

Abstract
Neuropathic pain is an intractable medical condition with few or no options for effective treatment. Emerging evidence shows a strong structure-function relationship between dendritic spine dysgenesis and the presence of neuropathic pain. Postmortem tissue analyses can only imply dynamic structural changes associated with injury-induced pain. Here, we profiled the in vivo dynamics of dendritic spines over time on the same superficial dorsal horn (lamina II) neurons before and after peripheral nerve injury-induced pain. We used a two-photon, whole-animal imaging paradigm that permitted repeat imaging of the same dendritic branches of these neurons in C57/Bl6 Thy1-YFP male mice. Our study demonstrates, for the first time, the ongoing, steady-state changes in dendritic spine dynamics in the dorsal horn associated with peripheral nerve injury and pain. Ultimately, the relationship between altered dendritic spine dynamics and neuropathic pain may serve as a structure-based opportunity to investigate mechanisms of pain following injury and disease.
SIGNIFICANCE STATEMENT This work is important because it demonstrates for the first time: (1) the powerful utility of intravital study of dendritic spine dynamics in the superficial dorsal horn; (2) that nerve injury-induced pain triggers changes in dendritic spine steady-state behavior in the spinal cord dorsal horn; and (3) this work opens the door to further investigations in vivo of spinal cord dendritic spine dynamics in the context of injury and disease.
Keywords: dendritic spines, dorsal horn, intravital imaging, nerve injury, pain, two-photon microscopy
Introduction
Neuropathic pain is a major health problem with limited medical options for effective treatment. Drug development for neuropathic pain has been especially challenging due in part to mechanistic uncertainty underlying the disease. Understanding how trauma leads to disability is a major challenge toward developing novel therapeutics for managing chronic pain. A common physiological sign of neuropathic pain is hyperexcitability within nociceptive circuits in the PNS and CNS pain axis (Ji and Strichartz, 2004; Waxman and Hains, 2006).
In the spinal cord, multiple factors contribute to dorsal horn hyperexcitability associated with neuropathic pain. Excessive and spontaneous nerve firing activity, loss of local or descending cortical inhibition, central or peripheral inflammation, ion channel mis-expression (e.g., sodium channels), and reactive fiber plasticity can contribute to adverse changes in nociceptive-sensory transmission after injury or disease (Kuner, 2010; Todd, 2010; Dib-Hajj et al., 2013; Potter et al., 2016; Bennett et al., 2019). Emerging evidence has further demonstrated a strong correlation between dendritic spine remodeling and injury- or disease-induced pain (Tan et al., 2009; Tan and Waxman, 2015; Zhao et al., 2016; Guo et al., 2018).
Our previous work showed a common structural motif of altered dendritic spine morphology strongly associated with nociceptive hyperexcitability and neuropathic pain in neurons located in the superficial and intermediate zone of the dorsal horn, laminae II or IV, respectively. Pain-associated dendritic spine morphologies include increased spine density (particularly, larger mushroom-shaped spines) and regional redistribution of spines along dendrite branches (Tan et al., 2011; Zhao et al., 2016; Cao et al., 2017). However, these previous studies relied on histologic analysis of postmortem tissue, which could only imply the dynamic dendritic spine changes associated with injury-induced pain.
Here we profiled the in vivo dynamics of dendritic spines over time on the same neurons in the superficial dorsal horn (lamina II) before and after peripheral nerve injury-induced pain. Because lamina II is known to be involved in pain processing, and dendritic spine remodeling events may contribute to hyperexcitability in deeper laminae (e.g., intermediate zone and ventral horn) (Tan et al., 2008, 2012; Tan and Waxman, 2015; Bandaru et al., 2015; Zhao et al., 2016), we hypothesized that neurons in lamina II would experience dynamic changes in spine shape and density after peripheral nerve injury. We used a two-photon, whole-animal imaging paradigm (Fenrich et al., 2012), which permitted repeat imaging of the same dorsal horn neurons in Thy1-YFP mice. In “paired-imaging” sessions, we measured three characteristics of dendritic spine dynamics before and after nerve injury and pain onset, including the following: (1) ongoing steady-state fluctuations (i.e., spine length and maximal head diameter); (2) de novo formation and elimination (i.e., spine turnover rate); and (3) spine density change. We chose the established spared nerve injury (SNI) model of pain because it produces a reliable and consistent increase in tactile and thermal hypersensitivity (Decosterd and Woolf, 2000; Shields et al., 2003; Samad et al., 2013). Moreover, the SNI model rapidly elicits a predictable pain profile with elevated pain-sensitivity occurring by 2 d following surgery, and reaching maximal pain sensitivity within a week (Cichon et al., 2018).
Consistent with previous findings, 3 and 7 d after SNI, animals exhibited progressively increased pain sensitivity in response to adverse mechanical and thermal stimuli. Concomitant to pain onset, we observed the following increases: (1) in mean steady-state fluctuations in spine length and spine head width, and (2) in dendritic spine turnover activity. Interestingly, attributes driving spine turnover activity coincided with the temporal progression of pain severity. Early after injury (3 d after SNI), a significant decrease in thin-shaped spine elimination was followed by a transition to increased mushroom-shaped formation 7 d after SNI (coinciding with the presentation of maximal pain). As a consequence, we observed significantly increased mushroom-shaped spine density in neurons of injured animals compared with the uninjured cohort. Together, our study reports, for the first time, the ongoing, steady-state changes in dendritic spine dynamics in the dorsal horn associated with nerve injury and pain. Ultimately, dendritic spine dynamics and neuropathic pain may provide a structural-based opportunity to investigate the structural mechanisms of pain following injury and in disease.
Materials and Methods
Animals
Experiments were performed in accordance with the National Institutes of Health's Guide for the care and use of laboratory animals. All animal protocols were approved by the Yale University/Veterans Affairs Institutional Animal Use Committee. Animals were housed under a 12 h light-dark cycle in a pathogen-free area with food and water provided ad libitum. To visualize spinal cord neurons, we used weight-matched male mice (8- to 10-week-old, C57/Bl6 Thy1-YFP HJRS; The Jackson Laboratory). Similar transgenic mice using Thy1-promoter reporter systems have been used in intravital cortical imaging studies of dendritic spines (H. T. Xu et al., 2007; Pan and Gan, 2008; Barretto et al., 2011).
Behavioral assays
All behavioral experiments were performed by blinded experimenters in a dedicated quiet room under invariant conditions as previously described. All behavioral testing was performed after “spinal cord window implantation” (see below for procedure details; see study design in Fig. 1). For pain behavioral testing at the 3 and 7 d post-SNI time points (see Fig. 1), mechanical sensory thresholds were determined by paw withdrawal away from a series of von Frey filaments (Stoelting) applied to the glabrous surface of the right hindpaw. To determine the applied force value, we used a modification of the Dixon “up-down” method, which indicates paw withdrawal occurring 50% of the time as the mechanical nociceptive threshold (Chaplan et al., 1994; Li et al., 2000). The Hargreaves test was used to assess the animal's response to noxious heat. The plantar surface of the left paw was exposed to radiant heat from a Hargreaves instrument (IITC). The withdrawal latency was recorded in seconds and was measured 3 times per session. A limit of 30 s was set to prevent tissue damage.
Figure 1.
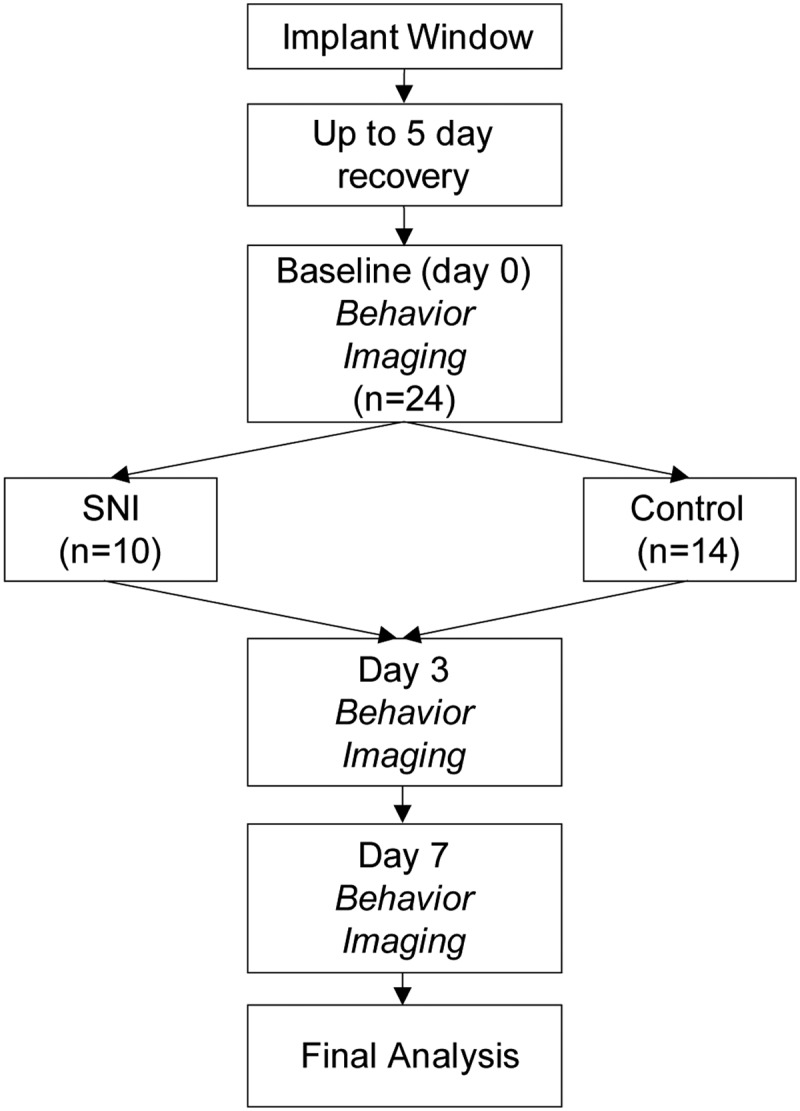
Study design. All animals were implanted with a spinal cord window and allowed to recover for up to 5 d. After the recovery period, animals underwent 3 d of baseline pain testing (von Frey hairs and Hargreaves test). Baseline imaging session was done before and on the same day as SNI. Pain testing and imaging occurred on 3 and 7 d after SNI. N values indicate the number of neurons sampled in each group. Neurons sampled for baseline to day 3, and baseline to day 7, were separate groups.
SNI model of pain
Following spinal cord window implantation (see below; Fig. 1), baseline imaging of sampled neurons, and pain-behavioral assessment, we performed surgical procedures to produce the SNI model of pain in mice. The SNI model is a well-established partial denervation model of neuropathic pain. In mice, the SNI surgery results in a detectable increase pain onset within 2 d after injury, and reaches maximal pain sensitivity at 7–10 d after the nerve injury (Decosterd and Woolf, 2000). Briefly, mice were anesthetized by exposure to ventilated isoflurane (1%–3% in ambient air; Kent Scientific SomnoSuite) The overlying skin on the left hindlimb was shaved and disinfected with cycles of alcohol and betadine (% 5 povidone iodine). An incision (∼10 mm in length) was made on the lateral mid-thigh, and the underlying muscles separated via blunt resection to expose the three branches of the sciatic nerve bundle (i.e., tibial, common peroneal, and sural nerve). The tibial and common peroneal were individually ligated with 6-0 silk sutures and completely transected (Decosterd and Woolf, 2000). The sural nerve was left intact. Distal to the site of ligation, we removed a 2 mm margin of nerve tissue to prevent the possibility of regeneration. The overlying muscle and skin were closed in layers using 6-0 monofilament nylon sutures (Ethicon).
Spinal cord window implantation
We used a modified spinal cord window implantation procedure, as described previously (Fenrich et al., 2012, 2013). Briefly, mice were anesthetized with ketamine and xylazine (100/10 mg/kg). We performed a dorsal midline incision over the T10-L1 spinal vertebrae columns (Harrison et al., 2013). We stabilized the dorsal surface of the spinal column with a spinal-fork stereotaxic apparatus. With surgical blunt-dissection technique, we resected the overlying muscles covering the lateral aspects of the transverse processes between T11 and T13. Custom-shaped sterile office staples were inserted under the lateral processes, along the caudal- and rostral-most vertebrae, and secured the staples with cyanoacrylate (Krazy Glue, Elmer's Products). To provide a clamping-handle for the “metal-frame,” we secured a “U-shaped” paperclip to the imbedded staples. To seal and protect the cyanoacrylate-metal framework (Fenrich et al., 2012), we covered this region of tissue and surrounding vertebrae with dental cement (Dental Mfg.).
To perform the laminectomy, we clamped the metal frame into a stereotaxic holder. We used a high-speed drill with a carbide burr to remove the overlying vertebral bone (T11-T13) overlying the L3-L4 spinal cord dorsal segments (i.e., revealing the dorsal root entry zones of the respective nerves). Glass windows were made from modified glass coverslips cut into the appropriate size to span the laminectomy opening (VWR, no. 1.5 thickness; ∼1.8–2.1 mm wide; 3.8–4.5 mm long; for window placement location; see Fig. 2). Following debris cleanup with sterile buffered saline, we applied a small amount of an optically clear sterile adhesive (Kwik-Sil, World Precision Instruments) over the dura matter, followed immediately by placement of the glass window. Gentle downward pressure was applied to the glass for up to 60 s while the Kwik-Sil cured. To further secure the window and protect the framework from damage over the experiment, we applied thin layers of cyanoacrylate and dental cement, sequentially around the edge of the glass. All exposed tissue and cyanoacrylate were ultimately covered in hardened layer of dental cement. Animals were returned to their cage and kept warm until they recovered from anesthesia. Animal were allowed to recovery for 3–5 d before beginning imaging experiments. Similar to previous reports using this methodology (Fenrich et al., 2012), following this recovery period, animals with window implants showed normal ambulatory behavior, gained significant weight compared with naive animals, and presented no symptoms of persistent pain (data not shown).
Figure 2.

Location of imaged dorsal horn neurons. A, Schematic diagram of 3D printed gimble imaging stage. The rotational mounts added to this stage allow fine adjustments to the window position to optimize image quality. Glass window was implanted over the L3-L4 spinal cord segments. B, Reconstruction of two-photon images of the spinal dorsal horn of a Thy1-YFP animal. 3D volume shows YFP-labeled neurons located 50–150 μm below the surface of the spinal cord. C, A composite image showing midline of the spinal cord and lateral location of Thy1-YFP-expressing neurons. D, Representative of in vivo captured image of a single dorsal horn neuron expressing Thy1-YFP. E, High-magnification image of the region denoted by *, showing dendritic spines. F, G, Cross sections of postmortem spinal cord tissue from beneath the window immunolabeled for (F) PKCγ and (G) CGRP and IB4 to delineate the inner and outer subregions of lamina II. H, YFP-labeled cell body locations are shown as a percentage of total sampled YFP-labeled cells that were topographically present in CGRP, IB4, or PKCγ-immunopositive laminae in the dorsal horn. YFP-positive cells are mostly present within the PKCγ and IB4 layers (i.e., 92.8% of sampled neurons are located in inner lamina II). Scale bars: C, 100 μm; D, E, 10 μm; F, G, 10 μm.
Intravital 2-photon imaging
For each imaging session, mice were anesthetized with ketamine/xylazine (100/10 mg/kg). The mice were stabilized during imaging by clamping the U-shaped paperclip into a 3D-printed stage gimble system. The stage was designed, and 3D modeled using Fusion 360 (Autodesk). The stage was 3D printed on an Ultimaker 2+ (Ultimaker BV) using polylactic acid plastic. Two rotation mounts (PRM1 and PRM05, Thor Labs) were built into the stage allowing fine adjustment of the window position to optimize image quality. This stage permitted us to move the suspended animal with a full degree of freedom in the x, y, and z axes. During initial testing of the design (data not shown), suspension of the animal with the paperclip clamp-holder minimized imaging artifacts as a result of breathing movement (e.g., the thoracic trunk of the animals did not contact any part of the stage during inspiration/expiration).
A two-photon microscope was used to collected images of identified dorsal horn neurons in the superficial dorsal horn (lamina II; depth up to 250 μm). An A1RMP+ (Nikon) equipped with gallium arsenide phosphide detectors and a Chameleon Vision II (Coherent) two-photon laser tuned to 940 nm to excite YFP, was used to acquire z-axis optical slices (0.5 µm steps; Nikon 25×/1.1 NA water immersion objective). Imaged sequences were composited into z stacks of up to 200 µm thick, which included the full volume of a neuron's cell body and extending dendrites. We collected optical sections with a pixel area of 512 × 512 (per pixel area ∼0.04 μm2 or voxel ∼0.02 μm3), which was close to the estimated optical diffraction limit of our equipment (∼0.25 μm). The location of large blood vessels and local cellular morphology was used as landmarks to confirm the same neuronal position across experimental days. The schedule of imaging sessions is shown in Figure 1.
Sampled sensory neurons in this study (n = 24 neurons tracked over time) were located between 70–160 µm (average depth: 96.8 µm) below the dorsal spinal cord surface and 435–720-µm lateral from spinal cord midline (average mediolateral distance: 582 µm) ipsilateral to SNI (see Fig. 2).
Longitudinal dendritic spine profiling
All analyses were performed by 2 investigators blinded to the experimental groups. Nikon Elements AR (version 5.1, Nikon) and ImageJ software (https://imagej.nih.gov/ij/download.html) were used for image data postprocessing and analyses. To identify excitatory superficial dorsal horn neurons for analysis, we sampled neurons based on four morphologic and topographical criteria used previously (Light and Kavookjian, 1988; Yasaka et al., 2010; Cao et al., 2017) as follows: (1) neurons must be located within lamina II in the superficial dorsal horn (up to 200 μm deep); (2) YFP-labeled neurons must have clearly visible dendrites at least 100 μm in length; (3) neurons must have at least three primary dendrites extending from the cell body, although branching was not required; and (4) the cell body diameter must fall between 15-30 μm. In total, we longitudinally studied 28 dorsal horn sensory neurons in lamina II from 9 animals over time. With our intravital imaging approach, we were able to study the same neurons over time, before and after injury. All animals were imaged following recovery from glass window implantation. We captured and analyzed “paired” images of neurons (e.g., same cell) captured at three different paired intervals: 1 h (n = 6 neurons in 3 uninjured, control animals), and 3 or 7 d following SNI surgery (vs baseline before injury) in a different animal cohort (n = 24 neurons from 6 animals). The 3 and 7 d post-SNI neuron datasets were not compared due to the lack of statistical power. Morphologic characteristics of the sampled neurons can be found in Table 1.
Table 1.
Location and cell body dimensions of sampled Thy1-YFP neurons
| Depth from surface of spinal cord (μm) | Distance from midline (μm) | Maximum cell body diameter (μm) | Minimum cell body diameter (μm) | Cell body area (μm2) | |
|---|---|---|---|---|---|
| Average | 96.8 ± 3.9 | 582.5 ± 20.2 | 18.8 ± 0.74 | 9.82 ± 0.4 | 146.5 ± 9.1 |
| Minimum | 70 | 435 | 12.2 | 7.5 | 98 |
| Maximum | 160 | 720 | 22.3 | 13.2 | 258 |
We quantified “dendritic spine dynamics” on dendritic segments ∼100–150 μm from the soma with structural data collected in “paired” imaging sessions (e.g., baseline to 3 d or baseline to 7 d). We measured three characteristics of dendritic spine dynamics before and after nerve injury and pain onset; including the following: (1) steady-state fluctuations (i.e., spine length and maximal spine head diameter); (2) de novo formation and elimination (i.e., turnover activity); and (3) spine density change (i.e., spines per dendritic branch length). We profiled steady-state fluctuations of spine length and head diameter using “mean absolute % change” as calculated from paired data of the same dorsal horn neurons captured over time. To normalize for variation across neurons, we calculated the percent change in spine length or head width. New or lost spines were given a value of 100%. Additionally, because we were interested in comparing the absolute change in these morphometries, we performed a nondirectional transformation (i.e., rectification), which provided the “absolute % change” unit for comparison across groups (see Figs. 3, 5, 6).
Figure 3.

Normal dendritic spine length and head width dynamics within the dorsal horn of the spinal cord. Absolute % change in (B–D) spine length and head width (between time intervals of 1 h, 3 d, and 7 d). A, Representative images of in vivo paired images with intervals of 1 h, 3 d, and 7 d. B, Absolute % change in length and head width of all spine types (total spines). Dynamics within the total spine population over a 1 h period were less compared with the dynamics observed over 3 and 7 d intervals. C, Absolute % change in length and head width of thin spines. There was a greater change in length and head width of thin spines over 3 and 7 d time intervals, which was not observed during a single hour. D, Absolute % change in length and head width of mushroom spines. Mushroom-shaped spines are stable over a 3 d interval. Over a 7 d interval, there was an increase in the absolute % change in length of mushroom spines. *p < 0.05 (one-way ANOVA, Kruskal–Wallis test).
Figure 5.

Spinal cord dorsal horn dendritic spine length and head width dynamics over the first 3 d after SNI. SNI did not change spine length or head width dynamics between baseline and 3 d following injury. A, Absolute % change in spine length and head width of total spines, which includes all spine shape categories. B, Change in spine length and head width of thin-shaped spines. C, Absolute % change in spine length and head width of mushroom spines.
Figure 6.

Dendritic spine length and head width dynamics over 7 d after SNI. A, Absolute % change in spine length and head width of total spines, which includes all spine shape categories. SNI induced greater % change in total spine length and width during a 7 d interval compared with controls. B, Absolute % change in spine length and head width of thin-shaped spines. After SNI, thin spines displayed a greater % change in spine length and head width over 7 d compared with controls. C, Absolute % change in spine length and head width in mushroom spines. SNI induced an increase in spine head width dynamics compared with controls. *p < 0.05 (Mann–Whitney test).
For de novo formation and elimination datasets, we only included dendritic spines that completely appeared or disappeared between the two imaging datasets of the same neuron. To normalize data for sampling variation, due to differing lengths of dendritic branches on individual neurons, we compared spine formation/elimination density (calculated as the number of spines gained/lost per micron dendritic branch length, or spines/micron dendrite) (see Figs. 8, 9). To calculate total turnover activity, we summed the formation and elimination densities for each group (3 or 7 d after SNI or control).
Figure 8.

SNI causes an increase in newly formed dendritic spines 7 d after injury. A, Gain and loss of total, thin, and mushroom spines over the first 3 d following SNI or control. From baseline to day 3 compared with control, SNI led to a reduction in the loss of total spines and thin-shaped spines. B, Gain and loss of total, thin, and mushroom spines over 7 d following SNI or control. From baseline to day 7 compared with control, SNI led to an increase in the gain in total and mushroom-shaped spines. C, D, Total, thin, and mushroom spine turnover per micron of dendrite (gain/µm + loss/µm). C, At day 3, SNI led to a decrease in thin spine turnover/µm compared with control. D, In contrast, at 7 d, SNI led an increase in the total and mushroom spine turnover. #p < 0.05 for total spines; *p < 0.05 for thin spines; §p < 0.05 for mushroom spines; Mann–Whitney test.
Figure 9.

SNI leads to an increase in the density of mushroom-shaped dendritic spines. A compared with uninjured controls, SNI does not alter total spine density. B, There is no statistical difference in thin spine density between uninjured controls and SNI groups. C, SNI increased the density of mushroom-shaped dendritic spines compared with uninjured controls. *p < 0.05 (unpaired t test).
To ensure that dendritic spines were accurately identified using YFP labeling in our intravital images, we use several spine characteristics defined previously (B. G. Kim et al., 2006; Zhao et al., 2016). Briefly, a dendritic spine neck is the structure juxtaposed between the parent dendrite branch and the base of the spine head, which appears as a distal swelling into a bulb-shaped structure. Thin- and mushroom-shaped spines were classified and analyzed separately: thin spines had head diameters that were less than or equal to the length of the spine neck, whereas mushroom spines had head diameters that were greater than the length of the spine neck. Although this approach precluded discrimination of other subtle variations in spine shape, we and others have shown that thin- and mushroom-shaped morphologies provide correlative power to interpret the physiological characteristics of circuit function and may predict functional outcome (e.g., including pain) (Bandaru et al., 2015; Zhao et al., 2016; Cao et al., 2017).
Immunohistochemistry
Mice were anesthetized using a mixture of ketamine/xylazine (100/10 mg/kg, i.p.) and intracardially perfused with ice-cold PBS followed by 4% PFA (0.01 M PBS). The spinal cord region (∼L4-L5) covered by the glass window was dissected and postfixed in 4% PFA overnight. To cryoprotect tissue for sectioning, spinal cords were submerged in 30% sucrose for 48 h. For immunohistochemistry, 20-µm-thick tissues were cut on a cryostat (Leica Microsystems) and mounted on Superfrost+ slides (Fisher Scientific). Briefly, sections were blocked for 1 h in 4% normal donkey serum, 2% BSA, 0.1% Triton X-100, 0.01 M PBS. Sections were incubated overnight in rabbit polyclonal CGRP (1:500, Abcam, catalog #ab36001), PKCγ (1:500, Proteintech, catalog #14364-1-AP), and IB4-568 (1:250, Thermo Fisher Scientific, catalog #I21412), washed in 0.01 M PBS, and incubated with donkey anti-rabbit 647 (Jackson ImmunoResearch Laboratories, catalog #711-496-152). Immunofluorescent images were captured using a laser scanning confocal microscope (A1R, Nikon).
Statistical analysis
All statistical tests were performed at the α level of significance of 0.05 by two-tailed analyses using parametric or nonparametric test, as appropriate. We used paired and unpaired t tests, Mann–Whitney U test, and Kruskal–Wallis one-way ANOVA on ranks, followed by Dunn's post hoc analysis. Normality assumptions of each dataset were determined using a Shapiro–Wilk test. As a standard measure of data “dispersion,” we calculated the coefficient of variation (CoV), which is a ratio of the SD to the mean (CoV = SD/mean). Data management and statistical analyses were performed using Prism Graphpad (version 8.0, GraphPad Software) and Microsoft Office Excel 2018. Graphs were rendered using Origin (OriginLab 2019). Data are presented as mean ± SEM.
Results
Intravital profiling of dendritic spines on lamina II neurons
To profile dendritic spine dynamics on superficial dorsal horn neurons, we implanted glass windows over the surface of the spinal cord (Fenrich et al., 2012). Animals were allowed to recover for up to 5 d following window implant (Fig. 1). Neuron-specific Thy1-promoter-driven YFP expression clearly revealed cells within the superficial laminae of the dorsal horn (L4 spinal segments) (Fig. 2E). Overlying white matter nerve roots and ascending tracts led to exposure oversaturation. We therefore imaged neurons located within a narrow lateral region ∼582.5 µm (range: 435-720 µm) from the spinal cord midline, near the L4 spinal roots and innervating segment associated with the sural nerve (Fig. 2B,C; Table 1) (Swett et al., 1991). As shown in Figure 2F, G, postmortem histology analysis shows that the majority of cell bodies of YFP-labeled neurons are located in the inner region of lamina II, which is delineated with immunoreactivity to isolectin IB4 and PKC-γ (Fig. 2F,G). Of the cells analyzed from spinal cord cross sections, 3 of 58 YFP-labeled cells (or 5.2%) resided in the CGRP layer corresponding to lamina I and II outer, 21 of 58 (or 32.8%) of these cells were in the IB4 layer, and 36 of 58 (or 62.0%) of YFP-labeled cells were located in the PKC-γ layer (Fig. 2H) (Todd, 2010; Duan et al., 2014). Sampled neurons had mean cell diameters of 18.9 µm, which is within the range expected for neurons in lamina II dorsal horn (Light and Kavookjian, 1988; Todd, 2010).
Ongoing, steady-state fluctuations in dendritic spine structure
To profile basal steady-state dynamics in dendritic spine morphology, we performed paired-imaging sessions over time (Fig. 3). Baseline imaging in animals with spinal window implants captured three time intervals: 1 h, 3 d, and 7 d (representative dendritic branch from the same neurons imaged over time is shown in Fig. 3A). We analyzed differences in dendritic spine length (i.e., distance from the base of the spine neck to the furthest distance on the spine head), and spine head width (i.e., maximal width of the bulb-like head structure) on lamina II dorsal horn sensory neurons (see also Materials and Methods). To normalize for variation across neurons, we calculated the percent change in spine length or head width. Additionally, because we were interested in comparing the absolute change in these morphometries, we performed a nondirectional transformation (i.e., rectified datasets). Together, we profile steady-state fluctuations of spine length and head diameter using “mean absolute % change” as calculated from paired-imaging data of the same neuron captured over time.
At the 1 h paired interval (imaging dendritic spine from the same neurons), we detected relatively low fluctuations in overall spine length and spine head width compared with the 3 or 7 d imaging intervals of the same neurons (total spines, 1 h vs 3 or 7 d: 1 h % Δ spine length 18.2 ± 1.8; % Δ head width 17.1 ± 1.6 vs 3 d % Δ spine length 45.5 ± 4.1; % Δ head width 39.3 ± 3.8; 7 d % Δ spine length 46.8 ± 3.6% Δ head width 39.5 ± 3.5, p < 0.05, Kruskal–Wallis one-way ANOVA) (Fig. 3B). Similarly, we observed the same lower fluctuations in thin-shaped dendritic spines at the 1 h imaging interval compared with 3 or 7 d interval (thin-shaped spines, 1 h vs 3 or 7 d: 1 h % Δ spine length 20.3 ± 2.5; % Δ head width 16.5 ± 2.1 vs 3 d % Δ spine length 49.3 ± 4.6; % Δ head width 41.4 ± 4.3; 7 d % Δ spine length 51.4 ± 4.3; % Δ head width 43.0 ± 4.3, p ≤ 0.05 Kruskal–Wallis one-way ANOVA) (Fig. 3C). As expected, mushroom-shaped spines were associated with more structural stability (Matsuzaki et al., 2004; Bourne and Harris, 2007; Tan et al., 2011). We were only able to detect a statistical increase in absolute % change in mushroom spine length between the 1 h interval and the 7 d interval (mushroom-shaped spines, 1 h vs 3 or 7 d: 1 h % Δ spine length 14.1 ± 2.3; % Δ head width 18.1 ± 2.6; 3 d % Δ spine length 24.6 ± 6.6; % Δ head width 27.4 ± 7.4; 7 d % Δ spine length 32.9 ± 5.9; % Δ head width 28.7 ± 4.5, p ≤ 0.05, Kruskal–Wallis one-way ANOVA) (Fig. 3D). We observed no difference in mean absolute % change in spine length or head diameter for any spine type in 3 versus 7 d comparisons (Fig. 3B,C).
To assess changes in dispersion (e.g., data spread from the mean), we calculated the CoV (SD/mean of spine length or head width). In general, CoV values of spine length increased by nearly 14%-20% at the 7 d imaging interval compared with the 1 h imaging interval (CoV for spine lengths of all spine types: 7 d, 0.95; 3 d, 0.89; 1 h, 0.81). The CoV for spine head diameter also increased by a similar amount (∼16%) between the 7 d and 1 h imaging intervals (CoV for spine head width of all spine types: 7 d, 0.92; 3 d, 0.80; 1 h, 0.77). Together, these values demonstrate that dorsal horn dendritic spines are more stable in the shorter imaging intervals (i.e., 1 h).
SNI leads to the development of neuropathic pain
If a relationship exists between abnormal dendritic spines and abnormal pain condition, then SNI-induced pain should coincide with changes in dendritic spine dynamics. We previously showed significant changes in dendritic spine morphology on neurons in the dorsal horn in a spectrum of preclinical pain models (e.g., spinal cord injury, diabetes, peripheral nerve injury) (Tan et al., 2008, 2009, 2011; Zhao et al., 2016; Cao et al., 2017; Guo et al., 2018). Disrupting dendritic spine remodeling with the Rac1 GTPase inhibitor, NSC23766, or the Pak1 inhibitor, romidepsin (FK268), prevented the development and ongoing maintenance of elevated pain sensitivity (Tan et al., 2012; Zhao et al., 2016; Cao et al., 2017; Guo et al., 2017). However, a caveat of these prior studies is the reliance on postmortem tissue analyses, which produces findings that only imply the relationship between spine remodeling and injury-induced pain.
Here, we sought to profile the in vivo dynamic behavior of dendritic spines over time before and after peripheral nerve injury and pain onset. After a recovery period following window implant surgeries (Fig. 1), we performed an SNI procedure on Thy1-YFP mice (n = 9). The SNI procedure produces a partial denervation model of neuropathic pain, which rapidly increases cutaneous pain sensitivity along the spared sural nerve receptive field located on the glabrous surface of ipsilateral hindpaw (Decosterd and Woolf, 2000; Shields et al., 2003). We observed a significant decrease in hindpaw withdrawal threshold in von Frey testing at 3 d after SNI compared with baseline (no injury), indicating the presence of significant punctate tactile allodynia (baseline vs 3 d after SNI: 0.8 ± 0.1 vs 0.2 ± 0.1 g, p < 0.05 paired t test) (Fig. 4A). By 7 d after SNI, tactile pain withdrawal thresholds decreased further compared with baseline control (baseline vs 7 d after SNI: 0.8 ± 0.1 vs 0.06 ± 0.04 g, p < 0.05, paired t test). Similarly, we observed an increase in neuropathic heat pain sensitivity (i.e., reduced hindpaw withdrawal latency) in Hargreaves testing following 3 and 7 d after SNI compared with baseline threshold (baseline vs 3 d after SNI: 11.5 ± 0.5 vs 9.8 ± 0.3 s; baseline vs 7 d after SNI: 11.5 ± 0.5 vs 8.1 ± 0.6 s, p < 0.05 paired t test) (Fig. 4B).
Figure 4.

SNI produces thermal and mechanical hypersensitivity 3 and 7 d after injury. A compared with baseline, SNI produced a significant reduction in von Frey withdrawal threshold at both 3 and 7 d after injury. B, Thermal nociception was measured using the Hargreaves assay. SNI reduced withdrawal latency at both 3 and 7 d after injury compared with baseline, indicating thermal hypersensitivity. *p < 0.05 (paired t test).
Peripheral nerve injury increases dendritic spine dynamics in the dorsal horn neurons
To investigate how peripheral nerve injury would change dendritic spine behavior over time, we imaged the same ipsilateral dorsal horn sensory neurons in lamina II before and after an SNI. Images taken at baseline were paired with their corresponding images taken at 3 or 7 d after SNI or without nerve injury and analyzed for absolute % change in dendritic spine length and head width (Figs. 5, 6). Three days after SNI, we observed no difference in the mean absolute % change in dendritic spine length compared with no injury (total spines, SNI vs no injury: % Δ spine length 48.3 ± 4.8 vs 45.5 ± 4.1; thin-shaped spines: % Δ spine length 57.7 ± 6.1 vs 49.3 ± 4.6; mushroom-shaped spines: % Δ spine length 29.9 ± 29.3 vs 24.6 ± 25.4, p > 0.05 Mann–Whitney test) (Fig. 5A-C). Similarly, we observed no % change in spine head width following SNI compared with no injury (total spines, SNI vs no injury: % Δ head width 44.0 ± 4.9 vs 39.3 ± 3.8; thin-shaped spines: % Δ head width 53.9 ± 6.4 vs 41.4 ± 4.3; mushroom-shaped spines: % Δ head width 24.5 ± 5.2 vs 27.4 ± 7.4, p > 0.05 Mann–Whitney test) (Fig. 4A–C). Overall, we observed no significant changes in steady-state fluctuations in spine length and head diameter on lamina II dorsal horn neurons 3 d after nerve injury.
In contrast, 7 d following SNI, we observed a significant difference in the mean absolute % change in spine length for total spines and thin-shaped spines (total spines, SNI vs no injury: % Δ spine length 62.0 ± 4.2 vs 46.8 ± 3.6; thin-shaped spines: % Δ spine length 70.0 ± 5.1 vs 51.4 ± 4.30 p < 0.05 Mann–Whitney test) (Fig. 6A,B). For mushroom-shaped spines, we found no difference in mean absolute % change in spine length (mushroom-shaped spines, SNI vs no injury: % Δ spine length 49.7 ± 6.6 vs 32.9 ± 5.9, p > 0.05 Mann–Whitney test) (Fig. 6C). A comparison of spine head width 7 d following SNI demonstrated a mean increase in the absolute % change in this parameter compared with no injury (total spines, SNI vs no injury: % Δ head width 57.6 ± 4.5 vs 39.5 ± 3.4; thin-shaped spines: % Δ head width 61.1 ± 6.0 vs 43.0 ± 4.3; mushroom-shaped spines: % Δ head width 52.2 ± 6.6 vs 28.8 ± 4.6, p < 0.05 Mann–Whitney test) (Fig. 6A-C).
Interestingly, data dispersion analysis of the 7 d imaging interval demonstrated that SNI results in a 25%–30% reduction in CoV values for overall spine length and head width compared with animals without nerve injury (total spine population, spine length: SNI, 0.59; no injury, 0.81; spine head width: SNI, 0.68; no injury, 0.92). SNI resulted in smaller reduction in CoV values, ∼17%–25%, for spine length and head width of mushroom-shaped spines compared with no injury (mushroom-shaped spines only, spine length: SNI, 0.72; no injury, 0.93; spine head width: SNI, 0.69; no injury, 0.84). In general, our data suggest that mushroom-shaped spines are more resilient to the effect of SNI, which is in agreement with other studies that demonstrate that mushroom-shaped spines are generally more stable structures (Matsuzaki et al., 2004; Bourne and Harris, 2007).
SNI triggers a transition from spine elimination to increased de novo spine formation
We investigated the effect of SNI on dendritic spine formation, elimination, and turnover activity (i.e., an additive function of spine formation and elimination; see Materials and Methods). As shown in Figure 7, we imaged the same dendritic branch regions on dorsal horn neurons over time before and 3 or 7 d after SNI. To normalize for variation in dendritic branch length, we analyzed data as a gain or loss of spines per dendrite branch length (i.e., spines/μm) (Fig. 8). From baseline to 3 d following SNI, we observed a reduction in spine loss per micron of total dendritic spines (total spines, SNI vs no injury: 0.04 ± 0.01 vs 0.06 ± 0.01 spine loss/μm dendrite length, p < 0.05 Mann–Whitney test) (Fig. 8A). This overall reduction in total spine loss was driven primarily by the elimination of thin-shaped spines following SNI (thin-shaped spines, SNI vs no injury: 0.03 ± 0.01 vs 0.06 ± 0.01 spine loss/μm dendrite length, p < 0.05 Mann–Whitney test) (Fig. 8A). Mushroom-shaped spines remained stable with no difference in gain or loss at 3 d after SNI (SNI vs no injury: p > 0.05 Mann–Whitney test) (Fig. 8A). Additionally, we did not find a significant increase in the de novo formation of dendritic spines of any type (SNI vs no injury; total spines 0.03 ± 0.01 vs 0.02 ± 0.01; thin spines 0.02 ± 0.01 vs 0.02 ± 0.01; mushroom spines 0.006 ± 0.01 vs 0.004 ± 0.004 spine gain/μm dendrite, p > 0.05 Mann–Whitney test) (Fig. 8A).
Figure 7.

Representative composite traces of dendritic segments before and following injury. White arrows indicate the location of a lost spine. Black arrows indicate the presence of a newly formed spine. A, Dendritic branch segments from baseline to 3 d following SNI or control. B, Dendritic branch segments from baseline to 7 d following SNI or control.
To assess the turnover activity of dendritic spines before injury compared with following SNI, we calculated the turnover activity (i.e., additive function of spine formation and elimination). From baseline to 3 d following SNI, turnover activity declined in thin-shaped dendritic spines (thin spines, SNI vs injury: 0.05 ± 0.01 vs 0.08 ± 0.004 spine turnover/μm dendrite, p < 0.05 Mann–Whitney test) (Fig. 8C). There was no significant difference in the turnover activity of total or mushroom spine at 3 d between SNI and no injury (SNI vs injury: total spines 0.07 ± 0.01 vs 0.08 ± 0.002; mushroom spines 0.01 ± 0.01 vs 0.004 ± 0.004 turnover/μm dendrite, p > 0.05 Mann–Whitney test).
From baseline to 7 d after SNI, total dendritic spine formation markedly increased (total spines, SNI vs no injury: 0.1 ± 0.04 vs 0.02 ± 0.01 spine gain/μm dendrite length) (Fig. 8B). Interestingly, at 7 d after SNI, we observed a significant increase in formation of new mushroom-shaped spines compared with no injury (mushroom-shaped spines, SNI vs no injury: 0.04 ± 0.02 vs 0.0 ± 0.0 spine gain/μm dendrite length) (Fig. 8B). We observed no difference in newly formed thin-shaped dendritic spines 7 d following SNI (thin-shaped spines, SNI vs no injury: 0.06 ± 0.03 vs 0.02 ± 0.01 spine gain/μm dendrite length) (Fig. 8A). Additionally, we observed no change in the elimination of any dendritic spine type (SNI vs no injury; total spines 0.06 ± 0.01 vs 0.04 ± 0.01; thin spines 0.06 ± 0.01 vs 0.04 ± 0.01; mushroom spines 0.007 ± 0.007 vs 0.003 ± 0.002 spine loss/μm dendrite, p > 0.05 Mann–Whitney test) (Fig. 8B).
In contrast to 3 d after injury, 7 d after SNI we observed a significant increase in the turnover activity of total and mushroom-shaped spines (SNI vs no injury; total spines 0.2 ± 0.04 vs 0.07 ± 0.01; mushroom spines 0.04 ± 0.02 vs 0.003 ± 0.002 turnover/μm dendrite; p < 0.05, Mann–Whitney test; Fig. 8D). In addition, 7 d after SNI, there was no change in the turnover activity of thin-shaped spines compared with no injury (thin spine, SNI vs injury: 0.1 ± 0.03 vs 0.06 ± 0.02 turnover/μm dendrite, p < 0.05, Mann–Whitney test) (Fig. 8D). These data demonstrate that, at the earlier time point after SNI, thin-shaped dendritic spines are eliminated at a higher rate than compared with later after injury. In contrast, at the later time point, there is an increased gain and turnover activity, particularly of mushroom-shaped spines.
As shown in previously published postmortem tissue analyses, increased mushroom spine density is strongly associated with the presence of neuropathic pain in several models of pain (Zhao et al., 2016; Cao et al., 2017; Guo et al., 2017, 2018). In agreement with these previous findings, our intravital study demonstrated a significant dynamic increase in the density of mushroom-shaped dendritic spines at 7 d following SNI compared with neurons in animals with no injury (mushroom-shaped spine density, SNI vs no injury: 0.1 ± 0.01 vs 0.07 ± 0.01 spines/μm; p < 0.05, unpaired t test; Fig. 9C). This likely occurred from the increase in de novo mushroom-shaped dendritic spine formation. We observed no change in total- or thin-shaped dendritic spine densities (SNI vs no injury; total spine density 0.3 ± 0.02 vs 0.2 ± 0.02: thin-shaped density 0.2 ± 0.03 vs 0.2 ± 0.01 spine/μm dendrite, p > 0.05 unpaired t test; Fig. 9A,B).
Discussion
Spinal cord pain circuits are highly plastic and undergo structural and functional reorganization in response to environmental conditions (Raisman, 1994; Sandkuhler and Liu, 1998; Ikeda et al., 2006; Sandkuhler, 2007; Beauparlant et al., 2013). Our work over the past decade and that of others demonstrate a common structural motif of dendritic spine dysgenesis associated with neuropathic pain. This dysgenesis includes an increase in mushroom-shaped spines, which are known to be more mature, stable structures that contribute to amplified excitatory transmission in nociceptive circuits, as well as significant alternations in spine length and head size (Tan et al., 2012; Zhao et al., 2016; Cao et al., 2017; Guo et al., 2017). Blocking molecular regulators of dendritic spine remodeling with pharmacological inhibitors attenuates both injury-induced spine dysgenesis and neuropathic pain. This emerging evidence supports the premise that dendritic spine profiling serves as a structural-based opportunity to study and treat pathologic pain.
To further understand the relationship between dendritic spine dynamics and injury-induced neuropathic pain, we performed a longitudinal intravital study of dendritic spines in the superficial dorsal horn. Previous anatomic studies have been limited by analyses of dendritic spines in postmortem spinal cord tissue. To bridge this gap, we modified the whole-animal imaging approach pioneered by Fenrich et al. (2012), which permitted us to repeatedly image the same superficial sensory neurons from lamina II in Thy1-YFP mice. Using this technique, we profiled the in vivo dynamics of dendritic spines on the same neurons before and at several time points after peripheral nerve injury-induced pain. In agreement with previous work (Cichon et al., 2018), our SNI model reproducibly elicited a consistent pain profile in mice with elevated pain within 3 d following surgery, which peaked at 1 week.
Our main findings demonstrate a time-dependent relationship between progressively increased pain sensitivity and mean steady-state fluctuations in spine length and head width after SNI. Additionally, dendritic spine turnover activity depended on the progression of pain severity: A decrease in thin spine elimination 3 d after SNI was followed by an increase in mushroom spine formation 7 d after SNI. The increase in the rate of mushroom spine formation and overall density coincided with the onset of maximal pain. Overall, our current study reports, for the first time, the ongoing, steady-state changes in dendritic spine dynamics in the dorsal horn in an established peripheral nerve injury model of pain.
Interestingly, malformed dendritic spines occur in a spectrum of chronic neurologic diseases, including epilepsy, stroke, post-traumatic stress disorder, autism spectrum disorder, mental retardation, dementia, and chronic substance abuse or addiction (Sigler and Murphy, 2010; Penzes et al., 2011). The strongest evidence for a relationship between malformed dendritic spines and abnormal function comes from a series of Fragile X syndrome (e.g., fmr1 KO) studies. Fmr1 KO mice have impaired LTP (Wilson and Cox, 2007) and malformed dendritic spines due to dysfunctional Rac1 signaling (Chen et al., 2010). Moreover, these mice displayed decreased neuropathic allodynia (Price et al., 2007). Although a relationship between LTP, dendritic spine behavior, and neuropathic pain has never been studied directly, electrical tetanus and inflammation can induce LTP within nociceptive circuits in the superficial dorsal horn (Sandkuhler and Liu, 1998; Ikeda et al., 2006; Sandkuhler, 2007). An important avenue of future study could include a mechanistic investigation of dendritic spine plasticity in the context of LTP and pain.
Our previous investigations of dendritic spines relied entirely on postmortem tissue. Although these studies provided evidence of a relationship between dendritic spine remodeling and abnormal pain, we were only able to provide a snapshot of dendritic spine behavior. Previous intravital imaging in the brain using two-photon microscopy has demonstrated dendritic spine dynamic behavior in response to varied inputs. For example, in vivo imaging of the adult mouse cortex reveals that thin-shaped dendritic spines are highly motile and short-lived or transient. In contrast, the presence of dendritic spines with a mature mushroom-shaped morphology tends to be highly stable (Grutzendler et al., 2002; Matsuzaki et al., 2004; Bourne and Harris, 2007; Tan et al., 2009; Cao et al., 2017). Importantly, these intravital brain imaging studies reveal that dendritic spine morphology as well as steady-state dynamics are dependent on the brain location studied, neuron type, topographical location, and age. Even dendrite branch regions on the same neuron can have different spine densities (B. G. Kim et al., 2006; Ma et al., 2016; Tjia et al., 2017). Moreover, in vivo cortical imaging has shown that dendritic spines respond to injury (e.g., spinal cord injury or sciatic nerve ligation) or fear-conditioning differently depending on brain region (e.g., motor or somatosensory cortex) (B. G. Kim et al., 2006; S. K. Kim and Nabekura, 2011; Z. Xu et al., 2019). Together, these cortical dendritic spine studies show that structural-related mechanisms are context-dependent to tissue region, time, and injury response.
Our current study profiles the dynamic dendritic spine plasticity in the superficial dorsal horn of the spinal cord. The superficial dorsal horn (i.e., lamina I-II) is a significant site of pain transmission and modulation in the spinal cord. Neurons in lamina II receive afferent signals from nociceptive C fibers and Aδ fibers innervating various tissues, including skin, muscle, and visceral organs (Todd, 2002). Notably, unmyelinated C fibers comprise ∼70% of nociceptive afferents and terminate primarily in lamina I and II (Nagy and Hunt, 1983; Todd, 2010). Since lamina II is known to be involved in pain processing, and our previous work suggests that dendritic spine remodeling contributes to pain, we hypothesized that neurons in lamina II would experience dynamic changes in spine shape and density after peripheral nerve injury.
Our implanted window approach provides notable advantages for studying dendritic spines in the spinal cord dorsal horn. Nonetheless, we do note several important caveats to our study. First, we only investigated cells labeled with Thy1-YFP, a specific marker for neurons. Although we cannot define these cells, the majority of spiny neurons in the CNS are glutamatergic (Keck et al., 2011) and have served as established structural proxies and synaptic activity (Majewska et al., 2006). Second, our imaging study was limited by the size of the dendritic spine structures, which was close to the estimated optical diffraction limit of our equipment (∼0.25 μm). It is possible in some cases that we did not have the resolution to confidently detect more subtle changes in dendritic spine structure; thus, our data may underestimate the overall dynamics. Finally, we observed the rapid onset of pain within 3 d, but no mean changes in steady-state spine length or head width from the same time span. This suggests that additional factors contribute to the pain behavioral outcomes that we observed. Several studies using similar models of nerve injury demonstrate the contribution of early inflammatory processes, such as microgliosis, in neuropathic pain (Ji and Strichartz, 2004; Scholz and Woolf, 2007; Bai et al., 2016; Liu et al., 2017). Future studies might leverage genetic tools to label inflammatory cells to determine the time course of events that may precede dendritic spine dysgenesis. Suffice it to say, our current report using intravital imaging extends our previous studies, and that of others, demonstrating the increased dendritic spine density and pain occur following SNI.
Our study raises the question of how dendritic spine structure contributes to pain and hyperexcitability. A core feature of neuropathic pain is hyperexcitability within nociceptive pathways in the PNS and CNS, including DRG and dorsal spinal cord. Dendritic spines are microscopic-sized postsynaptic structures that contribute directly to synaptic and circuit function (Segev and Rall, 1988; Tan et al., 2009). Dendritic spines act as bioelectrical transducers through which even small changes in geometry can powerfully influence excitatory postsynaptic potentials and alter the risk for generating an action potential (Araya et al., 2006; Tan et al., 2009). In our study following nerve injury, we observed a significant increase in mushroom-shaped dendritic spines, a morphology that may permit an increase in membrane expressed glutamatergic postsynaptic receptors (e.g., AMPA, NMDA) (Bourne and Harris, 2008). Dendritic spine size linearly correlates with postsynaptic density size, the number of presynaptic vesicles (Harris and Stevens, 1989), and synaptic strength (Matsuzaki et al., 2004; Oh et al., 2013). Together, the biophysical attributes of mature, mushroom-shaped structures contribute to enhancing input summation, frequency following ability, and could be a mechanistic factor in contributing to neuronal hyperexcitability associated with central sensitization (Araya et al., 2006; Tan et al., 2009).
We can only speculate regarding the mechanisms involved in the dendritic spine changes we observed. Dendritic spine reorganization depends in part on synaptic activity. For example, loss of activity leads to dendritic spine stabilization in cortical neurons (Holtmaat and Svoboda, 2009). In studies by several groups, sensory deprivation in mice through unilateral whisker trimming or monocular deprivation leads first to a reduced rate of spine elimination followed by an increase in newly formed persistent spines with mushroom-like structure in the affected cortex (Halpain et al., 2005; Zuo et al., 2005; Berry and Nedivi, 2017; Tjia et al., 2017). In our study, nerve injury-induced loss of peripheral afferent input may have triggered similar spine reorganization behavior. We report an initial loss of spines at 3 d after SNI followed by an increase in the formation of new mushroom spines at 7 d. These results extend our previous work using Golgi staining, which suggests a structure-function link between dendritic spine dysgenesis and injury-induced pain.
In conclusion, our findings demonstrate the powerful utility of intravital study of dendritic spine behavior in the superficial dorsal horn. We show, for the first-time, changes in dendritic spine steady-state behavior associated with nerve injury-induced chronic pain. Importantly, the dynamic increase in steady-state presence of more stable mushroom spines supports a conceptual “locking in of pain” through stabilization of abnormal structures within spinal nociceptive circuits. Ultimately, this work opens the door to further investigations in vivo of spinal cord dendritic spine dynamics in the context of injury and disease.
Footnotes
The authors declare no competing financial interests.
This work was supported by the Department of Veterans Affairs Rehabilitation R&D Service, Paralyzed Veterans of America, and the Nancy Taylor Foundation for Chronic Diseases.
References
- Araya R, Eisenthal KB, Yuste R (2006) Dendritic spines linearize the summation of excitatory potentials. Proc Natl Acad Sci USA 103:18799–18804. 10.1073/pnas.0609225103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Wang X, Li Z, Kong C, Zhao Y, Qian JL, Kan Q, Zhang W, Xu JT (2016) Upregulation of chemokine CXCL12 in the dorsal root ganglia and spinal cord contributes to the development and maintenance of neuropathic pain following spared nerve injury in rats. Neurosci Bull 32:27–40. 10.1007/s12264-015-0007-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru SP, Liu S, Waxman SG, Tan AM (2015) Dendritic spine dysgenesis contributes to hyperreflexia after spinal cord injury. J Neurophysiol 113:1598–1615. 10.1152/jn.00566.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto RP, Ko TH, Jung JC, Wang TJ, Capps G, Waters AC, Ziv Y, Attardo A, Recht L, Schnitzer MJ (2011) Time-lapse imaging of disease progression in deep brain areas using fluorescence microendoscopy. Nat Med 17:223–228. 10.1038/nm.2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauparlant J, van den Brand R, Barraud Q, Friedli L, Musienko P, Dietz V, Courtine G (2013) Undirected compensatory plasticity contributes to neuronal dysfunction after severe spinal cord injury. Brain 136:3347–3361. 10.1093/brain/awt204 [DOI] [PubMed] [Google Scholar]
- Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD (2019) The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99:1079–1151. 10.1152/physrev.00052.2017 [DOI] [PubMed] [Google Scholar]
- Berry KP, Nedivi E (2017) Spine dynamics: are they all the same? Neuron 96:43–55. 10.1016/j.neuron.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Harris KM (2007) Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol 17:381–386. 10.1016/j.conb.2007.04.009 [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31:47–67. 10.1146/annurev.neuro.31.060407.125646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XC, Pappalardo LW, Waxman SG, Tan AM (2017) Dendritic spine dysgenesis in superficial dorsal horn sensory neurons after spinal cord injury. Mol Pain 13:1744806916688016. 10.1177/1744806916688016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramar EA, Lynch G, Gall CM, Lauterborn JC (2010) Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci 30:10977–10984. 10.1523/JNEUROSCI.1077-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon J, Sun L, Yang G (2018) Spared nerve injury model of neuropathic pain in mice. Bio Protoc 8:e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87:149–158. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14:49–62. 10.1038/nrn3404 [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, Ross S, Lowell BB, Wang Y, Goulding M, Ma Q (2014) Identification of spinal circuits transmitting and gating mechanical pain. Cell 159:1417–1432. 10.1016/j.cell.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Hocine M, Zalc M, Rougon G, Debarbieux F (2012) Long-term in vivo imaging of normal and pathological mouse spinal cord with subcellular resolution using implanted glass windows. J Physiol 590:3665–3675. 10.1113/jphysiol.2012.230532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenrich KK, Weber P, Rougon G, Debarbieux F (2013) Implanting glass spinal cord windows in adult mice with experimental autoimmune encephalomyelitis. J Vis Exp. Advance online publication. Retrieved 21 December, 2013. doi: 10.3791/50826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB (2002) Long-term dendritic spine stability in the adult cortex. Nature 420:812–816. 10.1038/nature01276 [DOI] [PubMed] [Google Scholar]
- Guo Y, Effraim P, Benson C, Henry S, Waxman SG, Dib-Hajj SD, Tan AM (2017) Therapeutic targeting of Pak1 signaling alleviates neuropathic pain after traumatic 2nd degree burn injury. Soc Neurosci Abstr 676. [Google Scholar]
- Guo Y, Benson C, Hill M, Henry S, Effraim P, Waxman SG, Dib-Hajj S, Tan AM (2018) Therapeutic potential of Pak1 inhibition for pain associated with cutaneous burn injury. Mol Pain 14:1744806918788648. 10.1177/1744806918788648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S, Spencer K, Graber S (2005) Dynamics and pathology of dendritic spines. Prog Brain Res 147:29–37. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9:2982–2997. 10.1523/JNEUROSCI.09-08-02982.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison M, O'Brien A, Adams L, Cowin G, Ruitenberg MJ, Sengul G, Watson C (2013) Vertebral landmarks for the identification of spinal cord segments in the mouse. Neuroimage 68:22–29. 10.1016/j.neuroimage.2012.11.048 [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10:647–658. 10.1038/nrn2699 [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, Sandkuhler J (2006) Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science 312:1659–1662. 10.1126/science.1127233 [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G (2004) Cell signaling and the genesis of neuropathic pain. Sci STKE 2004:reE14. 10.1126/stke.2522004re14 [DOI] [PubMed] [Google Scholar]
- Keck T, Scheuss V, Jacobsen RI, Wierenga CJ, Eysel UT, Bonhoeffer T, Hubener M (2011) Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron 71:869–882. 10.1016/j.neuron.2011.06.034 [DOI] [PubMed] [Google Scholar]
- Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS (2006) Remodeling of synaptic structures in the motor cortex following spinal cord injury. Exp Neurol 198:401–415. 10.1016/j.expneurol.2005.12.010 [DOI] [PubMed] [Google Scholar]
- Kim SK, Nabekura J (2011) Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J Neurosci 31:5477–5482. 10.1523/JNEUROSCI.0328-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R. (2010) Central mechanisms of pathological pain. Nat Med 16:1258–1266. 10.1038/nm.2231 [DOI] [PubMed] [Google Scholar]
- Li Y, Dorsi MJ, Meyer RA, Belzberg AJ (2000) Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain 85:493–502. [DOI] [PubMed] [Google Scholar]
- Light AR, Kavookjian AM (1988) Morphology and ultrastructure of physiologically identified substantia gelatinosa (lamina II) neurons with axons that terminate in deeper dorsal horn laminae (III-V). J Comp Neurol 267:172–189. 10.1002/cne.902670203 [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG (2017) TNF-alpha differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci 37:871–881. 10.1523/JNEUROSCI.2235-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Qiao Q, Tsai JW, Yang G, Li W, Gan WB (2016) Experience-dependent plasticity of dendritic spines of layer 2/3 pyramidal neurons in the mouse cortex. Dev Neurobiol 76:277–286. 10.1002/dneu.22313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska AK, Newton JR, Sur M (2006) Remodeling of synaptic structure in sensory cortical areas in vivo. J Neurosci 26:3021–3029. 10.1523/JNEUROSCI.4454-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Hunt SP (1983) The termination of primary afferents within the rat dorsal horn: evidence for rearrangement following capsaicin treatment. J Comp Neurol 218:145–158. 10.1002/cne.902180203 [DOI] [PubMed] [Google Scholar]
- Oh WC, Hill TC, Zito K (2013) Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc Natl Acad Sci USA 110:E305–E312. 10.1073/pnas.1214705110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Gan WB (2008) Two-photon imaging of dendritic spine development in the mouse cortex. Dev Neurobiol 68:771–778. 10.1002/dneu.20630 [DOI] [PubMed] [Google Scholar]
- Penzes P, Woolfrey KM, Srivastava DP (2011) Epac2-mediated dendritic spine remodeling: implications for disease. Mol Cell Neurosci 46:368–380. 10.1016/j.mcn.2010.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter LE, Paylor JW, Suh JS, Tenorio G, Caliaperumal J, Colbourne F, Baker G, Winship I, Kerr BJ (2016) Altered excitatory-inhibitory balance within somatosensory cortex is associated with enhanced plasticity and pain sensitivity in a mouse model of multiple sclerosis. J Neuroinflammation 13:142. 10.1186/s12974-016-0609-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F (2007) Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci 27:13958–13967. 10.1523/JNEUROSCI.4383-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisman G. (1994) Plasticity of adult central fibre tracts. Prog Brain Res 100:195–201. 10.1016/s0079-6123(08)60786-4 [DOI] [PubMed] [Google Scholar]
- Samad OA, Tan AM, Cheng X, Foster E, Dib-Hajj SD, Waxman SG (2013) Virus-mediated shRNA knockdown of Na(v)1.3 in rat dorsal root ganglion attenuates nerve injury-induced neuropathic pain. Mol Ther 21:49–56. 10.1038/mt.2012.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J. (2007) Understanding LTP in pain pathways. Mol Pain 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J, Liu X (1998) Induction of long-term potentiation at spinal synapses by noxious stimulation or nerve injury. Eur J Neurosci 10:2476–2480. 10.1046/j.1460-9568.1998.00278.x [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ (2007) The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 10:1361–1368. 10.1038/nn1992 [DOI] [PubMed] [Google Scholar]
- Segev I, Rall W (1988) Computational study of an excitable dendritic spine. J Neurophysiol 60:499–523. 10.1152/jn.1988.60.2.499 [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, Basbaum AI (2003) Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain 4:465–470. 10.1067/S1526-5900(03)00781-8 [DOI] [PubMed] [Google Scholar]
- Sigler A, Murphy TH (2010) In vivo 2-photon imaging of fine structure in the rodent brain: before, during, and after stroke. Stroke 41:S117–S123. 10.1161/STROKEAHA.110.594648 [DOI] [PubMed] [Google Scholar]
- Swett JE, Torigoe Y, Elie VR, Bourassa CM, Miller PG (1991) Sensory neurons of the rat sciatic nerve. Exp Neurol 114:82–103. 10.1016/0014-4886(91)90087-S [DOI] [PubMed] [Google Scholar]
- Tan AM, Waxman SG (2015) Dendritic spine dysgenesis in neuropathic pain. Neurosci Lett 601:54–60. 10.1016/j.neulet.2014.11.024 [DOI] [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, Hains BC (2008) Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci 28:13173–13183. 10.1523/JNEUROSCI.3142-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Choi JS, Waxman SG, Hains BC (2009) Dendritic spine remodeling after spinal cord injury alters neuronal signal processing. J Neurophysiol 102:2396–2409. 10.1152/jn.00095.2009 [DOI] [PubMed] [Google Scholar]
- Tan AM, Chang YW, Zhao P, Hains BC, Waxman SG (2011) Rac1-regulated dendritic spine remodeling contributes to neuropathic pain after peripheral nerve injury. Exp Neurol 232:222–233. 10.1016/j.expneurol.2011.08.028 [DOI] [PubMed] [Google Scholar]
- Tan AM, Samad OA, Fischer TZ, Zhao P, Persson AK, Waxman SG (2012) Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J Neurosci 32:6795–6807. 10.1523/JNEUROSCI.1017-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjia M, Yu X, Jammu LS, Lu J, Zuo Y (2017) Pyramidal neurons in different cortical layers exhibit distinct dynamics and plasticity of apical dendritic spines. Front Neural Circuits 11:43. 10.3389/fncir.2017.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ. (2002) Anatomy of primary afferents and projection neurones in the rat spinal dorsal horn with particular emphasis on substance P and the neurokinin 1 receptor. Exp Physiol 87:245–249. 10.1113/eph8702351 [DOI] [PubMed] [Google Scholar]
- Todd AJ. (2010) Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 11:823–836. 10.1038/nrn2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Hains BC (2006) Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci 29:207–215. 10.1016/j.tins.2006.02.003 [DOI] [PubMed] [Google Scholar]
- Wilson BM, Cox CL (2007) Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci USA 104:2454–2459. 10.1073/pnas.0610875104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB (2007) Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci 10:549–551. 10.1038/nn1883 [DOI] [PubMed] [Google Scholar]
- Xu Z, Adler A, Li H, Perez-Cuesta LM, Lai B, Li W, Gan WB (2019) Fear conditioning and extinction induce opposing changes in dendritic spine remodeling and somatic activity of layer 5 pyramidal neurons in the mouse motor cortex. Sci Rep 9:4619. 10.1038/s41598-019-40549-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasaka T, Tiong SY, Hughes DI, Riddell JS, Todd AJ (2010) Populations of inhibitory and excitatory interneurons in lamina II of the adult rat spinal dorsal horn revealed by a combined electrophysiological and anatomical approach. Pain 151:475–488. 10.1016/j.pain.2010.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Hill M, Liu S, Chen L, Bangalore L, Waxman SG, Tan AM (2016) Dendritic spine remodeling following early and late Rac1 inhibition after spinal cord injury: evidence for a pain biomarker. J Neurophysiol 115:2893–2910. 10.1152/jn.01057.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Yang G, Kwon E, Gan WB (2005) Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature 436:261–265. 10.1038/nature03715 [DOI] [PubMed] [Google Scholar]