

Abstract
Angiogenesis is a hallmark of cancer. However, current antivascular therapies that primarily target angiogenic factors have faced difficulties and failures in treating most cancers. It is proposed that endothelial transformation acts as an alternative driver of excessive and abnormal vascularity that fuels cancer progression and immunosuppression, and also induces resistance to therapy. Thus, vascular detransformation may serve as a promising therapeutic strategy against cancer.
Aberrant vascularization is a fundamental hallmark of cancer. Malignant solid tumors are characterized by overgrown, topologically and structurally abnormal vasculature that fuels cancer progression and metastasis [1]. Newly formed blood vessels supply oxygen and diffusible nutrients to solid tumors and secrete soluble factors that promote tumorigenesis. Thus, antivascular therapy has emerged as a key approach against cancer. However, current antivascular therapies that primarily target angiogenic factors including vascular endothelial growth factor (VEGF), albeit initially groundbreaking, have faced major difficulties and failures in treating most cancers. The major therapeutic challenge encountered with antivascular treatment is insufficient eradication and inefficient functional inhibition of tumor-associated endothelial cells (ECs). Identification of additional mechanisms that control the formation of abnormal tumor vasculature is therefore vital for the development of next-generation antivascular therapies.
Endothelial transformation may represent a crucial cellular mechanism for aberrant tumor vascularization. Cellular plasticity in ECs has been well characterized during embryogenesis [2]. Likewise, previous work has documented the robust ability of ECs to transdifferentiate and transition into hematopoietic cells and stem cells during embryonic development. In pathological settings including cardiac, renal, and liver fibrosis, ossifying myositis, pulmonary hypotension, and cerebral cavernous malformation, ECs undergo mesenchymal transition de novo to generate fibroblasts and stem-like cells [3–6]. Notably, recent data show that tumor-associated ECs express mesenchymal genes in melanoma and Kaposi’s sarcoma, suggesting the potential existence of an endothelial–mesenchymal transition (EndMT) in cancer [7,8]. Strikingly, recent studies confirm the expression of mesenchymal genes in glioma-associated ECs [9,10]. Importantly, these studies also indicate that tumor ECs acquire mesenchymal phenotypes to enhance cell proliferation and migration, thereby promoting vessel sprouting and outgrowth in glioma because these cells maintain their endothelial identity in the cancer microenvironment, as indicated by their ability to uptake acetylated low-density lipoprotein (Ac-LDL) and to form tube-like structures on matrigel, as well as by their expression of the EC marker, von Willebrand factor (vWF ) but not the pericyte marker NG2 [9,10]. This process is accordingly termed EC mesenchymal transformation to distinguish it from mesenchymal transition because no cell fate transition occurs and key endothelial functions are retained. Thus, the major biological consequence of endothelial transformation in cancer is not the generation of tumor-associated mesenchymal cells de novo, but is instead the modulation of the functions of ECs themselves. For clarification, mesenchymal transformation refers to acquisition of mesenchymal phenotypes without a complete cell fate change into a mesenchymal cell, which emphasizes no loss of key endothelial functions and, therefore, is similar to the term ‘partial EndMT’, in contrast to the term ‘complete EndMT’ that indicates fully altered cell fate and functions.
The main functions of tumor-associated ECs are in vessel formation and altered vascular niche function, both of which significantly contribute to cancer progression and resistance to therapy. During endothelial transformation, ECs gradually downregulate their expression of the endothelial-specific genes PECAM1 (CD31), CDH5 (VE-cadherin), and KDR (VEGFR-2), while acquiring the expression of mesenchymal genes including S100A4 (FSP-1), ACTA2 [alpha-smooth muscle actin (α-SMA)], and CDH2 (N-cadherin), likely due to global genetic reprogramming. This process can be induced in tumors in vivo as well as in normal ECs exposed to tumor-derived conditioned medium in vitro [9,10]. After endothelial transformation, tumor-associated ECs acquire mesenchymal phenotypes including enhanced cell division and motility, and they also lose their cell–cell adhesion properties and dissociate from each other as a result of downregulated expression of the adhesion protein VE-cadherin, which together lead to the generation of excessive abnormal vasculature in the cancer microenvironment. As such, endothelial transformation induces ECs to enhance their ability to proliferate and thereby promotes vascularization, likely providing a better explanation for the excessive vascularity observed in tumors because the previously proposed mesenchymal transition can theoretically reduce the EC population after making a complete cell transition to fibroblasts. Furthermore, EC transformation may affect global gene expression and vascular niche function, thus regulating the survival, activation, and/or stemness of adjacent cells including, but not limited to, cancer stem cells, tumor cells, and immune cells by producing paracrine factors or through cell–cell interactions that eventually modulate niche-mediated therapy resistance and tumor immunity. Thus, EC transformation plays a multifaceted role in vessel abnormalities, resistance to therapy, and immunity regulation in cancer (Figure 1).
Figure 1. Endothelial Transformation Induces Vascular Abnormalities, Immune Suppression, and Resistance to Therapy in Cancer.
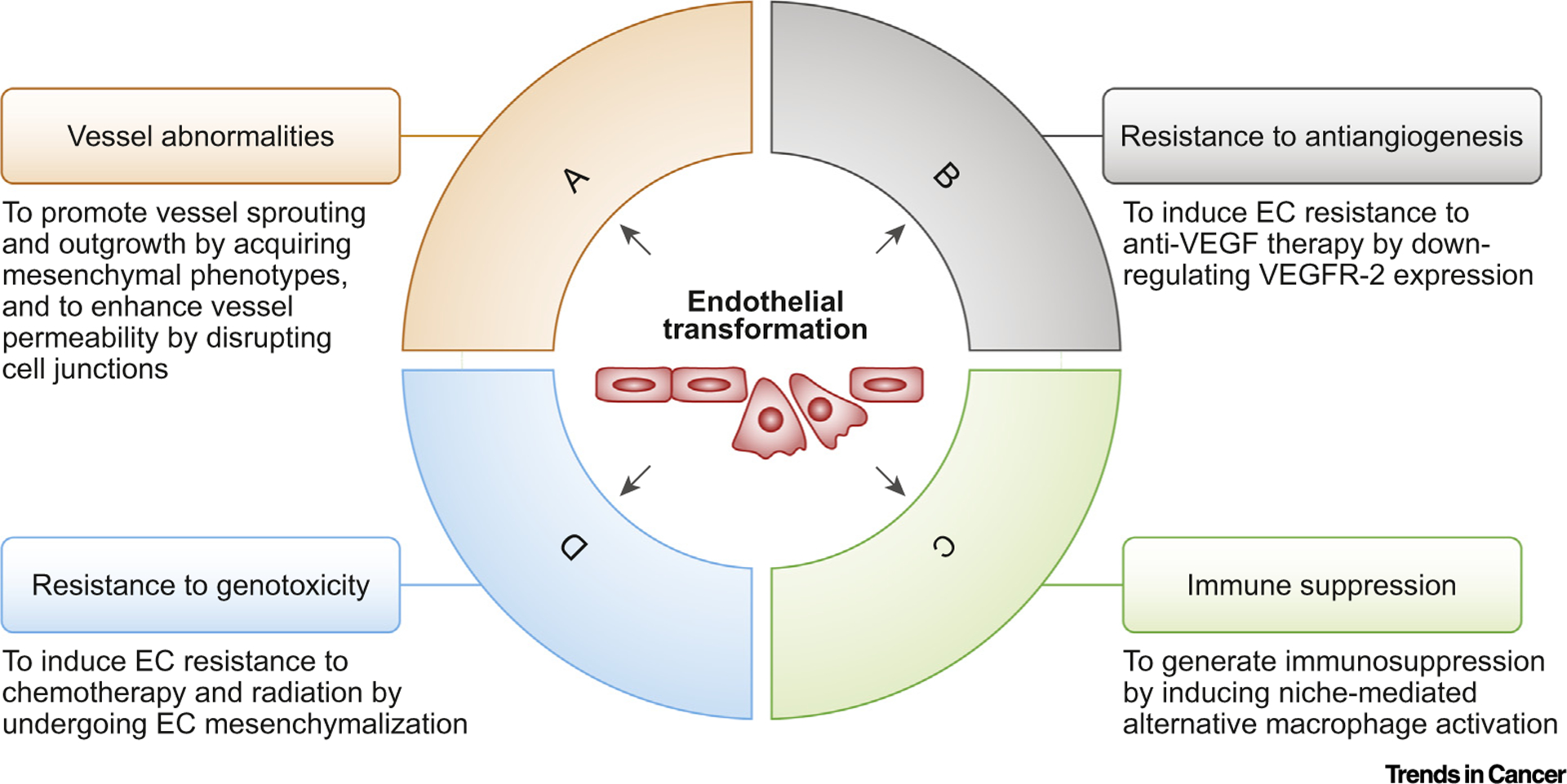
Endothelial transformation has a multifaceted role in cancer. (A) Endothelial cells (ECs) acquire mesenchymal phenotypes that enhance cell proliferation and migration, thereby promoting vessel sprouting and outgrowth. EC transformation also induces VE-cadherin degradation, resulting in increased vessel permeability. (B) Endothelial transformation downregulates vascular endothelial growth factor receptor (VEGFR)-2 expression, leading to EC resistance to anti-VEGF therapy. (C) Transformed ECs express interleukin (IL)-6, which induces alternative macrophage activation and immune suppression in the tumor microenvironment. (D) Mesenchymal transformation causes ECs to acquire resistance to chemotherapy and radiation therapy.
Vessel Abnormalities
Tumor-associated blood vessels are characterized by tortuous morphology and excessive vessel sprouting and outgrowth, and they are highly proliferative and leaky [1]. These abnormalities impede vessel-based functions by inducing heterogeneous hypoxia, reducing drug delivery, and inhibiting the egress of immune effector cells, including T and natural killer (NK) cells, from blood circulation and their infiltration into tumors. Vessel normalization has recently joined antiangiogenesis as an important strategy for cancer therapy, and may improve drug delivery and immune cell infiltration and also inhibit hypoxia-dependent tumorigenesis and radioresistance. It has been proposed that imbalanced expression of proangiogenic and antiangiogenic factors contributes to abnormal vascularization [1]. However, therapies that target proangiogenic factors show small, transient effects on vessel normalization in cancer patients [11], suggesting that additional mechanisms control aberrant vascularization. Notably, endothelial transformation may serve as an alternative driving force for vessel abnormalities in cancer. First, tumor-associated ECs acquire mesenchymal phenotypes that enhance cell proliferation and migration, leading to excessive vessel outgrowth and sprouting [9]. Second, endothelial transformation induces the degradation of VE-cadherin, the major endothelial adhesion molecule controlling cellular junctions, via a c-Met/ETS-1/ matrix metalloproteinase14 (MMP14)-mediated mechanism, and this disrupts cell junctions between ECs and leads to vessel hyper-permeability that also facilitates vessel sprouting [9]. Interestingly, mouse studies show that loss of c-Met expression specifically in ECs normalizes tumor-associated vasculature and sensitizes gliomas to chemotherapy, suggesting that endothelial transformation may serve as a promising target for vessel normalization therapy.
Resistance to Anti-VEGF Treatment
VEGF is the best-studied angiogenic factor and is believed to play a central role in the process of tumor vascularization. However, anti-VEGF therapy shows little overall survival benefits in most patients with malignant solid tumors. Multiple mechanisms contribute to the resistance of tumors to anti-VEGF treatment, including angiogenic pathway redundancy, compensatory activation of survival signals, and pericyte-and macrophage-mediated protection [12]. Notably, endothelial transformation may serve as a pivotal mechanism driving tumor resistance to anti-VEGF therapy. Endothelial transformation downregulates the expression of vascular endothelial growth factor receptor (VEGFR)-2, a receptor that mediates almost all the known cellular responses to VEGF-A, leading to primary resistance of glioma-associated ECs to anti-VEGF treatment [10]. This process is regulated by platelet-derived growth factor (PDGF)/nuclear factor (NF)-κB-mediated expression of Snail, which binds to the VEGFR-2 promoter and inhibits its expression [10]. Importantly, combination treatment with an anti-VEGF antibody together with pharmacological inhibition or with genetic ablation of platelet-derived growth factor receptor (PDGFR)-β inhibits glioma growth and improves mouse survival [10]. These findings suggest that targeting endothelial transformation may be useful for overcoming tumor resistance to anti-VEGF therapy.
Resistance to Genotoxic Treatment
Given the crucial role played by the perivascular niche in stemness maintenance and self-renewal in cancer stem cells, as well as in the activation of prosurvival and antiapoptotic pathways in tumor cells, the success of cytotoxic treatment relies heavily on the efficient eradication of tumor-associated ECs. The link between mesenchymalization and resistance to therapy is well established in tumor cells. Likewise, EC mesenchymalization by endothelial transformation may contribute to EC resistance to genotoxic treatments, including chemotherapy and radiation. In support, a recent report shows that radiotherapy enhances the expression of mesenchymal protein α-SMA in tumor-associated ECs and induces vascular abnormalities in mouse lung adenocarcinoma tumors [13], indicating that transformed ECs are retained after treatment and form aberrant vasculature in the tumor microenvironment. Endothelial transformation may therefore be an effective target for eradicating tumor-associated ECs, cancer stem cells, and tumor cells by radio/chemotherapy. In addition, endothelial mesenchymalization may induce greater deposition of extracellular matrix across the vessel capillaries, thereby serving as a physical barrier to chemodrug diffusion into the tumors. Thus, inhibition of endothelial transformation may sensitize tumor ECs and tumor cells to genotoxic therapy.
Immune Suppression
Immunotherapy holds great promise for cancer treatment. However, solid tumors remain a significant challenge for current immunotherapy approaches that primarily activate and utilize T cells by immune checkpoint inhibition and chimeric antigen receptor (CAR) T cell therapy, respectively. This is largely due to the immunosuppressive microenvironment that inhibits T cell infiltration, survival, and persistence in solid tumors. Reversal of cancer immunosuppression is therefore crucial for the success of immunotherapies directed against solid tumors. Myeloid cells, including macrophages and myeloid-derived suppressor cells (MDSCs), are key immunosuppressive cell types, particularly in immunologically cold tumors such as pancreatic and brain cancers that are characterized by the absence of a sufficient population of pre-existing T cells. Recent work shows that ECs provide an instructive niche for macrophage differentiation and alternative polarization that is known to induce immunosuppression [14]. Interestingly, our work shows that transformed ECs in the glioma microenvironment locally express and release interleukin (IL)-6, thereby acting as a vascular niche that promotes spatially proximate macrophages to undergo alternative activation, suggesting that endothelial transformation induces macrophage-mediated cancer immunosuppression [15]. Consistent with these findings, a recently published work shows that radiation-induced endothelial transformation promotes alternative polarization in tumor-associated macrophages [13]. We identified a peroxisome proliferator-activated receptor (PPAR)-γ/ hypoxia-inducible factor (HIF)-2α/ arginase-1-mediated axis that controls vascular niche-mediated macrophage polarization [15]. Interestingly, IL-6 deficiency in ECs reverses macrophage-mediated immunosuppression and improves mouse survival, suggesting that endothelial transformation plays a crucial role in vascular niche-mediated cancer immunosuppression in the microenvironment.
In sum, vascular detransformation may represent a promising therapeutic strategy for targeting cancer that normalizes and reduces tumor-associated blood vessels, induces re-expression of VEGFR-2 in tumor ECs, inhibits endothelial mesenchymalization, and activates perivascular niche immunity in the tumor microenvironment (Figure 2). The molecular mechanisms underlying endothelial transformation remain largely unknown, but this is likely to take place via stimulus-dependent genetic, epigenetic, and/or metabolic reprogramming. Identification of the regulatory node, such as the key kinase(s) and transcriptional factor(s), will aid in the discovery and development of novel therapeutic targets for inhibition of vascular detransformation treatment. Thus, new therapies using vascular detransformation either as a stand-alone treatment or as a combined treatment with radiotherapy, chemotherapy, molecular targeted therapy, antiangiogenic therapy, or immunotherapy may serve as effective next-generation antivascular approaches in cancer treatment.
Figure 2. Vascular Detransformation Serves as a Promising Therapeutic Strategy for Cancer.
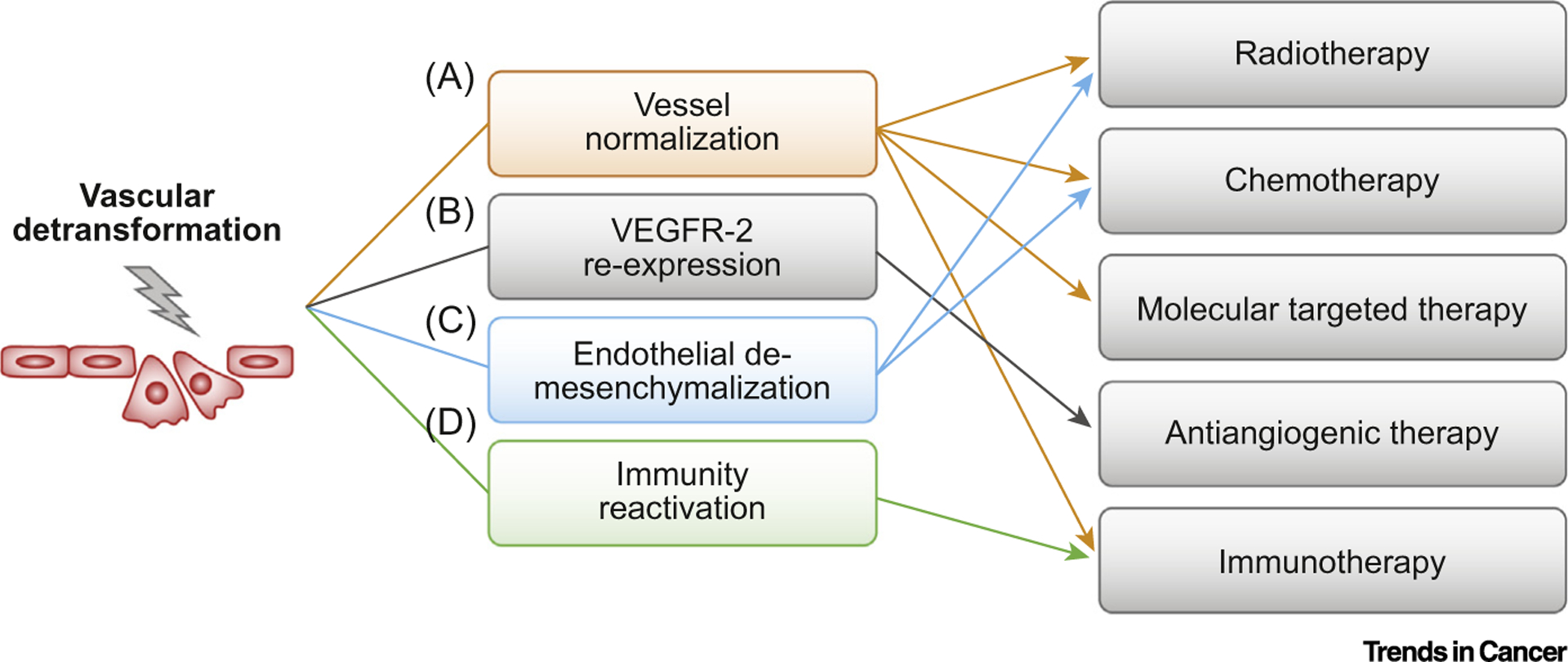
Vascular detransformation results in multiple beneficial consequences including vessel normalization, endothelial cell (EC) re-expression of vascular endothelial growth factor receptor (VEGFR)-2, EC de-mesenchymalization, and immunity reactivation in cancer. (A) Vessel normalization reduces intratumor hypoxia, increases drug delivery, and promotes T cell infiltration, thereby enhancing the effects of radiotherapy, chemotherapy, molecular targeted therapy, and immunotherapy. (B) EC re-expression of VEGFR-2 sensitizes tumor-associated ECs to antiangiogenic therapy that targets VEGF. (C) Inhibition of EC mesenchymalization sensitizes tumor-associated ECs to chemotherapy and radiation. (D) Reactivation of immunity in the perivascular niche promotes T cell survival and activation in the tumor microenvironment, thereby improving T cell-based immunotherapy outcomes.
Acknowledgments
This work was supported in part by National Institutes of Health grants R01NS094533 and R01NS106108, an American Association for Cancer Research Judah Folkman Award, an American Heart Association Innovative Project Award, a B*Cured Foundation Brain Cancer Investigator Award, a National Brain Tumor Society Sharpe Award, and a McCabe Award (to Y.F.).
References
- 1.Carmeliet P and Jain RK (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov 10, 417–427 [DOI] [PubMed] [Google Scholar]
- 2.Kovacic JC et al. (2012) Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation 125, 1795–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeisberg EM et al. (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med 952–961 [DOI] [PubMed] [Google Scholar]
- 4.Medici D et al. (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med 16, 1400–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maddaluno L et al. (2013) EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498, 492–496 [DOI] [PubMed] [Google Scholar]
- 6.Piera-Velazquez S and Jimenez SA (2019) Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol. Rev 99, 1281–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeisberg EM et al. (2007) Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 67, 10123–10128 [DOI] [PubMed] [Google Scholar]
- 8.Gasperini P et al. (2012) Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 72, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang M et al. (2016) c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J. Clin. Invest 126, 1801–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu T et al. (2018) PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun 9, 3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batchelor TT et al. (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11, 83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergers G and Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi SH et al. (2018) Tumour-vasculature development via endothelial-to-mesenchymal transition after radiotherapy controls CD44v6+ cancer cell and macrophage polarization. Nat. Commun 9, 5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He H et al. (2012) Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2–like macrophages. Blood 120, 3152–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q et al. (2018) Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2alpha. Nat. Commun 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]