

Abstract
Immunoglobulin (Ig) glycosylation is recognized for its influence on Ig turnover and effector functions. However, the large-scale profiling of Ig glycosylation in a biomedical setting is challenged by the existence of different Ig isotypes and subclasses, their varying serum concentrations, and the presence of multiple glycosylation sites per Ig. Here, a high-throughput nanoliquid chromatography (LC)- mass spectrometry (MS)-based method for simultaneous analysis of IgG and IgA glycopeptides was developed and applied on a serum sample set from 185 healthy donors. Sample preparation from minute amounts of serum was performed in 96-well plate format. Prior to trypsin digestion, IgG and IgA were enriched simultaneously, followed by a one-step denaturation, reduction, and alkylation. The obtained nanoLC-MS data were subjected to semiautomated, targeted feature integration and quality control. The combined and simplified protocol displayed high overall method repeatability, as assessed using pooled plasma and serum standards. Taking all samples together, 143 individual N- and O-glycopeptides were reliably quantified. These glycopeptides were attributable to 11 different peptide backbones, derived from IgG1, IgG2/3, IgG4, IgA1, IgA2, and the joining chain from dimeric IgA. Using this method, novel associations were found between IgA N- and O-glycosylation and age. Furthermore, previously reported associations of IgG Fc glycosylation with age in healthy individuals were confirmed. In conclusion, the new method paves the way for high-throughput multiprotein plasma glycoproteomics.
Immunoglobulins (Igs) are crucial to adaptive immunity, and their specific functions vary between isotypes and subclasses. In humans, immunoglobulin G (IgG) and A (IgA) are the first and second most abundant Igs in the circulation (approximately 12.5 and 2.2 mg/mL, respectively1,2) and IgA is the most abundant Ig in mucosal secretions (e.g., approximately 0.2 mg/mL in saliva3). Glycosylation is a prominent co- and post-translational modification of antibodies, with a large impact on their function. This is well described for IgG effector functions in vitro.4−7 For example, IgG Fc glycosylation influences binding of the antibody to the Fcγ receptor III and subsequent antibody-dependent cellular cytotoxicity, as well as binding to C1q and complement-dependent cytotoxicity.4−6 Furthermore, changes in human IgG glycosylation have been associated with, for example, autoimmune diseases and cancer.8 In contrast, our understanding of the role of serum IgA and its glycosylation in (patho-)physiological processes, as well as the factors affecting IgA glycosylation, is still limited. This is mainly due to the existing analytical challenges for analyzing IgA glycosylation in a subclass- and site-specific manner.
Human IgG occurs in four subclasses, which all carry a conserved N-glycosylation site in the Fc domain. This site is occupied by an N-glycan in more than 99% of the IgG molecules.9,10 IgG3 contains an additional N-glycosylation site in its constant domain and mucin-type O-glycosylation sites in the hinge region, which both have site occupancies of around 10%.10,11 Human IgA consists of two subclasses, IgA1 and IgA2, which in serum are mostly present as monomers. Dimeric IgA is present as a minor fraction and is formed when a joining chain (JC) connects two monomers via disulfide bridges in the tailpiece region.1 The JC itself is a glycoprotein with one N-glycosylation site. IgA2 has four highly occupied N-glycosylation sites in the constant region, whereas IgA1 has two highly occupied N-glycosylation sites in the constant region and six O-glycosylation sites in the hinge region.9,12 Although less is known about the structure–function relationship of IgA glycosylation as compared to IgG glycosylation, recent studies indicated serum IgA N- and O-glycosylation effects in a small cohort of breast cancer patients.13,14 Moreover, IgA1 O-glycans are thought to play a central role in the pathogenesis of IgA nephropathy.15
Since Ig glycosylation has been associated with numerous diseases, Ig glycosylation profiling has been suggested as a promising approach to screen for pathology-associated glycosylation changes with potential for biomarker development.8,16−18 To this end, the development of high-throughput workflows for multiprotein glycoprofiling is much-needed.19 Mass spectrometry (MS)-based glycosylation analysis at the glycopeptide level is particularly useful, as it provides site-specific and protein-specific information.20 MS-based approaches relying on electrospray ionization (ESI) allow the hyphenation between a liquid-mode separation and MS, enabling the differentiation between isomeric or isobaric glycopeptides.20 The use of reversed-phase (RP) liquid chromatography (LC) for glycopeptides results in separation mainly based on their peptide portion, allowing straightforward data processing, as reported for glycopeptides derived from different Ig isotypes.9,21−23 The integrated analysis of different Ig isotypes results in a higher sample throughput and gain in information density, as was shown for IgM, IgA, and IgG glycopeptide profiling via targeted tandem MS after the direct injection of a digest of the complete plasma proteome.24 Alternatively, a more in-depth characterization of Ig glycosylation microheterogeneity is expected when antibodies are enriched prior to glycopeptide generation.9,25 However, since the serum concentrations of the different Igs vary significantly during disease and between isotypes and subclasses, their simultaneous preparation and MS-based analysis is a great challenge. To date, only methods were reported for the preparation and profiling of glycopeptides from individual Ig isotypes.9,25
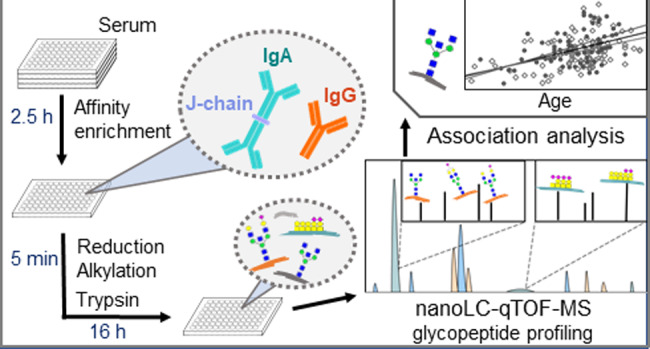
Here, we developed an integrated workflow for simultaneous, high-throughput glycosylation analysis of serum IgG and IgA by nanoRPLC-ESI-MS. Samples were prepared in 96-well plate format from minute amounts of serum employing affinity purification. Differences in serum Ig concentrations were overcome by the controlled, incomplete capturing of IgG, while aiming for a near-complete IgA capture. Three IgG and five IgA N-glycopeptide elution clusters (based on the peptide portions surrounding the glycosylation sites) were detected by LC-qTOF-MS analysis of the captured and proteolytically digested Igs, next to one cluster for the IgA hinge-region O-glycopeptides and two clusters from the JC. Implementation of the semiautomated data processing software LaCyTools provided fast and consistent data processing.23 The new method was applied on a medium-size sample set from healthy donors, which revealed novel IgA N- and O-glycosylation associations with age and confirmed IgG-Fc glycosylation associations with age.26
Experimental Section
Chemicals and Enzymes
CaptureSelect FcXL Affinity Matrix beads (binding capacity IgG 25–35 g/L; Thermo Fisher Scientific, Leiden, Netherlands) and CaptureSelect IgA Affinity Matrix (binding capacity IgA 8 g/L, Thermo Fisher Scientific) were used for affinity purification. Ammonium bicarbonate, formic acid, tris(hydroxymethyl)amino-methane, tris(2-carboxyethyl)phosphine (TCEP) hydrochloride, sodium deoxycholate (SDC), and 2-chloracetamide (CAA) were from Sigma-Aldrich (Zwijndrecht, The Netherlands). Water was purified via a Purelab Ultra, maintained at 18.2 MΩ (Veolia Water Technologies Netherlands B.V., Ede, Netherlands). Acetic acid was from Honeywell (Seelze, Germany), acetonitrile from Biosolve (Valkenswaard, The Netherlands), trifluoroacetic acid (TFA) from Merck (Darmstadt, Germany), and sequencing grade modified trypsin was from Promega (Madison, WI).
Samples
Serum samples were obtained from the Leiden University Medical Center (LUMC) Surgical Oncology Biobank. Samples were collected between October 2002 and March 2013 according to a standardized protocol.27 This study was approved by the Medical Ethics Committee of the LUMC and was performed in accordance to the Code of Conduct of the Federation of Medical Scientific Societies in The Netherlands. In-house available pooled human serum (N = 48 × 5.4 μL) and commercially available human plasma Visucon-F (Affinity Biologicals, Ancaster, ON, Canada) (N = 40 × 5.4 μL) were used as technical standards. Negative controls, consisting of PBS, were included at random locations in the sample plate layout (N = 20).
Immunoaffinity Enrichment of Immunoglobulins from Serum
Using a Microlab STAR liquid handling robot (Hamilton, Bonaduz, Switzerland), 5.4 μL of serum were pipetted into V-bottom 96-well plates (Greiner Bio-One B.V., Alphen a/d Rijn, The Netherlands) containing 130 μL 35 mM phosphate-buffered saline (PBS; pH 7.6, made in-house from 5.7 g Na2HPO4/L of water, 2H2O, 0.5 g KH2PO4/L of water, and 8.5 g NaCl/L of water). Ig enrichment was performed similar to Selman et al. with the following modifications.21 Per sample, 40 μL slurry was used, containing 0.2 μL CaptureSelect FcXL Affinity Matrix beads (absolute theoretical capacity for total IgG: 5 μg) and 5 μL CaptureSelect IgA Affinity Matrix beads (absolute theoretical capacity for IgA: 40 μg) in PBS. The bead slurry was applied to 96-well filter plates (10 μm pore size, Orochem, Naperville, IL) and washed three times with 200 μL PBS using a vacuum manifold (50 kPa pressure gradient). To prevent drying of the beads 30 μL of PBS were added to each well. For combined IgG and IgA capturing, 125 μL of diluted sample (corresponding to 5 μL serum) were added per well, followed by 1 h incubation at room temperature with agitation. Using a vacuum manifold, the beads were washed three times with 200 μL PBS and three times with 200 μL water, prior to centrifugation for 1 min at 911 g. For elution, 100 μL of 100 mM formic acid were added, followed by 5 min incubation with agitation. Eluates were collected into nonskirted 96-well PCR plates (Greiner Bio-One, Kremsmünster, Austria) by 2 min centrifugation at 1763 g and dried by vacuum centrifugation for 2.5 h at 60 °C.
Glycopeptide Preparation
Dried eluates were dissolved in 10 μL reduction-alkylation buffer (100 mM tris(hydroxymethyl)aminomethane, 1% (w/v) SDC, 10 mM TCEP and 40 mM CAA) and sealed with VersiCap Mat flat cap strips (Thermo Fisher Scientific) followed by 5 min of shaking at 500 rpm with 1.5 mm orbit. For one-step denaturation, reduction, and alkylation the samples were incubated for 5 min at 95 °C and cooled to room temperature in a 2720 Thermal Cycler (Thermo Fisher Scientific). For digestion, 50 μL of digestion buffer (0.004 μg/μL sequencing grade modified trypsin in 50 mM ammonium bicarbonate (pH 8.5) was added to each sample. The plate was closed with VersiCap Mat flat cap strips, followed by 5 min shaking at 1000 rpm and overnight incubation at 37 °C. On the next day, 1.2 μL of concentrated formic acid was added to each sample, followed by centrifugation for 45 min at 2800 g for the acid precipitation of SDC. Using a semiautomated pipetting system Liquidator 96 (Mettler Toledo, ’s-Hertogenbosch, Netherlands), 40 μL of supernatant containing Ig (glyco-)peptides were transferred to a V-bottom 96-well plate (Greiner). The plate was sealed and stored at −20 °C prior to MS analysis.
Protein and Glycopeptide Identification by MS/MS
(Glyco-)peptides were identified in one serum sample pooled from a subset of the clinical cohort described above. To this end, an Easy nLC 1200 gradient system (Thermo Fisher Scientific), and an Orbitrap Fusion Lumos Tribrid MS were used (Thermo Fisher Scientific; Supporting Information (SI) Tables S1 and S2 and Figure S1). The tryptic digest sample was injected into a homemade precolumn (15 mm × 100 μm; Reprosil-Pur C18-AQ 3 μm, Dr. Maisch, Ammerbuch, Germany). Separation was performed via an analytical nanoLC column (15 cm × 75 μm; Reprosil-Pur C18-AQ 3 μm; drawn to a spray tip of 5 μm at the end of the column) with a solvent B (20/80/0.1, water/acetonitrile/FA, v/v/v) gradient from 10 to 40% in 20 min (solvent A: 0.1% formic acid in water). After electrospray ionization, a data-dependent tandem MS analysis was performed where a full MS1 scan within m/z 400–1500 was collected with a resolution of 120 000, at an automatic gain control (AGC) target of 400 000 and with a maximum fill time of 50 ms. The n most abundant ions per 3 s were fragmented with higher-energy C trap dissociation (HCD, 32 normalized collision energy). Dynamic exclusion was applied after n = 1 with exclusion duration of 10 s. Charges 2–5 were included for MS2 using a quadrupole for precursor isolation with an isolation width of 1.2 Th and MS2 scan resolution of 30 000. The AGC target was 50 000, with a maximum fill time of 60 ms. Further fragmentation was focused on glycopeptides using the N-acetylhexosamine oxonium ion product at m/z 204.087 as trigger for a sequence of additional MS2 scans of the same precursor, that is, one collision-induced dissociation (CID; 35 normalized collision energy) spectrum and three spectra with different HCD collision energies (32, 37, and 42 normalized collision energy), at an AGC target of 500 000 with a maximum fill time of 200 ms. Protein identification was based on peptide comparison with the “Homo2018Canonical” database. The search for glycopeptides was performed using Byonic software version 3.2–38 2.13.2 with default settings, including a search for either tryptic or chymotryptic peptides. Carbamidomethylation (Cys) was set as fixed and oxidation (Met) as variable modifications using the following Byonic databases: “N-glycan 309 mammalian no sodium”, “O-glycan 78 mammalian”. Manual spectra exploration was performed with Xcalibur (v. 2.2 SP1.48, Thermo Fisher Scientific).
Glycosylation Profiling by NanoLC-qTOF-MS
All samples were thawed, mixed and centrifuged for 5 min at 1763 g. (Glyco-)peptides were analyzed using an Ultimate 3000 RSLCnano system (Dionex/Thermo Fisher Scientific, Sunnyvale, CA) coupled to an Impact qTOF-MS (Bruker Daltonics, Bremen, Germany) as described previously.28 Briefly, sample loading was performed using an autosampler equipped with a 2.4 μL needle and 5 μL PEEK sample loop. From each well, 0.5 μL of sample was loaded in 0.1% TFA onto a C18 PepMap100 trap column (300 μm i.d. × 5 mm, particle size 5 μm, pore size 100 Å, Thermo Fisher Scientific) at a flow of 25 μL/min. After 1 min the trap column was switched in-line with a nanoEase M/Z Peptide BEH C18 analytical column (75 μm x 100 mm, particle size 1.7 μm, pore size 130 Å, 1/PK, Waters) at a flow of 0.6 μL/min. A linear gradient of solvent A (0.1% TFA in water) and solvent B (95% acetonitrile) was applied: 3%B 0 min, 30%B 6.5 min, 95%B 10 min, 95%B 10–12 min, 3%B 13 min, 3%B 13−21 min. Positive-mode ESI was performed using a Captive Spray source equipped with nanoBooster technology (Bruker Daltonics), enriching the nitrogen dry gas (3 L/min, 180 °C) with acetonitrile. The collision energy was at 5 eV, the transfer time at 112 μs, and the prepulse storage at 21 μs. The acquisition window was set to m/z 400–1800 at a frequency of 1 Hz.
NanoLC-qTOF-MS Data Processing
Glycopeptides were identified in the profiling data, based on features described in literature25,29,30 and the Orbitrap tandem MS data of the pooled sample. For glycosylation profiling using nanoLC-qTOF-MS, 11 retention time clusters were defined based on the peptide portions surrounding the IgG and IgA glycosylation sites, i.e LAGy, LAGc, TPL, LSL, SES, HYT, ENI, IIV, IgG1, IgG2/3, IgG4 (Table 1, Figure 1A). For each run, calibration was performed in DataAnalysis 5.0 (build number 203.2.23586, Bruker Daltonics) on a sum spectrum (2.6–3.2 min) using 9 calibrants (SI Table S3). Calibrated LC-MS data files were converted to mzXML format with MSConvert (ProteoWizard 3.0 suite). Using LaCyTools version 1.0.1 build 8, chromatograms were aligned to a target chromatogram (SI Table S3).23 If the root-mean-square difference between targeted and resulting retention times was higher than 5, the sample was excluded. Sum spectra were generated per glycopeptide cluster, further used for area integration within a ± 0.05 Th window for each individual glycopeptide composition, covering a minimum of 80% of the theoretical isotopic envelope area. Local background subtraction was performed for all included isotopes. For quality control, analyte presence was evaluated per charge state based on the following quality criteria: isotopic pattern quality score <0.2, signal-to-noise ratio >9, and absolute mass error <20 ppm. For each glycopeptide cluster, the total area of all analytes passing the analyte quality criteria was summed over all included charge states. For spectral quality control, a specific cluster was excluded per sample, if either the respective total intensity was below the first percentile in the sample group (healthy individuals, pooled plasma, pooled serum) or if the number of analytes passing the criteria described above was below the 15th percentile. After the exclusion of low-quality clusters, only analytes measured with good quality, based on analyte criteria described above, in more than 20% of the samples per group were included. For analytes which passed quality control, the absolute areas for all included charges per glycopeptide were summed, corrected for the isotopic fraction integrated, and normalized to the total area per glycopeptide cluster to retrieve relative intensities of the validated glycopeptide compositions.
Table 1. IgG and IgA Glycosylation Sites Targeted for Glycopeptide-Based Glycosylation Analysis.
| protein | clustera | peptide sequenceb | glycosylation site | retention time (min) |
|---|---|---|---|---|
| IgA1 | HYT | (K)HYTNPSQDVTVPCPVPSTPPTPSPSTPPTPSPSCCHPR(L) | T106/T109/S111/S113/T114/T117 | 3.3–4.1 |
| IgA1/2 | LAGy | (R)LAGKPTHVNVSVVMAEVDGTCY | N340/327 | 5.3–5.6 |
| IgA1/2 | LAGc | (R)LAGKPTHVNVSVVMAEVDGTC | N340/327 | 5.0–5.3 |
| IgA1/2 | LSL | (R)LSLHRPALEDLLLGSEANLTCTLTGLR(D) | N144/131 | 6.3–6.6 |
| IgA2 | TPL | (K)TPLTANITK(S) | N205 | 2.7–3.1 |
| IgA2 | SES | (W)SESGQNVTAR(N) | N47 | 0.6–1.3 |
| JC | ENI | (R)ENISDPTSPLR(T) | N71 | 2.8–3.3 |
| JC | IIV | (R)IIVPLNNRENISDPTSPLR(T) | N71 | 4.8–5.1 |
| IgG1 | IgG1 | (R)EEQYNSTYR(V) | N180 | 1.3–1.7 |
| IgG2/3c | IgG2/3c | (R)EEQFNSTFR(V)c | N176/N227c | 2.7–3.1 |
| IgG4 | IgG4 | (R)EEQFNSTYR(V) | N177 | 2.0–2.3 |
Cluster names for IgA and JC are referring to the first three letters of the peptide sequence, while IgG clusters are named after their subclass.
Glycosylation sites are marked in bold within the peptide sequence, and the respective threonine, serine, and asparagine residues are numbered according to UniProtKB nomenclature.34 The N-glycosylation consensus motif is underlined.
The amino acid sequence of the IgG3 glycopeptide is, depending on the IgG3 allotype, either exactly the same as the IgG2 glycopeptide sequence (EEQFNSTFR; allotypes IGHG3*11 and *12), represents an isomer of the IgG4 glycopeptide (EEQYNSTFR), or has the unique sequence TKPWEEQYNSTFR.35 Based on the tandem MS data of the pooled subset of the studied population, no indications were found for the presence of IgG3 allotypes that would result in the EEQYNSTFR (IGHG3*01 to *10 and *13 to *17) or TKPWEEQYNSTFR (IGHG3*18 and *19) sequences, which is consistent with the predominant presence of IGHG3*11 or *12 allotypes in the Caucasian population.36 However, an overlap between IgG3 and IgG4 glycoforms in the individual samples cannot be excluded.
Figure 1.

NanoLC-qTOF-MS glycopeptide profiling of IgG and IgA. (A) Extracted ion chromatograms (EICs) for one major glycopeptide per cluster. Protein names and the first three letters of the amino acid sequence of the respective tryptic peptide (in parentheses) are given. Separation was based on the peptide backbones, clustering the analytes with the same peptide sequence, but varying glycan portions. The m/z window for the EICs was set as follows for the different clusters: ± 0.05 Th for SES, ± 0.002 Th for IgG4, ± 0.01 Th for ENI, and ±0.02 Th for all other clusters. The blue and red boxes indicate the time windows used for sum spectra generation in (B) and (C). (B) The 25 most abundant glycopeptide peaks from the IgA1 HYT O-glycopeptide cluster, their measured m/z and suggested monosaccharide compositions. (C) The 20 most abundant glycopeptide peaks from the IgA1/2 LSL N-glycopeptide cluster, their measured m/z and proposed N-glycan structures.25,37 Note: linkages were not determined. Asterisks mark signals not derived from glycopeptides.
Statistical Analysis
Microsoft Excel (2016) was used for data handling and quality control, while R Version 3.4.3 (R Core Team, 2014) and RStudio (Version 1.2.1335 Build 1379) were used for data analysis, and ggplot2 package and Microsoft Powerpoint (2016) for data visualization.31 Batch-effect correction was performed using the function ComBat from the sva package in R.32 Subsequently, extreme outliers (mean ±5 SD for the glycopeptide relative intensities) were removed. Spearman correlation analysis was performed to assess glycopeptide correlation between different glycopeptide clusters. Associations between glycopeptide relative intensities and age of the healthy donors were tested via linear regression analysis on standardized data (subtraction of the mean and division by the SD). After multiple testing correction via the Bonferroni procedure p-values below 3.6 × 10–4 were considered statistically significant.
Results and Discussion
Combined IgG and IgA Enrichment and Glycopeptide Generation
A high-throughput method for the combined IgG and IgA glycosylation profiling of serum and plasma samples was developed. To account for the large serum concentration difference between IgG and IgA,1,2 their simultaneous immunoaffinity enrichment was optimized by combining IgG and IgA affinity beads in a ratio allowing to capture only a fraction of the total IgG, while IgA capturing was almost complete. The optimized capturing conditions resulted in a theoretical capturing capacity of 5 μg of IgG and 40 μg of IgA. Assuming an absolute amount of 62.5 μg of IgG and 11 μg of IgA in 5 μL plasma,1,2 the final captured amount of IgG (approximately 5 μg) and IgA (approximately 11 μg) were in the same order of magnitude. This resulted in a method that allowed the detection of all of the highly occupied glycosylation sites from all subclasses of IgG and IgA in a single LC-qTOF-MS run (Figure 1). Previous reports on the combined analysis of IgG and IgA glycopeptides relied on the enrichment of IgA separately from IgG, and either performed separate LC-qTOF-MS analysis for the two proteins, or needed to adjust protein amounts after capturing. Both approaches introduced additional steps into the workflows and yet were not able to cover all expected glycosylation sites.9,25 Although incomplete capturing of IgG may come with the risk of introducing glycoform-dependent biases, this was not expected, as the CaptureSelect FcXL beads are binding the CH3 domain of IgG, while the conserved Fc glycosylation site is located on the CH2 domain. Furthermore, the IgG glycosylation profiles obtained using this method were similar to profiles obtained after near-complete capturing of plasma IgG (SI Figure S2).
The reported sample preparation procedure using TCEP and CAA for the reduction and alkylation of the proteins prior to trypsin digestion, resulted in a higher flexibility regarding sample concentration differences as compared to the often used dithiothreitol and iodoacetamide approaches, as no byproducts were formed by the unreacted reagents. In addition, it allowed a one-step denaturation, reduction and alkylation of the samples, considerably simplifying and shortening the sample preparation as compared to most other approaches.33
Protein and Glycopeptide Identification
Orbitrap tandem MS of a pooled serum sample confirmed the presence of the targeted proteins IgG1, 2, 3, and 4 as well as IgA1, 2 and the JC (SI Table S1). For 10 of the 11 glycopeptide elution clusters (based on the peptide portions; Table 1) the peptide sequence was confirmed by tandem MS (SI Table S2 and Figure S1). For the elution cluster representing glycosylation site N47 on IgA2, annotation was based on accurate mass and isotopic pattern matching. While all other glycopeptides were a result of specific trypsin digestion, this site was detected in the form of the chymotryptic peptide (W)SESGQNVTAR(N), which is in line with previous reports.25,37 Concerning the previously reported O-glycosylation in IgG3 hinge region, only nonglycosylated peptides were confirmed by tandem MS in our study (not shown).
Robust, High-Throughput Profiling of IgG and IgA Glycopeptides by NanoLC-qTOF-MS
A total of 129 and 139 glycopeptides were quantified in the pooled plasma (N = 40) and serum (N = 48) standard samples, respectively (SI Table S4). More than 50% of glycopeptides in pooled serum samples had relative standard deviations below 10% (Figure 2, SI Table S5). Using the optimized sample preparation, 384 samples can be prepared by one person in two working days, and 60 digested samples can be analyzed per 24 h on the nanoLC-qTOF-MS system, taking into account suitable numbers of LC-MS system suitability standards and blanks. Thus, the developed method is suited for high-throughput applications on large sets of clinical samples.
Figure 2.

Method repeatability. Two different standards, that is, pooled human serum (N = 48) and commercially available pooled human plasma (N = 40), were randomly distributed over eight 96-well plates and underwent the full sample preparation and analysis workflow. Filled bars show the mean and error bars show the SD of relative intensities for the three most abundant glycopeptides after total area normalization per each of the 11 different peptides (clusters). Monosaccharide symbols: blue square = N-acetylglucosamine; green circle = mannose; yellow circle = galactose; pink diamond = sialic acid; red triangle = fucose; yellow square = N-acetylgalactosamine (see also Figure 1).
IgA and IgG Glycosylation in Healthy Individuals
A total of 101 glycopeptides derived from IgA1, IgA2 and the JC were detected and reliably quantified (SI Table S4A and B). The LAGy and LAGc (truncated version, lacking tyrosine29,38) glycopeptide clusters cover the C-terminal part of both IgA1 (glycosylation site N340) and allotype IGHA2*01 of IgA2 (glycosylation site N327; for peptide nomenclature see Table 1). Other IgA2 allotype sequences covering this glycosylation site, as described in literature,9,12,25 were not detected in the present study. Both the LAGy and the LAGc glycopeptide species eluted at two retention times (Figure 1). This elution pattern is likely due to the isomerization of aspartic acid (IgA1 D348 and IgA2 D335) into isoaspartic acid.25,39 In healthy individuals, 10 and 9 different glycoforms were quantified for LAGy and LAGc, respectively. The LSL cluster (covering IgA1 N144 and IgA2 N131) consisted of 12 glycoforms, the TPL cluster (IgA2 N205) of 10 glycoforms, the SES cluster (IgA2 N47) of seven glycoforms and the HYT cluster contained 39 O-glycopeptides from the IgA1 hinge region.
All IgA N-glycosylation sites carried mainly complex-type glycans, including diantennary and triantennary, mostly fucosylated structures with a varying degree of bisection, galactosylation and sialylation as described before.25,29,37 In addition, hybrid and high-mannose type structures were registered in the LSL cluster, which was additionally characterized by the absence of measurable levels of fucosylation.25
For IgA1 hinge-region O-glycosylation, exclusively simple Core 1 structures were reported before.40 Assuming one N-acetylgalactosamine per O-glycan, a site occupancy of three to six O-glycans per heavy chain was found in the current study, with each glycan carrying zero or one galactose and zero to two sialic acids (SI Table S4).29
Finally, glycopeptides from JC, which is associated with dimeric IgA, were detected in two glycopeptide clusters (both covering N71). The ENI cluster represented glycopeptides with the expected tryptic peptide sequence. In this cluster, mainly afucosylated, fully galactosylated, diantennary glycans were found, next to hybrid-type glycoforms. The IIV cluster represented glycopeptides with a miscleaved peptide, (R)IIVPLNNREN71ISDPTSPLR(T), which carried almost exclusively fucosylated glycans. This miscleavage has been associated with core fucosylation of the glycan at N71.25
On IgG-Fc, mainly diantennary fucosylated glycans were observed, differing in the presence of terminal galactoses and sialylation, with optional bisection, which is in line with previous reports.26 The different allotypes of IgG3 result in the potential occurrence of three different amino acid sequences for the IgG3 glycopeptides: EEQYNSTFR, EEQFNSTFR, and TKPWEEQYNSTFR (Table 1).35 Based on the tandem MS data of the sample pool from a subset of the studied population, no indications were found for the presence of IgG3 allotypes that would result in the EEQYNSTFR sequence. Therefore, the tryptic glycopeptides derived from IgG2 and IgG3 Fc are assumed to be identical in our data and were here reported as a combined molecular species (IgG2/3).35,36 Of note, an overlap between IgG3 and IgG4 glycoforms in individual samples cannot be excluded. For IgG1, IgG2/3 and IgG4, 17, 11, and 14 different glycopeptides were detected, respectively (SI Table S4).
Similarly to the conserved IgG-Fc N-glycosylation site, both IgA subclasses contain a conserved N-glycosylation site in the CH2 domain of the antibody (N144 on IgA1 and N131 on IgA2; LSL cluster). In addition, IgA2 contains a glycosylation site at N205 (TPL cluster). Notably, whereas IgA2 TPL glycopeptides and IgG CH2 glycopeptides were mostly fucosylated, IgA1/2 LSL glycopeptides lacked detectable levels of fucosylation. However, a positive correlation was observed for a number of structures from the LSL and IgG clusters. Interestingly, the agalactosylated IgA glycoform H3N5F0S0 correlated positively with the most abundant agalactosylated glycoforms of IgG1, such as H3N3F1S0, H3N4F1S0 and H3N5F1S0 (all with p-values <1.6 × 10–3; Figure 3A), as did the monogalactosylated glycoforms of IgA (SI Figure S3). Accordingly, the fully galactosylated, fucosylated diantennary glycans on IgG1 correlated with their afucosylated counterparts in IgA (SI Figure S3).
Figure 3.

Selected IgG and IgA glycopeptide relative intensities and their association with age. Correlation between the relative intensities of IgG1 H3N5F1S0 and IgA1/2 N144/N131 H3N5F0S0 with Spearman correlation coefficient r and corresponding p-value (A); N-glycopeptide relative intensities from IgG1 H3N5F1S0 (B), IgA1/2 N144/N131 H3N5F0S0 (C), IgA2 N205 H4N5F1S1 (D), IgA1/2 N340/N327 H4N5F1S0 (E), and O-glycopeptides of IgA1 H3N4F0S4 (F) plotted vs age. Linear regression lines are depicted, which all had a p-value ≤1.64 × 10–4 (see SI Table S6). Color code for data points and regression lines: pink: female, blue: male, black and purple: both sexes combined. Nomenclature for glycan compositions: H = hexose, N = N-acetylhexosamine, F = fucose, and S = sialic acid.
The application of the combined IgG and IgA glycopeptide profiling confirmed the expected IgG galactosylation decrease with increasing age (SI Table S6).8,26 For example, the agalactosylated structures on IgG1 showed a positive association with age (e.g., H3N5F1S0: beta = 4.5, p-value = 1.2 × 10–9; Figure 3B, SI Table S6), while the opposite was observed for the fully galactosylated variants (e.g., H5N4F1S0: beta = −4.8, p-value = 9.4 × 10–11; SI Table S6). Similarly, the non- and monogalactosylated glycoforms on N-glycosylation site N144 on IgA1 and N131 on IgA2 in the CH2 domain, showed a positive association with age (e.g., H3N5F0S0: beta = 4.0, p-value = 1.9 × 10–7; SI Table S6, Figure 3C), while digalactosylated glycoforms were negatively associated with age (e.g., H5N4F0S2: beta = −3.5, p-value = 5.24 × 10–6; SI Table S6). Also for the additional CH2 domain glycosylation site on IgA2 (N205) a negative association was found between galactosylation and age (e.g., the only incompletely galactosylated glycoform H4N5F1S1: beta = 4.1, p-value = 3.6 × 10–8; Figure 3D, SI Table S6). Finally, we observed a similar association pattern at the tailpiece glycosylation site, i.e. a positive association with age for the monogalactosylated glycopeptides LAGy H4N5F1S0 (Figure 3E) and a negative association for the digalactosylated LAGy H5N4F1S2 glycopeptides (SI Table S6). Similarities in the processing of the IgG-Fc glycosylation between the different IgG subclasses were described before, which is most probably influenced by the comparable quaternary structures of the antibody molecules.41 Our data on the correlation between IgG and IgA CH2 domain glycosylation, together with the similar age associations of the two different antibody isotypes, suggest that comparable galactosylation mechanisms play a role in the biosynthesis of IgG and IgA CH2-domain glycans.
Finally, three of the detected O-glycopeptides associated with age in the studied population. H3N6S3 and H3N4S4 showed a negative association (beta = −3.1 and −3.3, respectively, p-values <7.03 × 10–5; Figure 3F, SI Table S6), while H4N4S2 was positively associated with age (beta = 3.1, p-value = 6.9 × 10–5; SI Table S6). Negative trends with age were observed for other compositions where the number of hexoses was lower than the number of N-acetylhexosamines, suggesting a higher galactosylation per O-glycan at higher age (SI Table S6). Alternative methods, for example using O-glycan proteases, may be used to further assess IgA1 site-specific O-glycosylation.42 This will likely provide a higher resolution of the associations of O-glycosylation with biological variables. Previous studies analyzing tryptic IgA1 O-glycopeptides reported higher levels of galactosylation during pregnancy (third trimester) and after delivery, as compared to the early stage of pregnancy.29 Furthermore, a higher overall sialylation was found in patients with rheumatoid arthritis compared to healthy controls.43 To our knowledge, this is the first report on associations of serum IgA1 O-glycosylation with age.
Conclusions
A high-throughput method for site-specific, simultaneous glycosylation analysis of IgG and IgA was developed and applied on a medium-sized set of serum samples from healthy individuals. We report on subclass-specific glycosylation profiles including all major glycan types and different structural features. Despite the challenge of a large dynamic range of IgG and IgA glycopeptide abundances, we were able to cover all common, expected glycosylation sites. This was achieved by adjusting the amounts of capturing beads which resulted in an efficient IgA capturing combined with a partial IgG capturing. We think that setting up capturing methods to adjust and nivellate protein amounts is a promising approach for multiplexed in-depth glycosylation profiling or, more generally, protein attribute monitoring. Furthermore, the combined capture of multiple proteins from the same sample saves precious patient material and allows faster data evaluation.
Our method can be applied in large clinical studies for biomarker development in various disease contexts. IgG and IgA are among the most abundant serum glycoproteins with central roles in the human immune system. The biomarker potential of IgG glycosylation profiling has been demonstrated in numerous publications during the past decade. In addition, the biological relevance of IgA glycosylation is becoming more apparent. In particular, inflammatory pathologies with mucosal involvement, such as inflammatory bowel diseases or colorectal cancer, may be attractive targets to study IgG and IgA glycosylation as well as their interaction. Furthermore, the current approach paves the way for the combined high-throughput glycomics of multiple serum glycoproteins.
Acknowledgments
A.M. was supported by the European Union (Marie Curie European Training Network), GlyCoCan project, grant number 676421. D.F. was funded by The Netherlands Organisation for Scientific Research (NWO; Vernieuwingsimpuls Veni Project No. 722.016.008). P.A.v.V. received financial support by an Investment Grant NWO Medium (project number 91116004), which is (partially) financed by ZonMw.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b05722.
Author Present Address
§ (A.B.) Biomolecular Mass Spectrometry and Proteomics, Universiteit Utrecht, Padualaan 8, 3584CH Utrecht, The Netherlands.
Author Present Address
∥ (L.A.d.N.) Netherlands Forensic Institute, Division of Chemical and Physical Traces, P.O. Box 24044, 2490 AA The Hague, The Netherlands.
Author Contributions
The manuscript was written through contributions of all authors. A.M., N.H., A.H., C.K., L.N., A.B., and A.R. optimized the method and collected the experimental data. A.M., N.H., D.F., A.R., and V.D. processed the data, which was further analyzed by A.M., N.H., M.W., and V.D. W.M. and R.T. collected clinical samples and were responsible for the ethical approval. A.M., N.H., and V.D. wrote the manuscript, which was critically revised and approved by all authors. P.V., M.W., and V.D. supervised the study. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Delacroix D. L.; Dive C.; Rambaud J. C.; Vaerman J. P. Immunology 1982, 47 (2), 383–5. [PMC free article] [PubMed] [Google Scholar]
- Plebani A.; Ugazio A. G.; Avanzini M. A.; Massimi P.; Zonta L.; Monafo V.; Burgio G. R. Eur. J. Pediatr. 1989, 149 (3), 164–7. 10.1007/BF01958271. [DOI] [PubMed] [Google Scholar]
- Brandtzaeg P.J. Oral Microbiol. 2013, 5.20401. 10.3402/jom.v5i0.20401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers G.; Treffers L.; Plomp R.; Bentlage A. E. H.; de Boer M.; Koeleman C. A. M.; Lissenberg-Thunnissen S. N.; Visser R.; Brouwer M.; Mok J. Y.; Matlung H.; van den Berg T. K.; van Esch W. J. E.; Kuijpers T. W.; Wouters D.; Rispens T.; Wuhrer M.; Vidarsson G. Front. Immunol. 2017, 8, 877. 10.3389/fimmu.2017.00877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke B.; Keller C. W.; Weber P.; Quast I.; Lunemann J. D. Front. Immunol. 2017, 8, 646. 10.3389/fimmu.2017.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkawa T.; Nakamura K.; Yamane N.; Shoji-Hosaka E.; Kanda Y.; Sakurada M.; Uchida K.; Anazawa H.; Satoh M.; Yamasaki M.; Hanai N.; Shitara K. J. Biol. Chem. 2003, 278 (5), 3466–73. 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- Varki A. Glycobiology 2017, 27 (1), 3–49. 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudelj I.; Lauc G.; Pezer M. Cell. Immunol. 2018, 333, 65–79. 10.1016/j.cellimm.2018.07.009. [DOI] [PubMed] [Google Scholar]
- Chandler K. B.; Mehta N.; Leon D. R.; Suscovich T. J.; Alter G.; Costello C. E. Mol. Cell. Proteomics 2019, 18 (4), 686–703. 10.1074/mcp.RA118.001185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavenhagen K.; Plomp R.; Wuhrer M. Anal. Chem. 2015, 87 (23), 11691–9. 10.1021/acs.analchem.5b02366. [DOI] [PubMed] [Google Scholar]
- Plomp R.; Dekkers G.; Rombouts Y.; Visser R.; Koeleman C. A.; Kammeijer G. S.; Jansen B. C.; Rispens T.; Hensbergen P. J.; Vidarsson G.; Wuhrer M. Mol. Cell. Proteomics 2015, 14 (5), 1373–84. 10.1074/mcp.M114.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M. P.; Giudicelli V.; Ginestoux C.; Jabado-Michaloud J.; Folch G.; Bellahcene F.; Wu Y.; Gemrot E.; Brochet X.; Lane J.; Regnier L.; Ehrenmann F.; Lefranc G.; Duroux P. Nucleic Acids Res. 2009, 37 (Database issue), D1006–12. 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welinder C.; Baldetorp B.; Blixt O.; Grabau D.; Jansson B. PLoS One 2013, 8 (4), e61749. 10.1371/journal.pone.0061749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax-Browne H. J.; Robertson C.; Antonopoulos A.; Leathem A. J. C.; Haslam S. M.; Dell A.; Dwek M. V. Interface Focus 2019, 9 (2), 20180079. 10.1098/rsfs.2018.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomana M.; Novak J.; Julian B. A.; Matousovic K.; Konecny K.; Mestecky J. J. Clin. Invest. 1999, 104 (1), 73–81. 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papista C.; Berthelot L.; Monteiro R. C. Cell. Mol. Immunol. 2011, 8 (2), 126–34. 10.1038/cmi.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp R.; Ruhaak L. R.; Uh H. W.; Reiding K. R.; Selman M.; Houwing-Duistermaat J. J.; Slagboom P. E.; Beekman M.; Wuhrer M. Sci. Rep. 2017, 7 (1), 12325. 10.1038/s41598-017-12495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simurina M.; de Haan N.; Vuckovic F.; Kennedy N. A.; Stambuk J.; Falck D.; Trbojevic-Akmacic I.; Clerc F.; Razdorov G.; Khon A.; Latiano A.; D’Inca R.; Danese S.; Targan S.; Landers C.; Dubinsky M.; McGovern D. P. B.; Annese V.; Wuhrer M.; Lauc G. Gastroenterology 2018, 154 (5), 1320–1333. 10.1053/j.gastro.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; e10
- Almeida A.; Kolarich D. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860 (8), 1583–95. 10.1016/j.bbagen.2016.03.012. [DOI] [PubMed] [Google Scholar]
- de Haan N.; Falck D.; Wuhrer M.. Glycobiology 2019. 10.1093/glycob/cwz048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman M. H.; Derks R. J.; Bondt A.; Palmblad M.; Schoenmaker B.; Koeleman C. A.; van de Geijn F. E.; Dolhain R. J.; Deelder A. M.; Wuhrer M. J. Proteomics 2012, 75 (4), 1318–29. 10.1016/j.jprot.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Plomp R.; Hensbergen P. J.; Rombouts Y.; Zauner G.; Dragan I.; Koeleman C. A.; Deelder A. M.; Wuhrer M. J. Proteome Res. 2014, 13 (2), 536–46. 10.1021/pr400714w. [DOI] [PubMed] [Google Scholar]
- Jansen B. C.; Falck D.; de Haan N.; Hipgrave Ederveen A. L.; Razdorov G.; Lauc G.; Wuhrer M. J. Proteome Res. 2016, 15 (7), 2198–210. 10.1021/acs.jproteome.6b00171. [DOI] [PubMed] [Google Scholar]
- Hong Q.; Ruhaak L. R.; Stroble C.; Parker E.; Huang J.; Maverakis E.; Lebrilla C. B. J. Proteome Res. 2015, 14 (12), 5179–92. 10.1021/acs.jproteome.5b00756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp R.; de Haan N.; Bondt A.; Murli J.; Dotz V.; Wuhrer M. Front. Immunol. 2018, 9, 2436. 10.3389/fimmu.2018.02436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakovic M. P.; Selman M. H.; Hoffmann M.; Rudan I.; Campbell H.; Deelder A. M.; Lauc G.; Wuhrer M. J. Proteome Res. 2013, 12 (2), 821–31. 10.1021/pr300887z. [DOI] [PubMed] [Google Scholar]
- de Vroome S. W.; Holst S.; Girondo M. R.; van der Burgt Y. E. M.; Mesker W. E.; Tollenaar R.; Wuhrer M. Oncotarget 2018, 9 (55), 30610–30623. 10.18632/oncotarget.25753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck D.; Jansen B. C.; de Haan N.; Wuhrer M. Methods Mol. Biol. 2017, 1503, 31–47. 10.1007/978-1-4939-6493-2_4. [DOI] [PubMed] [Google Scholar]
- Bondt A.; Nicolardi S.; Jansen B. C.; Stavenhagen K.; Blank D.; Kammeijer G. S.; Kozak R. P.; Fernandes D. L.; Hensbergen P. J.; Hazes J. M.; van der Burgt Y. E.; Dolhain R. J.; Wuhrer M. Sci. Rep. 2016, 6, 27955. 10.1038/srep27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Guerrero A.; Parker E.; Strum J. S.; Smilowitz J. T.; German J. B.; Lebrilla C. B. J. Proteome Res. 2015, 14 (3), 1335–49. 10.1021/pr500826q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H., In ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, 2016. [Google Scholar]
- Leek J. T.; Johnson W. E.; Parker H. S.; Fertig E. J.; Jaffe A. E.; Storey J. D.; Zhang Y.; Torres L. C., In sva: Surrogate Variable Analysis. R package version 3.34.0., 2019.
- Kulak N. A.; Pichler G.; Paron I.; Nagaraj N.; Mann M. Nat. Methods 2014, 11 (3), 319–24. 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- UniProt C. Nucleic Acids Res. 2019, 47 (D1), D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidarsson G.; Dekkers G.; Rispens T. Front. Immunol. 2014, 5, 520. 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbín M.; Grubb A.; de Lange G. G.; Grubb R. Immunogenetics 1994, 39 (3), 187–193. 10.1007/BF00241259. [DOI] [PubMed] [Google Scholar]
- Deshpande N.; Jensen P. H.; Packer N. H.; Kolarich D. J. Proteome Res. 2010, 9 (2), 1063–75. 10.1021/pr900956x. [DOI] [PubMed] [Google Scholar]
- Klapoetke S. C.; Zhang J.; Becht S. J. Pharm. Biomed. Anal. 2011, 56 (3), 513–20. 10.1016/j.jpba.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Yang H.; Zubarev R. A. Electrophoresis 2010, 31 (11), 1764–72. 10.1002/elps.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak R. P.; Royle L.; Gardner R. A.; Bondt A.; Fernandes D. L.; Wuhrer M. Anal. Biochem. 2014, 453, 29–37. 10.1016/j.ab.2014.02.030. [DOI] [PubMed] [Google Scholar]
- Sonneveld M. E.; Koeleman C. A. M.; Plomp H. R.; Wuhrer M.; van der Schoot C. E.; Vidarsson G. Front. Immunol. 2018, 9, 129. 10.3389/fimmu.2018.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Onigman P.; Wu W. W.; Sjogren J.; Nyhlen H.; Shen R. F.; Cipollo J. Anal. Chem. 2018, 90 (13), 8261–8269. 10.1021/acs.analchem.8b01834. [DOI] [PubMed] [Google Scholar]
- Bondt A.; Nicolardi S.; Jansen B. C.; Kuijper T. M.; Hazes J. M. W.; van der Burgt Y. E. M.; Wuhrer M.; Dolhain R. Arthritis Res. Ther. 2017, 19 (1), 160. 10.1186/s13075-017-1367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.