

Abstract
People with Down syndrome show signs of chronic immune dysregulation, including a higher prevalence of autoimmune disorders, increased rates of hospitalization during respiratory viral infections, and higher mortality rates from pneumonia and sepsis. At the molecular and cellular levels, they show markers of chronic autoinflammation, including interferon hyperactivity, elevated levels of many inflammatory cytokines and chemokines, and changes in diverse immune cell types reminiscent of inflammatory conditions observed in the general population. However, the impact of this immune dysregulation in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and CoV disease of 2019 (COVID-19) remains unknown. This Perspective outlines why individuals with Down syndrome should be considered an at-risk population for severe COVID-19. Specifically, the immune dysregulation caused by trisomy 21 may result in an exacerbated cytokine release syndrome relative to that observed in the euploid population, thus justifying additional monitoring and specialized care for this vulnerable population.
Graphical Abstract
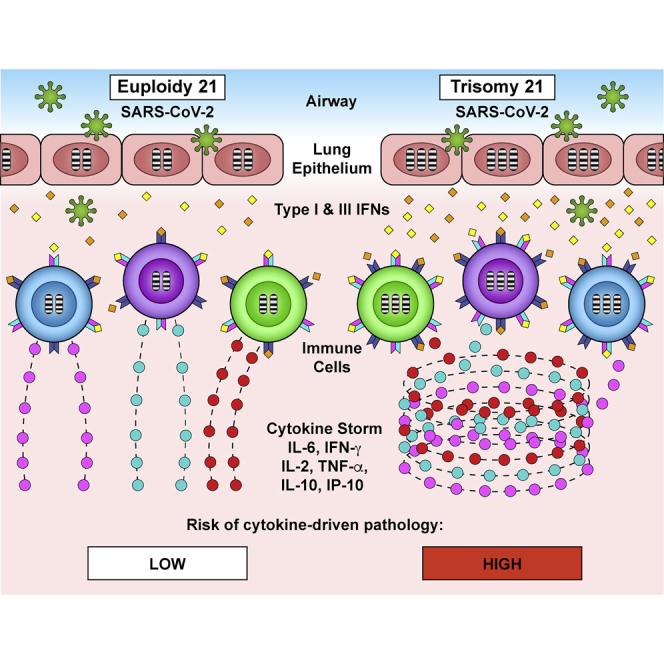
Individuals with Down syndrome display immune dysregulation associated with interferon hyperactivity, and in the context of SARS-CoV-2 infections, this could lead to a stronger cytokine storm and an increased risk of severe COVID-19 pathology. Espinosa proposes that individuals with trisomy 21 are a vulnerable population during the COVID-19 pandemic.
Main Text
Individuals with Down syndrome (DS) show widespread and chronic immune dysregulation. This population shows increased rates of diverse autoimmune conditions, including autoimmune thyroid disease,1, 2, 3 celiac disease,4, 5, 6, 7, 8, 9, 10 autoimmune skin conditions (e.g., alopecia areata, psoriasis, vitiligo, atopic dermatitis and/or eczema, hidradenitis suppurativa),11, 12, 13, 14 and type 1 diabetes.15, 16, 17 At the molecular and cellular levels, individuals with trisomy 21 show clear signs of inflammation in the absence of any detectable infections, such as elevated levels of potent inflammatory cytokines and chemokines,18,19 and changes in diverse immune cell types indicative of hyperactive, pro-inflammatory cellular states.20, 21, 22, 23, 24, 25, 26, 27, 28, 29 In addition, individuals with trisomy 21 show more severe consequences during lung viral infections, such as increased rates of hospitalization during respiratory syncytial virus (RSV) and H1N1 influenza A infections,30,31 as well as increased rates of mortality from bacterial pneumonia and sepsis.32,33 Despite this knowledge, in the context of the ongoing coronavirus disease of 2019 (COVID-19) pandemic, it is unclear how individuals with DS may respond to severe acute respiratory syndrome CoV 2 (SARS-CoV-2) infections, and it may take several months before enough epidemiological and clinical data are gathered to address this issue. Despite the clear limitations imposed by the lack of available data, I provide evidence that individuals with trisomy 21 should be considered at high risk of developing more severe symptoms and increased rates of hospitalization, intensive care, secondary bacterial infections, and mortality from SARS-CoV-2 infections relative to the general population, thus justifying increased monitoring and specialized care for those with COVID-19 and DS.
The Negative Impact of Cytokine Storms during Respiratory Infections
Mounting evidence supports the notion that morbidity and mortality during SARS-CoV-2 infections are driven by the exacerbated immune response to the virus, leading to a cascade of events involving a cytokine storm, acute respiratory distress syndrome (ARDS), and eventual myocardial damage and multi-organ failure.34,35 This pathological cascade is similar to that observed in other lethal lung viral infections, in which the presence of the virus in the lungs triggers a first wave of cytokines, including type I and III interferons (IFNs); activation and recruitment of immune cells, leading to further production of cytokines and chemokines; exacerbated immune activation; and progressive shutdown of respiratory function.36
Cytokine storms, also known as cytokine release syndrome (CRS) or hypercytokinemia, have been described as drivers of pathology in myriad infectious and non-infectious diseases.36 Among infectious diseases, cytokine storms have been postulated to drive mortality during severe viral infections, such as influenza,37 including the 1918 Spanish flu epidemic38 and the H5N1 bird flu,39 as well as the 2003 SARS epidemic,40 hantavirus,41 ebola,42 and smallpox.43 In the specific case of COVID-19, independent reports indicate that the magnitude of the cytokine storm correlates positively with the severity of pathology, likelihood of needing intensive care, and death. Many inflammatory markers, cytokines, and chemokines have been found to be significantly associated with worse prognosis, including C-reactive protein (CRP), interleukin-6 (IL-6), IL-2, IL-7, IL-10, granulocyte colony-stimulating factor (G-CSF), interferon γ-induced protein 10 (IP-10), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1A (MIP-1A), and tumor necrosis factor α (TNF-α).34,35 When integrated with the current understanding of the role of cytokine storms in other respiratory infections, these findings support the notion of combined antiviral treatments and targeted immunosuppression as a therapeutic strategy in COVID-19.44 There are now multiple clinical trials testing the impact of targeted immunosuppressants, such as inhibitors of IL-6 signaling (e.g., Tocilizumab, Sarilumab), TNF-α signaling (e.g., Humira), IL-1β signaling (e.g., Anakinra), and Janus kinase (JAK) inhibitors (e.g., Ruxolitinib, Baricitinib, Tofacitinib) in the hope that attenuating the cytokine storm will improve prognosis.
Interferon Hyperactivity in DS
The exact mechanisms by which trisomy 21 causes the immune dysregulation observed in people with DS remains to be elucidated. However, several genes encoded on chromosome 21 have established roles in immune control, and their overexpression could contribute to the general immune phenotype of DS. Most prominent among the immune regulators encoded on chromosome 21 are four of the six interferon receptors: the two type I IFN receptors IFNAR1 and IFNAR2, the type II IFN receptor IFNGR2, and IL10RB, which serves as a receptor subunit for both type III IFN ligands and the cytokines IL-10, IL-22, and IL-26.45
Several lines of evidence demonstrate the hyperactivation of IFN signaling in DS. First, the IFN receptors (IFNRs) encoded on chromosome 21 are overexpressed in multiple cell types of individuals with trisomy 21. This overexpression is obvious not only at the mRNA level in multiple immune and non-immune cell types (Figure 1)46 but also at the cell surface level, as demonstrated by a recent mass cytometry analysis of 100 different immune cell types.21 Second, immune and non-immune cell types of people with DS are hypersensitive to IFN stimulation, as demonstrated by the super-induction of downstream JAK/STAT signaling and IFN-stimulated genes (ISGs).20,21,47, 48, 49, 50, 51, 52, 53 Third, transcriptome analyses have revealed gene expression signatures indicative of hyperactive IFN signaling in diverse immune and non-immune cell types of people with DS,20,21,46 as well as brain tissue of diverse mouse models of DS carrying triplication of the IFNR cluster.54 In addition, many independent studies have revealed changes indicative of chronic autoinflammation in people with DS, including the elevation of many cytokines and chemokines known to act downstream of IFN signaling.18, 19, 20,47 Of critical importance to the topic of cytokine storms, people with DS show significantly elevated levels of key cytokines such as CRP, IL-6, IL-2, TNF-α, IP-10, IL-10, and MCP-1.19,20,47 To further strengthen this point, I include here data demonstrating that individuals with DS display signs of a mild cytokine storm even in the absence of any obvious infections (Figure 2) (http://explorer.trisome.org/proteome/). Lastly, metabolomics studies of both plasma and cerebrospinal fluid have revealed that individuals with DS display dysregulation of the IFN-inducible kynurenine pathway of tryptophan catabolism, leading to elevated levels of quinolinic acid, a neurotoxic tryptophan catabolite.47 The reduction of the copy number of IFNRs reversed this metabolic dysregulation in vitro, pointing to IFNR triplication as the driving event.
Figure 1.

Individuals with Down Syndrome Overexpress IFN Receptors
Box and whisker plots displaying mRNA expression for the type I IFNRs encoded on chromosome 21, IFNAR1 and IFNAR2. Data are presented in reads per kilobase of transcripts per million (RPKM) mapped reads. Data were generated via RNA sequencing (RNA-seq) transcriptome analysis as described by Araya et al.,20 Sullivan et al.,46 and Powers et al.47 p values were calculated with the Student’s t test. All of the boxplots show median, 25th, and 75th percentile values. Error bars are 1.5 times the interquartile range (IQR) or the maximum data point if <1.5 IQR. Data are publicly available in the research portal TrisomExplorer at http://explorer.trisome.org/transcriptome/.
Figure 2.

Elevated Baseline Cytokine Levels in Individuals with Down Syndrome
Box and whisker plots showing cytokine levels in typical people versus people with Down syndrome. Plasma cytokine levels were measured using the Meso Scale Discovery V-PLEX 54-PLEX Human Cytokine Kit and a U-PLEX custom array, as described in Powers et al.47 All p values were calculated using a Kolmogorov-Smirnov (KS) test. All of the boxplots show median, 25th, and 75th percentile values. Error bars are 1.5 times the IQR or the maximum data point if <1.5 IQR. Data are publicly available at http://explorer.trisome.org/proteome/.
This body of evidence indicates that the IFN response, which is key for both mounting antiviral responses and initiating and amplifying the cytokine storm, is much more active in people with DS. Given this knowledge, several questions then arise: what is the likely impact of SARS-CoV-2 infections on the immune response of individuals with DS? Are individuals with trisomy 21 more or less likely to develop a severe cytokine storm and downstream pathology?
Potential Benefits of a Stronger IFN Response
At first glance, IFN hyperactivity may be considered beneficial, in the sense that it may enable cells with trisomy 21 to mount a stronger antiviral response during the first contact with SARS-CoV-2. As the first viral particles enter lung epithelial cells of an individual with trisomy 21, the consequent production of type I and type III IFN ligands may elicit a stronger response in neighboring epithelial cells and resident immune cells overexpressing IFNRs. In theory, this stronger initial response could restrain viral spread. This potentially beneficial aspect would result in a lower rate of SARS-CoV-2 carriers among those with trisomy 21. However, CoVs have developed strategies to evade the antiviral effects of the IFN response. Studies of the SARS-CoV-1 and the Middle East Respiratory Syndrome CoV (MERS-CoV) have revealed that their genomes encode an unusual number of proteins that dampen or neutralize the IFN signaling cascade (reviewed in Kindler et al.55), which enables viral disguise, immune evasion, and rapid viral replication and spread. Since SARS-CoV-2 is a member of the Betacoronavirus clade, which also includes SARS-CoV-1 and MERS-CoV, it is likely that SARS-CoV-2 uses a similar suite of anti-IFN strategies.56 Therefore, the IFN hyperactivity observed in people with DS may not be sufficient in providing additional protection against SARS-CoV-2 infection. In the event that SARS-CoV-2 evades the initial immune response of a person with trisomy 21 by dampening IFN signaling, the lifelong chronic IFN hyperactivity experienced by these individuals is likely to prime the immune system for super-induction of the cytokine storm. In support of this notion, type I IFN signaling was found to be detrimental in a mouse model of SARS-CoV-1 infection and pathology,57 in which deletion of one of the type I IFNRs encoded on chromosome 21 (IFNAR1) prevented the development of SARS pathology. While there are no data on animal models of SARS-CoV-2 to address the role of IFNR copy number, and assuming the two related viruses provoke similar pathological cascades, it follows then that the exacerbated type I IFN signaling observed in DS would contribute to exacerbated COVID-19 pathology (Figure 3).
Figure 3.

Proposed Model of the Impact of Trisomy 21 on the COVID-19 Pathological Cascade
Trisomy 21 involves triplication of 4 IFN receptors, the type I IFN receptors IFNAR1 and IFNAR2, the type II IFN receptor IFNGR2, and the type III IFN receptor IL10RB. IFN receptor overexpression leads to hypersensitivity to type I and type III IFNs upon exposure of lung epithelial cells to SARS-CoV-2. Key immune cell types of people with Down syndrome, including dendritic cells and T cells, display changes indicative of hyperactivation and increased differentiation toward inflammatory states even before SARS-CoV-2 infection. This heightened immune activity could predispose the immune system of individuals with Down syndrome to cytokine overproduction and an increased risk of acute respiratory distress syndrome, myocardial damage, organ failure, and secondary bacterial infections.
Increased Risk of an Exacerbated Cytokine Storm in DS
There is evidence to support the notion that, upon confirmation of SARS-CoV-2 infection, individuals with DS are more likely to develop a stronger and more prolonged cytokine storm (Figure 3).
During respiratory viral infections, as viral particles enter the lung tissue, lung-resident respiratory dendritic cells (rDCs) acquire antigens from the epithelial cells or are infected themselves, thus being activated and draining to mediastinal and cervical lymph nodes, where they process viral antigens and present them to naive circulating T cells (reviewed in Channappanavar et al.58). Upon T cell receptor (TCR) engagement by peptide-major histocompatibility complex (MHC) complexes on the surface of rDCs, T cells become activated, proliferate, and migrate to the infected lung tissue. Once in the lung, virus-specific effector T cells contribute to the cytokine storm by producing several pro-inflammatory cytokines and chemokines (IFN-γ, TNF-α, IL-2, C-X-C motif chemokine ligand 9 [CXCL9], and IP-10), which in turn recruit more innate and adaptive immune cells. In a careful analysis of lymphocyte recruitment to the lungs of SARS-CoV-1-infected mice, three patterns of immune cell infiltrates were identified in lung tissue.59 Plasmacytoid dendritic cells (pDCs) infiltrated the lung early on (first wave). This was followed by a second wave of natural killer (NK) T cells, NK cells, macrophages, and CD4+ T cells. Finally, a third wave involved neutrophils and CD8+ T cells. In this experimental model, SARS-CoV-1 infection induced an enhanced virus-specific T cell response in the lungs, which coincided with the development of pneumonitis and viral clearance. Furthermore, the depletion of CD4+ T cells resulted in diminished virus-specific antibody responses and decreases in the production of Th1 and Th2 cytokines in the lung.
Within this framework, given the clear signs of pro-inflammatory changes in DCs, monocyte lineages, NK cells, and effector T cells isolated from individuals with DS,20,21 it is then predicted that the cascade of events described above will be exacerbated and driven toward a stronger cytokine storm.
First, when the whole blood of children with DS is exposed to live influenza A virus, the production of TNF-α, IL-1β, IL-6, IL-8, IFN-α, and IFN-γ is significantly elevated relative to their typical siblings.60 However, in this in vitro experiment, viral clearance was equivalent in both groups. The authors concluded that “the production of higher levels of pro-inflammatory cytokines may be responsible for a more severe clinical course of viral disease in these children.”
Second, deep mapping of the immune system of adults with DS revealed many changes indicative of an enhanced pro-inflammatory state in the monocyte lineage and among DCs.21 The monocyte lineage in DS is driven toward the intermediate and non-classical states associated with inflammatory conditions, and diverse DC subsets show alterations consistent with a heightened state of inflammation and cytotoxic potential.21 Thus, it is predicted that the pro-inflammatory state of these myeloid cell types in DS will contribute to enhanced cytokine production as SARS-CoV-2 particles cross the lung epithelium and encounter resident alveolar macrophages and DCs.
Third, within the T cell compartment, T cell lineages of adults with DS show clear signs of differentiation and hyperactivation, even in the absence of any obvious infections, which could be explained by chronic IFN hyperactivity.20 When the CD4 and CD8 T cells of adults with DS are activated ex vivo via stimulation of the TCR, akin to what occurs during a viral infection, they overproduce many key cytokines and chemokines.20 Upon activation, CD4 T cells with trisomy 21 overproduce IL-10, IL-17A, IL-22, and MIP-3α, and CD8 T cells overproduce TNF-α, IFN-γ, IL-2, MIP-1α, GM (granulocyte-macrophage)-CSF, IL-8, MIP-1β, and eotaxin.20 Thus, the T cells of people with DS are likely to fuel a stronger cytokine storm as they infiltrate the infected lung epithelia.
In addition, it has been shown that effector CD4 and CD8 T cells with trisomy 21 are resistant to suppression by regulatory T cells (Tregs)20. Tregs are key players during the resolution of an infection by dampening the action of effector CD4 and CD8 T cells once the virus has been cleared. However, effector T cells of people with DS were found to be refractory to dampening by Tregs after activation, which results in more proliferation of effector T cells relative to euploid counterparts.20 Although Tregs with trisomy 21 were found to be capable of suppressing effector T cells of typical people, they could not suppress effector T cells with trisomy 21, pointing again to a hyperactive, resilient state of effector T cells in DS.20 As the immune system of a person with DS attacks SARS-CoV-2-infected cells, it is predicted that the T cell response, including cytokine overproduction, will be stronger and last longer. This may be further exacerbated by additional pro-inflammatory changes in the NK cells of adults with DS.21
Overall, this evidence supports the notion that COVID-19 in DS will display a stronger cytokine storm and accelerated onset of cytokine-driven pathology (Figure 3).
Increased Burden of Secondary Bacterial Infections
It is well documented that pandemics of respiratory viral infections are followed by a surge in cases of bacterial pneumonia, supporting the notion of a post-viral state of bacterial pneumonia susceptibility.61,62 In fact, most influenza-related deaths are due to secondary infections, with bacteria commonly found in the nasopharyngeal tract, such as Streptococcus pneumoniae and Staphylococcus aureus.62,63 During the Spanish flu pandemic of 1918–1919, >90% of deaths were attributed to subsequent bacterial pneumonia.64,65 During later influenza pandemics in the 1950s and 1960s, the percentage of deaths due to bacterial lung infections was estimated at 50%–70%, with the decreases being explained by the broader use of antibiotics.66,67 In the COVID-19 pandemic, the impact of secondary bacterial infections is likely to be further decreased by the widespread use of broadly acting antibiotics such as azithromycin. However, secondary bacterial infections remain a concern, particularly for individuals with DS.
Of importance to secondary infections in DS, the production of type I IFNs during the antiviral response was shown to drive the increased risk of secondary bacterial infections in mouse models. Knockout of one of the type I IFNRs encoded on chromosome 21 improved the survival and clearance of S. pneumoniae.68 The harmful effects of type I IFN signaling seem to be driven by the impairment of macrophage and/or neutrophil function by IFN-induced cytokines, most prominently among them the anti-inflammatory cytokine IL-10.69,70
IL-10 is involved in the dampening and resolution of immune response and is consistently elevated at baseline in people with DS19 (Figure 3). Furthermore, one of the subunits of the IL-10 receptor, IL10RB, is encoded by one of the four genes in the IFNR cluster on chromosome 21. IL10RB serves as a subunit not only for the IL-10 receptor, but also for the type III IFNs, IL-22, and the IL-26 receptors.71 These findings beg the question: what would be the impact of elevated IL-10 signaling in DS? At this point, all of the evidence supports the notion that elevated IL-10 signaling in DS does not suffice to bring balance to what is clearly a hyperactive, overresponsive immune system. In our analysis of the T cells of people with DS, we observed that CD8+ T cells with trisomy 21 overproduce IL-10 upon stimulation, and they also overproduce many more pro-inflammatory cytokines such as TNF-α, IFN-γ, MIP-1α, IL-2, IL-8, MIP-1β, and eotaxin.20 However, the suppressive effects of IL-10 on the antibacterial branch of the immune system could increase the risk of secondary bacterial infections.62,70 The role of IL-10 in this phenomenon has been well investigated for pneumococcal pneumonia and tuberculosis. In a mouse model of influenza A, treatment with anti-IL-10 neutralizing antibodies before inoculation with S. pneumoniae resulted in reduced bacterial outgrowth and reduced lethality during secondary bacterial pneumonia.70 In the case of tuberculosis, type I IFN signaling was shown to actually promote Mycobacterium tuberculosis bacterial expansion and pathogenesis,72 which was explained by the IFN-dependent induction of IL-10 and consequent impairment in bacterial killing.73
These observations support the notion that type I IFN hyperactivity and increased downstream IL-10 signaling could be drivers of the known susceptibility to bacterial pneumonia in people with DS. Children with DS have a >60-fold higher rate of pneumonia than do typical children,74 and bacterial pneumonia is a leading cause of mortality in adults with DS.75 Several reports have documented impaired neutrophil function in DS.76, 77, 78 Therefore, it is predicted that individuals with COVID-19 and DS would be at a higher risk of secondary bacterial lung infections during the COVID-19 pandemic.
Other Potential Risk Factors for Exacerbated COVID-19 Pathology in DS
The evidence outlined above indicates that COVID-19 in DS would present accelerated progression toward dyspnea, ARDS, and myocardial damage, with a potential increased risk of secondary bacterial infections. In addition, the impact of immune dysregulation in DS could be modulated by other potential risk factors.
People with DS have a unique profile of cardiovascular and cardiopulmonary disease.79 Nearly 50% of newborns with DS are affected by some form of congenital heart disease (CHD), which in many cases requires repair via heart surgery.80 However, adults with DS seem to be protected from coronary heart disease.81 This duality in the cardiac phenotype makes it hard at this point to assess the impacts of CHD on the development of COVID-19 in DS. While it is likely that mild, viable, unrepaired CHD will be a risk factor for severe COVID-19, the assessment is less clear for those individuals who have undergone effective repair via cardiac surgery and have otherwise normal cardiac function.
In addition, diverse anatomic abnormalities of the upper airway are considered major risk factors for respiratory infections in DS.82, 83, 84 Dysphagia and aspiration, which are more common in DS, could also increase the predisposition to lung infection in this population.85,86 In addition to these structural factors, hypotonia can increase the likelihood of proximal airway obstruction and dysphagia in DS.83,86 Obstructive sleep apnea (OSA), which is very common in people with DS,87,88 can often cause chronic intermittent hypoxia and respiratory acidosis, which in turn can drive pulmonary hypertension, a condition that is also more common in DS.89,90 Therefore, OSA could indirectly create an additional risk factor for the severity of COVID-19 by accelerating hypoxemia, predisposing to pulmonary hypertension, and decreasing cardiopulmonary capacity. While some studies support the notion that OSA is a risk factor for ARDS in some settings,91,92 other studies suggest that obesity (which is associated with OSA) may be a higher risk factor for ARDS than OSA itself.93 In the context of COVID-19, chronic lung disease and obesity are recognized risk factors, but the autonomous impact of OSA specifically remains to be fully elucidated.
In summary, in addition to the risks imposed by the immune dysregulation caused by trisomy 21, other risk factors could contribute to a more severe form of COVID-19 in DS.
A Word of Caution
Overall, the combination of immune dysregulation and other potential risk factors suggest that individuals with DS and confirmed SARS-CoV-2 infection should undergo closer monitoring, including rapid, real-time monitoring of inflammatory markers (e.g., CRP, IL-6, TNF-α), myocardial damage (e.g., brain natriuretic peptide, cardiac troponin, myoglobin), and secondary bacterial infections. As more clinical trials for targeted immunosuppressants are performed around the world, individuals with DS are prime candidates for this intervention and other approaches to tone down the cytokine storm.
Clearly, in the absence of clinical and epidemiological data to define the impact of COVID-19 in the population with DS, this Perspective and the hypotheses stated herein should be approached with skepticism. Nevertheless, I hope that this analysis of the literature prompts physicians around the world to pay special attention to individuals with DS and adopt measures to counteract the effects of the cytokine storm and other potential risk factors in this population.
Acknowledgments
I am grateful to all of the students, post-doctoral fellows, and other scientists who have significantly advanced our understanding of immune dysregulation in DS, particularly Dr. Katherine Waugh, Dr. Kathryn Tuttle, Dr. Paula Araya, Dr. Michael Yeager, and Dr. Kelly Sullivan.
Author Contributions
J.M.E. wrote this Perspective with guidance from numerous conversations with colleagues, students, and post-doctoral fellows.
Declaration of Interests
The author is listed as a co-inventor in provisional patents for strategies to attenuate the cytokine storm in COVID-19.
References
- 1.Iughetti L., Predieri B., Bruzzi P., Predieri F., Vellani G., Madeo S.F., Garavelli L., Biagioni O., Bedogni G., Bozzola M. Ten-year longitudinal study of thyroid function in children with Down’s syndrome. Horm. Res. Paediatr. 2014;82:113–121. doi: 10.1159/000362450. [DOI] [PubMed] [Google Scholar]
- 2.Aversa T., Valenzise M., Salerno M., Corrias A., Iughetti L., Radetti G., De Luca F., Wasniewska M. Metamorphic thyroid autoimmunity in Down Syndrome: from Hashimoto’s thyroiditis to Graves’ disease and beyond. Ital. J. Pediatr. 2015;41:87. doi: 10.1186/s13052-015-0197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pierce M.J., LaFranchi S.H., Pinter J.D. Characterization of Thyroid Abnormalities in a Large Cohort of Children with Down Syndrome. Horm. Res. Paediatr. 2017;87:170–178. doi: 10.1159/000457952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlsson A., Axelsson I., Borulf S., Bredberg A., Forslund M., Lindberg B., Sjöberg K., Ivarsson S.A. Prevalence of IgA-antigliadin antibodies and IgA-antiendomysium antibodies related to celiac disease in children with Down syndrome. Pediatrics. 1998;101:272–275. doi: 10.1542/peds.101.2.272. [DOI] [PubMed] [Google Scholar]
- 5.Pueschel S.M., Romano C., Failla P., Barone C., Pettinato R., Castellano Chiodo A., Plumari D.L. A prevalence study of celiac disease in persons with Down syndrome residing in the United States of America. Acta Paediatr. 1999;88:953–956. doi: 10.1080/08035259950168432. [DOI] [PubMed] [Google Scholar]
- 6.Zachor D.A., Mroczek-Musulman E., Brown P. Prevalence of celiac disease in Down syndrome in the United States. J. Pediatr. Gastroenterol. Nutr. 2000;31:275–279. doi: 10.1097/00005176-200009000-00014. [DOI] [PubMed] [Google Scholar]
- 7.Mackey J., Treem W.R., Worley G., Boney A., Hart P., Kishnani P.S. Frequency of celiac disease in individuals with Down syndrome in the United States. Clin. Pediatr. (Phila.) 2001;40:249–252. doi: 10.1177/000992280104000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cogulu O., Ozkinay F., Gunduz C., Cankaya T., Aydogdu S., Ozgenc F., Kutukculer N., Ozkinay C. Celiac disease in children with Down syndrome: importance of follow-up and serologic screening. Pediatr. Int. 2003;45:395–399. doi: 10.1046/j.1442-200x.2003.01755.x. [DOI] [PubMed] [Google Scholar]
- 9.Hansson T., Dahlbom I., Rogberg S., Nyberg B.I., Dahlström J., Annerén G., Klareskog L., Dannaeus A. Antitissue transglutaminase and antithyroid autoantibodies in children with Down syndrome and celiac disease. J. Pediatr. Gastroenterol. Nutr. 2005;40:170–174, discussion 125–127. doi: 10.1097/00005176-200502000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Mårild K., Stephansson O., Grahnquist L., Cnattingius S., Söderman G., Ludvigsson J.F. Down syndrome is associated with elevated risk of celiac disease: a nationwide case-control study. J. Pediatr. 2013;163:237–242. doi: 10.1016/j.jpeds.2012.12.087. [DOI] [PubMed] [Google Scholar]
- 11.Madan V., Williams J., Lear J.T. Dermatological manifestations of Down’s syndrome. Clin. Exp. Dermatol. 2006;31:623–629. doi: 10.1111/j.1365-2230.2006.02164.x. [DOI] [PubMed] [Google Scholar]
- 12.Sureshbabu R., Kumari R., Ranugha S., Sathyamoorthy R., Udayashankar C., Oudeacoumar P. Phenotypic and dermatological manifestations in Down Syndrome. Dermatol. Online J. 2011;17:3. [PubMed] [Google Scholar]
- 13.Daneshpazhooh M., Nazemi T.M., Bigdeloo L., Yoosefi M. Mucocutaneous findings in 100 children with Down syndrome. Pediatr. Dermatol. 2007;24:317–320. doi: 10.1111/j.1525-1470.2007.00412.x. [DOI] [PubMed] [Google Scholar]
- 14.Bilgili S.G., Akdeniz N., Karadag A.S., Akbayram S., Calka O., Ozkol H.U. Mucocutaneous disorders in children with down syndrome: case-controlled study. Genet. Couns. 2011;22:385–392. [PubMed] [Google Scholar]
- 15.Anwar A.J., Walker J.D., Frier B.M. Type 1 diabetes mellitus and Down’s syndrome: prevalence, management and diabetic complications. Diabet. Med. 1998;15:160–163. doi: 10.1002/(SICI)1096-9136(199802)15:2<160::AID-DIA537>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Bergholdt R., Eising S., Nerup J., Pociot F. Increased prevalence of Down’s syndrome in individuals with type 1 diabetes in Denmark: a nationwide population-based study. Diabetologia. 2006;49:1179–1182. doi: 10.1007/s00125-006-0231-6. [DOI] [PubMed] [Google Scholar]
- 17.Rohrer T.R., Hennes P., Thon A., Dost A., Grabert M., Rami B., Wiegand S., Holl R.W., DPV Initiative Down’s syndrome in diabetic patients aged <20 years: an analysis of metabolic status, glycaemic control and autoimmunity in comparison with type 1 diabetes. Diabetologia. 2010;53:1070–1075. doi: 10.1007/s00125-010-1686-z. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Che M., Yuan J., Yu Y., Cao C., Qin X.Y., Cheng Y. Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: a meta-analysis. Oncotarget. 2017;8:84489–84496. doi: 10.18632/oncotarget.21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan K.D., Evans D., Pandey A., Hraha T.H., Smith K.P., Markham N., Rachubinski A.L., Wolter-Warmerdam K., Hickey F., Espinosa J.M., Blumenthal T. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 2017;7:14818. doi: 10.1038/s41598-017-13858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Araya P., Waugh K.A., Sullivan K.D., Núñez N.G., Roselli E., Smith K.P., Granrath R.E., Rachubinski A.L., Enriquez Estrada B., Butcher E.T. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl. Acad. Sci. USA. 2019;116:24231–24241. doi: 10.1073/pnas.1908129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waugh K.A., Araya P., Pandey A., Jordan K.R., Smith K.P., Granrath R.E., Khanal S., Butcher E.T., Estrada B.E., Rachubinski A.L. Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome. Cell Rep. 2019;29:1893–1908.e4. doi: 10.1016/j.celrep.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spina C.A., Smith D., Korn E., Fahey J.L., Grossman H.J. Altered cellular immune functions in patients with Down’s syndrome. Am. J. Dis. Child. 1981;135:251–255. doi: 10.1001/archpedi.1981.02130270043015. [DOI] [PubMed] [Google Scholar]
- 23.Cossarizza A., Monti D., Montagnani G., Ortolani C., Masi M., Zannotti M., Franceschi C. Precocious aging of the immune system in Down syndrome: alteration of B lymphocytes, T-lymphocyte subsets, and cells with natural killer markers. Am. J. Med. Genet. Suppl. 1990;7:213–218. doi: 10.1002/ajmg.1320370743. [DOI] [PubMed] [Google Scholar]
- 24.Barrena M.J., Echaniz P., Garcia-Serrano C., Cuadrado E. Imbalance of the CD4+ subpopulations expressing CD45RA and CD29 antigens in the peripheral blood of adults and children with Down syndrome. Scand. J. Immunol. 1993;38:323–326. doi: 10.1111/j.1365-3083.1993.tb01733.x. [DOI] [PubMed] [Google Scholar]
- 25.Roat E., Prada N., Lugli E., Nasi M., Ferraresi R., Troiano L., Giovenzana C., Pinti M., Biagioni O., Mariotti M. Homeostatic cytokines and expansion of regulatory T cells accompany thymic impairment in children with Down syndrome. Rejuvenation Res. 2008;11:573–583. doi: 10.1089/rej.2007.0648. [DOI] [PubMed] [Google Scholar]
- 26.Bloemers B.L., van Bleek G.M., Kimpen J.L., Bont L. Distinct abnormalities in the innate immune system of children with Down syndrome. J. Pediatr. 2010;156:804–809. doi: 10.1016/j.jpeds.2009.12.006. 809.e1–809.e5. [DOI] [PubMed] [Google Scholar]
- 27.Cetiner S., Demirhan O., Inal T.C., Tastemir D., Sertdemir Y. Analysis of peripheral blood T-cell subsets, natural killer cells and serum levels of cytokines in children with Down syndrome. Int. J. Immunogenet. 2010;37:233–237. doi: 10.1111/j.1744-313X.2010.00914.x. [DOI] [PubMed] [Google Scholar]
- 28.Pellegrini F.P., Marinoni M., Frangione V., Tedeschi A., Gandini V., Ciglia F., Mortara L., Accolla R.S., Nespoli L. Down syndrome, autoimmunity and T regulatory cells. Clin. Exp. Immunol. 2012;169:238–243. doi: 10.1111/j.1365-2249.2012.04610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carsetti R., Valentini D., Marcellini V., Scarsella M., Marasco E., Giustini F., Bartuli A., Villani A., Ugazio A.G. Reduced numbers of switched memory B cells with high terminal differentiation potential in Down syndrome. Eur. J. Immunol. 2015;45:903–914. doi: 10.1002/eji.201445049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beckhaus A.A., Castro-Rodriguez J.A. Down Syndrome and the Risk of Severe RSV Infection: A Meta-analysis. Pediatrics. 2018;142:e20180225. doi: 10.1542/peds.2018-0225. [DOI] [PubMed] [Google Scholar]
- 31.Pérez-Padilla R., Fernández R., García-Sancho C., Franco-Marina F., Aburto O., López-Gatell H., Bojórquez I. Pandemic (H1N1) 2009 virus and Down syndrome patients. Emerg. Infect. Dis. 2010;16:1312–1314. doi: 10.3201/eid1608.091931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bloemers B.L., Broers C.J., Bont L., Weijerman M.E., Gemke R.J., van Furth A.M. Increased risk of respiratory tract infections in children with Down syndrome: the consequence of an altered immune system. Microbes Infect. 2010;12:799–808. doi: 10.1016/j.micinf.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Garrison M.M., Jeffries H., Christakis D.A. Risk of death for children with down syndrome and sepsis. J. Pediatr. 2005;147:748–752. doi: 10.1016/j.jpeds.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 34.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruan Q., Yang K., Wang W., Jiang L., Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020 doi: 10.1007/s00134-020-05991-x. Published online March 3, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tisoncik J.R., Korth M.J., Simmons C.P., Farrar J., Martin T.R., Katze M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012;76:16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Q., Zhou Y.H., Yang Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016;13:3–10. doi: 10.1038/cmi.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osterholm M.T. Preparing for the next pandemic. N. Engl. J. Med. 2005;352:1839–1842. doi: 10.1056/NEJMp058068. [DOI] [PubMed] [Google Scholar]
- 39.Haque A., Hober D., Kasper L.H. Confronting potential influenza A (H5N1) pandemic with better vaccines. Emerg. Infect. Dis. 2007;13:1512–1518. doi: 10.3201/eid1310.061262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang K.J., Su I.J., Theron M., Wu Y.C., Lai S.K., Liu C.C., Lei H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005;75:185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mori M., Rothman A.L., Kurane I., Montoya J.M., Nolte K.B., Norman J.E., Waite D.C., Koster F.T., Ennis F.A. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 1999;179:295–302. doi: 10.1086/314597. [DOI] [PubMed] [Google Scholar]
- 42.Paessler S., Walker D.H. Pathogenesis of the viral hemorrhagic fevers. Annu. Rev. Pathol. 2013;8:411–440. doi: 10.1146/annurev-pathol-020712-164041. [DOI] [PubMed] [Google Scholar]
- 43.Stanford M.M., McFadden G., Karupiah G., Chaudhri G. Immunopathogenesis of poxvirus infections: forecasting the impending storm. Immunol. Cell Biol. 2007;85:93–102. doi: 10.1038/sj.icb.7100033. [DOI] [PubMed] [Google Scholar]
- 44.Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration, UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:P1033–P1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Weerd N.A., Nguyen T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012;90:483–491. doi: 10.1038/icb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sullivan K.D., Lewis H.C., Hill A.A., Pandey A., Jackson L.P., Cabral J.M., Smith K.P., Liggett L.A., Gomez E.B., Galbraith M.D. Trisomy 21 consistently activates the interferon response. eLife. 2016;5:e16220. doi: 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Powers R.K., Culp-Hill R., Ludwig M.P., Smith K.P., Waugh K.A., Minter R., Tuttle K.D., Lewis H.C., Rachubinski A.L., Granrath R.E. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat. Commun. 2019;10:4766. doi: 10.1038/s41467-019-12739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan Y.H., Schneider E.L., Tischfield J., Epstein C.J., Ruddle F.H. Human chromosome 21 dosage: effect on the expression of the interferon induced antiviral state. Science. 1974;186:61–63. doi: 10.1126/science.186.4158.61. [DOI] [PubMed] [Google Scholar]
- 49.Chany C., Vignal M., Couillin P., Van Cong N., Boué J., Boué A. Chromosomal localization of human genes governing the interferon-induced antiviral state. Proc. Natl. Acad. Sci. USA. 1975;72:3129–3133. doi: 10.1073/pnas.72.8.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cupples C.G., Tan Y.H. Effect of human interferon preparations on lymphoblastogenesis in Down’s syndrome. Nature. 1977;267:165–167. doi: 10.1038/267165a0. [DOI] [PubMed] [Google Scholar]
- 51.Slate D.L., Shulman L., Lawrence J.B., Revel M., Ruddle F.H. Presence of human chromosome 21 alone is sufficient for hybrid cell sensitivity to human interferon. J. Virol. 1978;25:319–325. doi: 10.1128/jvi.25.1.319-325.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maroun L.E. Interferon effect on ribosomal ribonucleic acid related to chromosome 21 ploidy. Biochem. J. 1979;179:221–225. doi: 10.1042/bj1790221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Epstein C.J., Weil J., Epstein L.B. Abnormalities in the interferon response and immune systems in Down syndrome: studies in human trisomy 21 and mouse trisomy 16. Prog. Clin. Biol. Res. 1987;246:191–208. [PubMed] [Google Scholar]
- 54.Aziz N.M., Guedj F., Pennings J.L.A., Olmos-Serrano J.L., Siegel A., Haydar T.F., Bianchi D.W. Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of Down syndrome. Dis. Model. Mech. 2018;11:dmm031013. doi: 10.1242/dmm.031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kindler E., Thiel V., Weber F. Interaction of SARS and MERS Coronaviruses with the Antiviral Interferon Response. Adv. Virus Res. 2016;96:219–243. doi: 10.1016/bs.aivir.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang C., Liu Z., Chen Z., Huang X., Xu M., He T., Zhang Z. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J. Med. Virol. 2020 doi: 10.1002/jmv.25762. Published online March 13, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., Perlman S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Channappanavar R., Zhao J., Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014;59:118–128. doi: 10.1007/s12026-014-8534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen J., Lau Y.F., Lamirande E.W., Paddock C.D., Bartlett J.H., Zaki S.R., Subbarao K. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 2010;84:1289–1301. doi: 10.1128/JVI.01281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Broers C.J., Gemke R.J., Weijerman M.E., van der Sluijs K.F., van Furth A.M. Increased pro-inflammatory cytokine production in Down Syndrome children upon stimulation with live influenza A virus. J. Clin. Immunol. 2012;32:323–329. doi: 10.1007/s10875-011-9625-4. [DOI] [PubMed] [Google Scholar]
- 61.Metersky M.L., Masterton R.G., Lode H., File T.M., Jr., Babinchak T. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int. J. Infect. Dis. 2012;16:e321–e331. doi: 10.1016/j.ijid.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 62.van der Sluijs K.F., van der Poll T., Lutter R., Juffermans N.P., Schultz M.J. Bench-to-bedside review: bacterial pneumonia with influenza - pathogenesis and clinical implications. Crit. Care. 2010;14:219. doi: 10.1186/cc8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ballinger M.N., Standiford T.J. Postinfluenza bacterial pneumonia: host defenses gone awry. J. Interferon Cytokine Res. 2010;30:643–652. doi: 10.1089/jir.2010.0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chien Y.W., Klugman K.P., Morens D.M. Bacterial pathogens and death during the 1918 influenza pandemic. N. Engl. J. Med. 2009;361:2582–2583. doi: 10.1056/NEJMc0908216. [DOI] [PubMed] [Google Scholar]
- 65.Morens D.M., Fauci A.S. The 1918 influenza pandemic: insights for the 21st century. J. Infect. Dis. 2007;195:1018–1028. doi: 10.1086/511989. [DOI] [PubMed] [Google Scholar]
- 66.Bisno A.L., Griffin J.P., Van Epps K.A., Niell H.B., Rytel M.W. Pneumonia and Hong Kong influenza: a prospective study of the 1968-1969 epidemic. Am. J. Med. Sci. 1971;261:251–263. doi: 10.1097/00000441-197105000-00004. [DOI] [PubMed] [Google Scholar]
- 67.Morens D.M., Taubenberger J.K., Fauci A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shahangian A., Chow E.K., Tian X., Kang J.R., Ghaffari A., Liu S.Y., Belperio J.A., Cheng G., Deng J.C. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 2009;119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pittet L.A., Hall-Stoodley L., Rutkowski M.R., Harmsen A.G. Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am. J. Respir. Cell Mol. Biol. 2010;42:450–460. doi: 10.1165/rcmb.2007-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van der Sluijs K.F., van Elden L.J., Nijhuis M., Schuurman R., Pater J.M., Florquin S., Goldman M., Jansen H.M., Lutter R., van der Poll T. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J. Immunol. 2004;172:7603–7609. doi: 10.4049/jimmunol.172.12.7603. [DOI] [PubMed] [Google Scholar]
- 71.Pestka S., Krause C.D., Walter M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 72.Moreira-Teixeira L., Mayer-Barber K., Sher A., O’Garra A. Type I interferons in tuberculosis: foe and occasionally friend. J. Exp. Med. 2018;215:1273–1285. doi: 10.1084/jem.20180325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McNab F.W., Ewbank J., Howes A., Moreira-Teixeira L., Martirosyan A., Ghilardi N., Saraiva M., O’Garra A. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-γ for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J. Immunol. 2014;193:3600–3612. doi: 10.4049/jimmunol.1401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.So S.A., Urbano R.C., Hodapp R.M. Hospitalizations of infants and young children with Down syndrome: evidence from inpatient person-records from a statewide administrative database. J. Intellect. Disabil. Res. 2007;51:1030–1038. doi: 10.1111/j.1365-2788.2007.01013.x. [DOI] [PubMed] [Google Scholar]
- 75.Ram G., Chinen J. Infections and immunodeficiency in Down syndrome. Clin. Exp. Immunol. 2011;164:9–16. doi: 10.1111/j.1365-2249.2011.04335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rascón Trincado M.V., Lorente Toledano F., Villalobos V.S. A study of the functions of polymorphonuclear neutrophil in patients with Down’s syndrome. Allergol. Immunopathol. (Madr.) 1988;16:339–345. [PubMed] [Google Scholar]
- 77.Cocchi P., Silenzi M., Ravina A. Neutrophil viability in Down’s syndrome. JAMA. 1978;240:737. [PubMed] [Google Scholar]
- 78.Izumi Y., Sugiyama S., Shinozuka O., Yamazaki T., Ohyama T., Ishikawa I. Defective neutrophil chemotaxis in Down’s syndrome patients and its relationship to periodontal destruction. J. Periodontol. 1989;60:238–242. doi: 10.1902/jop.1989.60.5.238. [DOI] [PubMed] [Google Scholar]
- 79.Colvin K.L., Yeager M.E. What people with Down Syndrome can teach us about cardiopulmonary disease. Eur. Respir. Rev. 2017;26:160098. doi: 10.1183/16000617.0098-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bull M.J., Committee on Genetics Health supervision for children with Down syndrome. Pediatrics. 2011;128:393–406. doi: 10.1542/peds.2011-1605. [DOI] [PubMed] [Google Scholar]
- 81.Sobey C.G., Judkins C.P., Sundararajan V., Phan T.G., Drummond G.R., Srikanth V.K. Risk of Major Cardiovascular Events in People with Down Syndrome. PLoS One. 2015;10:e0137093. doi: 10.1371/journal.pone.0137093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDowell K.M., Craven D.I. Pulmonary complications of Down syndrome during childhood. J. Pediatr. 2011;158:319–325. doi: 10.1016/j.jpeds.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 83.Pandit C., Fitzgerald D.A. Respiratory problems in children with Down syndrome. J. Paediatr. Child Health. 2012;48:E147–E152. doi: 10.1111/j.1440-1754.2011.02077.x. [DOI] [PubMed] [Google Scholar]
- 84.Watts R., Vyas H. An overview of respiratory problems in children with Down’s syndrome. Arch. Dis. Child. 2013;98:812–817. doi: 10.1136/archdischild-2013-304611. [DOI] [PubMed] [Google Scholar]
- 85.Jackson A., Maybee J., Moran M.K., Wolter-Warmerdam K., Hickey F. Clinical Characteristics of Dysphagia in Children with Down Syndrome. Dysphagia. 2016;31:663–671. doi: 10.1007/s00455-016-9725-7. [DOI] [PubMed] [Google Scholar]
- 86.Jackson A., Maybee J., Wolter-Warmerdam K., DeBoer E., Hickey F. Associations between age, respiratory comorbidities, and dysphagia in infants with down syndrome. Pediatr. Pulmonol. 2019;54:1853–1859. doi: 10.1002/ppul.24458. [DOI] [PubMed] [Google Scholar]
- 87.Marcus C.L., Keens T.G., Bautista D.B., von Pechmann W.S., Ward S.L. Obstructive sleep apnea in children with Down syndrome. Pediatrics. 1991;88:132–139. [PubMed] [Google Scholar]
- 88.Trois M.S., Capone G.T., Lutz J.A., Melendres M.C., Schwartz A.R., Collop N.A., Marcus C.L. Obstructive sleep apnea in adults with Down syndrome. J. Clin. Sleep Med. 2009;5:317–323. [PMC free article] [PubMed] [Google Scholar]
- 89.King P., Tulloh R. Management of pulmonary hypertension and Down syndrome. Int. J. Clin. Pract. Suppl. 2011;(174):8–13. doi: 10.1111/j.1742-1241.2011.02823.x. [DOI] [PubMed] [Google Scholar]
- 90.Bush D., Galambos C., Ivy D.D., Abman S.H., Wolter-Warmerdam K., Hickey F. Clinical Characteristics and Risk Factors for Developing Pulmonary Hypertension in Children with Down Syndrome. J. Pediatr. 2018;202:212–219.e2. doi: 10.1016/j.jpeds.2018.06.031. [DOI] [PubMed] [Google Scholar]
- 91.Kaw R., Chung F., Pasupuleti V., Mehta J., Gay P.C., Hernandez A.V. Meta-analysis of the association between obstructive sleep apnoea and postoperative outcome. Br. J. Anaesth. 2012;109:897–906. doi: 10.1093/bja/aes308. [DOI] [PubMed] [Google Scholar]
- 92.Memtsoudis S., Liu S.S., Ma Y., Chiu Y.L., Walz J.M., Gaber-Baylis L.K., Mazumdar M. Perioperative pulmonary outcomes in patients with sleep apnea after noncardiac surgery. Anesth. Analg. 2011;112:113–121. doi: 10.1213/ANE.0b013e3182009abf. [DOI] [PubMed] [Google Scholar]
- 93.Karnatovskaia L.V., Lee A.S., Bender S.P., Talmor D., Festic E., US Critical Illness and Injury Trials Group Lung Injury Prevention Study Investigators (USCIITG-LIPS) Obstructive sleep apnea, obesity, and the development of acute respiratory distress syndrome. J. Clin. Sleep Med. 2014;10:657–662. doi: 10.5664/jcsm.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]