

Abstract
Glutathione-mediated biotransformation in the liver is a well-known detoxification process to eliminate small xenobiotics but its impacts on nanoparticle retention, targeting and clearance are much less understood than liver macrophage uptake even though both processes are involved in the liver detoxification. By designing a thiol-activatable fluorescent gold nanoprobe that can bind to serum protein and be transported to the liver, we noninvasively imaged this biotransformation kinetics in vivo at high specificity and examined this process at the chemical level. Our results show that glutathione efflux from hepatocytes resulted in high local concentrations of both glutathione and cysteine in liver sinusoids, which transformed the nanoparticle surface chemistry, reduced its affinity to serum protein and significantly altered its blood retention, targeting and clearance. With this biotransformation, liver detoxification, a long-standing barrier in nanomedicine translation, can be turned into a bridge toward maximizing targeting and minimizing nanotoxicity.
Liver detoxification is a natural defense response that the body uses to remove foreign materials; however, due to rapid uptake by mononuclear phagocyte system (MPS) in the liver1, 2, 3, it often dramatically shortens the blood retention of engineered nanoparticles, prevents them from efficiently targeting diseases and retains them in the body for a long time, which can induce long-term nanotoxicity and hamper their clinical translation, particularly for those non-degradable ones composed of toxic elements or heavy metals4, 5. However, liver detoxification also plays an important role in minimizing toxicities of small xenobiotics. For instance, glutathione (GSH)-mediated biotransformation is one of the most common liver detoxification strategies to eliminate lipophilic molecules and heavy metals6. As the most abundant biothiol (~10 mM) in the liver, glutathione is synthesized inside hepatocytes and constantly effluxes into liver sinusoids through the fenestrated sinusoidal endothelium7 (Fig. 1a), followed by being transported to other compartments of the body8, 9. Within the liver, the nucleophilic cysteinyl residue of glutathione is highly reactive to electrophilic metabolites or heavy metals, reducing their toxicity, increasing their hydrophilicity and enhancing their clearance through either hepatobiliary pathway or renal system10, 11. However, it is still largely unknown how this well-known glutathione-mediated biotransformation impacts nanoparticle transport such as blood retention, disease targeting, and clearance even though this physiological process occurs constantly in our body.
Figure 1. Interactions of ICG4-GS-Au25 with sinusoidal glutathione efflux in the liver.
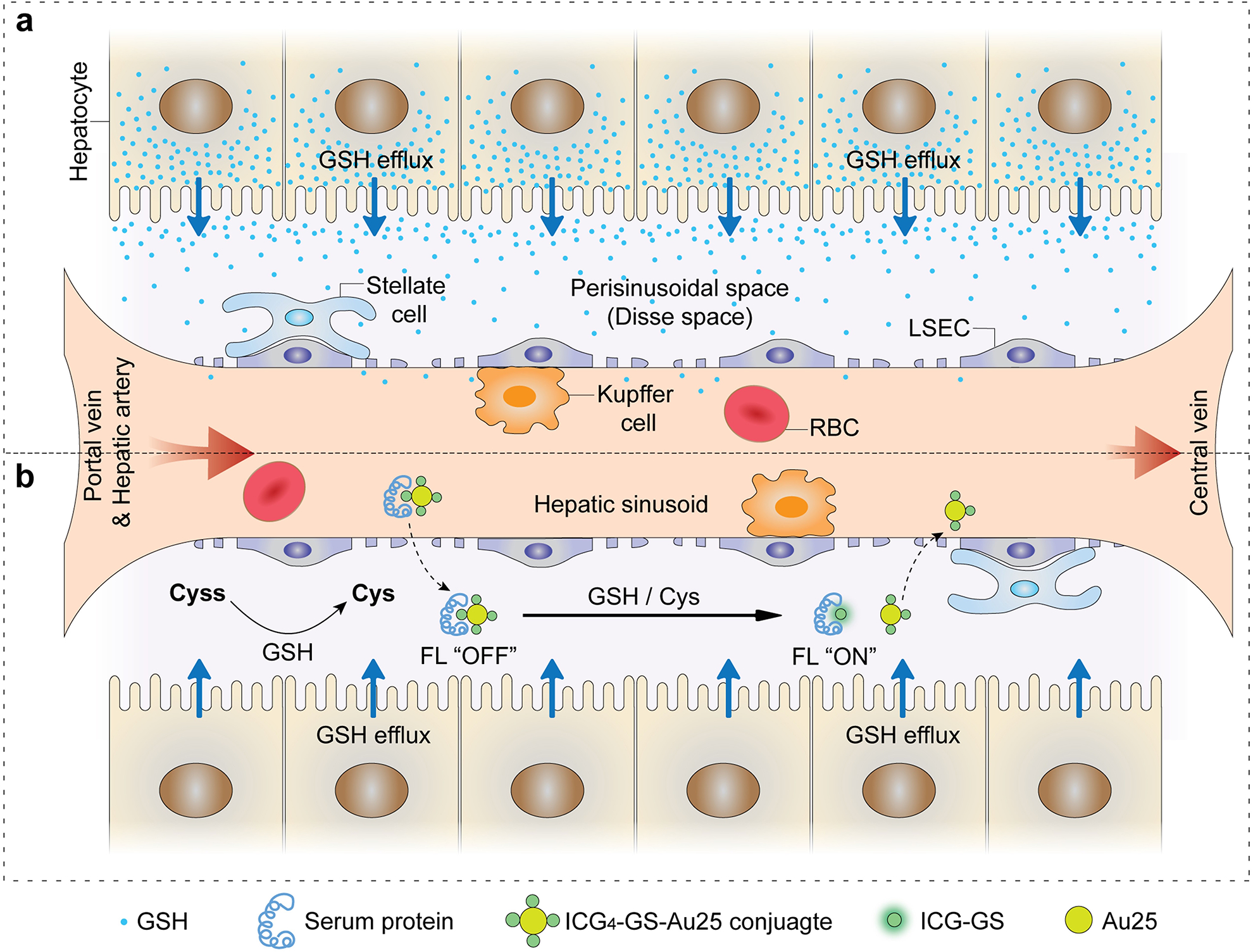
a, Sinusoidal glutathione efflux in the liver: reduced glutathione (GSH) is efficiently synthesized inside hepatocytes and consistently transported to the perisinusoidal space (Disse space), which then diffuses into the sinusoids through the well-fenestrated liver sinusoidal endothelial cells and joins systematic circulation. b, When ICG4-GS-Au25 conjugates enter liver sinusoids, they can diffuse across endothelial fenestrations due to the significantly reduced blood velocity in liver sinusoids as compared to that in arteries and veins. Glutathione efflux from the hepatocytes reduces extracellular cystine (Cyss) to cysteine (Cys), which, along with the glutathione itself, react with ICG4-GS-Au25 conjugates by displacing protein-binding ICG-GS from the surface of Au25, recovering the fluorescence of ICG and transforming serum-protein bound non-renal clearable ICG4-GS-Au25 nanoclusters to renal clearable ones.
Compared to liver MPS-mediated detoxification of engineered nanoparticles, glutathione one is much less understood because of the following challenges: 1) liver detoxifications mediated by MPS and glutathione are often entangled, making it hard to exactly pinpoint the role of glutathione-mediated biotransformation in the nanoparticle transport; 2) there is a lack of imaging techniques for real-time noninvasive monitoring of glutathione-mediated biotransformation in the liver; 3) it is very challenging to recover engineered nanoparticles for detailed chemical analysis after in vivo biotransformation. Therefore, to unambiguously unravel this process, we designed and constructed a specific nanoprobe, ICG4-GS-Au25, that can reach liver sinusoids, escape MPS uptake, report glutathione-mediated biotransformation kinetics noninvasively and be recovered from the urine afterwards for further analysis at the chemical level.
Interestingly, serum protein-bound and non-renal clearable ICG4-GS-Au25 became renal clearable as ICG-GS was gradually displaced by not only glutathione but also cysteine derived from sinusoidal glutathione efflux (Fig. 1b). Because of this biotransformation, the clearance pathways of ICG and GS-Au25 were not sacrificed after conjugation while tumor targeting of both ICG and GS-Au25 were greatly enhanced. This hepatic glutathione-mediated biotransformation is not limited to Au25 cluster but also observed from larger AuNPs ranging from 5nm to 100nm in core size. Moreover, renal clearance of 100nm nanoparticles and tumor targeting of small molecule drugs can be enhanced with this biotransformation strategy by designing them to efficiently react with glutathione or cysteine in the liver. These findings clearly demonstrate that hepatic glutathione-mediated biotransformation could be used to modulate in vivo transport and nano-bio interactions of many nanomedicines.
Design and synthesis of ICG-GS-Au25 nanoprobes
Glutathione-coated Au25 nanoclusters, GS-Au25 (Au25(SG)18), were chosen as the model because they are well-defined and their ultrasmall size allows them to clear out through kidneys12. In addition, gold nanoparticles are thiol (e.g. glutathione) reactive due to strong Au-S bonding. However, since GS-Au25 nanoclusters have little affinity to serum proteins and minimal hepatic accumulation12, near-infrared (NIR) dye indocyanine green (ICG) was conjugated onto the GS-Au25 to enhance their hepatic delivery because ICG has high affinity to serum proteins and is rapidly transported to the liver and taken up by hepatocytes rather than by liver MPS cells13. Thus, conjugation of ICG onto GS-Au25 nanoclusters is expected to enhance their transport to the liver while minimizing liver MPS uptake. Moreover, strong distance-dependent photoinduced electron transfer between ICG and Au25 enables facile monitoring of thiol conjugation in vivo: once ICG is on the particle surface, ICG NIR emission is completely quenched; whereas once ICG is released from the particle surface, the NIR emission could be fully recovered (Fig. 2a).
Figure 2. Characterization of ICG-GS-Au25 nanoprobes.
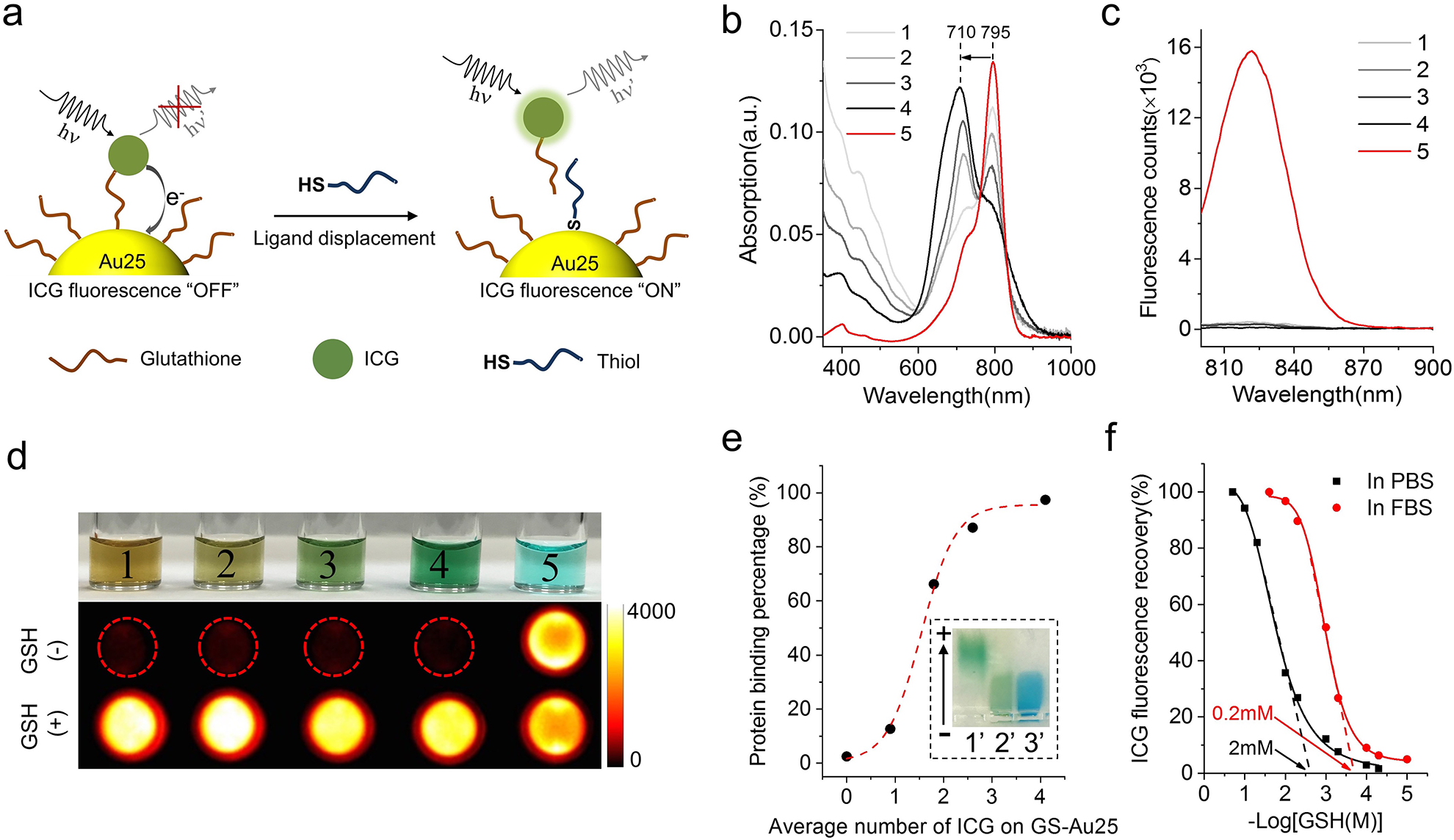
a, Schematic of thiol-activatable ICG-GS-Au25 nanoprobe: when ICG is conjugated onto glutathione ligands on Au25, the NIR fluorescence of ICG is severely quenched due to photoinduced electron transfer; once the ICG-GS ligand is displaced by biothiol molecules and detached from the Au25 surface, the electron transfer process is disrupted and NIR fluorescence of ICG is recovered concurrently. b, Absorption profiles of GS-Au25 conjugated with an average number of 0.9 (1), 1.8 (2), 2.6 (3), 4.1 (4) ICG molecules and free ICG (5). c, Fluorescence spectra of species 1–5 in PBS with the same amount of ICG (excited at 760nm), indicating that GS-Au25 can efficiently quench the fluorescence of ICG regardless of the number of ICG on each Au25. d, Color picture of species 1–5 dissolved in PBS (top) and the corresponding ICG fluorescence signals (middle) taken with an in vivo imaging system. The bottom shows fluorescence signals of the same amount of species 1–5 dissolved in PBS containing 10mM GSH (pH adjusted to 7.4). e, serum protein binding percentage of GS-Au25 and GS-Au25 conjugated with an average number of 0.9, 1.8, 2.6 and 4.1 ICG molecules after 15 min incubation in 50% FBS at 37°C. Protein bound and non-bound portions were separated by gel electrophoresis and quantified by ICP-MS. Inset is a color picture showing gel electrophoresis results of GS-Au25 conjugated with an average number of 4.1 ICG molecules (ICG4-GS-Au25), (1’) ICG4-GS-Au25 in PBS, (2’) ICG4-GS-Au25 in 50% FBS, (3’) 50% FBS stained with 1% CBB dye. f, Percentage of ICG fluorescence recovery after incubating ICG4-GS-Au25 in PBS or 50% FBS with different concentrations of GSH (pH adjusted to 7.4) for 10 min at 37°C. The threshold GSH concentration was estimated by finding the x-axis intercept of the tangent line at the maximal first derivative (slope) point of the respective curve. All the measurements were repeated twice with similar results.
As shown in Fig. 2b, the characteristic absorption of ICG blue shifted from 795 nm to 710 nm with increasing number of ICG on Au25 surface due to dipole-dipole coupling of multiple ICG on the same particle14. Unlike the absorption, ICG emission was completely quenched regardless of the number of ICG on the Au25 (Fig. 2c), indicating efficient electron transfer between each ICG and Au25. However, this electron transfer process was disrupted as soon as ICG was detached from Au25, resulting in an instantaneous “turn-on” of ICG fluorescence (Fig. 2d). Serum protein binding of ICG-GS-Au25 also depends on the number of ICG on particle surface. For GS-Au25 with ~1 ICG, it has low serum protein adsorption efficiency (~13% bound to serum protein). However, the protein adsorption efficiency reached ~97% once GS-Au25 were conjugated with ~4 ICG molecules (ICG4-GS-Au25) (Fig. 2e and Supplementary Fig. 4). ICG4-GS-Au25 was therefore chosen for the following studies because of its high affinity to serum proteins. The “turn-on” fluorescence of ICG4-GS-Au25 was glutathione-concentration dependent and the threshold glutathione concentration for activating ICG emission of ICG4-GS-Au25 is ~ 2 mM in phosphate buffered saline (PBS). Interestingly, in the presence of serum proteins, the threshold concentration is lowered to ~ 0.2 mM (Fig. 2f). The one-order decrease of glutathione threshold-concentration is fundamentally due to the binding of ICG4-GS-Au25 to serum protein, which likely weakened Au-S bond strength, making ICG-GS more easily to be released from Au25. Consistent with the glutathione-dependent activation of ICG fluorescence, incubating ICG4-GS-Au25 in different concentrations of glutathione reduced its protein binding affinity as ICG on the particle surface was gradually displaced (Supplementary Fig. 5). These results clearly indicate that interaction of ICG4-GS-Au25 with serum protein strongly depends on local thiol concentration and glutathione conjugation can be in situ monitored through the “turn-on” of ICG fluorescence.
Hepatic glutathione-mediated biotransformation of ICG4-GS-Au25
While GS-Au25 was rapidly eliminated out of the body through the urine with ~65% ID at 30 min p.i., only ~33% ID of gold was found in the urine in the same period for ICG4-GS-Au25 (Fig. 3a). However, the amount of gold cleared into the urine for ICG4-GS-Au25 increased by 73% at 24 h p.i. (~57% ID), becoming more comparable to that of GS-Au25 (~71% ID at 24 h p.i.), which only increased by 9% from 30 min to 24 h p.i.. This dramatic decrease in renal clearance during the early elimination phase is fundamentally because that serum protein binding of ICG4-GS-Au25 significantly increased its hydrodynamic diameter (HD) from ~3.4 nm (in PBS) to ~8.2 nm (in BSA) in vivo without inducing aggregation (Supplementary Fig. 6), which is above the kidney filtration threshold (~6 nm)15. Therefore, ICG4-GS-Au25 was no longer immediately eliminated through the kidneys but retained in blood longer than GS-Au25. As shown in Fig. 3b, the 24h blood retention (AUC: ~96.7 h×% ID/g) and clearance (CL: ~1.0 mL/h) of ICG4-GS-Au25 were nearly 2 times longer and 2 times slower than those (~49.3 h×% ID/g, ~2.0 mL/h) of GS-Au25, respectively. Serum protein binding not only prevented GS-Au25 from rapid renal clearance but also enhanced its hepatic transport. Liver accumulation of ICG4-GS-Au25 is ~7% ID/g, nearly 20 times higher than that of GS-Au25 (~0.35% ID/g) at 1h p.i. However, after 24 hours the liver accumulation of Au25 dropped to ~4% ID/g, only 6 times higher than that (~0.65% ID/g) of non-conjugated GS-Au25 (Supplementary Fig. 7). Long-term studies revealed ICG4-GS-Au25 accumulation in liver and other major organs continued to decrease over time, suggesting it can eventually clear out of the body (Supplementary Fig. 8). These kidney and liver clearance kinetics are distinct from those of engineered nanoparticles that go through liver MPS-mediated detoxification, which accumulate predominantly and persistently in the liver5, 16, 17. While Au25 nanoclusters were eliminated mainly through the urinary system, ICG cleared primarily via the hepatobiliary system with ~60% ID in feces at 24 h p.i. (Fig. 3c). This unique bi-pathway clearance of ICG4-GS-Au25 implied that ICG4-GS-Au25 were dissociated in the body.
Figure 3. Effect of sinusoidal glutathione efflux on the in vivo behavior of ICG4-GS-Au25.
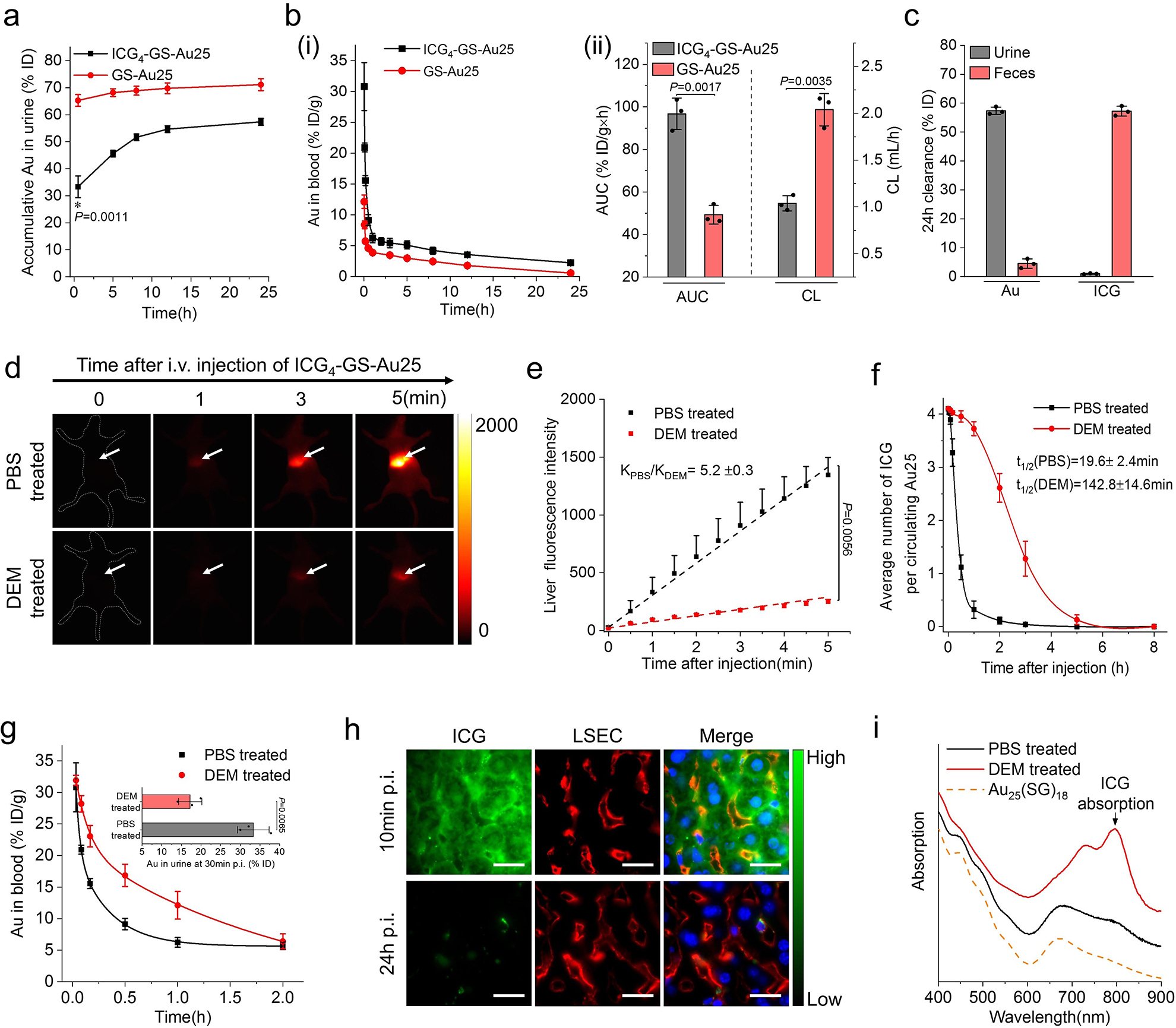
a, Renal clearance kinetics of GS-Au25 (n=3 mice) and ICG4-GS-Au25 (n=3 mice) after intravenous injection in BALB/c mice. b, (i) Pharmacokinetics of GS-Au25 (n=3 mice) and ICG4-GS-Au25 (n=3 mice) after intravenous injection. (ii) Area under the pharmacokinetics curve (AUC) and clearance parameters of GS-Au25 (n=3 mice) and ICG4-GS-Au25 (n=3 mice) derived from their pharmacokinetics measurements. c, ICG and Au clearance in urine and feces at 24 h after intravenous injection of ICG4-GS-Au25 (n=3 mice). d, Representative time-series noninvasive fluorescence imaging of PBS-treated (control) and DEM-treated mice intravenously injected with ICG4-GS-Au25. The white arrow indicates liver area. e, Liver ICG fluorescence kinetics of PBS-treated (n=3) mice and DEM-treated (n=3) mice in the first 5 min after intravenous injection of the same ICG4-GS-Au25. f, Average number of ICG molecules on each circulating Au25 in PBS-treated (n=3) mice and DEM-treated (n=3) mice after intravenous injection of ICG4-GS-Au25 (see Supplementary Fig. 13 for quantification methods). g, The first 2 h Au pharmacokinetics of PBS-treated (n=3) and DEM-treated (n=3) mice following intravenous injection of the same ICG4-GS-Au25. Inset is the Au renal clearance of PBS-treated (n=3) mice and DEM-treated (n=3) mice at 30min post intravenous injection of ICG4-GS-Au25. h, Fluorescence imaging of liver tissue slides of normal mice at 10 min and 24 h after intravenous injection of ICG4-GS-Au25. Liver sinusoidal endothelial cells (LSECs) were immunostained by anti-CD16/CD32 antibodies (red channel) and cell nuclei were counterstained with DAPI (blue channel). Scale bar, 15μm. i, Absorption profiles of the Au nanoclusters excreted in urine from either PBS-treated (n=3) mice or DEM-treated (n=3) mice intravenously injected with ICG4-GS-Au25. Absorption of Au25(SG)18 is included as a reference. Experiments of figure 3d and 3e were repeated 3 times with similar results. Experiments of figure 3f and 3g were performed once with corresponding controls. Experiments of other figures were repeated twice with similar results. Statistical significance is evaluated using a two-sample unequal variances (Welch’s) t-test. Data points are reported as mean values with error bar denoting the standard deviation.
To further unravel the dissociation mechanism, we investigated the integrity of ICG4-GS-Au25 in vivo by noninvasively monitoring the time-dependent ICG fluorescence after systemic administration. While ICG4-GS-Au25 is almost non-fluorescent during extra-hepatic blood circulation, strong ICG emission was immediately observed from the liver region after intravenous injection of the particles (Supplementary Fig. 9), indicating ICG was quickly dissociated from Au25 after entering liver, which was likely induced by sinusoidal glutathione. To confirm the pivotal role of sinusoidal glutathione efflux in the in vivo dissociation of ICG from Au25, we pretreated BALB/c mice with diethyl maleate (DEM) to temporarily inhibit GSH efflux in the liver18, 19. While DEM treatment did not significantly affect the liver uptake of ICG (Supplementary Fig. 11), the ICG emission signals from the liver were dramatically reduced in DEM-treated mice compared to that of PBS-treated mice injected with the same ICG4-GS-Au25 (Fig. 3d and supplementary movie). Quantitative comparison of the liver fluorescence kinetics revealed that inhibition of glutathione efflux with DEM slowed down the ICG4-GS-Au25 dissociation kinetics for more than 5 times (kPBS/kDEM=5.2±0.3) (Fig. 3e). In agreement with the in vivo fluorescence imaging, the ICG dissociation half-life of circulating ICG4-GS-Au25 in DEM-treated mice was ~7 times longer than that in PBS-treated mice (142.8±14.6 min vs 19.6±2.4 min) (Fig. 3f). The significant reduction of ICG releasing rate in DEM-treated mice dramatically increased the blood retention of Au25 and slowed down its renal clearance as compared to that in PBS-treated mice (Fig. 3g). Moreover, depletion of liver macrophage did not affect the dissociation of ICG4-GS-Au25 in vivo, indicating that liver MPS uptake was not involved in the observed dissociation (Supplementary Fig. 12). The distribution and clearance of ICG4-GS-Au25 in liver sinusoids were further investigated by immunohistochemistry (Fig. 3h). ICG fluorescence mainly accumulated in hepatocytes at 10 min p.i., indicating that the dissociated ICG-GS moieties were quickly taken up by the hepatocytes. At 24h p.i., ICG fluorescence signals in hepatocytes almost completely disappeared, consistent with the observed efficient hepatobiliary clearance of ICG after dissociation. These results further confirmed that glutathione mediated the dissociation of ICG4-GS-Au25 in liver sinusoids and hepatobiliary route was the major clearance pathway for the dissociated ICG-GS. Noteworthily, some bright and punctate ICG fluorescence distributed near the walls of sinusoids were observed both 10min and 24h p.i.. These long-lasting ICG fluorescence dots colocalized with either liver macrophages (Kupffer cells, etc.) or LSECs (Supplementary Fig. 14), suggesting that a very small portion of ICG4-GS-Au25 were sequestered by those scavenger cells even though the majority of them was eliminated through the hepatic glutathione-mediated biotransformation.
To gain more insights into this biotransformation process in vivo, the Au nanoclusters excreted in the urine were extracted and characterized (Fig. 3i). The absorption spectrum of Au nanoclusters in the urine of PBS-treated mice was almost identical to that of GS-Au25 and negligible ICG absorption was observed, indicating that ICG was completely removed before Au25 reached the urine and the Au25 structure remained intact after in vivo biotransformation. In contrast, Au nanoclusters excreted by DEM-treated mice exhibited an evident ICG absorption peak in addition to Au25 absorption even though the average number of ICG per Au25 is only ~0.5, which further validated the important role of hepatic glutathione efflux in the in vivo dissociation of ICG4-GS-Au25 and their renal clearance as well.
Cysteine involved in hepatic biotransformation of ICG4-GS-Au25
While glutathione-mediated biotransformation in liver sinusoids undoubtedly played a key role in modulating in vivo transport of ICG4-GS-Au25, similar to its function in enhancing the elimination of small xenobiotics, whether other biothiols might be involved in the displacement deserves a thorough investigation. Since the biotransformed Au25 can be recovered from the urine, we further analyzed the surface chemistries of excreted Au25 in the urine. The surface ligands of excreted Au25 were obtained by a two-phase ligand-exchange reaction (Fig. 4a) and then analyzed by high-performance liquid chromatography (HPLC) according to the established method20, 21. Surprisingly, more than half of the original ligands were replaced by cysteine (Cys, ~42%) and cysteinylglycine (Gly-Cys, ~17%) rather than just glutathione (Fig. 4b), suggesting that cysteine and cysteinylglycine were also involved in the biotransformation of ICG4-GS-Au25 in vivo. Since GSH exported from hepatocytes is known to readily reduce the abundant extracellular (plasma) cystine (~200 μM) to cysteine through a thiol-disulfide exchange reaction22, 23, the locally generated high concentration of cysteine was also very likely involved in the displacement. Indeed, incubating GSH and cystine at 1:1 molar ratio in PBS for just 5 min resulted in the rapid conversion of ~60% cystine to cysteine (Supplementary Fig. 16), which in turn could displace ICG-GS from the surface of Au25. To further quantitatively verify the significance of sinusoidal cysteine in modifying nanoparticle surface, we compared the surface ligands of the well-defined Au25(SG)18 after 10 min in vivo circulation and 10 min in vitro incubation in freshly acquired blood. Taking advantage of the defined chemical formula of Au25(SG)18, we found that the average number of cysteine ligand per Au25 was ~1.3 after in vivo circulation, 6.5-fold more than that (~0.2) of Au25 clusters incubated in in vitro blood, consolidating that locally high concentration of cysteine generated through the glutathione-cystine reaction in liver sinusoids was also responsible for the ICG-GS displacement (Fig. 4c). The primary role of sinusoidal glutathione and cysteine in biotransforming surface chemistry of Au25 was further verified using our previous polyethylene glycol (PEG) coated AuNPs24 as probes (Supplementary Fig. 17), where both glutathione and cysteine were observed on the particle surface at a molar ratio of 1:3 after 10 min in vivo circulation. While cysteinylglycine was also observed from Au25 in the urine, the absence of cysteinylglycine ligand on Au25(SG)18 obtained in the plasma indicated that cysteinylglycine ligand was not conjugated onto the particles in the liver or during the blood circulation. Since cysteinylglycine is extensively generated from the enzymatic (γ-glutamyltransferase, GGT) degradation of GSH within kidney proximal tubules25, 26, kidney slides were prepared from ICG4-GS-Au25 injected mice and imaged (Fig. 4d). The ICG fluorescence “turn-on” also occurred predominantly at the brush-border of kidney proximal tubules, where the highest GGT level in kidney is presented27, while the glomeruli, where blood filtration takes place, remain to be relatively dark. These results indicated that the liver detoxified ICG-GS-Au25 nanoclusters (with reduced number of ICG on Au25 and no longer bind to serum proteins) were filtered through glomeruli but underwent additional ligand displacement by cysteinylglycine in the lumen of kidney proximal tubule, where the left ICG was further removed from Au25.
Figure 4. Analysis of the Au25 surface chemistry after in vivo biotransformation.
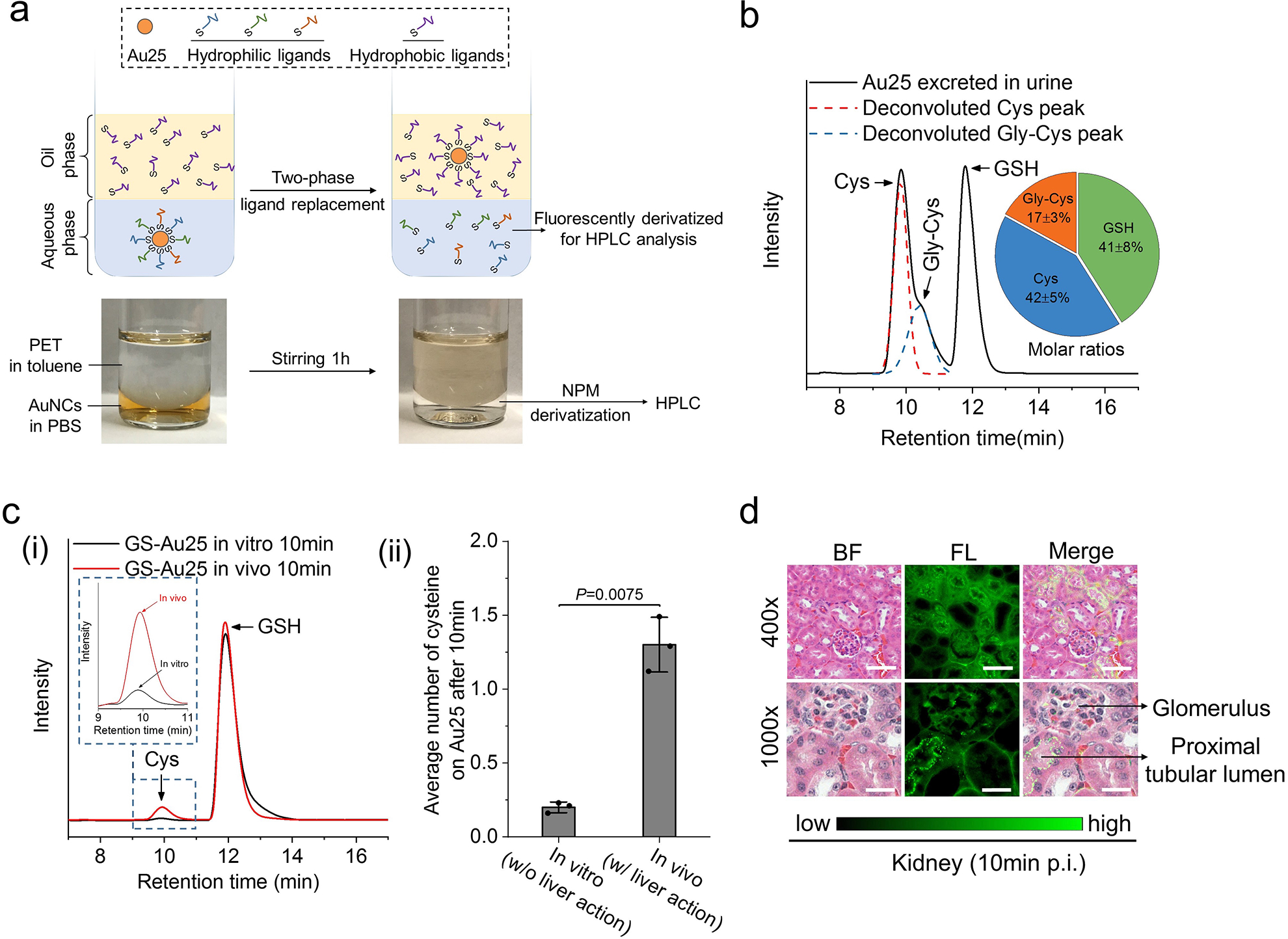
a, Illustration of the two-phase ligand-exchange method for the analysis of surface ligands on Au25 after biotransformation. Excess amount of hydrophobic thiol ligand, 2-phenylethanethiol (PET), was used to displace those hydrophilic ligands on the surface of Au25 and transfer Au25 to the organic phase while leaving the original hydrophilic ligands in aqueous phase, which were then fluorescently derivatized by N-(1-pyrenyl)maleimide (NPM). The color pictures below show Au25 purified from the urine of ICG4-GS-Au25 injected mice before and after the two-phase ligand-exchange reaction. b, Representative HPLC results of the surface ligands of Au25 excreted in the urine of ICG4-GS-Au25 injected mice (n=3). Molar ratios of the three surface ligands were presented as the inserted pie chart. The detailed HPLC peak assignment and quantification can be found in Supplementary Fig.15. c, (i) Representative HPLC results of the surface ligands of GS-Au25 (Au25(SG)18) after 10 min in vitro incubation in freshly-acquired mouse blood at 37°C and 10 min in vivo circulation with both renal arteries temporarily clamped to prevent rapid renal clearance of GS-Au25. The inset is a zoom-in figure of the cysteine signal peaks. (ii) Average number of cysteine ligands on each Au25 surface after in vitro incubation or in vivo circulation for 10 min (n=3 mice). d, Fluorescence imaging of H&E stained kidney tissues of normal mice at 10 min after intravenous injection of ICG4-GS-Au25 (see Supplementary Fig. 18 for additional images). Images of two magnifications (400x and 1000x) show that ICG fluorescence light-up is mainly at the wall of proximal tubular lumens (characterized by the dense microvilli) rather than the glomeruli. Scale bar, 50μm in 400x images; 20 μm in 1000x images. Experiments of figure 4b were repeated twice with similar results. Experiments of figure 4c were performed once with corresponding controls. Experiments of figure 4d were repeated 3 times with similar results. Statistical significance is evaluated by two-sample unequal variances (Welch’s) t-test. Data points are reported as mean values with error bar denoting the standard deviation.
Enhanced tumor targeting and retention of ICG4-GS-Au25
Although both free ICG and GS-Au25 have low tumor targeting efficiency, integration of ICG and GS-Au25 significantly enhanced tumor targeting for both ICG and GS-AuNPs while retaining their respective clearance pathways due to the hepatic biotransformation. Although only 4 ICG molecules were conjugated onto GS-Au25, the accumulation of Au25 in the tumor was improved to ~5.4% ID/g, 2.3 times higher than non-conjugated ones (Fig. 5a). Such improvement mainly originated from the enhanced blood retention of Au25 due to the serum protein adsorption, which prevented Au25 from rapid renal clearance without inducing significant MPS uptake. In addition, integration of ICG with Au25 also enhanced the tumor targeting of ICG. While both free ICG and ICG4-GS-Au25 passively targeted tumors, ICG4-GS-Au25 had 27 times higher tumor targeting efficiency (~4.1% ID/g vs. ~0.15% ID/g) than free ICG at 24 h p.i. This enhancement is mainly because zwitterionic GS-Au25 slowed down the liver uptake of ICG and greatly increased ICG blood retention (AUC) to ~23.7 h×% ID/g, more than 12 times longer than that (~1.9 h×% ID/g) of free ICG (Fig. 5b). More interestingly, not only tumor targeting efficiency but tumor imaging contrast and time window of ICG were also significantly enhanced (Fig. 5c). For ICG4-GS-Au25 injected mice, tumor contrast index reached the threshold (CI=2.5) 24 h p.i. and kept increasing for the rest 2 weeks with the maximum contrast index of ~7.6, 4 times higher than that (maximum CI= ~1.8) of free ICG (Fig. 5d). This prolonged tumor-imaging time window originates from that ICG could be retained in tumor with a half-life of ~310.3 h after conjugation with Au25, which is over 80 times longer than that (~3.8 h) of free ICG (Fig. 5e). Meanwhile, ICG signals of ICG4-GS-Au25 in the background tissues continued decreasing at a rate only slightly slower than that of free ICG (half-life: 13 h vs. 3.5 h, Supplementary Fig.19). The greatly enhanced tumor retention of ICG was fundamentally because ICG-Au25 conjugates effectively entered the tumor microenvironment, which was confirmed by the immediate turn-on of ICG fluorescence in tumor microenvironment after injection of glutathione into the tumor tissues ex vivo (Fig. 5f and Supplementary Fig.20). At the in vivo level, ICG-Au25 conjugates were taken up by the cells in the tumor microenvironment, turned on fluorescently inside cells due to high concentration of intracellular glutathione and lighted up the tumor for prolonged time. These results were consistent with the efficient endocytosis of ICG4-GS-Au25 and releasing of ICG from Au25 in tumor cells in vitro (Supplementary Fig. 22 and 23) as well as tumor tissue imaging (Fig. 5g, see Supplementary Fig. 24 for silver staining of tumor tissues).
Figure 5. Tumor targeting of ICG4-GS-Au25.
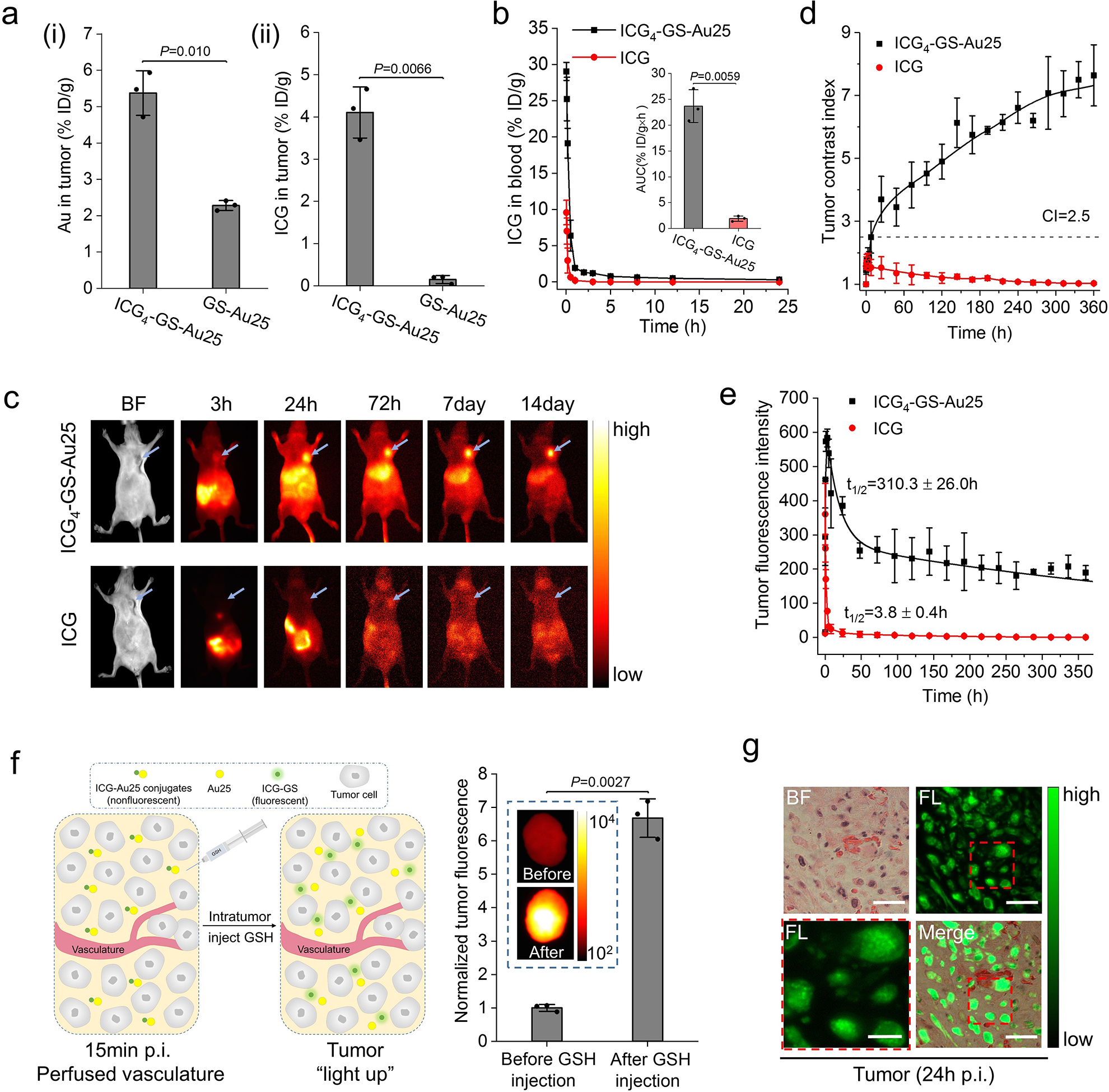
a, MCF-7 tumor targeting efficiency of ICG4-GS-Au25 (n=3 mice) in terms of Au (i) and ICG (ii) at 24 h post injection as compared to those of GS-Au25 (n=3 mice) and free ICG (n=3 mice), respectively. b, Comparison of ICG blood pharmacokinetics between mice intravenously injected with free ICG (n=3 mice) and ICG4-GS-Au25 (n=3 mice). The inserted is the area under the ICG pharmacokinetics curve (AUC) for free ICG and ICG4-GS-Au25. c, Representative in vivo fluorescence images of MCF-7 tumor bearing nude mice at various time points after intravenous injection of either ICG4-GS-Au25 or equal amount of free ICG. Blue arrows indicate the tumors. d, Time-dependent tumor contrast index (CI= mean fluorescence intensity of tumor/ mean fluorescence intensity of contralateral background tissue) after intravenous injection with ICG4-GS-Au25 (n=3 mice) and free ICG (n=3 mice). e, Time-dependent tumor fluorescence intensity for ICG4-GS-Au25 (n=3 mice) and free ICG (n=3 mice). f, On the left is an illustration of the experiment. At 15 min post intravenous injection of ICG4-GS-Au25, the mice were sacrificed and thoroughly perfused with PBS to remove ICG-Au25 conjugates in blood vessels, and then tumors were resected and intratumorally injected with 10mM GSH (pH 7.4) to induce the dissociation of ICG-Au25 in tumor microenvironment. On the right is the tumor ICG fluorescence intensity (normalized) before and after introducing GSH (n=3 tumors). The significantly enhanced ICG fluorescence after GSH injection proves that ICG-Au25 conjugates as a whole effectively entered the tumor microenvironment. Inserted is the fluorescence image of a representative tumor before and after intratumor injection of GSH. g, Fluorescence imaging of H&E stained tumor tissue at 24 h after intravenous injection of ICG4-GS-Au25 (see Supplementary Fig.20 for additional images). ICG was efficiently taken up by the tumor cells and located in endosome-like membrane-bound compartments (see the zoom-in image at lower left), consistent with the observations at the in vitro level (Supplementary Fig.21). Scale bar, 6 μm for the zoom-in image at lower left and 20 μm for the rest. Experiments of figure 5c and 5g were repeated 3 times with similar results. Experiments of other figures were repeated twice with similar results. Statistical significance is evaluated by two-sample unequal variances (Welch’s) t-test. Data points are reported as mean values with error bar denoting the standard deviation.
Conclusions
Using ICG4-GS-Au25 as a model, we unraveled how glutathione efflux from the hepatocytes transformed the nanoparticle in vivo. By gradually displacing ICG-GS from the Au25 surface with glutathione or cysteine in the liver sinusoids, this biotransformation process alters not only the interactions of the nanoparticle with serum proteins but also its clearance and targeting profiles (Fig. 6). While serum protein adsorption and liver uptake have been long-standing barriers in the clinical translation of nanomedicines, combining hepatic glutathione-mediated biotransformation and temporary serum protein binding prolongs blood retention of both ICG and Au25, enables them to target tumors much more efficiently, whereas “off-target” ICG and Au25 are still eliminated through the hepatic and renal pathways, respectively.
Figure 6. Liver glutathione-mediated biotransformation impacting in vivo transport of ICG4-GS-Au25.
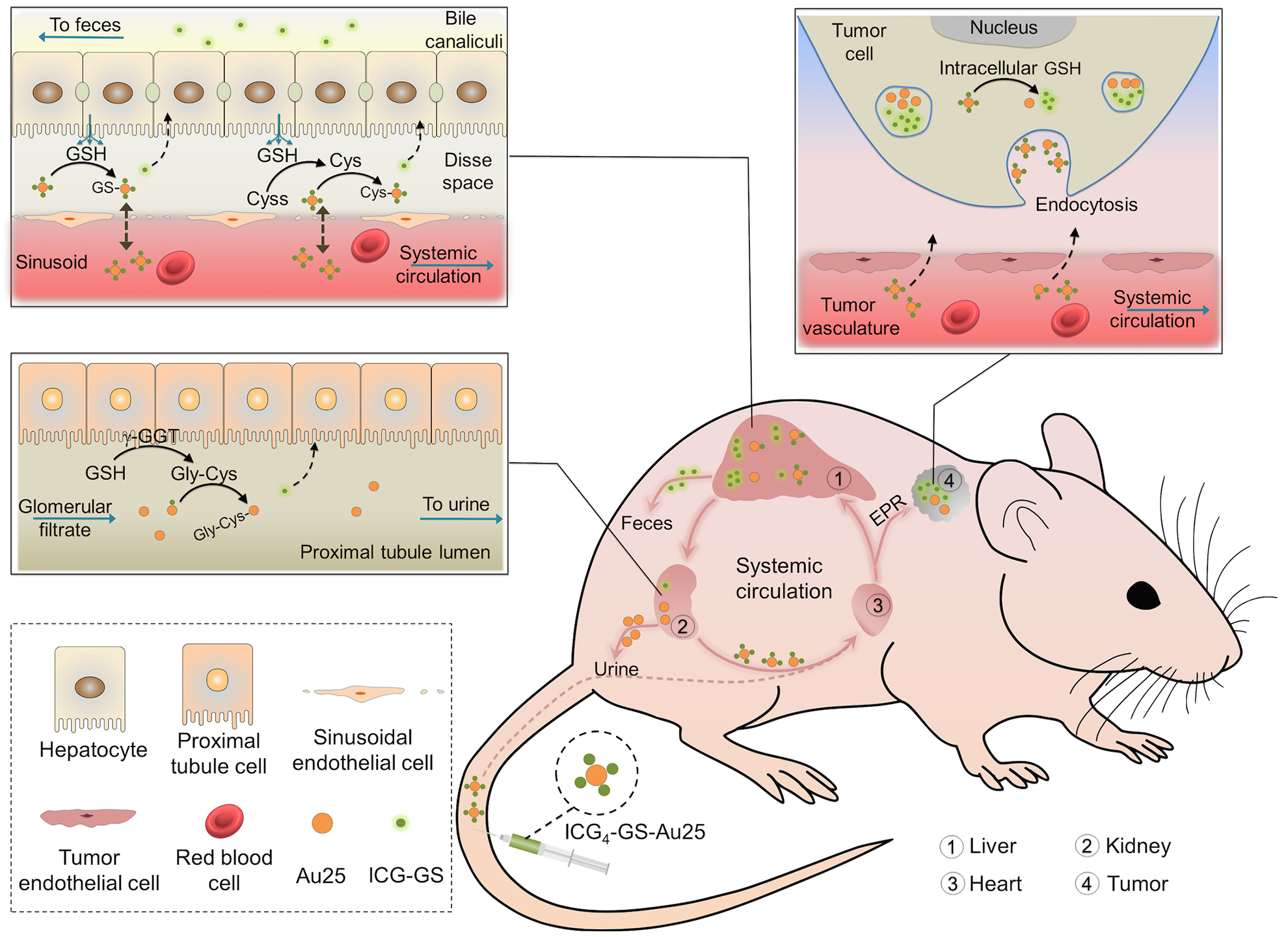
After intravenous administration, ICG4-GS-Au25 nanoclusters immediately bound to serum proteins. The protein-bound ICG4-GS-Au25 had an overall hydrodynamic size larger than kidney filtration threshold and thus was prevented from rapid renal elimination but was in part transported to the liver sinusoids, where the local high concentrations of glutathione and cysteine resulting from sinusoidal glutathione efflux displaced some or all of the ICG-GS from the surface of Au25, reducing the protein-binding affinity of ICG4-GS-Au25. The displaced ICG-GS was then taken up by hepatocytes and eliminated through the hepatobiliary pathway whereas the biotransformed ICG-GS-Au25 nanoclusters were back to the blood circulation and target tumor along with ICG4-GS-Au25 through EPR effect. When the biotransformed ICG-GS-Au25 nanoclusters circulated to the kidneys, those with low affinity to serum proteins passed the glomerular filtration and underwent additional surface modifications in kidney proximal tubules, where the left ICG-GS on Au25 nanoclusters were further displaced by cysteinylglycine, the extracellular metabolite of glutathione in proximal tubules. For those biotransformed ICG-GS-Au25 that still bound to serum proteins, they remained in the blood stream and continued to target the tumor. In the tumor, ICG4-GS-Au25 along with its biotransformed derivatives entered the tumor microenvironment and were internalized by the cells in tumor. The high concentration of intracellular glutathione then induced the dissociation of ICG-GS from Au25 within the cells and the tumor was lighted up for prolonged time. Some cell types such as Kupffer cells in liver and many stromal cells in tumor are omitted in this scheme for clarity.
This glutathione-mediated biotransformation is not limited to ultrasmall Au25 but also observed from a series of PEGylated AuNPs ranging from 5nm to 100nm in core sizes, though the biotransformation rate was found to exponentially decrease with the increase in their core sizes due to more reactive surface gold atoms on smaller AuNPs (Supplementary Fig. 25). Noteworthily, for engineered nanoparticles that adsorb serum proteins, liver MPS uptake and glutathione-mediated biotransformation are coupled together to impact their in vivo transport and nanoparticle size plays an important role. For example, we found that 3nm 4-mercaptobenzoic acid (MBA) coated AuNPs cleared into urine 10-fold more efficiently than that of 6nm MBA-AuNPs (26.5% vs 2.5% ID at 24h p.i.), despite both of them strongly bind to serum proteins (Supplementary Figure 26). For small nanoparticles, even though they bind to serum proteins, glutathione-mediated biotransformation can enhance their renal clearance significantly; however, for large nanoparticles, serum protein binding will promote macrophage uptake though we believe their surface chemistries are also partially modified by the biotransformation process.
Although we did observe size-dependency in the glutathione-mediated biotransformation of PEGylated AuNPs, large nanoparticles can still go through the biotransformation effectively once they are designed to efficiently react with glutathione or cysteine in the liver. For example, ~100nm thiol-degradable GS-Au polymeric nanoparticles can be disintegrated into 2–3nm renal-clearable oligomers by the hepatic glutathione-mediated biotransformation, resulting in nearly 35% ID of such polymeric nanoparticles being cleared out into urine at 48h p.i. (Supplementary Fig. 27), more than 20 times higher than that of large non-renal clearable GS-AuNPs28. Such biotransformation can also be utilized to enhance the tumor targeting of small anticancer drugs. Conjugating protein-binding ICG onto the cisplatin prodrug (see Supplementary Fig. 28) through a thiol-cleavable disulfide linker slowed down the rapid renal clearance of the prodrug, prolonged its blood retention more than 4 times and improved tumor targeting more than 2.5-fold; meanwhile, the “off-target” prodrug was still efficiently eliminated through the kidney with ~ 76% ID in urine at 24h p.i..
Combining these results clearly indicates that hepatic glutathione-mediated biotransformation, a well-known detoxification process for the body to remove small xenobiotics, can be used to modulate the in vivo transport of nanoparticles; so that disease targeting of nanoparticles can be maximized while nonspecific accumulation and health hazards of “off-target” ones are minimized. Moreover, these fundamental understandings of nano-bio interactions at the chemical level and in vivo transport of nanoparticles will further advance physiology and toxicology on the nanoscale, opening up more opportunities for clinical translation of nanomedicines.
Methods
Materials and equipment.
ICG-NHS was purchased from Intrace Medical (Switzerland) while all the other chemicals were obtained from Sigma-Aldrich (USA) and used as received unless specified. Four distinct-sized bare AuNPs (citrate protected) and Clodronate liposomes (Clophosome) were purchased from NanoComposix, Inc (USA) and FormuMax Scientific, Inc (USA), respectively. The related absorption spectra were measured with a Virian 50 Bio UV-Vis spectrophotometer. Fluorescence spectra were acquired with a PTI QuantaMasterTM 30 fluorometer. The core size and hydrodynamic diameter of nanoparticles were measured 200kV JEOL 2100 transmission electron microscope and Malvern ZS90 particle size analyzer. In vivo fluorescence images were recorded using a Carestream In-Vivo FX Pro imaging system. Optical imaging of cultured cells and tissue slides was obtained with an Olympus IX-71 inverted fluorescence microscope coupled with Photon Max 512 CCD camera (Princeton Instruments). Electron microscopic images of cultured cells were acquired using a 120 kV Tecnai G2 spirit transmission electron microscope (FEI) equipped with a LaB6 source. Shimadzu Prominence Modular HPLC equipped with UV-Vis detector (SPD-20A) and fluorescence detector (RF-20A) was used for the separation of derivatized thiol ligands. Agarose gel electrophoresis was carried out in a Bio-Rad Mini-Sub Cell GT system. Agilent 7900 inductively coupled plasma mass spectrometry (ICP-MS) was used for the quantitative analysis of Au in biological samples. Human breast cancer MCF-7 cell line was used for in vitro cell study and in vivo xenograft model as well. Animal studies were performed according to the guidelines and ethical regulations of the University of Texas System Institutional Animal Care and Use Committee. BALB/c mice (strain code 047, 6–8 weeks old, weighing 20–25 g) were purchased from Envigo. All mice were randomly allocated and housed under standard environmental conditions (23±1°C, 50±5% humidity and a 12/12h light/dark cycle) with free access to water and standard laboratory food.
Synthesis of GS-Au25 (Au25(SG)18) and ICG-GS-Au25 conjugates.
Atomically monodisperse Au25(SG)18 nanoclusters were synthesized according to the reported method29. Characterization of the synthesized Au25(SG)18 can be found in Supplementary Fig. 1. For the synthesis of ICG4-GS-Au25, 4mg ICG-NHS (dissolved in DMSO) was added into 6mg GS-Au25 aqueous solution and the mixture was vortexed for 3h. Then ICG-GS-Au25 conjugates were purified after removing unconjugated ICG dye through centrifugation in the presence of ethanol. The conjugates were again re-dispersed in 1X PBS buffer and purified by Amicon Ultra centrifuge filters to remove any unconjugated GS-Au25 nanoclusters. For the synthesis of GS-Au25 conjugated with fewer number of ICG molecules, reduced feeding ratios of ICG-NHS/GS-Au25 were adopted and the products were purified following the same protocol as that of ICG4-GS-Au25. Additional characterization of the synthesized ICG4-GS-Au25 is in Supplementary Fig. 2.
Synthesis of different-sized ICG/PEG-AuNPs, MBA-AuNPs and GS-Au polymeric nanoparticles.
Different-sized ICG/PEG-AuNPs were obtained by surface-ligand exchange of different-sized bare AuNPs with ICG-GSH and PEG-SH (average molecular weight ~2000) in water for 1h at 55°C under vigorous stirring. The resulting ICG/PEG-AuNPs were purified by gel filtration column to remove free ICG-GSH and PEG-SH. The 4-mercaptobenzoic acid (MBA) coated AuNPs (MBA-AuNPs) were synthesized by adding 1M HAuCl4 aqueous solution to 10mM MBA solution containing 50% (v/v) methanol at a molar ratio of 1:3. The mixture was first stirred at room temperature for 15 min and then brought to 0°C in ice cold water followed by introducing ice cold 150mM NaBH4 aqueous solution with a NaBH4: HAuCl4 molar ratio of 2:1. The reaction was further proceeded for another 30min and dark MBA-AuNPs were collected by centrifugation. Large and small MBA-AuNPs were obtained by 25% native polyacrylamide gel electrophoresis (PAGE) separation of the synthesized MBA-AuNPs. The ~100 nm (HD) GS-Au polymeric nanoparticles were synthesized by adding 1M HAuCl4 aqueous solution to GSH aqueous solution (~12mM) at a molar ratio of 1:3 with gentle stirring. Colorless GS-Au polymer precipitates were formed subsequently and collected by centrifugation. The precipitates were redispersed in water and freeze dried to yield GS-Au polymeric nanoparticles. Conjugation of ICG to GS-Au polymeric nanoparticles were performed in a similar way to that of GS-Au25 using amine-reactive ICG-NHS.
Synthesis of ICG-GSSG, ICG-GSH, ICG-Cis-Pt prodrug.
ICG-GSSG conjugates were synthesized by mixing ICG-NHS (dissolved in DMSO) and GSSG (glutathione oxidized, dissolved in water) at a molar ratio of 1:5 and at pH~8. The reaction proceeded for 2h under agitation and ICG-GSSG could be purified by acetone-induced precipitation and subsequent dialysis against ultrapure water. ICG-GSH was facilely obtained by reducing ICG-GSSG with TCEP (tris(2-carboxyethyl)phosphine) in aqueous solution. ICG-Cis-Pt prodrug was synthesized by linking the primary amine group of ICG-GSSG with the carboxyl group of Cis-Pt prodrug through a typical EDC/NHS coupling reaction. The mono-carboxylated Cis-Pt prodrug (cis,cis,trans-[Pt(NH3)2Cl2(OH)(O2CCH2CH2COOH)]) was synthesized according to the reported protocol30.
Quantification of the number of ICG molecules per GS-Au25 nanocluster.
The purified ICG-GS-Au25 conjugates were dissolved in ultrapure water containing 20mM cysteine (pH adjusted to 7.4 by NaOH) and incubated for 30 min (protected from light exposure) to completely release ICG from Au25 surface (see Supplementary Fig. 3), then ICG concentration was quantified by its characteristic absorption peak at 780nm and a pre-established concentration vs. absorption standard curve while the Au25 concentration was quantified by ICP-MS analysis of the Au content.
Depletion of tissue GSH in vivo.
The temporary depletion of tissue GSH was achieved by a single injection of diethyl maleate (DEM). DEM was intraperitoneally administered into BALB/c mice at a dose of 1mL/kg body weight ~40 min prior to the injection of ICG4-GS-Au25 or other probes. The success of tissue GSH depletion was verified by the rapid decrease in plasma GSH level following DEM administration (Supplementary Fig. 10). A single injection of DEM decreased plasma GSH level down to ~12% of the initial value (~30 μM) and maintained this low GSH level for ~2–3 h before the gradual recovery of plasma GSH, consistent with the literatures31,32. Quantification of GSH was carried out by a modified Tietze enzymatic recycling assay established previously33.
In vivo fluorescence imaging of ICG4-GS-Au25.
Hair-removed BALB/c mice (~25 g/mouse) were pretreated with PBS, DEM (1mL/kg body weight), Clophosome (7mg/mL clodronate disodium, 200uL/mouse as recommended by the manufacturer) and control liposomes, respectively. Then under 3% isoflurane anesthesia, mouse was tail-vein catheterized and prone-positioned on the imaging stage. 150μL ICG4-GS-Au25 (20μM, in PBS) was tail vein injected following by a sequential time-series imaging collection. The fluorescence imaging parameters were set as follow: EX760nm/EM830nm; 10s exposure time; 2×2 binning.
Quantification of the average number of ICG molecules per circulating Au25 nanocluster.
Blood samples (~30μL) from BALB/c mice were collected retro-orbitally at different time points after i.v. injection of ICG4-GS-Au25. Immediately after each collection, blood sample was mixed with 100μL ice cold PBS buffer containing 2% EDTA and centrifuged at 1200g for 5min to precipitate blood cells. Then the supernatant was equally divided into two aliquots and 100μL PBS or dithiothreitol (DTT, 20mM) PBS solution were added to each aliquot, respectively. After 15 min incubation, the ICG fluorescence of each aliquot was measured and the increase (dithiothreitol treated vs. PBS treated) in fluorescence intensity was normalized to Au amount (quantified by ICP-MS) for each time point. The percentage of ICG fluorescence increment per Au at each time point with respect to that of ICG4-GS-Au25 dissolved directly in plasma (simulated as the 0 min time point) was used to calculate the average number of ICG molecules per circulating Au25. A scheme of these experiment procedure is included in Supplementary Fig. 13.
Pharmacokinetics and biodistribution study of ICG4-GS-Au25.
PBS or DEM pretreated BALB/c mice were i.v. injected with 150 μL ICG4-GS-Au25 (~20μM, in PBS) per mouse. At certain time point post injection, blood sample (~30μL) was retro-orbitally collected and weighed, followed by the addition of 500μL lysis buffer containing 20mM dithiothreitol (pH~8) to liberate ICG from Au25 nanocluster and recover the quenched fluorescence of ICG. Then fluorescence of dithiothreitol treated blood sample was measured by the in vivo imaging system to quantify ICG in blood. Afterwards, the blood sample was completely dissolved in freshly made aqua regia and the Au amount in blood was analyzed by ICP-MS. Biodistribution of ICG4-GS-Au25 in different organs/tissues was quantified in a similar way. Organs/tissues were collected and weighed following the sacrifice of mice and then completely digested in aqua regia to determine the Au content via ICP-MS. The ICG content in different organs/tissues was quantified by its NIR fluorescence after dissociation from Au25 by homogenizing organs/tissues in lysis buffer containing 20mM dithiothreitol (pH~8). Standard curves of ICG fluorescence vs. concentration were constructed in the respective control organ/tissue lysate.
Tissue slide imaging with optical microscopy.
BALB/c mice were sacrificed at 10min following the i.v administration of 150μL/mouse ICG4-GS-Au25, ICG-GSH, ICG or PBS, respectively. The liver and kidneys were then collected and fixed immediately in 10% neutral buffered formalin, followed by standard dehydration and paraffin embedding. The embedded tissues were then sectioned into 4 μm slices and H&E stained. The final slides were visualized under Olympus IX-71 fluorescence microscope equipped with ICG filters set (Chroma). Tumor tissues were obtained from BALB/c nude mice bearing MCF-7 xenograft tumors 24h after i.v. injection of ICG4-GS-Au25 and processed with the same procedures as those for liver and kidney tissues. To visualize the gold nanoclusters in tumor tissues under bright-field optical microscope, silver staining was used to enhance their size and contrast. Tumor slides were first dewaxed in xylene and incubated in silver staining solution containing 0.05M AgNO3 and 1mM hydroquinone for half an hour. Afterwards, tumor slides were washed with copious ultrapure water and dried in lab oven (65°C) for imaging under bright-field of Olympus IX-71 microscope.
Fluorescent immunohistochemistry of liver tissue.
Liver tissues from BALB/c mice i.v. injected with ICG4-GS-Au25 were harvested at 10 min as well as 24 h p.i. and immediately fixed with 4% freshly prepared paraformaldehyde PBS solution for 2 h. Then the fixed liver tissues were immersed in 20% sucrose PBS solution overnight at 4°C before being embedded in freezing medium (OTC). The embedded liver tissues were sectioned into 5 μm-thickness slides in cryostat and blocked with PBS containing 5% normal goat serum for 1h at RT. Afterwards, liver slides were incubated with either rat anti-mouse F4/80 (Invitrogen) or 2.4G2 (CD16/CD32, BD Biosciences) primary antibodies in PBS containing 1% goat serum overnight at 4°C to stain macrophage or liver sinusoidal endothelial cell (LSEC), respectively. Primary antibody binding was visualized using goat anti-rat IgG secondary antibody conjugated with Alexa Fluor 647 (Invitrogen). Cell nuclei were counterstained with DAPI for 10 min before slides were mounted and subject to fluorescent microscopy.
Extract gold nanoparticles from the urine and blood samples for surface ligand analysis.
The excreted AuNPs in the urine were purified first by adding 10% (m/v) 5-sulfosalicylic acid to precipitate urine proteins and followed by 10KDa centrifuge filter to separate AuNPs from the small molecules (urea etc.) presented in the urine. The separated AuNPs were further purified by 2% agarose gel electrophoresis and gel filtration column (Sephadex LH-20, GE Healthcare). The extraction of AuNPs in blood was similar to that in the urine. The blood from AuNPs injected mice was collected by cardiac puncture and centrifuged to obtain the plasma, then plasma protein was precipitated by adding 5-sulfosalicylic acid and the remaining AuNPs in supernatant were purified following the same protocol as we did for the urine. To extract sufficient AuNPs from the blood after circulation, renal arteries were clamped right before the i.v. injection of AuNPs in order to prevent the rapid blood elimination of ultrasmall AuNPs through the kidney filtration.
HPLC analysis of gold nanoparticle surface ligands.
Toluene of 1mL was added to 500μL PBS containing purified AuNPs, then 2- pheylethanethiol was added to the top toluene layer and the mixture was vigorously stirred for 1–2h at room temperature to transfer gold cores to the organic phase while leaving the original hydrophilic ligands in the aqueous phase. Afterwards, TCEP was added to the PBS solution to reduce any disulfides formed in this process before HPLC analysis of the thiol ligands. A well-established NPM derivatization method34 was used to quantitatively analyze the thiol ligands in PBS. The derivatives were separated by Shimadzu HPLC equipped with TSKgel column (Tosoh Bioscience) and fluorescence detector. The HPLC conditions were as follow: mobile phase 20/80 H2O/MeCN containing 1ml/L acetic acid and 1ml/L phosphoric acid; isocratic flow 0.6ml/min; column temperature 25°C; detector wavelength EX330nm/EM380nm.
Statistics and reproducibility.
Welch’s t-test was used to compare two groups of data and analysis of variance (ANOVA) was used for the comparison of multiple data points among data sets. Data were reported as mean value with error bar representing the standard deviation. Differences were considered statistically significant when P < 0.05. Unless otherwise specified, all the experiments were repeated at least twice with similar results to ensure reproducibility.
Supplementary Material
Acknowledgements
We would like to acknowledge in part the financial support from National Institutes of Health (NIH) (R01DK103363 and R01DK115986), Cancer Prevention Research Instituted of Texas (CPRIT) (RP160866), Welch Research Foundation (AT-1974-20180324) and Cecil H. and Ida Green Professorship in System Biology of J.Z. from the University of Texas at Dallas. We also acknowledge E. Hernandez and Dr. J.T. Hsieh from The University of Texas Southwestern Medical Center for tissue slides preparation.
Footnotes
Competing interests
The authors declare no competing financial interests.
Supplementary information is available in the online version of the paper. Reprints and permission information is available online at www.nature.com/reprints. Correspondence and requests for materials should be addressed to J.Z.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Tsoi KM, MacParland SA, Ma X-Z, Spetzler VN, Echeverri J, Ouyang B, et al. Mechanism of hard-nanomaterial clearance by the liver. Nature materials 2016, 15(11): 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumours. Nature reviews materials 2016, 1(5): 16014. [Google Scholar]
- 3.Fischer HC, Liu L, Pang KS, Chan WC. Pharmacokinetics of nanoscale quantum dots: in vivo distribution, sequestration, and clearance in the rat. Advanced Functional Materials 2006, 16(10): 1299–1305. [Google Scholar]
- 4.Ye L, Yong K-T, Liu L, Roy I, Hu R, Zhu J, et al. A pilot study in non-human primates shows no adverse response to intravenous injection of quantum dots. Nature Nanotechnology 2012, 7(7): 453–458. [DOI] [PubMed] [Google Scholar]
- 5.Balasubramanian SK, Jittiwat J, Manikandan J, Ong C-N, Liya EY, Ong W-Y. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials 2010, 31(8): 2034–2042. [DOI] [PubMed] [Google Scholar]
- 6.Gu X, Manautou JE. Molecular mechanisms underlying chemical liver injury. Expert Rev Mol Med 2012, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comparative hepatology 2002, 1(1): 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplowitz N, Aw TY, Ookhtens M. The regulation of hepatic glutathione. Annu Rev Pharmacool Toxicol 1985, 25(1): 715–744. [DOI] [PubMed] [Google Scholar]
- 9.Ballatori N, Krance SM, Marchan R, Hammond CL. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol Aspects Med 2009, 30(1–2): 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochem Pharmacol 2002, 64(5–6): 1019–1026. [DOI] [PubMed] [Google Scholar]
- 11.Singhal RK, Anderson ME, Meister A. Glutathione, a first line of defense against cadmium toxicity. The FASEB Journal 1987, 1(3): 220–223. [DOI] [PubMed] [Google Scholar]
- 12.Du B, Jiang X, Das A, Zhou Q, Yu M, Jin R, et al. Glomerular barrier behaves as an atomically precise bandpass filter in a sub-nanometre regime. Nature Nanotechnology 2017, 12: 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shinohara H, Tanaka A, Kitai T, Yanabu N, Inomoto T, Satoh S, et al. Direct measurement of hepatic indocyanine green clearance with near‐infrared spectroscopy: Separate evaluation of uptake and removal. Hepatology 1996, 23(1): 137–144. [DOI] [PubMed] [Google Scholar]
- 14.Sun S, Ning X, Zhang G, Wang YC, Peng C, Zheng J. Dimerization of organic dyes on luminescent gold nanoparticles for ratiometric pH sensing. Angew Chem 2016, 128(7): 2467–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, et al. Renal clearance of quantum dots. Nat Biotechnol 2007, 25(10): 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dreaden EC, Austin LA, Mackey MA, El-Sayed MA. Size matters: gold nanoparticles in targeted cancer drug delivery. Therapeutic delivery 2012, 3(4): 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirn S, Semmler-Behnke M, Schleh C, Wenk A, Lipka J, Schäffler M, et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur J Pharm Biopharm 2011, 77(3): 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ookhtens M, Hobdy K, Corvasce M, Aw TY, Kaplowitz N. Sinusoidal efflux of glutathione in the perfused rat liver. Evidence for a carrier-mediated process. J Clin Invest 1985, 75(1): 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plummer JL, Smith BR, Sies H, Bend JR. [8] Chemical depletion of glutathione in vivo. Methods Enzymol 1981, 77: 50–59. [DOI] [PubMed] [Google Scholar]
- 20.Winters RA, Zukowski J, Ercal N, Matthews RH, Spitz DR. Analysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl) maleimide. Anal Biochem 1995, 227(1): 14–21. [DOI] [PubMed] [Google Scholar]
- 21.Parmentier C, Leroy P, Wellman M, Nicolas A. Determination of cellular thiols and glutathione-related enzyme activities: versatility of high-performance liquid chromatography–spectrofluorimetric detection. Journal of Chromatography B: Biomedical Sciences and Applications 1998, 719(1–2): 37–46. [DOI] [PubMed] [Google Scholar]
- 22.Jocelyn P The Standard Redox Potential of Cysteine‐Cystine from the Thiol‐Disulphide Exchange Reaction with Glutathione and Lipoic Acid. The FEBS Journal 1967, 2(3): 327–331. [DOI] [PubMed] [Google Scholar]
- 23.Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. The Journal of nutrition 2004, 134(3): 489–492. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Yu M, Ning X, Zhou C, Yang S, Zheng J. PEGylation and Zwitterionization: Pros and Cons in the Renal Clearance and Tumor Targeting of Near‐IR‐Emitting Gold Nanoparticles. Angewandte Chemie 2013, 125(48): 12804–12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tate SS, Meister A. γ-Glutamyl transpeptidase: catalytic, structural and functional aspects The Biological Effects of Glutamic Acid and Its Derivatives. Springer, 1981, pp 357–368. [DOI] [PubMed] [Google Scholar]
- 26.Paolicchi A, Sotiropuolou M, Perego P, Daubeuf S, Visvikis A, Lorenzini E, et al. γ-Glutamyl transpeptidase catalyses the extracellular detoxification of cisplatin in a human cell line derived from the proximal convoluted tubule of the kidney. Eur J Cancer 2003, 39(7): 996–1003. [DOI] [PubMed] [Google Scholar]
- 27.Hanigan MH, Pitot HC. Gamma-glutamyl transpeptidase–its role in hepatocarcinogenesis. Carcinogenesis 1985, 6(2): 165–172. [DOI] [PubMed] [Google Scholar]
- 28.Peng C, Gao X, Xu J, Du B, Ning X, Tang S, et al. Targeting orthotopic gliomas with renal-clearable luminescent gold nanoparticles. Nano research 2017, 10(4): 1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Z, Suhan J & Jin R One-pot synthesis of atomically monodisperse, thiol-functionalized Au 25 nanoclusters. J. Mater. Chem 19, 622–626 (2009). [Google Scholar]
- 30.Dhar S, Daniel WL, Giljohann DA, Mirkin CA & Lippard SJ Polyvalent oligonucleotide gold nanoparticle conjugates as delivery vehicles for platinum (IV) warheads. Journal of the American Chemical Society 131, 14652–14653 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber CA, Duncan CA, Lyons MJ & Jenkinson SG Depletion of tissue glutathione with diethyl maleate enhances hyperbaric oxygen toxicity. American Journal of Physiology-Lung Cellular and Molecular Physiology 258, L308–L312 (1990). [DOI] [PubMed] [Google Scholar]
- 32.Adams J, Lauterburg B & Mitchell J Plasma glutathione and glutathione disulfide in the rat: regulation and response to oxidative stress. J. Pharmacol. Exp. Ther 227, 749–754 (1983). [PubMed] [Google Scholar]
- 33.Rahman I, Kode A & Biswas SK Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nature protocols 1, 3159 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Winters RA, Zukowski J, Ercal N, Matthews RH & Spitz DR Analysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl) maleimide. Anal. Biochem 227, 14–21 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.