, and chemical formula confirmation analysis.
Cellular thermal shift assay (CETSA).
For intact-cell CETSA experiments, cells were pretreated with 10 μM compound or DMSO control (0.1%, v/v) for 1 h at 37 °C. Media was then aspirated and cells were washed with PBS (pH 7.4). Adherent cells were detached from the flask with trypsin-EDTA and pelleted by centrifugation (500 × g, 5 min). Cells were aliquoted into PCR tubes (50 μL volume, ~1 million cells/condition) for heating at different temperatures (typically 40–67 °C in 3 °C increments) in a thermocycler for 3 minutes. Samples were allowed to cool to room temperature for an additional 3 minutes. Cells were lysed by either three freeze-thaw cycles in liquid nitrogen, or by the addition of Triton X-100 solution (1% final TX-100 volume, PBS pH 7.4) and subsequent incubation on ice for 20 minutes with occasional vortexing. After lysis, cells were centrifuged (20 minutes at 20,000 rcf, 4 °C) to remove insoluble material. The soluble fraction was carefully separated and diluted with 6x SDS loading buffer for SDS-PAGE and western blotting analysis.
For lysate CETSA experiments, lysate from untreated cells was prepared as described in the intact-cell CETSA protocol and diluted with PBS (pH 7.4) to a total protein concentration of 1 mg/mL. Samples were treated with 10 μM compound (or 0.1% v/v DMSO control) for 1.5 h at 37 °C. After compound treatment, samples were aliquoted (50 μL per sample) into PCR tubes processed as described above for intact-cell CETSA experiments.
Cell treatment for GPX4 pulldown and ABPP experiments.
LOX-IMVI cells were seeded in 6-well plates in RPMI media supplemented with 10% FBS. Alkyne affinity probes were added to the cells and incubated for 1 h at 37 °C (10 mM stocks in DMSO, final alkyne concentration = 10 μM). Media was removed and cells were washed once with ice-cold PBS (pH 7.4). Cells were then lysed in PBS containing 1% Triton X-100 and protease inhibitors (Roche Complete) for 20 min on ice. Samples were cleared by centrifugation (20,000 × g, 4 °C). Total protein content of lysates was assessed with a Pierce Coomassie Plus Bradford assay kit (ThermoFisher) and adjusted to 2 mg/mL.
For lysate pulldown experiments, LOX-IMVI cell lysates were prepared as described above from untreated cells and adjusted to a total protein concentration of 2 mg/mL. Lysates were treated with the indicated alkyne probe (10 μM, 1 h) or DMSO at ambient temperature and then used immediately for subsequent click chemistry reactions.
Sample processing for GPX4 pulldown assay.
Lysates were subjected to copper-catalyzed azide-alkyne cycloaddition (CuAAC) conditions with azide-PEG3-biotin conjugate (Sigma Aldrich). Typical reactions were performed with a final volume of 120 μL consisting of 100 μL lysate (2 mg/mL; final concentration = 1.67 mg/mL), 2.4 μL SDS (10%; final concentration = 0.2%), 2.4 μL azide-PEG3-biotin conjugate (Sigma-Aldrich; 5 mM in DMSO; final concentration = 100 μM), and 15.2 μL of catalyst mix (final concentration = 1.3 mM Cu2SO4, 1.3 mM TCEP, and 75 μM TBTA). The catalyst mix stock was prepared by mixing 3 parts tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]-amine (TBTA; Sigma-Aldrich; 1 mM in 1:4 DMSO/tBuOH), 1 part 50 mM Cu2SO4 in water, and 1 part tris(2-carboxyethyl)phosphine (TCEP; Sigma-Aldrich; 50 mM in water, pH 7.0). After addition of all components, CuAAC reactions were vortexed and allowed to react at ambient temperature for 1 h and then diluted with 120 μL 0.2% SDS in PBS. A 40 μL aliquot was removed and quenched with 6x SDS sample buffer as an input control. Pierce highcapacity streptavidin agarose beads (ThermoFisher) were added to the remaining sample and rotated overnight at 4 °C. Beads were then separated by centrifugation and washed sequentially with 1% SDS (3 × 1 mL) and PBS (2 × 1 mL). Proteins were eluted by boiling the beads in 75 μL of 2x SDS sample buffer for 10 minutes and analyzed by SDS-PAGE and western blotting.
Sample processing for ABPP experiments.
Lysates from LOX-IMVI cells treated with alkyne probes were prepared as described above for the GPX4 pulldown assay. The CuAAC protocol described for pulldown assays was used to conjugate samples with IRDye 680RD azide (LI-COR Biosciences, Lincoln, NE). Typical reactions consisted of 100 μL lysate (2 mg/mL; final concentration = 1.67 mg/mL), 2.4 μL SDS (10%; final concentration = 0.2%), 3.6 μL IRDye 680RD azide (1 mM in DMSO; final concentration = 30 μM), and 14 μL of catalyst mix (final concentration = 1.2 mM Cu2SO4, 1.2 mM TCEP, and 70 μM TBTA). After addition of all components, CuAAC reactions were vortexed and allowed to react at ambient temperature for 1 h. The reaction was quenched by addition of 1.2 mL acetone (precooled to −20 °C) and incubated overnight at −20 °C. The sample was then centrifuged (20,000 × g, 10 min, 4 °C) to pellet precipitated proteins. The supernatant was removed and the light blue pellets were washed twice with 600 μL of ice-cold methanol. After addition of methanol, the pellets were resuspended in an ultrasonic bath and centrifuged as described above. After the final wash, pellets were allowed to air dry to remove excess methanol. Pellets were suspended in SDS sample buffer (1x), heated at 95 °C for 5 min, and separated by SDS-PAGE. In-gel fluorescence was measured using an Odyssey imaging system (LI-COR Biosciences). Gels were then stained with InstantBlue Coomassie stain (Expedeon) and imaged to assess total protein loading.
Mass spectrometry-based proteomics.
LOX-IMVI cells (T75 flask, ~90% confluent) were treated with ML210-yne (10 μM, 1 h) or DMSO. Sample preparation and protein enrichment was performed as described above. Enriched proteins were identified by LC-MS/MS as described previously34.
Plasmid, expression and purification of recombinant GPX4 proteins.
The described GPX4_WT_FLAG DNA construct was synthesized (Life Technologies) and encodes: 5’ (Pst I) and 3’ (Bcl I) restriction sites for subcloning, a Kozak sequence with a start codon preceding a N-terminal Flag tag fused in-frame to the GPX4 region M1 to F170 (Uniprot P36969, DNA sequence X71973). A stop codon and SECIS element (selenocysteine insertion sequence) follow codon F170. Three different SECIS elements were tested in the expression cassette: a previously described chimeric element54, the human element from GPX4 X71973–1, and the element from SelN from NM_206926–1. All were found to be equally efficient in expression of GPX4 (U46) when co-expressed with SBP2 (SECIS-binding protein2) in HEK293–6E cells as described below. The chimeric SECIS was used for subsequent experiments. Additionally, a DNA construct was synthesized encoding full length rat SBP2 (Q9QX72) with a Kozak sequence as well as EcoRI and BamHI sites at the 5’ and 3’ regions, respectively. Subcloning of both DNA constructs into the mammalian expression vector pTT555 was achieved using restriction endonuclease sites and resulted in the final expression vectors GPX4_WT_FLAG–pTT5 and SBP2-pTT5. For protein expression, the plasmid GPX4_WT_FLAG-pTT5 together with the SBP2-pTT5 (in a plasmid ratio 4:1, w/w) was co-expressed transiently in HEK293–6E cells in the presence of 1 μM sodium selenite in culture vessels with various volumes up to 10 L according to a described procedure55. For preparative protein purification the cleared lysate supernatant (prepared in lysis buffer 50 mM sodium phosphate, pH 7.4, 300 mM NaCl, 1 mM DTT and 0.1% NP-40 supplemented with Roche Complete protease inhibitor cocktail) from transfected cells was applied to a column with anti-FLAG M2 Agarose (Sigma) and subsequently the relevant eluate fractions were concentrated and further purified by size exclusion chromatography (50 mM sodium phosphate (pH 7.4), 300 mM NaCl, 1 mM DTT)). The resulting pooled monomeric GPX4 fractions were concentrated and stored at −80 °C
The expression and purification of GPX4U46C allCys(−) protein was accomplished as described previously34.
In-cell GPX4 mass spectrometry binding assay.
HEK293–6E cells were transfected with GPX4_WT_FLAG-pTT5/SPB2-pTT5 in 24-well plates. Cells were harvested 72 h following seeding and compounds were added 1, 4 or 24 h before cell harvest. For each time-point, compounds were added at the following concentrations: 1, 10, or 20 μM. Viability of transfected and compound-treated cells was monitored. Cells were harvested by centrifugation and lysed in lysis buffer (a pH 7.4 solution of 50 mM sodium phosphate, 300 mM NaCl, and 0.1% NP-40 supplemented with Roche Complete protease inhibitor cocktail). GPX4 was purified by anti-FLAG chromatography as described for the large-scale preparation of FLAG-GPX4WT. Denaturing MS analysis of purified GPX4 samples was performed with a SYNAPT G2-S quadrupole time-of-flight mass spectrometer connected to a nanoAcquity UPLC system (Waters). Samples were loaded on a 2.1 × 5 mm mass prep C4 guard column and desalted with a short gradient (3 min) of increasing concentrations of acetonitrile at a flow rate of 100 μL/min. Spectra were analyzed by using MassLynx v4.1 software (Waters) and deconvoluted with the MaxEnt1 algorithm.
GPX4 mass spectrometry binding assay with purified GPX4.
Recombinant FLAG-GPX4WT protein (10 μM in pH 7.4 buffer containing 50 mM sodium phosphate and 300 mM NaCl) was incubated with 100 μM compound (1% v/v final DMSO concentration) at room temperature for 2 h. The reaction was quenched by adding 1 μL 5% v/v TFA to 15 μL reaction volume and was subjected to LC-MS analysis as described for the in-cell GPX4 MS binding assay. Binding experiments with GPX4U46C allCys(−) were performed as described previously34.
Generation of cell lines expressing 3xFLAG-GPX4 mutants.
Replacement of the GFP sequence in the pBabe-puro GFP-GPX4-cyto expression vector (gift from the Stockwell lab, Columbia University)4 with a 3xFLAG sequence (MDYKDHDGDYKDHDIDYKDDDDK) was performed with Q5 Site-Directed Mutagenesis Kit (New England Biolabs). Cysteine (U46C) and alanine (U46A) active-site mutants were obtained by site-directed mutagenesis of codon TGA to TGC and GCC, respectively. The 3x-FLAGGPX4U46C and 3xFLAG-GPX4U46A constructs were stably introduced into LOXIMVI cells using retroviral delivery and selection with puromycin.
Metabolite identification.
LOX-IMVI cells were cultured in Williams’ medium E without FCS in the presence of ferrostatin-1 (1 μM) for up to 48 h. Reactions were started by adding ML210 (10 μM) to the cells. Aliquots were stopped at pre-determined time points (0, 2, 6, 24 and 48 h) by addition of acetonitrile and subsequent centrifugation of precipitated material. Supernatants were analyzed by HPLC-DAD-HRMS to generate metabolite profiles. Metabolite structures were elucidated by HPLC-MS/MS. Metabolite identification experiments with cells treated with JKE-1674 (10 μM) and JKE-1708 (10 μM) were performed as described for ML210.
Physiochemical characterization of GPX4 inhibitors.
Compound solubility, distribution coefficient (logD) values, and pH stability were determined as described previously56,57. Stability of compounds to small-molecule thiols was determined according to reported procedures using LC-UV-MS34 or HPLC-UV58.
Pharmacokinetic characterization of GPX4 inhibitors.
The metabolic stabilities of compounds in plasma, microsomes, and hepatocytes were assessed by LC-MS/MS or HPLC-UV according to reported procedures56,57. Membrane permeability of compounds was measured in Caco-2 cells as described previously56,57. Plasma samples were purchased from Sigma-Aldrich and Charles River Laboratories and microsomes were purchased from Sekisui XenoTech. Rat hepatocytes were isolated freshly from Han/Wistar rats via a two-step perfusion method56.
Pharmacokinetic (PK) studies in mice.
To determine the pharmacokinetic properties of JKE-1674 in SCID mice (Janvier Labs) after oral administration, blood samples for analysis of plasma were taken from three animals/time point at 1, 3, 6 and 24 h after single oral dose of JKE-1674 at 50 mg/kg formulated in PEG400/Ethanol (90/10, v/v). Blood was collected into tubes containing lithium heparin, centrifuged to isolate plasma, precipitated with acetonitrile (1/5, v/v) and analyzed by LC-MS/MS.
Toxicity assessment in mice.
Tolerability of JKE-1674 was assessed preceding pharmacokinetic measurements. Over a period of 7 consecutive dosing days, average body weight loss in mice which received 50 mg/kg active compound did not exceed 10%. Doses higher than 50 mg/kg were not tolerated in this experimental setting.
Data availability.
The data that support the findings of this study are available from the corresponding authors upon request.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Virendar Kaushik for assistance with intact protein mass spectrometry experiments; Michael Palte for assistance with preparing phosphatidylcholine hydroperoxide; Bogdan Budnik and Renee Robinson for assistance with proteomics; William Ho and Suvruta Iruvanti for helpful comments on this manuscript. This work was supported by grants from the National Institutes of Health (R01GM038627 and R35GM127045) to S.L.S. It was also supported in part through a collaboration between the Broad Institute and Bayer AG.
Footnotes
COMPETING INTERESTS STATEMENT
S.L.S. declares no conflicts associated with this research. A complete accounting of his outside professional activities, including a disclosure statement and links to the governing conflict of interest policies, are available at https://chemistry.harvard.edu/people/stuart-l-schreiber. P.A.C. is an advisor to Pfizer, Inc. D.M., A.H., K.Z., M.N., V.B., R.C.H., S.G., S.Ch., R.N., and A.L.E. are employed by Bayer AG.
REFERENCES
- 1.Viswanathan VS et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hangauer MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsoi J et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang WS et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas JP, Geiger PG, Maiorino M, Ursini F & Girotti AW Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim. Biophys. Acta - Lipids Lipid Metab 1045, 252–260 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Kühn H & Borchert A Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic. Biol. Med 33, 154–172 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Scheerer P et al. Structural Basis for Catalytic Activity and Enzyme Polymerization of Phospholipid. Biochemistry 46, 9041–9049 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Borchert A et al. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1863, 1095–1107 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Yang WS et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci 113, E4966–E4975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimada K et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol 12, 497–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang WS & Stockwell BR Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol 15, 234–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakamoto K et al. Discovery of GPX4 inhibitory peptides from random peptide T7 phage display and subsequent structural analysis. Biochem. Biophys. Res. Commun 482, 195–201 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Jiang C, Chen R, Pandey A, Kalita B & Duraiswamy AJ US 2019/0263802 A1. 1–292 (2019).
- 14.Weïwer M et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorganic Med. Chem. Lett 22, 1822–1826 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seashore-Ludlow B et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rees MG et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol 12, 109–116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu A et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaschler MM et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol 14, 507–515 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon SJ et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skouta R et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc 136, 4551–4556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zilka O et al. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci 3, 232–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molina DM et al. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 341, 84–87 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Gao J et al. Selenium-Encoded Isotopic Signature Targeted Profiling. ACS Cent. Sci 4, 960–970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedmann Angeli JP et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trefzer C et al. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-beta-D-ribose 2’-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc 132, 13663–13665 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Patterson S & Wyllie S Nitro drugs for the treatment of trypanosomatid diseases: Past, present, and future prospects. Trends Parasitol. 30, 289–298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J et al. Elucidation of a novel bioactivation pathway of a 3,4-unsubstituted isoxazole in human liver microsomes: Formation of a glutathione adduct of a cyanoacrolein derivative after isoxazole ring opening. Drug Metab. Dispos 39, 302–311 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Duranti E, Balsamini C, Spadoni G & Staccioli L Reaction of Secondary Acetylenic Bromides with Sodium Nitrite: Synthesis of 3,5-Alkyl(aryl)-4-nitroisoxazoles. J. Org. Chem 2870–2872 (1988). [Google Scholar]
- 29.Ray S, Kreitler DF, Gulick AM & Murkin AS The Nitro Group as a Masked Electrophile in Covalent Enzyme Inhibition. ACS Chem. Biol 13, 1470–1473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curini M et al. Alumina promoted cyclization of α-nitro-oximes: A new entry to the synthesis of 1,2,5-oxadiazoles N-oxides (furoxans). Tetrahedron Lett. 41, 8817–8820 (2000). [Google Scholar]
- 31.Zhao JQ et al. Synthesis of furoxan derivatives: DABCO-mediated cascade sulfonylation/cyclization reaction of α-nitro-ketoximes. Tetrahedron 71, 1560–1565 (2015). [Google Scholar]
- 32.Burakevich JV, Butler RS & Volpp GP Phenylfurazan Oxide. Chemistry. J. Org. Chem 37, 593–596 (1972). [Google Scholar]
- 33.Kalinina MI & Mosiev IK Properties of Furoxans Monosubstituted with Adamantanes. Chem. Heterocycl. Compd 24, 217–220 (1988). [Google Scholar]
- 34.Eaton JK, Ruberto RA, Kramm A, Viswanathan VS & Schreiber SL Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J. Am. Chem. Soc 141, 20407–20415 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Shelton BR, Howe R & Liu KC A Particularly Convenient Preparation of Benzohydroximinoyl Chlorides (Nitrile Oxide Precursors). J. Org. Chem 45, 3916–3918 (1980). [Google Scholar]
- 36.Egan C, Clery M, Hegarty AF & Welch AJ Mechanism of Reaction of Isomeric Nitrolic Acids to Nitrile Oxides in Aqueous Solution. J. Chem. Soc. Perkin Trans 2 249–256 (1991). [Google Scholar]
- 37.Matt C, Gissot A, Wagner A & Mioskowski C Nitrolic acids: Efficient precursors of nitrile oxides under neutral conditions. Tetrahedron Lett. 41, 1191–1194 (2000). [Google Scholar]
- 38.Matt C, Wagner A & Mioskowski C Novel Transformation of Primary Nitroalkanes and Primary Alkyl Bromides to the Corresponding Carboxylic Acids. J. Org. Chem 62, 234–235 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zilka O et al. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci 3, 232–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah R, Shchepinov MS & Pratt DA Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci 4, 387–396 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Backus KM et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bar-Peled L et al. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allimuthu D & Adams DJ 2-Chloropropionamide As a Low-Reactivity Electrophile for Irreversible Small-Molecule Probe Identification. ACS Chem. Biol 12, 2124–2131 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Shindo N et al. Selective and reversible modification of kinase cysteines with chlorofluoroacetamides. Nat. Chem. Biol 15, 250–258 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Zaro BW, Whitby LR, Lum KM & Cravatt BF Metabolically Labile Fumarate Esters Impart Kinetic Selectivity to Irreversible Inhibitors. J. Am. Chem. Soc 138, 15841–15844 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martín-Gago P et al. Covalent Protein Labeling at Glutamic Acids. Cell Chem. Biol 24, 589–597.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Geu-Flores F et al. Glucosinolate engineering identifies a γ-glutamyl peptidase. Nat. Chem. Biol 5, 575–577 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Mutlib AE et al. P450-Mediated Metabolism of 1-[3-(Aminomethyl)phenyl]- N -[3-fluoro-2’-(methylsulfonyl)- [1,1’-biphenyl]-4-yl]-3-(trifluoromethyl)-1 H -pyrazole- 5-carboxamide (DPC 423) and Its Analogues to Aldoximes. Characterization of Glutathione Conjugates of Postula. Chem. Res. Toxicol 15, 63–75 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Roveri A, Maiorino M & Ursini F Myoglobin Which Possesses a Very Slow Rate of Autoreduction; (3) Its Formation Is Relatively Easy, Requiring Only Metmyoglobin and. Methods Enzymol. 233, 202–212 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Kato S et al. Preparation of 13 or 9-hydroperoxy-9Z,11E (9E,11E) or 10E,12Z (10E,12E)-octadecadienoic phosphatidylcholine hydroperoxide. J. Oleo Sci 63, 431–437 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Kriska T & Girotti AW A thin layer chromatographic method for determining the enzymatic activity of peroxidases catalyzing the two-electron reduction of lipid hydroperoxides. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 827, 58–64 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Novoselov SV et al. A highly efficient form of the selenocysteine insertion sequence element in protozoan parasites and its use in mammalian cells. Proc. Natl. Acad. Sci 104, 7857–62 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Durocher Y, Perret S & Kamen A High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, 9e–9 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen D et al. Discovery and Characterization of the Potent and Highly Selective (Piperidin-4-yl)pyrido[3,2- d]pyrimidine Based in Vitro Probe BAY-885 for the Kinase ERK5. J. Med. Chem 62, 928–940 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Werner S et al. Discovery and Characterization of the Potent and Selective P2X4 Inhibitor N-[4-(3-Chlorophenoxy)-3-sulfamoylphenyl]-2-phenylacetamide (BAY-1797) and Structure-Guided Amelioration of Its CYP3A4 Induction Profile. J. Med. Chem 8, (2019). [DOI] [PubMed] [Google Scholar]
- 58.Cee VJ et al. Systematic Study of the Glutathione (GSH) Reactivity of N -Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem 58, 9171–9178 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon request.