

Abstract
Human Phenotype Ontology (HPO) terms are increasingly used in diagnostic settings to aid in the characterization of patient phenotypes. The HPO annotation database is updated frequently and can provide detailed phenotype knowledge on various human diseases, and many HPO terms are now mapped to candidate causal genes with binary relationships. To further improve the genetic diagnosis of rare diseases, we incorporated these HPO annotations, gene–disease databases and gene–gene databases in a probabilistic model to build a novel HPO-driven gene prioritization tool, Phen2Gene. Phen2Gene accesses a database built upon this information called the HPO2Gene Knowledgebase (H2GKB), which provides weighted and ranked gene lists for every HPO term. Phen2Gene is then able to access the H2GKB for patient-specific lists of HPO terms or PhenoPacket descriptions supported by GA4GH (http://phenopackets.org/), calculate a prioritized gene list based on a probabilistic model and output gene–disease relationships with great accuracy. Phen2Gene outperforms existing gene prioritization tools in speed and acts as a real-time phenotype-driven gene prioritization tool to aid the clinical diagnosis of rare undiagnosed diseases. In addition to a command line tool released under the MIT license (https://github.com/WGLab/Phen2Gene), we also developed a web server and web service (https://phen2gene.wglab.org/) for running the tool via web interface or RESTful API queries. Finally, we have curated a large amount of benchmarking data for phenotype-to-gene tools involving 197 patients across 76 scientific articles and 85 patients’ de-identified HPO term data from the Children’s Hospital of Philadelphia.
INTRODUCTION
Rapid and accurate genetic diagnosis of Mendelian diseases is necessary to optimize both treatment and management strategies and implement precision medicine. Compared to traditional single-gene tests or gene panels, recent efforts have utilized next-generation sequencing (NGS) technologies, such as whole exome sequencing and whole genome sequencing. The intent of the NGS effort is to improve diagnostic rates, enhance time efficiency and decrease overall financial burdens (1–5). However, due to the substantially larger pool of candidate genes created by NGS data, sequence interpretation has become a major hurdle in diagnostic settings. Computational approaches that streamline the diagnostic workflow and shorten the analytical turnaround time are needed.
The Human Phenotype Ontology (HPO) database (6) associates human diseases with phenotypic abnormalities. These terms possess ever-increasing interoperability with other ontologies (7–11) and allow for computational deep phenotyping, making it the prevailing standard terminology for human phenotypes. We have developed a few computational tools (12–14) that use phenotype data for gene prediction and prioritization. Although these methods are useful, they cannot provide real-time decision support in clinical settings; for example, users may need to add or remove one phenotype term for a patient and immediately observe how the candidate gene list changes.
Recent studies have shown the utility of incorporating phenotype data such as HPO terms in identifying causal genes from NGS data, which increases time efficiency and diagnostic yields (15). There are phenotype analysis tools that use HPO terms to prioritize candidate causal genes, some of which take genes, variants or both: Phevor (16), VarElect (17), OVA (18), Phen-Gen (19), Exomiser (20), AMELIE 2 (21), DeepPVP (22), GADO (23), VarSight (24) and Xrare (25). These HPO terms can be supplied by diagnostic labs, clinical geneticists, doctors or natural language processing (NLP) algorithms that parse doctors’ notes such as Doc2HPO (14). The prioritized list of genes can then be combined with NGS data to identify potential disease genes. The downside of the gene-based tools, however, is that they require gene lists (or variant files) beforehand, some of them take longer to prioritize genes and some of them can only be used via a web interface with no open source code. Thus, such tools cannot be implemented on a large scale and cannot be integrated into existing clinical diagnostic workflows, which are often protected within institutional firewalls.
We present a new rapid, accurate, phenotype-based gene prioritization tool called Phen2Gene. Phen2Gene takes HPO terms for a patient and generates a patient-specific ranked list of candidate genes using our precomputed database, the HPO2Gene Knowledgebase (H2GKB), in a median time of 0.94 s. The H2GKB is built on Enhanced Phenolyzer (v0.4.0), and then Phen2Gene defines weights for each HPO term. Unlike existing binary HPO–gene annotations in the HPO annotation database, the H2GKB is a new database that links each HPO term to its own ranked list of candidate genes, each with a confidence score. These scores represent a substantial accuracy improvement over the previous version of Phenolyzer. We also provide open source code under the MIT license, together with a web server for downloading and accessing the H2GKB and a RESTful API web service for automated queries of phenotype terms with JSON output.
METHODS
Acquiring patient-specific gene lists with Phen2Gene
Physicians can manually curate HPO terms for their patients, which is becoming more common, or feed the patients’ notes into Doc2HPO to discover the relevant and negated HPO terms for the patient’s phenotype. Doc2HPO is a public tool that uses multiple NLP tools and algorithms to parse patient notes into HPO terms. HPO terms act as the sole form of input into Phen2Gene, which then searches the H2GKB, generated by Enhanced Phenolyzer, for each term’s ranked gene list.
All HPO terms under the root term ‘Phenotypic abnormality’ (HP:0000118) are recognizable by Phen2Gene. By default, Phen2Gene weights the inputted HPO terms by skewness of gene scores, as some HPO terms possess greater information content than others (26). Then, Phen2Gene searches the H2GKB for each input term’s gene list and sorts and ranks all the genes based on their ranks in each term’s list and the weight of each HPO term to produce a final, ranked candidate gene list (Figure 1).
Figure 1.
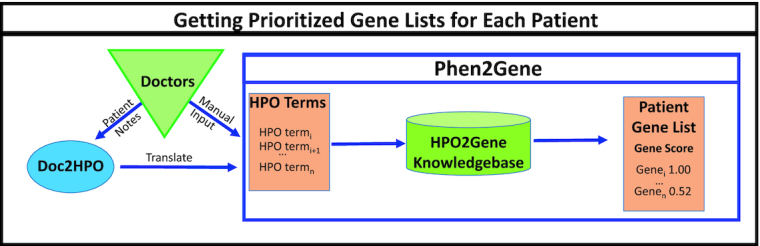
How to use Phen2Gene. Physicians or clinical geneticists can curate HPO terms themselves or provide patient notes to Doc2HPO to generate HPO terms in semi-automated fashion, and these terms will help create a candidate disease gene list using Phen2Gene.
HPO2Gene Knowledgebase construction
In order to construct the H2GKB, we first extract every term from the HPO database, underneath the root term ‘Phenotypic abnormality’ (HP:0000118) (Figure 2A). For each HPO term, we run an enhanced version of Phenolyzer (ver. 0.4.0), dubbed Enhanced Phenolyzer, which incorporates HPO–gene annotations from the Jackson Laboratory (6) and gene–disease annotations from OMIM (27), ClinVar (28), Orphanet (29) and GeneReviews (30). It then adds information from gene–gene databases HPRD (31), NCBI’s Biosystems Database (32), HGNC Gene Family (33) and HTRI (34), and prioritizes and outputs the associated genes.
Figure 2.
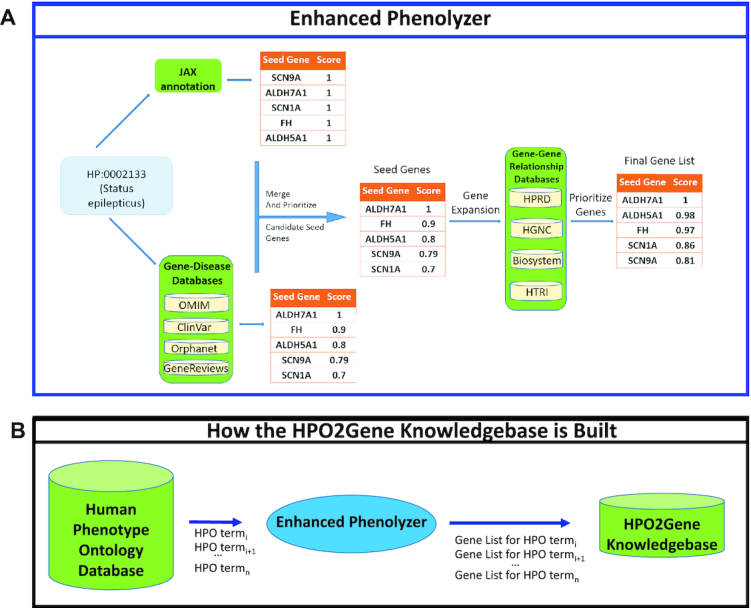
The construction of the H2GKB. HPO terms are extracted one by one from the HPO database and passed into an enhanced version of Phenolyzer (dubbed Enhanced Phenolyzer) to create a database of ranked gene lists for all HPO terms. (A) The workflow of Enhanced Phenolyzer. (B) Construction of the H2GKB.
This generates a ranked list of candidate causal genes for each HPO term, which are then consolidated into the H2GKB (Figure 2B). This precomputed H2GKB can then be rapidly accessed by Phen2Gene and used to rank lists of genes for individual patients. The H2GKB is also freely available online and downloadable from the Phen2Gene web server.
Enhanced Phenolyzer
The original version of Phenolyzer (ver. 0.2.2) processed free-text terms supplied by users. We updated the databases inside Phenolyzer, incorporated new HPO–gene annotations from the Jackson Laboratory database, fixed some bugs and released it as Enhanced Phenolyzer (ver. 0.4.0). Enhanced Phenolyzer contains a new function to turn HPO terms into a list of genes, by generating a prioritized gene list for each HPO term. Unlike the original Phenolyzer, Enhanced Phenolyzer first generates two seed gene sets. Seed Gene Set 1 is built on HPO–gene annotation files downloaded from the Jackson Laboratory for Genomic Medicine available at https://hpo.jax.org/app/download/annotation, while Seed Gene Set 2 construction follows the method outlined in the original Phenolyzer paper, which translates phenotype terms to disease names and incorporates the five precompiled gene–disease databases to search for seed genes.
Candidate gene prioritization
In Enhanced Phenolyzer, we generate a seed gene list of candidate genes for each HPO term in order to create the H2GKB. For seed genes in Set 1, we gave an equal score to each gene and HPO term pair,
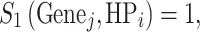 |
since JAX annotation only lists genes for each individual HPO term, but without quantitative scores representing the strength of associations.
In Set 2, we followed the calculation method in the original Phenolyzer for each seed gene using gene–disease databases associated with each individual HPO term, and noted as
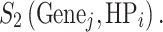 |
We sum up the two scores,
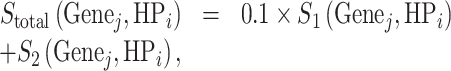 |
and normalized it to a range between 0 and 1 as the final seed gene score,
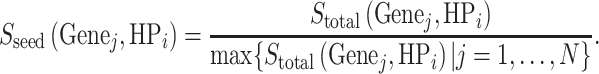 |
We created some arbitrary weights for the JAX associations to test like the original Phenolyzer method did for OMIM and Orphanet. We came to the conclusion that downweighting the JAX HPO–gene associations by a factor of 0.1 was the optimal solution after testing performance on a variety of different factors from 0.1 to 1.0 (Supplementary Figures S1 and S2). In the following steps, we used the original Phenolyzer’s method for expanding the list of candidate genes and reprioritizing the seed gene list using gene–gene databases. Then, we generated the H2GKB with Enhanced Phenolyzer.
Weighting by skewness
Phen2Gene defines weights to add to each HPO term’s gene list generated by Enhanced Phenolyzer in the H2GKB. We calculated the skewness value for the distribution of all gene scores for each HPO term, and used it multiplicatively to adjust the weights of HPO terms individually. The gene score distributions vary widely from term to term. The gene score distributions of ‘Seizures’ (HP:0001250) and ‘Cleft palate’ (HP:0000175) demonstrate the difference in the specificity of HPO terms (Supplementary Figure S3). ‘Cleft palate’ has a positively skewed gene score distribution compared to ‘Seizures’. For ‘Cleft palate’, most genes have a near-zero raw score value, but for ‘Seizures’ the mean and standard deviation are much larger. In fact, ‘Seizures’ occurs 74 times in the benchmark dataset, and ‘Cleft palate’ occurs only 5 times in the benchmark set. In the JAX database, ‘Seizures’ has 1465 gene associations and 2265 disease associations, while ‘Cleft palate’ has 453 gene associations and 747 disease associations. These numbers demonstrate that ‘Seizures’ is a far less specific HPO term than ‘Cleft palate’.
We assume that the more skewed the gene score distribution, the greater the difference between high- and low-ranking genes. This discrepancy provides HPO terms with better information for their associated genes. Thus, we used Pearson’s moment coefficient of skewness to represent the skew and weight HPO terms’ gene weights:
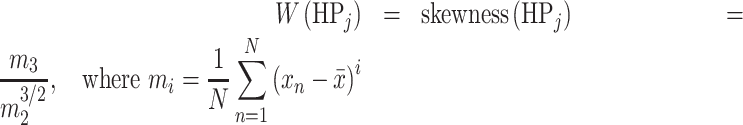 |
and where skewness(HPj) is the skewness of the gene score distribution of HPj, which we calculated with Python 3.8 and the SciPy 1.3.1 stats module. We also created alternative weighting schemes involving no weight or informational content, and our choice of skewness was due to its greater performance over the other methods (Supplementary Figures S4 and S5).
Gene score computation with weighted HPO terms
In this weighting, Phen2Gene defines weights to add to each HPO term’s gene list generated by Enhanced Phenolyzer in the H2GKB. Given a set of HPO terms, 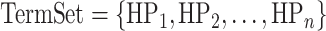 , each HPO term is assigned a weight representing the granularity of phenotypic information given by the HPO term. In each HPj’s candidate gene list, every candidate gene has a score calculated by Enhanced Phenolyzer. It is a quantitative representation of how genei is associated with HPj. Phen2Gene gives a weighted score to genei, if genei is in HPj’s candidate gene list,
, each HPO term is assigned a weight representing the granularity of phenotypic information given by the HPO term. In each HPj’s candidate gene list, every candidate gene has a score calculated by Enhanced Phenolyzer. It is a quantitative representation of how genei is associated with HPj. Phen2Gene gives a weighted score to genei, if genei is in HPj’s candidate gene list,
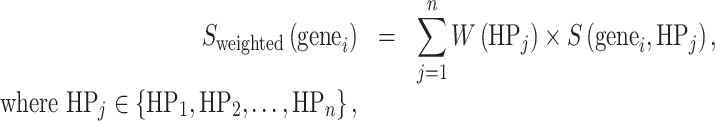 |
where W(HPj) is the assigned weight as illustrated in the previous section, S(genei, HPj) is genei’s score in HPj’s candidate gene list. S(genei, HPj) = 0, if genei is not a candidate gene of HPj. All of genes are sorted by their scores in descending order.
RESULTS
General use
Since the H2GKB is precomputed, the results for Phen2Gene are instant. The weight given to the HPO terms can be chosen or defined by the end user. The terms can be unweighted, weighted by ontology-based information content or the skewness of gene scores for each HPO term, which is the recommended default. No prior gene list knowledge is required, and if a physician has no candidate genes, or if whole exome or whole genome sequencing cannot be performed for practical reasons (such as insurance reimbursement issues), it could help select a targeted sequencing gene panel to find variants causal for the phenotype. This process can be performed case by case on the web server or using the Phen2Gene python script and thus can be scaled up massively to thousands of patients without prior gene knowledge.
Accuracy evaluation with collected expert-curated phenotype data
For our benchmark testing of Phen2Gene, we used 281 de-identified patients who were diagnosed with single-gene diseases as our study subjects. Their study data were from five different sources but three were manually curated by different curators from different institutions. Considering the curation methods—manual and semi-autonomous—the different curation styles and levels of expertise of the curators, they were hence divided into four groups. Group 1 only contained one disease gene (TAF1), but the other three groups contained numerous known and previously validated disease genes (Table 1). The phenotypes in these three groups were chosen because the diseases were monogenic and the causal genes were known, but the phenotypes are not meant to be related in any way: symptoms, genes or pathophysiology.
Table 1.
Curation of benchmark dataset
| Set | Data curation | Where HPO terms are derived |
|---|---|---|
| 1 | 14 cases, with 1 unique causal gene (TAF1), from 1 American Journal of Human Genetics article (35) | Doctor-curated HPO terms |
| 2 | 27 cases from Columbia University Medical Center, with 24 unique causal genes, from 1 article (13) | Manually curated HPO terms from doctor-defined phenotypes |
| 3 | 85 cases from the Department of Genomic Diagnostics at the Children’s Hospital of Philadelphia, with 75 unique causal genes (36) | Doctor-curated HPO terms |
| 4 | 72 cases, with 59 unique causal genes, from 61 Cold Spring Harbor Molecular Case Studies articles (37–97), and 83 cases from with 13 unique genes, from 13 American Journal of Human Genetics articles (98–111) | Aho–Corasick algorithm embedded in Doc2HPO with manual review removing negated and duplicated terms |
Each dataset comes from different literature sources, except the third set, which comes directly from the Children’s Hospital of Philadelphia. Some HPO term sets have been curated by the Aho–Corasick algorithm embedded in Doc2HPO and others were manually curated by expert physicians.
An effective way to understand how well a phenotype-based gene prioritization tool performs is to have experts curate HPO terms and phenotype information for single-gene diseases. These experts also know the causal genes for these diseases, thus aiding in assessing whether a tool is able to properly rank the causal gene highly.
Each patient case in the benchmark dataset has only a single causal gene that is known beforehand by the physicians who curated the patient data. These data were used to create the benchmark test between Phen2Gene and the original version of Phenolyzer (Figure 3).
Figure 3.

Accuracy test for Phen2Gene and the original version of Phenolyzer, AMELIE 2 and GADO. The accuracy of the tool is determined by the proportion of patient cases where the causal gene was successfully identified in the top 10, 50, 100 and 250 genes for the respective tool. (A) Set 1 of patient cases for TAF1 syndrome as described in Table 1. (B) Set 2 of patient cases from Columbia University as described in Table 1. (C) Set 3 of patient cases from the Division of Genomic Diagnostics at the Children’s Hospital of Philadelphia as described in Table 1. (D) Set 4 of patient cases from 61 Cold Spring Harbor Molecular Case Studies articles and patient cases from 13 American Journal of Human Genetics articles as described in Table 1.
We attempted to use Phevor, OVA and VarElect for a more direct comparison but they did not have APIs, and running each patient case one by one on their websites is a very unrealistic use case for hundreds or thousands of patients. Running multiple CLI instances in parallel on a cluster or the cloud is ubiquitously faster than using a website, and in general a keyboard is faster than using a mouse when done optimally (112–114). Luckily, we were able to run GADO and AMELIE 2 via their web server APIs and obtain speed and accuracy results.
However, AMELIE 2’s web server only accepts a maximum of a 1000-gene input list for gene prioritization. Hence, we randomly created 10 gene sets, each of which consists of the causal gene and 999 random genes. In the accuracy evaluation, we first filtered the outputs from Phen2Gene, original Phenolyzer and GADO down to the 1000 genes in those 10 gene sets, and then took the median rankings of the causal genes from the four tools.
The performance of the four tools varies from set to set. Overall, Phen2Gene is more accurate than Phenolyzer. There is a trade-off in accuracy from set to set between AMELIE 2 and Phen2Gene, and Phen2Gene is more accurate than GADO. The test for accuracy constitutes each tool’s ability to rank the known causal gene in the top 10, 50, 100 and 250 genes, respectively, for each patient case, for each benchmark set. Phen2Gene represents a step forward in accuracy compared to Phenolyzer and GADO, and future improvements to Phenolyzer will improve the H2GKB and thus the performance of Phen2Gene even further.
We performed two-tailed sign tests for each tool compared with Phen2Gene for each set for the performance comparisons in Figure 3. Using a P-value cutoff of 0.5, the difference in performance between Phen2Gene and Phenolyzer (ver. 0.2.0) was statistically significant for all sets; however, for AMELIE 2 and GADO, it was only significantly better for Set 4 (AJHG + CSH) (Supplementary Table S4).
Since Phen2Gene leverages the precomputed H2GKB, the speedup in using Phen2Gene over Phenolyzer is substantial (Table 2). Of course, if a H2GKB were computed with the original Phenolyzer, the speedup would be the same. The Phen2Gene API is the fastest on average as the scripts are saved in the cache—which explains the speedup for the GADO API over its CLI as well. AMELIE 2’s API is extremely slow on average compared to the other tools and would be pretty impractical to scale up to thousands of patients at a time. The speed with which Phen2Gene can both access and rank gene information from the H2GKB compared to competitive tools speaks to its scalability in future large-scale phenotype analysis studies.
Table 2.
Speed benchmark test for Phen2Gene and Phenolyzer
| Tool | Phen2Gene (API) | Phen2Gene (CLI) | Phenolyzer (ver. 0.2.0, CLI) | AMELIE 2 (API) | GADO (API) | GADO (CLI) |
|---|---|---|---|---|---|---|
| Median time (s) | 0.94 | 0.96 | 504.54 | 519.97 | 1.52 | 5.89 |
| Minimum time (s) | 0.17 | 0.51 | 187.97 | 198.35 | 0.74 | 3.43 |
| Maximum time (s) | 2.96 | 1.92 | 1021.54 | 852.70 | 4.15 | 10.3 |
These represent the average, minimum and maximum run-times of these tools in seconds and were taken from all 281 patient case runs.
Interestingly, noise terms do not affect performance significantly. Some phenotypic descriptions may have a few noisy terms in the four benchmark datasets due to different curation criteria, methods, styles or background experience. To test how noise affects Phen2Gene performances, we performed two noise tests by adding a single HPO term to all benchmark cases. In the first test, ‘Seizure’ (HP:0001250) was the noise term added, and it has a low skewness value (1.97) in the H2GKB. In the second test, ‘Macrodontia’ (HP:0001572) was the noise term added, possessing a high skewness value (33.56) in the H2GKB. Based on our results, it appears that one noisy HPO term, no matter how specific and skewed, will not adversely affect Phen2Gene’s performance (Supplementary Figure S6).
General use case: narrowing down candidate genes for undiagnosed diseases
To demonstrate the real-world usage of phenotype-driven gene prioritization in clinical diagnostic settings, we performed a retrospective analysis on a previously published case (Figure 4). We were previously presented with a proband possessing a suspected Mendelian disease, and we performed whole exome sequencing on the proband and the parents. In a previous study, we identified a de novo, single-nucleotide insertion in ankyrin repeat domain 11 (ANKRD11) as the disease causal variant, and reached a genetic diagnosis of KBG syndrome, an extremely rare disease. In the current study, we evaluated whether Phen2Gene and Phenolyzer can facilitate automated gene finding from the exome data, by analyzing the proband only (i.e. without parental information).
Figure 4.

General use case. Proband has a condition with unknown genetic cause but several candidate variants annotated and filtered using ANNOVAR (115). Clinical notes on the proband’s condition are used by Doc2HPO to generate a list of HPO terms, which act as input for Phen2Gene or Phenolyzer. These tools rank several thousand genes, and by intersecting them with the candidate list of genes overlapping the variants, we obtain a list of likely candidate genes for KGB syndrome, which is known to be caused by variants in ANKRD11, shown here.
We used the proband’s HPO terms as input for Phen2Gene and the proband’s disease and symptom terms as input for Phenolyzer. The causal gene, ANKRD11, was initially ranked second and fifth by Phen2Gene and Phenolyzer, respectively, among all the genes in the genome. We intersected these gene lists with the list of candidate genes derived from genes that harbor at least one rare, protein-altering variant in the patient. ANKRD11 was ranked first by both Phen2Gene and Phenolyzer. This example shows how Phen2Gene, Phenolyzer, Exomiser and DeepPVP can be used in practice to rank a causal gene in the top 10 genes based on disease and symptom information and the list of candidate genes extracted from exome sequencing. It is crucial to note this is but one example and both speed and performance will vary greatly from case to case.
However, one item worthy of note is that the speed of Phen2Gene is much greater than DeepPVP. It took over a day to download the database necessary to run DeepPVP and unzip the files necessary to run it. It seems pretty impractical to deploy such a software on the cloud as it would require a large amount of space to deploy (1 TB+) and a large amount of memory to run (60 GB+), which would be an expensive computation across multiple machines.
We were unable to find other freely accessible data containing both HPO terms/patient notes and corresponding VCF files. Nonetheless, the expectation is that human reviewers, such as clinical geneticists or genetic counselors, can review the top 10 or 50 genes and reach a genetic diagnosis with great expedition, perform targeted sequencing on top candidate genes or combine the top 1000 genes with variant information to shorten their lists of candidate genes.
DISCUSSION
Phen2Gene represents a look at the cloud-based future of phenotype-to-gene software. Currently, to the best of our knowledge, very few tools allow for scalability to thousands of patients. Tools that have APIs are quite slow as we have shown, and the website-based tools require manual copy-and-paste input, for one patient at a time. In addition, while some tools like DeepPVP and Exomiser have open source licenses, many tools that rank genes based on HPO terms have no open source code available—which means their work cannot be easily checked or improved upon by the community. Some tools have license restrictions, do not work as advertised or scaling to thousands of patients is not realistic. In comparison, Phen2Gene is extremely fast and open source, does not require prior gene or variant knowledge and does not need to be run on a web server, though we do provide a server for those with less computational backgrounds. We further provide the H2GKB and the benchmark data as freely downloadable files. Compared to the annotation file that documents ∼20 binary relationships between HPO terms and genes on average from Jackson Laboratory’s HPO website, the H2GKB we provide here contains weighted relationships between each HPO term and hundreds or even thousands of genes. Phen2Gene shows marked improvement over the original version of Phenolyzer, and in our future work we plan to greatly improve Phenolyzer and expand upon the H2GKB, increasing performance.
Another benefit of Phen2Gene is that it is variant agnostic. Structural variants (SVs) and repeat expansions in intronic regions are known to cause disease (116,117), and on average, there are >20 000 SVs in the human genome (118). Based on our calculation using the gold standard SV call set from HG002 (119) and the gene annotation file from GENCODE (v25), more than half of the SVs overlap with genes and most overlap intronic regions. The list of tools that can score SVs (120) or repeat expansions (121) is extremely small, but Phen2Gene and tools like it (AMELIE, GADO, etc.) could be used to narrow down a candidate variant list containing repeat expansions or SVs.
In the future, there are several concepts that we hope to address, not the least of which is a double-counting bias ubiquitous to all such HPO-to-gene tools. Some doctors’ notes may contain terms like myoclonic seizures, epilepsy and absence seizures, all of which represent three different HPO terms (HP:0002123, HP:0001250 and HP:0002121, respectively) for what is essentially the same combined condition. As a result, it may be biased toward terms mentioned more often in doctors’ notes. This redundancy can be eliminated through manual HPO term input by human experts, but is still a common issue that needs to be addressed, perhaps by downweighting similar HPO terms.
Another issue we need to handle is the issue of negated terms such as ‘no seizures’. Obviously, if experts input HPO terms manually, this is not a difficult issue to address, but for NLP algorithms that extract terms from doctors’ notes, we could be adding false-positive HPO terms if negation is not properly detected. Conversely, using negated HPO terms to lower the ranking of negated-term-associated genes is another useful incorporation of negated term data. Integrating algorithms like DEEPEN (122) and NegEx (123) into tools such as Doc2HPO may help us solve this problem.
We could improve the granularity of the scoring algorithm by incorporating corpus-based information content. Phen2Gene could still be improved further, and one method for more properly assessing information content is to use HPO terms in tandem with a large body of clinical literature. This could enable us to give the proper weight to HPO terms or perhaps incorporate other terminology not covered by HPO, like UMLS, or NLP-derived classifications or clusters. There is a need for a more widely applicable terminology in the medical field, especially for diseases requiring deeper phenotyping, and this would become a useful resource for researchers doing similar work.
Finally, we can combine Phen2Gene with variant prioritization software or disease gene discovery tools such as CADD (124), REVEL (125) or CCR (126), to further narrow down potential disease gene candidates. If a diagnostician has a list of genetic variants, they are more likely to use one of these tools first. Some tools like VarSight or Exomiser already combine some of these elements of variant prioritization, but we would like to incorporate them into our tool, while maintaining its current fast speed. In the future, we hope to create a hybrid score that combines computationally derived variant scores with phenotype-derived gene prioritization, much like DeepPVP or AMELIE 2, but faster.
In summary, the H2GKB provides a better alternative for linking standardized phenotype terms to genes with weighted scores with expeditiousness, and our hope is that it may facilitate or inspire the development of more accurate, faster, novel computational tools that link HPO terms to genetic information, especially where whole exome/genome sequencing data are available. The Phen2Gene tool provided in this paper can rapidly access and rank this information. It has been implemented in Dx29 (www.dx29.ai) and Doc2HPO’s pipeline so far, and we hope to deploy it in other similar web services. Through command line tools, web servers and RESTful API web services, we believe that Phen2Gene will facilitate and expedite phenotype-driven gene prioritization for rare diseases.
DATA AVAILABILITY
The current version of Phen2Gene is 1.1.0. The source code and scripts for figures are available at https://github.com/WGLab/Phen2Gene. Additionally, we built a Phen2Gene web server available at https://phen2gene.wglab.org, to facilitate users who prefer to use web interface for gene prioritization. The current version of H2GKB is also downloadable at https://github.com/WGLab/Phen2Gene/releases/download/1.1.0/H2GKBs.zip. All the benchmark datasets and code to run the other tools are available in the Supplementary Data.
WEB RESOURCES
HPO website: https://hpo.jax.org/app/; JAX annotations: https://hpo.jax.org/app/download/annotation; Phenolyzer: https://phenolyzer.wglab.org/; Doc2HPO: https://impact2.dbmi.columbia.edu/doc2hpo/; Dx29: https://www.dx29.ai/
Supplementary Material
ACKNOWLEDGEMENTS
We thank the affected individuals and their family members who participated in published genetic studies to improve the diagnostic rates in clinical exome testing. We thank the original authors of the studies to provide detailed phenotype information in published manuscripts, to help us benchmark performance of different software tools. We thank the developers of the HPO for continuous development of this ontology over the past few years, which greatly facilitated and standardized clinical diagnosis of affected individuals with suspected genetic disorders.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Institutes of Health [LM012895 to C.W. and K.W.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z. et al.. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013; 369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eldomery M.K., Coban-Akdemir Z., Harel T., Rosenfeld J.A., Gambin T., Stray-Pedersen A., Kury S., Mercier S., Lessel D., Denecke J. et al.. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017; 9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trujillano D., Bertoli-Avella A.M., Kumar Kandaswamy K., Weiss M.E., Koster J., Marais A., Paknia O., Schroder R., Garcia-Aznar J.M., Werber M. et al.. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 2017; 25:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Retterer K., Juusola J., Cho M.T., Vitazka P., Millan F., Gibellini F., Vertino-Bell A., Smaoui N., Neidich J., Monaghan K.G. et al.. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016; 18:696–704. [DOI] [PubMed] [Google Scholar]
- 5. Sawyer S.L., Hartley T., Dyment D.A., Beaulieu C.L., Schwartzentruber J., Smith A., Bedford H.M., Bernard G., Bernier F.P., Brais B. et al.. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 2016; 89:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kohler S., Carmody L., Vasilevsky N., Jacobsen J.O.B., Danis D., Gourdine J.P., Gargano M., Harris N.L., Matentzoglu N., McMurry J.A. et al.. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019; 47:D1018–D1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al.. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bello S.M., Shimoyama M., Mitraka E., Laulederkind S.J.F., Smith C.L., Eppig J.T., Schriml L.M.. Disease Ontology: improving and unifying disease annotations across species. Dis. Model. Mech. 2018; 11:dmm032839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haendel M.A., Balhoff J.P., Bastian F.B., Blackburn D.C., Blake J.A., Bradford Y., Comte A., Dahdul W.M., Dececchi T.A., Druzinsky R.E. et al.. Unification of multi-species vertebrate anatomy ontologies for comparative biology in Uberon. J. Biomed. Semant. 2014; 5:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bard J., Rhee S.Y., Ashburner M.. An ontology for cell types. Genome Biol. 2005; 6:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith C.L., Goldsmith C.-A.W., Eppig J.T.. The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005; 6:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang H., Robinson P.N., Wang K.. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat. Methods. 2015; 12:841–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Son J.H., Xie G., Yuan C., Ena L., Li Z., Goldstein A., Huang L., Wang L., Shen F., Liu H. et al.. Deep phenotyping on electronic health records facilitates genetic diagnosis by clinical exomes. Am. J. Hum. Genet. 2018; 103:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu C., Peres Kury F.S., Li Z., Ta C., Wang K., Weng C.. Doc2Hpo: a web application for efficient and accurate HPO concept curation. Nucleic Acids Res. 2019; 47:W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aerts S., Lambrechts D., Maity S., Van Loo P., Coessens B., De Smet F., Tranchevent L.C., De Moor B., Marynen P., Hassan B. et al.. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006; 24:537–544. [DOI] [PubMed] [Google Scholar]
- 16. Singleton M.V., Guthery S.L., Voelkerding K.V., Chen K., Kennedy B., Margraf R.L., Durtschi J., Eilbeck K., Reese M.G., Jorde L.B. et al.. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am. J. Hum. Genet. 2014; 94:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stelzer G., Plaschkes I., Oz-Levi D., Alkelai A., Olender T., Zimmerman S., Twik M., Belinky F., Fishilevich S., Nudel R. et al.. VarElect: the phenotype-based variation prioritizer of the GeneCards suite. BMC Genomics. 2016; 17:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Antanaviciute A., Watson C.M., Harrison S.M., Lascelles C., Crinnion L., Markham A.F., Bonthron D.T., Carr I.M.. OVA: integrating molecular and physical phenotype data from multiple biomedical domain ontologies with variant filtering for enhanced variant prioritization. Bioinformatics. 2015; 31:3822–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Javed A., Agrawal S., Ng P.C.. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat. Methods. 2014; 11:935–937. [DOI] [PubMed] [Google Scholar]
- 20. Smedley D., Jacobsen J.O.B., Jäger M., Köhler S., Holtgrewe M., Schubach M., Siragusa E., Zemojtel T., Buske O.J., Washington N.L. et al.. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015; 10:2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birgmeier J., Haeussler M., Deisseroth C.A., Steinberg E.H., Jagadeesh K.A., Ratner A.J., Guturu H., Wenger A.M., Diekhans M.E., Stenson P.D. et al.. AMELIE 2 speeds up Mendelian diagnosis by matching patient phenotype & genotype to primary literature. 2019; 14 November 2019, preprint: not peer reviewed 10.1101/839878. [DOI] [PMC free article] [PubMed]
- 22. Boudellioua I., Kulmanov M., Schofield P.N., Gkoutos G.V., Hoehndorf R.. DeepPVP: phenotype-based prioritization of causative variants using deep learning. BMC Bioinformatics. 2019; 20:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deelen P., van Dam S., Herkert J.C., Karjalainen J.M., Brugge H., Abbott K.M., van Diemen C.C., van der Zwaag P.A., Gerkes E.H., Zonneveld-Huijssoon E. et al.. Improving the diagnostic yield of exome-sequencing by predicting gene–phenotype associations using large-scale gene expression analysis. Nat. Commun. 2019; 10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holt J.M., Wilk B., Birch C.L., Brown D.M., Gajapathy M., Moss A.C., Sosonkina N., Wilk M.A., Anderson J.A., Harris J.M. et al.. VarSight: prioritizing clinically reported variants with binary classification algorithms. BMC Bioinformatics. 2019; 20:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Q., Zhao K., Bustamante C.D., Ma X., Wong W.H.. Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet. Med. 2019; 21:2126–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sánchez D., Batet M., Isern D.. Ontology-based information content computation. Knowl. Based Syst. 2011; 24:297–303. [Google Scholar]
- 27. McKusick V.A. Mendelian inheritance in man and its online version, OMIM. Am. J. Hum. Genet. 2007; 80:588–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rath A., Olry A., Dhombres F., Brandt M.M., Urbero B., Ayme S.. Representation of rare diseases in health information systems: the Orphanet approach to serve a wide range of end users. Hum. Mutat. 2012; 33:803–808. [DOI] [PubMed] [Google Scholar]
- 30. Adam M.P. GeneReviews. 1993; Seattle: University of Washington. [Google Scholar]
- 31. Peri S., Navarro J.D., Kristiansen T.Z., Amanchy R., Surendranath V., Muthusamy B., Gandhi T.K., Chandrika K.N., Deshpande N., Suresh S. et al.. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 2004; 32:D497–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geer L.Y., Marchler-Bauer A., Geer R.C., Han L., He J., He S., Liu C., Shi W., Bryant S.H.. The NCBI BioSystems database. Nucleic Acids Res. 2010; 38:D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seal R.L., Gordon S.M., Lush M.J., Wright M.W., Bruford E.A.. genenames.org: the HGNC resources in 2011. Nucleic Acids Res. 2011; 39:D514–D519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bovolenta L.A., Acencio M.L., Lemke N.. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics. 2012; 13:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Rawe J.A., Wu Y., Dorfel M.J., Rope A.F., Au P.Y., Parboosingh J.S., Moon S., Kousi M., Kosma K., Smith C.S. et al.. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am. J. Hum. Genet. 2015; 97:922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu C., Devkota B., Evans P., Zhao X., Baker S.W., Niazi R., Cao K., Gonzalez M.A., Jayaraman P., Conlin L.K. et al.. Rapid and accurate interpretation of clinical exomes using Phenoxome: a computational phenotype-driven approach. Eur. J. Hum. Genet. 2019; 27:612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Swaminathan M., Bannon S.A., Routbort M., Naqvi K., Kadia T.M., Takahashi K., Alvarado Y., Ravandi-Kashani F., Patel K.P., Champlin R. et al.. Hematologic malignancies and Li–Fraumeni syndrome. Cold Spring Harb. Mol. Case Stud. 2019; 5:a003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanaka A.J., Bai R., Cho M.T., Anyane-Yeboa K., Ahimaz P., Wilson A.L., Kendall F., Hay B., Moss T., Nardini M. et al.. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb. Mol. Case Stud. 2015; 1:a000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang H., Douglas G., Monaghan K.G., Retterer K., Cho M.T., Escobar L.F., Tucker M.E., Stoler J., Rodan L.H., Stein D. et al.. De novo truncating variants in the AHDC1 gene encoding the AT-hook DNA-binding motif-containing protein 1 are associated with intellectual disability and developmental delay. Cold Spring Harb. Mol. Case Stud. 2015; 1:a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zimmerman E., Maron J.L.. FOXP2 gene deletion and infant feeding difficulties: a case report. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanaka A.J., Cho M.T., Retterer K., Jones J.R., Nowak C., Douglas J., Jiang Y.H., McConkie-Rosell A., Schaefer G.B., Kaylor J. et al.. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joshi M., Anselm I., Shi J., Bale T.A., Towne M., Schmitz-Abe K., Crowley L., Giani F.C., Kazerounian S., Markianos K. et al.. Mutations in the substrate binding glycine-rich loop of the mitochondrial processing peptidase-alpha protein (PMPCA) cause a severe mitochondrial disease. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu H.C., Coughlin C.R., Geiger E.A., Salvador B.J., Elias E.R., Cavanaugh J.L., Chatfield K.C., Miyamoto S.D., Shaikh T.H.. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leinoe E., Nielsen O.J., Jonson L., Rossing M.. Whole-exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Griffin L.B., Farley F.A., Antonellis A., Keegan C.E.. A novel FGD1 mutation in a family with Aarskog–Scott syndrome and predominant features of congenital joint contractures. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pierce S.B., Gulsuner S., Stapleton G.A., Walsh T., Lee M.K., Mandell J.B., Morales A., Klevit R.E., King M.C., Rogers R.C.. Infantile onset spinocerebellar ataxia caused by compound heterozygosity for Twinkle mutations and modeling of Twinkle mutations causing recessive disease. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moskowitz A.M., Belnap N., Siniard A.L., Szelinger S., Claasen A.M., Richholt R.F., De Both M., Corneveaux J.J., Balak C., Piras I.S. et al.. A de novo missense mutation in ZMYND11 is associated with global developmental delay, seizures, and hypotonia. Cold Spring Harb. Mol. Case Stud. 2016; 2:a000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smedemark-Margulies N., Brownstein C.A., Vargas S., Tembulkar S.K., Towne M.C., Shi J., Gonzalez-Cuevas E., Liu K.X., Bilguvar K., Kleiman R.J. et al.. A novel de novo mutation in ATP1A3 and childhood-onset schizophrenia. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Malcolmson J., Kleyner R., Tegay D., Adams W., Ward K., Coppinger J., Nelson L., Meisler M.H., Wang K., Robison R. et al.. SCN8A mutation in a child presenting with seizures and developmental delays. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kleyner R., Malcolmson J., Tegay D., Ward K., Maughan A., Maughan G., Nelson L., Wang K., Robison R., Lyon G.J.. KBG syndrome involving a single-nucleotide duplication in ANKRD11. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Webster E., Cho M.T., Alexander N., Desai S., Naidu S., Bekheirnia M.R., Lewis A., Retterer K., Juusola J., Chung W.K.. De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Colby S., Yehia L., Niazi F., Chen J., Ni Y., Mester J.L., Eng C.. Exome sequencing reveals germline gain-of-function EGFR mutation in an adult with Lhermitte–Duclos disease. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu A.C., Chan A.Y., Au W.C., Shen Y., Chan T.F., Chan H.E.. Whole-genome sequencing of two probands with hereditary spastic paraplegia reveals novel splice-donor region variant and known pathogenic variant in SPG11. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Polfus L.M., Boerwinkle E., Gibbs R.A., Metcalf G., Muzny D., Veeraraghavan N., Grove M., Shete S., Wallace S., Milewicz D. et al.. Whole-exome sequencing reveals an inherited R566X mutation of the epithelial sodium channel beta-subunit in a case of early-onset phenotype of Liddle syndrome. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Delpire E., Wolfe L., Flores B., Koumangoye R., Schornak C.C., Omer S., Pusey B., Lau C., Markello T., Adams D.R.. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb. Mol. Case Stud. 2016; 2:a001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bourne S.C., Townsend K.N., Shyr C., Matthews A., Lear S.A., Attariwala R., Lehman A., Wasserman W.W., van Karnebeek C., Sinclair G. et al.. Optic atrophy, cataracts, lipodystrophy/lipoatrophy, and peripheral neuropathy caused by a de novo OPA3 mutation. Cold Spring Harb. Mol. Case Stud. 2017; 3:a001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Patel R.M., Liu D., Gonzaga-Jauregui C., Jhangiani S., Lu J.T., Sutton V.R., Fernbach S.D., Azamian M., White L., Edmond J.C. et al.. An exome sequencing study of Moebius syndrome including atypical cases reveals an individual with CFEOM3A and a TUBB3 mutation. Cold Spring Harb. Mol. Case Stud. 2017; 3:a000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morton S.U., Prabhu S.P., Lidov H.G.W., Shi J., Anselm I., Brownstein C.A., Bainbridge M.N., Beggs A.H., Vargas S.O., Agrawal P.B.. AIFM1 mutation presenting with fatal encephalomyopathy and mitochondrial disease in an infant. Cold Spring Harb. Mol. Case Stud. 2017; 3:a001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caglayan A.O., Sezer R.G., Kaymakcalan H., Ulgen E., Yavuz T., Baranoski J.F., Bozaykut A., Harmanci A.S., Yalcin Y., Youngblood M.W. et al.. ALPK3 gene mutation in a patient with congenital cardiomyopathy and dysmorphic features. Cold Spring Harb. Mol. Case Stud. 2017; 3:a001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Inlora J., Sailani M.R., Khodadadi H., Teymurinezhad A., Takahashi S., Bernstein J.A., Garshasbi M., Snyder M.P.. Identification of a novel mutation in the APTX gene associated with ataxia-oculomotor apraxia. Cold Spring Harb. Mol. Case Stud. 2017; 3:a002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnston J.J., Lee C., Wentzensen I.M., Parisi M.A., Crenshaw M.M., Sapp J.C., Gross J.M., Wallingford J.B., Biesecker L.G.. Compound heterozygous alterations in intraflagellar transport protein CLUAP1 in a child with a novel Joubert and oral-facial-digital overlap syndrome. Cold Spring Harb. Mol. Case Stud. 2017; 3:a001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dardour L., Roelens F., Race V., Souche E., Holvoet M., Devriendt K.. SPG20 mutation in three siblings with familial hereditary spastic paraplegia. Cold Spring Harb. Mol. Case Stud. 2017; 3:a001537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Whitford W., Hawkins I., Glamuzina E., Wilson F., Marshall A., Ashton F., Love D.R., Taylor J., Hill R., Lehnert K. et al.. Compound heterozygous SLC19A3 mutations further refine the critical promoter region for biotin-thiamine-responsive basal ganglia disease. Cold Spring Harb. Mol. Case Stud. 2017; 3:a001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rohanizadegan M., Abdo S.M., O’Donnell-Luria A., Mihalek I., Chen P., Sanders M., Leeman K., Cho M., Hung C., Bodamer O.. Utility of rapid whole-exome sequencing in the diagnosis of Niemann–Pick disease type C presenting with fetal hydrops and acute liver failure. Cold Spring Harb. Mol. Case Stud. 2017; 3:a002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaiwar C., Zimmermann M.T., Ferber M.J., Niu Z., Urrutia R.A., Klee E.W., Babovic-Vuksanovic D.. Novel NR2F1 variants likely disrupt DNA binding: molecular modeling in two cases, review of published cases, genotype–phenotype correlation, and phenotypic expansion of the Bosch–Boonstra–Schaaf optic atrophy syndrome. Cold Spring Harb. Mol. Case Stud. 2017; 3:a002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sailani M.R., Chappell J., Jingga I., Narasimha A., Zia A., Lynch J.L., Mazrouei S., Bernstein J.A., Aryani O., Snyder M.P.. WISP3 mutation associated with pseudorheumatoid dysplasia. Cold Spring Harb. Mol. Case Stud. 2018; 4:a001990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tanaka A.J., Cho M.T., Willaert R., Retterer K., Zarate Y.A., Bosanko K., Stefans V., Oishi K., Williamson A., Wilson G.N. et al.. De novo variants in EBF3 are associated with hypotonia, developmental delay, intellectual disability, and autism. Cold Spring Harb. Mol. Case Stud. 2017; 3:a002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lu J.G., Bishop J., Cheyette S., Zhulin I.B., Guo S., Sobreira N., Brenner S.E.. A novel PRRT2 pathogenic variant in a family with paroxysmal kinesigenic dyskinesia and benign familial infantile seizures. Cold Spring Harb. Mol. Case Stud. 2018; 4:1621–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koboldt D.C., Mihalic Mosher T., Kelly B.J., Sites E., Bartholomew D., Hickey S.E., McBride K., Wilson R.K., White P.. A de novo nonsense mutation in ASXL3 shared by siblings with Bainbridge–Ropers syndrome. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miller K.E., Kelly B., Fitch J., Ross N., Avenarius M.R., Varga E., Koboldt D.C., Boue D.R., Magrini V., Coven S.L. et al.. Genome sequencing identifies somatic BRAF duplication c.1794_1796dupTAC;p.Thr599dup in pediatric patient with low-grade ganglioglioma. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sanford E., Watkins K., Nahas S., Gottschalk M., Coufal N.G., Farnaes L., Dimmock D., Kingsmore S.F. RCIGM Investigators . Rapid whole-genome sequencing identifies a novel AIRE variant associated with autoimmune polyendocrine syndrome type 1. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berland S., Toft-Bertelsen T.L., Aukrust I., Byska J., Vaudel M., Bindoff L.A., MacAulay N., Houge G.. A de novo Ser111Thr variant in aquaporin-4 in a patient with intellectual disability, transient signs of brain ischemia, transient cardiac hypertrophy, and progressive gait disturbance. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Miller C.A., Dahiya S., Li T., Fulton R.S., Smyth M.D., Dunn G.P., Rubin J.B., Mardis E.R.. Resistance-promoting effects of ependymoma treatment revealed through genomic analysis of multiple recurrences in a single patient. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bodian D.L., Schreiber J.M., Vilboux T., Khromykh A., Hauser N.S.. Mutation in an alternative transcript of CDKL5 in a boy with early-onset seizures. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Velez G., Bassuk A.G., Schaefer K.A., Brooks B., Gakhar L., Mahajan M., Kahn P., Tsang S.H., Ferguson P.J., Mahajan V.B.. A novel de novo CAPN5 mutation in a patient with inflammatory vitreoretinopathy, hearing loss, and developmental delay. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sweeney N.M., Nahas S.A., Chowdhury S., Campo M.D., Jones M.C., Dimmock D.P., Kingsmore S.F. RCIGM Investigators . The case for early use of rapid whole-genome sequencing in management of critically ill infants: late diagnosis of Coffin–Siris syndrome in an infant with left congenital diaphragmatic hernia, congenital heart disease, and recurrent infections. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cotter J.A., Szymanski L., Karimov C., Boghossian L., Margol A., Dhall G., Tamrazi B., Varaprasathan G.I., Parham D.M., Judkins A.R. et al.. Transmission of a TP53 germline mutation from unaffected male carrier associated with pediatric glioblastoma in his child and gestational choriocarcinoma in his female partner. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Antwi P., Hong C.S., Duran D., Jin S.C., Dong W., DiLuna M., Kahle K.T.. A novel association of campomelic dysplasia and hydrocephalus with an unbalanced chromosomal translocation upstream of SOX9. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Murry J.B., Machini K., Ceyhan-Birsoy O., Kritzer A., Krier J.B., Lebo M.S., Fayer S., Genetti C.A., VanNoy G.E., Yu T.W. et al.. Reconciling newborn screening and a novel splice variant in BTD associated with partial biotinidase deficiency: a BabySeq Project case report. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schwartz J.R., Walsh M.P., Ma J., Lamprecht T., Wang S., Wu G., Raimondi S., Triplett B.M., Klco J.M.. Clonal dynamics of donor-derived myelodysplastic syndrome after unrelated hematopoietic cell transplantation for high-risk pediatric B-lymphoblastic leukemia. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fomchenko E.I., Duran D., Jin S.C., Dong W., Erson-Omay E.Z., Antwi P., Allocco A., Gaillard J.R., Huttner A., Gunel M. et al.. De novo MYH9 mutation in congenital scalp hemangioma. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grant A.R., Hemphill S.E., Vincent L.M., Rehm H.L.. Reclassification of the BRAF p.Ile208Val variant by case-level data sharing. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tan Q.K., Cope H., Spillmann R.C., Stong N., Jiang Y.H., McDonald M.T., Rothman J.A., Butler M.W., Frush D.P., Lachman R.S. et al.. Further evidence for the involvement of EFL1 in a Shwachman–Diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koboldt D.C., Kastury R.D., Waldrop M.A., Kelly B.J., Mosher T.M., McLaughlin H., Corsmeier D., Slaughter J.L., Flanigan K.M., McBride K.L. et al.. In-frame de novo mutation in BICD2 in two patients with muscular atrophy and arthrogryposis. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dubard Gault M., Mandelker D., DeLair D., Stewart C.R., Kemel Y., Sheehan M.R., Siegel B., Kennedy J., Marcell V., Arnold A. et al.. Germline SDHA mutations in children and adults with cancer. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Erdrich J., Schaberg K.B., Khodadoust M.S., Zhou L., Shelton A.A., Visser B.C., Ford J.M., Alizadeh A.A., Quake S.R., Kunz P.L. et al.. Surgical and molecular characterization of primary and metastatic disease in a neuroendocrine tumor arising in a tailgut cyst. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zech M., Lam D.D., Weber S., Berutti R., Polakova K., Havrankova P., Fecikova A., Strom T.M., Ruzicka E., Jech R. et al.. A unique de novo gain-of-function variant in CAMK4 associated with intellectual disability and hyperkinetic movement disorder. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Haskell G.T., Mori M., Powell C., Amrhein T.J., Rice G.I., Bailey L., Strande N., Weck K.E., Evans J.P., Berg J.S. et al.. Combination of exome sequencing and immune testing confirms Aicardi–Goutieres syndrome type 5 in a challenging pediatric neurology case. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. David M.P., Venkatramani R., Lopez-Terrada D.H., Roy A., Patil N., Fisher K.E.. Multimodal molecular analysis of an atypical small cell carcinoma of the ovary, hypercalcemic type. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Khurana M., Edwards D., Rescorla F., Miller C., He Y., Sierra Potchanant E., Nalepa G.. Whole-exome sequencing enables correct diagnosis and surgical management of rare inherited childhood anemia. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Okur V., Ganapathi M., Wilson A., Chung W.K.. Biallelic variants in VARS in a family with two siblings with intellectual disability and microcephaly: case report and review of the literature. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Martignetti J.A., Pandya D., Nagarsheth N., Chen Y., Camacho O., Tomita S., Brodman M., Ascher-Walsh C., Kolev V., Cohen S. et al.. Detection of endometrial precancer by a targeted gynecologic cancer liquid biopsy. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Briggs B., James K.N., Chowdhury S., Thornburg C., Farnaes L., Dimmock D., Kingsmore S.F. RCIGM Investigators . Novel factor XIII variant identified through whole-genome sequencing in a child with intracranial hemorrhage. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tanaka A.J., Okumoto K., Tamura S., Abe Y., Hirsch Y., Deng L., Ekstein J., Chung W.K., Fujiki Y.. A newly identified mutation in the PEX26 gene is associated with a milder form of Zellweger spectrum disorder. Cold Spring Harb. Mol. Case Stud. 2019; 5:a003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Qian Y., Wu B., Lu Y., Dong X., Qin Q., Zhou W., Wang H.. Early-onset infant epileptic encephalopathy associated with a de novo PPP3CA gene mutation. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sanford E., Farnaes L., Batalov S., Bainbridge M., Laubach S., Worthen H.M., Tokita M., Kingsmore S.F., Bradley J.. Concomitant diagnosis of immune deficiency and Pseudomonas sepsis in a 19 month old with ecthyma gangrenosum by host whole-genome sequencing. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Claassen D., Boals M., Bowling K.M., Cooper G.M., Cox J., Hershfield M., Lewis S., Wlodarski M., Weiss M.J., Estepp J.H.. Complexities of genetic diagnosis illustrated by an atypical case of congenital hypoplastic anemia. Cold Spring Harb. Mol. Case Stud. 2018; 4:a003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Windpassinger C., Piard J., Bonnard C., Alfadhel M., Lim S., Bisteau X., Blouin S., Ali N.B., Ng A.Y.J., Lu H. et al.. CDK10 mutations in humans and mice cause severe growth retardation, spine malformations, and developmental delays. Am. J. Hum. Genet. 2017; 101:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lessel D., Schob C., Kury S., Reijnders M.R.F., Harel T., Eldomery M.K., Coban-Akdemir Z., Denecke J., Edvardson S., Colin E. et al.. De novo missense mutations in DHX30 impair global translation and cause a neurodevelopmental disorder. Am. J. Hum. Genet. 2017; 101:716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Paul A., Drecourt A., Petit F., Deguine D.D., Vasnier C., Oufadem M., Masson C., Bonnet C., Masmoudi S., Mosnier I. et al.. FDXR mutations cause sensorial neuropathies and expand the spectrum of mitochondrial Fe–S-synthesis diseases. Am. J. Hum. Genet. 2017; 101:630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Watson L.M., Bamber E., Schnekenberg R.P., Williams J., Bettencourt C., Lickiss J., Jayawant S., Fawcett K., Clokie S., Wallis Y. et al.. Dominant mutations in GRM1 cause spinocerebellar ataxia type 44. Am. J. Hum. Genet. 2017; 101:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Habarou F., Hamel Y., Haack T.B., Feichtinger R.G., Lebigot E., Marquardt I., Busiah K., Laroche C., Madrange M., Grisel C. et al.. Biallelic mutations in LIPT2 cause a mitochondrial lipoylation defect associated with severe neonatal encephalopathy. Am. J. Hum. Genet. 2017; 101:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lake N.J., Webb B.D., Stroud D.A., Richman T.R., Ruzzenente B., Compton A.G., Mountford H.S., Pulman J., Zangarelli C., Rio M. et al.. Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and leigh syndrome. Am. J. Hum. Genet. 2017; 101:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Boudin E., de Jong T.R., Prickett T.C.R., Lapauw B., Toye K., Van Hoof V., Luyckx I., Verstraeten A., Heymans H.S.A., Dulfer E. et al.. Bi-allelic loss-of-function mutations in the NPR-C receptor result in enhanced growth and connective tissue abnormalities. Am. J. Hum. Genet. 2018; 103:288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lamers I.J.C., Reijnders M.R.F., Venselaar H., Kraus A. DDD Study DDD Study Jansen S., de Vries B.B.A., Houge G., Gradek G.A., Seo J. et al.. Recurrent de novo mutations disturbing the GTP/GDP binding pocket of RAB11B cause intellectual disability and a distinctive brain phenotype. Am. J. Hum. Genet. 2017; 101:824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Reijnders M.R.F., Ansor N.M., Kousi M., Yue W.W., Tan P.L., Clarkson K., Clayton-Smith J., Corning K., Jones J.R., Lam W.W.K. et al.. RAC1 missense mutations in developmental disorders with diverse phenotypes. Am. J. Hum. Genet. 2017; 101:466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bayram Y., White J.J., Elcioglu N., Cho M.T., Zadeh N., Gedikbasi A., Palanduz S., Ozturk S., Cefle K., Kasapcopur O. et al.. REST final-exon-truncating mutations cause hereditary gingival fibromatosis. Am. J. Hum. Genet. 2017; 101:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. De Mori R., Romani M., D’Arrigo S., Zaki M.S., Lorefice E., Tardivo S., Biagini T., Stanley V., Musaev D., Fluss J. et al.. Hypomorphic recessive variants in SUFU impair the sonic hedgehog pathway and cause Joubert syndrome with cranio-facial and skeletal defects. Am. J. Hum. Genet. 2017; 101:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ivanova E.L., Mau-Them F.T., Riazuddin S., Kahrizi K., Laugel V., Schaefer E., de Saint Martin A., Runge K., Iqbal Z., Spitz M.A. et al.. Homozygous truncating variants in TBC1D23 cause pontocerebellar hypoplasia and alter cortical development. Am. J. Hum. Genet. 2017; 101:428–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Skraban C.M., Wells C.F., Markose P., Cho M.T., Nesbitt A.I., Au P.Y.B., Begtrup A., Bernat J.A., Bird L.M., Cao K. et al.. WDR26 haploinsufficiency causes a recognizable syndrome of intellectual disability, seizures, abnormal gait, and distinctive facial features. Am. J. Hum. Genet. 2017; 101:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Guella I., McKenzie M.B., Evans D.M., Buerki S.E., Toyota E.B., Van Allen M.I., Epilepsy Genomics S., Suri M., Elmslie F. Deciphering Developmental Disorders Study et al.. De novo mutations in YWHAG cause early-onset epilepsy. Am. J. Hum. Genet. 2017; 101:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Miller C.S., Denkov S., Omanson R.C.. Categorization costs for hierarchical keyboard commands. Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, CHI’11. 2011; Vancouver: Association for Computing Machinery; 2765–2768. [Google Scholar]
- 113. Lane D.M., Napier H.A., Peres S.C., Sandor A.. Hidden costs of graphical user interfaces: failure to make the transition from menus and icon toolbars to keyboard shortcuts. Int. J. Hum. Comput. Interact. 2005; 18:133–144. [Google Scholar]
- 114. Omanson R.C., Miller C.S., Young E., Schwantes D.. Comparison of mouse and keyboard efficiency. Proc. Hum. Factors Ergon. Soc. Annu. Meet. 2010; 6:600–604. [Google Scholar]
- 115. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zeng S., Zhang M., Wang X., Hu Z., Li J., Li N., Wang J., Liang F., Yang Q., Liu Q. et al.. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J. Med. Genet. 2019; 56:265–270. [DOI] [PubMed] [Google Scholar]
- 117. Ishiura H., Doi K., Mitsui J., Yoshimura J., Matsukawa M.K., Fujiyama A., Toyoshima Y., Kakita A., Takahashi H., Suzuki Y. et al.. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 2018; 50:581–590. [DOI] [PubMed] [Google Scholar]
- 118. Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L. et al.. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019; 10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Zook J.M., Hansen N.F., Olson N.D., Chapman L.M., Mullikin J.C., Xiao C., Sherry S., Koren S., Phillippy A.M., Boutros P.C. et al.. A robust benchmark for germline structural variant detection. 2019; 9 June 2019, preprint: not peer reviewed 10.1101/664623. [DOI]
- 120. Ganel L., Abel H.J., Hall I.M.. SVScore: an impact prediction tool for structural variation. Bioinformatics. 2017; 33:1083–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Costa I.P.D., Almeida B.C., Sequeiros J., Amorim A., Martins S.. A pipeline to assess disease-associated haplotypes in repeat expansion disorders: the example of MJD/SCA3 locus. Front. Genet. 2019; 10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mehrabi S., Krishnan A., Sohn S., Roch A.M., Schmidt H., Kesterson J., Beesley C., Dexter P., Schmidt C.M., Liu H. et al.. DEEPEN: a negation detection system for clinical text incorporating dependency relation into NegEx. J. Biomed. Inform. 2015; 54:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chapman W.W., Bridewell W., Hanbury P., Cooper G.F., Buchanan B.G.. A simple algorithm for identifying negated findings and diseases in discharge summaries. J. Biomed. Inform. 2001; 34:301–310. [DOI] [PubMed] [Google Scholar]
- 124. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D. et al.. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016; 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Havrilla J.M., Pedersen B.S., Layer R.M., Quinlan A.R.. A map of constrained coding regions in the human genome. Nat. Genet. 2019; 51:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The current version of Phen2Gene is 1.1.0. The source code and scripts for figures are available at https://github.com/WGLab/Phen2Gene. Additionally, we built a Phen2Gene web server available at https://phen2gene.wglab.org, to facilitate users who prefer to use web interface for gene prioritization. The current version of H2GKB is also downloadable at https://github.com/WGLab/Phen2Gene/releases/download/1.1.0/H2GKBs.zip. All the benchmark datasets and code to run the other tools are available in the Supplementary Data.