

Abstract
Introduction
The quest to identify an effective therapeutic strategy for neurodegenerative diseases, such as mild congitive impairment (MCI) and Alzheimer's disease (AD), suffers from the lack of good human‐based models. Animals represent the most common models used in basic research and drug discovery studies. However, safe and effective compounds identified in animal studies often translate poorly to humans, yielding unsuccessful clinical trials.
Methods
A functional in vitro assay based on long‐term potentiation (LTP) was used to demonstrate that exposure to amyloid beta (Aβ42) and tau oligomers, or brain extracts from AD transgenic mice led to prominent changes in human induced pluripotent stem cells (hiPSC)‐derived cortical neurons, notably, without cell death.
Results
Impaired information processing was demonstrated by treatment of neuron‐MEA (microelectrode array) systems with the oligomers and brain extracts by reducing the effects of LTP induction. These data confirm the neurotoxicity of molecules linked to AD pathology and indicate the utility of this human‐based system to model aspects of AD in vitro and study LTP deficits without loss of viability; a phenotype that more closely models the preclinical or early stage of AD.
Discussion
In this study, by combining multiple relevant and important molecular and technical aspects of neuroscience research, we generated a new, fully human in vitro system to model and study AD at the preclinical stage. This system can serve as a novel drug discovery platform to identify compounds that rescue or alleviate the initial neuronal deficits caused by Aβ42 and/or tau oligomers, a main focus of clinical trials.
Keywords: Alzheimer's disease, human induced pluoripotent stem cell, human‐on‐a‐chip, long‐term potentiation, oligomers
1. BACKGROUND
Alzheimer's disease (AD) is a chronic, progressive condition characterized by deterioration of memory and other cognitive domains leading to death within 10 years of diagnosis. However, the onset of AD is thought to begin well in advance of disease diagnosis, ≈20 years before symptoms manifest clinically, with accumulating protein deposits in the brain (preclinical stage) giving rise to subtle behavioral changes and impaired episodic memory not yet having reached the level of impairment for clinical diagnosis. 1 The early clinical stage, referred to as mild cognitive impairment (MCI), has gained significant attention as a focus for therapeutic intervention with several international trials under way. 2 Previously, most research and clinical trials have focused on the mild to moderate dementia stage of the disease, but increasingly studies are examining this pre‐MCI or preclinical stage as secondary prevention opportunities.
Over the past 20 years, brain‐related diseases, such as stroke and AD, have risen from not listed in the top 10 causes of death to the 5th and 6th leading cause of death (in the case of AD) globally and in the United States, respectively, in 2018. 3 , 4 AD and other forms of dementia affect ≈ 50 million individuals worldwide and the costs associated with treatment are close to 1 trillion U.S. dollars/year. 5 , 6 Moreover, the number of projected dementia cases is expected to grow to about 150 million by 2050, barring any major advancement in prevention or treatment. 7 Apart from the global rise in the aged population, the increase in AD cases and related deaths can be attributed to the paucity of effective treatment options.
To date, there are only four approved drugs used to treat AD symptoms. 8 In contrast, over 100 different potential therapeutics, across several drug classes, were abandoned or failed during clincal trials. 9 , 10 , 11 Animals are the most commonly used models for disease modeling and play a large role in preclinical drug development for safety and efficacy investigations. However, despite their ubiquitous role in drug discovery, lead compounds identified during animal studies often fail during the clinical phase of drug development. There are many reasons for this failure. Transgenic animal models of disease fail to accurately capture the full spectrum of the human disease phenotype. Murine orthologues of human proteins may not respond the same as the human version. Moreover, animal drug safety data are often not reproducible in humans due to differences in drug metabolism and excretion. 12 Additionally, the established treatment options are symptomatic in nature, attempting to counter neurotransmitter imbalances caused by neuronal dysfunction and cell death; existing therapeutics are not disease‐modifying. 13 Consequently, the development of diagnostic criteria that identify patients in the very early stages of disease progression, ie, MCI or preclinical stages, is critical to effective therapeutic intervention approaches. 1 Furthermore, there is a need to develop human‐based models of the preclinical phase of AD and develop disease‐modifying strategies that can be adiministered prior to extensive neuronal cell death to enable recovery from the effects of this disease.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (eg, PubMed) sources. There is a lack of good human‐based in vitro models for Alzheimer's disease (AD), espeically for reproducing cognitive functional deficits at the preclinical stage. Relevant work reporting the generation of cortical neurons from patient human induced pluripotent stem cells (hiPSCs) has been cited.
Interpretation: In this human‐on‐a‐chip model, long‐term potentiation (LTP) was induced in hiPSC‐derived cortical neuronal cultures on microelectrode arrays and the treatment of amyloid beta, tau oligomers, or brain extracts from AD transgenic mice caused deficits in LTP and other neuronal electrical and morphological characteristics, but not cell viability. This work presented a human‐based phenotypic model recapturing pre‐mild cognitive impairment AD pathology.
Future direction: This model can be utilized in investigating AD etiology and therapeutic testing to understand genetic mutations or the role of inflammation on AD initiation and progression, and testing drugs for mechansim and efficacy in a preclinical model of AD.
Highlights
Cortical neurons derived from human induced pluripotent stem cells were used
Functional human in vitro model based on long‐term potentiation (LTP)
Utilized LTP to evaluate functional toxicity of amyloid beta (Aβ42) and tau oligomers
Neuronal viability is not affected so is a correlate for pre‐mild cognitive impairment
Functional screen for drug candidates to reverse cognitive deficits
Due to the poor disease reproducibility and lack of translatable compound efficacy and safety findings from current preclinical animal models there has been growing interest in adding the use of human‐based, microphysiological systems (MPS) or human‐on‐a‐chip (HoaC) systems for disease modeling and drug discovery. 14 , 15 These systems offer several advantages over traditional preclinical drug discovery models. For example, by combining a human liver module with efficacy and relevant safety/toxicity targets they can be used to investigate the pharmacodynamics and pharmacokinetics of potential therapeutics in the same system. 16 , 17 , 18 Additionally, when integrated with functional readouts such as microelectrode arrays (MEAs) and devices to measure mechanical function, these systems can provide physiological data in response to drug treatment that can be more sensitive than viability or biomarker data alone and mimic aspects of clinical measurements. 19 , 20 Finally, establishing MPS systems using human induced pluripotent stem cells (hiPSCs) faciliates the investigation of the role of specific gene mutations on drug metabolism or disease progression and lays the foundation for personalized, precision medicine. Specifically, in the area of brain research, several MPS systems have been developed using organoids and iPSCs. In fact, there is now emerging evidence from many studies in support of using hiPSCs for AD and other neurodegenerative disorders research. 21 , 22 , 23 Human iPSCs combined with MPS offer a more relevant cell‐based model to investigate AD pathology and drug discovery. 21 , 24
Alzheimer's disease is characterized by two main pathological features, the deposition of toxic amyloid beta (Aβ), leading to extracellular plaque formation, and tau hyperphosphorylation, which leads to tau aggregation and neurofibrillary tangle formation. The oligomerization or aggregation of these proteins is postulated to trigger the activation or inactivation of various downstream pathways, which leads to neuronal cell dysfunction and eventual death in the brain. 25 Recent findings, both in vivo and in vitro, have indicated that the oligomeric forms of Aβ and tau are required for this neurotoxicity in AD. 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 For example, extracelluar tau oligomers were shown to impair long‐term potentiation (LTP) in mouse hippocampal neurons. 26 In an embryonic mouse cell–MEA model, Charkhkar et al. showed that Aβ42 treatment reduced spontaneous network activity in primary neuronal cultures. 36 Additionally, in previous works done in this laboratory, Berry et al. showed that treatment of hiPSC‐derived neurons with Aβ42 significantly suppressed neurons’ electrophysiological activity without concomit neural death, whereas Varghese et al. demonstrated that the addition of Aβ42 to an embryonic rat hippocampal neurons–MEA system greatly diminished spontaneous firing in the neuronal cells but was reversed by curcumin treatment. 37 , 38 While those findings highlight the pathological mechanisms of Aβ‐ and tau‐induced pathology in AD, the use of animal cells hinders the translation of those models for human drug development. 39 , 40 , 41 , 42 , 43
In this study, a coculture of primary human astrocytes and hiPSCs‐derived cortical neurons was treated with Aβ42 oligomers, tau oligomers (the two main pathological features of AD), or brain extracts from AD transgenic mice. This human‐based approach represents the early stages of AD in the human brain. 21 , 22 , 44 , 45 The astrocyte‐cortical neuron coculture was established on MEAs facilitating the long‐term monitoring of populational neuron physiology, something not possible with patch clamp electrophysiology. 46 , 47 The findings presented here provide strong support for the use of this model to study AD pathology at the pre‐MCI or preclinical stage with the potential to be a drug discovery platform for the current focus on AD clinical studies. 26 , 27 , 38 , 48 , 49 , 50 , 51
2. METHODS
2.1. Neuronal cells
Cortical neurons derived from hiPSC cells were used to study the effects of both Aβ and tau in inducing neuronal cell dysfunction. The cells were purchased from Cellular Dynamics International (CDI, iCell GlutaNeurons, Cat. #: C1033, Madison, WI, USA) and are derived from hiPSCs from healthy individuals. They consisted of a mixed population of post‐mitotic cells containing both GABAergic and glutamatergic neurons. These cells were previously characterized in the lab for their suitability, including their viability and function and considered suitable for research purpose, including the study of neurodegenerative conditions, such as AD, and for drug screening. 21 , 52
2.2. Cell culture
Cortical neurons were seeded on both coverslips and on MEAs. For morphology analysis and patch‐clamp electrophysiology, the cells were plated initially at a density of 160 cells/mm2 on acid washed, poly‐l‐ornithine and laminin (PLO/LM, 20 µg/mL and 10 µg/mL, respectively) coated coverslips and incubated for 14 to 28 days prior to treatment. To assess cell function extracellularly, cortical neurons were cocultured on fibronectin and laminin (FB/LM, 10 µg/mL each for 2 hours at room temperature [RT]) coated MEAs with human astrocytes. The neuronal cells were plated at a density of 2000 cells/mm2 and the astrocytes at 1000 cells/mm2. Initially, the cells were incubated in the manufacturer's (CDI) recommended medium (BrainPhys Neuronal Medium supplemented with N‐2 supplement, laminin, pen/strep, and CDI supplemented kit) for ≈ 24‐hours at 37°C, 5% CO2. On the ensuing day, half of the CDI medium was replaced with an equal (1×) volume of neuromuscular junction (NMJ) media. 53 After 48 hours in the mixed CDI/NMJ medium, a full medium change was peformed with NMJ medium only and the cells were maintained in that medium thereafter. Prior to cell plating on the MEAs, hiPSC‐derived astrocytes were plated and cultured for 2 days. On the day of cell plating, the astrocytes were harvested using acutase for ≈15 minutes at room temperature.
2.3. Aβ and tau oligomer preparation
The oligomerization of Aβ and tau peptides are considered to be the primary neurotoxic species of these proteins and perhaps the catalysts in the development and progression of AD. 54 , 55 , 56 Therefore, to study the effects of these proteins, the oligomeric forms were used in this study. The synthetic Aβ and tau oligomers were obtained from Dr. David Morgan, MSU, and were prepared as described previously. 57 The toxic Aβ oligomers were prepared using peptides from rPeptide (Aβ42 catalog number A‐1002‐2; Aβ‐scrambled A‐1004‐2). First, the peptides were resuspended in 500 µL of hexafluoroisopropanol (HFIP; catalog number AC445820100; Fisher Scientific) and left to dry overnight under a ventilated hood. The next day the samples were spun in a SpeedVac until dry (≈30 minutes). They were then stored desiccated at −20°C until use. Recombinant human tau (hT40 or 2N4R isoform, with a C‐terminal His tag) was produced as described. 58 The tau‐O were made by incubating 8 µM tau with 150 µM arachidonic acid (catalog number 90010.1; Cayman Chemicals) for 15 minutes and stopping the reaction by freezing and then stored at 80°C until use. 59 , 60 The aggregation buffer solution (tau BC, without tau oligomers) was used as control. Prior to using, the solution was sonicated for 5 minutes and centrifuged at 1400 × g for 5 minutes.
2.4. Mouse brain extracts preparation
Brain extracts from two different AD mouse models and their age‐, background‐matched wild‐type mice were used in this study, APP (Tg2576, 24‐month‐old) transgenic mice and tau (Tg4510, 12‐month‐old) transgenic mouse model. The APP mice express the human APP gene, which harbors the Swedish mutation (K670N/M671L). The tau transgenic mice carry a mutant human tau gene harboring the P301L mutation. 61 After the brains were harvested and dissected, the cortices were homogenized in Dulbecco's Modified Eagle Medium (DMEM) using a pestle gun for 10 seconds twice, followed by sonication on ice with a 2‐second pulse three times. The suspensions were then spun down for 10 minutes at 700 × g. The supernatant was collected and stored at –80°C until the day of the experiment. Prior to use, the samples were centrifuged at 1400 × g for 5 minutes and the supernatant was used for cell dosing.
2.5. Aβ, tau oligomers and brain extract dosing
On the day of treatment, a full medium change was performed, followed by the administration of either tau or beta amyloid oligomers, or mouse brain extracts. For oligomeric Aβ or tau treatment, the cells were dosed with a final concentration of either 1 µM or 5 µM of Aβ42, or Aβscrambled (Aβscr), or 100 nM of Tau oligomers or tau‐BC in the culture, or left untreated. For brain extract dosing, the cells were dosed with a final concentration of either 10 or 20 µg/mL of brain extracts from transgenic and wild‐type mice in the culture. To examine the neurotoxic effects of Aβ and tau on cell morphology and electrophysiological function, the cells were either incubated for 1 hour (MEAs), 24 hours (patch‐clamp), or 3 to 6 days (immunocytochemistry) after treatment.
2.6. Cell viability assessment
To assess the effects of Aβ and/or tau oligomers on cell survivability, the cells were fluorescence‐activated cell (FAC) sorted at 3 days post Aβ or tau oligomers dosing using a live and dead cell assay kit from ThermoFisher (LIVE/DEAD Viability/Cytotoxicity Kit, L3224).
2.7. Immunocytochemistry and confocal microscopy
For morphological analysis, the cells were fixed in 4% paraformaldehyde (PFA) for 10 minutes at room temperature followed by three rinses in phosphate buffered saline (PBS). The cells were then permeabilized in 0.25% Triton X‐100 for 10 minutes at RT, followed by three washes in PBS, and incubation in a blocking solution of 3% bovine serum albumin (BSA), 3% normal goat serum (NGS), 0.1% Tween 20 (T20), for 1 hour at RT. Soon after, the cells were incubated with primary antibodies for 1 hour at RT or overnight at 4°C. Thereafter, the cells were again washed three times with PBS, followed by incubation with fluorescently conjugated secondary antibodies (diluted in BSA/NGS/T20). To stain the nuclei, the cells were counterstained with DAPI (4′,6‐diamidino‐2‐phenylindole) and mounted on glass slides for analysis. To measure axon length and compute neurite densities, images were taken using a confocal microscope (Zeiss, Axioskop 2, Germany) followed by quantification analyses using ImageJ software. The following primary antibodies (at 1/1000 dilution) were used: Rabbit Anti‐Microtubule‐Associated Protein 2 (Millipore, Cat. #: AB5622), Mouse Anti‐Tau‐1 (Millipore, Cat. # MAB3420). The secondary antibodies used: Alexa‐Fluor 488 goat anti‐rabbit (ThermoFisher, Cat. # A11034), and AlexFluor 568 Donkey anti‐mouse (ThermoFisher Cat. # A10037).
2.8. Evaluation of cortical neuron physiology by patch‐clamp electrophysiology
Neurons physiological activity was evaluated at day 28. For whole cell patch‐clamp electrophysiology, the recording was carried out using a Zeiss, upright microscope (Axioscope, FS2, Carl Zeiss, Germany) equiped with a multiclamp 700B amplifier as described previously. 21 An intracellular solution consisting of 140 mM K‐gluconate, 4 mM NaCl, 0.5 mM CaCl2, 1 mM MgCl2, 1 mM EGTA, 5 mM HEPES Acid, 5 mM HEPES base, and 5 mM Na2ATP was used for recording. Depolarization‐evoked inward and outward currents were examined in voltage‐clamp mode. Depolarization‐evoked action potentials (APs) were recorded in current‐clamp mode. Spontaneous activity was recorded at gap‐free condition. The data were analyzed using pClamp 10 software (Axon Instrument, Foster City, CA, USA) and quantified using Microsoft Excel.
2.9. Long‐term potentiation induction on MEAs
To induce LTP on cortical neurons cultured on MEAs, a high frequency stimulation (HFS) protocol was used as described previously. 37 , 62 Test stimuli were delivered to all the electrodes as follows: 10 repetitions of 4× biphasic voltage pulses at 500 mV and 5 ms duration. The evoked response or recorded cell activity was then analyzed using Anaconda with Python software. The waveforms (eg, action potential spikes and frequency) were high‐pass filtered at 200 Hz and five standard deviations. Any electrodes or cells with firing frequency <0.1 Hz were excluded from the data analysis.
2.10. Evaluation of cortical neuron physiology on MEAs
The MEAs system consists of the MEA chips, which are comprised of a silicon base outfitted with several electrodes made of titanium nitride 37 and the rig that is comprised of a temperature controller, a stimulator for LTP induction, an amplifier, and MC_Rack data acquisition software. The cells were maintained in culture for 14 to 21 days before acute treatment with Aβ or tau oligomers, or brain extracts. Prior to treatment, spontaneous activities (baseline) of the cells were measured followed immediately by stimulation (LTP induction). Immediately after LTP induction, the cells were treated with either 5 µM of Aβ or 100 nM of tau oligomers or their controls, or 20 µg/mL of brain extracts, followed by incubation for 1 hour. Next, the neuron's action potential firing frequency was recorded and the data were analyzed using Anaconda with Python software.
2.11. Statistical analysis
Comparison of the mean of at least three replicates or more between groups was performed. For computational analyses, Microsoft Excel software was used. Student t‐tests was used for statistical comparison analysis betweeen two experimental groups, whereas one‐way analysis of variance (ANOVA) followed by Tukey's post hoc (multiple comparison) test was used for multiple experimental groups. The standard error of the mean (SEM) was used for statistical significance at P ≤.05. For axon length measurements, a minimum of 20 cells were counted.
3. RESULTS
3.1. Morphological defects in Aβ oligomer or tau oligomer‐treated hiPSC‐cortical neurons
Initially, evaluation of neurons for AD‐related phenotypes in the presence or absence of Aβ oligomers or tau oligomers (tau‐O) was performed. hiPSC‐derived cortical neurons were treated with different concentrations of Aβ42 and Aβscrambled, or tau oligomers (tau‐O) and tau buffer (tau‐BC). After 3 to 4 days of treatment and 18 days total in culture, pronounced defects were observed in cell morphology in the 5 µM Aβ42‐treated samples compared to Aβscrambled‐treated and untreated controls (Figure S1A, S1B in supporting information). To characterize and quantify these changes, the cells were fixed and incubated with antibodies to MAP2, followed by analysis of various AD‐associated neuropathologies, including axon outgrowths and neurite density for each group (Figure S1C). As expected, the results indicated that exposure to Aβ42 oligomers at 5 µM (≈22.5 µg/mL) led to prominent cell damage, including a decrease in the number of neurites per cell (Figure S1D). Also, neurite outgrowth was strongly inhibited by 5 µM treatment of Aβ42 oligomers, leading to shorter axon length in the Aβ42‐treated samples compared to control groups (Aβ scrambled and untreated groups; Figure S1E). Similar to the Aβ42 results, 100 nm (≈4.59 µg/mL) tau oligomer‐treated neurons showed a significant deficit in morphology (Figure S2A, S2B in supporting information) and a reduction in the average number of neurites per cell (Figure S2C) and a decrease in axon length compared to tau‐BC treated samples (Figure S2D). These results are consistent with data from studies using rodent models or other cell types. 27 Having demonstrated significant cell morphology defects using 5 µM Aβ42, and 100 nM tau‐O, the remaining experiments were conducted using 5 µM Aβ42 oligomers and 100 nM tau. All concentrations reported here represent final concentration in culture.
3.2. Electrophysiological dysfunction in hiPSC‐cortical neurons treated with Aβ oligomers and tau aggregates
Multiple studies have implicated altered synaptic function and plasticity in AD pathogenesis. 63 , 64 We investigated the effects of exposure to Aβ or tau oligomers on electrical activity of the hiPSC‐derived cortical neurons by whole cell patch‐clamp electrophysiology. The cells were maintained in culture for 22 to 28 days and after 24‐hours incubation with either 5 µM Aβ42, 5 µM Aβscrambled, 100 nM tau oligomers, or 100 nM tau‐BC, the electrical activity of the neurons was evaluated. Analysis of the data revealed a prominent loss in cell function caused by both Aβ42 oligomers and tau oligomers (Figures 1 and 2). More specifically, a marked decrease in sodium (inward) currents was observed for Aβ42 and tau oligomer‐treated hiPSC‐derived cortical neurons (Figures 1A, B and 2A, B). Exposure to either Aβ42 oligomers or tau oligomers also led to a marked reduction in induced APs under depolarization in comparison to the Aβscrambled or tau‐BC (Figures 1A, C and 2A, C). Furthermore, the cells displayed a significant reduction in the firing rate and peak amplitudes for spontaneous firing in the presence of Aβ42 oligomers and tau oligomers compared to samples treated with Aβscrambled oligomers and tau‐BC, respectively (Figures 1A, D and 2A, D). However, no significant changes in cell viability was observed after Aβ42 treatment for 3 days (Figure S3 in supporting information).
FIGURE 1.
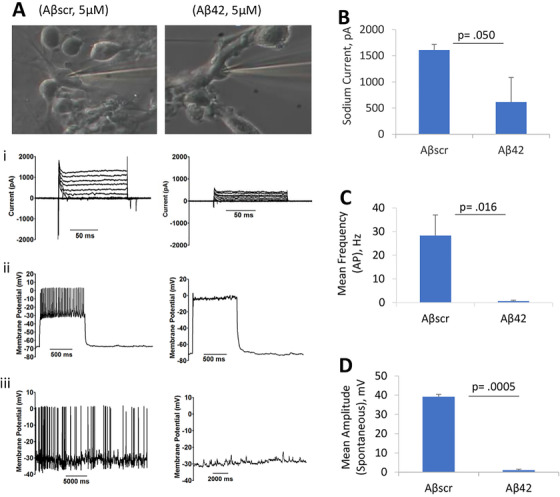
Amyloid beta (Aβ42) oligomer‐induced human induced pluripotent stem cell‐cortical neuron electrophysiological dysfunction. Representative images of patched cells (22‐28 DIV) after Aβscrambled (Aβscr) or Aβ42 oligomers treatment. Application of Aβ42 oligomers (at 5 µM, final concentration) led to defects in cortical neuron function (firing potential) at 24 hours post‐treatment, including cell current, induced action potentials under depolarization, and spontaneous firing (A). Importantly, the cells indicated a significant reduction in peak inward current (A‐i, B), absence of repetitive firing (action potential) (A‐ii, C), and spontaneous activity (A‐iii, D) in the presence of Aβ42 treatment compared to the control samples that were treated with Aβscr. Statistical analysis was computed using student's t‐test. Error bars represent standard error of the mean and P ≤ .05 is considered statistically significant
FIGURE 2.
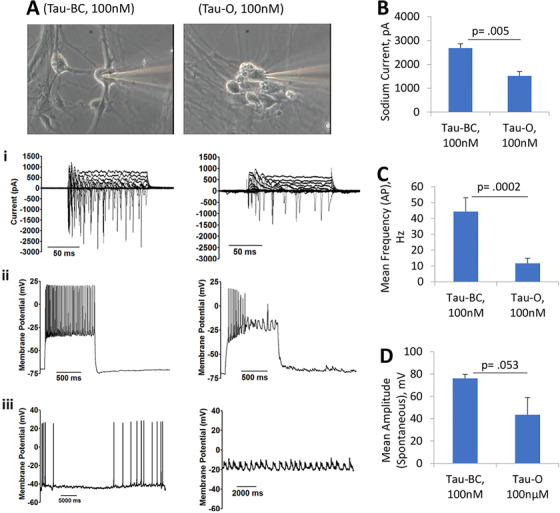
Tau oligomer‐induced human induced pluripotent stem cell‐cortical neuron electrophysiological dysfunction. Application of tau oligomers (tau‐O) at 100 nM (final concentration) led to altered physiology in neurons (22‐28 DIV) after 24 hour of treatment. In particular, a pronounced decrease in current, induced repetitive firing under depolarization, and spontaneous firing (A) was observed. Moreover, quantitative analysis revealed a significant reduction in peak inward current (A‐i, B), a sharp decrease in the firing frequency of action potentials (A‐ii, C), and spontaneous activity (A‐i, D) in the samples treated with tau oligomers compared to controls (ie, untreated and tau‐BC). Statistical analysis was computed using student's t‐test. Error bars represent standard error of the mean and P ≤.05 is considered statistically significant
3.3. Aβ42 or tau treatment of hiPSC‐cortical neurons abolishes HFS‐induced LTP
MEA electrophysiological recordings enable measurement of neuron electrophysiological activity extracellularly. To evaluate hiPSC‐derived cortical neurons physiological activity on MEAs, baseline spontaneous activity was measured followed by HFS. LTP induction was determined by measuring the spontaneous activity at 1 hour post‐stimulation compared to baseline activity. The pathophysiological effects of 5 µM Aβ42 oligomers and 100 nM tau oligomers on induced cortical neuron LTP was measured and compared to Aβscrambled, or tau‐BC. A significant increase in spontaneous firing frequency was observed compared to baseline levels after LTP induction (Figures 3A, B and 4A, B). This change in cell activity persisted at 1 hour post‐treatment with scrambled Aβ oligomers or tau‐BC; however, the LTP‐induced increases in cell activity were abolished after treatment with 5 µM of Aβ42 oligomers or 100 nM of tau oligomers at 1 hour post‐dose measurement (Figures 3C and 4C). In summary, these results indicate the deleterious effects of Aβ42 oligomers and tau oligomers on the hiPSC‐derived cortical neurons after HFS‐mediated LTP induction.
FIGURE 3.
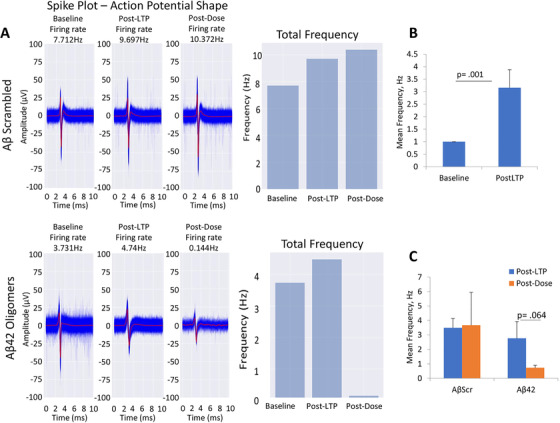
Effects of amyloid beta (Aβ42) oligomers on long‐term potentiation (LTP) maintenance in cortical neurons (A). The LTP induction protocol increased spontaneous firing frequency in hiPSC‐derived cortical neurons (14‐21 DIV) on microelectrode arrays (MEAs) (B). The LTP effects continued 1 hour post‐Aβscr (control) dosing while the LTP effects were abolished by 1 hour of Aβ42 dosing (C). Representative MEA measured action potential traces and frequency for the Aβoligomers dosed systems indicated LTP induction and no change in LTP after AβScr dosing and representative MEA measured action potential traces and frequency for the Aβ42 dosed systems indicated LTP induction and reduction of LTP after Aβ42 dosing (A). Alpha level was set at P ≤.05
FIGURE 4.
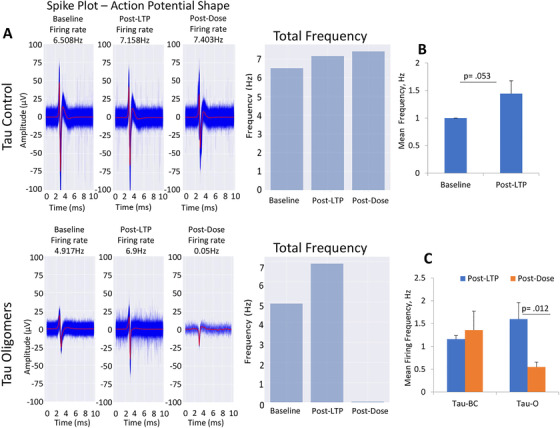
Effects of tau oligomers on long‐term potentiation (LTP) maintenance in cortical neurons (A). The LTP induction protocol increased hiPSC‐derived cortical neuron spontaneous firing frequency on microelectrode arrays (MEAs; 14‐21 DIV) (B), this increase in LTP was maintained at 1 hour post‐dosing in the samples treated with the buffer control solution, whereas LTP was completely abolished by 1 hour of tau oligomer dosing (C). Representative MEA measured action potential traces and firing frequency bar graphs showed an increase in LTP after LTP induction and no significant changes in the samples dosed with the buffer control whereas tau oligomer dosing led to a significant reduction in LTP after 1 hour of dosing (A). Error bars represent standard error of the mean and alpha level was set at P ≤.05
3.4. Morphological defects in hiPSC‐cortical neurons treated with APP or tau transgenic mice brain extract
The neurotoxic effects of Aβ and tau from different sources (eg, brain extracts prepared from APP or tau transgenic [Tg] mice) were also examined to further evaluate the effectiveness of the system. Both APP and TAU transgenic animal models have been used extensively to reproduce typical AD pathologies and therefore provide invaluable models in deciphering AD etiology as well as for drug evaluation. To investigate the neurotoxic effects of brain extracts on human brain cells and determine whether the results with the synthetic oligomers were reproduceable, hiPSC‐cortical neurons (at 14 DIV) were dosed with final concentration of 10 or 20 µg/mL of brain extract solution and harvested at 3 days post‐treatment. Compared to untreated cells or cells treated with brain extracts from wild‐type mice (WT‐be), cells treated with 20 µg/mL of APP or tau transgenic mice brain extracts (APP‐be and tau‐be, respectively) showed a significantly higher percentage of cells with broken neurites and/or axonal vericosities, and shorter axon length (Figure S4 in supporting information). Similar to the oligomer data, the brain extracts caused an adverse effect on cell morphology, including changes in neurites complexity, changes in axon outgrowth, and the presence of either axonal varicosities or broken axons (Figure S4A–C). In particular, the quantified data demonstrated that treatment with the APP‐be led to a slight but significant reduction in neurite branches in the APP‐be treated samples relative to controls (untreated and WT‐be; Figure S4D). In addition, a significant decrease in axon length was observed in the APP‐be and tau‐be treated samples compared to controls (Figure S4E). These results also indicated a significantly higher percentage of cells with broken neurites when exposed to 20 µg/mL APP‐be or tau‐be compared to WT‐be (Figure S4F). Taken together, these results indicate that Aβ and tau, whether in the synthetic form or prepared from brain tissue, are neurotoxic and confirm what has been suggested previously in the literature regarding Aβ and tau toxicity.
3.5. Electrohysiological dysfunction in hiPSC‐cortical neurons treated with brain extracts from APP Tg mice
The effects of APP‐be on hiPSC cortical neuron intracellular activity was analyzed by patch‐clamp electrophysiology after a 24‐hour treatment with 20 µg/mL brain extracts from APP Tg mice or wild‐type mice. As shown in Figure 5, the addition of brain extracts from APP Tg mice (APP‐be) to the hiPSC neuronal cultures (28 DIV) led to a marked decrease, and sometimes a complete lack, of cell activity. Notably, a significant decrease in both sodium and potassium currents was observed in cells exposed to APP‐be compared to WT‐be treated cells (Figure 5A‐i, 5B). Furthermore, the APP‐be treatment also blocked action potential firing compared to WT‐be, including an elimination in repetitive firing of induced action potential (Figures 5A‐ii and 5C). Additionally, exposure to APP‐be almost completely abolished spontaneous firing in the hiPSC cortical neurons, resulting in significantly reduced peak amplitudes (Figures 5A‐iii and 5D).
FIGURE 5.
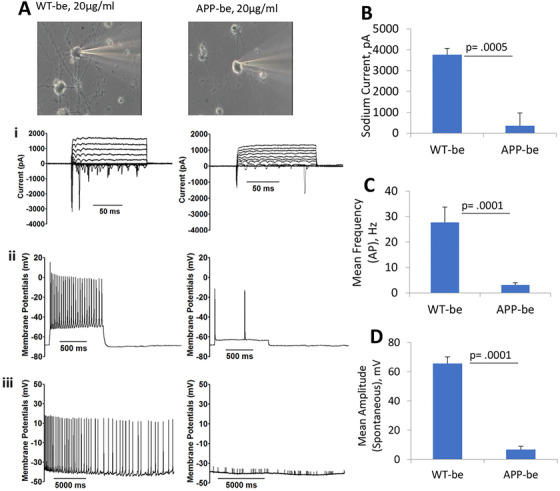
Inhibition of cortical neuron electrophysiological function after brain extract dosing. The addition of brain extracts from APP transgenic mice (APP‐be) at 20 µg/mL to cultures of human induced pluripotent stem cell‐derived cortical neurons (28 DIV) resulted in a significant decrease in cell electrophysiological function at 24 hours post‐treatment. Specifically, the application of APP‐be significantly reduced the voltage induced currents (A‐i, B) of cortical neurons in comparison to the brain extracts from wild‐type (WT‐be) mice. Additionally, both the repetitive firing frequency (A‐ii, C) and the peak amplitude of the cell spontaneous firing (A‐iii, D) were drastically diminished in the presence of APP‐be at 24‐hour post‐dosing compared to when the cells were treated with brain extracts from WT‐be mice. Error bars represent standard error of the mean. Statistical significance was computed using student's t‐test and the alpha level was set at P ≤ .05
3.6. Effects of APP or tau Tg mouse brain extract treatment on iPSC‐cortical neurons utilizing HFS‐induced LTP
To evaluate the effects of brain extracts from wild‐type, APP, or tau Tg mice on hiPSC‐derived cortical neurons, cortical‐HoaC systems were established and maintained for 22 to 30 days and then treated with the brain extracts after LTP induction. HFS‐induced LTP resulted in significant increase in cell firing frequency as indicated in Figures 6A, C, E, G. APP‐be treatment of hiPSC‐cortical neurons reduced post‐LTP spontaneous firing frequency within 1 hour compared to WT‐be treated systems (Figures 6B, D). Similarly, tau Tg‐be treatment of hiPSC‐cortical neurons significantly reduced the post‐LTP spontaneous firing frequency at 1 hour compared to WT‐be treated systems (Figures 6F, H). Taken together, these data indicate that exposure to Aβ (whether the synthetic form or from brain extracts) can interfere with LTP maintenance after HFS.
FIGURE 6.
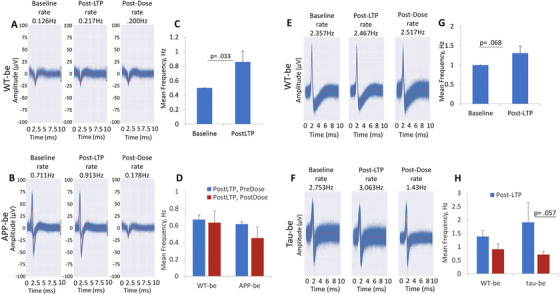
Effects of transgenic mice brain extracts on long‐term potentiation (LTP) maintenance in cortical neurons. Representative images showing cell baseline activities in iPSC‐derived cortical neurons and an increased activity thereafter after LTP induction, and at 1 hour post‐treatment with brain extracts from wild‐type (WT‐be; A) and APP transgenic (APP‐be) (B) mice as indicated by their action potential traces. The LTP induction protocol generated a significant increase in neuron firing frequency (C, G); however, while LTP persisted 1 hour after WT brain extract treatment, application of APP brain extract (APP‐be) led to a marked decrease in spontaneous activity at 1‐hour post LTP induction (D). Similarly, brain extracts from tau transgenic mice (tau‐be; F) adversely affected human induced pluripotent stem cell firing ability as indicated by a reduction in the amplitude of the action potential traces (F) compared to WT‐be (E). The firing frequency was quantified, and the results revealed a significant decrease in cell firing frequency at 1 hour post tau‐be treatment relative to cells treated with brain extracts from WT‐be (H). The results represent the mean values from three to four replicates and the error bars indicate standard error of the mean. Student's t‐test was used for comparison analyses and the alpha level was set at P ≤ .05
4. DISCUSSION
The paucity of treatment options for AD and many other neurological diseases can be attributed, in part, to a lack of effective translational models for drug discovery. Animal models represent the standard and most commonly used models in drug discovery. But, due to species differences, data from animal models oftentimes are not transferable to humans, which usually results in a low success rate of new therapeutic agents in human clinical trials. It is imperative to develop translational models that are capable of reproducing aspects of AD‐associated pathology and accelerate the drug screening process for disease‐modifying therapies especially in the early stages of AD. In this study, we combined two important concepts in the study of neurodegenerative disorders and drug discovery, hiPSC‐derived cortical neurons and MEAs, to study the functions of human cortical neurons in the presence of Aβ and tau oligometric peptides, and evaluate whether this system can become a key component of the drug evaluation process for AD treatment.
Synthetic Aβ oligomers and brain extracts from APP transgenic mice were used to investigate Aβ‐induced neurotoxicity on hiPSC‐derived cortical neurons in vitro. This study indicated that the addition of Aβ oligomers and brain extracts to the cultures caused prominent neuronal dysfunctions in the cortical neurons in vitro. In particular, treatment with Aβ42 oligomers, or APP brain extracts from transgenic mice, led to a marked reduction in axon length and neurite complexity in human cortical neurons (Figures S1 and S4). These results were in line with previous findings by other groups who have shown altered cell morphology in rat primary cultures. 65 , 66 In addition to causing morphological damage, Aβ42 has also been implicated in numerous other molecular processes, including disruption of axonal transport, blocking of synaptic recruitment of receptors (eg, NMDAR, AMPAR), and altered gene expression, all of which have been implicated in AD pathogenesis and are expected to alter cell function. 63 , 64 In this study, a noticeable reduction in spontaneous neuronal activity was observed, where both the peak amplitude and firing frequency were greatly reduced in Aβ42 and APP‐be treated samples compared to controls (Figures 1, 5, 6 and Figure S3). These results are consistent with findings from other studies in which rodent neuronal cultures, animal models, or iPS cells derived from AD patients were used. 38 , 64
In addition to Aβ deposition and plaque formation, AD is also characterized by tau hyperphosphorylation and aggregation, which then leads to tangle formation. 67 , 68 In fact, Aβ‐induced tau phosphorylation, resulting in neurofibrillary tangle formation and accumulation, is considered to be one of the key events in AD pathogenesis. 69 To investigate the deleterious effects of tau oligomers on cortical neurons in vitro, in parallel experiments with the Aβ studies, hiPSC‐derived cortical neurons were dosed with tau oligomers and brain extracts from tau Tg mice. Similar to the Aβ results, tau treatment led to significant morphological alterations in the hiPSC‐derived cortical neurons (Figure S2). After further analysis, we observed a slight decrease in neuritic complexity and an increase in the number of cells with axonal varicosities caused by the addition of brain extracts from tau transgenic mice. Furthermore, the analysis of tau oligomers’ deleterious actions on cortical neuronal activity, after acute treatment, as noted from the MEA recordings, revealed a significant reduction in cell activity (ie, reduced rate of firing frequency) but without significant loss in cellular viability (Figures 2, 4, and 6). This is especially important as it is now hypothesized that treatment of AD after the MCI stage of the disease may be too late to rescue the phenotype. Clinical trials are now moving to recruitment of patients in the preclinical and MCI stages of the disease and a large effort is under way to identify biomarkers that can predict AD pathology even before the MCI stage.[ 1 ] It is now becoming more apparent that the preclinical/pre‐MCI stage, without extensive cell death, offers the best option for treating and potentially recovering healthy brain physiology. Thus, preclinical models using human neurons with loss of function, but without cell death, are needed to develop the next generation of therapeutics for AD. In summary, the results, in cortical neurons derived from human induced pluoripotent stem cells, offer strong support for the use of hiPSCs and a HoaC system as an alternative and relevant model for AD research and for drug testing.
5. CONCLUSION
Given the prevelance, severity, and cost of treating AD, plus the projected increase in the number of cases by the year of 2050, it is important that we not only clarify the etiology of AD, but also develop effective human‐based models for all stages of the disease to improve the drug development process. In this study, we developed a HoaC sytem composed of hiPSC‐derived cortical neurons cocultured with human astrocytes on MEAs to examine the effects of Aβ oligomers, tau oligomers, and brain extracts from AD transgenic mice on neuron physiology and structure. We showed that treatment with oligomers or brain extracts rapidly (within 1 hour) decreased LTP‐mediated increases in spontaneous action potential firing frequencey. Further, these treatments led to dysfunctional neuronal electrical characteristics as determined by patch‐clamp electrophysiology and changes to axonal length and branching compared to control‐treated cultures but without commensurate losses in cell viability. While more studies are needed to evaluate the system's ability to reproduce the effects of known therapeutic agents to improve AD symptoms, the findings presented here confirm the model's ability to reproduce important aspects of AD pathology, especially LTP disruption. These findings highlight this HoaC system's potential as a drug discovery platform for early stages of AD.
FUNDING INFORMATION
This study was conducted with funding by NIH SBIR grant numbers: 1R44AG058330 and 1R3AG060886.
AUTHOR CONTRIBUTIONS
Experimental design: James J. Hickman, Julbert Caneus, Xiufang Guo, John W. Rumsey, David Morgan, Nicholas M. Kanaan; performed the experiments: Julbert Caneus, Frank Sommerhage, Nesar Akanda, Sanya Georgieva, data analysis: Julbert Caneus, John W. Rumsey, Max Jackson, Christopher J. Long, Sanya Georgieva, Xiufang Guo; prepared the manuscript: Julbert Caneus, John W. Rumsey, James J. Hickman, David Morgan.
CONFLICT OF INTEREST
James J. Hickman has ownership interest and is Chief Scientist and member of the Board of Directors in a company that may benefit financially as a result of the outcomes of the research or work reported in this publication.
Supporting information
Supplemental Information
ACKNOWLEDGMENTS
The authors thank Dr. Arindon Goswami who prepared the surfaces (ie, coverslips and MEA surfaces) used to plate the cells and Sarah Lindquist who assisted with the cell cultures used for the experiments.
Caneus J, Akanda N, Rumsey JW, et al. A human induced pluripotent stem cell‐derived cortical neuron human‐on‐a chip system to study Aβ42 and tau‐induced pathophysiological effects on long‐term potentiation. Alzheimer's Dement. 2020;6:e12029 10.1002/trc2.12029
Julbert Caneus, Nesar Akanda, and John W. Rumsey have contributed equally to the work.
REFERENCES
- 1. Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow‐up study. The Lancet Neurology. 2006;5(3):228‐234. [DOI] [PubMed] [Google Scholar]
- 2. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. JAMA Neurology. 2001;58(12):1985‐1992. [DOI] [PubMed] [Google Scholar]
- 3. WHO, World Health Organization . The top 10 causes of death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. 2018.
- 4. CDC, Center for Disease Control . FastStat: Leading Causes of Death. https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm. 2016.
- 5. World‐Alzheimer‐Report , World Alzheimer Report 2018, The state of the art of demential research: new frontiers. https://www.alz.co.uk/research/world-report-2018. 2018.
- 6. Alzheimer's‐Association , 2019 Alzheimer's disease facts and figures. Alzheimers Dement 2019;15(3):321‐387. https://alz.org/media/Documents/alzheimers-facts-and-figures-2019-r.pdf. [DOI] [PubMed] [Google Scholar]
- 7. WHO, World Health Organization. Dementia: Key Facts . https://www.who.int/news-room/fact-sheets/detail/dementia. 2019.
- 8. Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer's disease. Therapeutic advances in neurological disorders. 2013;6(1):19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mehta D, Jackson R, Paul G, Shi J, Sabbagh M, et al. Why do trials for Alzheimer's disease drugs keep failing? A discontinued drug perspective for 2010‐2015. Expert Opin Investig Drugs. 2017;26(6):735‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alzheimer‐Association , Medications for Memory. 2019. https://www.alz.org/alzheimers-dementia/treatments/medications-for-memory. [Google Scholar]
- 12. McGonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol. 2014;87(1):162‐171. [DOI] [PubMed] [Google Scholar]
- 13. Eckert A, Keil U, Marques CA, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochemical Pharmacology. 2003;66(8):1627‐1634. [DOI] [PubMed] [Google Scholar]
- 14. Puzzo D, Gulisano W, Palmeri A, Arancio O, et al. Rodent models for Alzheimer's disease drug discovery. Expert Opin Drug Discov. 2015;10(7):703‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ronaldson‐Bouchard K, Vunjak‐Novakovic G. Organs‐on‐a‐Chip: a fast track for engineered human tissues in drug development. Cell Stem Cell. 2018;22(3):310‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Esch MB, Smith AS, Prot JM, Oleaga C, Hickman JJ, huler ML. How multi‐organ microdevices can help foster drug development. Adv Drug Deliv Rev. 2014;69(70):158‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coppeta JR, Mescher MJ, Isenberg BC, et al. A portable and reconfigurable multi‐organ platform for drug development with onboard microfluidic flow control. Lab Chip. 2016;17(1):134‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McAleer CW, Pointon A, Long JC, et al. On the potential of in vitro organ‐chip models to define temporal pharmacokinetic‐pharmacodynamic relationships. Sci Rep. 2019;9(1):9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oleaga C, Bernabini C, , Smith AS, et al. Multi‐Organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci Rep. 2016;6:20030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oleaga C, Riu A, Rothemund S, et al. Investigation of the effect of hepatic metabolism on off‐target cardiotoxicity in a multi‐organ human‐on‐a‐chip system. Biomaterials. 2018;182:176‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berry BJ, Nesar Akanda, Smith STA, et al. Morphological and functional characterization of human induced pluripotent stem cell‐derived neurons (iCell Neurons) in defined culture systems. Biotechnol Prog. 2015;31(6):1613‐1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ochalek A, Mihalik B, Avci XH, et al. Neurons derived from sporadic Alzheimer's disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res Ther. 2017;9(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang J, Li Song, Xi‐Biao He, Cheng C, Weidong Le, et al. Induced pluripotent stem cells in Alzheimer's disease: applications for disease modeling and cell‐replacement therapy. Mol Neurodegener. 2016;11(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vazin T, K AureliaBall, Hui Lu, et al. Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: a model system to study neurotoxicity in Alzheimer's disease. Neurobiol Dis. 2014;62:62‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295‐e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fa M, Puzzo D, Piacentini R, et al. Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep. 2016;6:19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Usenovic M, Niroomand S, Drolet RE, et al. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in Human Neurons Derived from induced pluripotent stem cells. J Neurosci. 2015;35(42):14234‐14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maity BK, Das AK, Dey S, et al. Ordered and Disordered segments of amyloid‐beta drive sequential steps of the toxic pathway. ACS Chem Neurosci. 2019;10(5):2498‐2509. [DOI] [PubMed] [Google Scholar]
- 29. Bate C, Williams A. Monomeric amyloid‐beta reduced amyloid‐beta oligomer‐induced synapse damage in neuronal cultures. Neurobiol Dis. 2018;111:48‐58. [DOI] [PubMed] [Google Scholar]
- 30. Gulisano W, Melone M, Li Puma DD, et al. The effect of amyloid‐beta peptide on synaptic plasticity and memory is influenced by different isoforms, concentrations, and aggregation status. Neurobiol Aging. 2018;71:51‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomic JL, et al. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35(3):352‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koss DJ, Jones G, Cranston A, et al. Soluble pre‐fibrillar tau and beta‐amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol. 2016;132(6):875‐895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kondo T, Asai M, Tsukita K, et al. Modeling Alzheimer's disease with iPSCs reveals stress phenotypes associated with intracellular abeta and differential drug responsiveness. Cell Stem Cell. 2013;12(4):487‐496. [DOI] [PubMed] [Google Scholar]
- 34. Campioni S, Mannini B, Zampagni M, et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6(2):140‐147. [DOI] [PubMed] [Google Scholar]
- 35. Tai HC, Serrano‐Pozo A, Hashimoto T, et al. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin‐proteasome system. Am J Pathol. 2012;181(4):1426‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Charkhkar H, , Meyyappan S, Matveeva E, et al. Amyloid beta modulation of neuronal network activity in vitro. Brain Res. 2015;1629:1‐9. [DOI] [PubMed] [Google Scholar]
- 37. Varghese K, Molnar P, Das M, et al. A new target for amyloid beta toxicity validated by standard and high‐throughput electrophysiology. PLoS One. 2010;5(1):e8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berry BJ, Smith AST, Long CJ, Martin CC, Hickman JJ, et al. Physiological abeta concentrations produce a more biomimetic representation of the Alzheimer's Disease Phenotype in iPSC derived Human Neurons. ACS Chem Neurosci. 2018;9(7):1693‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forny‐Germano L, Lyra e Silva NM, Batista AF. Alzheimer's disease‐like pathology induced by amyloid‐beta oligomers in nonhuman primates. J Neurosci. 2014;34(41):13629‐13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chabrier MA, Cheng D, Castello NA, Green KN, et al. Synergistic effects of amyloid‐beta and wild‐type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer's disease. Neurobiol Dis. 2014;64:107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Christensen DZ, Kraus SL, Flohr A, et al. Transient intraneuronal a beta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008;116(6):647‐655. [DOI] [PubMed] [Google Scholar]
- 42. Wong RS, Cechetto DF, Whitehead SN. Assessing the effects of acute amyloid beta oligomer exposure in the Rat. Int J Mol Sci. 2016;17(9):E1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zussy C, Brureau A, Keller E, et al. Alzheimer's disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid‐beta peptide injection in rats. PLoS One. 2013;8(1):e53117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wing C, Komatsu M, Delaney SM, et al. Application of stem cell derived neuronal cells to evaluate neurotoxic chemotherapy. Stem Cell Res. 2017;22:79‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482(7384):216‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hutzler M, Lambacher A, Eversmann B, Jenkner M, Thewes R, Fromherz P. High‐resolution multitransistor array recording of electrical field potentials in cultured brain slices. J Neurophysiol. 2006;96(3):1638‐1645. [DOI] [PubMed] [Google Scholar]
- 47. Obien ME, Deligkaris K,ullmann T, , et al. Revealing neuronal function through microelectrode array recordings. Front Neurosci. 2014;8:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Balducci C, Forloni G. In vivo application of beta amyloid oligomers: a simple tool to evaluate mechanisms of action and new therapeutic approaches. Curr Pharm Des. 2014;20(15):2491‐2505. [DOI] [PubMed] [Google Scholar]
- 49. Puzzo D, Piacentini R, Fá M, et al. LTP and memory impairment caused by extracellular abeta and tau oligomers is APP‐dependent. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, Clos AL, , Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild‐type mice. Mol Neurodegener. 2011;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sengupta U, Nilson AN, Kayed R. The role of Amyloid‐beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang W, Jiao B, Zhou M, Zhou T, Shen L. Modeling Alzheimer's Disease with Induced Pluripotent Stem Cells: current Challenges and Future Concerns. Stem Cells Int. 2016;2016:7828049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Santhanam N, Kumanchik L, Guo X, et al. Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials. 2018;166:64‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palop JJ, Mucke L. Amyloid‐beta‐induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13(7):812‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ma T, Klann E. Amyloid beta: linking synaptic plasticity failure to memory disruption in Alzheimer's disease. J Neurochem. 2012;120(Suppl 1):140‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta‐induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener. 2014;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stine WB , Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid‐beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278(13):11612‐11622. [DOI] [PubMed] [Google Scholar]
- 58. Combs B, Tiernan CT, Hamel C, Kanaan NM. Production of recombinant tau oligomers in vitro. Methods Cell Biol. 2017;141:45‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. 2012;40(4):667‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: characterization of a selective oligomeric tau antibody. J Alzheimers Dis. 2013;37(3):593‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abraham WC, Huggett A. Induction and reversal of long‐term potentiation by repeated high‐frequency stimulation in rat hippocampal slices. Hippocampus. 1997;7(2):137‐145. [DOI] [PubMed] [Google Scholar]
- 63. Hu NW, et al. Extracellular forms of abeta and tau from iPSC models of Alzheimer's disease disrupt synaptic plasticity. Cell Rep. 2018;23(7):1932‐1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nieweg K, Andreyeva A, van SB, Tanriöver G, Gottmann K. Alzheimer's disease‐related amyloid‐beta induces synaptotoxicity in human iPS cell‐derived neurons. Cell Death Dis. 2015;6:e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Korotzer AR, Pike CJ, Cotman CW. beta‐Amyloid peptides induce degeneration of cultured rat microglia. Brain Res. 1993;624(1‐2):121‐125. [DOI] [PubMed] [Google Scholar]
- 66. Nguyen L, Wright S, Lee M, et al. Quantifying amyloid beta (Abeta)‐mediated changes in neuronal morphology in primary cultures: implications for phenotypic screening. J Biomol Screen. 2012;17(6):835‐842. [DOI] [PubMed] [Google Scholar]
- 67. Busciglio J, Lorenzo A, Yeh J, Yankner BA. beta‐amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14(4):879‐888. [DOI] [PubMed] [Google Scholar]
- 68. Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S, et al. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115(1):201‐211. [DOI] [PubMed] [Google Scholar]
- 69. De Paula VJR, Guimarães FM, Diniz BS, Forlenza OV. Neurobiological pathways to Alzheimer's disease: amyloid‐beta, TAU protein or both? Dement Neuropsychol. 2009;3(3):188‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information