

SUMMARY
Disease-causing mutations in many neurodegenerative disorders lead to proteinopathies that trigger endoplasmic reticulum (ER) stress. However, few therapeutic options exist for patients suffering from these diseases. Using an in vitro screening platform to identify compounds that protect human motor neurons from ER-stress-mediated degeneration, we discovered that compounds targeting the mitogen-activated protein kinase kinase kinase kinase (MAP4K) family are neuroprotective. The kinase inhibitor URMC-099 (compound 1) stood out as a promising lead compound for further optimization. We coupled structure-based compound design with functional activity testing in neurons subjected to ER stress to develop a series of analogs with improved MAP4K inhibition and concomitant increases in potency and efficacy. Further structural modifications were performed to enhance the pharmacokinetic profiles of the compound 1 derivatives. Prostetin/12k emerged as an exceptionally potent, metabolically stable, and blood-brain-barrier (BBB)-penetrant compound that is well-suited for future testing in animal models of neurodegeneration.
Graphical Abstract

INTRODUCTION
Many neurodegenerative disorders, including Amyotrophic Lateral Sclerosis (ALS), Alzheimer’s disease, and Parkinson’s disease, are characterized by the accumulation of misfolded proteins in the affected neuronal subtypes and the subsequent induction of ER stress pathways (Roussel et al., 2013). These pathways initially engage protective mechanisms that slow protein synthesis and promote re-folding or clearance of misfolded proteins. If these efforts are unsuccessful, the unfolded protein response (UPR) activates cell death pathways.
ER stress and the UPR have been implicated in both familial and sporadic forms of ALS, a neurodegenerative disorder that selectively targets motor neurons and progresses rapidly once patients have been diagnosed, with a mean survival time of <5 years (Medinas et al., 2019). Markers of ER stress also are amongst the earliest pathological features to appear in in vitro and in vivo models of ALS (Kiskinis et al., 2014; Saxena et al., 2009). Furthermore, while the expression of ALS-causing mutant proteins is not restricted to motor neurons, we have found that motor neurons are significantly more sensitive than other spinal neuronal subtypes to ER stress-inducing compounds (Thams et al., 2019), which may explain their selective vulnerability in ALS.
We harnessed these findings to build a screening platform to identify compounds that could protect against ER-stress-mediated neurodegeneration. In this model, stem cell-derived motor neurons are treated with cyclopiazonic acid (CPA), a mycotoxin that inhibits the transport of calcium from the cytoplasm into the ER by blocking sarcoendoplasmic reticulum calcium transport ATPase (SERCA) pumps (Thams et al., 2019). Because many protein folding chaperones in the ER require calcium as a co-factor, CPA treatment leads to the accumulation of misfolded proteins, the induction of ER stress pathways, and apoptosis in spinal motor neurons in vitro (Thams et al., 2019).
Using this platform, we identified broad-spectrum kinase inhibitors, including Gö6976, sunitinib, K252a, and kenpaullone, as compounds that protect motor neurons from ER stress (Thams et al., 2019). These compounds were largely unsuitable for clinical development, either because of poor pharmacokinetic properties or a lack of target specificity.
Here, we identified MAP4Ks as the shared functional targets of these kinase inhibitors and performed a secondary screen for MAP4K inhibitors that could be tested in in vivo models of neurodegeneration. URMC-099 (compound 1), a MAP3K inhibitor that also strongly inhibits MAP4Ks (Goodfellow et al., 2013), emerged as a promising lead, but required further optimization to increase its potency and efficacy in motor neurons subjected to ER stress, and to overcome its relatively poor metabolic stability, oral bioavailability, and blood-brain barrier penetration.
By combining structure-based small molecule design using computational chemistry tools with rapid feedback on the activity of newly-synthesized compounds in vitro in motor neurons subjected to ER stress, we generated a series of potent compound 1 analogs. Compound 12k (termed Prostetin for promoting neuronal viability via Ste20 inhibition), which inhibits MAP4Ks at sub-nanomolar concentrations, was the most promising based on its neuroprotective and pharmacokinetic profiles. Furthermore, we found that Prostetin/12k retains the anti-inflammatory properties of 1, which may provide additional therapeutic benefit in neurodegenerative disorders such as ALS where inflammation may exacerbate disease progression (Boillée et al., 2006; Trias et al., 2016). Prostetin/12k is therefore primed for future testing in animal models of neurodegenerative disease.
RESULTS
HGK (MAP4K4) and NUAK1 are common targets of neuroprotective kinase inhibitors
In our original screen for compounds that protect against ER-stress-mediated neurodegeneration, libraries of small molecules were combined with the ER-stress-inducing agent CPA and added to mouse stem cell-derived motor neurons (Fig. 1A) (Thams et al., 2019). Four kinase inhibitors emerged as neuroprotective hits – Gö6976, sunitinib, K252a, and kenpaullone – but none were viable therapeutic options: Gö6976 and K252a are insufficiently selective (Anastassiadis et al., 2011); sunitinib does not readily cross the blood-brain barrier (Tang et al., 2012); and - paullone derivatives are highly insoluble in aqueous biofluids (Greenwald et al., 2004).
Figure 1. HGK inhibitors are neuroprotective and act through the JNK pathway.

(A) Schematic of motor neuron CPA survival assay. (B) Hierarchical clustering of the kinase targets of neuroprotective hits from previous survival screens. HGK and NUAK1 are shared targets of these compounds. Red indicates that a given kinase is strongly inhibited by a hit compound (0–20% activity remaining); blue indicates that a kinase is poorly inhibited (>20% activity remaining). (C) Maximum efficacy of compounds that rescue motor neurons from ER stress-induced neurodegeneration (expressed as the maximum motor neuron survival recorded across all concentrations tested, % untreated controls). Hits from the initial screen are in blue; HGK and NUAK1 inhibitors are in green and maroon, respectively. Compounds were tested in motor neurons derived from the stem cells of a healthy patient. Bars represent means ± SEM of wild-type motor neurons treated with CPA + test compound normalized to vehicle controls. n = 3 replicate wells from a 96-well plate. See also Fig. S1. (D) Western blots of motor neuron cultures treated with vehicle, CPA, or CPA + URMC-099. n = 2 separate motor neuron differentiations. (E) Quantification of western blots. Band intensities for all phospho-isoforms were normalized to their respective total protein, and are represented as a percentage of CPA band intensities. Bars represent means ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001.
Nevertheless, we reasoned that identifying their shared functional targets could guide the selection of new compounds that would be suitable for in vivo use. We mined a publicly available database (Anastassiadis et al., 2011) to compare the catalytic activity of >300 kinases in the presence of the four hit compounds. We also included alsterpaullone in this analysis, as it is a structural analog of kenpaullone that we found to be more potent than kenpaullone in rescuing CPA-treated motor neurons (Fig. 1C). We then performed two-way hierarchical clustering analysis to rank the shared targets of these compounds (Fig. 1B). Kinases whose activity was strongly inhibited by all five compounds included the AMPK-related kinase NUAK1 and the MAP4K/STE20 kinase HGK (MAP4K4; Fig. 1B).
HGK inhibitors, but not NUAK1 inhibitors, are strongly neuroprotective
To evaluate the relative contributions of NUAK1 and HGK to ER stress-mediated neurodegeneration, and to identify compounds that would be better-suited than the original hits for in vivo administration, we asked whether known NUAK1 and HGK inhibitors could counteract the effects of the ER stress-inducing compound CPA. We humanized our screening platform (Fig. 1A) by incorporating motor neurons differentiated from human induced pluripotent stem cells derived from a healthy patient. Cells were treated with CPA ± NUAK1 and HGK inhibitors under conditions where motor neuron survival in CPA-treated cells drops to ~40% compared to vehicle-treated controls.
We found that the NUAK1 inhibitors WZ4003 (Banerjee et al., 2014) and HTH 01–015 (Banerjee et al., 2014) did not rescue CPA toxicity at any of the concentrations tested (Fig. 1C and Fig. S1A). At concentrations above 2 μM, both WZ4003 and HTH 01–015 were toxic to motor neurons even in the absence of CPA (Fig. S1A). NUAK1 inhibitors were therefore not suitable for the treatment of ER stress-mediated neurodegeneration.
We also tested three compounds that had previously been shown to inhibit HGK activity: PF-6260933 (Ammirati et al., 2015), GNE-495 (Ndubaku et al., 2015), and URMC-099 (Goodfellow et al., 2013). All three of the HGK inhibitors tested were neuroprotective, with URMC-099 as the most potent and effective in this assay, restoring survival to 90% at 0.6 μM (Fig. 1C and Fig. S1B). These findings pointed to a role for HGK in ER-stress-mediated neurodegeneration, and suggested that URMC-099 warranted further study.
URMC-099 inhibits ER stress-mediated apoptosis via the JNK pathway
Unlike PF-6260933 and GNE-495, URMC-099 is not a selective HGK inhibitor, and its mode of action in stressed neurons was not established. Using published data (Ndubaku et al., 2015), we calculated that GNE-495 was exceptionally selective, with a selectivity score of 0.01 out of 1 (ratio of the number of kinases with less than 20% of activity remaining in the presence of 1 μM GNE-495 to the total number of kinases assayed in cell-free conditions). Meanwhile, URMC-099 was originally optimized to inhibit the MAP3K kinase MLK3 but was shown to bind HGK and inhibit >99% of its activity at 1 μM (Goodfellow et al., 2013). Accordingly, URMC-099 had a weaker selectivity score (0.36) than GNE-495 under similar conditions, based on data from Goodfellow et al. (2013).
To assess whether URMC-099 was acting through the same pathways as HGK-selective compounds in stressed neurons, we evaluated its effects on the c-Jun N-terminal kinase (JNK) signaling pathway. HGK is a well-established regulator of the JNK pathway (Yao et al., 1999), and selective HGK inhibitors have been shown to inhibit JNK pathway activation in the context of neurodegeneration. Yang et al. (2013) showed that kenpaullone, one of the hit compounds from our original screen that strongly and selectively inhibits HGK, prevents motor neuron degeneration in the context of neurotrophic factor withdrawal by inhibiting the TAK1 (MAP3K7) / MKK7 / JNK axis of the JNK pathway. Meanwhile, URMC-099 has been shown to prevent MLK3-mediated activation of JNK in microglia in in vitro and in vivo models of neuroinflammation ((Goodfellow et al., 2013).
Through a series of western blots, we observed that URMC-099 prevented the activation of the JNK pathway in CPA-treated motor neurons. We found that URMC-099 attenuated the phosphorylation of the MAP2K MKK4 at Ser257, a site that is associated with cell stress and is essential to the regulation of downstream kinases such as JNK1–3 (MAPK8–10; Fig. 1D and E) (Deacon and Blank, 1997). Using an antibody that recognizes JNK1, 2, and 3 when phosphorylated at sites known to regulate stress responses (Thr183/Tyr185), we found that URMC-099 significantly decreased JNK activation compared to CPA treatment alone (Fig. 1D and E). Neither CPA nor URMC-099 treatment influenced basal levels of MKK4 or JNK expression (Fig. 1D), indicating that CPA and URMC-099 were acting in a post-translational, kinase-dependent manner to activate or suppress phosphorylation, respectively.
JNKs phosphorylate and activate transcription factors such as c-Jun to regulate the transcription of apoptosis-related genes (Pulverer et al., 1991), and we have shown that CPA treatment induces rapid c-Jun phosphorylation at Ser63 in motor neurons (Thams et al., 2019). Interestingly, URMC-099 treatment not only suppressed c-jun phosphorylation (Fig. 1E), but also decreased c-Jun expression (Fig. 1D), perhaps because activated c-Jun perpetuates its own expression under conditions of stress (Hayakawa et al., 2005).
Finally, we investigated the effects of URMC-099 on the cleavage and activation of caspase 3, a critical step in apoptotic cell death pathways (Robertson et al., 2000). We have previously shown that CPA treatment induces caspase 3 cleavage in motor neurons (Thams et al., 2019), and we found here that URMC-099 attenuated CPA-mediated caspase 3 cleavage (Fig. 1D and E), suggesting that it acts on apoptotic pathways to promote motor neuron survival. Taken together, these findings suggest that the neuroprotective effects of URMC-099 stem from the inhibition of JNK-mediated apoptotic pathways, and are in line with previous studies that have linked HGK inhibitors with JNK pathway attenuation, though they do not exclude the involvement of MLK3, a MAP3K that can directly phosphorylate MKK4 (Deacon and Blank, 1997).
Structure-based compound design yields potent and efficacious derivatives of URMC-099
Considering that URMC-099 (compound 1) was originally optimized for MLK3 inhibition, but that HGK inhibition was the likely driving force behind the neuroprotective effects of the kinase inhibitors identified in our previous screen (Fig. 1B), we speculated that more potent and efficacious derivatives might be obtained by optimizing their capacity to inhibit HGK. Furthermore, we reasoned that increasing the potency of compound 1 analogs could lower their effective dose and thereby mitigate the potential for off-target effects in vivo given the low selectivity score calculated for compound 1.
To aid in the design of analogs, we docked compound 1 in a crystal structure of HGK (PDB ID: 5DI1) using Glide (Schrödinger Maestro Suite 2017–1) (Fig. 2A). From the docking pose and the binding site interaction (Fig. 2B), it appeared that: (1) the 7-azaindole moiety binds to the hinge region of the kinase and is essential for activity, (2) the piperazine moiety extends into the solvent-exposed region and could be modified to improve the stability and the physicochemical properties of the molecule, and (3) the indole sidechain could be modified to optimize interactions in the binding pocket and increase kinase specificity.
Figure 2. Docking pose of compound 1 in HGK and summary of analog properties.
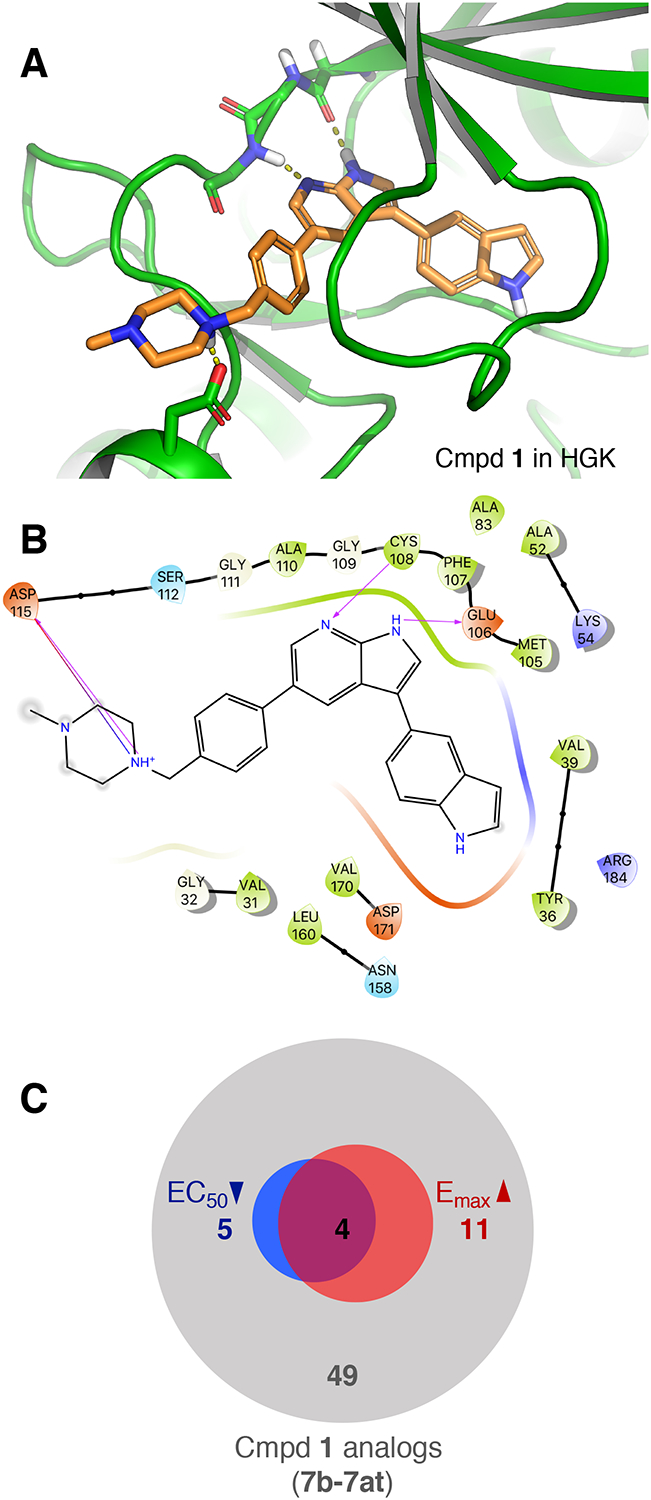
(A) Docking pose of compound 1 in a crystal structure of HGK. Dashed lines indicate hydrogen bonds with the hinge region (Cys108, Glu106) and Asp115. (B) Ligand interaction diagram of compound 1 with HGK corresponding to the docking pose shown in Fig 2A (arrows indicating hydrogen bonds). (C) Summary of improvements in efficacy (red) and potency (blue) in the CPA survival assay amongst the first 49 compound 1 analogs that were generated (7b – 7at).
Based on these observations, we set out to generate a series of compound 1 analogs that had a high docking score in HGK and at the same time represented a diverse set of structures. However, the published synthetic route was not easily scalable: the sidechain (Scheme 1, R-group) is introduced in the first step, and has to be carried through the entire synthesis, which is not efficient for the generation of sidechain analogs (Goodfellow et al., 2013). We therefore developed an improved synthetic route with increased flexibility (Scheme 1). In the first step, a Suzuki coupling of 5-bromo-7-azaindole (2) with 4-formylphenylboronic acid in a pressure tube provided aldehyde 3. Reductive amination with 1-methylpiperazine at room temperature in dichloromethane yielded compound 4. In the next two steps, compound 4 was iodinated at the 3-position to form compound 5, followed by the introduction of a Boc-group on the azaindole to yield compound 6. Initial attempts to forego this protecting group led to significantly decreased yields in the subsequent Suzuki coupling. Compound 6 was used as the substrate for the final Suzuki coupling with a number of boronic acids and boronic pinacol esters. Conveniently, the Boc-group was deprotected under these reaction conditions, obviating an additional deprotection step. Compound 1 and the desired analogs, 7a – 7az, were isolated in reasonable yields (40 – 88%) after purification by preparatory HPLC.
Scheme 1.
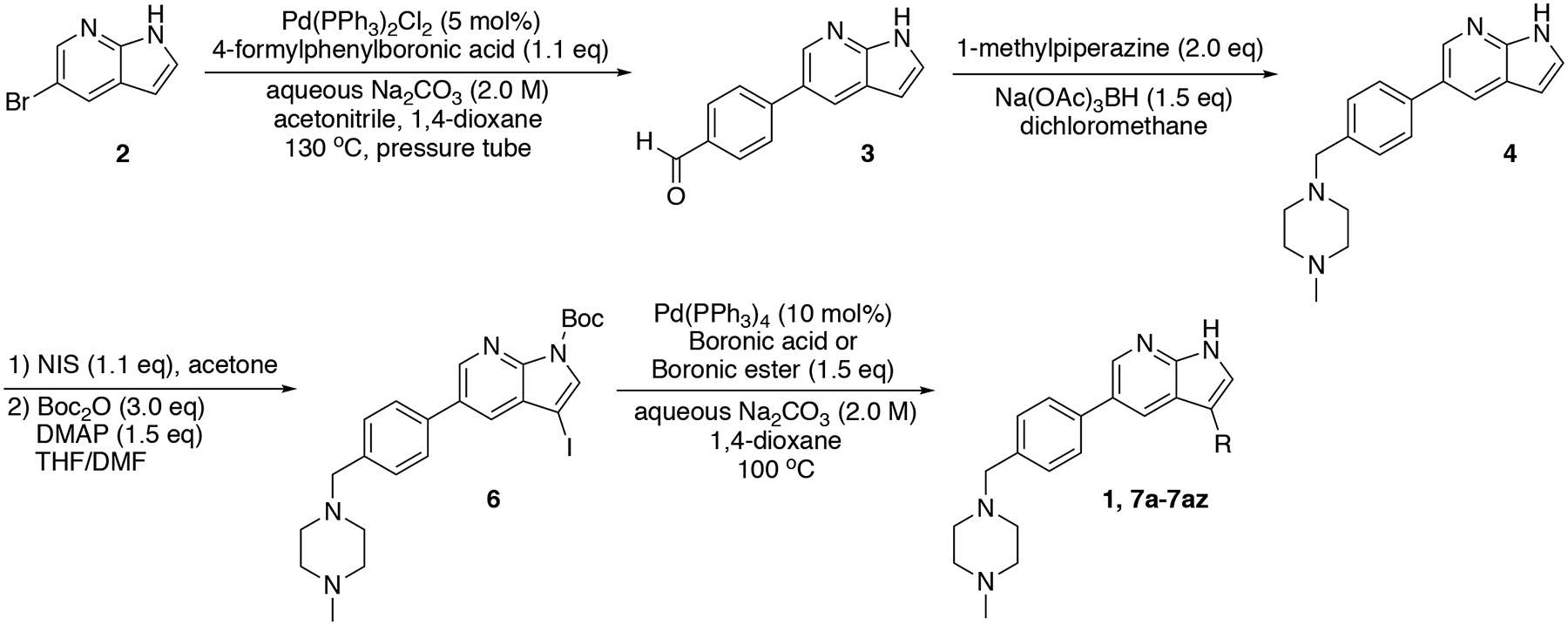
Flexible synthesis route for the synthesis of compound 1 and analogs 7a – 7az.
Once this synthetic route was established, we set out to generate a series of structurally diverse compound 1 analogs that met basic HGK docking criteria. We evaluated the analogs for their ability to rescue CPA-mediated toxicity in motor neurons on a rolling basis, providing us with real-time feedback on analog design and allowing us to circumvent structural modifications that decreased the efficacy of the compounds or were overtly toxic. Through this iterative process, we initially developed and tested 49 analogs of compound 1 (7b–7at; Fig. 2C and Table S1).
For these and all subsequent survival experiments, we used human stem-cell-derived motor neurons carrying the ALS-causing SOD1A4V mutation to maximize the translational potential of these analogs (Thams et al., 2019). Survival in this ALS line dropped to ~10% of untreated controls after CPA treatment (compared to ~40% in the healthy human motor neuron line used in Fig. 1C and Fig. S1), consistent with our previous findings that CPA exacerbates basal levels of ER stress in ALS motor neurons (Thams et al., 2019). Under these conditions, compound 1 achieved maximum efficacy at 1 μM and completely prevented motor neuron death (102% motor neuron survival compared to untreated controls; Table S1). Out of the 49 analogs, 11 analogs (7e, 7h, 7l, 7p, 7v, 7y, 7aa, 7ac, 7ak, 7al, and 7am) demonstrated greater maximum efficacy than compound 1 (Fig. 2C and Table S1), indicating that some analogs were also counteracting the basal levels of cell death that occurred across the 72 h assay independently of CPA. Furthermore, 5 analogs (7h, 7l, 7m, 7v, and 7aa) were more potent than compound 1 (EC50 < 0.22 μM), and 4 analogs (7h, 7l, 7v, and 7aa) were both more efficacious and more potent than compound 1 (Fig. 2C and Table S1). Only 2 analogs (7c and 7ay) were completely ineffective (survival ≤CPA alone; Tables S1 and S2).
HGK inhibition correlates strongly with motor neuron survival
The battery of compound 1 analogs generated and tested here offered a unique opportunity to investigate whether improved potency in the motor neuron survival assays corelated with increased inhibition of HGK or MLK3, given that compound 1 was originally optimized to inhibit MLK3. The neuroprotective effects of compound 1 and 53 analogs (7b–7at and 12b–12e, which are further described below) were assessed concurrently in CPA-treated human ALS motor neuron cultures (Table S2). Meanwhile, the same analogs were analyzed in a cell-free radiolabeled ATP assay (Reaction Biology Corp.) to compare the extent to which they could inhibit the activity of HGK, MLK3, or MLK1, a homolog of MLK3 that has also been shown to activate the JNK pathway (Gallo and Johnson, 2002) (Table S3 and S4).
We then used regression analysis to compare the ability of a given analog to inhibit HGK, MLK1, or MLK3 with its ability to rescue human ALS motor neurons in the CPA survival assay. Overall, we observed a significant correlation between the inhibition of MLK3 activity and neuroprotection (R2 = 0.52; p < 0.0001; Fig. 3A). We performed the same analysis for MLK1 and found the correlation to be weaker than that for MLK3 (R2 = 0.39; p < 0.0001; Fig. 3B). Meanwhile, we observed a more robust correlation between the inhibition of HGK activity and neuroprotection than for either MLK (R2 = 0.84; p < 0.0001; Fig. 3C). These findings point to HGK as an important biological target of compound 1 and its analogs. The fact that some of the analogs tested were highly neuroprotective despite their inability to inhibit MLK3 and MLK1 (e.g. compound 12e (Tables S1 and S3)) indicates that direct MLK inhibition is not a requirement for motor neuron protection.
Figure 3. Inhibition of HGK and related MAP4Ks correlates strongly with motor neuron rescue.
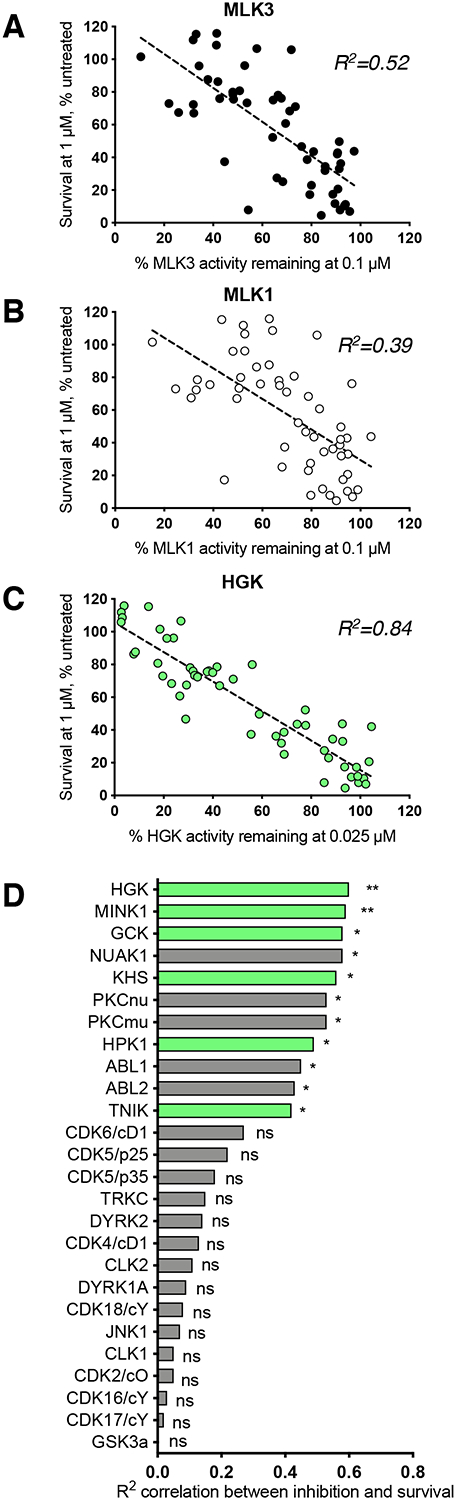
(A – C) Plots of the associations between motor neuron survival and the inhibition of MLK3, MLK1, or HGK for 53 compound 1 analogs (Table S2). Each data point represents one compound; dashed lines indicate linear regression. HGK inhibition correlates more strongly with motor neuron survival than MLK inhibition. (D) Ranked R2 values (from Pearson’s r) describing the correlation between kinase inhibition and motor neuron survival for ten compound 1 analogs across 60 different kinases. MAP4Ks (highlighted in green) are amongst the most highly-ranked kinases. *p < 0.05; **p < 0.01.
The inhibition of related MAP4Ks correlates with motor neuron survival
To further explore the biologically relevant targets of compound 1 and its analogs, we selected 10 analogs ranging from weakly to very strongly protective in CPA-treated motor neurons and evaluated their ability to inhibit a panel of 60 human kinases in the radiolabeled ATP assay (Table S5). The kinases in the expanded panel were selected from known targets of compound 1 (Goodfellow et al., 2013), known targets of other kinase inhibitors that were protective in the CPA survival assay (Thams et al., 2019), and relevant targets from the literature. RNAseq data were used to confirm the expression of each kinase in ES motor neurons (Thams et al., 2019).
We found that the inhibition of HGK, TNIK, MINK1 (MAP4K6), GCK (MAP4K2), and KHS (MAP4K5) all correlated strongly with motor neuron survival (Fig. 3D). These findings were also consistent with our analyses of the shared targets of the hit compounds from our preliminary screen (Fig. 1B), where HGK, MINK1, GCK, and KHS were all among the most highly ranked targets. (TNIK did not appear in our initial analysis of shared targets because its activity was not assessed in the existing publicly available database (Anastassiadis et al., 2011).) Because the catalytic domains of all MAP4K kinases are highly conserved (Read et al., 2019), it is possible that neuroprotection by compound 1 and its analogs hinges on the inhibition of several MAP4K family members simultaneously (Larhammar et al., 2017).
Compound 12k exhibits increased metabolic stability
Among the HGK inhibitors that we tested in CPA-treated motor neurons in vitro, compound 1 is unique in that it exhibits CNS penetrance in vivo without any overt toxicity. Both PF-6260933 and GNE-495 had been modified to prevent CNS penetrance because previous brain-penetrant iterations had been poorly tolerated in multi-dose assays in mice (Dow et al., 2018; Ndubaku et al., 2015). By contrast, compound 1 has previously been shown to be well-tolerated over long-term dosing regimens and to modulate CNS phenotypes in mice: in a study by Marker et al. (2013), compound 1 was administered intraperitoneally (IP) at 10 mg/kg twice a day for 28 days, and was found to attenuate Human Immunodeficiency Virus (HIV)-mediated microgliosis in the brain.
Though the study by Marker et al. (2013) highlights the safety and efficacy of compound 1 in vivo, it also sheds light on the limitations of testing this drug in mouse models of neurodegenerative disease. One of the major drawbacks of compound 1 is the fact that it must be delivered twice-daily to achieve sustained therapeutic levels in the CNS, both because of its low metabolic stability and because of its relatively limited brain exposure (Goodfellow et al., 2013). Dosing strategies involving multiple daily injections increase the risk of handling stress in treated animals (Ryabinin et al., 1999), which can, in turn, lead to the induction of MAPK pathways in the CNS (Liu et al., 2004) and may confound the effects of kinase inhibitors in the context of neurodegeneration. We therefore set out to enhance the metabolic stability of the potent and neuroprotective compound 1 analogs described above.
To evaluate the metabolic stability of compound 1 derivatives, we incubated them with mouse liver microsomes and analyzed samples collected over 2 h by LC-MS/MS. Under these conditions, compounds with a half-life of less than 30 min were considered to have a high clearance rate and were not suitable therapeutic candidates. In accordance with previous findings (Goodfellow et al., 2013), we found that compound 1 had a half-life of 15 min (Table S7). Initially, we selected 11 analogs for further analysis based on their structural variety and their activity profiles in the motor neuron survival assay. Only two of these analogs (7y and 7ad) exhibited a half-life ≥30 min (Table S7).
Next, we set out to improve the half-life of the analogs through further structural modification. Computational docking experiments indicated that the N-methylpiperazine moiety is solvent-exposed, suggesting that its modifications might alter pharmacokinetic properties of compounds, while maintaining their potency in the ER stress assay (Scheme 2A and 2B, 12a–12l).
Scheme 2.
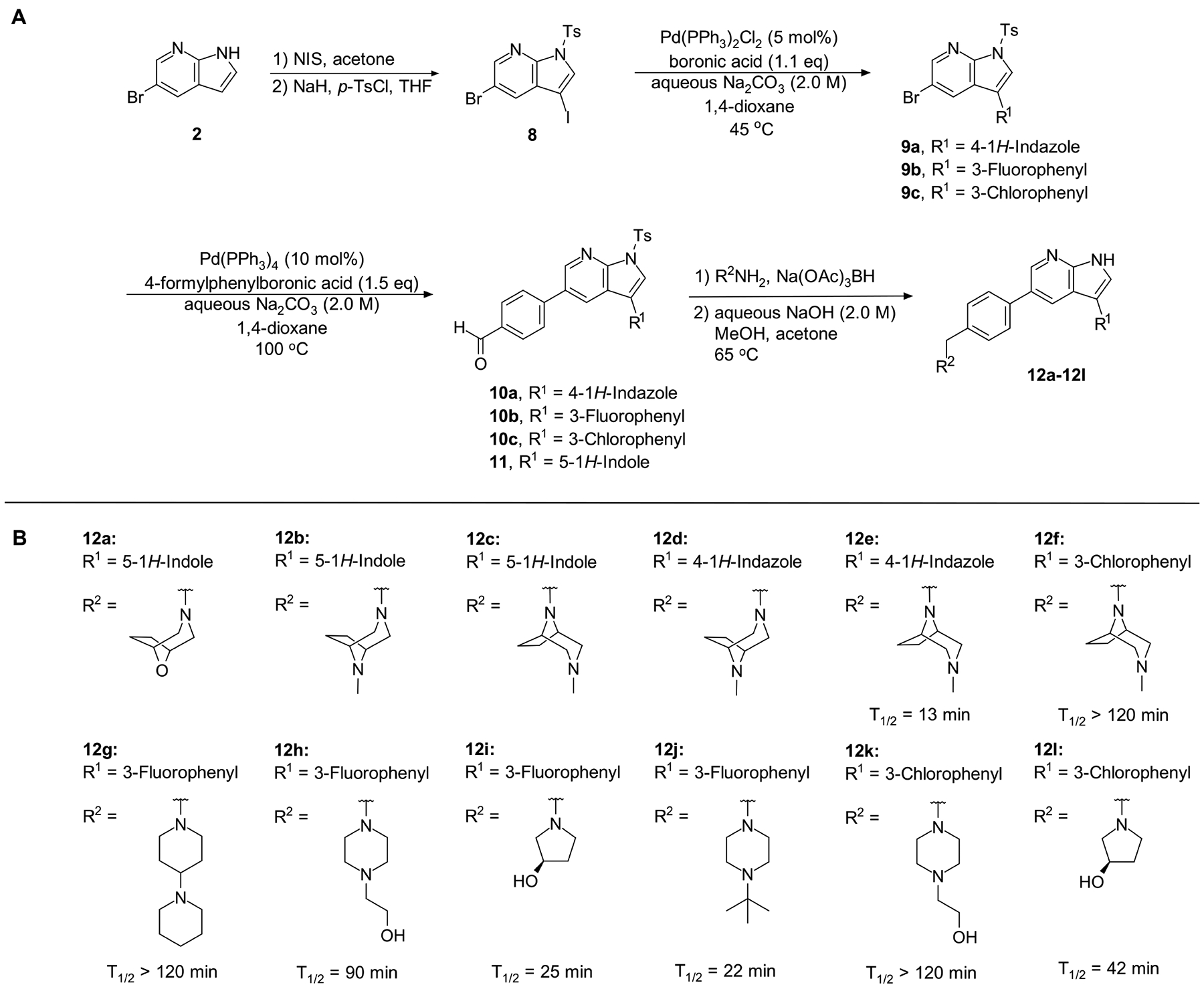
(A) Synthesis of compounds 12a – 12l; (B) Structures of analogs 12a – 12l and half-life in mouse liver microsomes (CD-1) of analogs 12e – 12l.
Introduction of a bicyclic N-methylpiperazine to one of the most potent analogs (Scheme 2) only slightly increased the half-life from 9 min to 13 min (compound 12e; Table S7). To further enhance the microsomal stability of these analogs, we introduced the same bicyclic piperazine to one of the analogs from the initial series that had a half-life of ≥30 min (compound 7ad; Table S7). In this case, we were able to considerably increase the half-life from 38 min to >120 min (compound 12f; Table S7).
Compound 12f was more potent than compound 1 in the motor neuron survival assay, with an EC50 of 0.11 μM (Table S1) but was toxic at concentrations above 0.3 μM. To mitigate this increase in toxicity, while maintaining potency and microsomal stability, we synthesized a new series of analogs (Scheme 2; 12g – 12l) containing the m-chlorophenyl or m-fluorophenyl substituent on the 3-position of the 7-azaindole scaffold. We evaluated the potency of these compounds in the ER stress assay, and their stability in the microsomal stability assay. Two of these compounds, 12g and 12j, either had a shorter half-life than desired or showed a decrease in potency in the ER stress assay (Tables S1 and S7). The remaining 4 compounds (12h, 12i, 12k, and 12l) completely blocked the effects of CPA treatment at concentrations <0.3 μM (Table S1). We also confirmed by western blot that they effectively inhibit the same JNK pathway components as compound 1 (Fig. S2). One of the resulting compounds, 12k (Fig. 4B), had a half-life of >120 min in the microsome assay (Fig. 4C) and rescued CPA-induced neurodegeneration with an improved EC50 of 0.10 μM (Table S1; Fig. 4D – H). We termed this compound Prostetin/12k (Fig. 4B).
Figure 4. In vitro and in vivo characterization of Prostetin/12k.
(A – B) Structures of compound 1 and Prostetin/12k. (C) In vitro microsomal stability curves for compound 1 and Prostetin/12k. Prostetin/12k has a markedly longer half-life in this assay than compound 1. Data are means ± SEM; n = 3 replicates per compound per timepoint. (D) Survival curves for SOD1A4V mutant ALS motor neurons treated with CPA and compound 1 or Prostetin/12k. Prostetin/12k shows improved potency and efficacy. Data are means ± SEM of motor neurons treated with CPA + test compounds normalized to vehicle controls. n = 3 replicate wells from a 96-well plate for each test compound dose. (E – H) Representative fields from whole-well images of SOD1A4V motor neurons treated with vehicle (DMSO) alone (E); CPA alone (F); CPA + Compound 1 (G); or CPA + Compound 12k (H). (I – J) IC50 determination for HGK (I) and MLK3 (J) in cell-free assays. Compared to compound 1, Prostetin 12k shows a reduced IC50 for HGK but little change in IC50 for MLK3. See also Fig. S3. (K) Docking pose of Prostetin/12k in HGK. Prostetin/12k has a similar binding mode as compound 1, but forms an additional hydrogen bond through the hydroxyl group with Asn119. (L – M) Concentration of compounds 1 vs. Prostetin/12k in brain and plasma samples over 24 h following IP or OG administration. Prostetin/12k is more effective than 1 at reaching the CNS. Pharmacokinetic parameters are further described in Table 1. Data are means ± SEM of plasma (μg/L) or brain (μg/kg) concentrations. n = 3 mice per treatment per time point. (N) Ranked fold-changes in kinase inhibition for Prostetin 12k vs. compound 1 for all kinases assayed in Table S6, where fold-change >1.5. MAP4Ks (green) are amongst the most enhanced targets of Prostetin/12k. (O) Suppression of cytokine release in LPS-stimulated microglia. Microglial N9 cells were treated compound 1, Prostetin/12k, or 12e ± LPS. TNFα release was measured by ELISA. Prostetin/12k was ~5x less potent than compound 1, but significantly attenuated TNFα release at concentrations of 0.5 μM and above. Compound 12e, which strongly inhibits HGK but has little activity at MLK3, did not inhibit TNFα release at 0.5 μM. Data are mean ± SEM. n = 2 replicate wells of a 96-well plate. ***p < 0.001, ****p < 0.0001.
To determine whether the computationally- and functionally-guided structural modifications made to Prostetin/12k corresponded to a decrease in the IC50 of HGK, we measured HGK activity in cell-free radiolabeled ATP assays for each compound. The IC50 of HGK was 5.93 nM for compound 1 and 0.30 nM for Prostetin/12k (Fig. 4A and B; Fig. 4I). Meanwhile, the IC50 of MLK3 was 18.60 nM for compound 1 and 23.70 for Prostetin/12k (Fig. 4J), indicating that the improvement in EC50 observed for Prostetin/12k in CPA-treated motor neurons was independent of MLK3. Similar trends were observed for MLK1, a homolog of MLK3, and MINK1, a homolog of HGK (Fig. S3). Together, these findings provided further support for the idea that MAP4K inhibition drives neuroprotection. The docking pose of Prostetin/12k in HGK (PDB ID: 5DI1; Fig. 4K) revealed that it had a similar binding mode to compound 1 (Fig. 2A), but formed an additional hydrogen bond through the hydroxyl group that may underlie its reduced IC50.
Prostetin/12k is stable, orally bioavailable, and brain penetrant in vivo
In additional to its low metabolic stability, another critical hurdle in the administration of compound 1 in translational models of neurodegeneration is its low oral bioavailability, which is compounded by its low brain-to-plasma ratio. Goodfellow et al. (2013) reported that the bioavailability (%F) of compound 1 as measured in the plasma was 41. The %F of compound 1 in the brain was not evaluated in that study, but given the brain-to-plasma ratios that were observed for intravenous administration (<1 at all time points measured; (Goodfellow et al., 2013)), the %F in the brain would be expected to be even lower than that observed in the plasma. The limited oral bioavailability of compound 1 in the CNS precludes the possibility of implementing more translational routes of administration, such as oral gavage, in preclinical assessments of the drug.
To evaluate the brain exposure and oral bioavailability of Prostetin 12k vs. compound 1, each drug was administered to mice at 10 mg/kg through intraperitoneal (IP) injection or oral gavage (OG). The pharmacokinetic properties of Prostetin/12k were improved relative to 1 across several measures. The area under the curve (AUC) was greatly increased for Prostetin/12k in the brain after both IP and OG administration, consistent with the decreased rate of clearance predicted by the in vitro microsomal stability assay (Table 1). The concentration of Prostetin/12k in the brain was increased across all time points and routes of administration assessed, as were brain-to-plasma ratios (Fig. 4L and M; Table 1), indicating improved brain penetrance. The brain Cmax of Prostetin/12k in IP-injected animals was 1162 ng/g (2.60 μM; Fig. 4L and M; Table 1), which exceeds the concentration at which Emax was observed in CPA-treated motor neurons (0.5 μM; Fig. 4D). Oral bioavailability (%F) based on plasma measurements in IP vs. OG-treated animals was 46 for compound 1, which is in line with the %F of 41 reported by Goodfellow et al. (2013). Meanwhile, the %F for Prostetin/12k was 73, a marked increase compared to 1. Similar results were obtained for Prostetin/12k by comparing brain measurements (%F=73). Taken together, these results indicate that Prostetin/12k is well suited to future use in vivo, particularly in the study of CNS disorders.
Table 1. Pharmacokinetic parameters for compound 1 vs Prostetin/12k administered by IP injection or by OG.
Error is ± standard error of the mean (SEM).
| Parameter | Cmpd 1, IP | Cmpd 1, OG | Prostetin/12k, IP | Prostetin/12k, OG |
|---|---|---|---|---|
| Plasma AUC (μg/L•h) | 7328 (±1000) | 3401 (±908.8) | 3899 (±539.3) | 2785 (±797.0) |
| Brain AUC (μg/L•h) | 1568 (±147.1) | 567.2 (±110.5) | 11570 (±1655) | 8429 (±1458) |
| Plasma mean Cmax (μg/L) | 971.7 (±138.2) | 476.2 (±165.3) | 454.0 (±48.62) | 184.1 (±57.26) |
| Brain mean Cmax (μg/kg) | 208.8 (±10.21) | 83.35 (±21.40) | 1162 (±91.99) | 658.9 (±257.7) |
| Mean brain/plasma ratio at 3h | 0.23 (±0.014) | 0.19 (±0.019) | 3.0 (±0.59) | 3.3 (±0.53) |
Identification of additional kinase targets of Prostetin/12k
The increased potency and favorable pharmacokinetic profile of Prostetin/12k make it a strong candidate for future studies in vivo, where off-target effects will be a concern. To obtain global kinase inhibition profiles, compound 1 and Prostetin/12k were tested in cell-free radiolabeled ATP assays across a panel of 371 human kinases (Table S6). We found that the MAP4Ks MINK1, KHS, HGK, GLK, and GCK exhibited the greatest degree of change in activity following Prostetin/12k exposure compared to 1 (Fig. 4N). However, several unrelated kinases were inhibited more effectively (>1.5-fold) by Prostetin/12k than by compound 1, including kinases that have previously been implicated in neurodegeneration such as NUAK1 (Lasagna-Reeves et al., 2016) and LRRK2 (Islam and Moore, 2017). These observations raised the possibility that additional kinase targets beyond the MAP4Ks might contribute to the enhanced potency of Prostetin/12k in ER stress-mediated neurodegeneration.
Prostetin/12k maintains the anti-inflammatory properties of compound 1
Compound 1 was originally validated in in vitro and in vivo models of HIV-associated neuroinflammation, where it was shown to attenuate inflammatory cytokine production by microglia (Marker et al., 2013). Our aim in developing the compound 1 analogs described herein was primarily to generate potent, metabolically stable drugs that could prevent neuronal death, but given the fact that neurodegenerative disorders such as ALS are also accompanied by aggressive microgliosis (Hall et al., 1998), and that drug-mediated inhibition of microgliosis is protective in a rodent model of ALS (Trias et al., 2016), we asked whether the optimized compound could also modulate microglial cytokine release. This question was also prompted by the fact that, according to Marker et al., the anti-inflammatory effects of compound 1 are orchestrated by MLK3 in microglia (Marker et al., 2013); meanwhile, all of the analogs tested here showed diminished activity at MLK3 compared to 1 (Table S3).
To model microglial activation in vitro, we stimulated mouse N9 microglial cells with E. coli-derived lipopolysaccharides (LPS) and measured the release of the pro-inflammatory cytokine TNFα into cell culture media. The LPS concentration selected here strongly induced TNFα secretion (Fig. 4O), but did not induce N9 cell death (data not shown). We then compared TNFα secretion in cultures co-treated with LPS and compound 1 or the optimized analog Prostetin/12k, which showed increased inhibition of MAP4Ks (Fig. 4N), but slightly decreased inhibition of MLK3 (Table S6). We also tested compound 12e, which strongly inhibited HGK activity (Table S4) and was highly neuroprotective (Table S1) but had little activity at MLK3 (Table S3).
We found that compound 1 significantly and dose-dependently decreased TNFα secretion in LPS-stimulated microglia, consistent with its previously reported anti-inflammatory effects (EC50 = 0.20 μM; Fig. 4O). Prostetin/12k also significantly decreased TNFα secretion, though was less potent in this assay than compound 1 (EC50 ~0.47 μM; Fig. 4L). Meanwhile, compound 12e did not significantly decrease TNFα levels (Fig. 4O). Taken together, these findings suggest that MAP4K inhibition does not mitigate TNFα release in this context, and that the retention of non-MAP4K targets such as MLK3 may confer anti-inflammatory activity to compound 1 analogs.
DISCUSSION
Our previous work in human motor neurons subjected to ER stress pointed to broad-spectrum kinase inhibitors as potent neuroprotective agents. Here we determined that one of the shared targets of these protective compounds was HGK, a MAP4K. Using compound 1 as a scaffold, we developed an array of neuroprotective, pharmacologically stable compounds that potently inhibit HGK, culminating in Prostetin/12k.
While our initial focus was on HGK, a kinase previously implicated in motor neuron degeneration (Wu et al., 2019; Yang et al., 2013), we found that the efficacy of compound 1 derivatives also correlated with other closely-related MAP4Ks, including MINK1, GCK, KHS, and TNIK, raising the possibility that multiple MAP4Ks may be involved in ER stress-mediated neurodegeneration. In support of this idea, Larhammar et al. (2017) showed that siRNA-mediated knockdown of HGK, MINK1, and TNIK simultaneously – but not individually – was neuroprotective in dorsal root ganglia neurons subjected to neurotrophic factor withdrawal. They argue that the roles of related MAP4Ks in their neurodegeneration assay are redundant, and that the effects of inhibiting a single kinase can be overcome by the activity of the remaining kinases. It is therefore possible that the combined inhibition of multiple MAP4Ks underlies the robust neuroprotective effects of compound 1 and its analogs in CPA-treated motor neurons.
At the same time, kinome-wide profiles of 1 and Prostetin/12k (Fig. 4N; Table S6) make it clear that neither compound is perfectly selective for MAP4Ks. It is possible that some of the additional targets of compound 1 and its analogs may enhance their neuroprotective effects. Other kinases whose inhibition correlated strongly with neuroprotection included PRKD3, ABL1, and ABL2 (Fig. 3D). Accordingly, we have observed that kinase inhibitors targeting PKC or ABL kinases in a non-subtype-specific manner (e.g., Ro-318220 mesylate and bosutinib, respectively) are mildly protective in CPA-treated motor neurons (data not shown). Furthermore, both the addition of bosutinib and the genetic ablation of ABL1 have been shown to be neuroprotective in a human stem cell-derived motor neuron model of ALS (Imamura et al., 2017). Though it is unclear how these might be activated in the context of CPA, the combined inhibition of multiple kinase subtypes by compound 1 and its analogs may be key to their success in the in vitro motor neuron survival assay.
Furthermore, the anti-inflammatory properties of compound 1 analogs, which may be linked to MLK3 inhibition (Marker et al., 2013), could prove to be an additional benefit. Microgliosis is a hallmark of many neurodegenerative disorders and is readily observed in murine models of ALS (Hall et al., 1998). There is an ongoing debate as to whether microglia are helpful or harmful in the context of ALS (Chiu et al., 2013; Gowing et al., 2008; Spiller et al., 2018), but treatment with the kinase inhibitor masitinib has been shown to attenuate microgliosis and slow disease progression in late-stage SOD1G93A mutant rats (Trias et al., 2016). Thus, Prostetin/12k and the other compound 1 analogs generated here that target both neuroinflammatory and neurodegenerative pathways may be particularly strong therapeutic candidates for ALS.
Finally, MAP4Ks have been directly implicated in multiple, diverse disorders outside of the CNS, including obesity (Danai et al., 2015), insulin resistance (Ammirati et al., 2015; Bouzakri and Zierath, 2007; Chuang et al., 2014; Flach et al., 2016; Tang et al., 2006; Tesz et al., 2007), atherosclerosis (Aouadi et al., 2009; Virbasius and Czech, 2016), hepatocellular carcinoma (Han et al., 2010), lung adenocarcinoma (Qiu et al., 2012), colorectal cancer (Lin et al., 2018; Salem et al., 2018), and general transformation and metastatic processes in many types of human tumors (Wright et al., 2003). The compounds generated here may therefore have broad therapeutic potential across a wide range of human diseases.
SIGNIFICANCE
ALS and other neurodegenerative diseases are characterized by the accumulation of misfolded proteins, which trigger ER stress and, if left unchecked, ultimately lead to neuronal death. Given that the therapeutic options for these diseases are currently limited, we used a human motor-neuron-based screening platform to identify compounds that could reverse the neurotoxic effects of ER stress. We found that the hits resulting from this screen shared the MAP4 kinase HGK as their target. We selected an orally bioavailable HGK inhibitor with a strong safety profile for further optimization, and developed analogs with enhanced efficacy, potency, metabolic stability, and blood-brain barrier penetration. These compounds, including Prostetin/12k, are now primed for further investigation into their therapeutic potential in animal models of neurodegeneration.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for resources, reagents, and further information should be directed to Brent Stockwell (bstockwell@columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of human and mouse stem cell lines
Preliminary survival assays to identify lead compounds were performed in human motor neurons derived from a wild-type male iPS cell line, NCRM-1 (RRID:CVCL_1E71).
Survival assays to characterized compound 1 analogs were performed in human motor neurons derived from an HB9:GFP motor neuron reporter embryonic stem cell line carrying a heterozygous SOD1A4V point mutation (HUES3 Hb9::GFP-hSOD1A4V) that was generated as described previously (Thams et al., 2019).
Western blot experiments were carried out in ES-derived mouse motor neurons (HB9::GFP-hSOD1G93A) generated by Thams et al. (2019) from crosses of heterozygous Tg(Hlxb9-GFP)1Tmj motor neuron reporter mice (RRID:IMSR_JAX:005029) and mice expressing wild-type human SOD1 (B6SJL-Tg(SOD1)2Gur/J, RRID:IMSR_JAX:002726).
All stem cell lines were routinely tested for mycoplasma.
Human motor neuron differentiation
Human motor neurons were differentiated from the above human ES or iPS cells as previously described (Maury et al., 2014; Thams et al., 2019). Stem cells were maintained in humidified incubators at 37°C on irradiated CF-1 mouse embryonic fibroblast (MEF) feeder layers (Thermo Fisher) in serum-free media (DMEM/F12, Life Technologies) supplemented with knockout serum replacement (20%, Life Technologies), Glutamax (1%, Life Technologies), non-essential amino acids (1%, Millipore), beta-mercaptoethanol (0.1%, Sigma), and human recombinant fibroblast growth factor 2 (FGF2, 20 ng/mL, Life Technologies).
On day 0, stem cells were dissociated into single cells using Accutase (Thermo Fisher) and differentiated into motor neurons over the course of 16 days as embryoid bodies cultured in suspension at 37°C. Differentiations were carried out in N2B27 media, which was comprised of a 1:1 mixture of Advanced DMEM/F12 and Neurobasal media (Life Technologies), Glutamax (1%), beta-mercaptoethanol (0.1%), N-2 (1%, Thermo Fisher), B-27 (2%, Thermo Fisher), and ascorbic acid (10 μM, Sigma). From day 0–1, differentiation medium was supplemented with FGF2 (10 ng/mL), Y-27632 dihydrochloride (10 μM, Abcam), SB 431542 hydrate (SB, 20 μM, Sigma), LDN193189 (LDN, 0.1 μM, Stemgent) and CHIR 99021 (CHIR, 3 μM, Tocris). From days 2–7, differentiation medium was supplemented with SB (20 μM), LDN (0.1 μM), CHIR (3 μM), retinoic acid (100 nM, Sigma), and sonic hedgehog agonist (SAG, 500 nM; Millipore). From days 7–9, differentiation medium was supplemented with retinoic acid (100 nM), and SAG (500 nM), and brain-derived neurotrophic factor (BDNF, 10 ng/mL, Peprotech). From days 9–14, differentiation medium was supplemented with retinoic acid (100 nM), SAG (500 nM), BDNF (10 ng/mL, Peprotech), and DAPT (10 μM, Tocris). From days 14–6, differentiation medium was supplemented with retinoic acid (100 nM), SAG (500 nM), BDNF (10 ng/mL), DAPT (10 μM), and glial-derived neurotrophic factor (GDNF, 10 ng/mL; R&D Systems).
On day 16 of differentiation, embryoid bodies were dissociated using 0.05% trypsin (Life Technologies) and mechanical trituration, and motor neurons were plated at 2000 cells / well in 96-well plates (Greiner) coated with poly-ornithine (100 μg/mL, Sigma) and mouse laminin (3 μg/mL, Thermo Fisher). Cultures were maintained at 37°C in serum-free neurobasal media (Life Technologies) supplemented with N-2 (1%), B-27 (2%), a 1:1 cocktail of uridine and fluorodeoxyuridine as anti-mitotics (1 μM, Sigma), and the following recombinant human neurotrophic factors: GDNF, BDNF, ciliary neurotrophic factor (CNTF; R&D Systems), and insulin-like growth factor 1 (IGF-1; R&D systems), all at 10 ng/mL.
Mouse motor neuron differentiation
We used mouse rather than human motor neurons for biochemistry assays because mouse motor neurons can be rapidly generated in the quantities required for protein extraction. Differentiations were carried out as described (Thams et al., 2019; Wichterle et al., 2002). In brief, ES cells were maintained at 37°C on CF-1 MEF feeder layers, then dissociated and differentiated for 6 days using retinoic acid (1 μM) and SAG (0.5 μM). Motor neurons were maintained at 37°C in media containing 2% fetal bovine serum (FBS, Fisher Scientific), GDNF (400 pg/mL), Forskolin (10 μM, Sigma-Aldrich), and IBMX (100 μM, Tocris).
N9 microglia culture details
Immortalized N9 mouse microglia cells (Corradin et al., 1993) were kindly provided by Serge Przedborski, Columbia University. Cultures were maintained at 37°C in DMEM (Life Technologies) containing FBS (10%, Fisher Scientific) and Glutamax (1%, Life Technologies) on gelatinized plates. This cell line was not authenticated upon receipt, but appeared and behaved as expected.
Mouse maintenance
All animal procedures were approved by the Columbia University Institutional Animal Care and Use committee under protocol number AC-AAAU6461, and were performed in accordance with all relevant regulatory standards. Mice were housed 4 per cage under 12h light/dark cycle conditions. Food and water were provided ad libitum. All experiments described here involved 8-week-old male wild-type B6SJLF1/J mice (The Jackson Laboratory; RRID_IMSR_JAX:100012). This strain was chosen because it shares the same background as the most commonly used ALS mouse model (B6SJL-Tg(SOD1*G93A)1Gur/J; RRID:IMSR_JAX:002726). The preliminary pharmacokinetic studies described here only involved male mice in anticipation of future studies in ALS mice where male and female phenotypes diverge (Scott et al., 2009), and where single-sex studies in males are common. All mice were drug-naïve prior to the initiation of the pharmacokinetic studies. Mice used in this study were bred by Jackson laboratories, and all were born within ± 3 days of each other. Experimental groups (n = 3 mice per treatment per time point) were balanced by average mouse weight on the day before treatment.
METHOD DETAILS
Human motor neuron survival assays
48 h after dissociation and plating (DIV2), motor neurons were treated simultaneously with CPA (33 μM, Tocris) and test compounds. DMSO (Sigma) was used as a vehicle control. For all test compounds, stock solutions were prepared in DMSO, and working solutions were prepared in motor neuron media. WZ4003 (Tocris) and HTH 01–015 (Selleckchem) were tested in 8-point, 2-fold dilution series from 0.25–32 μM in 3 replicate wells. PF-6260933 (Tocris), GNE-495 (Cayman Chemical), and URMC-099 (Selleckchem) were tested in 8-point, 2-fold dilution series from 0.075–9.6 μM in 3 replicate wells. Compound 1 analogs were evaluated at 0.1, 0.3, 0.5, 1, and 3 μM in 3 replicate wells. The EC50 of compound 1 analogs were determined using Prism 8 software (GraphPad) from experiments where compound 1 and 53 analogs were assayed simultaneously.
Motor neurons were incubated with CPA and test compounds for 72 h. Live cells were visualized using CellTrace Calcein AM (1.3 μM, Life Technologies) and background fluorescence was quenched with a solution of 10% brilliant black (Sigma) in PBS. Whole-well images were acquired using a Plate RunnerHD system (Trophos). Cells were counted using Metamorph software (Molecular Devices). All cell counts are expressed as a percentage of surviving vehicle (DMSO)-treated cells.
Biochemical analysis of JNK pathway regulation
Whole mouse embryoid bodies at day 6 of differentiation were treated with CPA (10 μM) and test compounds (0.5 μM) for 4 h, then lysed and sonicated in ice-cold buffer containing HEPES (50 mM), NaCl (100 mM), Triton-X (1%), SDS (0.1%), DTT (1 mM), EDTA (1 mM), plus protease and phosphatase inhibitor cocktails (Sigma-Aldrich). Total protein content was analyzed using a BCA kit according to the manufacturer’s instructions (Pierce). For each sample, 30 μg total protein was reduced with dithiothreitol (DTT, 100 mM) for 10 mins at 70°C and loaded into a NuPAGE Novex 4–12% bis-tris gel (Thermo Fisher) with LDS loading buffer (Thermo Fisher). Samples were run for 4h at 100V at 4°C, then transferred to 0.2 μ nitrocellulose membranes overnight at 15V at 4°C. Membranes were blocked with tris-buffered saline (TBS) containing 5% bovine serum albumen (BSA, Cell Signaling Technologies), then incubated with primary antibodies in a buffer containing 5% BSA and 0.1% Tween-20 (Sigma) in TBS overnight at 4°C. Primary antibodies against cleaved caspase 3 (Asp175, Cell Signaling Technology) were added at 1:1000; primary antibodies against c-Jun (Cell Signaling Technology) were added at 1:1000; primary antibodies against phospho-c-Jun (Ser63, Cell Signaling Technology) were added at 1:1000; primary antibodies against JNK (Cell Signaling Technology) were added at 1:1000; primary antibodies against phospho-JNK (Thr183/Tyr185, Cell Signaling Technology) were added at 1:1000; primary antibodies against MKK4 (Cell Signaling Technology) were added at 1:1000; primary antibodies against phospho-MKK4 (Ser257, Cell Signaling Technology) were added at 1:1000; primary antibodies against GAPDH (Santa Cruz Biotechnologies) were added at 1:3000 as a loading control. Following washes in TBS containing 0.1% Tween, membranes were incubated in goat anti-rabbit HRP-conjugated secondary antibody solutions (1:5000; Cell Signaling Technology) for 1h. Membranes were washed again in TBS containing 0.1% Tween, then incubated in SuperSignal West Femto reagent (Thermo Fisher) for 5 min. Chemiluminescence was visualized using a LiCor Odyssey. Band intensities were quantified using ImageStudio Lite (LiCor). Cleaved caspase 3, c-Jun, phospho-c-Jun, JNK, phospho-JNK, MKK4, and phospho-MKK4 intensities were normalized to GAPDH. Intensities for phospho-c-Jun, phospho-JNK, and phospho-MKK4 were then normalized to c-Jun, JNK, and MKK4. Treatment-mediated changes in normalized intensities were expressed as a percentage of CPA-treated samples.
General experimental details for the synthesis of Compound 1 analogs and intermediates
Starting materials were purchased form Sigma-Aldrich, Fisher Scientific, Ark Pharm, Oakwood Chemical, Cambridge Isotope Laboratory, or AK Scientific and were used as received unless stated otherwise. All solvents were reagent grade. Column chromatography was performed on a Teledyne ISCO CombiFlash® Rf+ using RediSep® Normal-phase silica flash columns. Thin layer chromatography (TLC) was performed on Silicycle SiliaPlate™ Glass TLC Plates (250 μm, 20 × 20 cm). Where indicated, compounds were purified by preparatory HPLC on a Phenomenex Gemini NX-C18 column (250 × 21.2 mm, particle size: 5 μm, pore size: 110 Å) using a Gilson HPLC with GX-271 liquid handler. 1H NMR spectra were recorded at ambient temperature using 400 MHz, or 500 MHz spectrometers as indicated. Chemical shifts are reported in ppm relative to the residual solvent peaks (1H NMR: DMSO-d6, δ 2.50; chloroform-d, δ 7.26; methanol-d4, δ 3.31). The following abbreviations are used to indicate multiplicity: s (singlet), d (doublet), t (triplet), q (quartet), hept (heptuplet), m (multiplet), br (broad). High resolution mass spectra (HRMS) were acquired on a time-of-flight spectrometer with atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI), as indicated, and were obtained by peak matching. All reactions were run under an atmosphere of nitrogen or argon in glassware that was flame-dried under argon unless otherwise stated. Aqueous solutions were prepared from nanopure water with a resistivity over 18 MΩ·cm. Unless otherwise noted, all reagents were commercially available.
Synthesis Route for the Synthesis of Piperazine Analogs (see Scheme 1)
4-(1H-Pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (3)

5-Bromo-1H-pyrrolo[2,3-b]pyridine (2, 3.94 g, 20 mmol, 1.0 eq) and 4-formylphenylboronic acid (3.30 g, 22 mmol, 1.1 eq) were added to a pressure tube and acetonitrile (80 mL) and 1,4-dioxane (20 mL) were added. The reaction mixture was degassed and kept under argon. Pd(PPh3)2Cl2 (0.70 g, 1.0 mmol, 5 mol%) was added followed by addition of aqueous Na2CO3 (2.0 M, 50 mL). The reaction mixture was stirred for 5 min, transferred to an oil bath and stirred at 130 °C overnight. After cooling to room temperature, the reaction mixture was partitioned between ethyl acetate and brine, separated and the aqueous layer extracted with ethyl acetate (3x). The combined organic layers were dried with anhydrous sodium sulfate and filtered over Celite. The solvent was evaporated, and the crude product purified by column chromatography (0–100% EtOAc in hexanes) to give 4-(1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (3, 2.15 g, 48% yield) as a colorless solid. 1H NMR (400 MHz, DMSO-d6) δ 11.80 (s, 1H), 10.06 (s, 1H), 8.63 (d, J = 2.2 Hz, 1H), 8.35 (d, J = 2.2 Hz, 1H), 8.05 – 7.94 (m, 4H), 7.55 (d, J = 3.5 Hz, 1H), 6.54 (d, J = 3.4 Hz, 1H) ppm. HRMS (APCI+, m/z): calcd. for C14H11N2O [M+H+]: 223.0872, found: 223.0872.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (4)
4-(1H-Pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (3, 2.15 g, 9.66 mmol, 1.0 eq) was suspended in dichloromethane (~0.1 M), and 1-methylpiperazine (1.93 g, 2.14 mL, 19.3 mmol, 2.0 eq), and Na(OAc)3BH (3.07 g, 14.5 mmol, 1.5 eq) were added and the reaction mixture was stirred overnight at room temperature. The reaction mixture was partitioned between dichloromethane and brine, the layers separated, and the aqueous layer was extracted with dichloromethane. The combined organic layer was washed with brine, dried with sodium sulfate, filtered over Celite, and the solvent evaporated to give 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (4, 2.2 gram, 74% yield) as a colorless solid. The product was used in the next step without further purification. 1H NMR (400 MHz, Methanol-d4) δ 8.43 (d, J = 2.1 Hz, 1H), 8.21 (d, J = 2.1 Hz, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 3.5 Hz, 1H), 6.55 (d, J = 3.5 Hz, 1H), 3.64 (s, 2H), 2.71 (bs, 4H), 2.63 (bs, 4H), 2.44 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C19H23N4 [M+H+]: 307.1923, found: 307.1918.
3-Iodo-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (5)
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (4, 2.2 g, 7.18 mmol, 1.0 eq) was suspended in acetone (0.015 M, 480 mL) and N-iodosuccinimide (1.78 g, 7.9 mmol, 1.1 eq) was added and the reaction mixture was stirred at room temperature overnight. The solvent was evaporated and the crude product was purified by column chromatography (0–10% MeOH in DCM containing 1% Et3N) to give 3-iodo-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (5, 2.3 g, 74% yield) as a tan-colored solid. 1H NMR (400 MHz, Methanol-d4) δ 8.46 (d, J = 2.1 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.57 (s, 1H), 7.47 (d, J = 8.2 Hz, 2H), 3.71 (s, 2H), 3.35 (s, 1H), 3.21 (bs, 8H), 2.80 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C19H22N4I [M+H+]: 433.0889, found: 433.0883.
tert-Butyl 3-iodo-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-1-carboxylate (6)

3-Iodo-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (5, 2.3 g, 5.32 mmol, 1.0 eq) was dissolved in a mixture of THF (250 mL) and DMF (20 mL). Boc2O (3.48 g, 15.96 mmol, 3.0 eq) and DMAP (0.97 g, 7.98 mmol, 1.5 eq) were added and the mixture was stirred at room temperature overnight. Water was added and the reaction mixture was extracted with ethyl acetate (3x). The combined organic layers were washed with brine, dried with sodium sulfate, filtered and the solvent evaporated. The crude material was purified by column chromatography on silica (0–10% MeOH in DCM containing 1% Et3N) to give tert-butyl 3-iodo-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-1-carboxylate as a tan-colored solid (6, 2.0 g, 71% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.73 (d, J = 2.1 Hz, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.82 (s, 1H), 7.59 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 3.58 (s, 2H), 2.53 (bs, 4H), 2.47 (bs, 4H), 2.30 (s, 3H), 1.69 (s, 9H) ppm. HRMS (APCI+, m/z): calcd. for C24H30N4O2I [M+H+]: 533.1413, found: 533.1423.
General Procedure for the Suzuki Coupling and in situ Deprotection

tert-butyl 3-iodo-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine-1-carboxylate (6, 26.2 mg, 0.05 mmol, 1.0 eq) was dissolved in 1,4-dioxane (2.0 mL) in a 1 dram glass vial with Teflon lined cap. The boronic acid or boronic ester (0.075 mmol, 1.5 eq) was added, followed by addition of Tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4, 5.8 mg, 5 mol%). The reaction mixture was degassed, aqueous Na2CO3 (2.0 M, 0.5 mL) was added, and was stirred overnight at 100 °C. The reaction mixture was filtered over Celite and the Celite was washed with ethyl acetate. The solvent was evaporated and the crude product was purified by preparatory HPLC purification to give the pure material.
3-(1H-Indol-5-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (1)

Compound 1 was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 5-indolylboronic acid (12.1 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(1H-indol-5-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (1, 14.8 mg, 70% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 2.0 Hz, 1H), 7.86 (dd, J = 1.7, 0.7 Hz, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.67 (s, 1H), 7.50 (m, 3H), 7.44 (dd, J = 8.4, 1.7 Hz, 1H), 7.28 (d, J = 3.2 Hz, 1H), 6.51 (dd, J = 3.2, 0.9 Hz, 1H), 3.84 (s, 2H), 3.00 – 2.80 (bs, 8H), 2.87 (s, 3H) ppm. The spectroscopic data matched those reported in literature (Goodfellow et al., 2013).
3-(1H-Indol-2-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7a)
Compound 7a was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using indole-2-boronic acid pinacol ester (18.2 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(1H-Indol-2-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7a, 14.1 mg, 67% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.54 (d, J = 2.1 Hz, 1H), 8.49 (d, J = 2.1 Hz, 1H), 7.82 (s, 1H), 7.65 (d, J = 8.2 Hz, 2H), 7.54 (dt, J = 7.8, 1.0 Hz, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.42 – 7.35 (m, 1H), 7.07 (ddd, J = 8.0, 7.2, 1.1 Hz, 1H), 7.00 (ddd, J = 8.0, 7.1, 1.1 Hz, 1H), 6.78 (d, J = 1.0 Hz, 1H), 3.56 (s, 2H), 2.75 – 2.40 (bs, 8H), 2.34 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H28N5 [M+H+]: 422.2339, found: 422.2336.
3-(6-Ethoxypyridin-3-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7b)

Compound 7b was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 6-ethoxy-3-pyridinylboronic acid (12.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(6-ethoxypyridin-3-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7b, 16.2 mg, 76% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.48 (d, J = 2.1 Hz, 1H), 8.42 (dd, J = 2.6, 0.8 Hz, 1H), 8.33 (d, J = 2.1 Hz, 1H), 7.99 (dd, J = 8.6, 2.5 Hz, 1H), 7.66 (s, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 6.88 (dd, J = 8.6, 0.8 Hz, 1H), 4.34 (q, J = 7.0 Hz, 2H), 2.80 – 2.45 (bs, 8H), 2.40 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H30N5O [M+H+]: 428.2450, found: 428.2449.
3-(Dibenzo[b,d]furan-4-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7c)

Compound 7c was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-(dibenzofuranyl)boronic acid (15.9 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(dibenzo[b,d]furan-4-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7c, 12.6 mg, 58% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.57 (s, 2H), 8.18 (s, 1H), 8.12 (dt, J = 7.7, 1.2 Hz, 1H), 8.00 (dd, J = 7.7, 1.2 Hz, 1H), 7.94 (dd, J = 7.6, 1.2 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.70 – 7.65 (m, 2H), 7.57 – 7.48 (m, 4H), 3.69 (s, 2H), 3.05 – 2.75 (bs, 4H), 2.85 – 2.55 (bs, 4H), 2.59 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C31H29N4O [M+H+]: 473.2341, found: 473.2345.
3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7d)

Compound 7d was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1,4-benzodioxane-6-boronic acid (13.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7d, 10.2 mg, 46% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.45 (d, J = 2.1 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.55 (s, 1H), 7.43 (d, J = 8.2 Hz, 2H), 7.17 – 7.10 (m, 2H), 6.91 (d, J = 8.8 Hz, 1H), 4.27 (s, 4H), 3.60 (s, 2H), 2.77 – 2.46 (bs, 8H), 2.39 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H29N4O2 [M+H+]: 441.2291, found: 441.2287.
4-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7e)
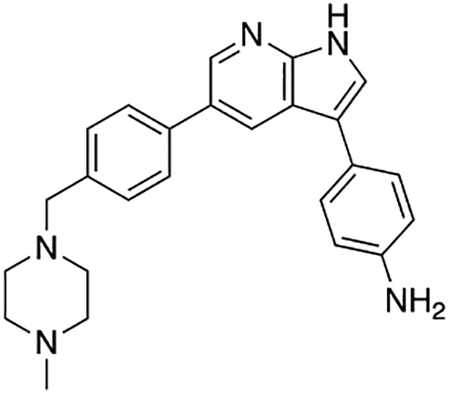
Compound 7e was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-aminophenylboronic acid pinacol ester (17.0 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7e, 14.4 mg, 73% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.43 (d, J = 2.1 Hz, 1H), 8.34 (d, J = 2.1 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.50 (s, 1H), 7.46 −7.39 (m, 4H), 6.84 (d, J = 8.5 Hz, 2H), 3.62 (s, 2H), 2.79 (bs, 4H), 2.63 (bs, 4H), 2.49 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H28N5 [M+H+]: 398.2345, found: 398.2344.
3-(Benzo[b]thiophen-3-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7f)

Compound 7f was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using benzo[b]thien-3-ylboronic acid (14.0 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(benzo[b]thiophen-3-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7f, 16.0 mg, 73% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.50 (d, J = 2.1 Hz, 1H), 8.17 (d, J = 2.1 Hz, 1H), 7.97 – 7.87 (m, 2H), 7.71 (s, 1H), 7.63 (s, 1H), 7.55 (d, J = 8.2 Hz, 2H), 7.44 – 7.33 (m, 4H), 3.55 (s, 2H), 2.71 – 2.45 (bs, 8H), 2.37 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H27N4S [M+H+]: 439.1956, found: 439.1955.
3-(Benzo[b]thiophen-2-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7g)

Compound 7g was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using benzo[b]thien-2-ylboronic acid (14.0 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(benzo[b]thiophen-2-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7g, 15.9 mg, 73% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.52 (d, J = 2.1 Hz, 1H), 8.50 (d, J = 2.1 Hz, 1H), 7.83 – 7.74 (m, 3H), 7.64 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 0.7 Hz, 1H), 7.43 (d, J = 8.2 Hz, 2H), 7.32 (ddd, J = 7.9, 7.2, 1.2 Hz, 1H), 7.26 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 3.59 (s, 2H), 2.72 – 2.48 (bs, 8H), 2.38 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H27N4S [M+H+]: 439.1956, found: 439.1959.
3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7h)

Compound 7h was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-aminophenylboronic acid hydrochloride (13.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7h, 14.9 mg, 75% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.47 (d, J = 2.1 Hz, 1H), 8.45 (d, J = 2.1 Hz, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.61 (s, 1H), 7.44 (d, J = 8.2 Hz, 2H), 7.19 (t, J = 7.8 Hz, 1H), 7.11 (t, J = 2.0 Hz, 1H), 7.03 (ddd, J = 7.6, 1.7, 1.0 Hz, 1H), 6.68 (ddd, J = 7.8, 2.3, 1.0 Hz, 1H), 3.60 (s, 2H), 2.58 (bs, 8H), 2.34 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H28N5 [M+H+]: 398.2345, found: 398.2337.
2-Fluoro-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid (7i)
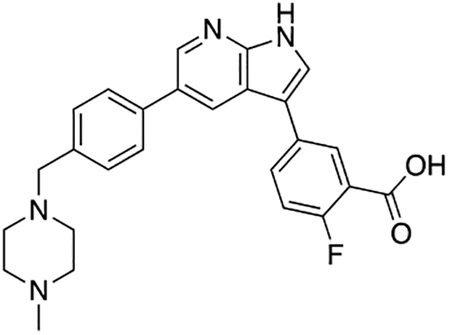
Compound 7i was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-carboxy-4-fluorophenylboronic acid (13.8 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 2-fluoro-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid (7i, 8.9 mg, 40% yield). 1H NMR (500 MHz, Methanol-d4) δ 8.56 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 2.0 Hz, 1H), 8.24 (dd, J = 6.8, 2.5 Hz, 1H), 7.95 (ddd, J = 8.6, 4.5, 2.5 Hz, 1H), 7.81 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 7.32 (dd, J = 10.6, 8.6 Hz, 1H), 3.96 (s, 2H), 3.38 (bs, 4H), 3.05 (bs, 4H), 2.91 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H26FN4O2 [M+H+]: 445.2040, found: 445.2035.
3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid (7j)
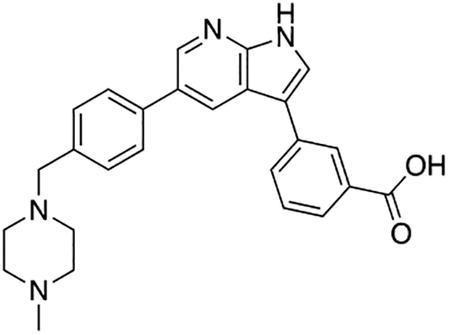
Compound 7j was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-carboxyphenylboronic acid (12.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid (7j, 12.0 mg, 56% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.62 – 8.56 (m, 2H), 8.36 (s, 1H), 8.02 – 7.94 (m, 2H), 7.85 (s, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 8.0 Hz, 2H), 3.97 (s, 2H), 3.39 (bs, 4H), 3.05 (bs, 4H), 2.91 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H27N4O2 [M+H+]: 427.2134, found: 427.2137.
5-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-2-amine (7k)

Compound 7k was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2-aminopyrimidine-5-boronic acid (11.0 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-2-amine (7k, 16.4 mg, 82%). 1H NMR (400 MHz, Methanol-d4) δ 8.87 (s, 2H), 8.58 (d, J = 2.1 Hz, 1H), 8.46 (d, J = 2.1 Hz, 1H), 7.87 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 3.93 (s, 2H), 3.37 (bs, 4H), 3.01 (bs, 4H), 2.90 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C23H26N7 [M+H+]: 400.2250, found: 400.2250.
3-Methyl-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyridin-2-amine (7l)

Compound 7l was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2-amino-3-methylpyridine-5-boronic acid pinacol ester (18.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-methyl-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyridin-2-amine (7l, 8.2 mg, 40% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.55 (s, 1H), 8.42 (d, J = 2.0 Hz, 1H), 8.24 (dd, J = 2.2, 1.1 Hz, 1H), 8.14 – 8.08 (m, 1H), 7.83 (s, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 3.79 (s, 2H), 3.04 – 2.68 (bs, 8H), 2.87 (s, 3H), 2.37 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H29N6 [M+H+]: 413.2454, found: 413.2457.
5-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyridin-2-amine (7m)
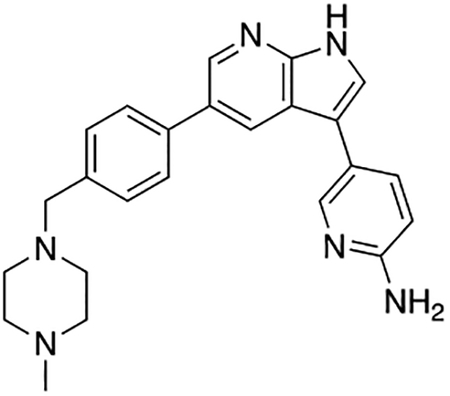
Compound 7m was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2-aminopyridine-5-boronic acid pinacol ester (16.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyridin-2-amine (7m, 15.2 mg, 76% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.60 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 2.0 Hz, 1H), 8.35 (dd, J = 9.3, 2.2 Hz, 1H), 8.22 (dd, J = 2.2, 0.7 Hz, 1H), 7.87 (s, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 7.16 (dd, J = 9.2, 0.7 Hz, 1H), 4.10 (s, 2H), 3.46 (bs, 4H), 3.20 (s, 4H), 2.93 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C24H27N6 [M+H+]: 399.2297, found: 399.2307.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(pyridin-4-yl)-1H-pyrrolo[2,3-b]pyridine (7n)
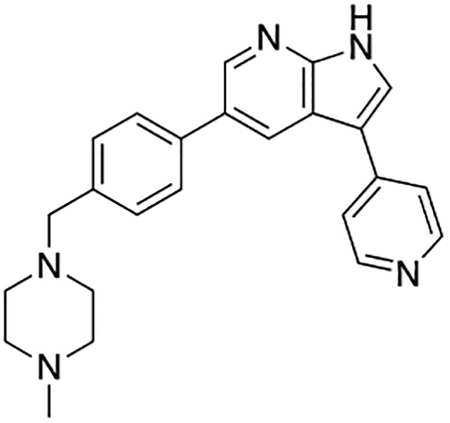
Compound 7n was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-pyridinylboronic acid (10.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(pyridin-4-yl)-1H-pyrrolo[2,3-b]pyridine (7n, 8.7 mg, 45% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.74 (d, J = 2.0 Hz, 1H), 8.68 – 8.64 (m, 3H), 8.57 (s, 1H), 8.43 (d, J = 7.1 Hz, 2H), 7.83 (d, J = 8.2 Hz, 2H), 7.60 (d, J = 8.2 Hz, 2H), 4.04 (s, 2H), 3.43 (bs, 4H), 3.13 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C24H26N5 [M+H+]: 384.2188, found: 384.2194.
4-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzonitrile (7o)

Compound 7o was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-cyanophenylboronic acid (11.0 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzonitrile (7o, 14.4 mg, 71% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.56 (d, J = 2.1 Hz, 1H), 8.54 (d, J = 2.1 Hz, 1H), 7.97 – 7.91 (m, 3H), 7.83 – 7.77 (m, 2H), 7.75 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 3.89 (s, 2H), 3.35 (bs, 4H), 2.96 (bs, 4H), 2.89 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H26N5 [M+H+]: 408.2188, found: 408.2198.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(3-nitrophenyl)-1H-pyrrolo[2,3-b]pyridine (7p)

Compound 7p was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-nitrophenylboronic acid (12.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(3-nitrophenyl)-1H-pyrrolo[2,3-b]pyridine (7p, 15.4 mg, 72% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.59 (d, J = 2.0 Hz, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.54 (t, J = 2.0 Hz, 1H), 8.15 (dddd, J = 7.7, 4.3, 2.1, 1.0 Hz, 2H), 7.96 (s, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.71 (t, J = 8.0 Hz, 1H), 7.58 (d, J = 8.2 Hz, 2H), 4.08 (s, 2H), 3.44 (bs, 4H), 3.17 (bs, 4H), 2.93 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H26N5O2 [M+H+]: 428.2087, found: 428.2081.
3-(4-Isopropoxyphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7q)
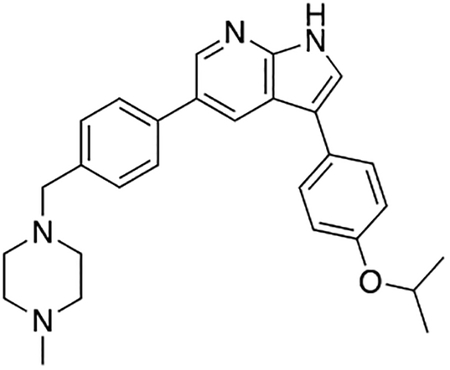
Compound 7q was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-isopropoxyphenylboronic acid (13.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(4-isopropoxyphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7q, 18.2 mg, 83% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 2.0 Hz, 1H), 8.56 (d, J = 2.0 Hz, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.69 (s, 1H), 7.64 – 7.58 (m, 2H), 7.55 (d, J = 8.2 Hz, 2H), 7.08 – 6.98 (m, 2H), 4.65 (hept, J = 6.0 Hz, 1H), 3.94 (s, 2H), 3.37 (bs, 4H), 3.02 (bs, 4H), 2.90 (s, 3H), 1.35 (d, J = 6.0 Hz, 6H) ppm. HRMS (APCI+, m/z): calcd. for C28H33N4O [M+H+]: 441.2654, found: 441.2646.
3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenol (7r)
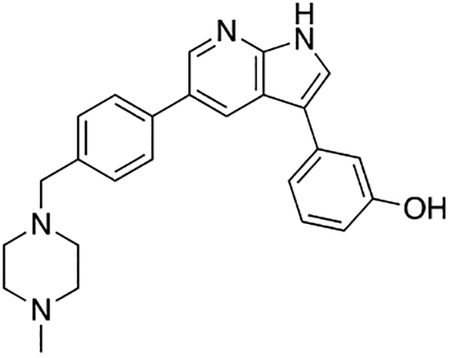
Compound 7r was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-hydroxyphenylboronic acid (10.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenol (7r, 9.8 mg, 49% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.68 (d, J = 1.9 Hz, 1H), 8.60 (s, 1H), 7.80 – 7.73 (m, 3H), 7.58 (d, J = 8.2 Hz, 2H), 7.30 (t, J = 7.8 Hz, 1H), 7.24 – 7.17 (m, 1H), 7.16 (t, J = 2.0 Hz, 1H), 6.78 (ddd, J = 8.1, 2.5, 1.0 Hz, 1H), 4.04 (s, 2H), 3.42 (bs, 4H), 3.13 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H27N4O [M+H+]: 399.2185, found: 399.2180.
3-(1-Methyl-1H-pyrazol-4-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7s)

Compound 7s was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1-methylpyrazole-4-boronic acid pinacol ester (16.1 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(1-methyl-1H-pyrazol-4-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7s, 14.8 mg, 77% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.62 (d, J = 2.0 Hz, 1H), 8.31 (d, J = 2.0 Hz, 1H), 7.80 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.64 (d, J = 2.0 Hz, 1H), 7.58 (d, J = 8.2 Hz, 2H), 6.60 (d, J = 2.1 Hz, 1H), 4.14 (s, 2H), 3.95 (s, 3H), 3.48 (bs, 4H), 3.25 (bs, 4H), 2.94 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C23H27N6 [M+H+]: 387.2297, found: 387.2303.
4-((5-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)thiophen-2-yl)methyl)morpholine (7t)

Compound 7t was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 5-(morpholinomethyl)-2-thiopheneboronic acid pinacol ester (24.4 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 4-((5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)thiophen-2-yl)methyl)morpholine (7t, 10.0 mg, 41% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.57 (d, J = 2.1 Hz, 1H), 8.52 (d, J = 2.1 Hz, 1H), 7.84 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 3.7 Hz, 1H), 7.37 (d, J = 3.7 Hz, 1H), 4.64 (s, 2H), 4.06 (bs, 4H), 4.01 (s, 2H), 3.80 (bs, 4H), 3.41 (bs, 4H), 3.09 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C28H34N5OS [M+H+]: 488.2484, found: 488.2475.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(6-(methylsulfonyl)pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine (7u)
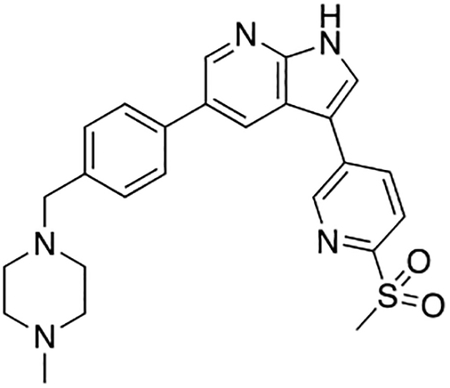
Compound 7u was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 6-(methylsulfonyl)pyridine-3-boronic acid (15.0 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(6-(methylsulfonyl)pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine (7u, 11.0 mg, 48% yield). 1H NMR (400 MHz, Methanol-d4) δ 9.14 (dd, J = 2.2, 0.8 Hz, 1H), 8.59 (q, J = 2.1 Hz, 2H), 8.46 (dd, J = 8.2, 2.2 Hz, 1H), 8.15 (dd, J = 8.2, 0.8 Hz, 1H), 8.08 (s, 1H), 7.78 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 4.02 (s, 2H), 3.41 (bs, 4H), 3.27 (s, 3H), 3.11 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H28N5O2S [M+H+]: 462.1964, found: 462.1968.
5-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (7v)

Compound 7v was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1H-indazole-5-boronic acid (12.1 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (7v, 8.4 mg, 40% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.67 (d, J = 1.9 Hz, 1H), 8.58 (d, J = 1.9 Hz, 1H), 8.13 – 8.09 (m, 2H), 7.79 (s, 1H), 7.80 – 7.73 (m, 3H), 7.67 (dt, J = 8.7, 0.9 Hz, 1H), 7.55 (d, J = 8.3 Hz, 2H), 3.95 (s, 2H), 3.38 (bs, 4H), 3.03 (bs, 4H), 2.90 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H27N6 [M+H+]: 423.2297, found: 423.2298.
3-(Furan-3-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7w)
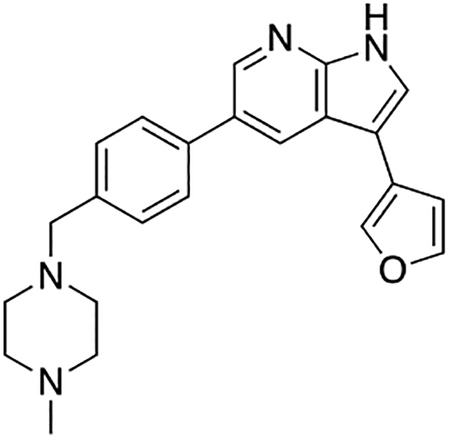
Compound 7w was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-furanylboronic acid (7w, 8.4 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(furan-3-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12.9 mg, 69% yield). 1H NMR (500 MHz, Methanol-d4) δ 8.58 (s, 2H), 8.08 (t, J = 1.1 Hz, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.76 (s, 1H), 7.62 (t, J = 1.7 Hz, 1H), 7.58 (d, J = 8.2 Hz, 2H), 6.87 (dd, J = 1.8, 1.1 Hz, 1H), 4.00 (s, 2H), 3.41 (bs, 4H), 3.07 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C23H25N4O [M+H+]: 373.2028, found: 373.2036.
1-(4-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)ethan-1-one (7x)
Compound 7x was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-acetylphenylboronic acid (12.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 1-(4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)ethan-1-one(7x, 15.1 mg, 71% yield). 1H NMR (500 MHz, Methanol-d4) δ 8.63 (d, J = 2.0 Hz, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.11 (d, J = 8.4 Hz, 2H), 7.95 (s, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 4.01 (s, 2H), 3.41 (bs, 4H), 3.09 (bs, 4H), 2.92 (s, 3H), 2.65 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H29N4O [M+H+]: 425.2341, found: 425.2347.
3-(3-Fluorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7y)

Compound 7y was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-fluorophenylboronic acid (10.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(3-fluorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7y, 12.8 mg, 64% yield). 1H NMR (500 MHz, Methanol-d4) δ 8.59 (d, J = 2.0 Hz, 1H), 8.58 (d, J = 2.2 Hz, 1H), 7.85 (s, 1H), 7.78 (d, J = 8.2 Hz, 2H), 7.61 – 7.55 (m, 3H), 7.52 – 7.44 (m, 2H), 7.11 – 7.02 (m, 1H), 4.02 (s, 2H), 3.41 (bs, 4H), 3.09 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H26N4F [M+H+]: 401.2142, found: 401.2146.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(4-(trifluoromethoxy)phenyl)-1H-pyrrolo[2,3-b]pyridine (7z)

Compound 7z was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-(trifluormethoxy)phenylboronic acid (15.4 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(4-(trifluoromethoxy)phenyl)-1H-pyrrolo[2,3-b]pyridine (7z, 17.4 mg, 75% yield). 1H NMR (500 MHz, Methanol-d4) δ 8.62 (d, J = 2.0 Hz, 1H), 8.59 (d, J = 2.0 Hz, 1H), 7.85 (s, 1H), 7.86 – 7.80 (m, 2H), 7.79 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 4.05 (s, 2H), 3.43 (bs, 4H), 3.13 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H26F3N4O [M+H+]: 467.2059, found: 467.2062.
4-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (7aa)

Compound 7aa was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1H-Indazole-5-boronic acid (12.5 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (7aa, 15.6 mg, 74% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.62 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 2.0 Hz, 1H), 8.18 (s, 1H), 7.94 (s, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.58 – 7.51 (m, 4H), 7.44 (dd, J = 6.2, 1.7 Hz, 1H), 3.97 (s, 2H), 3.38 (bs, 4H), 3.05 (bs, 4H), 2.90 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H27N6 [M+H+]: 423.2297, found: 423.2303.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(3-(trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ab)
Compound 7ab was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-(trifluoromethyl)phenylboronic acid (14.2 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(3-(trifluoromethyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ab, 17.0 mg, 75% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.59 (d, J = 2.0 Hz, 1H), 8.54 (d, J = 2.0 Hz, 1H), 8.00 (d, J = 7.8 Hz, 1H), 7.97 (s, 1H), 7.89 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.68 (t, J = 7.7 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H), 7.57 (d, J = 8.2 Hz, 2H), 4.03 (s, 2H), 3.42 (bs, 4H), 3.12 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H26F3N4 [M+H+]: 451.2110, found: 451.2115.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-phenyl-1H-pyrrolo[2,3-b]pyridine (7ac)

Compound 7ac was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using phenylboronic acid (9.6 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-phenyl-1H-pyrrolo[2,3-b]pyridine (7ac, 16.2 mg, 85% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.69 (d, J = 1.9 Hz, 1H), 8.61 (d, J = 1.9 Hz, 1H), 7.82 (s, 1H), 7.78 (d, J = 8.2 Hz, 2H), 7.76 – 7.69 (m, 2H), 7.59 (d, J = 8.3 Hz, 2H), 7.53 – 7.44 (m, 2H), 7.39 – 7.29 (m, 1H), 4.08 (s, 2H), 3.45 (bs, 4H), 3.17 (bs, 4H), 2.93 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H27N4 [M+H+]: 383.2236, found: 383.2236.
3-(3-Chlorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ad)
Compound 7ad was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-chlorophenylboronic acid (11.7 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(3-chlorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ad, 18.4 mg, 88% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.60 (d, J = 1.3 Hz, 2H), 7.85 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.71 (t, J = 1.9 Hz, 1H), 7.66 (dt, J = 7.8, 1.3 Hz, 1H), 7.61 (d, J = 8.2 Hz, 2H), 7.46 (t, J = 7.9 Hz, 1H), 7.32 (ddd, J = 8.1, 2.1, 1.0 Hz, 1H), 4.17 (s, 2H), 3.49 (bs, 4H), 3.29 (bs, 4H), 2.94 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H26ClN4 [M+H+]: 417.1846, found: 417.1843.
3-(2,4-Dichlorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ae)

Compound 7ae was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2,4-dichlorophenylboronic acid (14.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(2,4-dichlorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ae, 14.9 mg, 66% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 2.0 Hz, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.77 (s, 1H), 7.75 (d, J = 8.3 Hz, 2H), 7.64 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 8.3, 1.4 Hz, 3H), 7.44 (dd, J = 8.3, 2.2 Hz, 1H), 4.16 (s, 2H), 3.49 (bs, 4H), 3.27 (bs, 4H), 2.94 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H25Cl2N4 [M+H+]: 451.1456, found: 451.1447.
3-(2,5-Dimethylphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7af)
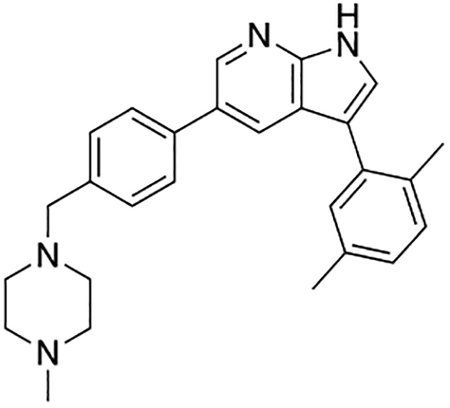
Compound 7af was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2,5-dimethylphenylboronic acid (11.2 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(2,5-dimethylphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7af, 14.0 mg, 68% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.65 (d, J = 1.9 Hz, 1H), 8.36 (d, J = 1.9 Hz, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.63 (s, 1H), 7.60 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 7.8 Hz, 1H), 7.21 (s, 1H), 7.13 (dd, J = 7.4, 1.7 Hz, 1H), 4.18 (s, 2H), 3.50 (bs, 4H), 3.30 (bs, 4H), 2.94 (s, 3H), 2.35 (s, 3H), 2.26 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H31N4 [M+H+]: 411.2549, found: 411.2556.
1-Methyl-6-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (7ag)
Compound 7ag was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1-methyl-1H-indazole-6-boronic acid (13.2 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 1-methyl-6-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (7ag, 13.2 mg, 60% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.67 (d, J = 2.0 Hz, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.01 (d, J = 0.9 Hz, 1H), 7.88 (s, 1H), 7.84 (dd, J = 8.4, 0.9 Hz, 1H), 7.81 (d, J = 1.2 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.60 – 7.52 (m, 3H), 4.11 (s, 3H), 3.99 (s, 2H), 3.40 (bs, 4H), 3.09 (bs, 4H), 2.91 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H29N6 [M+H+]: 437.2454, found: 437.2449.
3-(1-Benzyl-1H-pyrazol-4-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ah)
Compound 7ah was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1-benzyl-1H-pyrazole-4-boronic acid (15.2 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(1-benzyl-1H-pyrazol-4-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ah, 14.3 mg, 62% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 2.0 Hz, 1H), 8.57 (d, J = 2.0 Hz, 1H), 8.19 (d, J = 0.8 Hz, 1H), 7.94 (d, J = 0.8 Hz, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.75 (s, 1H), 7.57 (d, J = 8.2 Hz, 2H), 7.39 – 7.26 (m, 5H), 5.42 (s, 2H), 4.01 (s, 2H), 3.41 (bs, 4H), 3.09 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C29H31N6 [M+H+]: 463.2610, found: 463.2612.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(1,3,5-trimethyl-1H-pyrazol-4-yl)-1H-pyrrolo[2,3-b]pyridine (7ai)

Compound 7ai was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 1,3,5-trimethyl-1H-pyrazole-4-boronic acid pinacol ester (17.7 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(1,3,5-trimethyl-1H-pyrazol-4-yl)-1H-pyrrolo[2,3-b]pyridine (7ai, 7.6 mg, 37% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.59 (d, J = 2.0 Hz, 1H), 8.16 (d, J = 2.0 Hz, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.56 – 7.50 (m, 3H), 3.93 (s, 2H), 3.88 (s, 3H), 3.37 (bs, 4H), 3.01 (bs, 4H), 2.90 (s, 3H), 2.28 (s, 3H), 2.22 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H31N6 [M+H+]: 415.2610, found: 415.2604.
3-(3,5-Bis(trifluoromethyl)phenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7aj)
Compound 7aj was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3,5-bis(trifluoromethyl)phenylboronic acid (19.3 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(3,5-bis(trifluoromethyl)phenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7aj, 11.2 mg, 43% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.58 (d, J = 2.0 Hz, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.29 – 8.23 (m, 2H), 8.02 (s, 1H), 7.86 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 3.96 (s, 2H), 3.38 (bs, 4H), 3.04 (bs, 4H), 2.90 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H25F6N4 [M+H+]: 519.1983, found: 519.1983.
1-Methyl-3-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7ak)
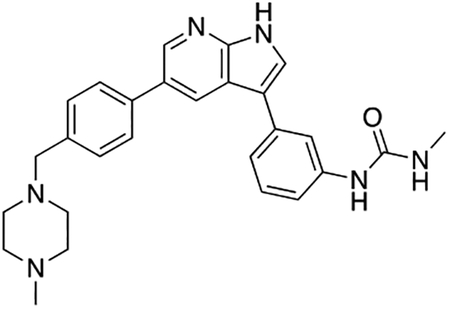
Compound 7ak was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using (3-(3-methylureido)phenyl)boronic acid (13a, 14.6 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 1-methyl-3-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7ak, 6.4 mg, 14% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.70 (d, J = 2.0 Hz, 1H), 8.57 (d, J = 2.0 Hz, 1H), 7.96 (t, J = 1.8 Hz, 1H), 7.81 – 7.75 (m, 3H), 7.54 (d, J = 8.2 Hz, 2H), 7.39 – 7.31 (m, 2H), 7.18 (dt, J = 7.3, 2.0 Hz, 1H), 3.91 (s, 2H), 3.35 (bs, 4H), 2.98 (bs, 4H), 2.89 (s, 3H), 2.80 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H31N6O [M+H+]: 455.2559, found: 455.2559.
Methyl (3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)carbamate (7al)

Compound 7al was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using (3-((methoxycarbonyl)amino)phenyl)boronic acid (13b, 14.6 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford methyl (3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)carbamate (7al, 8.5 mg, 19% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.74 (d, J = 2.0 Hz, 1H), 8.59 (d, J = 2.0 Hz, 1H), 7.98 (s, 1H), 7.83 – 7.78 (m, 3H), 7.56 (d, J = 8.2 Hz, 2H), 7.41 – 7.37 (m, 2H), 7.36 – 7.27 (m, 1H), 3.97 (s, 2H), 3.77 (s, 3H), 3.38 (bs, 4H), 3.04 (bs, 4H), 2.91 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H30N5O2 [M+H+]: 456.2400, found: 456.2400.
1-Methyl-3-(4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7am)
Compound 7am was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using (4-(3-methylureido)phenyl)boronic acid (13c, 14.6 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 1-methyl-3-(4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7am, 5.1 mg, 22% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.63 (d, J = 2.0 Hz, 1H), 8.56 (d, J = 2.0 Hz, 1H), 7.76 (s, 1H), 7.74 (d, J = 4.6 Hz, 2H), 7.65 – 7.60 (m, 2H), 7.55 (d, J = 8.1 Hz, 2H), 7.52 – 7.45 (m, 2H), 3.92 (s, 2H), 3.36 (bs, 4H), 2.99 (bs, 4H), 2.90 (s, 3H), 2.80 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H31N6O [M+H+]: 455.2559, found: 455.2566.
Methyl (4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)carbamate (7an)

Compound 7an was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using (4-((methoxycarbonyl)amino)phenyl)boronic acid (13d, 14.6 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford methyl (4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)carbamate (7an, 7.6 mg, 33% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.68 (d, J = 1.9 Hz, 1H), 8.59 (d, J = 2.0 Hz, 1H), 7.82 – 7.72 (m, 3H), 7.65 (d, J = 8.7 Hz, 2H), 7.61 – 7.51 (m, 4H), 4.04 (s, 3H), 3.76 (s, 2H), 3.43 (bs, 4H), 3.13 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H30N5O2 [M+H+]: 456.2400, found: 456.2397.
3-(4-((4-Methoxybenzyl)oxy)phenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ao)

Compound 7ao was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-(4-methoxybenzyloxy)phenylboronic acid (19.4 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(4-((4-methoxybenzyl)oxy)phenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ao, 20.4 mg, 79% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.69 (dd, J = 7.3, 1.9 Hz, 2H), 8.59 (d, J = 1.8 Hz, 1H), 7.77 (dd, J = 8.3, 1.6 Hz, 2H), 7.72 (d, J = 8.2 Hz, 1H), 7.66 – 7.51 (m, 3H), 7.37 (dd, J = 8.7, 6.9 Hz, 2H), 7.28 – 7.03 (m, 1H), 6.98 – 6.77 (m, 3H), 5.05 (s, 2H), 4.06 (s, 2H), 3.80 (s, 3H), 3.44 (bs, 4H), 3.16 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C33H35N4O2 [M+H+]: 519.2760, found: 519.2755.
5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-3-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine (7ap)

Compound 7ap was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3,4,5-trimethoxyphenylboronic acid (15.9 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-3-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine (7ap, 19.2 mg, 81% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.67 (d, J = 1.9 Hz, 1H), 8.60 (d, J = 1.9 Hz, 1H), 7.81 (s, 1H), 7.78 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 8.2 Hz, 2H), 6.96 (s, 2H), 4.10 (s, 2H), 3.92 (s, 6H), 3.82 (s, 3H), 3.46 (bs, 4H), 3.20 (bs, 4H), 2.93 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C28H33N4O3 [M+H+]: 473.2553, found: 473.2554.
3-(4-Methoxy-3-methylphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7aq)

Compound 7aq was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 4-methoxy-3-methylphenylboronic acid (12.4 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(4-methoxy-3-methylphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7aq, 17.2 mg, 81% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.69 (d, J = 1.9 Hz, 1H), 8.59 (d, J = 1.9 Hz, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.72 (s, 1H), 7.59 (d, J = 8.2 Hz, 2H), 7.51 (dd, J = 8.4, 2.3 Hz, 1H), 7.48 – 7.44 (m, 1H), 7.03 (d, J = 8.4 Hz, 1H), 4.03 (s, 2H), 3.88 (s, 3H), 3.43 (bs, 4H), 3.12 (bs, 4H), 2.92 (s, 3H), 2.28 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H31N4O [M+H+]: 427.2498, found: 427.2501.
3-(3-(Benzyloxy)phenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ar)
Compound 7ar was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 3-(benzyloxy)phenylboronic acid (17.1 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(3-(benzyloxy)phenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7ar, 15.4 mg, 63% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.58 (s, 2H), 7.79 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.2 Hz, 2H), 7.49 – 7.43 (m, 2H), 7.43 – 7.22 (m, 6H), 7.06 – 6.96 (m, 1H), 5.17 (s, 2H), 4.06 (s, 2H), 3.43 (bs, 4H), 3.15 (bs, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C32H33N4O [M+H+]: 489.2654, found: 489.2657.
3-(2-Chlorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7as)
Compound 7as was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2-chlorophenylboronic acid (17.1 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(2-chlorophenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7as, 16.6 mg, 80% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.60 (d, J = 2.0 Hz, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.74 (s, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.59 (ddd, J = 7.7, 4.0, 1.7 Hz, 2H), 7.55 (d, J = 8.2 Hz, 2H), 7.43 (td, J = 7.5, 1.7 Hz, 1H), 7.38 (td, J = 7.5, 2.0 Hz, 1H), 4.01 (s, 2H), 3.41 (bs, 4H), 3.10 (bs, 4H), 2.91 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C25H26N4Cl [M+H+]: 417.1846, found: 417.1849.
3-(2-Methoxyphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7at)

Compound 7at was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using 2-methoxyphenylboronic acid (11.4 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 3-(2-methoxyphenyl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (7at, 17.1 mg, 83% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 1.9 Hz, 1H), 8.58 (d, J = 1.9 Hz, 1H), 7.80 (s, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.61 – 7.54 (m, 3H), 7.38 (ddd, J = 8.3, 7.4, 1.7 Hz, 1H), 7.16 (dd, J = 8.3, 1.1 Hz, 1H), 7.08 (td, J = 7.4, 1.1 Hz, 1H), 4.02 (s, 2H), 3.86 (s, 3H), 3.42 (s, 4H), 3.11 (s, 4H), 2.92 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C26H29N4O [M+H+]: 413.2341, found: 413.2337.
1-(3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)-3-phenylurea (7au)
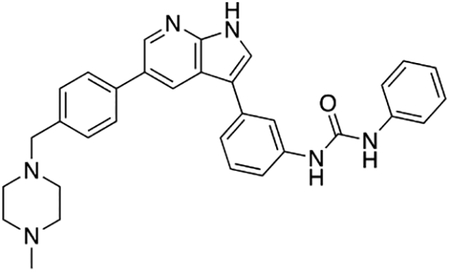
Compound 7au was prepared according to the general procedure for the Suzuki coupling and in situ deprotection as described above using (3-(3-phenylureido)phenyl)boronic acid (13e, 19.2 mg, 0.075 mmol, 1.5 eq). The crude product was purified by preparatory HPLC purification to afford 1-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)-3-phenylurea (7au, 5.4 mg, 21% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 2.0 Hz, 1H), 8.51 (d, J = 2.0 Hz, 1H), 8.08 (s, 1H), 7.78 – 7.71 (m, 3H), 7.60 (d, J = 6.9 Hz, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.46 – 7.40 (m, 1H), 7.41 – 7.36 (m, 2H), 7.36 – 7.27 (m, 2H), 7.24 – 7.17 (m, 1H), 3.76 (s, 2H), 3.13 (bs, 8H), 2.85 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C32H33N6O [M+H+]: 517.2716, found: 517.2713.
2-Fluoro-N-(furan-2-ylmethyl)-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzamide (7av)

2-Fluoro-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzoic acid (7i, 3.3 mg, 7.4 μmol), EDC×HCl (2.8 mg, 14.8 μmol, 2.0 eq), and HOBT (2.0 mg, 14.8 μmol, 2.0 eq) were dissolved in DMF (1.0 mL). Furfurylamine (1.4 mg, 1.3 μL, 14.8 μmol, 2.0 eq) and Et3N (1.5 mg, 2.0 μL, 14.8 μmol, 2.0 eq) were added and the reaction mixture was stirred at room temperature overnight. The solvent was evaporated and the crude mixture was purified by preparatory HPLC purification to afford 2-fluoro-N-(furan-2-ylmethyl)-5-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)benzamide (7av, 3.8 mg, 98% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.54 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 2.0 Hz, 1H), 8.08 (dd, J = 6.9, 2.4 Hz, 1H), 7.88 (ddd, J = 8.6, 4.8, 2.4 Hz, 1H), 7.78 (s, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.44 (dd, J = 1.9, 0.9 Hz, 1H), 7.32 (dd, J = 10.7, 8.6 Hz, 1H), 6.37 (dd, J = 3.2, 1.9 Hz, 1H), 6.33 (dd, J = 3.2, 0.9 Hz, 1H), 4.61 (s, 2H), 3.84 (s, 2H), 2.97 – 2.84 (m, 8H), 2.88 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C31H31FN5O2 [M+H+]: 524.2462, found: 524.2452.
1-Cyclohexyl-3-(4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7aw)

4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7e, 10 mg, 0.025 mmol, 2.5 eq) was dissolved in DCM (0.5 mL) and triethylamine (10 mg, 14 μL, 0.1 mmol, 10 eq) was added. The solution was added dropwise to a solution of triphosgene (3.0 mg, 0.01 mmol, 1.0 eq) in DCM (0.5 mL) and stirred for 5 min at room temperature. The reaction mixture was added to a solution of cyclohexylamine (19.8 mg, 23 μL, 0.2 mmol, 20.0 eq) in DCM (0.5 mL) and stirred overnight. The solvent was evaporated and the crude mixture was purified by preparatory HPLC purification to afford 1-cyclohexyl-3-(4-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7aw, 2.8 mg, 54% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.53 (m, 2H), 7.72 (d, J = 8.2 Hz, 2H), 7.68 (s, 1H), 7.64 – 7.59 (m, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.49 – 7.44 (m, 2H), 3.82 (s, 2H), 3.65 – 3.54 (m, 1H), 3.00 – 2.74 (bs, 8H), 2.88 (s, 3H), 2.00 – 1.90 (m, 2H), 1.84 – 1.71 (m, 2H), 1.65 (d, J = 12.9 Hz, 1H), 1.48 – 1.34 (m, 2H), 1.36 – 1.16 (m, 3H) ppm. HRMS (APCI+, m/z): calcd. for C32H39N6O [M+H+]: 523.3185, found: 523.3182.
(R)-1-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)-3-(1-phenylethyl)urea (7ax)

3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7h, 16 mg, 0.04 mmol, 2.5 eq) was dissolved in DCM (1.0 mL) and triethylamine (16.2 mg, 22 μL, 0.16 mmol, 10 eq) was added. The solution was added dropwise to a solution of triphosgene (4.75 mg, 0.016 mmol, 1.0 eq) in DCM (1.0 mL) and stirred for 5 min at room temperature. The reaction mixture was added to a solution of (R)-(+)-α-methylbenzylamine (38.8 mg, 41 μL, 0.32 mmol, 20.0 eq) in DCM (1.0 mL) and stirred overnight. The solvent was evaporated and the crude mixture was purified by preparatory HPLC purification to afford (R)-1-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)-3-(1-phenylethyl)urea (7ax, 6.7 mg, 31% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.65 (d, J = 2.0 Hz, 1H), 8.54 (d, J = 2.1 Hz, 1H), 7.94 (s, 1H), 7.78 – 7.72 (m, 3H), 7.51 (d, J = 8.0 Hz, 2H), 7.42 – 7.28 (m, 6H), 7.23 (t, J = 7.0 Hz, 1H), 7.18 – 7.11 (m, 1H), 4.95 (q, J = 7.0 Hz, 1H), 3.83 (s, 2H), 3.14 – 2.50 (bs, 8H), 2.88 (s, 3H), 1.50 (d, J = 7.0 Hz, 3H) ppm. HRMS (APCI+, m/z): calcd. for C34H37N6O [M+H+]: 545.3029, found: 545.3027.
1-(Furan-2-ylmethyl)-3-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7ay)

3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7h, 16 mg, 0.04 mmol, 2.5 eq) was dissolved in DCM (1.0 mL) and triethylamine (16.2 mg, 22 μL, 0.16 mmol, 10 eq) was added. The solution was added dropwise to a solution of triphosgene (4.75 mg, 0.016 mmol, 1.0 eq) in DCM (1.0 mL) and stirred for 5 min at room temperature. The reaction mixture was added to a solution of furfurylamine (31.1 mg, 28.3 μL, 0.32 mmol, 20.0 eq) in DCM (1.0 mL) and stirred overnight. The solvent was evaporated and the crude mixture was purified by preparatory HPLC purification to afford 1-(furan-2-ylmethyl)-3-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7ay, 8.6 mg, 41% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.74 (d, J = 2.0 Hz, 1H), 8.69 – 8.66 (m, 2H), 7.82 – 7.78 (m, 2H), 7.74 (s, 1H), 7.65 (d, J = 7.9 Hz, 2H), 7.41 – 7.34 (m, 4H), 6.35 (dd, J = 3.2, 1.9 Hz, 1H), 6.28 (dd, J = 3.2, 0.8 Hz, 1H), 4.41 (s, 2H), 3.89 (s, 2H), 3.21 (bs, 4H), 3.11 (bs, 4H), 2.89 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C31H33N6O2 [M+H+]: 521.2665, found: 521.2670.
1-(2-Methoxyethyl)-3-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7az)
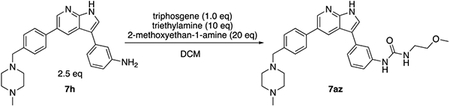
3-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)aniline (7h, 16 mg, 0.04 mmol, 2.5 eq) was dissolved in DCM (1.0 mL) and triethylamine (16.2 mg, 22 μL, 0.16 mmol, 10 eq) was added. The solution was added dropwise to a solution of triphosgene (4.75 mg, 0.016 mmol, 1.0 eq) in DCM (1.0 mL) and stirred for 5 min at room temperature. The reaction mixture was added to a solution of 2-methoxyethan-1-amine (24.0 mg, 27.8 μL, 0.32 mmol, 20.0 eq) in DCM (1.0 mL) and stirred overnight. The solvent was evaporated and the crude mixture was purified by preparatory HPLC purification to afford 1-(2-methoxyethyl)-3-(3-(5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)phenyl)urea (7az, 4.0 mg, 20% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.78 (d, J = 2.0 Hz, 1H), 8.60 (d, J = 2.0 Hz, 1H), 8.48 (d, J = 2.0 Hz, 1H), 7.81 (s, 1H), 7.76 (s, 1H), 7.61 – 7.53 (m, 4H), 7.45 – 7.25 (m, 4H), 3.54 (s, 2H), 3.50 (s, 2H), 3.47 (s, 3H), 3.27 – 3.12(bs, 8H), 2.87 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C29H35N6O2 [M+H+]: 499.2821, found: 499.2827.
General Procedure for the Synthesis of 9a, 9b, and 9c
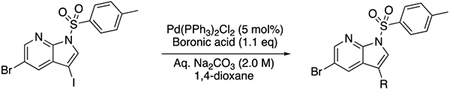
5-Bromo-3-iodo-1-tosyl-1H-pyrrolo[2,3-b]pyridine1 (8, 1.0 g, 2.1 mmol, 1.0 eq) was dissolved in 1,4-dioxane (20 mL). The boronic acid (2.3 mmol, 1.1 eq) was added, followed by Pd(PPh3)2Cl2 (70 mg, 0.1 mmol, 5 mol%). The reaction mixture was degassed by sonication under argon. Aqueous Na2CO3 was added and the reaction mixture was stirred at 45 °C until full completion (typically 8h). The reaction mixture was partitioned between EtOAc and brine, the layers separated, and the aqueous layer extracted with EtOAc (2x). The combined organic layers were dried with Na2SO4, filtered, and the solvent evaporated. The crude material was purified by column chromatography on silica (0 – 50% EtOAc in hexanes) to yield the desired product (9a–c).
4-(5-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (9a)

Following the general procedure described above, 4-(5-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (9a) was isolated as a tan-colored solid (461 mg, 47% yield). 1H NMR (400 MHz, Chloroform-d) δ 10.29 (bs, 1H), 8.53 (d, J = 2.2 Hz, 1H), 8.19 – 8.13 (m, 2H), 8.13 – 8.10 (m, 2H), 8.05 (s, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.49 (dd, J = 8.4, 6.9 Hz, 1H), 7.37 – 7.31 (m, 2H), 7.29 (dd, J = 6.9, 1.0 Hz, 1H), 2.41 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C21H16N4O2SBr [M+H+]: 467.0177, found: 467.0176.
5-Bromo-3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (9b)
Following the general procedure described above, 5-bromo-3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (9b) was isolated as a colorless solid (617 mg, 67% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.50 (d, J = 2.1 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.90 (s, 1H), 7.44 (td, J = 8.0, 5.9 Hz, 1H), 7.36 – 7.28 (m, 3H), 7.27 – 7.21 (m, 1H), 7.08 (tdd, J = 8.4, 2.6, 1.0 Hz, 1H), 2.39 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C20H15N2O2FSBr [M+H+]: 445.0022, found: 445.0020.
5-Bromo-3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (9c)

Following the general procedure described above, 5-bromo-3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (9c) was isolated as a colorless solid (850 mg, 88% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.50 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 2.1 Hz, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.89 (s, 1H), 7.56 – 7.50 (m, 1H), 7.46 – 7.40 (m, 2H), 7.40 – 7.34 (m, 1H), 7.31 (d, J = 7.8 Hz, 2H), 2.39 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C20H15N2O2SBrCl [M+H+]: 460.9726, found: 460.9715.
General Procedure for the Synthesis of 10a, 10b, and 10c

The substrate (9a–c, 1.0 mmol, 1.0 eq) was dissolved in 1,4-dioxane (40 mL) in a pressure tube, and 4-formylphenylboronic acid (225 mg, 1.5 mmol, 1.5 eq) was added. The reaction mixture was degassed by sonication under argon, and Pd(PPh3)4 (116 mg, 0.1 mmol, 10 mol%) was added. After addition of aqueous Na2CO3 (12.5 mL, 2.0 M), the reaction mixture was stirred at 100 °C overnight. The crude mixture was partitioned between EtOAc and brine, the layers separated and the aqueous layer extracted with EtOAc (3x). The combined organic layers were dried with Na2SO4, and filtered over Celite. The solvent was evaporated and the crude material purified by column chromatography on silica (0 – 100% EtOAc in hexanes) to afford the desired product (10a–c).
4-(3-(1H-Indazol-4-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10a)
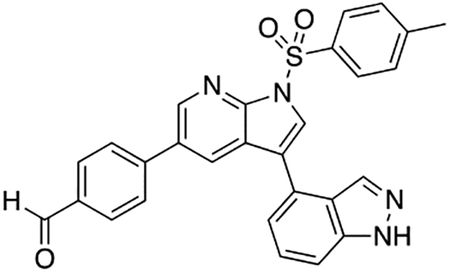
Following the general procedure described above, 4-(3-(1H-indazol-4-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10a) was isolated as a tan-colored solid (350 mg, 71% yield).
1H NMR (400 MHz, Chloroform-d) δ 10.37 (bs, 1H), 10.06 (s, 1H), 8.78 (d, J = 2.2 Hz, 1H), 8.23 – 8.17 (m, 3H), 8.14 (d, J = 1.1 Hz, 1H), 8.10 (s, 1H), 7.96 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.2 Hz, 2H), 7.59 – 7.51 (m, 2H), 7.40 – 7.32 (m, 3H), 2.41 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C28H21N4O3S [M+H+]: 493.1334, found: 493.1329.
4-(3-(3-Fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10b)

Following the general procedure described above, 4-(3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10b) was isolated as a colorless solid (466 mg, 99% yield). 1H NMR (400 MHz, Chloroform-d) δ 10.08 (s, 1H), 8.74 (d, J = 2.1 Hz, 1H), 8.26 (d, J = 2.1 Hz, 1H), 8.16 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 7.96 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.47 (td, J = 7.9, 5.8 Hz, 1H), 7.40 (dt, J = 7.7, 1.3 Hz, 1H), 7.36 – 7.28 (m, 3H), 7.10 (tdd, J = 8.4, 2.6, 1.1 Hz, 1H), 2.40 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H20N2O3FS [M+H+]: 471.1179, found: 471.1185.
4-(3-(3-Chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c)

Following the general procedure described above, scaled up to 2.0 mmol starting material, 4-(3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c) was isolated as a colorless solid (730 mg, 87% yield). 1H NMR (400 MHz, Chloroform-d) δ 10.08 (s, 1H), 8.74 (d, J = 2.1 Hz, 1H), 8.24 (d, J = 2.1 Hz, 1H), 8.16 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.2 Hz, 2H), 7.95 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.59 (t, J = 1.8 Hz, 1H), 7.50 (dt, J = 7.6, 1.5 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 7.38 (ddd, J = 7.9, 2.0, 1.3 Hz, 1H), 7.36 – 7.29 (m, 2H), 2.40 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C27H20N2O3ClS [M+H+]: 487.0883, found: 487.0878.
General Procedure for the Synthesis of 12a–l

The aldehyde (10a–c, or 11 (Goodfellow et al., 2013)) (0.5 mmol, 1.0 eq) was dissolved in dichloromethane and the corresponding amine (1.0 mmol, 2.0 eq) and Na(OAc)3BH (159 mg, 0.75 mmol, 1.5 eq) were added. The reaction mixture was stirred at room temperature overnight, partitioned between dichloromethane and brine. The aqueous layer was extracted with dichloromethane (2x), the combined organic layers dried with Na2SO4, filtered, and the solvent evaporated. The crude product was used in the deprotection step without further purification. The intermediate was dissolved in a mixture of acetone (20 mL), methanol (30 mL) and aqueous NaOH (2.0 M, 15 mL) and stirred at 65 °C for 3 h. The reaction mixture was partitioned between ethyl acetate and aqueous NaOH (1.0 M). The layers were separated and the aqueous layer was extracted with ethyl acetate (2x). The combined organic layers were washed with brine, dried with Na2SO4, filtered and the solvent evaporated. The crude material was purified by preparatory HPLC purification to afford the product (12a–l).
3-(4-(3-(1H-Indol-5-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)-8-oxa-3-azabicyclo[3.2.1]octane (12a)

Following the general procedure described above, with 4-(3-(1H-Indol-5-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (11 (Goodfellow et al., 2013), 100 mg, 0.2 mmol) and 8-oxa-3-azabicyclo[3.2.1]octane (45 mg, 0.4 mmol, 2.0 eq) as the starting materials, 3-(4-(3-(1H-indol-5-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)-8-oxa-3-azabicyclo[3.2.1]octane (12a) was isolated as a light-brown solid (37.2 mg, 43% yield over two steps). 1H NMR (500 MHz, Methanol-d4) δ 8.42 (d, J = 2.1 Hz, 1H), 8.40 (d, J = 2.1 Hz, 1H), 7.83 (d, J = 1.5 Hz, 1H), 7.55 (s, 1H), 7.52 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 8.4 Hz, 1H), 7.46 – 7.39 (m, 1H), 7.33 (d, J = 7.9 Hz, 2H), 7.24 (d, J = 3.1 Hz, 1H), 6.49 (dd, J = 3.1, 0.8 Hz, 1H), 4.20 (dd, J = 4.7, 2.3 Hz, 2H), 3.41 (s, 2H), 2.52 (d, J = 11.5 Hz, 2H), 2.25 (dd, J = 11.4, 2.1 Hz, 2H), 2.00 – 1.90 (m, 3H), 1.86 – 1.73 (m, 2H) ppm. HRMS (APCI+, m/z): calcd. for C32H33N6O [M+H+]: 517.2716, found: 517.2713.
3-(1H-Indol-5-yl)-5-(4-((8-methyl-3,8-diazabicyclo[3.2.1]octan-3-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12b)
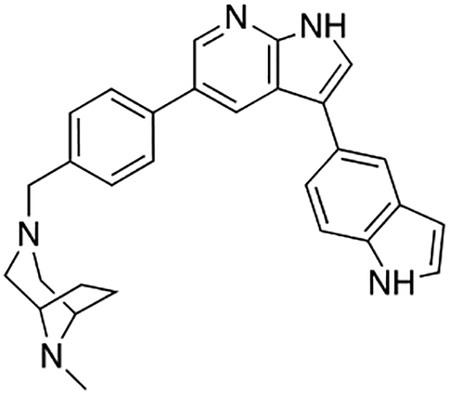
Following the general procedure described above, with 4-(3-(1H-Indol-5-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (11 (Goodfellow et al., 2013), 67.1 mg, 0.137 mmol) and 8-methyl-3,8-diazabicyclo[3.2.1]octane (16b, 55.5 mg, 0.44 mmol, 3.2 eq) as the starting materials, 3-(1H-indol-5-yl)-5-(4-((8-methyl-3,8-diazabicyclo[3.2.1]octan-3-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12b) was isolated as a tan-colored solid (2.6 mg, 4% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.57 (d, J = 2.0 Hz, 1H), 8.51 (d, J = 2.0 Hz, 1H), 7.87 (dd, J = 1.6, 0.7 Hz, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.65 (s, 1H), 7.56 – 7.38 (m, 5H), 7.27 (d, J = 3.2 Hz, 1H), 3.88 (s, 2H), 3.70 (s, 2H), 2.95 (d, J = 12.6 Hz, 2H), 2.80 (s, 3H), 2.57 (d, J = 13.0 Hz, 2H), 2.23 (s, 4H) ppm. HRMS (APCI+, m/z): calcd. for C29H30N5 [M+H+]: 448.2501, found: 448.2505.
3-(1H-Indol-5-yl)-5-(4-((3-methyl-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12c)
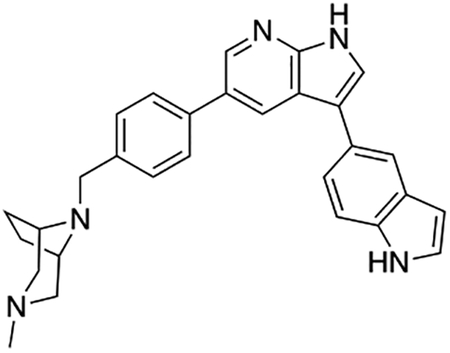
Following the general procedure described above, with 4-(3-(1H-Indol-5-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (11 (Goodfellow et al., 2013), 60.2 mg, 0.122 mmol) and 3-methyl-3,8-diazabicyclo[3.2.1]octane (16a, 36.6 mg, 0.29 mmol, 2.4 eq) as the starting materials, 3-(1H-indol-5-yl)-5-(4-((3-methyl-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12c) was isolated as a tan-colored solid (9.6 mg, 18% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.74 (d, J = 2.0 Hz, 1H), 8.60 (d, J = 2.0 Hz, 1H), 7.88 (dd, J = 1.7, 0.8 Hz, 1H), 7.86 – 7.79 (m, 2H), 7.74 (s, 1H), 7.68 – 7.64 (m, 3H), 7.54 – 7.50 (m, 1H), 7.48 – 7.43 (m, 2H), 4.12 (s, 2H), 3.84 (bs, 4H), 3.29 (d, J = 2.7 Hz, 2H), 3.10 (bs, 4H), 2.69 (s, 3H) ppm. HRMS (APCI+, m/z): calcd. for C29H30N5 [M+H+]: 448.2501, found: 448.2503.
4-(5-(4-((8-Methyl-3,8-diazabicyclo[3.2.1]octan-3-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (12d)
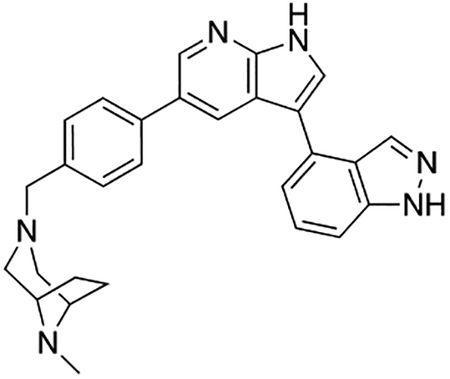
Following the general procedure described above, with 4-(3-(1H-indazol-4-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10a, 49.2 mg, 0.1 mmol) and 8-methyl-3,8-diazabicyclo[3.2.1]octane (16b, 39.1 mg, 0.31 mmol, 3.1 eq) as the starting materials, 4-(5-(4-((8-methyl-3,8-diazabicyclo[3.2.1]octan-3-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (12d) was isolated as a light-brown solid (11.7 mg, 26% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.61 (d, J = 2.0 Hz, 1H), 8.55 (d, J = 2.0 Hz, 1H), 8.18 (d, J = 0.9 Hz, 1H), 7.95 (s, 1H), 7.67 (d, J = 8.2 Hz, 2H), 7.55 – 7.50 (m, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.43 (dd, J = 6.5, 1.4 Hz, 1H), 3.88 (s, 2H), 3.70 (s, 2H), 2.94 (dd, J = 13.1, 2.8 Hz, 2H), 2.80 (s, 3H), 2.67 – 2.62 (m, 2H), 2.27 – 2.19 (bs, 4H) ppm. HRMS (APCI+, m/z): calcd. for C28H29N6 [M+H+]: 449.2454, found: 449.2462.
4-(5-(4-((3-Methyl-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (12e)
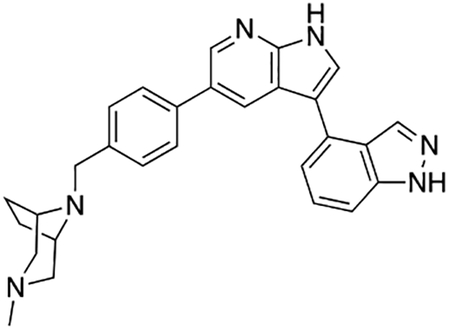
Following the general procedure described above, with 4-(3-(1H-indazol-4-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10a, 49.2 mg, 0.1 mmol) and 3-methyl-3,8-diazabicyclo[3.2.1]octane (16a, 34.0 mg, 0.27 mmol, 2.7 eq) as the starting materials, 4-(5-(4-((3-methyl-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-indazole (12e) was isolated as a light-brown solid (12.0 mg, 27% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.60 (d, J = 2.1 Hz, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 0.7 Hz, 1H), 7.90 (s, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.62 (d, J = 8.2 Hz, 2H), 7.54 – 7.51 (m, 2H), 7.43 (dd, J = 5.6, 2.3 Hz, 1H), 4.09 (s, 2H), 3.80 (s, 2H), 3.18 (d, J = 11.9 Hz, 2H), 2.94 (d, J = 12.6 Hz, 2H), 2.61 (s, 3H), 2.45 – 2.36 (m, 2H), 2.14 (d, J = 8.6 Hz, 2H) ppm. HRMS (APCI+, m/z): calcd. for C28H29N6 [M+H+]: 449.2454, found: 449.2455.
3-(3-Chlorophenyl)-5-(4-((3-methyl-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12f)

Following the general procedure described above, with 4-(3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c, 48.6 mg, 0.1 mmol) and 3-methyl-3,8-diazabicyclo[3.2.1]octane (16a, 25.2 mg, 0.2 mmol, 2.0 eq) as the starting materials, 3-(3-chlorophenyl)-5-(4-((3-methyl-3,8-diazabicyclo[3.2.1]octan-8-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (12f) was isolated as a pale-yellow solid (18.0 mg, 41% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.54 (d, J = 2.1 Hz, 1H), 8.46 (d, J = 2.1 Hz, 1H), 7.81 – 7.75 (m, 3H), 7.70 (t, J = 1.9 Hz, 1H), 7.68 – 7.61 (m, 3H), 7.45 (t, J = 7.9 Hz, 1H), 7.30 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 4.11 (s, 2H), 3.82 (s, 2H), 3.25 – 3.16 (m, 2H), 3.04 – 2.94 (m, 2H), 2.62 (s, 3H), 2.42 (dd, J = 9.6, 4.7 Hz, 2H), 2.17 (t, J = 7.3 Hz, 2H) ppm. HRMS (APCI+, m/z): calcd. for C27H28N4Cl [M+H+]: 443.2002, found: 443.2005.
5-(4-([1,4′-Bipiperidin]-1′-ylmethyl)phenyl)-3-(3-fluorophenyl)-1H-pyrrolo[2,3-b]pyridine (12g)

Following the general procedure described above, with 4-(3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10b, 26.0 mg, 0.055 mmol) and 4-piperidinopiperidine (18.6 mg, 0.11 mmol, 2.0 eq) as the starting materials, 5-(4-([1,4′-bipiperidin]-1′-ylmethyl)phenyl)-3-(3-fluorophenyl)-1H-pyrrolo[2,3-b]pyridine (12g) was isolated as an off-white solid (6.2 mg, 24% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.58 (d, J = 2.0 Hz, 1H), 8.56 (d, J = 2.1 Hz, 1H), 7.86 (d, J = 8.2 Hz, 2H), 7.82 (s, 1H), 7.66 (d, J = 8.2 Hz, 2H), 7.57 – 7.53 (m, 1H), 7.52 – 7.42 (m, 2H), 7.04 (tdd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.43 (s, 2H), 3.72 (d, J = 12.9 Hz, 2H), 3.63 – 3.47 (m, 4H), 3.23 – 3.12 (m, 4H), 3.03 (t, J = 11.3 Hz, 2H), 2.41 (d, J = 13.5 Hz, 2H), 2.20 – 2.08 (m, 3H), 2.04 – 1.94 (m, 2H), 1.86 – 1.73 (m, 2H), 1.64 – 1.46 (m, 1H), 1.42 – 1.26 (m, 2H) ppm. HRMS (APCI+, m/z): calcd. for C30H34N4F [M+H+]: 469.2768, found: 469.2771.
2-(4-(4-(3-(3-Fluorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (12h)

Following the general procedure described above, with 4-(3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10b, 318 mg, 0.675 mmol) and 1-(2-hydroxyethyl)piperazine (176 mg, 166 μL, 1.35 mmol, 2.0 eq) as the starting materials, 2-(4-(4-(3-(3-fluorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (12h) was isolated as an off-white solid (153.7 mg, 53% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.62 (d, J = 2.0 Hz, 1H), 8.60 (d, J = 2.0 Hz, 1H), 7.85 (s, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.62 (d, J = 8.2 Hz, 2H), 7.55 (dt, J = 7.8, 1.3 Hz, 1H), 7.51 – 7.42 (m, 2H), 7.05 (dddd, J = 9.0, 8.2, 2.6, 1.1 Hz, 1H), 4.22 (s, 2H), 3.95 – 3.85 (m, 2H), 3.58 (bs, 4H), 3.34 (bs, 6H) ppm. HRMS (APCI+, m/z): calcd. for C26H28N4OF [M+H+]: 431.2247, found: 431.2250.
(R)-1-(4-(3-(3-Fluorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)pyrrolidin-3-ol (12i)

Following the general procedure described above, with 4-(3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10b, 318 mg, 0.675 mmol) and (R)-3-pyrrolidinol (118 mg, 109 μL, 1.35 mmol, 2.0 eq) as the starting materials, (R)-1-(4-(3-(3-fluorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)pyrrolidin-3-ol (12i) was isolated as an off-white solid (113 mg, 43% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.56 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 2.0 Hz, 1H), 7.84 – 7.77 (m, 3H), 7.68 – 7.61 (m, 2H), 7.52 (dt, J = 7.8, 1.3 Hz, 1H), 7.50 – 7.38 (m, 2H), 7.02 (dddd, J = 9.0, 8.2, 2.6, 1.1 Hz, 1H), 4.66 – 4.34 (m, 3H), 3.82 – 3.42 (m, 2H), 3.41 – 3.20 (m, 2H), 2.49 – 1.92 (m, 2H) ppm. HRMS (APCI+, m/z): calcd. for C24H23N3OF [M+H+]: 388.1825, found: 388.1824.
5-(4-((4-(tert-Butyl)piperazin-1-yl)methyl)phenyl)-3-(3-fluorophenyl)-1H-pyrrolo[2,3-b]pyridine (12j)
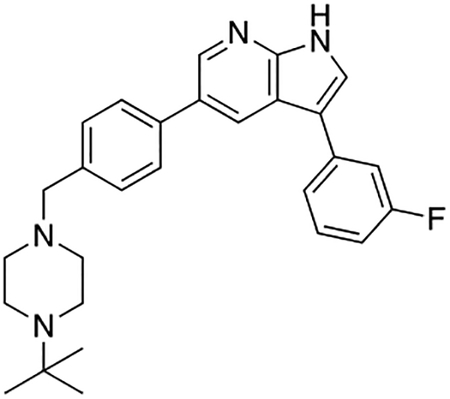
Following the general procedure described above, with 4-(3-(3-fluorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10b, 32.8 mg, 0.055 mmol) and 1-tert-butylpiperazine (15.7 mg, 0.11 mmol, 2.0 eq) as the starting materials, 5-(4-((4-(tert-butyl)piperazin-1-yl)methyl)phenyl)-3-(3-fluorophenyl)-1H-pyrrolo[2,3-b]pyridine (12j) was isolated as an off-white solid (9.8 mg, 40% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.63 (d, J = 1.9 Hz, 1H), 8.60 (bs, 1H), 7.85 (s, 1H), 7.83 – 7.78 (m, 2H), 7.65 – 7.60 (m, 2H), 7.56 (dt, J = 7.8, 1.3 Hz, 1H), 7.52 – 7.42 (m, 2H), 7.05 (dddd, J = 9.0, 8.2, 2.6, 1.0 Hz, 1H), 4.22 (s, 2H), 3.89 – 3.04 (m, 8H), 1.44 (s, 9H) ppm. HRMS (APCI+, m/z): calcd. for C28H32N4F [M+H+]: 443.2611, found: 443.2610.
2-(4-(4-(3-(3-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (Protstetin/12k)

Following the general procedure described above, with 4-(3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c, 418 mg, 0.86 mmol) and 1-(2-hydroxyethyl)piperazine (224 mg, 211 μL, 1.72 mmol, 2.0 eq) as the starting materials, 2-(4-(4-(3-(3-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (Prostetin/12k) was isolated as an off-white solid (139.9 mg, 36% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.57 (d, J = 2.0 Hz, 1H), 8.54 (d, J = 2.0 Hz, 1H), 7.80 (s, 1H), 7.76 (d, J = 8.3 Hz, 2H), 7.65 (t, J = 1.9 Hz, 1H), 7.64 – 7.57 (m, 3H), 7.42 (t, J = 7.9 Hz, 1H), 7.29 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 4.27 (s, 2H), 3.93 – 3.86 (m, 2H), 3.62 (s, 4H), 3.41 (s, 4H), 3.35 – 3.31 (m, 2H) ppm. HRMS (APCI+, m/z): calcd. for C26H28N4OCl [M+H+]: 447.1952, found: 447.1954.
(R)-1-(4-(3-(3-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)pyrrolidin-3-ol (12l)

Following the general procedure described above, with 4-(3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c, 177 mg, 0.86 mmol) and (R)-3-pyrrolidinol (150 mg, 139 μL, 1.72 mmol, 2.0 eq) as the starting materials, (R)-1-(4-(3-(3-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)pyrrolidin-3-ol (12l) was isolated as an off-white solid (177 mg, 51% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.52 (d, J = 2.0 Hz, 1H), 8.42 (d, J = 2.0 Hz, 1H), 7.78 – 7.71 (m, 3H), 7.64 – 7.60 (m, 3H), 7.58 (ddd, J = 7.7, 1.7, 1.1 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.25 (ddd, J = 8.0, 2.2, 1.0 Hz, 1H), 4.58 (s, 1H), 4.54 – 4.32 (m, 2H), 3.82 – 3.39 (m, 4H), 2.50 – 1.96 (m, 2H) ppm. HRMS (APCI+, m/z): calcd. for C24H23N3OCl [M+H+]: 404.1530, found: 404.1529.
General Procedure for the Synthesis of Boronic Acids 13a–e

The boronic acid (3-aminophenylboronic acid or 4-aminophenylboronic acid, 87 mg, 0.5 mmol, 2.5 eq) was suspended in DCM (4 mL) and triethylamine (202 mg, 277 μL, 2.0 mmol, 10 eq) was added. The resulting solution was added dropwise to a solution triphosgene (59 mg, 0.2 mmol, 1.0 eq) in DCM (6 mL) and stirred for 5 min at room temperature. The amine (0.6 mmol, 3 eq) or alcohol (excess) was added dropwise and the mixture was stirred overnight at room temperature. The solvent was evaporated, toluene was added and the solvent was evaporated again. The crude material was analyzed by NMR and used in the next step without further purification.
(3-(3-Methylureido)phenyl)boronic acid (13a)

Following the general procedure described above, with 3-aminophenylboronic acid and methylamine, (3-(3-methylureido)phenyl)boronic acid (13a) was isolated as an off-white solid and used in the next step without further purification. 1H NMR (400 MHz, Methanol-d4) δ 7.49 (bs, 1H), 7.38 (bs, 1H), 7.22 (d, J = 5.3 Hz, 2H), 2.77 (s, 3H).
(3-((Methoxycarbonyl)amino)phenyl)boronic acid (13b)

Following the general procedure described above, with 3-aminophenylboronic acid and methanol, (3-((methoxycarbonyl)amino)phenyl)boronic acid (13b) was isolated as an off-white solid and used in the next step without further purification. 1H NMR (400 MHz, Methanol-d4) δ 7.59 (bs, 1H), 7.48 (bs, 1H), 7.26 (d, J = 5.2 Hz, 2H), 3.73 (s, 3H).
(4-(3-Methylureido)phenyl)boronic acid (13c)
Following the general procedure described above, with 4-aminophenylboronic acid and methylamine, (4-(3-methylureido)phenyl)boronic acid (13c) was isolated as an off-white solid and used in the next step without further purification. 1H NMR (400 MHz, Methanol-d4) δ 7.62 (d, J = 8.5 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 2.77 (s, 3H).
(4-((Methoxycarbonyl)amino)phenyl)boronic acid (13d)

Following the general procedure described above, with 4-aminophenylboronic acid and methanol, (4-((methoxycarbonyl)amino)phenyl)boronic acid (13d) was isolated as an off-white solid and used in the next step without further purification. 1H NMR (400 MHz, Methanol-d4) δ 7.65 (d, J = 8.6 Hz, 1H), 7.43 (d, J = 8.6 Hz, 2H), 3.74 (s, 3H).
(3-(3-Phenylureido)phenyl)boronic acid (13e)

Following the general procedure described above, with 3-aminophenylboronic acid and aniline, (3-(3-phenylureido)phenyl)boronic acid (13e) was isolated as an off-white solid and used in the next step without further purification. 1H NMR (400 MHz, Methanol-d4) δ 7.62 (bs, 1H), 7.51 (d, J = 7.4 Hz, 1H), 7.34 – 7.23 (m, 2H), 7.08 (dd, J = 8.5, 7.4 Hz, 2H), 6.71 (dt, J = 7.7, 1.1 Hz, 2H), 6.67 (tt, J = 7.4, 1.1 Hz, 1H).
General Procedure for the Synthesis of Bicyclic Piperazines 16a–b

The Boc-protected bicyclic piperazine (14a–b; 250 mg, 1.18 mmol, 1.0 eq) was dissolved in a mixture of THF/MeOH (1:1). Formaldehyde (37% in H2O, 287 μL, 3.53 mmol, 3.0 eq) was added, followed by addition of Na(OAc)3BH (274.5 mg, 1.30 mmol, 1.1 eq), and the reaction mixture was stirred overnight. The solvent was evaporated and the crude product purified by column chromatography on silica (0 – 20% MeOH in DCM). The product was visualized on the TLC plate using a KMnO4 stain.
In order to remove the Boc-group, the product (15a–b) was dissolved in DCM, an excess of TFA was added, and the reaction mixture was stirred at room temperature overnight. The solvent was evaporated, toluene was added and the solvent was evaporated again. The crude products (16a–b) were used in the next step without further purification and the spectroscopic data matched those reported in literature (Paliulis et al., 2013).
In vitro microsomal stability
The microsomal stability assay was performed in round-bottom 96-well plates (Perkin-Elmer). To each well was added phosphate buffer (182.2 μL, pH 7.4, 100 mM) followed by addition of NADPH regenerating system solution A (10 μL), and NADPH regenerating system solution B (2 μL) (Corning). A stock solution of the compound to be analyzed (0.8 μL. 5 mM) or ethoxycoumarin (positive control, 0.8 μL, 5 mM) was added and the mixture was warmed to 37 °C for 5 min. The mouse microsomes (CD-1, 5 μL, thawed in a 37 °C water bath before use, 20 mg/mL, Thermo Fisher) were added. At selected time points (0, 15, 30, 45, 60 and 120 min) aliquots (15 μL) were withdrawn from the plate and quenched upon addition to cold acetonitrile (60 μL), containing an internal standard (5 μM) in a 96-well plate. The samples were centrifuged at 13,000 rpm for 10 min at 4 °C. The supernatant (40 μL) was withdrawn and transferred to a sample vial with insert. The samples were analyzed by LC-MS.
LC-MS analysis was performed on a platform comprising a Thermo Scientific Dionex Ultimate 3000 and a Bruker amaZon SL equipped with an electrospray ionization source controlled by Bruker Hystar 3.2. Chromatographic separation was performed by injecting 5 μL of the sample onto an Agilent Eclipse Plus C18 column (2.1 × 50 mm, 3.5 μm) maintained at 20 °C. The flow rate was maintained at 400 μL/min. The initial flow conditions were 60% solvent A (water containing 0.1% acetic acid) and 40% solvent B (methanol containing 0.1% acetic acid). Solvent B was raised to 60% over 0.25 min and to 70% by 6.75 min. Solvent B was raised to 95% by 7.00 min and lowered back to initial conditions (40%) by 8.00 min with a total run time of 9.00 min.
In vivo pharmacokinetics
For in vivo administration, 40 mg/mL DMSO stocks of compound 1 and Prostetin/12k were further diluted to 2 mg/mL in a vehicle containing 40% PEG-400 (Sigma) and 55% of a (2-hydroxypropyl)-beta-cyclodextrin solution (20% in nanopure water, Sigma). Compounds were delivered at 10 mg/kg by intraperitoneal injection or oral gavage to 8-week old wild-type mice. After 15 min, 3 h, 8 h or 24 h, animals were decapitated to collect trunk blood and dissect fresh brain tissue. Blood samples were treated with EDTA solution (50 mM final concentration) to prevent coagulation and centrifuged for 10 min at 845 rcf at 4°C to separate plasma. Fresh brains were lysed in 5 volumes of ddH2O.
Plasma samples were transferred to Eppendorf tubes (50 μL per sample) and diluted with acetonitrile (200 μL). The samples were sonicated for 1 min followed by centrifugation at 16,000 rpm for 10 min. The supernatant was transferred to another Eppendorf tube and the solvent evaporated overnight. The samples were reconstituted in a mixture of 40 μL acetonitrile and 10 μL water transferred to a sample vial with insert, and analyzed by LC-MS.
Homogenized brain samples (250 μL) were transferred to Eppendorf tubes and diluted with acetonitrile (750 μL). The samples were sonicated for 1 min followed by centrifugation at 16,000 rpm for 10 min. The supernatant (900 μL) was transferred to another Eppendorf tube and the solvent evaporated overnight. The samples were reconstituted in a mixture of 80 μL acetonitrile and 20 μL water, transferred to a sample vial with insert, and analyzed by LC-MS.
LC-MS analysis was performed on a platform comprising a Thermo Scientific Dionex Ultimate 3000 and a Bruker amaZon SL equipped with an electrospray ionization source controlled by Bruker Hystar 3.2. The compound concentration in each plasma sample was determined using a calibration curve. Chromatographic separation was performed by injecting 10 μL of the sample onto an Agilent Eclipse Plus C18 column (2.1 × 50 mm, 3.5 μm) maintained at 20 °C. The flow rate was maintained at 400 μL/min. The initial flow conditions were 60% solvent A (water containing 0.1% acetic acid) and 40% solvent B (methanol containing 0.1% acetic acid). Solvent B was raised to 60% over 0.25 min and to 70% by 6.75 min. Solvent B was raised to 95% by 7.00 min and lowered back to initial conditions (40%) by 8.00 min with a total run time of 9.00 min.
Commercial In Vitro Kinase Inhibition Assays
Kinase inhibition assays were carried out by Reaction Biology Corp. using the “HotSpot” assay platform as described (Anastassiadis et al., 2011). In brief, recombinant kinases and their appropriate substrates were combined at 25°C with test compound and a mixture of ATP and 33P ATP to a final concentration of 10 μM. For IC50 determination, 10 concentrations of each compound were tested for each kinase (0.0002 – 0.1 μM, 2-fold serial dilution for HGK and MINK1; 0.002 – 1 μM, 2-fold serial dilution for MLK1 and MLK3); otherwise, single compound concentrations were used, as indicated (Tables S3 – S6). After 120 min, reactions were spotted onto P81 ion exchange filter paper, and unbound phosphate was removed through washing. Background signal was assessed from control reactions containing inactive enzymes. Kinase activity data were expressed as the percent remaining kinase activity in samples containing test compound vs. samples containing DMSO alone.
Microglia LPS treatment and TNFα ELISAs
Immortalized N9 mouse microglia cells (Corradin et al., 1993) were plated at 8000 cells / well in gelatin-coated (0.1%, EMD Millipore) 96-well plates in DMEM containing FBS (10%, Fisher Scientific) and Glutamax (1%, Life Technologies). 24 h after plating, cells were treated with LPS from E. coli O111:B4 (Sigma) plus test compounds at the indicated concentrations. 24 h after the initiation of LPS treatment, 50 μL of cell culture media (25% of the final culture volume) were used to perform quantitative ELISAs for TNFα release according to the manufacturer’s instructions (R&D Systems). Absorbance was assessed at 450 nm on a spectrophotometer (TECAM), and readings at 540 nm were used for wavelength correction. N9 cells remaining in the culture dishes were treated with AlamarBlue (Thermo Fisher) to assess viability. 5 μL AlamarBlue was added to each well (containing 50 μL culture media) and incubated for 4h at 37 °C. Absorbance was assessed at 570 nm, and readings at 600 nm were used for wavelength correction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-way hierarchical clustering analysis was performed using Spotfire software (version 2017, TIBCO) to rank the shared targets of hit compounds (Fig. 1B). Strong inhibition of kinase activity was defined as <20% kinase activity remaining at 0.5 μM inhibitor concentrations. All statistical analyses were performed with Prism 8 (GraphPad Software). Data are represented as means ± SEM, and were assessed for normal distribution. Plots of means from motor neuron survival assays represent triplicate wells from single assays. Unpaired, two-tailed t-tests were used to compare the effects of treatment on motor neurons in western blot assays. Regression analyses (Pearson’s r) were used to evaluate the degree to which the inhibition of the activity of a given kinase correlated with neuroprotection in the CPA assay for the listed compound 1 analogs (Table S2) at the indicated concentrations. In Fig. 3D, kinases whose activity was poorly inhibited by all 10 of the analogs tested (>20% activity remaining) were excluded from analysis. Two-way ANOVAs were used to compare the effects of LPS vs. compound 1 analog treatment on N9 microglia. ANOVAs were supplemented with Dunnett’s multiple comparisons tests to assess interactions between conditions.
DATA AND CODE AVAILABILITY
This study did not generate any datasets or code.
Supplementary Material
Table S6. Inhibition profile for 371 wild-type human kinases using radioligand binding assay performed by Reaction Biology Corp for compound 1 and Prostetin/12k analogs, Related to Figure 4. Compounds were tested in single dose mode at a concentration of 0.1 μM. Reactions were carried out at 10 μM ATP.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Cleaved caspase 3 (Asp175) | Cell Signaling Technology | Cat # 9661; RRID:AB_2341188 |
| Rabbit monoclonal anti-c-Jun | Cell Signaling Technology | Cat # 9165; RRID:AB_213065 |
| Rabbit polyclonal anti-Phospho-c-Jun (Ser63) | Cell Signaling Technology | Cat # 9261; RRID:AB_2130162 |
| Rabbit polyclonal anti-JNK | Cell Signaling Technology | Cat # 9252; RRID:AB_2250373 |
| Rabbit monoclonal anti-Phospho-JNK (Thr183/Tyr185) | Cell Signaling Technology | Cat # 4668; RRID:AB_823588 |
| Rabbit polyclonal anti-MKK4 | Cell Signaling Technology | Cat# 9152; RRID:AB_330905 |
| Rabbit monoclonal anti-Phospho-MKK4 (Ser257) | Cell Signaling Technology | Cat # 4514; RRID:AB_2140946 |
| Rabbit polyclonal GAPDH (FL-335) | Santa Cruz Biotechnologies | Cat # sc-25778; RRID:AB_10167688 |
| Goat anti-rabbit HRP-linked | Cell Signaling Technology | Cat # 7074; RRID:AB_2099233 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Mouse CD-1 microsomes | Thermo Fisher | Cat # MSMCPL |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Knockout serum replacement | Life Technologies | Cat # 10828–028 |
| Human recombinant fibroblast growth factor 2 | Peprotech | Cat # PHG0263 |
| N-2 supplement | Thermo Fisher | Cat # 17502048 |
| B-27 supplement | Thermo Fisher | Cat # 17504044 |
| Ascorbic acid | Sigma-Aldrich | Cat # A4403 |
| Y-27632 dihydrochloride | Abcam | Cat # ab120129 |
| SB 431542 hydrate | Sigma-Aldrich | Cat # S4317 |
| LDN193189 | Stemgent | Cat # 04–0074 |
| CHIR 99021 | Tocris | Cat # 4423 |
| Retinoic acid (all trans) | Sigma-Aldrich | Cat # R2625 |
| Sonic Hedgehog agonist | Millipore | Cat # 566660 |
| Human recombinant brain-derived neurotrophic factor | Peprotech | Cat # 450–02 |
| DAPT | Tocris | Cat # 2643 |
| Human recombinant glial-derived neurotrophic factor | R&D Systems | Cat # 212-GD-050 |
| Poly-Ornithine | Sigma-Aldrich | Cat # 3655 |
| Mouse laminin | Thermo Fisher | Cat # 23017–015 |
| Uridine | Sigma-Aldrich | Cat # U3750 |
| Fluorodeoxyuridine | Sigma-Aldrich | Cat # F0503 |
| Human recombinant ciliary neurotrophic factor | Peprotech | Cat # 257-NT-050 |
| Human recombinant insulin-like growth factor 1 | Peprotech | Cat # 291-G1 |
| Cyclopiazonic acid | Tocris | Cat # 1235 |
| WZ4003 | Tocris | Cat # 5177 |
| HTH 01–015 | Selleckchem | Cat # S7318 |
| PF-6260933 | Tocris | Cat # 5752 |
| GNE-495 | Cayman Chemical | Cat # 21808 |
| URMC-099 | Selleckchem | Cat # S7343 |
| Brilliant Black | Sigma | Cat # 211842 |
| Fetal Bovine Serum, embryonic stem cell grade | Fisher Scientific | Cat # SH3007003E |
| Forskolin | Sigma-Aldrich | Cat # F6886 |
| IBMX | Tocris | Cat # 2845 |
| Lipopolysaccharides from E. coli O111:B4 | Sigma-Aldrich | Cat # L2640 |
| PhosSTOP phosphatase inhibitor tablets | Sigma-Aldrich | Cat # 4906845001 |
| Complete protease inhibitor cocktail | Sigma-Aldrich | Cat # 1183617001 |
| 7-Ethoxycoumarin | Sigma-Aldrich | Cat # 1379 |
| PEG-400 | Sigma-Aldrich | Cat # 202398 |
| (2-hydroxypropyl)-beta-cyclodextrins | Sigma-Aldrich | Cat # 332607 |
| 5-Bromo-1H-pyrrolo[2,3-b]pyridine | AK Scientific | 183208-35-7 |
| 4-formylphenylboronic acid | Sigma-Aldrich | 87199-17-5 |
| trans-Dichlorobis(triphenylphosphine)palladium(II), 99% | Strem | 13965-03-2 |
| 1-methylpiperazine | Sigma-Aldrich | 109-01-3 |
| N-iodosuccinimide | Sigma-Aldrich | 516-12-1 |
| Di-tert-butyl dicarbonate 99% (GC) | AK Scientific | 24424-99-5 |
| 4-(Dimethylamino)pyridine | Sigma-Aldrich | 1122-58-3 |
| Tetrakis(triphenylphosphine)palladium(0) | Strem | 14221-01-3 |
| 5-indolylboronic acid | Sigma-Aldrich | 144104-59-6 |
| indole-2-boronic acid pinacol ester | Sigma-Aldrich | 476004-81-6 |
| 6-ethoxy-3-pyridinylboronic acid | Sigma-Aldrich | 612845-44-0 |
| 4-(dibenzofuranyl)boronic acid | Sigma-Aldrich | 100124-06-9 |
| 1,4-benzodioxane-6-boronic acid | Sigma-Aldrich | 164014-95-3 |
| 4-aminophenylboronic acid pinacol ester | Sigma-Aldrich | 214360-73-3 |
| benzo[b]thien-3-ylboronic acid | Sigma-Aldrich | 113893-08-6 |
| benzo[b]thien-2-ylboronic acid | Sigma-Aldrich | 98437-23-1 |
| 3-Aminophenylboronic acid hydrochloride | Sigma-Aldrich | 85006-23-1 |
| 3-carboxy-4-fluorophenylboronic acid | AK Scientific | 872460-12-3 |
| 3-carboxyphenylboronic acid | Sigma-Aldrich | 25487-66-5 |
| 2-aminopyrimidine-5-boronic acid | Sigma-Aldrich | 936250-22-5 |
| 2-amino-3-methylpyridine-5-boronic acid pinacol ester | AK Scientific | 1111637-91-2 |
| 2-aminopyridine-5-boronic acid pinacol ester | Sigma-Aldrich | 827614-64-2 |
| 4-pyridinylboronic acid | Sigma-Aldrich | 1692-15-5 |
| 4-cyanophenylboronic acid | Sigma-Aldrich | 126747-14-6 |
| 3-nitrophenylboronic acid | Sigma-Aldrich | 13331-27-6 |
| 4-isopropoxyphenylboronic acid | Sigma-Aldrich | 153624-46-5 |
| 3-hydroxyphenylboronic acid | Sigma-Aldrich | 87199-18-6 |
| 1-methylpyrazole-4-boronic acid pinacol ester | Sigma-Aldrich | 761446-44-0 |
| 5-(morpholinomethyl)-2-thiopheneboronic acid pinacol ester | Sigma-Aldrich | 950603-39-1 |
| 6-(methylsulfonyl)pyridine-3-boronic acid | Sigma-Aldrich | 1088496-41-6 |
| 1H-indazole-5-boronic acid | Sigma-Aldrich | 338454-14-1 |
| 3-furanylboronic acid | Sigma-Aldrich | 55552-70-0 |
| 4-acetylphenylboronic acid | Sigma-Aldrich | 149104-90-5 |
| 3-fluorophenylboronic acid | Sigma-Aldrich | 768-35-4 |
| 4-(trifluoromethoxy)phenylboronic acid | Sigma-Aldrich | 139301-27-2 |
| 1H-Indazole-5-boronic acid | Sigma-Aldrich | 338454-14-1 |
| 3-(trifluoromethyl)phenylboronic acid | Sigma-Aldrich | 1423-26-3 |
| phenylboronic acid | Sigma-Aldrich | 98-80-6 |
| 3-chlorophenylboronic acid | AK Scientific | 63503-60-6 |
| 2,4-dichlorophenylboronic acid | Sigma-Aldrich | 68716-47-2 |
| 2,5-dimethylphenylboronic acid | Sigma-Aldrich | 85199-06-0 |
| 1-methyl-1H-indazole-6-boronic acid | Sigma-Aldrich | 1150114-80-9 |
| 1-benzyl-1H-pyrazole-4-boronic acid | Sigma-Aldrich | 852362-22-2 |
| 1,3,5-trimethyl-1H-pyrazole-4-boronic acid pinacol ester | Sigma-Aldrich | 844891-04-9 |
| 3,5-bis(trifluoromethyl)phenylboronic acid | Sigma-Aldrich | 73852-19-4 |
| 4-(4-methoxybenzyloxy)phenylboronic acid | Sigma-Aldrich | 156635-90-4 |
| 3,4,5-trimethoxyphenylboronic acid | Sigma-Aldrich | 182163-96-8 |
| 4-methoxy-3-methylphenylboronic acid | Sigma-Aldrich | 175883-62-2 |
| 3-(benzyloxy)phenylboronic acid | Sigma-Aldrich | 156682-54-1 |
| 2-chlorophenylboronic acid | Sigma-Aldrich | 3900-89-8 |
| 2-methoxyphenylboronic acid | Sigma-Aldrich | 5720-06-9 |
| N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC.HCl) | Sigma-Aldrich | 25952-53-8 |
| 1-Hydroxybenzotriazole hydrate (HOBT) | Sigma-Aldrich | 123333-53-9 |
| Furfurylamine | Sigma-Aldrich | 617-89-0 |
| Triphosgene | Sigma-Aldrich | 32315-10-9 |
| cyclohexylamine | Sigma-Aldrich | 108-91-8 |
| (R)-(+)-α-methylbenzylamine | Sigma-Aldrich | 3886-69-9 |
| 2-methoxyethan-1-amine | Sigma-Aldrich | 109-85-3 |
| 8-oxa-3-azabicyclo[3.2.1]octane | Sigma-Aldrich | 280-13-7 |
| 4-piperidinopiperidine | Sigma-Aldrich | 4897-50-1 |
| 1-(2-hydroxyethyl)piperazine | Sigma-Aldrich | 103-76-4 |
| (R)-3-pyrrolidinol | Sigma-Aldrich | 2799-21-5 |
| 1-tert-butylpiperazine | Ark Pharm | 38216-72-7 |
| t-Butyl-3,8-diazabicyclo[3.2.1]octane-3-carboxylate | Ark Pharm | 201162-53-0 |
| t-Butyl-3,8-diazabicyclo[3.2.1]octane-8-carboxylate | Ark Pharm | 149771-44-8 |
| Formaldehyde (37% in H2O) | Sigma-Aldrich | 50-00-0 |
| Sodium triacetoxyborohydride | Sigma-Aldrich | 56553-60-7 |
| Critical Commercial Assays | ||
| CellTrace Calcein AM | Life Technologies | Cat # C3100MP |
| SuperSignal West Femto | Thermo Fisher | Cat # 34096 |
| BCA Protein Assay | Pierce | Cat # PI23227 |
| Mouse TNF-alpha DuoSet ELISA | R&D Systems | Cat # DY410-05 |
| AlamarBlue Cell Viability Reagent | Thermo Fisher | Cat # DAL1025 |
| Gentest 3P NADPH Regenerating System | Corning | Cat # 451220 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: NCRM-1 (male) | NIH Center for Regenerative Medicine (CRM); Bethesda; USA. | RRID:CVCL_1E71 |
| Human: HUES3 Hb9::GFP-hSOD1A4V (male) | Thams et al., 2018 | N/a |
| Mouse: HB9::GFP-hSODG93A (male) | Thams et al., 2018 | N/a |
| Mouse: Immortalized N9 Microglia (male) | Corradin et al., 1993 | RRID:CVCL_0452 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6SJLF1/J | The Jackson Laboratory | Cat #100012; RRID:IMSR_JAX:10001 2 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Metamorph (version 2010) | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging/acquisition-and-analysis-software/metamorph-microscopy#gref |
| Spotfire (version 2017) | TIBCO | https://www.tibco.com/spotfire-trial?lp=y&utm_medium=3Q_cpc&utm_source=3Q_google&utmcontent=s&utm_campaign=ggl_s_us_en_spt_brand_alpha&utm_term=spotfire%20software&bt=309951201993&bm=e&bn=g&gclid=CjwKCAjwxrzoBRBBEiwAbtX1ntLuGHFVl9ABZePIWVSY7zp8ytQw9wyxyYC68KCS8wwawwTl2MoChoCDv8QAvD_BwE |
| Glide (versions 2012-2016) | Schrödinger | https://www.schrodinger.com/glide |
| Schrödinger Maestro (version 2017) | Schrödinger | https://www.schrodinger.com/maestro |
| ChemDraw Professional (version 16.0) | Perkin Elmer | https://www.perkinelmer.com/category/chemdraw |
| ImageStudio Lite (version 2018) | LiCor | https://www.licor.com/bio/image-studio-lite/ |
| Other | ||
ACKNOWLEDGEMENTS
We thank V. Jackson-Lewis and H. Melikyan for their assistance in animal tissue collection. This research was funded by a grant from the U.S. DOD to B.R.S. and H.W. (CDMRP/USAMRAA GG012467), and by Project ALS. E.R.L. is a Project ALS Women and the Brain fellow. H.W. holds an endowed chair from Jerry and Emily Spiegel. Work performed in the Columbia Chemistry Department NMR facility was supported by a 500 Ascend grant (S10RR025431). The graphical abstract was created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
B.R.S., H.W., A.Z., P.H.B., E.R.L., and S.T. have filed an application for a patent for the use of compound 1 analogs to suppress ER stress-related toxicity and inflammation. B.R.S. holds equity in and serves as a consultant to Inzen Therapeutics.
REFERENCES
- Ammirati M, Bagley SW, Bhattacharya SK, Buckbinder L, Carlo AA, Conrad R, Cortes C, Dow RL, Dowling MS, El-Kattan A, et al. (2015). Discovery of an in Vivo Tool to Establish Proof-of-Concept for MAP4K4-Based Antidiabetic Treatment. Acs Med Chem Lett 6, 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, and Peterson JR (2011). Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 29, 1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, and Czech MP (2009). Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 458, 1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Buhrlage SJ, Huang H-T, Deng X, Zhou W, Wang J, Traynor R, Prescott AR, Alessi DR, and Gray NS (2014). Characterization of WZ4003 and HTH-01–015 as selective inhibitors of the LKB1-tumour-suppressor-activated NUAK kinases. Biochem J 457, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, and Cleveland DW (2006). Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 312, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Bouzakri K, and Zierath JR (2007). MAP4K4 Gene Silencing in Human Skeletal Muscle Prevents Tumor Necrosis Factor-α-induced Insulin Resistance. J Biol Chem 282, 7783–7789. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Morimoto E, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, et al. (2013). A Neurodegeneration-Specific Gene-Expression Signature of Acutely Isolated Microglia from an Amyotrophic Lateral Sclerosis Mouse Model. Cell Reports 4, 385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H-C, Sheu WH, Lin Y-T, Tsai C-Y, Yang C-Y, Cheng Y-J, Huang P-Y, Li J-P, Chiu L-L, Wang X, et al. (2014). HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat Commun 5, 4602–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin S, Mauël J, Donini S, Quattrocchi E, and Ricciardi-Castagnoli P (1993). Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia 7, 255–262. [DOI] [PubMed] [Google Scholar]
- Danai LV, Flach RJ, Virbasius JV, Menendez L, Jung D, Kim J, Kim JK, and Czech MP (2015). Inducible Deletion of Protein Kinase Map4k4 in Obese Mice Improves Insulin Sensitivity in Liver and Adipose Tissues. Mol Cell Biol 35, 2356–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon K, and Blank JL (1997). Characterization of the Mitogen-activated Protein Kinase Kinase 4 (MKK4)/c-Jun NH2-terminal kinase 1 and MKK3/p38 Pathways Regulated by MEK Kinases 2 and 3 MEK KINASE 3 ACTIVATES MKK3 BUT DOES NOT CAUSE ACTIVATION OF p38 KINASE IN VIVO. J Biol Chem 272, 14489–14496. [DOI] [PubMed] [Google Scholar]
- Dow RL, Ammirati M, Bagley SW, Bhattacharya SK, Buckbinder L, Cortes C, El-Kattan AF, Ford K, Freeman GB, Guimaraes CR, et al. (2018). 2-Aminopyridine-Based Mitogen-Activated Protein Kinase Kinase Kinase Kinase 4 (MAP4K4) Inhibitors: Assessment of Mechanism-Based Safety. J Med Chem 7, 3114–3125. [DOI] [PubMed] [Google Scholar]
- Flach RJ, Danai LV, DiStefano MT, Kelly M, Menendez L, Jurczyk A, Sharma RB, Jung D, Kim J, Kim JK, et al. (2016). Protein Kinase Mitogen-activated Protein Kinase Kinase Kinase Kinase 4 (MAP4K4) Promotes Obesity-induced Hyperinsulinemia. J Biol Chem 291, 16221–16230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow VS, Loweth CJ, Ravula SB, Wiemann T, Nguyen T, Xu Y, Todd DE, Sheppard D, Pollack S, Polesskaya O, et al. (2013). Discovery, Synthesis, and Characterization of an Orally Bioavailable, Brain Penetrant Inhibitor of Mixed Lineage Kinase 3. J Med Chem 56, 8032–8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowing G, Philips T, Wijmeersch B, Audet J-N, Dewil M, Bosch L, Billiau AD, Robberecht W, and Julien J-P (2008). Ablation of Proliferating Microglia Does Not Affect Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis Caused by Mutant Superoxide Dismutase. J Neurosci 28, 10234–10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RB, Zhao H, Xia J, Wu D, Nervi S, Stinson SF, Majerova E, Bramhall C, and Zaharevitz DW (2004). Poly(ethylene glycol) Prodrugs of the CDK Inhibitor, Alsterpaullone (NSC 705701): Synthesis and Pharmacokinetic Studies. Bioconjugate Chem 15, 1076–1083. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, and Gurney ME (1998). Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23, 249–256. [DOI] [PubMed] [Google Scholar]
- Han S-X, Zhu Q, Ma J-L, Zhao J, Huang C, Jia X, and Zhang D (2010). Lowered HGK expression inhibits cell invasion and adhesion in hepatocellular carcinoma cell line HepG2. World J Gastroentero 16, 4541–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa J, Mittal S, Wang Y, Korkmaz KS, Adamson E, English C, Ohmichi M, McClelland M, and Mercola D (2005). Identification of Promoters Bound by c-Jun/ATF2 during Rapid Large-Scale Gene Activation following Genotoxic Stress. Mol Cell 16, 521–535. [DOI] [PubMed] [Google Scholar]
- Imamura K, Izumi Y, Watanabe A, Tsukita K, Woltjen K, Yamamoto T, Hotta A, Kondo T, Kitaoka S, Ohta A, et al. (2017). The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci Transl Med 9, eaaf3962. [DOI] [PubMed] [Google Scholar]
- Islam M, and Moore DJ (2017). Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation. Biochem Soc T 45, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, et al. (2014). Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1. Cell Stem Cell 14, 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larhammar M, Huntwork-Rodriguez S, Rudhard Y, Sengupta-Ghosh A, and Lewcock JW (2017). The Ste20 Family Kinases MAP4K4, MINK1, and TNIK Converge to Regulate Stress-Induced JNK Signaling in Neurons. J Neurosci 37, 11074–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, de Haro M, Hao S, Park J, Rousseaux M, Al-Ramahi I, Jafar-Nejad P, Vilanova-Velez L, See L, De Maio A, et al. (2016). Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron 92, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J-C, Lee Y-C, Tan T-H, Liang Y-C, Chuang H-C, Fann YC, Johnson KR, and Lin Y-J (2018). RBM4-SRSF3-MAP4K4 splicing cascade modulates the metastatic signature of colorectal cancer cell. Biochimica Et Biophysica Acta Bba - Mol Cell Res 1865, 259–272. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bertram K, Perides G, McEwen BS, and Wang D (2004). Stress induces activation of stress-activated kinases in the mouse brain. J Neurochem 89, 1034–1043. [DOI] [PubMed] [Google Scholar]
- Marker DF, Tremblay M-È, Puccini JM, Barbieri J, Marker MA, Loweth CJ, Muly CE, Lu S-M, Goodfellow VS, Dewhurst S, et al. (2013). The New Small-Molecule Mixed-Lineage Kinase 3 Inhibitor URMC-099 Is Neuroprotective and Anti-Inflammatory in Models of Human Immunodeficiency Virus-Associated Neurocognitive Disorders. J Neurosci 33, 9998–10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury Y, Côme J, Piskorowski RA, Salah-Mohellibi N, Chevaleyre V, Peschanski M, Martinat C, and Nedelec S (2014). Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol 33, 89–96. [DOI] [PubMed] [Google Scholar]
- Medinas DB, Cabral-Miranda F, and Hetz C (2019). ER stress links aging to sporadic ALS. Aging 11, 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndubaku CO, Crawford TD, Chen H, Boggs JW, Drobnick J, Harris SF, Jesudason R, McNamara E, Nonomiya J, Sambrone A, et al. (2015). Structure-Based Design of GNE-495, a Potent and Selective MAP4K4 Inhibitor with Efficacy in Retinal Angiogenesis. Acs Med Chem Lett 6, 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, and Woodgett JR (1991). Phosphorylation of c-jun mediated by MAP kinases. Nature 353, 670–674. [DOI] [PubMed] [Google Scholar]
- Qiu M-H, Qian Y-M, Zhao X-L, Wang S-M, Feng X-J, Chen X-F, and Zhang S-H (2012). Expression and prognostic significance of MAP4K4 in lung adenocarcinoma. Pathology - Res Pract 208, 541–548. [DOI] [PubMed] [Google Scholar]
- Read J, Collie IT, Nguyen-McCarty M, Lucaj C, Robinson J, Conway L, Mukherjee J, McCall E, Donohoe G, Flavell E, et al. (2019). Tool inhibitors and assays to interrogate the biology of the TRAF2 and NCK interacting kinase. Bioorg Med Chem Lett 29, 1962–1967. [DOI] [PubMed] [Google Scholar]
- Robertson GS, Crocker SJ, Nicholson DW, and Schulz JB (2000). Neuroprotection by the Inhibition of Apoptosis. Brain Pathol 10, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, and Marciniak SJ (2013). Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurology 12, 105–118. [DOI] [PubMed] [Google Scholar]
- Ryabinin AE, Wang Y-M, and Finn DA (1999). Different Levels of Fos Immunoreactivity After Repeated Handling and Injection Stress in Two Inbred Strains of Mice. Pharmacol Biochem Be 63, 143–151. [DOI] [PubMed] [Google Scholar]
- Salem SM, Hamed AR, Fayez AG, and Eldeen G (2018). Non-target Genes Regulate miRNAs-Mediated Migration Steering of Colorectal Carcinoma. Pathol Oncol Res 25, 559–566. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, and Caroni P (2009). A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12, 627–636. [DOI] [PubMed] [Google Scholar]
- Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, Bostrom A, Theodoss J, Al-Nakhala BM, Vieira FG, et al. (2009). Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Sc 9, 4–15. [DOI] [PubMed] [Google Scholar]
- Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, Roberts CJ, Miller KR, Ransohoff RM, Trojanowski JQ, et al. (2018). Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci 21, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Lagas JS, Lankheet N, Poller B, Hillebrand MJ, Rosing H, Beijnen JH, and Schinkel AH (2012). Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer 130, 223–233. [DOI] [PubMed] [Google Scholar]
- Tang X, Guilherme A, Chakladar A, Powelka AM, Konda S, Virbasius JV, Nicoloro SM, Straubhaar J, and Czech MP (2006). An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARγ, adipogenesis, and insulin-responsive hexose transport. P Natl Acad Sci Usa 103, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesz GJ, Guilherme A, Guntur KV, Hubbard AC, Tang X, Chawla A, and Czech MP (2007). Tumor Necrosis Factor α (TNFα) Stimulates Map4k4 Expression through TNFα Receptor 1 Signaling to c-Jun and Activating Transcription Factor 2. J Biol Chem 282, 19302–19312. [DOI] [PubMed] [Google Scholar]
- Thams S, Lowry ER, Larraufie M-H, Spiller KJ, Li H, Williams DJ, Hoang P, Jiang E, Williams LA, Sandoe J, et al. (2019). A stem cell-based screening platform identifies compounds that desensitize motor neurons to endoplasmic reticulum stress. Mol Ther 27, 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trias E, Ibarburu S, Barreto-Núñez R, Babdor J, Maciel TT, Guillo M, Gros L, Dubreuil P, Díaz-Amarilla P, Cassina P, et al. (2016). Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflamm 13, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virbasius JV, and Czech MP (2016). Map4k4 Signaling Nodes in Metabolic and Cardiovascular Diseases. Trends Endocrinol Metabolism 27, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed Differentiation of Embryonic Stem Cells into Motor Neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wright JH, Wang X, Manning G, LaMere BJ, Le P, Zhu S, Khatry D, Flanagan PM, Buckley SD, Whyte DB, et al. (2003). The STE20 Kinase HGK Is Broadly Expressed in Human Tumor Cells and Can Modulate Cellular Transformation, Invasion, and Adhesion. Mol Cell Biol 23, 2068–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Watts ME, and Rubin LL (2019). MAP4K4 Activation Mediates Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Cell Reports 26, 1143–1156. [DOI] [PubMed] [Google Scholar]
- Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, et al. (2013). A Small Molecule Screen in Stem-Cell-Derived Motor Neurons Identifies a Kinase Inhibitor as a Candidate Therapeutic for ALS. Cell Stem Cell 12, 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Zhou G, Wang X, Brown A, Diener K, Gan H, and Tan T-H (1999). A Novel Human STE20-related Protein Kinase, HGK, That Specifically Activates the c-Jun N-terminal Kinase Signaling Pathway. J Biol Chem 274, 2118–2125. [DOI] [PubMed] [Google Scholar]
- Yoshida H (2007). ER stress and diseases. Febs J 274, 630–658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S6. Inhibition profile for 371 wild-type human kinases using radioligand binding assay performed by Reaction Biology Corp for compound 1 and Prostetin/12k analogs, Related to Figure 4. Compounds were tested in single dose mode at a concentration of 0.1 μM. Reactions were carried out at 10 μM ATP.
Data Availability Statement
This study did not generate any datasets or code.