

ABSTRACT
In this brief review, the reader will find a timely update regarding some of the most commonly encountered glomerular diseases. The review will include an update on the etiology with a focus on new genetic and molecular discoveries. New classifications will be elucidated, and management will be updated in broad strokes. Illustrative pathology slides will be used as appropriate. It is critical for the reader to realize from the outset that terminology such as focal segmental glomerulosclerosis and crescentic glomerulonephritis represent a pattern of injury rather than a specific disease. Whenever possible, the specific etiology and pathogenesis of a given pattern should be sought. It is also important to know that the same disease or mechanism can cause multiple patterns of injury, whereas the same pattern of injury can be the result of multiple disease or mechanisms.
Key words: C3 nephropathy, glomerular disease, glomerulonephritis
INTRODUCTION
The past few years have witnessed a paradigm change in glomerular diseases. New genetic discoveries have been made, old classifications were abandoned, new terminology has emerged, and novel therapeutic agents have made their way into everyday clinical practice.
This is a timely, yet brief update not only for the practicing nephrologist but also for any practicing clinician and medical student. The review will focus on the major diagnostic entities.
The length of this review does not permit discussion of all primary and secondary glomerulonephritides.
A consensus report by the Mayo Clinic and the Renal Pathology Society in 2016[1] emphasized the importance of including the pattern of injury, the etiology or pathogenesis, and the specific disease entity with the glomerular disease diagnosis. For example, rather than membranous nephropathy (MN) the diagnosis would be anti-M-type phospholipase A2 receptor (PLA2R) MN.
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS
Membranoproliferative glomerulonephritis (MPGN) is arguably the most confusing of all glomerular diseases. It is a pattern of injury rather than a specific disease. This pattern of injury has multiple etiologies. The old classification has been abandoned in favor of a new system that incorporates the pathogenesis and the etiology resulting in MPGN pattern of injury. Light microscopy reveals lobulation, capillary wall thickening, and hypercellularity. Doubling of the glomerular basement membrane (reduplication or splitting) due to immune complex interposition may be also seen.
According to the old classification, there are three types of MPGN depending on the immune deposits seen on the renal biopsy: type I (subendothelial immune complex deposits), type II or dense deposit disease (DDD) (intramembranous immune deposits), and type III (subendothelial with subepithelial or intramembranous deposits).
The new classification divides MPGN based on immunohistochemical studies into two broad categories, see diagram 1:[2]
Diagram 1.
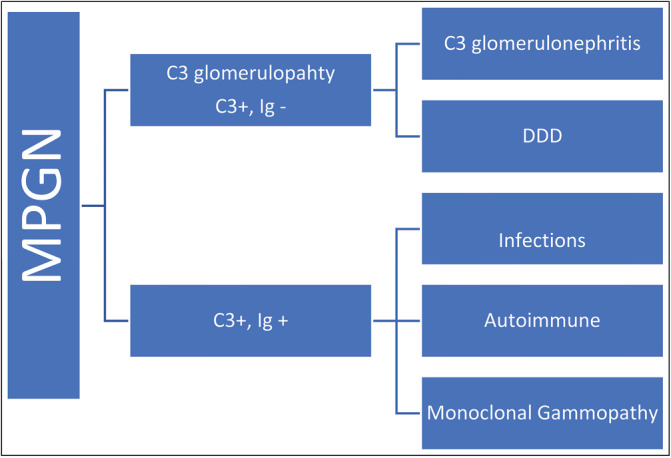
MPGN new classification
1-C3 glomerulopathy: It has C3 only or C3-dominant positive deposits.
This category is complement mediated and is due to a variety of acquired and genetic abnormalities such as mutations in complement regulators: Factor H, Factor I, or antibodies to C3 convertase (C3 nephritic factor).
C3 glomerulopathy is further divided into C3 glomerulonephritis and DDD characterized by intramembranous C3 deposits [Figures 1–3].
Figure 1.
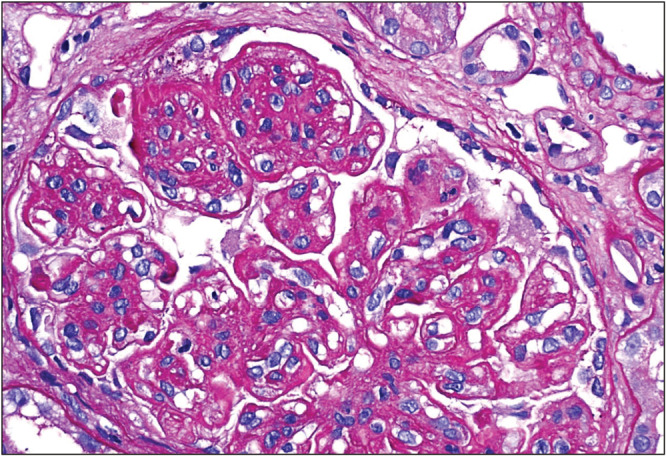
Membranoproliferative glomerulonephritis (MPGN) pattern in C3 glomerulonephritis. Periodic acid–Schiff (PAS) staining. Diagnosis can be ascertained only with immunofluorescence microscopy (IF) (Courtesy of Arkana Laboratories)
Figure 3.
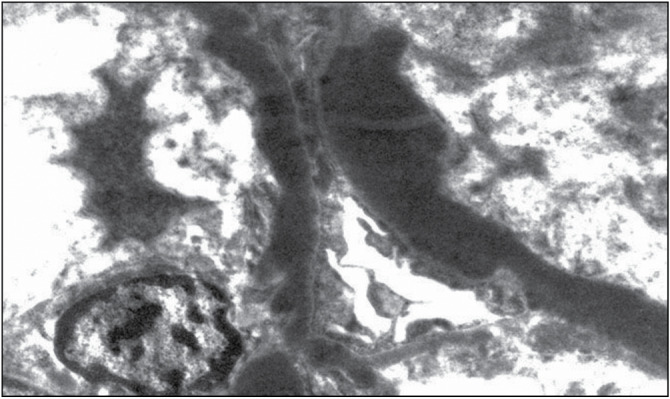
C3 deposit in dense deposit disease. Electron microscopy (Courtesy of Arkana Laboratories)
Figure 2.
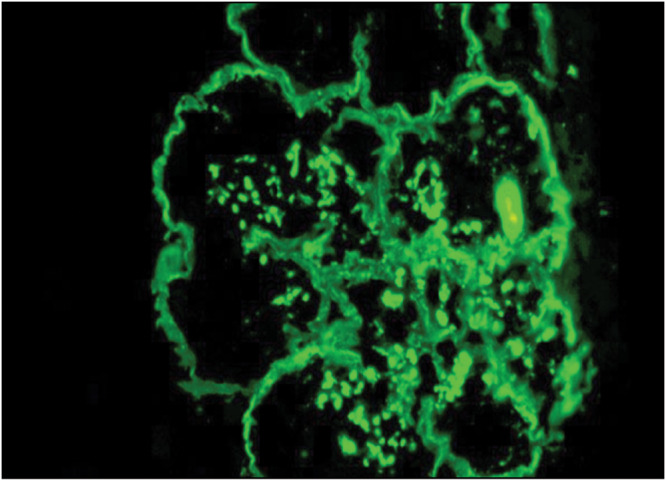
C3 deposits in dense deposit disease. Immunofluorescence microscopy (IF) microscopy (Courtesy of Arkana Laboratories)
C3 glomerulonephritis is a newly described condition that can mimic acute postinfectious glomerulonephritis and presents with hematuria and proteinuria. As it is due to persistent activation of the alternate complement pathway, C3 is low whereas C4 is normal.
Treatment for C3 glomerulopathy is unclear. Possible options include low-dose steroids plus mycophenolate mofetil (MMF) or eculizumab, which is a monoclonal antibody that binds to C5.
Ravulizumab is a humanized monoclonal antibody and a novel C5 inhibitor. It was approved by the Food and Drug Administration (FDA) in December 2018 for the treatment of atypical hemolytic uremic syndrome. The role of complement inhibitors in glomerular diseases will be defined in ongoing clinical trials.[3] No trials have directly compared eculizumab with ravulizumab in glomerular disease to date.
2-Immunoglobulin (Ig) and C3 positive MPGN: This category may be due to a variety of infectious (bacterial endocarditis, hepatitis B or C, and malaria), and autoimmune (lupus nephritis [LN] and rheumatoid arthritis) entities, in addition to monoclonal gammopathy (light-chain deposition and heavy-chain deposition diseases).
In this category, both C3 and C4 may be low.
MINIMAL CHANGE DISEASE
Minimal change disease (MCD) [Figure 4] is seen most commonly in children. It is characterized by an abrupt onset of the nephrotic syndrome. The etiology is likely a deficiency of regulatory T cells (Tregs or suppressor T cells). Corticosteroids are the main treatment as most patients are steroid-sensitive. Other agents have been used in patients who are steroid-dependent or with frequent relapses. Cyclophosphamide was used in the past; however, it has significant adverse effects. More commonly used agents include calcineurin inhibitors (cyclosporine and tacrolimus) and rituximab. Relapse is high when calcineurin inhibitors are stopped. Treatment with rituximab is well tolerated and seems promising.[4] Rituximab is effective in both children and adults with frequently relapsing nephrotic syndrome. In a large case series, 14 of 41 adults with MCD remained in remission after rituximab treatment with a median follow-up of 39 months.[5] Patients required 1–2 courses of rituximab with each course consisting of 2–4 doses.
Figure 4.
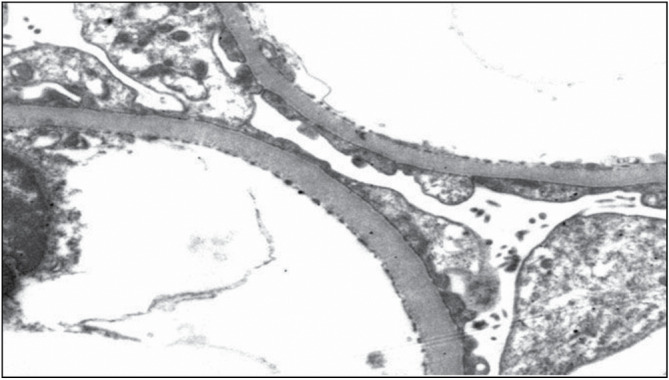
Minimal change disease (MCD). Foot process effacement on electron microscopy (Courtesy of Arkana Labs)
FOCAL SEGMENTAL GLOMERULOSCLEROSIS
Focal segmental glomerulosclerosis (FSGS) [Figure 5] is not a disease but a pattern of injury. It can be primary (idiopathic), genetic, or secondary (associated with another glomerular disease).
Figure 5.
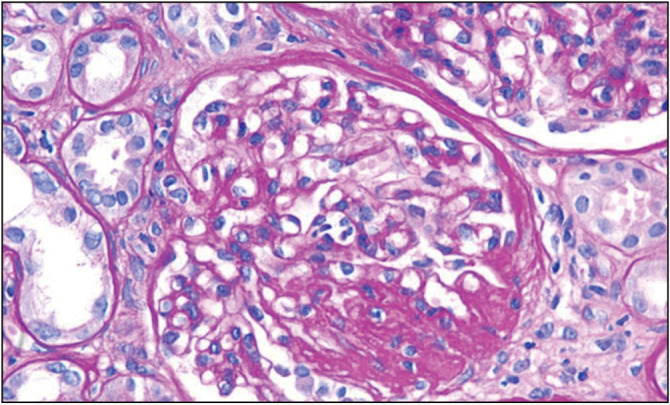
Enlarged glomerulus with focal segmental glomerulosclerosis (FSGS) on periodic acid–Schiff (PAS) staining (Courtesy of Arkana Labs)
All children and young adults should have genetic testing because genetic forms of FSGS respond poorly to steroids. Genetic testing is reasonable in adults with family history of FSGS.
Apolipoprotein L1 is a protein encoded by APOL1 gene. It is critical to note that more than 30% of people of West African ancestry have high-risk APOL1 gene variants predisposing them to primary FSGS (especially the collapsing variant), HIV nephropathy, and hypertensive nephrosclerosis.[6]
Corticosteroids remain the cornerstone of treatment. For patients who are steroids resistant, a calcineurin inhibitor is often added to a low-dose corticosteroid. In a case series of eight adults with FSGS, rituximab was ineffective.[7] Only one patient showed improvement in renal function and significant reduction in proteinuria.
MEMBRANOUS NEPHROPATHY
In a landmark study, Beck et al.[8] observed that the majority of patients with idiopathic MN [Figures 6 and 7] have antibodies against PLA2R in the glomeruli. Anti-PLA2R antibodies are mainly immunoglobulin G4 (IgG4) and can be measured in the serum. Kidney biopsy specimens show hyperexpression of PLA2R antigen in the glomeruli.
Figure 6.
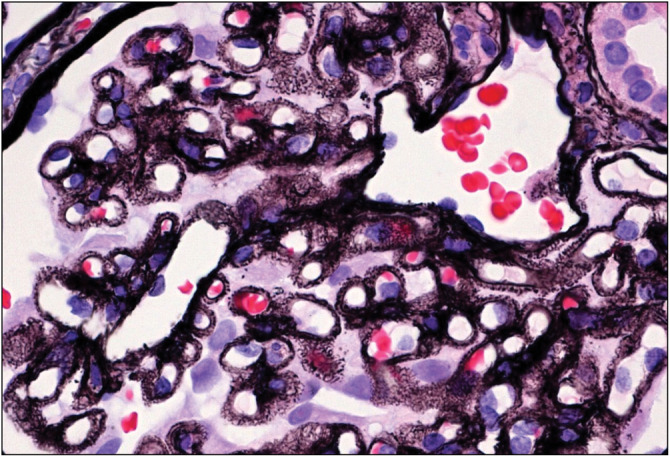
Membranous nephropathy on silver stain (Courtesy of Arkana Labs)
Figure 7.
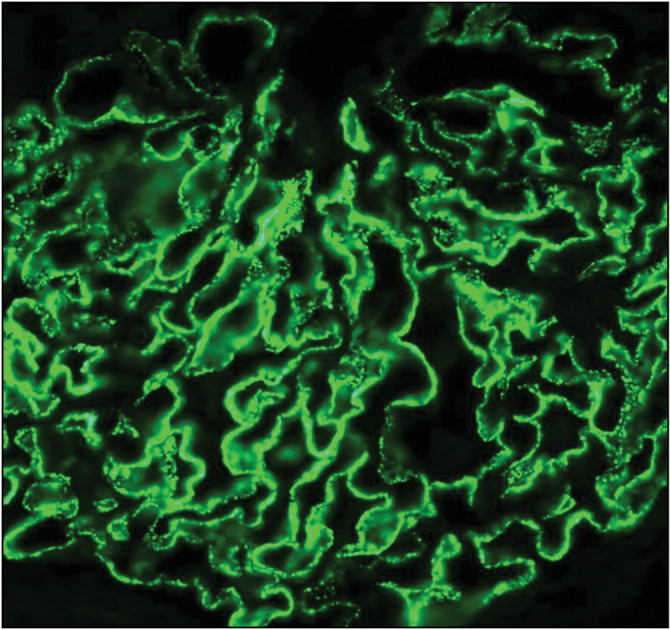
Positive M-type phospholipase A2 receptor (PLA2R) immunofluorescence microscopy (IF) stain, membranous nephropathy (Courtesy of Arkana Labs)
Serum measurement of these antibodies helps in differentiating idiopathic from secondary MN. Therefore, a negative test requires aggressive cancer screening. Moreover, serial measurements may help in following disease course in patients and identification of remission.
For decades, corticosteroids were used for treatment of MN in conjunction with cyclophosphamide or chlorambucil. These regimens were successful in reducing end-stage renal disease risk but have significant adverse reactions. Currently many patients are treated with corticosteroid and a calcineurin inhibitor (cyclosporine or tacrolimus). More recently, Rituximab has enjoyed success in the treatment of MN and may become the standard treatment.[9]
Results from the recently published Membranous Nephropathy Trial of Rituximab (MENTOR) showed equal efficacy of rituximab and cyclosporine in inducing partial or complete remission of proteinuria during the first year of treatment.[10] Rituximab was superior in maintaining remission up to 24 months.
Adrenocorticotropic hormone (ACTH) gel is a repository corticotropin injection that has been used to induce proteinuria remission in patients with treatment-resistant nephrotic syndrome. A multicenter retrospective case series[11] included 44 patients with nephrotic syndrome of diverse etiologies such as IgA nephropathy (IgAN), MN, minimal change nephropathy (MCN), and FSGS. Most patients had received prior treatments. ACTH gel showed ≥50% proteinuria reduction in about 60% of patients. The role of ACTH gel needs to be defined further in larger trials.
IgA NEPHROPATHY
The mechanism of IgAN [Figure 8] was elucidated over the past few years.[12] IgAN is attributed to immunoglobulin A1 (IgA1) with galactose-deficient O-linked glycans in the hinge region, which act as an autoantigen. Autoantibodies such IgG recognize this autoantigen resulting in circulating immune complexes. The immune complexes are deposited in the mesangium of the glomeruli resulting in mesangial proliferation.
Figure 8.
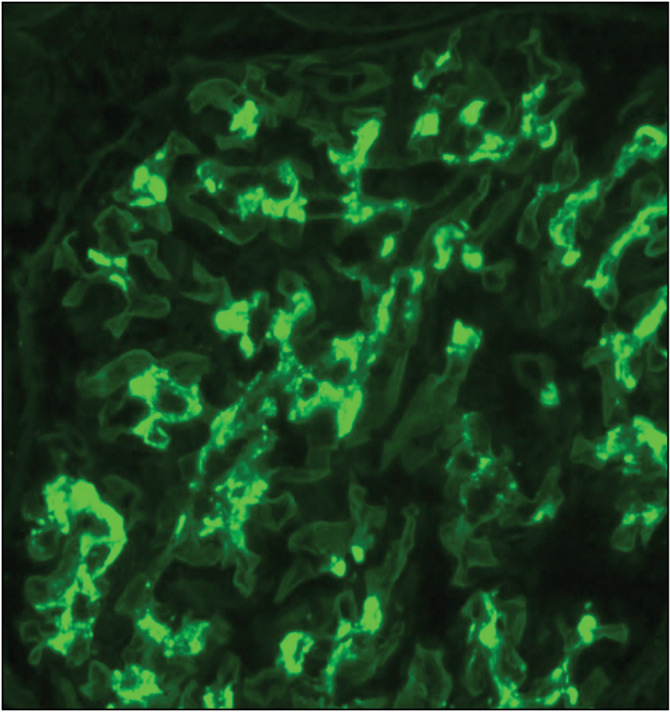
Mesangial immunoglobulin A (IgA) deposits on immunofluorescence microscopy (IF). Diagnosis of IgA nephropathy (IgAN) can only be made with immunofluorescence microscopy (IF) microscopy (Courtesy of Arkana Labs)
An important development in the field of IgAN is the publication of Oxford Classification of IgAN.[13] It uses MEST scores to predict clinical outcome. MEST stands for: mesangial hypercellularity (M), endocapillary proliferation (E), segmental sclerosis (S), and intestinal fibrosis/tubular atrophy (T).
The treatment of IgAN remains largely supportive with the use of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers to reduce proteinuria. In the past, corticosteroids were commonly used in patients with persistent proteinuria.
In the Supportive Versus Immunosuppressive Therapy for the Treatment of Progressive IgA Nephropathy (STOP-IgAN) trial,[14] corticosteroids did not slow progression of chronic kidney disease in high-risk IgAN (proteinuria >0.75g daily) and they were associated with significant adverse effects compared to supportive care.
RAPIDLY PROGRESSIVE (CRESCENTIC) GLOMERULONEPHRITIS
Rapidly progressive (crescentic) GN (RPGN) [Figures 9 and 10] is traditionally divided into three categories, see diagram 2:[15]
Figure 9.
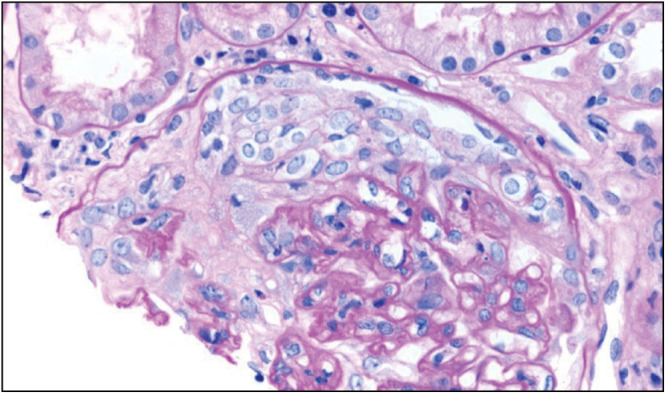
Cellular crescent in antineutrophil cytoplasmic antibody (ANCA) vasculitis. Periodic acid–Schiff (PAS) staining (Courtesy of Arkana Labs)
Figure 10.
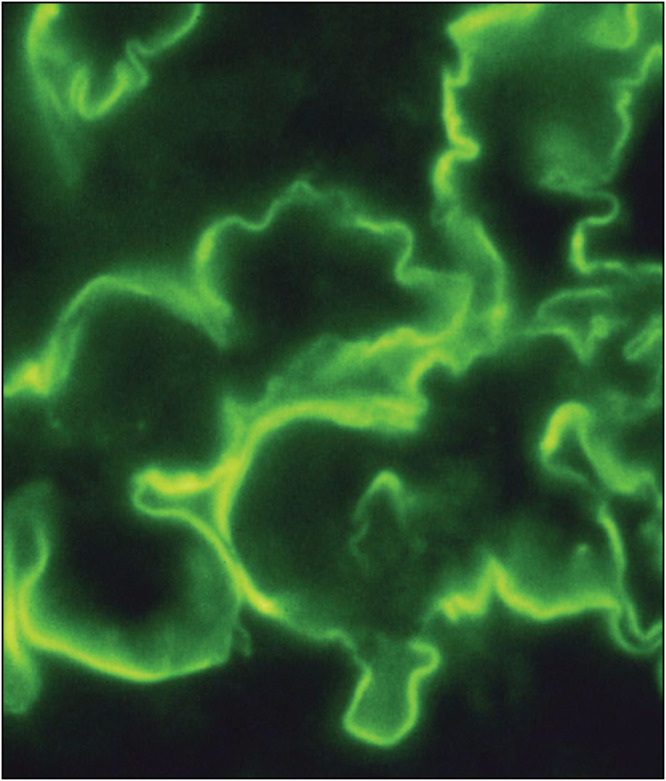
Linear immunoglobulin G (IgG) anti-glomerular basement membrane disease (anti-GBM disease), immunofluorescence microscopy (IF) (Courtesy of Arkana Labs)
Diagram 2.
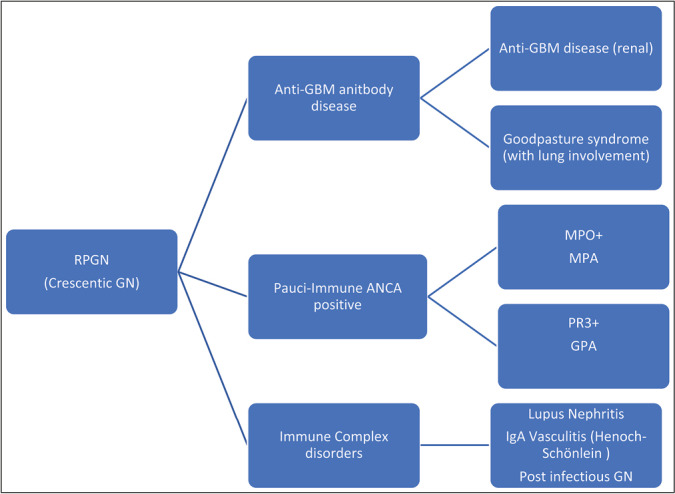
Categories of RPGN (Crescentic GN)
Anti-glomerular basement membrane antibody disease (anti-GBM), which is restricted to the kidneys and Goodpasture syndrome when there is pulmonary involvement as well.
-
Systemic small vessel vasculitides with circulating antineutrophil cytoplasmic antibodies (ANCA). There are no or few immune deposits seen upon examination with immunofluorescence microscopy (IF), hence the name pauci-immune. It is important for the reader here to note the new terminology. This group is divided into three syndromes:
-
A.Granulomatosis with polyangiitis (GPA, formerly Wegener granulomatosis). In this syndrome, ANCAs is usually directed against proteinase-3 (PR3). Granulomas in respiratory tract can be seen.
-
B.Microscopic polyangiitis (MPA). In this syndrome, ANCA is usually directed against myeloperoxidase (MPO).
-
C.Eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg–Strauss syndrome). In this syndrome, asthma and eosinophilia are encountered with the kidneys involved less often.
-
A.
Immune complex disorders: In this third category, RPGN is due to entities such as lupus nephritis, IgAN, mixed cryoglobulinemia, and postinfectious glomerulonephritis.
Crescents may be seen in any of the above disorders, therefore, crescentic GN is a pattern of injury, and the mechanism should be added to the name such as PR3-ANCA crescentic GN or mixed cryoglobulinemia crescentic GN.
Corticosteroids and cyclophosphamide have been used for decades in the management of anti-GBM disease, GPA, and MPA. More recently, rituximab is being used for induction and maintenance therapy of GPA and MPA.[16] Plasmapheresis is routinely used for Goodpasture disease, whereas its role is less well defined in GPA and MPA.[17]
LUPUS NEPHRITIS
A classification system was developed by the Renal Pathology Society/International Society of Nephrology.[18] There are six classes of LN representing different patterns of injury:
Class I (minimal mesangial), Class II (mesangial proliferative), Class III (focal), Class IV (proliferative), Class V (membranous), and Class IV (advanced sclerosing). A renal biopsy is required to confirm the diagnosis, determine the classification, and define the course of therapy.
Proliferative LN (Class IV) [Figure 11] was traditionally treated with corticosteroids and IV cyclophosphamide (the National Institutes of Health [NIH] protocol) to induce remission; this is followed by maintenance treatment with azathioprine.
Figure 11.
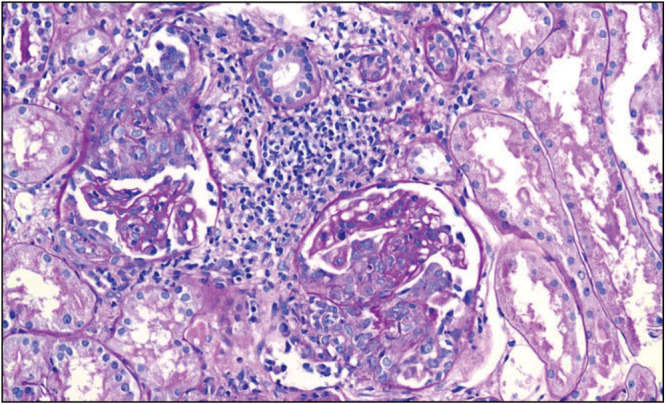
Class IV lupus nephritis with endocapillary proliferation and crescents formation (Courtesy of Arkana Labs)
Many patients are now induced with a tapering course of corticosteroids in addition to MMF then kept on MMF for maintenance.[19] Recently, there is a growing interest in multitargeted therapy where a calcineurin inhibitor (especially tacrolimus) is added to MMF and corticosteroids to induce remission.[20]
CONCLUSION
There has been a recent paradigm shift in the understanding of glomerular diseases.
Characterizing diseases as idiopathic or primary is inadequate, and the new terminology should reflect the pathogenesis of the disease. This has led to changes in the terminology. Advances in management have come about as well. The above changes will continue to accelerate over the upcoming years. The main points made in this review are summarized in Tables 1 and 2.
Table 1.
Major advance relevant to common glomerular diseases
| Glomerular disease | Major recent advances |
|---|---|
| Membranoproliferative glomerulonephritis | MPGN is divided into C3 glomerulopathy (including C3 glomerulonephritis and dense deposit disease) and immunoglobulin and C3 positive MPGN due to infections, autoimmune disease or monoclonal gammopathy. |
| Minimal change disease | MCD is likely due to deficiency of regulatory T cells (Tregs). Rituximab is playing an increasing role in treatment. |
| Focal segmental glomerulosclerosis | Genetic testing should be done in children because genetic FSGS responds poorly to corticosteroids. High risk APOL1 gene variants is the cause for the increased incidence of primary FSGS in African Americans |
| Membranous nephropathy | The majority of patients with idiopathic membranous nephropathy have antibodies against M-type phospholipase A2 receptors (PLA2R) in the glomeruli. These antibodies have diagnostic and prognostic value. Rituximab is gradually becoming the standard of care for treatment of MN. |
| IgA nephropathy | IgAN is attributed to galactose deficient IgA1 which act as an autoantigen. IgAN on renal biopsy is now classified using the Oxford classification system. Corticosteroids are playing a lesser role in the management of IgAN. |
| Rapidly progressive (crescentic) GN | Pauci-immune ANCA positive RPGN is divided into granulomatosis with polyangiitis (GPA, formerly Wegener granulomatosis) in which ANCAs is directed against proteinase-3 (PR3), and microscopic polyangiitis (MPA) in which ANCA is directed against myeloperoxidase (MPO). Rituximab is playing in increasing role in the induction and maintenance therapy of ANCA positive RPGN. |
| Lupus nephritis | Renal Pathology Society/International Society of Nephrology classification is used to determine the class of LN on renal biopsy. Mycophenolate mofetil (MMF) is now used as an induction and maintenance therapy of LN. A calcineurin inhibitor is sometimes added to MMF to induce remission. |
GN = glomerulonephritis, MPGN = membranoproliferative glomerulonephritis, MCD = minimal change disease, FSGS = focal segmental glomerulosclerosis, MN = membranous nephropathy, IgAN = IgA nephropathy, IgA1 = immunoglobulin A1, ANCA = antineutrophil cytoplasmic antibodies, RPGN = rapidly progressive (crescentic) glomerulonephritis, LN = lupus nephritis
Table 2.
Use of specific terminology in reporting of glomerular diseases
| Old terminology | Examples of new more specific terminology |
|---|---|
| Idiopathic membranous nephropathy | Membranous nephropathy due to PLA2R antibodies |
| MPGN | C3 glomerulonephritis secondary to factor I deficiency |
| Idiopathic FSGS | Genetic FSGS due to nephrin deficiency |
| ANCA vasculitis | Anti-PR3 granulomatosis with polyangiitis |
MPGN = membranoproliferative glomerulonephritis, PLA2R = M-type phospholipase A2 receptor, FSGS = focal segmental glomerulosclerosis, ANCA = antineutrophil cytoplasmic antibodies, PR3 = proteinase-3
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Sethi S, Haas M, Markowitz GS, D’Agati VD, Rennke HG, Jennette JC, et al. Mayo clinic/renal pathology society consensus report on pathologic classification, diagnosis, and reporting of GN. J Am Soc Nephrol. 2016;27:1278–87. doi: 10.1681/ASN.2015060612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis–a new look at an old entity. N Engl J Med. 2012;366:1119–31. doi: 10.1056/NEJMra1108178. [DOI] [PubMed] [Google Scholar]
- 3.Zipfel PF, Wiech T, Rudnick R, Afonso S, Person F, Skerka C. Complement inhibitors in clinical trials for glomerular diseases. Front Immunol. 2019;10:2166. doi: 10.3389/fimmu.2019.02166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King C, Logan S, Smith SW, Hewins P. The efficacy of rituximab in adult frequently relapsing minimal change disease. Clin Kidney J. 2017;10:16–9. doi: 10.1093/ckj/sfw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guitard J, Hebral AL, Fakhouri F, Joly D, Daugas E, Rivalan J, et al. Rituximab for minimal-change nephrotic syndrome in adulthood: Predictive factors for response, long-term outcomes and tolerance. Nephrol Dial Transplant. 2014;29:2084–91. doi: 10.1093/ndt/gfu209. [DOI] [PubMed] [Google Scholar]
- 6.Kruzel-Davila E, Wasser WG, Skorecki K. APOL1 nephropathy: A population genetics and evolutionary medicine detective story. Semin Nephrol. 2017;37:490–507. doi: 10.1016/j.semnephrol.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Roccatello D, Sciascia S, Rossi D, Alpa M, Naretto C, Radin M, et al. High-dose rituximab ineffective for focal segmental glomerulosclerosis: A long-term observation study. Am J Nephrol. 2017;46: 108–13. doi: 10.1159/000477944. [DOI] [PubMed] [Google Scholar]
- 8.Beck LH, Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi: 10.1056/NEJMoa0810457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruggenenti P, Fervenza FC, Remuzzi G. Treatment of membranous nephropathy: Time for a paradigm shift. Nat Rev Nephrol. 2017;13:563–79. doi: 10.1038/nrneph.2017.92. [DOI] [PubMed] [Google Scholar]
- 10.Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, et al. MENTOR Investigators Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med. 2019;381:36–46. doi: 10.1056/NEJMoa1814427. [DOI] [PubMed] [Google Scholar]
- 11.Madan A, Mijovic-Das S, Stankovic A, Teehan G, Milward AS, Khastgir A. Acthar gel in the treatment of nephrotic syndrome: A multicenter retrospective case series. BMC Nephrol. 2016;17:37. doi: 10.1186/s12882-016-0241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mestecky J, Novak J, Moldoveanu Z, Raska M. IgA nephropathy enigma. Clin Immunol. 2016;172:72–7. doi: 10.1016/j.clim.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, et al. Oxford classification of IgA nephropathy 2016: An update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017;91:1014–21. doi: 10.1016/j.kint.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Rauen T, Eitner F, Fitzner C, Sommerer C, Zeier M, Otte B, et al. STOP-IgAN Investigators Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med. 2015;373:2225–36. doi: 10.1056/NEJMoa1415463. [DOI] [PubMed] [Google Scholar]
- 15.Jennette JC. Overview of the 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Clin Exp Nephrol. 2013;17:603–6. doi: 10.1007/s10157-013-0869-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jennette JC, Nachman PH. ANCA glomerulonephritis and vasculitis. Clin J Am Soc Nephrol. 2017;12:1680–91. doi: 10.2215/CJN.02500317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geetha D, Jin Q, Scott J, Hruskova Z, Hanouneh M, Little MA, et al. Comparisons of guidelines and recommendations on managing antineutrophil cytoplasmic antibody-associated vasculitis. Kidney Int Rep. 2018;3:1039–49. doi: 10.1016/j.ekir.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. International Society of Nephrology Working Group on the Classification of Lupus Nephritis; Renal Pathology Society Working Group on the Classification of Lupus Nephritis The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65:521–30. doi: 10.1111/j.1523-1755.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 19.Hahn BH, McMahon MA, Wilkinson A, Wallace WD, Daikh DI, Fitzgerald JD, et al. American College of Rheumatology. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken) 2012;64:797–808. doi: 10.1002/acr.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Zhang H, Liu Z, Xing C, Fu P, Ni Z, et al. Multitarget therapy for induction treatment of lupus nephritis: A randomized trial. Ann Intern Med. 2015;162:18–26. doi: 10.7326/M14-1030. [DOI] [PubMed] [Google Scholar]