

Abstract
Adjusting to a wide range of light intensities is an essential feature of retinal rod bipolar cell (RBC) function. While persuasive evidence suggests this modulation involves phosphorylation by protein kinase C-alpha (PKCα), the targets of PKCα phosphorylation in the retina have not been identified. PKCα activity and phosphorylation in RBCs was examined by immunofluorescence confocal microscopy using a conformation-specific PKCα antibody and antibodies to phosphorylated PKC motifs. PKCα activity was dependent on light and expression of TRPM1, and RBC dendrites were the primary sites of light-dependent phosphorylation. PKCα-dependent retinal phosphoproteins were identified using a phosphoproteomics approach to compare total protein and phosphopeptide abundance between phorbol ester-treated wild type and PKCα knockout (PKCα-KO) mouse retinas. Phosphopeptide mass spectrometry identified over 1100 phosphopeptides in mouse retina, with 12 displaying significantly greater phosphorylation in WT compared to PKCα-KO samples. The differentially phosphorylated proteins fall into the following functional groups: cytoskeleton/trafficking (4 proteins), ECM/adhesion (2 proteins), signaling (2 proteins), transcriptional regulation (3 proteins), and homeostasis/metabolism (1 protein). Two strongly differentially expressed phosphoproteins, BORG4 and TPBG, were localized to the synaptic layers of the retina, and may play a role in PKCα-dependent modulation of RBC physiology. Data are available via ProteomeXchange with identifier PXD012906.
Significance:
Retinal rod bipolar cells (RBCs), the second-order neurons of the mammalian rod visual pathway, are able to modulate their sensitivity to remain functional across a wide range of light intensities, from starlight to daylight. Evidence suggests that this modulation requires the serine/threonine kinase, PKCα, though the specific mechanism by which PKCα modulates RBC physiology is unknown. This study examined PKCα phosophorylation patterns in mouse rod bipolar cells and then used a phosphoproteomics approach to identify PKCα-dependent phosphoproteins in the mouse retina. A small number of retinal proteins showed significant PKCα-dependent phosphorylation, including BORG4 and TPBG, suggesting a potential contribution to PKCα-dependent modulation of RBC physiology.
Keywords: Retina, Rod bipolar cell, Protein kinase C-alpha, Quantitative phosphoproteomics, TPBG, BORG4
1. Introduction
Rod bipolar cells (RBCs) are key retinal interneurons in the rod visual pathway that receive light-driven synaptic input from rod photoreceptors and drive retinal output via synapses onto AII amacrine cells. RBCs serve different visual functions depending on luminance conditions. When dark adapted, they are able to transmit single photon responses [1,2], allowing for useful vision in starlight. RBCs are also able to transmit contrast changes against dim background light [3,4], and have recently been shown to influence vision in daylight [5]. Little is known about how RBCs adjust their sensitivity and gain to transition between these modes, but compelling evidence suggests phosphorylation by protein kinase C-alpha (PKCα) may play a role.
PKCα is a serine/threonine protein kinase that undergoes calcium-dependent translocation from the cytosol to the plasma membrane, where it is activated upon binding to diacylglycerol (DAG). It is a powerful modulator of signal transduction pathways and is so abundant in RBCs that it is used as a cell marker to identify RBCs with retinal immunofluorescence [6–8]. The light response of RBCs is reflected in the b-wave of the dark-adapted electroretinogram (ERG), and comparison of ERGs from wild type (WT) and PKCα knockout (PKCα-KO) mice reveal that genetic deletion of PKCα results in increases in both amplitude and duration of the scotopic b-wave [9,10]. This effect is particularly dramatic at brighter light intensities, suggesting that PKCα modulates the light response in an intensity-dependent manner. If the effect of PKCα on the light response is due to its kinase activity, then RBC proteins phosphorylated by PKCα are also likely to be involved in the modulation of RBC activity.
We used a multiplexed tandem mass tag (TMT; [11]) mass spectrometry-based phosphoproteomics approach to identify proteins that were differentially phosphorylated between WT and PKCα-KO retinas in order to gain insight into the biochemical mechanisms and pathways that mediate the effect of PKCα in RBCs. Phosphopeptide abundance is expected to be dynamic, so TMT acquisition methods that have improved accuracy and wider dynamic ranges are necessary [12]. The larger number of replicates available with high-resolution instruments and TMT tags require improved data normalization and statistical testing methods, and we have successfully applied analysis techniques developed for large-scale protein expression studies [13] to phosphopeptide abundance data. Since PKCα-dependent changes in protein phosphorylation may be due to changes in total protein abundance, we also identified proteins that were differentially expressed between WT and PKCα-KO retinas.
2. Experimental procedures
2.1. Experimental design and statistical rationale
For quantification of immunofluorescence (Fig. 2C), we used retina sections from 4 WT and 3 TRPM-1-KO mice, each with 4 technical replicates. For the multiplexed TMT mass spectrometry experiments, we used 4 WT and 5 PKCα-KO mice. The 5 × 5 study design (one WT sample was lost) was the maximum number of samples accommodated by 10-plex TMT. For both total protein and phosphopeptide experiments, all nine samples were pooled after isobaric labeling and run simultaneously to reduce variability between samples. The statistical tests used are described within the Statistical analysis of differential expression section.
Fig. 2.
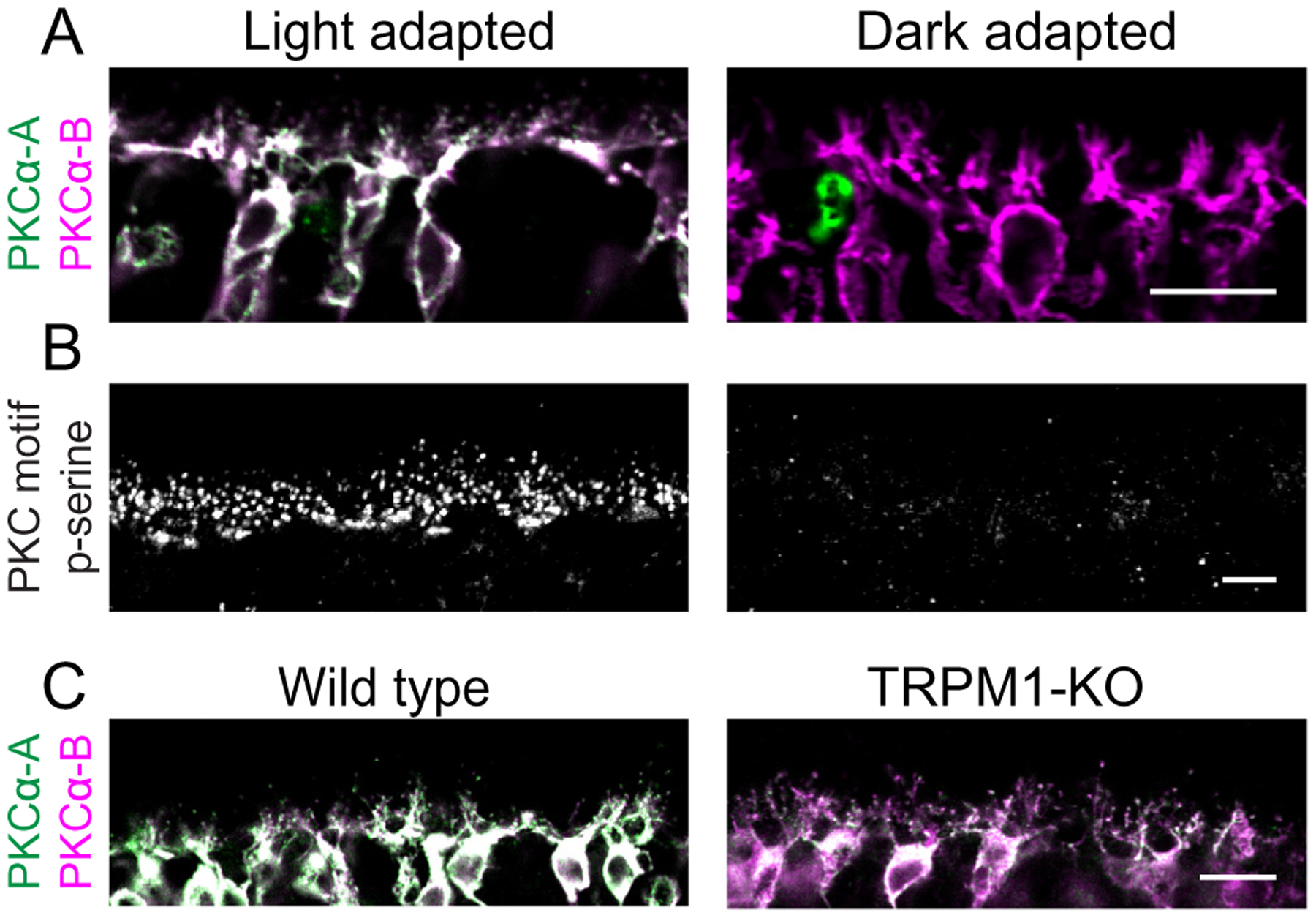
PKCα is active in light-adapted RBCs. (A) Light- and dark-adapted mouse retina sections double-labeled with two antibodies against PKCα; conformation-specific PKCα-A (green) binds only active PKCα, while PKCα-B (magenta) binds to both active and inactive PKCα. (B) Sections from light-adapted and dark-adapted mouse retina labeled with an antibody mixture against PKC motif phosphoserines (PKC motif p-serine). (C) WT and TRPM1-KO mouse retina sections double-labeled with conformation-specific PKCα-A (green) and conformation non-specific PKCα-B (magenta). Scale bars: 10 μm. OPL: outer plexiform layer.
2.1.1. Animals
All studies were approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University. Wild type mice used were C57BL/6 J (Jackson Laboratory; Bar Harbor, ME, USA; Cat# 000664). The PKCα-KO mouse strain was B6;129-Pkrcatm1Jmk/J (Jackson Laboratory; Cat# 009068; [10]). The TRPM1-KO mouse strain was TRPM1tm1Lex (Texas A&M Institute of Genomic Medicine; College Station, TX, USA; [14]).
2.1.2. PKCα activation by PMA treatment for immunoblotting or immunofluorescence
Freshly dissected retinas from WT and PKCα-KO mice were incubated at 37 °C in 1 μM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich; St. Louis, MO, USA; Cat# P8139) diluted in bicarbonate buffered Ames medium (US Biological Life Sciences; Salem, MA, USA; Cat# A1372–25) for 0 min, 15 min, 30 min, or 60 min. The retinas were then washed three times with Ames medium before being either fixed for cryo-sectioning or processed for western blotting.
2.1.3. Immunoblotting
Retinas were suspended in chilled lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% deoxycholate, 0.1% SDS) with 1× protease/phosphatase inhibitor cocktail (Cell Signaling Technology; Danvers, MA, USA; Cat# 5872). The retinas were then homogenized and incubated on ice for 1 h. Lysates were centrifuged for 15 min at 28,000 × g and 4 °C, and the pellets discarded. Lysates were stored at −20 °C. Equal quantities of WT and PKCα-KO retinal proteins were subjected to electrophoresis on precast NuPAGE 1 mm 4–12% Bis-Tris gels (Thermo Scientific; Waltham, MA, USA; Cat# NP0322BOX). The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes and then blocked with Odyssey Blocking Buffer (LI-COR Biosciences; Lincoln, NE, USA; Cat# 927–50,003) for 1 h. The blots were probed with primary antibody for 1 h at room temperature. The membranes were washed 3 × 5 min with TBST (Tris-buffered saline with 0.1% Tween-20) and then incubated for 1 h at room temperature with secondary antibody and washed 3 × 5 min with TBST. Immunoreactive bands were visualized with a LI-COR Odyssey CLx Imaging System at 700 or 800 nm.
Primary antibodies used for immunoblotting were rabbit anti-PKC motif phosphoserine [(R/KXpSX(R/K)] MultiMab mAb mix (1:250; Cell Signaling Technology; Cat# 6967), mouse anti-Cdc42EP4 (Borg4, 1:1000; Thermo Fisher Scientific; Cat# MA5–21509), rabbit anti-Borg4 (1:200; Bethyl Laboratories; Montgomery, TX, USA; Cat# A302–379A), rabbit anti-5 T4 (1:5000; Abcam; Cambridge, UK; Cat# ab129058), rabbit anti-NHERF1 (1:5000; Abcam; Cambridge, UK; Cat# ab3452). Secondary antibodies used were 680RD anti-rabbit (1:10,000; LI-COR Biosciences; Cat# 925–68,071) and 800CW anti-mouse (1:10,000; LI-COR Biosciences; Cat# 925–32,212).
2.1.4. Retinal immunofluorescence
Mouse eyecups were prepared by cutting the sclera behind the ora serrata and removing the cornea and lens. Eyecups were fixed for 30 min by immersion in 4% paraformaldehyde, followed by washing in PBS. The fixed tissue was cryoprotected via sequential immersion in 10, 20, and 30% sucrose, and then embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek; Torrance, CA, USA; Cat# 4583) and frozen. Sections were cut at 25 μm thickness on a cryostat and mounted onto glass slides, then air dried and stored at −20 °C. Thawed retina sections were blocked at room temperature for 1 h in Antibody Incubation Solution (AIS: 3% normal horse serum, 0.5% Triton X-100, 0.025% NaN3 in PBS). The sections were then incubated in primary antibody diluted in AIS for 1 h at room temperature. After washing with 3× with PBS, the sections were incubated for 1 h at room temperature in secondary antibody diluted 1:1000 in AIS. The slides were washed again 3× in PBS and coverslips applied with Lerner Aqua-Mount (Thermo Scientific; Cat# 13800).
Primary antibodies used for immunofluorescence were the same as used for immunoblotting unless indicated: rabbit anti-PKC motif phosphoserine [(R/KXpSX(R/K)] MultiMab mAb mix; 1:250), rabbit anti-PKCα (1:5000; Sigma-Aldrich; Cat# P4334), mouse anti-PKCα clone MC5 (1:5000; Sigma-Aldrich; Cat# P5704), rabbit anti-Borg4 (1:500), rabbit anti-5 T4 (1:500;), rabbit anti-NHERF1 (1:100). Secondary antibodies used were anti-rabbit-AF488 (1:1000; Jackson ImmunoResearch Labs; West Grove, PA, USA; Cat# 11–545–144) and anti-mouse-Cy3 (1:1000 Jackson ImmunoResearch Labs; Cat# 115–165–003).
2.1.5. Scanning confocal imaging
Confocal immunofluorescence images were taken with a Leica TCS SP8 X confocal microscope (Leica; Wetzlar, Germany) using a Leica HC PL APO CS2 63×/1.40 oil immersion objective (Leica; Cat#15506350) and Leica HyD hybrid detectors, or with an Olympus Fluoview 1000 microscope (Olympus; Tokyo, Japan) using a 60×/1.42 oil immersion objective. Laser lines used were AF488 (499 nm), and Cy3 (554 nm). Detection windows used were AF488 (509–544 nm) and Cy3 (564–758) nm. Brightness and contrast were adjusted using Olympus Fluoview software, Leica LAS X software, or ImageJ [15,16].
2.1.6. Preparation of retinas for TMT analysis
To maximize the difference between groups, retinas from wild type (n = 4) and PKCα-KO (n = 5) mice were extracted and treated for 1 h at 37 °C with 1 μM PMA diluted in bicarbonate buffered Ames medium. Following treatment, retinas were washed three times with Ames medium before being placed in chilled lysis buffer (50 mM HEPES, pH 8.5, 8 M urea, 1 mM NaF, 1 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 1 mM beta-glycerophosphate), and lysed by probe sonication (Sonic Dismembrator 60; Thermo Scientific) 3 × 15 s at a setting of 4 with cooling on ice for 30 s between treatments. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific; Cat# 223227) and approximately 500 μg of protein in 250 μL of lysis buffer was used for further processing. Protein disulfides were reduced with 5 μL of 1.25 M dithiothreitol at 55 °C for 30 min, then alkylated by adding 22 μL of 1 M iodoacetamide and incubation at room temperature in the dark for 15 min, followed by an additional 5 μL of 1.25 M dithiothreitol. Water was added to dilute the urea concentration to 2 M. Sequence grade modified trypsin (Promega; Madison, WI, USA; Cat# V5111) was added at a 25:1 protein:trypsin ratio and samples were incubated overnight at 37 °C before being acidified with trifluoroacetic acid (TFA) to a final concentration of 1%. Remaining particulates were removed by centrifugation at 16,000 xg for 10 min.
Peptides were purified by solid phase extraction using 1 cc (50 mg) Waters Sep-Pak Vac tC18 cartridges (Waters Corporation; Milford, MA, USA; Cat# WAT054960). Briefly, the cartridges were conditioned twice with 1 mL acetonitrile (ACN) and twice with 300 μL of 50% ACN/0.5% acetic acid, then equilibrated twice with 1 mL 0.1% TFA. The samples were loaded and passed through the bed, then were washed twice with 1 mL 0.1% TFA followed by 200 μL of 0.5% acetic acid. Finally, the samples were eluted twice with 500 μL of 50% ACN/0.5% acetic acid. Peptide concentrations were determined using the Pierce Quantitative Colorimetric Peptide Assay (Thermo Scientific; Cat# 23275). Approximately 300 μg of peptide was recovered from each digest. Fifteen μg of each sample was reserved for TMT analysis of total protein abundance, and the remainder was used for the phosphopeptide enrichment experiment.
2.1.7. Phosphopeptide enrichment
Phosphopeptides were enriched following previously described methods [17,18] with small modifications. Titanosphere TiO2 5 μm beads (GL Biosciences; Tokyo, Japan; Cat# 5020–75,000) were washed three times in 2 M lactic acid/50% ACN, then resuspended in the same solution at 24 mg/mL. Approximately 285 μg of dried peptide for each sample was resuspended in 950 μL of 2 M lactic acid/50% ACN and 100 μL (2.4 mg) of the bead suspension was added to each sample, ensuring an 8:1 ratio of beads:peptide. The bead:peptide mixture was rotated at room temperature for 1 h, then washed twice with 500 μL 2 M lactic acid/50% ACN, 0.1% TFA/50% ACN, then 0.1% TFA/25% ACN. The enriched phosphopeptides were eluted from the beads by vortexing in 100 μL of 50 mM K2HPO4 at pH 10 for 5 min. The elution step was repeated once and the 200 μL of eluate was dried by vacuum centrifugation.
The enriched phosphopeptides were then purified by solid phase extraction using UltraMicro Spin columns (The Nest Group Inc.; Southborough, MA, USA). The dried phosphopeptides were resuspended in 60 μL 1% TFA and the pH was tested to ensure the samples were acidic. The columns were conditioned three times with 100 μL 80% ACN/0.1% TFA and equilibrated three times with 50 μL of 0.1% TFA. The samples were loaded and passed through the columns three times, washed three times with 25 μL of 0.1% TFA, eluted three times with 50 μL 80% ACN/0.1% formic acid, and dried by vacuum centrifugation before TMT labeling.
2.1.8. TMT labeling and mass spectrometric analysis
In preparation for TMT labeling, nine dried unfractionated peptide samples (4 WT and 5 KO) and nine phosphopeptide enriched samples (4 WT and 5 KO) were dissolved in 25 μL of 100 mM triethylammonium bicarbonate buffer, and TMT 10-plex reagents (Thermo Scientific; Cat# 90110) were dissolved at a concentration of 15 μg/μL in anhydrous ACN. Each of the samples was then labeled by adding 12 μL (180 μg) of an individual TMT reagent, followed by shaking at room temperature for 1 h. Two μL of each of the nine labeled samples in each group were pooled, and 2 μL of 5% hydroxylamine was added. The samples were incubated for 15 min, dried by vacuum centrifugation, dissolved in 21 μL of 5% formic acid, and peptides were analyzed by a single 2-h LC-MS/MS method using an Orbitrap Fusion as described below. The run was performed to normalize the total reporter ion intensity of each multiplexed sample and to check labeling efficiency. After the normalization and efficiency run, the remaining unmixed samples were then quenched by the addition of 2 μL 5% hydroxylamine, then combined in adjusted volumes to yield equal summed reporter ion intensities during the subsequent two-dimensional LC/MS.
Following volume-based normalization, the combined samples were dried by vacuum centrifugation, and TMT-labeled samples were reconstituted in 20 μL water. The reconstituted peptides were separated by two-dimensional nano reverse-phase liquid chromatography (Dionex NCS-3500 UltiMate RSLCnano UPLC) EasySpray NanoSource (Thermo Scientific), ionized using an EasySpray NanoSource (Thermo Scientific), and SPS MS3 data acquired with an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific). The liquid chromatography details and mass spectrometer settings were as previously described [13] with the modification that non-enriched peptides were eluted from the first dimension high pH column using sequential injections of 20 μL volumes of 14, 17, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 50, and 90% ACN in 10 mM ammonium formate, pH 9, and enriched phosphopeptides were eluted by sequential 20 μL injections of 4, 6, 8, 10, 12, 18, 20, 22, 25, 30, and 60% ACN in 10 mM ammonium formate, pH 9.
2.1.9. TMT data analysis
The binary instrument files were processed with Proteome Discoverer version 1.4 (Thermo Scientific) to extract fragment ion spectra, precursor information (m/z values and peptide charge state), and TMT reporter ion peak intensities. The fragment ion spectra were searched against a canonical Uniprot Swiss-Prot mouse protein database (downloaded 07/07/2016 from https://www.uniprot.org) with 16,794 entries. There were 179 common contaminant entries appended for a total of 16,973 sequences.
The SEQUEST [19] search engine was used with a parent ion mass tolerance of 1.25 Da and fragment ion tolerance of 1.0005 Da. Trypsin cleavage with up to two missed cleavages was specified. A static modification of +57.0215 Da was applied to all cysteine residues and a variable modification of +15.9949 was applied to methionine. Confident peptide-to-spectrum matches (PSMs) were obtained using the percolator [20] node, and only peptides with q values < 0.05 were accepted. Parsimonious protein inference was used in Proteome Discoverer to produce final protein lists, and the results were exported to tab-delimited files for post processing using in-house Python scripts (https://www.github.com/pwilmart/PAW_pipeline.git). For the protein expression analysis of the total protein preparations, the reporter ion intensities from all unique (matching to just one protein) peptides were summed into protein reporter ion intensities. Any contaminant proteins were excluded from further analysis. Reporter ion data from PSMs where the trimmed average reporter ion intensity did not exceed 500 were excluded. Any final protein reporter ion sums of zero were replaced with a value of 150 (the smallest non-zero reporter ion intensities observed were approximately 350) to avoid mathematical errors during visualizations and statistical testing.
The data from the phosphopeptide enrichment experiment was searched with additional variable modifications of +79.9799 on serine, threonine, or tyrosine residues, and only peptides with q < 0.01 were accepted. The phosphorylation site localization node phosphoRS [21] was configured after the search node in Proteome Discoverer. Phosphorylation enrichment experiments are peptide centric, so data aggregation was done differently than for protein expression. Peptide sequences were aggregated by summing reporter ion intensities within each channel to reduce variance and increase statistical power. All PSMs assigned to the same base peptide sequence were aggregated in the same modification state, which was determined by integral peptide MH+ mass (the peptide in a 1+ charge state). Localization information from phosphoRS was simplified to the same number of top probabilities as the number of phosphorylation modifications present in the peptides. The reporter ion intensities from the combined reports were used for differential expression (DE) testing as described below. Any contaminant protein matches were excluded from further analysis. Minimum intensity filtering and zero replacement was done similarly to the total protein analysis.
2.1.10. Statistical analysis of differential expression
For immunofluorescence quantification, data are represented in text as the mean ± SEM and p-values were calculated using an unpaired t-test.
For TMT data, the table of non-contaminant reporter ion intensities for proteins or for aggregated phosphopeptides were exported to tab-delimited files and imported into R (https://www.r-project.org) for statistical analysis using the edgeR [22–24] Bioconductor package. Normalization was done using a “library size” factor and the trimmed mean of M-values normalization [25] function in edgeR was used to correct for sample loading and compositional bias. DE testing was performed pairwise using the exact test with Benjamini-Hochberg multiple testing corrections [26]. The results from edgeR analyses were exported and added to the combined data summaries. Annotation information was added from Uniprot using a script available at https://github.com/pwilmart/annotations.
2.1.11. Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository [27], with the dataset identifier PXD012906.
3. Results
3.1. RBC dendrites are major sites of light-dependent PKCα phosphorylation
To visualize sites of PKCα phosphorylation in the retina, we used a monoclonal antibody mix that binds to canonical PKC substrate motifs containing a phosphorylated serine (PKC motif p-serine). Anti-PKC motif phosphoserine immunofluorescence between wild type and PKCα-KO retina sections indicates that RBC dendrites are the main sites of PKCα phosphorylation in the mouse retina (Fig. 1A and B). The small immunofluorescent puncta in the outer plexiform layer (OPL) are presumptive RBC dendrites, whereas the larger patches of phosphoserine immunofluorescence (arrows) are associated with cone pedicles. The small immunofluorescent puncta are visible throughout the wild type OPL but are greatly reduced in intensity in the PKCα-KO retina, while immunofluorescence corresponding to cone pedicles is unchanged. These results suggest that PKCα phosphorylates targets in RBC dendrites of wild type adult mice, and that a different PKC isoform phosphorylates targets associated with cone pedicles.
Fig. 1.
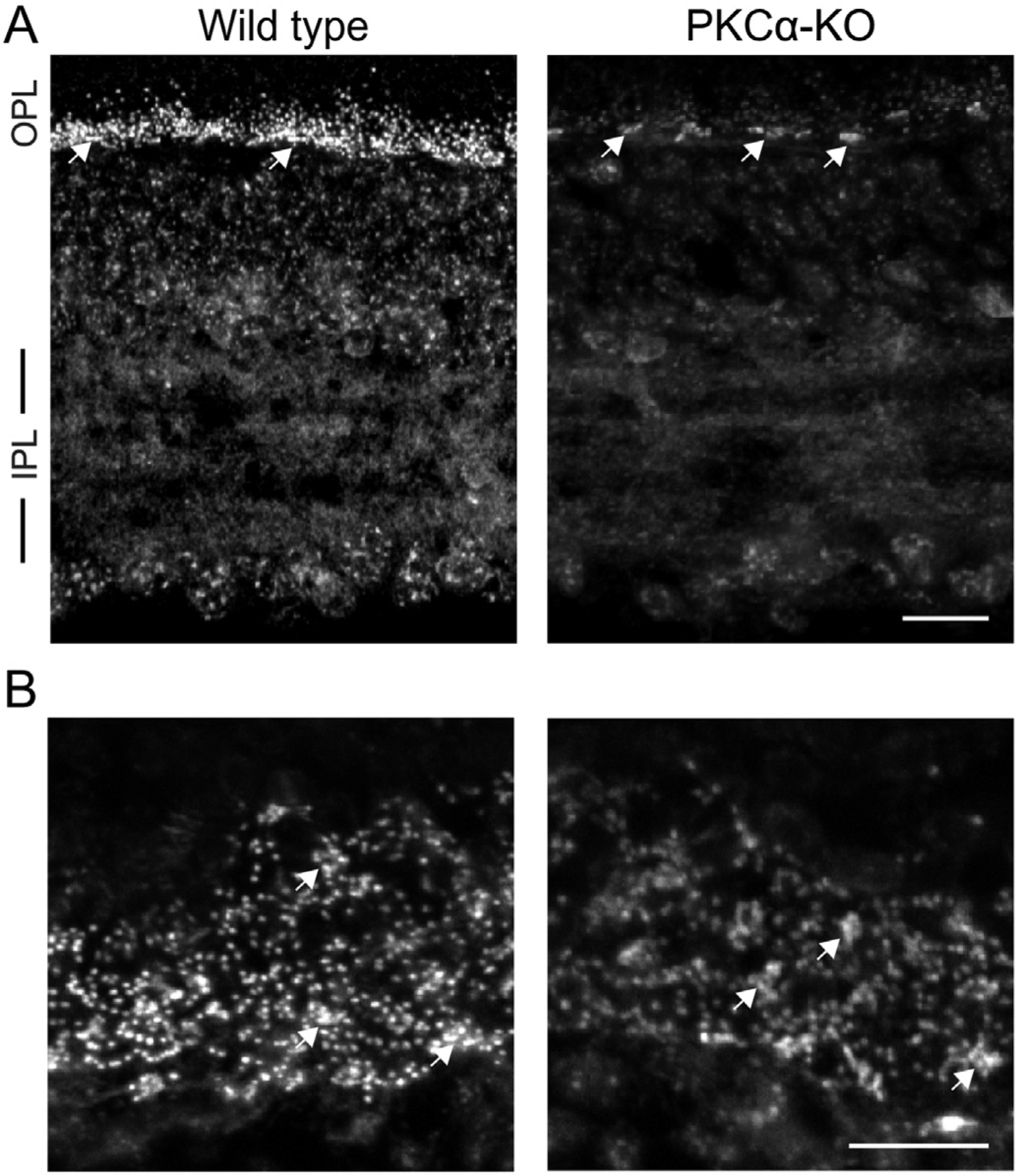
Phosphoserine labeling in the OPL is reduced in PKCα-KO retina. (A) Immunofluorescence confocal images of mouse retina sections from wild type and PKCα-KO retinas labeled with an antibody against phosphoserine residues within canonical PKC motif phosphoserine (PKC motif p-serine). (B) Images of PKC motif p-serine immunofluorescence in the outer plexiform layer of wild type and PKCα knockout (KO) retinas in obliquely cut sections. In both A and B, white arrows indicate labeling associated with presumptive cone pedicles. Scale bars: 20 μm. OPL: outer plexiform layer; IPL: inner plexiform layer.
To examine whether PKCα activity is light-dependent, we used a conformation-dependent monoclonal PKCα antibody which binds an epitope in the hinge region of PKCα that is inaccessible in the inactive state [28]. Retina sections from light-adapted and dark-adapted mice were double-labeled with the conformation-specific antibody (anti-PKCα-A) and a non-conformation-specific PKCα antibody (anti-PKCα-B; Fig. 2). Both antibodies strongly label RBC cell bodies and dendrites in sections from light-adapted retina, with co-localization of the two secondary antibodies appearing white (Fig. 2A, left). By contrast, only anti-PKCα-B labels RBCs in sections from dark adapted retina (Fig. 2A, right). These results suggest that PKCα is active in RBC dendrites in the light-adapted state. This was supported by labeling of light- and dark-adapted retina sections for phosphorylated PKC motifs. The anti-PKC motif phosphoserine antibody labeled puncta in the OPL of light-adapted retina (Fig. 2B, left), similar to the wild type immunofluorescence seen in Fig. 1. Anti-PKC motif phosphoserine immunofluorescence was absent in the OPL of the dark-adapted retina (Fig. 2B, right). Together, the results from Fig. 1 and Fig. 2 indicate that PKCα is active in RBC dendrites in the light-adapted state.
Conventional PKC isoforms, including PKCα, require calcium for activation [29]. In RBC dendrites, a likely source of calcium is the Transient Receptor Potential cation channel subfamily M 1 (TRPM1) cation channel, which mediates an influx of sodium and calcium to generate the RBC light response [14,30,31]. The dependence of PKCα activation on TRPM1 was assessed by double-labeling retina sections from wild type and TRPM1 knockout (TRPM1-KO) mice with anti-PKCα-A and anti-PKCα-B to detect active and total PKCα, respectively, in RBC cell bodies and dendrites (Fig. 2C), and the intensities of the immunofluorescence obtained with the two antibodies was compared. The average ratio of anti-PKCα-A (active) to anti-PKCα-B (total) immunofluorescence in wild type RBCs was 1.12 ± 0.12 (n = 4 mice, each with 4 technical replicates) compared to 0.70 ± 0.15 (n = 3 mice, each with 4 technical replicates) in TRPM1-KO RBCs (P < 0.0001), indicating that PKCα is less active in RBCs in the absence of TRPM1.
PKCα is a DAG-sensitive PKC isoform, and therefore PKCα phosphorylation can be potentiated by the DAG analogue phorbol 12-myristate 13-acetate (PMA). To confirm that PKCα is phosphorylating proteins in the retina ex vivo, we incubated freshly dissected wild type and PKCα-KO retinas in PMA before analyzing changes in protein phosphorylation by immunoblotting for PKC motif phosphoserines (PKC motif p-serine). PMA activation of other DAG-sensitive PKC isoforms expressed in the retina, such as PKCβ, should be relatively equivalent between WT and PKCα-KO samples and result in no difference in phosphorylation. PMA treatment resulted in a significant increase in intensity of several phosphoserine immunoreactive bands in the wild type samples at the 15, 30, and 60 min time-points compared to the PKCα-KO samples (Fig. 3A) demonstrating that PMA treatment increases differential phosphorylation between WT and PKCα-KO retinas. To identify sites of PMA-activated PKC phosphorylation in the wild type retina, PKC motif phosphoserine immunofluorescent labeling was performed on retina sections made from PMA-treated, light-adapted WT retinas. PMA incubation resulted in an increase in PKC motif phosphoserine immunofluorescence in presumptive RBC dendritic tips (Fig. 3B).
Fig. 3.
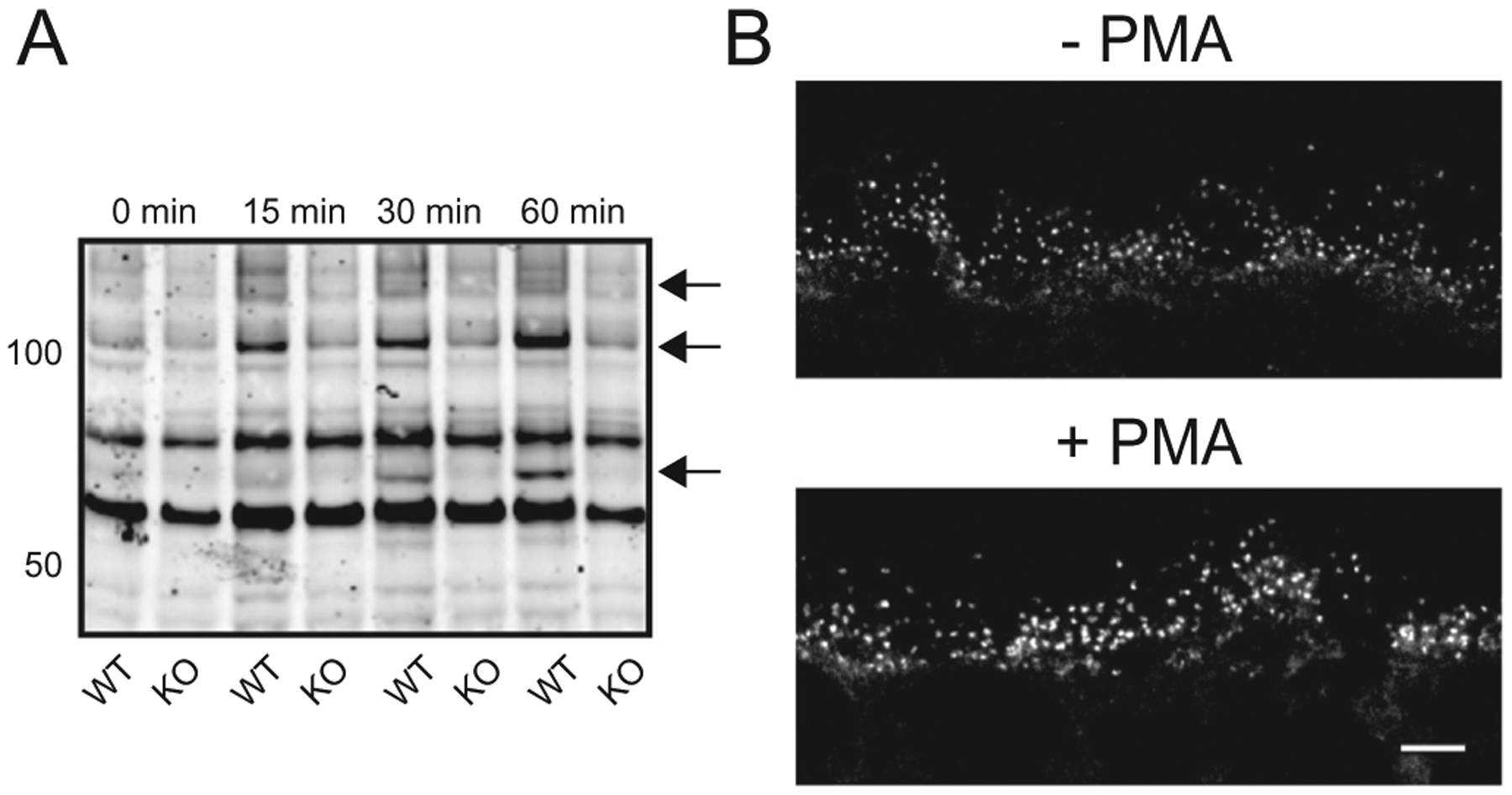
PMA increases phosphorylation by PKC isoforms in the mouse retina. (A) Wild type (WT) and PKCα knockout (PKCα-KO) retinas were incubated in PMA for 0, 15, 30, and 60 min followed by western blotting with an antibody against PKC motif phosphoserines. Arrows indicate candidate PKCα phosphorylation targets. (B) Immunofluorescent PKC motif phosphoserine labeling of wild type mouse OPL from retinas that were incubated with and without PMA for 1 h. Scale bar: 10 μm. PMA: phorbol 12-myristate 13-acetate; OPL: outer plexiform layer.
3.2. Differential protein abundance in wild type and PKCα-KO retina
To identify retinal proteins whose expression is dependent on PKCα, four wild type and five PKCα-KO retinas were incubated with PMA for an hour immediately after dissection and then processed for multiplexed TMT mass spectrometry (Fig. 4). Retinas were lysed and proteins digested with trypsin before TMT labeling and LCMS/MS. Peptide identification was performed with Proteome Discoverer using SEQUEST and Percolator. From 38,384 confidently identified PSMs, there were 34,969 unique peptides corresponding to 4435 proteins (excluding contaminants; Fig. 5, S1 – Total Protein and Phosphopeptide Abundance Analysis). Differential expression (DE) statistical testing was done with edgeR using the trimmed mean of M-values normalization and an exact pairwise test. After Benjamini-Hochberg p-value corrections for multiple comparisons to establish DE false discovery rates (FDRs), we grouped proteins with significant differential expression into significance groups based on FDR thresholds: not significant (FDR > 0.10 [4412 proteins]) low significance (0.10 > FDR > 0.05 [2 proteins]), medium significance (0.05 > FDR > 0.01 [5 proteins]), and high significance (FDR < 0.01 [16 proteins]).
Fig. 4.
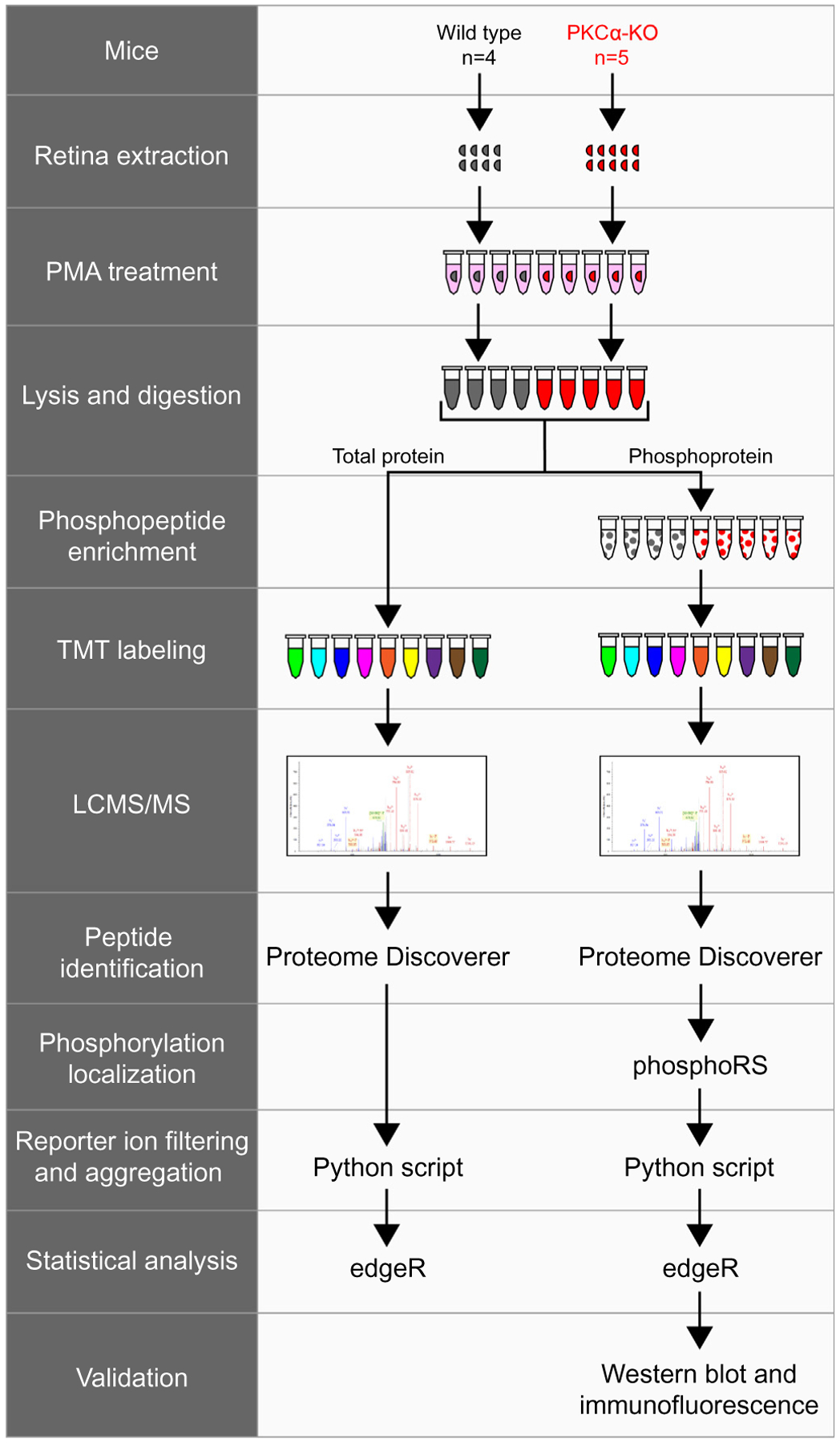
Experimental workflow of total protein and phosphopeptide identification. Wild type (n = 4) and PKCα knockout (KO) (n = 5) retinas were extracted and treated with PMA before lysis and trypsin digestion. A small fraction of each sample was removed for total protein analysis, while the rest of the samples underwent phosphopeptide enrichment. Following TMT labeling, samples were combined and analyzed by LCMS/MS. Tandem mass spectrometry data was collected on an Orbitrap Fusion and proteins were identified using Proteome Discoverer (SEQUEST and Percolator). Phosphorylation site localization was scored using phosphoRS, and reporter ion intensities were filtered and aggregated with an in-house Python script. TMT reporter ion intensities from total proteins or from phosphopeptides were tested for differential expression using the Bioconductor package edgeR. The presence of representative phosphoproteins was validated in the retina by western blot and confocal immunofluorescence microscopy. PMA: phorbol 12-myristate 13-acetate.
Fig. 5.
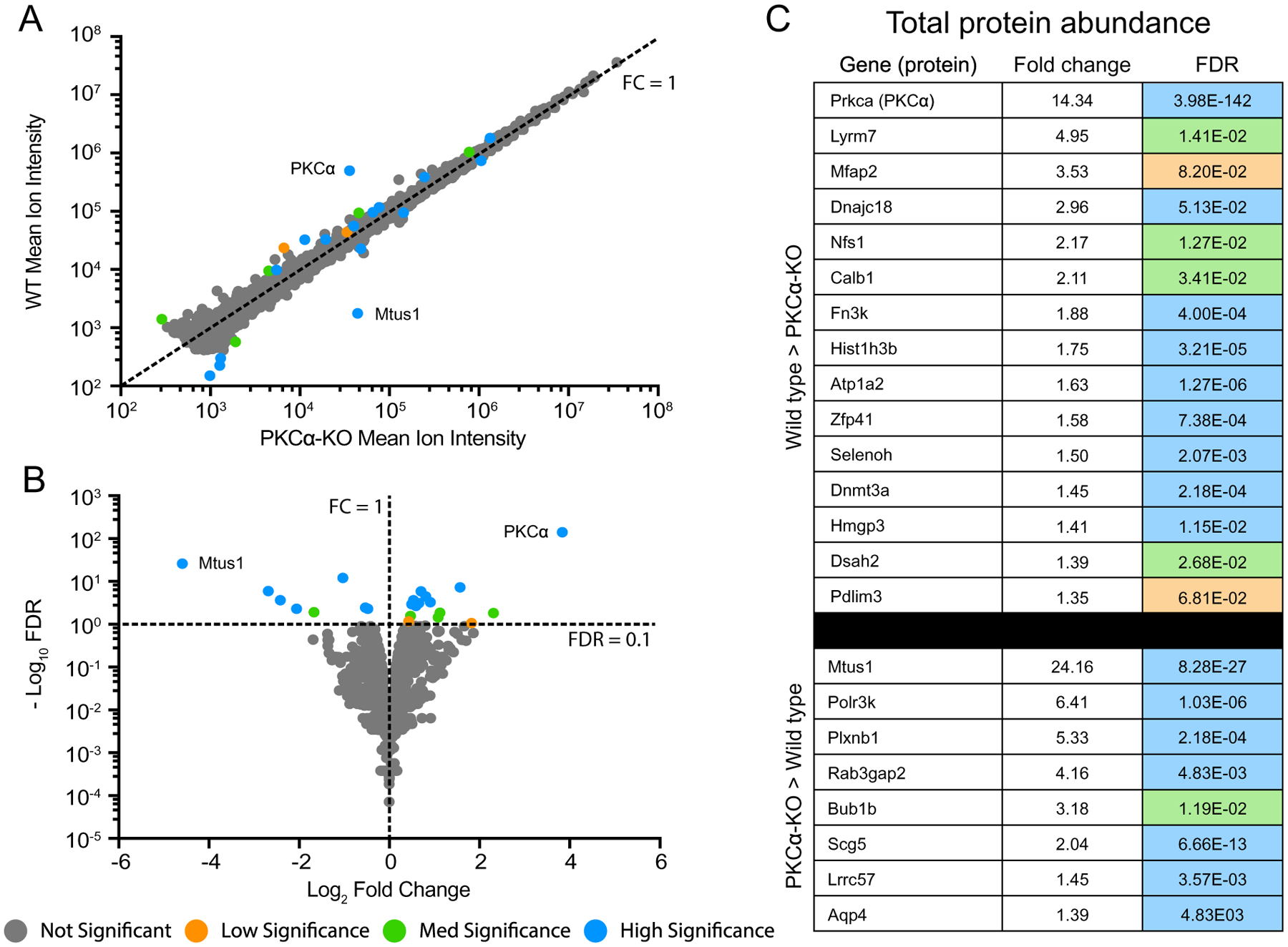
Identification of differentially expressed total proteins. (A) Scatter plot of peak reporter ion intensities from wild type (WT) and PKCα knockout (KO) total protein abundance samples. The dotted line corresponds to an FC (WT/KO) of 1, with FC > 1 corresponding to increased abundance in WT vs KO, and FC < 1 corresponding to increased abundance in KO vs WT. (B) Volcano plot of log2 FC and −log10 FDR. The dotted lines correspond to FC = 1 and FDR = 0.1. FDR < 0.1 corresponds to proteins passing the low significance threshold, and FDR > 0.1 corresponds to proteins failing the low significance threshold. The low significance (orange) threshold was 0.1, the med significance threshold (green) was 0.05, and the high significance threshold (blue) was 0.01. (C) Table of all differentially abundant proteins with an FDR lower than the low significance threshold of 0.1. Fold change was calculated by dividing the mean reporter ion intensities of each protein from the genotype with higher abundance by that with lower abundance. Colors correspond to DE FDR thresholds. DE: differential expression; FC: fold change; FDR: false discovery rate.
Plotting the mean reporter ion intensities between wild type and PKCα-KO samples (Fig. 5A) revealed a small number of significantly different protein abundances in both WT (15 proteins) and KO (8 proteins). The proteins with the largest changes in abundance were KPCA (PKCα; 14-fold decrease in KO), and MTUS1 (Microtubule Associated Scaffold Protein 1; 24-fold increase in KO). A volcano plot comparing log2 fold change (WT / KO) with −log10 FDR (Fig. 5B) depicts the 23 proteins passing the FDR < 0.1 cutoff for low significance. We did not attempt any isotopic corrections for reporter ions, so the degree of downregulation for PKCα is consistent with an absence of the kinase in the knockout. A list of all significant differentially expressed proteins can be seen in Fig. 5C. The sample-to-sample reproducibility of the protein abundance experiment was excellent: the WT samples had a median coefficient of variance (CV) of 11.4%, the KO samples had a median CV of 15.5%, and the independent CV was 13.5%. Ninety-five percent of the expression changes were < 1.25-fold different (S2 – Total Protein and Phosphopeptide Statistical Testing).
3.3. Differential protein phosphorylation between wild type and PKCα-KO retina
To examine differences in protein phosphorylation between wild type and PKCα-KO retinas, phosphopeptides were enriched from the same retinal extracts used above (described in the Methods), and phosphopeptide abundance was analyzed by multiplexed TMT mass spectrometry. Peptide identification was performed with Proteome Discoverer using SEQUEST and Percolator, and phosphorylation localization was performed using phosphoRS. Results files were exported for post processing (filtering and aggregation) using an in-house Python script (PD1.4 TMT phospho processer.py available at https://github.com/pwilmart/PAW pipeline). The aggregated phosphopeptide reporter ion data was tested for differential expression using edgeR with multiple testing corrections, and phosphopeptides were grouped into significance groups based on differential expression FDR as described for total proteins.
We identified 1137 distinct phosphopeptides in wild type or PKCα-KO retina lysates (Fig. 6, S1 – Total Protein and Phosphopeptide Abundance Analysis) 1113 were not significant (FDR > 0.10), seven showed low significance (0.10 > FDR > 0.05), four medium significance (0.05 > FDR > 0.01), and thirteen were highly significant (FDR < 0.01). Of the 24 significant phosphoproteins, only two showed differential expression in the total protein abundance analysis: Dnmt3a (DNA (Cytosine-5)-Methyltransferase 3A), which was significantly more abundant in the WT samples (1.5fold), and Scg5 (Neuroendrocrine Protein 7B2), which was significantly more abundant in the KO samples (2.04-fold). Plotting WT and KO peak ion intensities (Fig. 6A) highlights five phosphopeptides with much greater expression in WT than KO. A volcano plot comparing log2 FC (WT / KO) with −log10 FDR (Fig. 6B) shows the 24 phosphopeptides passing the FDR < 0.1 threshold (14 increased in WT and 10 increased in KO). A full list of all significant differentially expressed phosphopeptides, along with their total protein abundance changes, can be seen in Fig. 6C. The reproducibility of the peptide-centric experiment was strong: the WT samples had a median CV of 13.9%, the KO samples had a median CV of 18.7%, and the median CV independent of condition was 16.2%. Ninety-five percent of the expression changes were < 1.4-fold different (S2 – Total Protein and Phosphopeptide Statistical Testing). Annotated fragment ion spectra for the phosphorylated peptides are shown in S3 – Annotated Phosphopeptide Spectra.
Fig. 6.
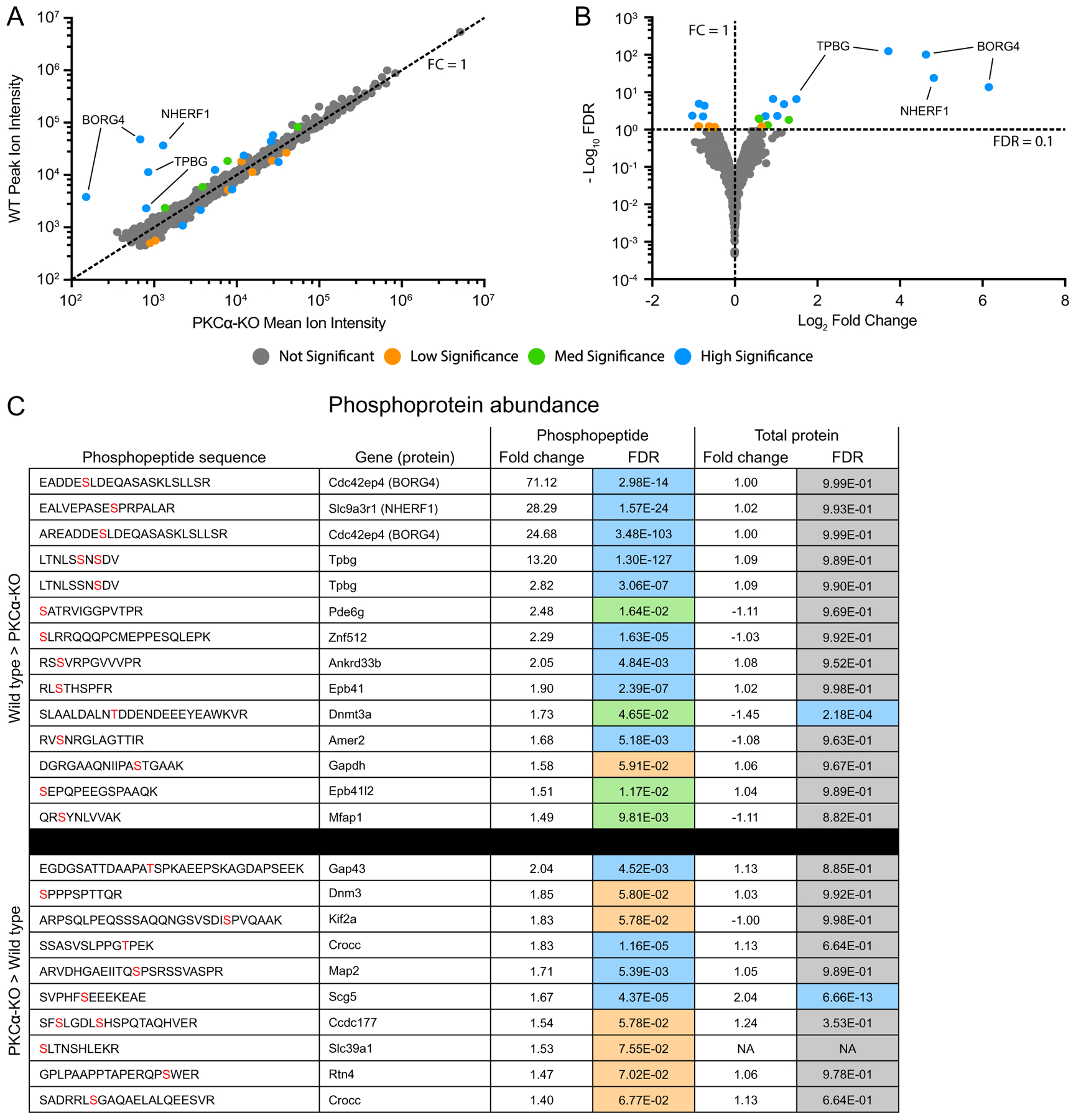
Identification of differentially expressed phosphopeptides. (A) Scatter plot of peak reporter ion intensities from wild type (WT) and PKCα knockout (KO) phosphopeptide abundance samples. The dotted line corresponds to an FC (WT / KO) of 1, with FC > 1 corresponding to increased abundance in WT vs KO, and FC < 1 corresponding to increased abundance in KO vs WT. (B) Volcano plot of log2 FC and −log10 FDR. The dotted lines correspond to FC = 1 and FDR = 0.1. FDR < 0.1 corresponds to proteins passing the low significance threshold, and FDR > 0.1 corresponds to proteins failing the low significance threshold. The low significance (orange) threshold was 0.1, the med significance threshold (green) was 0.05, and the high significance threshold (blue) was 0.01. (C) Table of all differentially abundance phosphopeptides with an FDR lower than the low significance threshold of 0.1. In the phosphopeptide sequence column, phosphorylated residues are in red. In the Total Protein Fold Change column, negative values indicate an increased abundance in the KO samples. Fold change was calculated by dividing the mean reporter ion intensities of each protein from the genotype with higher abundance by that with lower abundance. Fold change and DE FDR values were taken from the phosphopeptide abundance experiment and the total protein abundance experiment. Colors correspond to DE FDR thresholds. DE: differential expression; FC: fold change; FDR: false discovery rate.
Five phosphopeptides belonging to three proteins showed the largest and most significant differential abundance between the wild type and PKCα-KO samples (Fig. 7). Two similar phosphopeptides were identified corresponding to BORG4 (Binder of Rho-GTPase, also called CDC42EP4) with phosphoserine residues observed at the peptide sites S6 (71-fold increase) and S8 (25-fold increase). Both peptides were generated by slightly different cleavage patterns of the same amino acid sequence with the same phosphorylated serine residue corresponding to site S64 in full-length BORG4 (Fig. 7A). One phosphopeptide fragment was identified from NHERF1 (Na+/H+ Exchanger Regulatory Factor 1, also called SLC9A3R1 and EBP50) with a phosphorylated serine residue at the peptide S10 position (28-fold increase) corresponding to S275 in the full-length protein (Fig. 7B). The last two major significant phosphopeptides correspond to the same 10 amino acid sequence of TPBG (Trophoblast Glycoprotein, also called the 5 T4 antigen and WAIF1) with two identified species: a doubly-phosphorylated peptide with phosphoserines at peptide S6 and S8 (13-fold increase), and a singly-phosphorylated peptide with just phosphorylated peptide S8 (2.8-fold increase). These two serine residues correspond to S422 and S424 in the C-terminal intracellular tail of TPBG (Fig. 7C).
Fig. 7.
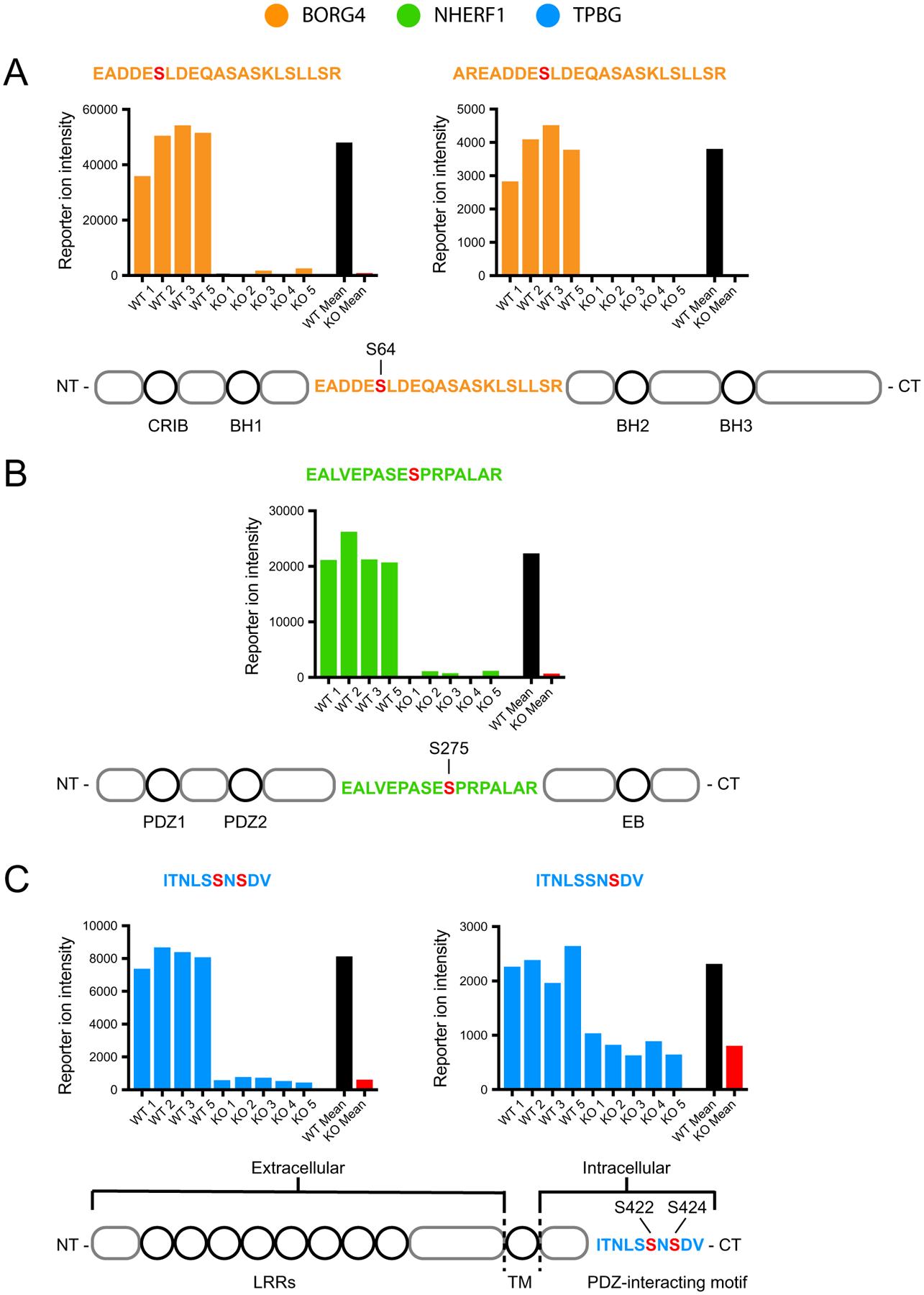
TMT data from representative phosphoproteins. Reporter ion intensity values from each TMT channel for the five phosphopeptide fragments with the largest differential expression between WT (n = 4) and PKCα-KO (n = 5): two from BORG4 (orange), one from NHERF1 (green), and two from TPBG (blue). For statistical significance of differential expression analysis of mean WT (black) and mean KO (red) reporter ion intensities, see S1 – Total Protein and Phosphopeptide Abundance Analysis. (A) Two phosphopeptide fragments from an overlapping region of BORG4, each containing a phosphorylated serine corresponding to S64 on the full-length protein. (B) One phosphopeptide fragment from NHERF1 with a phosphorylated serine corresponding to S275 on the full-length protein. (C) Two phosphopeptide fragments from the C-terminal tail of TPBG with two similar phosphorylation patterns: one with two phosphoserines corresponding to S422 and S424, and one with a single phosphoserine corresponding to S424 of the full-length protein.
3.4. Grouping of significant protein and phosphoprotein results by biological function
Proteins identified in the total protein abundance (Fig. 8A) and phosphoprotein abundance (Fig. 8B) experiments were grouped into broad categories based on general biological function annotations added from Uniprot (https://github.com/pwilmart/annotations.git). PKCα-KO resulted in differential expression of proteins and phosphoproteins involved in many aspects of cellular physiology, particularly cytoskeletal rearrangement and vesicle trafficking (15 proteins), transcriptional regulation (8 proteins), and homeostasis and metabolism (5 proteins).
Fig. 8.
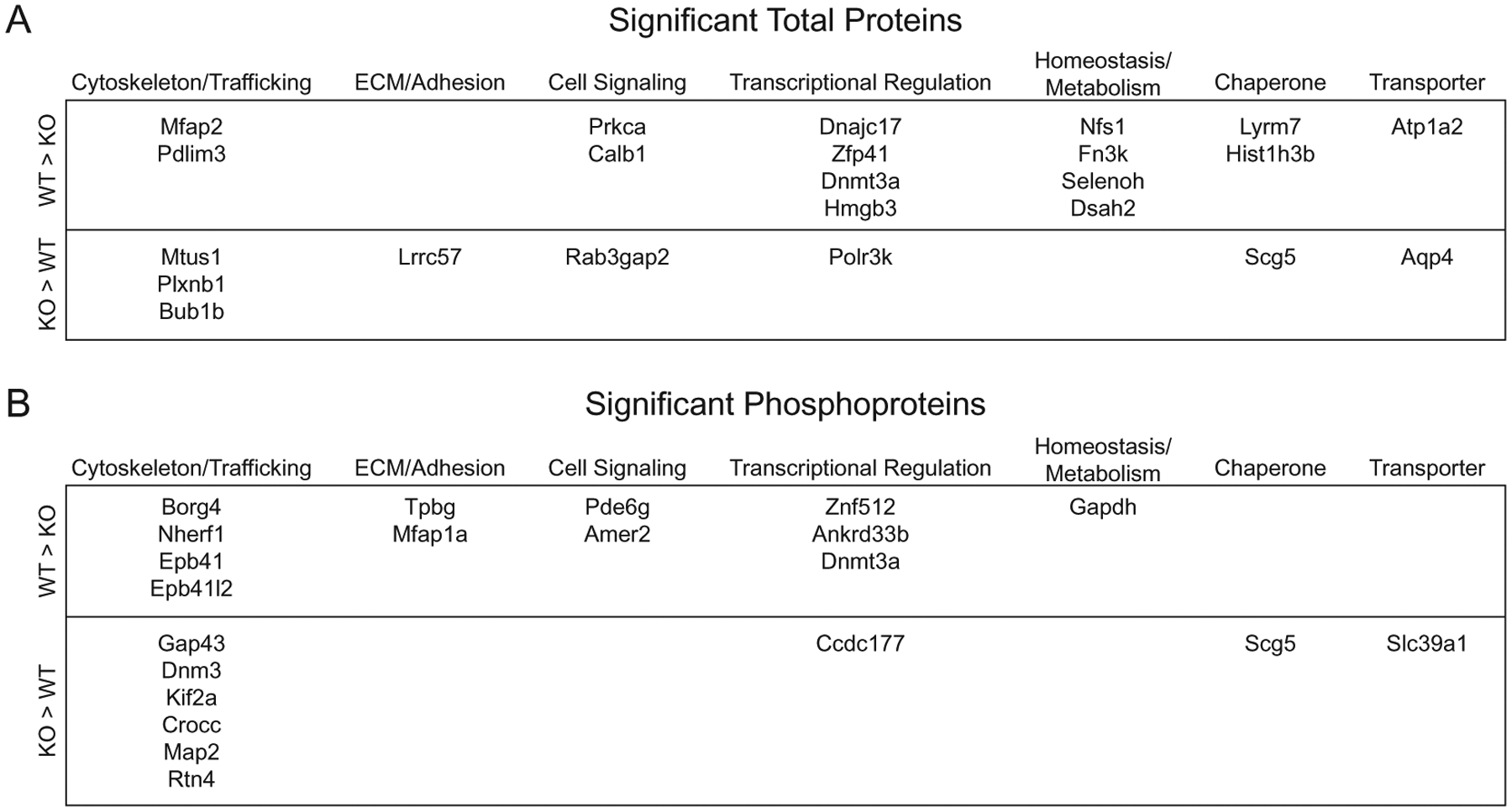
Significant total proteins and phosphoproteins grouped by biological function. Table of genes of identified total proteins (A) and phosphoproteins (B) with significant differential abundance between wild type and PKCα-KO samples grouped into broad categories based on general biological function gathered from Uniprot protein annotations.
3.5. Localization of the major PKCα-dependent phosphoproteins in the mouse retina
We used immunoblotting and immunofluorescence confocal microscopy to examine the presence of the three most prominent PKCα-dependent phosphoprotein hits in the wild type retina: BORG4, NHERF1, and TPBG. Immunoblotting for BORG4 shows a distinct band at 38 kDa (Fig. 9A) in agreement with the predicted molecular weight of BORG4. Immunofluorescence double-labeling of retina sections for BORG4 and PKCα shows punctate BORG4 labeling in the outer plexiform layer (OPL), but the BORG4 puncta are not strongly co-localized with PKCα (Fig. 9B). This is consistent with labeling of either RBC or horizontal cell dendritic tips, as they are closely apposed to one another within the rod spherule invagination [32,33].
Fig. 9.
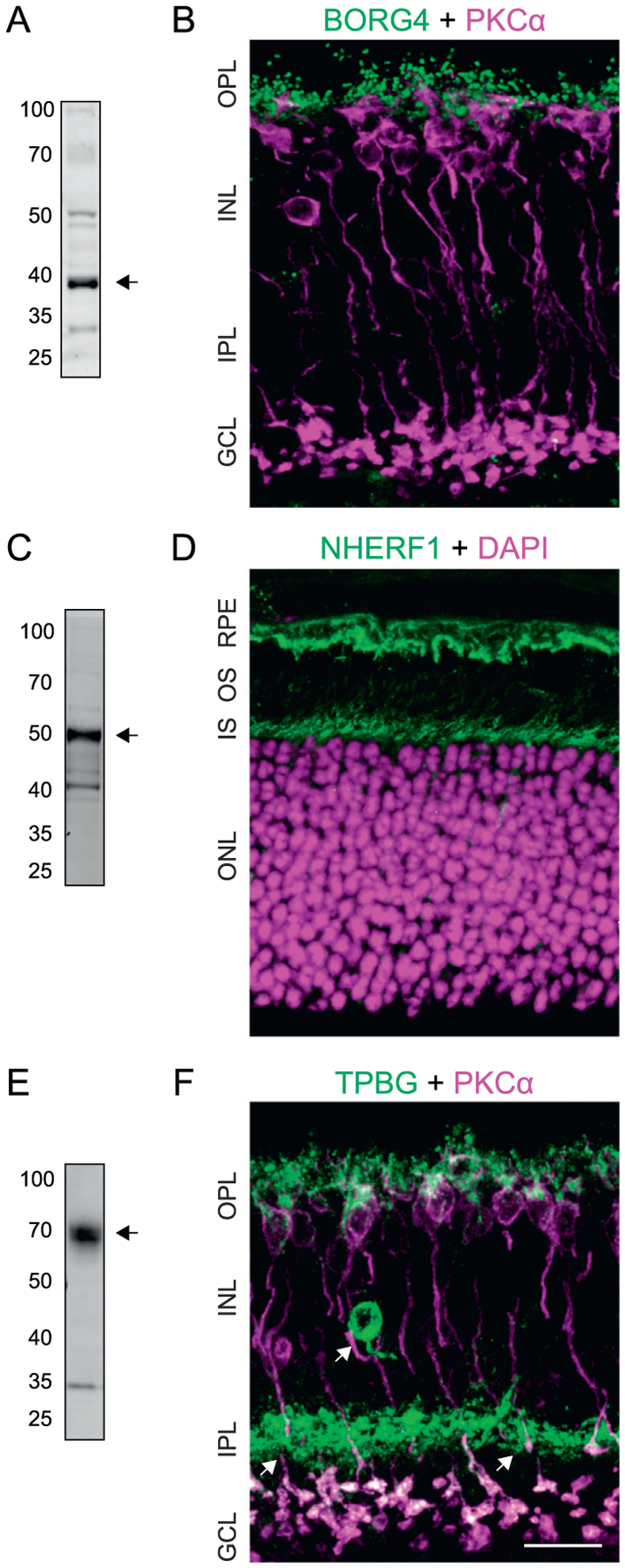
Validation of representative phosphoproteins in the mouse retina. (A) Immunoblot of retinal lysate labeled with rabbit anti-BORG4 shows a band corresponding to BORG4 at ~38 kDa. (B) Confocal microscopy analysis of BORG4 (also called Cdc42EP4) and PKCα immunoreactivity in the retina using mouse anti-Cdc42EP2. (C) Immunoblot of retinal lysate shows a band corresponding to NHERF1 at ~50 kDa. (D) Confocal microscopy analysis of NHERF1 immunoreactivity in the retina with retinal layers labeled with DAPI. (E) Immunoblot of retinal lysate shows a smear corresponding to glycosylated TPBG at ~72 kDa. (F) Confocal microscopy analysis of TPBG and PKCα immunoreactivity in the retina. Scale bars: 20 μm. RPE: retinal pigment epithelium; OS: outer segments; IS: inner segments; ONL: outer nuclear layer; OPL: outer plexiform layer; INL: inner nuclear layer; IPL: inner plexiform layer; GCL: ganglion cell layer.
Immunoblotting of retinal proteins for NHERF1 detected a strong band at ~50 kDa, the predicted molecular weight of NHERF1 (Fig. 9C). Immunofluorescent labeling of retina sections revealed NHERF1 immunoreactivity at the level of the photoreceptor inner segments and in the retinal pigment epithelium (RPE; Fig. 9D). It was not possible to determine whether NERF1 at the level of the inner segments is due to its presence in the photoreceptors themselves or the apical microvilli of the RPE, which surround the photoreceptor outer and inner segments. No NHERF1 immunofluorescence was seen in the synaptic layers of the retina.
Immunoblotting of retina lysate for TPBG labels a broad band centered around 72 kDa (Fig. 9E). This is higher than the predicted molecular weight of 42 kDa, but is consistent with extensive glycosylation of TPBG [34]. Immunofluorescent localization of TPBG revealed strong immunoreactivity in the OPL as well as in large synaptic terminals at sublamina 5 of the inner plexiform layer (IPL; Fig. 9F). In the OPL, TPBG labeling co-localizes with PKCα in RBC dendrites and cell bodies. In the IPL, TPBG immunoreactivity overlaps with all PKCα-positive RBC synaptic terminals. The immunofluorescence localization of TPBG to RBCs corroborates previous findings that TPBG mRNA is expressed primarily in RBCs [35]. The TPBG antibody also labeled a population of amacrine cells with cell bodies in the inner INL and dense dendritic projections to the middle of the IPL (Fig. 9F arrows). The existence of a TPBG-positive amacrine cell population in the retina was observed by Imamura et al. in 2006 [36], but its identity has not been established.
4. Discussion
RBCs are able to modulate their responses to changing light conditions and evidence suggests this process is regulated in part by PKCα [9,10,37]. The transient physiological effect of PKCα on the RBC dendrites is presumably mediated by its kinase activity, whereby PKCα-dependent phosphorylation changes the activity of downstream proteins. We show that RBC dendrites are the main sites of light- and PKCα-dependent phosphorylation (Fig. 1). Using a conformation-specific PKCα antibody and an antibody mix that recognizes phosphorylated PKC substrate motifs, our results suggest that PKCα is active in light-adapted RBC dendrites. In the dark-adapted retina, PKCα is inactive and PKC substrate phosphorylation in the OPL is significantly reduced (Fig. 2A and B). Comparison of light-adapted wild type and PKCα-KO retinas confirmed that phosphorylation in RBC dendrites requires PKCα, and also indicated that phosphorylation in presumed cone bipolar cell dendrites is mediated by a different kinase. Deletion of TRPM1, a calcium-permeable cation channel responsible for signal transduction in ON-bipolar cell dendrites, also results in a significant reduction in RBC labeling with the conformation-specific PKCα antibody, suggesting that TRPM1-mediated calcium influx during the RBC light response is a primary source of calcium required for PKCα activation in RBC dendrites.
Differential abundance of retinal phosphopeptides between WT and PKCα-KO retinas could potentially be due to altered expression of proteins caused by the deletion of PKCα, and not due to changes in PKCα kinase activity. To identify proteins whose phosphorylation status is dependent on PKCα, but whose expression levels are unaffected, we compared both phosphoprotein and total protein abundance between WT and PKCα-KO mouse retinas using isobaric tagging and mass spectrometry. We identified over 4000 total proteins in either WT or PKCα-KO samples with 23 showing significantly different expression between groups. These proteins can be clustered into several general categories corresponding to biological ontology, most notably proteins involved in regulating cell shape and vesicle transport, transcriptional regulation, and homeostasis and metabolism. PKCα has been implicated in a variety of diverse cell signaling processes, including cell proliferation and morphology, inflammation, and tumorgenesis. Furthermore, PKCα activity has a major impact on gene expression by modulating transcription factors such as CREB, NF-κB, and c-REL [38]. Identifying proteins that show altered expression levels in PKCα-KO mouse retina could be valuable to the examination of many PKCα-dependent functions in RBCs, such as regulation of RBC morphology and development.
To identify RBC proteins that display PKCα-dependent phosphorylation, total retinal phosphopeptides were compared between wild type and PKCα-KO mice using isobaric tagging. Normalizations and differential expression statistical testing for isobaric tagged phosphopeotides differs from traditional phosphoproteomic experiments where replicate numbers are smaller. When replicate numbers are greater than one or two, ratios are no longer an appropriate framework for the analysis. We extended our approach of working directly with aggregated reporter ion intensities for differential protein expression to phosphopeptide datasets. Our approach enables the use of robust statistical testing from genomics packages such as edgeR [23] or Limma [39]. The background of unchanged phosphorylation levels in the enrichment experiment, like that of the total protein experiment, was sufficient in these samples to use standard normalization approaches. Our normalizations and statistical testing for both experiments demonstrates that the same analysis steps used for total protein abundance can be successfully applied to the phosphopeptide datasets (S2 – Total Protein and Phosphopeptide Statistical Testing).
Of over 1100 distinct phosphopeptides identified by multiplex TMT mass spectrometry, 14 displayed significantly greater phosphorylation in wild type compared to PKCα-KO samples (Fig. 6), suggesting their phosphorylation state is dependent on PKCα. These putative PKCα-dependent phosphoproteins may be phosphorylated directly by PKCα or may be phosphorylated by a different kinase whose activity is dependent on PKCα. Only one phosphoprotein, Dnmt3a, showed a significant decrease in abundance in PKCα-KO compared to WT in both the total protein and phosphopeptide experiments (Fig. 5 and Fig. 6), indicating that reduced abundance of the Dnmt3a phosphopeptide may be due to downregulation of the protein in the KO. Three proteins, BORG4, NHERF1, and TPBG, displayed a particularly striking increase in phosphorylation in wild type compared to PKCα-KO samples (Fig. 6 and Fig. 7). Of these proteins, BORG4 and NHERF1 are known substrates of PKCα [40,41].
Surprisingly, several of the major PKCα-dependent phosphoproteins identified in this study, including TPBG, do not conform to a strong consensus PKCα substrate motif, in which a phosphorylated serine is flanked by positively charged arginine and lysine residues (Kinexus Database; Vancouver, Canada; http://www.kinexusnet.ca). However, kinase substrate motifs are not always linear, contiguous sequences, but can also be formed structurally by bending of flexible loops that brings positively charged amino acids into proximity to the phosphorylated serine or threonine residues [42]. In the case of TPBG, the cytoplasmic domain is predicted to be unstructured and flexible, possibly allowing the phosphorylated serines at the C-terminal tail (S422 and S424) to be brought close to upstream pairs of lysines and arginines (R384 K385 and K388 and K389) to form a structural PKCα substrate motif. Alternatively, TPBG and other PKCα-dependent non-consensus phosphopeptides may be phosphorylated by a downstream kinase that is dependent on PKCα activity. For example, casein kinase 2 (CK2) is a serine/threonine kinase that is activated by PKCα [43,44], and whose consensus substrate motif is a serine flanked by acidic residues (Kinexus Database). Two of the phosphopeptides that showed increased abundance in wild type samples compared to PKCα-KO contain a CK2 substrate motif (TPBG and Mfap2). The same site in Mfap2 has previously been demonstrated to be phosphorylated in vitro. The C-terminals of the NR2B subunit of the NMDA receptor is similar in sequence to the C-terminus of TPBG (LSSIESDV compared to LSSNSDV), and CK2 phosphorylation of the C-terminal serine in NR2B has been demonstrated to regulate trafficking of the receptor [45]. Ten phosphopeptides were more abundant in PKCα-KO samples than in wild type. These are likely to be phosphorylated by kinases that are inhibited by PKCα, such as GSK3 [46,47]. The consensus substrate motif for GSK3 kinase is a serine residue with a neighboring proline (Kinexus Database), and several of the phosphopeptides that are increased in the PKCα-KO samples fit this motif (examples: Dyn3, Crocc, Map2, and Rtn4).
We used immunofluorescence to localize three phosphoproteins that displayed the greatest differential phosphorylation between wild type and PKCα-KO samples. Immunofluorescence labeling of BORG4 resulted in bright puncta in the OPL (Fig. 9). BORG4 belongs to a protein family (BORG1–5) that bind to the Rho GTPase CDC42 as well as to septins, a family of GTP-binding cytoskeletal proteins that are involved in regulation of cell morphology through modulation of cytoskeletal rearrangement. BORG4 contains an N-terminal CCD42/Rac Interactive Binding Motif (CRIB) and three BORG Homology (BHs) domains that are conserved across all BORG family proteins [48]. Our proteomics data indicates that the serine residue S64 in BORG4, which is located between BH1 and BH2, is phosphorylated in a PKCα-dependent manner. Phosphorylation of BORG4 S64 has been previously recognized in large-scale analyses experiments of tissue-specific phosphorylation patterns [49,50] though not in retina. BORG4 and Septin-4 mRNAs have been previously found to be expressed in horizontal cells [51]. Our immunofluorescence labeling of BORG4 in retina resulted in bright puncta in the OPL consistent with BORG4 localization to either RBC or horizontal cell dendrites.
We detected NHERF1 immunofluorescence at the level of the photoreceptor inner segments in the region of the connecting cilia and in the retina pigment epithelium (RPE, Fig. 9). NHERF1 is a scaffolding protein containing tandem PDZ domains and an Ezrin/Radixin/Moesin Binding (EB) domain. Our detection of strong NHERF1 immunofluorescence in the RPE consistent with previous reports localizing NHERF1 to the RPE apical microvilli [52]. NHERF1 interacts with ezrin to maintain the structure of apical microvilli on epithelia. In the RPE, NHERF1 has been implicated in retinoid recycling [52] through its interactions with CRALBP [53,54]. PKCα is also expressed in the RPE [55] where it is involved in proliferation and migration [56], phagocytosis [57], and melanin production [58]; however, the specific role of PKCα-mediated phosphorylation of NHERF1 in the RPE is unknown.
Immunofluorescence confocal microscopy localized TPBG immunoreactivity to RBC dendrites and synaptic terminals, as well as to a class of amacrine cells (Fig. 9). TPBG is a heavily glycosylated type-1 transmembrane protein with a large extracellular N-terminal domain and a short C-terminal intracellular tail. The N-terminal domain contains eight leucine-rich repeats (LRRs) and seven N-linked glycosylation sites. The C-terminal tail ends with the class-1 PDZ-interacting motif (S/T X Φ) SDV [59], which our proteomics data suggests is phosphorylated in a PKCα-dependent manner. Since phosphorylation of a PDZ-interacting motif typically prevents binding of a PDZ protein [60], PKCα might be regulating interactions between TPBG and PDZ proteins by stimulating phosphorylation of its C-terminal tail. As an oncofetal antigen, TPBG is present primarily during embryonic development [61,62], but is also expressed in many carcinomas [63,64]. In the adult, TPBG is expressed in the brain, retina, and ovaries [65,66]. In the embryo and in cancer tissue, TPBG is involved in regulating actin polymerization [67] filopodia formation [68], and chemotaxis [69,70], and its expression in tumors is linked to increased metastatic malignancy and poor survival outcomes in cancer patients [71,72]. In the adult olfactory bulb, TPBG is required to stimulate the development of input-dependent dendritic arborization and synaptogenesis of newborn granule cells [36,73–75]. The role of TPBG in the retina is not yet understood; however, a recent transcriptomic classification of retinal cell types identified TPBG mRNA as being highly enriched in RBCs [35]. This is consistent with our immunofluorescent analysis which localized TPBG to RBC dendrites and synaptic terminals.
5. Conclusions
The molecular mechanisms of PKCα-mediated modulation of the RBC light response have not been thoroughly explored. In this study, we have shown that PKCα phosphorylation in the retina occurs pre-dominately in RBC dendrites in the light. Using a phosphoproteomics approach, we have identified a small number of phosphoproteins with significantly increased PKCα-dependent phosphorylation in the PMA-treated retina. These differentially phosphorylated proteins fall into several broad functional groups, including cytoskeleton/trafficking (4 proteins), structure and adhesion (2 proteins), cell signaling (2 proteins), transcriptional regulation (3 proteins), and homeostasis/metabolism (1 protein). Two strongly differentially expressed phosphoproteins, BORG4 and TPBG, are localized to the synaptic layers of the retina, and may play a role in PKCα-dependent modulation of RBC function.
Supplementary Material
Acknowledgments
The authors would like to thank Tammie L. Haley for producing mouse retina sections for immunofluorescence experiments. This work was supported by the National Institutes of Health grants R01EY022369 and 5P30EY010572 and an OHSU University Shared Resources Core Pilot Fund grant.
Abbreviations:
- BORG4
Binder of Rho GTPase 4
- DAG
Diacylglycerol
- ERG
Electroretinogram
- DE
Differential expression
- FC
Fold change
- FDR
False discovery rate
- GCL
Ganglion cell layer
- INL
Inner nuclear layer
- IPL
Inner plexiform layer
- KO
Knockout
- LC-MS/MS
Liquid chromatography tandem mass spectrometry
- NHERF1
Na+/H+ Exchange Regulatory Factor 1
- OPL
Outer plexiform layer
- PKCα/Prkca
Protein Kinase C-alpha
- PMA
Phorbol 12-myristate 13-acetate
- PSM
Peptide spectrum match
- RBC
Rod bipolar cell
- RPE
Retinal pigment epithelium
- TMT
Tandem mass tag
- TPBG
Trophoblast Glycoprotein
- TRPM1
Transient Receptor Potential cation channel subfamily M member 1
Footnotes
Conflict of interest statement
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jprot.2019.103423.
References
- [1].Berntson A, Smith RG, Taylor WR, Transmission of single photon signals through a binary synapse in the mammalian retina, Vis. Neurosci 21 (2004) 693–702. [DOI] [PubMed] [Google Scholar]
- [2].Sampath AP, Rieke F, Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse, Neuron 41 (2004) 431–443. [DOI] [PubMed] [Google Scholar]
- [3].Abd-El-Barr MM, Pennesi ME, Saszik SM, Barrow AJ, Lem J, Bramblett DE, Paul DL, Frishman LJ, Wu SM, Genetic dissection of rod and cone pathways in the dark-adapted mouse retina, J. Neurophysiol 102 (2009) 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ke J-B, Wang YV, Borghuis BG, Cembrowski MS, Riecke H, Kath WL, Demb JB, Singer JH, Adaptation to background light enables contrast coding at rod bipolar cell synapses, Neuron 81 (2014) 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Szikra T, Trenholm S, Drinnenberg A, Jüttner J, Raics Z, Farrow K, Biel M, Awatramani G, Clark DA, Sahel J-A, da Silveira RA, Roska B, Rods in daylight act as relay cells for cone-driven horizontal cell–mediated surround inhibition, Nat. Publ. Gr 17 (2014) 1728–1735. [DOI] [PubMed] [Google Scholar]
- [6].Greferath U, Grünert U, Wässle H, Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity, J. Comp. Neurol 301 (1990) 433–442. [DOI] [PubMed] [Google Scholar]
- [7].Haverkamp S, Haeseleer F, Hendrickson A, A comparison of immunocytochemical markers to identify bipolar cell types in human and monkey retina, Vis. Neurosci 20 (2003) 589–600. [DOI] [PubMed] [Google Scholar]
- [8].Haverkamp S, Ghosh KK, Hirano AA, Wässle H, Immunocytochemical description of five bipolar cell types of the mouse retina, J. Comp. Neurol 455 (2002) 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ruether K, Feigenspan A, Pirngruber J, Leitges M, Baehr W, Strauss O, PKC{alpha} is essential for the proper activation and termination of rod bipolar cell response., Investig. Ophthalmol. &, Vis. Sci 51 (2010) 6051–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xiong W-H, Pang J-J, Pennesi ME, Duvoisin RM, Wu SM, Morgans CW, The effect of PKCα on the light response of rod bipolar cells in the mouse retina, Investig. Ophthalmol. Vis. Sci 56 (2015) 4961–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C, Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS, Anal. Chem 75 (2003) 1895–1904. [DOI] [PubMed] [Google Scholar]
- [12].Erickson BK, Jedrychowski MP, McAlister GC, Everley RA, Kunz R, Gygi SP, Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer, Anal. Chem 87 (2015) 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Plubell DL, Wilmarth PA, Zhao Y, Fenton AM, Minnier J, Reddy AP, Klimek J, Yang X, David LL, Pamir N, Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse Epididymal adipose tissue, Mol. Cell. Proteomics 16 (2017) 873–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Morgans CW, Zhang J, Jeffrey BG, Nelson SM, Burke NS, Duvoisin RM, Brown RL, TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 19174–19178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW, ImageJ2: ImageJ for the next generation of scientific image data, BMC Bioinformatics 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schindelin J, Rueden CT, Hiner MC, Eliceiri KW, The ImageJ ecosystem: an open platform for biomedical image analysis, Mol. Reprod. Dev 82 (2015) 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Paulo JA, Mcallister FE, Everley RA, Beausoleil SA, Banks AS, Gygi SP, Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics, Proteomics 15 (2015) 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kettenbach AN, Gerber SA, Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: application to general and phosphotyrosine-specific phosphoproteomics experiments, Anal. Chem 83 (2011) 7635–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Eng JR, McCormack JK, Yates AL, An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database, J. Am. Soc. Mass Spectrom 5 (1994) 976–989. [DOI] [PubMed] [Google Scholar]
- [20].Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ, Semi-supervised learning for peptide identification from shotgun proteomics datasets, Nat. Methods 4 (2007) 923–925. [DOI] [PubMed] [Google Scholar]
- [21].Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K, Universal and confident phosphorylation site localization using phosphoRS, J. Proteome Res 10 (2011) 5354–5362. [DOI] [PubMed] [Google Scholar]
- [22].Agrawal N, Akbani R, Aksoy BA, Ally A, Xing M, Bioconductor: open software development for computational biology and bioinformatics, Genome Biol. 11 (2010) 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics 26 (2009) 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Horthon T, W H, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini G, Sawitzki AJ, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J, Bioconductor: open software development for computational biology and bioinformatics, Genome Biol. 5 (2004) R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Robinson MD, Oshlack A, A scaling normalization method for differential expression analysis of RNA-seq data, Genome Biol. 11 (2010) R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hochberg B, Controlling the false discovery rate: a practical and powerful approach to multiple testing, J. R. Stat. Soc 57 (1995) 289–300. [Google Scholar]
- [27].Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yılmaz Ş, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaíno JA, The PRIDE database and related tools and resources in 2019: improving support for quantification data, Nucleic Acids Res. 47 (2019) D422–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gabriel R, Lesauter J, Silver R, Garcia-Espaa A, Witkovsky P, Diurnal and circadian variation of protein kinase C immunoreactivity in the rat retina, J. Comp. Neurol 439 (2001) 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nishizuka Y, Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C, Science (80) 258 (1992) 607–614. [DOI] [PubMed] [Google Scholar]
- [30].Shen Y, Rampino MAF, Carroll RC, Nawy S, G-protein-mediated inhibition of the Trp channel TRPM1 requires the G dimer, Proc. Natl. Acad. Sci 109 (2012) 8752–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Koike C, Numata T, Ueda H, Mori Y, Furukawa T, TRPM1: a vertebrate TRP channel responsible for retinal ON bipolar function, Cell Calcium 48 (2010) 95–101. [DOI] [PubMed] [Google Scholar]
- [32].Boycott BB, Dowling JE, Kolb H, Organization of the primate Retina: light microscopy, Philos. Trans. R. Soc. B Biol. Sci 255 (1969) 109–184. [Google Scholar]
- [33].Dowling JE, Boycott BB, Organization of the primate Retina : electron microscopy, Proc. R. Soc. Lond. Ser. B Biol. Sci 166 (1966) 80–111. [DOI] [PubMed] [Google Scholar]
- [34].Shaw DM, Woods AM, Myers KA, Westwater C, Rahi-Saund V, Davies MJ, Renouf DV, Hounsell EF, Stern PL, Glycosylation and epitope mapping of the 5T4 glycoprotein oncofoetal antigen, Biochem. J 363 (2002) 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A, Sanes JR, Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics, Cell 166 (2016) 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Imamura F, Nagao H, Naritsuka H, Murata Y, Taniguchi H, Mori K, A leucine-rich repeat membrane protein, 5T4, is expressed by a subtype of granule cells with dendritic arbors in specific strata of the mouse olfactory bulb, J. Comp. Neurol 495 (2006) 754–768. [DOI] [PubMed] [Google Scholar]
- [37].Rampino MAF, Nawy SA, Relief of Mg2+-dependent inhibition of TRPM1 by PKCα at the rod bipolar cell synapse, J. Neurosci 31 (2011) 13596–13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Garg R, Caino MC, Kazanietz MG, Regulation of transcriptional networks by PKC isozymes: identification of c-Rel as a key transcription factor for PKC-regulated genes, PLoS ONE 8 (2013) e67319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, Limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res. 43 (2015) e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen X, Zhao X, Abeyweera TP, Rotenberg SA, Analysis of substrates of protein kinase C isoforms in human breast cells by the traceable kinase method, Biochemistry 51 (2012) 7087–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Garbett D, LaLonde DP, Bretscher A, The scaffolding protein EBP50 regulates microvillar assembly in a phosphorylation-dependent manner, J. Cell Biol 191 (2010) 397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Duarte ML, Pena DA, Ferraz FAN, Berti DA, Sobreira TJP, Costa-Junior HM, Baqui MMA, Disatnik MH, Xavier-Neto J, De Oliveira PSL, Schechtman D, Protein folding creates structure-based, noncontiguous consensus phosphorylation motifs recognized by kinases, Sci. Signal 7 (2014). [DOI] [PubMed] [Google Scholar]
- [43].Malkani N, Biggar K, Shehab MA, Li SSC, Jansson T, Gupta MB, Increased IGFBP-1 phosphorylation in response to leucine deprivation is mediated by CK2 and PKC, Mol. Cell. Endocrinol 425 (2016) 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lee Y-H, Park J-W, Bae Y-S, Regulation of protein kinase CK2 catalytic activity by protein kinase C and phospholipase D2, Biochimie 121 (2016) 131–139. [DOI] [PubMed] [Google Scholar]
- [45].Chung HJ, Huang YH, Lau L-F, Huganir RL, Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand, J. Neurosci 24 (2004) 10248–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moore SF, Van Den Bosch MTJ, Hunter RW, Sakamoto K, Poole AW, Hers I, Dual regulation of glycogen synthase kinase 3 (GSK3)α/β by protein kinase C (PKC)α and Akt promotes thrombin-mediated integrin α∥bβ3activation and granule secretion in platelets, J. Biol. Chem 288 (2013) 3918–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu Z-C, Chen X-H, Song H-X, Wang H-S, Zhang G, Wang H, Chen D-Y, Fang R, Liu H, Cai S-H, Du J, Snail regulated by PKC/GSK-3β pathway is crucial for EGF-induced epithelial-mesenchymal transition (EMT) of cancer cells, Cell Tissue Res. 358 (2014) 491–502. [DOI] [PubMed] [Google Scholar]
- [48].Farrugia AJ, Calvo F, The Borg family of Cdc42 effector proteins Cdc42EP1–5, Biochem. Soc. Trans 44 (2016) 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Villen J, Beausoleil SA, Gerber SA, Gygi SP, Large-scale phosphorylation analysis of mouse liver, Proc. Natl. Acad. Sci 104 (2007) 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP, A tissue-specific atlas of mouse protein phosphorylation and expression, Cell. 143 (2010) 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Blackshaw S, Harpavat S, Trimarchi J, Cai L, Huang H, Kuo WP, Weber G, Lee K, Fraioli RE, Cho SH, Yung R, Asch E, Ohno-Machado L, Wong WH, Cepko CL, Genomic analysis of mouse retinal development, PLoS Biol. 2 (2004) E247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Huang J, Possin DE, Saari JC, Localizations of visual cycle components in retinal pigment epithelium, Mol. Vis 15 (2009) 223–234. [PMC free article] [PubMed] [Google Scholar]
- [53].Bonilha VL, Bhattacharya SK, West KA, Crabb JS, Sun J, Rayborn ME, Nawrot M, Saari JC, Crabb JW, Support for a proposed retinoid-processing protein complex in apical retinal pigment epithelium, Exp. Eye Res 79 (2004) 419–422. [DOI] [PubMed] [Google Scholar]
- [54].Nawrot M, West K, Huang J, Possin DE, Bretscher A, Crabb JW, Saari JC, Cellular Retinaldehyde-binding protein interacts with ERM-binding phosphoprotein 50 in retinal pigment epithelium, Investig. Ophthalmol. Vis. Sci 45 (2004) 393–401. [DOI] [PubMed] [Google Scholar]
- [55].Yu K, Ma P, Ge J, Willey CD, Yang P, Wang Z, Gao Q, Expression of protein kinase C isoforms in cultured human retinal pigment epithelial cells, Graefes Arch. Clin. Exp. Ophthalmol 254 (2007) 993–999. [DOI] [PubMed] [Google Scholar]
- [56].Kishi H, Mishima HK, Yamashita U, Growth regulation of retinal pigment epithelial (RPE) cells in vitro, Curr. Eye Res 13 (1994) 661–668. [DOI] [PubMed] [Google Scholar]
- [57].Sheu SJ, Sakamoto T, Osusky R, Wang HM, Ogden TE, Ryan SJ, Hinton DR, Gopalakrishna R, Transforming growth factor-β regulates human retinal pigment epithelial cell phagocytosis by influencing a protein kinase C-dependent pathway, Graefes Arch. Clin. Exp. Ophthalmol 232 (1994) 695–701. [DOI] [PubMed] [Google Scholar]
- [58].Kishi H, Mishima HK, Yamashita U, Involvement of the protein kinase pathway in melanin synthesis by chick retinal pigment epithelial cells, Cell Biol. Int 24 (2000) 79–83. [DOI] [PubMed] [Google Scholar]
- [59].Zhao Y, Malinauskas T, Harlos K, Jones EY, Structural insights into the inhibition of Wnt signaling by cancer antigen 5T4/Wnt-activated inhibitory factor 1, Structure. 22 (2014) 612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lee HJ, Zheng JJ, PDZ domains and their binding partners: structure, specificity, and modification, Cell Commun. Signal 8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ward CM, Eastham AM, Stern PL, Cell surface 5T4 antigen is transiently up-regulated during early human embryonic stem cell differentiation: effect of 5T4 phenotype on neural lineage formation, Exp. Cell Res 312 (2006) 1713–1726. [DOI] [PubMed] [Google Scholar]
- [62].Ward CM, Barrow K, Woods AM, Stern PL, The 5T4 oncofoetal antigen is an early differentiation marker of mouse ES cells and its absence is a useful means to assess pluripotency, J. Cell Sci 116 (2003) 4533–4542. [DOI] [PubMed] [Google Scholar]
- [63].Southall PJ, Boxer GM, Bagshawe KD, Hole N, Bromley M, Stern PL, Immunohistological distribution of 5T4 antigen in normal and malignant tissues, Br. J. Cancer 61 (1990) 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hole N, Stern PL, A 72 kD trophoblast glycoprotein defined by a monoclonal antibody, Br. J. Cancer 57 (1988) 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Barrow KM, Ward CM, Rutter J, Ali S, Stern PL, Embryonic expression of murine 5T4 oncofoetal antigen is associated with morphogenetic events at implantation and in developing epithelia, Dev. Dyn 233 (2005) 1535–1545. [DOI] [PubMed] [Google Scholar]
- [66].King KW, Sheppard FC, Westwater C, Stern PL, Myers KA, Organisation of the mouse and human 5T4 oncofoetal leucine-rich glycoprotein genes and expression in foetal and adult murine tissues, Biochim. Biophys. Acta Gene Struct. Expr 1445 (1999) 257–270. [DOI] [PubMed] [Google Scholar]
- [67].Awan A, Lucic MR, Shaw DM, Sheppard F, Westwater C, Lyons SA, Stern PL, 5T4 interacts with TIP-2/GIPC, a PDZ protein, with implications for metastasis, Biochem. Biophys. Res. Commun 290 (2002) 1030–1036. [DOI] [PubMed] [Google Scholar]
- [68].Carsberg CJ, Myers KA, Evans GS, Allen TD, Stern PL, Metastasis-associated 5T4 oncofoetal antigen is concentrated at microvillus projections of the plasma membrane, J. Cell Sci 108 (1995) 2905–2916. [DOI] [PubMed] [Google Scholar]
- [69].McGinn OJ, Marinov G, Sawan S, Stern PL, CXCL12 receptor preference, signal transduction, biological response and the expression of 5T4 oncofoetal glycoprotein, J. Cell Sci 125 (2012) 5467–5478. [DOI] [PubMed] [Google Scholar]
- [70].Southgate TD, McGinn OJ, Castro FV, Rutkowski AJ, Al-Muftah M, Marinov G, Smethurst GJ, Shaw D, Ward CM, Miller CJ, Stern PL, CXCR4 mediated chemotaxis is regulated by 5T4 Oncofetal glycoprotein in mouse embryonic cells, PLoS ONE 5 (2010) e9920–e9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pukrop T, Binder C, The complex pathways of Wnt 5a in cancer progression, J. Mol. Med 86 (2008) 259–266. [DOI] [PubMed] [Google Scholar]
- [72].Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM, Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma, Cancer Cell 1 (2002) 279–288. [DOI] [PubMed] [Google Scholar]
- [73].Yoshihara S, Takahashi H, Nishimura N, Naritsuka H, Shirao T, Hirai H, Yoshihara Y, Mori K, Stern PL, Tsuboi A, 5T4 glycoprotein regulates the sensory input-dependent development of a specific subtype of newborn interneurons in the mouse olfactory bulb, J. Neurosci 32 (2012) 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Takahashi H, Ogawa Y, i Yoshihara S, Asahina R, Kinoshita M, Kitano T, Kitsuki M, Tatsumi K, Okuda M, Tatsumi K, Wanaka A, Hirai H, Stern PL, Tsuboi A, A subtype of olfactory bulb interneurons is required for odor detection and discrimination behaviors, J. Neurosci 36 (2016) 8210–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yoshihara S, Takahashi H, Tsuboi A, Molecular mechanisms regulating the dendritic development of newborn olfactory bulb interneurons in a sensory experience-dependent manner, Front. Neurosci 9 (2016) 134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository [27], with the dataset identifier PXD012906.