

FLARE enables rapid, feature-rich stains of chemical groups in biological samples for super-resolution and cleared tissue imaging.
Abstract
Fluorescence microscopy is a workhorse tool in biomedical imaging but often poses substantial challenges to practitioners in achieving bright or uniform labeling. In addition, while antibodies are effective specific labels, their reproducibility is often inconsistent, and they are difficult to use when staining thick specimens. We report the use of conventional, commercially available fluorescent dyes for rapid and intense covalent labeling of proteins and carbohydrates in super-resolution (expansion) microscopy and cleared tissue microscopy. This approach, which we refer to as Fluorescent Labeling of Abundant Reactive Entities (FLARE), produces simple and robust stains that are modern equivalents of classic small-molecule histology stains. It efficiently reveals a wealth of key landmarks in cells and tissues under different fixation or sample processing conditions and is compatible with immunolabeling of proteins and in situ hybridization labeling of nucleic acids.
INTRODUCTION
Two of the most important developments in fluorescence microscopy over the past one to two decades are super-resolution microscopy, for imaging small features beneath the ~250-nm diffraction limit of visible light, and cleared tissue microscopy, for deep imaging of intact specimens (1–5). Researchers are now able to routinely measure nanoscale molecule distributions, protein oligomerization, or protein-protein interactions and to determine the large-scale organization of biological specimens. Central to these efforts are a wide range of probes to fluorescently label the sample, with one of the most important classes being fluorescently labeled antibodies. While antibodies are powerful in their ability to specifically label specimens without need for genetic manipulation, they often suffer slow penetration in thick tissues, poor binding in specimens that have been heavily fixed or processed [e.g., formalin-fixed paraffin-embedded (FFPE) tissue], high cost, and inconsistent lot-to-lot reproducibility or commercial availability (6, 7).
Small-molecule histology stains such as H&E (hematoxylin and eosin) (8) are attractive potential alternative labels for super-resolution and cleared tissue microscopy, but they pose several challenges (9). First, many histology stains are affinity based, rather than covalent, and produce uneven staining particularly for relatively thick specimens, whereas covalent labeling renders the labels compatible with a wider range of sample processing or clearing techniques. Second, nonfluorescent histology stains are poorly suited to volumetric imaging and fluorescence-based super-resolution microscopy methods. Third, classic histology stains have limited flexibility in their spectral properties, whereas modern fluorescent dyes are available in many forms across the visible and near infrared with high quantum yields and good resistance to photobleaching.
To partly address the limitations of antibodies and small-molecule histology stains, we developed simple procedures for using commercially available small-molecule fluorophores to brightly label a wide variety of major organelles and landmark structures in cells and tissues for super-resolution microscopy and cleared tissue microscopy. The idea is to use small molecules to covalently label abundant chemical functional groups on biological samples, instead of specific biomolecules, to reveal the general physiology of the sample. These include the use of amine-reactive fluorophores for measuring distributions of proteins and aldehyde-reactive fluorophores for measuring distributions of oxidized carbohydrates, although other abundant reactive groups (thiols, carboxylates, etc.) may also be suitable. We termed this approach Fluorescent Labeling of Abundant Reactive Entities (FLARE). While the underlying chemical reactions are well known (10–16), for instance, when labeling purified proteins (12), specimen surfaces (17), or isolated live cells for subsequent tracking (18), the great utility of this simple labeling approach in super-resolution and cleared tissue microscopy has not been previously recognized. For super-resolution microscopy, we used expansion microscopy (ExM), a recently developed technique that physically expands biological specimens embedded in a swellable polymer (19–21). The physical expansion enables features closer than the ~250-nm diffraction limit of traditional light microscopy to be resolved in the expanded state on a conventional microscope. For cleared tissue microscopy with unexpanded specimens, we used a variant of the solvent-based tissue clearing method immunolabeling-enabled three-dimensional imaging of solvent-cleared organs (iDISCO) (22) that uses ethyl cinnamate (EC) for index matching (23).
RESULTS
FLARE reveals key landmarks in hydrogel-expanded specimens
We first tested the ability of FLARE to stain cultured human retinal pigment epithelial (RPE) cells that had been expanded fivefold using ExM. Using the labeling schematic in Fig. 1, we stained oxidized carbohydrates on the specimen with ATTO 565 hydrazide, amines with ATTO 647N NHS (N-hydroxysuccinimidyl) ester, and DNA (noncovalently) with SYBR Green (Fig. 2 and fig. S1). Mitochondria were densely labeled by the amine stain, while lysosomes and the plasma membrane were densely labeled by the carbohydrate stain, and perinuclear structures including portions of the Golgi were labeled by both amine and carbohydrate stains (Fig. 2, A to C, and figs. S1 and S2). The nucleus and nuclear envelope of interphase cells were evident as were nuclear pores in both the amine and carbohydrate channels (Fig. 2C and fig. S2, I to L). In dividing cells (Fig. 2, D to F, and fig. S1), kinetochores and centrosomes were also labeled well. Thus, the chemical stains efficiently report on many major landmark structures and components of the cell.
Fig. 1. Schematic illustration of FLARE staining of amines and carbohydrates, together with DNA staining, for super-resolution and cleared tissue microscopy.
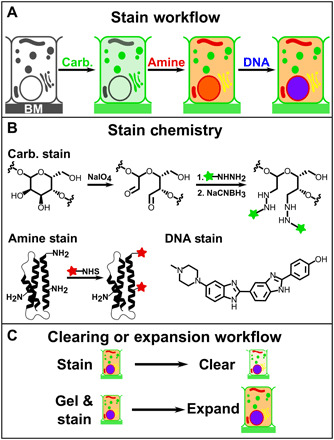
(A) Carbohydrates, amines, and DNA on cells or tissues are stained as the order shown. (B) Vicinal diols on carbohydrates are oxidized to aldehydes using sodium periodate, coupled to hydrazide-functionalized fluorophores (green), and then stabilized by reduction with sodium cyanoborohydride. Amines are labeled by reaction with an NHS-functionalized fluorophore (red). DNA is labeled noncovalently by incubation with a conventional DNA stain such as Hoechst or SYBR Green (blue). (C) Specimen processing workflow with respect to clearing and expansion.
Fig. 2. FLARE staining of hydrogel-expanded cultured cells.
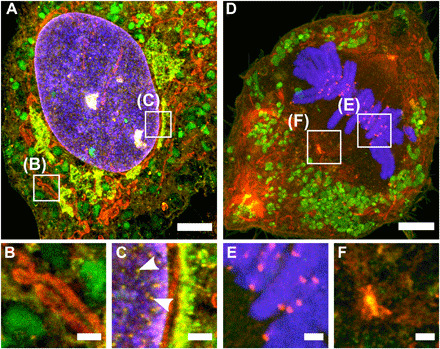
Confocal microscopy images of hydrogel-expanded RPE cells that have been covalently stained for amine (red) and carbohydrate (green) using FLARE along with a conventional noncovalent stain for DNA (blue). (A) Interphase cell showing various subcellular organelles including mitochondria (red), lysosomes (green), and perinuclear structures (yellow). (B and C) Zoomed-in views of boxed regions in (A). Arrowheads in (C) indicate nuclear pores. (D) Dividing cell showing various subcellular organelles. (E and F) Zoomed-in views of boxed region in (D) showing kinetochores and centrosomes, respectively. All scale bars are in preexpansion units. Scale bars, 3 μm (A and D) and 500 nm (B, C, E, and F).
FLARE can be straightforwardly combined with antibody labeling, which we used here to confirm the identities of some subcellular organelles or structures such as mitochondria, lysosomes, and the golgi apparatus (fig. S2). However, we also realized that some features observed in the images are not easily identified, and some known structural features are difficult to detect in the images. For instance, in dividing RPE cells, we frequently observed a swirling tuft of unknown amine-rich cytoskeletal filaments toward one end of the cell, while at the same time, we were surprised that microtubules of the mitotic spindle were unlabeled (Fig. 2D). Nonetheless, results with cultured cells indicate that these relatively nonspecific covalent stains together with the improved spatial resolution (~65 nm) attained using specimen expansion with confocal microscopy (19–21) can reveal a wealth of details but that more work will be required to better understand and possibly tune the specificity of labeling.
Beyond cultured cells, we found that FLARE also works well in hydrogel-expanded tissues, as we demonstrate using mouse kidney, a model specimen containing a diverse range of structures and cell types (Fig. 3). Within glomeruli, the filtration units of the kidney, the carbohydrate stain brightly labeled the basement membranes of the capillary loops and the mesangial matrix, while the amine stain efficiently outlined cell boundaries and other structures, including the intricate details of interdigitated podocyte epithelial cells that form a major component of the glomerular filtration barrier (Fig. 3, A to C, and movie S1). In proximal convoluted tubules of the kidney, the basement membrane was also labeled by the carbohydrate stain, and the amine stain revealed mitochondria, the nuclear envelope, and brush border microvilli (Fig. 3D). While we focus in this paper on a variation of ExM termed MAP (magnified analysis of the proteome) (20) that uses heat and detergent to dissociate tissue-hydrogel hybrids rather than enzymatic digestion as in the original ExM method (21), we found that enzymatically digested mouse kidney tissue could also be covalently stained (fig. S3, A to D), indicating that the possible loss of material due to digestion was not prohibitive. In addition, we found that FLARE is compatible with in situ hybridization for detection of nucleic acids (fig. S3, E to H).
Fig. 3. FLARE staining of hydrogel-expanded mouse kidney tissue.
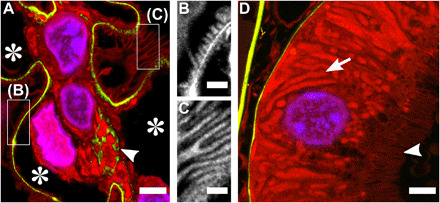
Confocal microscopy images of hydrogel-expanded mouse kidney that have been covalently stained for amines (red) and carbohydrates (green) using FLARE along with a conventional noncovalent stain for DNA (blue). (A) Region of a mouse kidney glomerulus in which carbohydrates are intensely labeled along the basement membrane of the capillary loops (stars) and within the mesangial matrix (arrowhead). (B and C) Zoomed-in views of amine channel for boxed regions in (A) showing interdigitated podocyte foot processes. (D) Region of a proximal convoluted tubule in which carbohydrates highlight tubular basement membrane and amines intensely label mitochondria (arrow) and microvilli (arrowhead). All scale bars are in pre-expansion units. Scale bars, 3 μm (A and D) and 500 nm (B and C).
FLARE labels thick tissue samples rapidly and uniformly
We next tested FLARE in various unexpanded specimens that were subsequently optically cleared using dehydration in a graded series of tetrahydrofuran (THF), followed by immersion in EC, a variant of the iDISCO clearing method (22, 23). The labeling protocol was approximately the same as for the labeling of tissue-hydrogel specimens with two important adjustments to help enhance the uniformity of the stains throughout the depth of the tissue. First, during both covalent labeling reactions, we dissolved dyes in a cosolvent mixture consisting of a 1:1 ratio of aqueous buffer to THF to increase the rate of diffusion through the specimen (Fig. 4, A to E, and fig. S4). Second, for the amine-labeling reaction, we adjusted the pH to 6.0 to reduce the rate of the coupling reaction while also reducing the rate of hydrolysis of the ester that would render the dye unreactive (24, 25). These measures greatly improved the uniformity (Fig. 4, A to E). For example, approximately uniform covalent amine labeling of a ~500-μm-thick section of mouse kidney could be achieved in ~2 hours, whereas the same tissue section when labeled with antibodies for 6 days was only stained at a depth of <50 μm at each surface of the tissue section (Fig. 4E). We then used this procedure to stain and clear a relatively large volume of mouse kidney (~4 mm by ~3.5 mm by ~1 mm) and imaged it using a recently published open-top light sheet microscope (26) at a resolution of ~2 μm (Fig. 4, F to H, and fig. S5, A to D). We were able to observe renal tubules over long distances with characteristic segment-specific stain distributions for proximal tubules, distal tubules, loop of Henle, etc., that provide bearing on the location of the tubule within the nephron. We validated the identities of these key features in mouse kidney using covalent amine and carbohydrate labeling concurrent with a triple immunostain against cytokeratin (CK8+18, enriched on collecting ducts), aquaporin (AQP-1, enriched on proximal tubules), and podocalyxin (PODXL, enriched on glomeruli) (fig. S6).
Fig. 4. FLARE staining of thick, cleared tissue specimens.
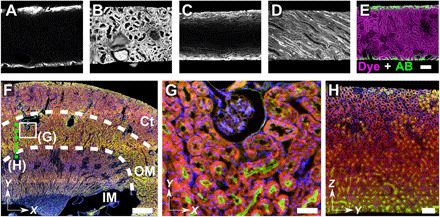
(A to E) Kidney sections (500-μm thick) were covalently stained, cut perpendicular to the section face, and imaged to measure stain intensity as a function of depth. Staining using an aqueous buffered solution produced uneven surface stains for (A) ATTO 647N NHS ester and (C) ATTO 565 hydrazide, but labeling with an aqueous/THF cosolvent mixture greatly improved the uniformity, as shown in (B) and (D), respectively. (E) Comparison of stain uniformity for immunolabeling of collagen (AB, green) for 6 days and amine labeling (dye, magenta) with ATTO 647N NHS ester for 2 hours. (F) Open-top light sheet microscopy images of ~1-mm-thick mouse kidney sections in which amines (red) and carbohydrates (green) show distinct distributions among the cortex (CT), outer medulla (OM), and inner medulla (IM) regions. (G) Zoom-in view of (F) taken from outer medullar region which contains various tubular architectures. (H) Cross-sectional view of the optically cleared mouse kidney sections along the green dashed line in (F). Scale bars, 100 μm (A to E and H), 50 μm (G), and 0.5 mm (F).
FLARE is applicable to a wide range of tissues
Besides kidney tissue, FLARE also revealed an abundance of physiological details in a diverse range of other specimens (Fig. 5, and fig. S5, E to X). For instance, mouse intestine exhibited clear staining of intestinal villi, crypts, subepithelial cells, specialized cells (goblet cells and Paneth cells), lymphatic vessels, and muscle cell boundaries; mouse liver revealed glycogen-rich hepatocytes with a strongly anticorrelated subcellular distribution of amine- and carbohydrate-reactive domains; mouse sperm exhibited distinct developmental patterns of carbohydrates within seminiferous tubules of the testis; and human prostate gland and connective tissue from stroma are distinctly labeled and well distinguished from each other. Even FFPE human kidney tissue was straightforwardly labeled using FLARE, a notable capability because FFPE tissue processing often leads to loss of antigenicity for immunolabeling, whereas primary amines and carbohydrates remain quite reactive. We further compared FLARE with traditional histological staining by treating consecutive 10-μm-thick mouse kidney FFPE sections to either H&E or FLARE (fig. S7). By converting the DNA and amine channels of the FLARE-stained sections to a simulated H&E image using the procedure of Giacomelli et al. (27), we found that the FLARE and H&E tissues are qualitatively quite similar, although the standard fluorescence display of the DNA, amine, and carbohydrate signals of the FLARE data provided much greater detail (fig. S7).
Fig. 5. FLARE staining of a variety of cleared tissue specimens.
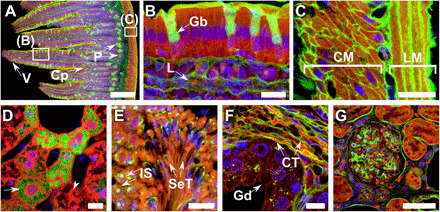
Fluorescence microscopy images of optically cleared tissue specimens that were covalently stained for amines (red) and oxidized carbohydrates (green) along with a conventional noncovalent stain for DNA (blue). (A to C) Mouse intestine, in which villi (V), crypt (Cp), Paneth cells (P), goblet cells (Gb), lymphatic vessels (L), circular muscle (CM), and longitudinal muscle (LM) layers are evident in the carbohydrate channel. (D) Mouse liver where carbohydrate- and amine-rich regions are indicated with an arrow and an arrowhead, respectively. (E) Mouse testis, in which immature spermatocytes (IS) and mature spermatocytes (MS) are evident and contain distinct carbohydrate distributions in the carbohydrate channel. (F) human prostate, where gland (Gd) and connective tissue (CT) from stroma are distinctly labeled. (G) Formalin-fixed, paraffin-embedded human kidney. Scale bars, 100 μm (A and G) and 10 μm (B to F).
Order of processing for FLARE is flexible
While there is considerable flexibility in the order of processing for FLARE in both expanded and nonexpanded samples, some care is required (see Materials and Methods and table S1). First, the sodium periodate oxidation reaction used in carbohydrate labeling bleaches fluorophores already on the sample and tended to produce cracks in the expanded kidney tissue specimen if the oxidation step was performed before sample gelation. As a result, we performed carbohydrate labeling before amine labeling, in general, and after expansion for kidney specimens [Figs. 3 and 4 (F to H), and figs. S1 (I to P), S5 (A to D), S6, and S7 (E and F)]. Second, although covalent modification of the sample via FLARE has the potential to perturb epitope antigenicity necessary for antibody binding, we found that antibody labeling generally works well after FLARE. Of a panel of 16 antibodies, 14 antibodies were able to effectively stain mouse kidney tissue (fig. S8) or RPE cells (fig. S9) after covalent labeling of carbohydrates and amines. Among the two impacted antibody stains, we found that the carbohydrate staining step partially blocked antibody staining of agrin on mouse kidney and fully blocked the antibody staining GM130 on RPE cells but that both of these antibodies bound their targets well after covalent labeling of amines. This may indicate that we only labeled a small fraction of available amines, such that most amines on epitopes are unmodified, but that carbohydrate oxidation and/or fluorophore labeling may be more complete. We found that it was straightforward to change the order of labeling in cases like anti-GM130 (fig. S2, M to Q) to immunolabel before covalent staining, although it is possible that a milder oxidation step may help better preserve antigenicity.
DISCUSSION
One might initially expect that the indiscriminate labeling of primary amines and carbohydrates in cells and tissues would yield little contrast because of the high abundance of these species. However, the resulting feature-rich labeling we obtained likely results from an interplay between the uneven distribution of these groups throughout the specimen (28, 29), the reactivity of those groups [which can vary depending on the amine pKa (30) or other local factors], and earlier reactions with the specimen, such as may have occurred during aldehyde fixation and would consume some available reactive groups. Some known structures that were difficult to detect in expanded RPE cells, such as the endoplasmic reticulum or kinetochore fibers of the mitotic spindle, likely result from the above considerations and from the possibility that relatively weak labeling of these structures may be difficult to detect due to the presence of many other brightly labeled structures within the specimen.
Here, we have shown that simple amine- and carbohydrate-reactive covalent stains, together with established DNA stains, are versatile tools in super-resolution fluorescence microscopy and cleared tissue fluorescence microscopy. Although we recently published a preliminary application of the use of amine-reactive fluorophores for rapid (<5 min) surface labeling of unexpanded and uncleared clinical specimens for rapid diagnosis (31, 32), the versatility of FLARE for super-resolution microscopy and cleared tissue microscopy was not explored. In addition, FLARE can be easily combined with different staining modalities [e.g., fluorescence in situ hybridization (FISH) labeling of nucleic acids and immunolabeling of proteins (figs. S3, E to H, and S8 to S9)], is compatible with a large selection of excellent fluorophores across the visible and near infrared, and reveals abundant details in a wide range of samples and sample processing methods. While the contrast provided by these nonspecific stains allows researchers to identify a range of features and key landmarks, the highlighted features vary between types of specimens, and the utility of the stains are therefore somewhat dependent on the specific application. In the future, we look toward expanding the palette of covalent probes for FLARE by developing stains that target a wider range of chemical groups and by developing probes that target more specific subsets of proteins or carbohydrates. FLARE, together with newly developed custom probes against lipids (33) or other groups, will provide a versatile toolset that complements molecule specific probes such as antibodies and FISH probes.
MATERIALS AND METHODS
Chemicals and reagents
Primary antibodies were purchased as follows: goat anti-podocalyxin (R&D Systems Inc., AF1556), rabbit anti-collagen IV (Abcam, ab6586), mouse anti-alpha smooth muscle actin (BioLegend Inc., 904601), rabbit anti-aquaporin 1 (Abcam, ab15080), guinea pig anti-cytokeratin 8+18 (Abcam, ab194130), mouse anti-agrin (DSHB Inc., 6D2), rabbit anti-podocin (Sigma- Aldrich, P0372), rabbit anti-TOMM20 (Santa Cruz Biotechnology, sc-11415), rabbit anti–LAMP-1 (Abcam, 24170), anti-GM130 (BD Biosciences, 610822), rabbit anti–GFP (green fluorescent protein) conjugated with Alexa Fluor 488 (Thermo Fisher Scientific, A-21311), rabbit anti-GFP conjugated with Alexa Fluor 647 (Thermo Fisher Scientific, A-31852), rabbit anti-H3K27ac (Active Motif , 39133), rabbit anti-H3K4me3 (Active Motif, 39159), rat anti-alpha tubulin (Invitrogen, MA1-80017), goat anti-nogo N-18 (Santa Cruz Biotechnology, sc-11027), rabbit anti-PMP70 (Thermo Fisher Scientific, PA1-650), and anti-vimentin V9 (Invitrogen, 18-0052). Unconjugated secondary antibodies were purchased as follows: donkey anti-goat (Jackson ImmunoResearch, 705-005-151), donkey anti-rabbit (Jackson ImmunoResearch, 712-545-150), donkey anti-mouse (Jackson ImmunoResearch, 715-005-151), donkey anti-guinea pig (Jackson ImmunoResearch, 706-001-003), and donkey anti-rat (Jackson ImmunoResearch, 712-005-153). Donkey anti-rabbit conjugated with Alexa Fluor 488 (711-545-152) and donkey anti-mouse conjugated with Alexa Fluor 488 (715-545-150) were purchased from Jackson ImmunoResearch. Fluorescent dyes were purchased as follows: Alexa Fluor 750 NHS ester (Thermo Fisher Scientific, A20011), Alexa Fluor 546 NHS ester (Thermo Fisher Scientific, A2002), ATTO 647N NHS ester (Sigma-Aldrich, 18373-1MG-F), ATTO 565 NHS ester (Sigma-Aldrich, 72464-1MG-F), ATTO 565 hydrazide (ATTO-TEC GmbH, AD 565-121), AFdye 405 NHS ester (Fluoroprobes, 1061-1), Hoechst 33258 (Sigma-Aldrich, B2883-25MG), and SYBR Green I (Invitrogen, S7563). Glutaraldehyde (50%; GA; 16320) was purchased from Electron Microscopy Sciences. Reagents for polymerization were purchased as follows: 32% paraformaldehyde (PFA) aqueous solution (Electron Microscopy Sciences, RT15714), 40% acrylamide (AA) aqueous solution (Bio-Rad Laboratories, 1610140), 2% bis-acrylamide (BA) aqueous solution (Bio-Rad Laboratories, 14101420), 97% sodium acrylate (SA) powder (Sigma-Aldrich, 408220), VA-044 initiator (Thermo Fisher Scientific, NC0471397), ammonium persulfate (APS; Thermo Fisher Scientific, 17874), tetramethylethylenediamine (TEMED; Thermo Fisher Scientific, 17919), and 97% 4-hydroxy-2,2,6,6-tetramethylpiperadin-1-oxyl (TEMPO; Sigma-Aldrich, 176141). Reagents for enzymatic digestion were purchased as follows: proteinase K (Thermo Fisher Scientific, EO0491), 10× tris-acetate-EDTA (TAE buffer; Fisher Bioreagents, BP2434-4), 10× phosphate-buffered saline (PBS buffer; Fisher Bioreagents, BP399-1), guanidine hydrochloride (Sigma-Aldrich, G3272), and collagenase (F type blend, Sigma-Aldrich, C7926), Hanks’ balanced salt solution (HBSS; 1×, Corning). Tris base (BP-152), calcium chloride (CaCl2; S25223), sodium chloride (NaCl; S271500), d-glucose (D16-1), and sodium acetate (NaOAc; BP333-500) powders were purchased from Thermo Fisher Scientific. SDS (L3771), sodium periodate (NaIO4; 311448), sodium cyanoborohydride (NaCNBH3; 156159), EC (112372), methacrylic acid NHS (MA-NHS; 730300), 4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol (Triton X-100, X100), 4-morpholineethanesulfonic acid (M3671), piperazine-1,4-bis(2-ethanesulfonic acid) disodium salt (PIPES disodium salt; P3768), EGTA (E4378), magnesium chloride (MgCl2; M8266), sodium borohydride (NaBH4; 213462), sodium azide (NaN3; S2002), poly-l-lysine (P8920), Trolox (238813), glucose oxidase (G2133-50KU), and catalase (C100) were purchased from Sigma-Aldrich. Sodium bicarbonate (NaHCO3; 470302) was purchased from VWR Scientific. Bovine serum albumin (BSA; BAS-50) was purchased from Rockland Immunochemicals Inc. Reagents for DNA FISH were purchased as follows: formamide (Fisher Chemical, F84-1), 20× saline sodium citrate (SSC buffer; Sigma-Aldrich, S6639), 50% OmniPur Dextran Sulfate (EMD Millipore, 3730), and Tween 20 (Sigma-Aldrich, P9416). Major satellite and ATTO 647N conjugated reporter oligonucleotides were obtained from Integrated DNA Technologies.
Cell culture
H-tert RPE cells were obtained and authenticated from ATCC and were used without further authentication. RPE cells were tested negative for mycoplasma with 4′,6-diaminodino-2-phenylindole dihydrochloride. RPE cells were cultured in Dulbecco’s modified eagle medium (Gibco, 11995-065) supplemented with 100 U/mL penicillin and streptomycin (Gibco, 11140-50), 1% nonessential amino acids (Gibco, 11140-050), and 10% fetal bovine serum (Gibco, 26140079), and grown to ~70 to 90% confluency. The cells were then trypsinized with 0.25% trypsin-EDTA (Gibco, 25200056) and seeded (~50,000 cells per well) on no. 1.5 round coverslip (~12 mm) in 24-well culture plates. Twenty-four hours later, the cells were fixed with either 3.2% PFA and 0.1% GA or 4% PFA in PEM buffer [100 mM aqueous Pipes, 1 mM EGTA, and 1 mM MgCl2 (pH 7)] for 10 min at room temperature. In some cases, cells were extracted with 0.5% Triton X-100 in PEM buffer for 30 s before fixation with 3.2% PFA and 0.1% GA in PEM buffer (see table S1 for details). Fixed cells were stored in 1× PBS with 3 mM sodium azide (1× PBS azide) at 4°C before use.
Transfection of cultured cells
Trypsinized RPE cultured cells (~1 to 2 × 106 cells) were pelleted down by centrifugation (150g for 5 min), resuspended in nucleofection solution (Lonza Kit V, VACA-1003) with 2 μg of plasmid mEmerald-Golgi-7, and electroporated following manufacturer’s (Lonza Amaxa Nucleofector I/II) X-001 pulse program. mEmerald-Golgi-7 was a gift from M. Davidson’s Lab (Addgene plasmid #54108; http://n2t.net/addgene:54108; RRID:Addgene_54108). pAc-GFPC1-Sec61β was a gift from T. Rapoport (Addgene plasmid #15108; http://n2t.net/addgene:15108; RRID:Addgene_15108). Transformed cells (transfection efficiency, ~70%) were then seeded (~80,000 cells per well) on no. 1.5 round coverslips (~12 mm) in 24-well culture plates and allowed to recover for 24 hours before fixation with 3.2% PFA and 0.1% GA in PEM buffer for 10 min at room temperature. Fixed cells were stored at 4°C in 1× PBS azide until use.
Preparation of fluorophore-labeled secondary antibodies
NHS ester functionalized dyes were used to conjugate with secondary antibodies. Briefly, 40 μl of secondary antibody, 5 μl of 1 M NaHCO3, and 1 to 2 μg of fluorophore were mixed. The reaction mixture was protected from light and was completed in 30 min. The fluorophore-conjugated secondary antibody was purified and collected from the crude reaction mixture via a disposable NAP-5 column (GE Healthcare Life Sciences, 17085301) and further characterized by ultraviolet/visible absorption spectroscopy.
Mouse organ dissection and preparation
All protocols and methods involving animals in this work were approved by the Institutional Animal Care and Use Committee at University of Washington.
Two-month old C57BL/6 male mice were anesthetized by isoflurane/oxygen mixture followed by cardiovascular perfusion with 1× PBS for 3 min followed by 4% PFA solution in 1× PBS for 5 min. Kidneys were then collected, and the renal capsules were removed. Other organs such as intestine and testis were also collected. Organs were fixed for 1 to 6 hours in 4% PFA solution in 1× PBS (see table S1 for details). Then, they were washed by 1× PBS solution three times and sliced by a vibratome to 100-μm thick. Because of the softness of testis and intestine, agarose gel was used to embed them while slicing. All slices were stored in 1× PBS azide at 4°C until use.
For the comparison of H&E and FLARE stains (fig. S7), mouse kidney tissue was collected from healthy Balb/c male mice at 12 weeks of age. Kidneys were perfused with PBS to remove blood cells, fixed in 10% buffered formalin, and then embedded in paraffin. FFPE kidney tissue was sliced into sections of ~10 μm, deparaffinized, and stained with H&E by Pathology Research Services Laboratory at the University of Washington. The stained tissue sections were then imaged with an Aperio ScanScope AT2 digital whole slide scanner at the Harborview Medical Center Digital Pathology Facility.
Human kidney and prostate preparation
A deidentified FFPE human kidney tissue block was obtained from NW BioTrust under approval from the University of Washington Institutional Review Board with deidentification. Sections of ~60-μm thickness were prepared using a microtome. To deparaffinize the section, the section was soaked in xylene solution for 10 min. Then, the section was rehydrated by incubation in a series of ethanol/water mixtures with descending ethanol concentration (100, 95, 85, 70, 50, and 0%). Eventually, rehydrated slices were stored in 1× PBS at 4°C until further use. Deidentified, freshly fixed human prostate samples were received from the University of Washington Genitourinary Biorepository with patient consent and stored at 4°C, and 100-μm sections were prepared with a vibratome. Approval was obtained from the University of Washington Institutional Review Board.
Cell gelation, denaturation, and expansion
The MAP (magnified analysis of the proteome) sample preparation used here for cells was adapted from Ku et al. (20). Fixed cells on 12-mm-round coverslips were treated with 10 mM aqueous NaBH4 solution for 10 min, washed with 1× PBS, and then incubated in monomer solution [20% (wt/wt) AA, 10% (wt/v) SA, 0.05% (wt/wt) BA, 4% (v/v) PFA, and 0.67% (v/v) TEMED] overnight at room temperature before gelation with 0.2% (wt/v) APS at room temperature for at least 30 min. After polymerization, the cell-embedded hydrogel on the coverslip was cleared in denaturing solution [200 mM SDS, 200 mM NaCl, and 50 mM tris base (pH 9.3)] for 1 hour at 90°C. The denatured cell-embedded hydrogel was then sequentially washed by PBST [1× PBS, 0.1% (wt/v) Triton X-100 and 0.02% (wt/v) NaN3] and 1× PBS before placing into deionized (DI) water for sample expansion.
Tissue gelation, denaturation/digestion, and expansion
The MAP sample preparation used here for expanded mouse kidney tissues was adapted from Unnersjö et al. (34). Fixed kidney sections were soaked in monomer solution [20% (wt/wt) AA, 10% (wt/v) SA, 0.05% (wt/wt) BA, 4% (v/v) PFA, and 0.1% (wt/v) VA-44] overnight at 4°C. Gelation was performed at 45°C for 2 hours. The polymerized gel was carefully peeled off, trimmed, and then transferred into denaturing solution [200 mM SDS, 200 mM NaCl, and 50 mM tris (pH 9.3)]. The expanded mouse kidney tissue was processed in a similar way but with denaturation at 70°C for 24 hours followed by 90°C for another 24 hours. The sample was then thoroughly washed by PBST before placing into DI water to expand.
The ExM sample preparation for fig. S3 was adapted from Chozinski et al. (21). Immunostained fixed mouse kidney sections were first incubated in 1 mM MA-NHS in 1× PBS at room temperature, followed by incubation in monomer solution [1× PBS, 2 M NaCl, 2.5% (wt/wt) AA, 0.15% (wt/wt) BA, and 8.625% (wt/wt) SA] for at least an hour in 4°C. Gelation of the kidney section was then performed at 37°C for 1.5 to 2.5 hours with monomer solution containing 0.2% (wt/wt) APS, 0.2% (wt/wt) TEMED, and 0.01% (wt/wt) TEMPO. The hydrogel-embedded kidney sample was digested overnight with proteinase K (8 U/ml) in proteinase digestion buffer (1× TAE buffer, 0.5% Triton X-100, and 0.8 M guanidine hydrochloride) at 37°C. The sample was then further digested with collagenase (5 mg/ml) in collagenase digestion buffer (1× HBSS with 0.7 mM CaCl2) overnight at 37°C.
General covalent staining procedure for hydrogel samples and cleared tissues
All samples were stained in the same sequence: carbohydrates, amines, and lastly, DNA. For carbohydrate staining, the samples were first oxidized with NaIO4 and then incubated with fluorescent dye ATTO 565 hydrazide after washing off excess NaIO4. NaCNBH3 was then used to reduce the hydrazone bond formed for better stability. The samples were then incubated with fluorescent dye ATTO 647N NHS ester for the staining of amines in proteins after washing off excess NaCNBH3. Last, fluorescent DNA dye (Hoechst 33258 or SYBR Green I) was added for nuclear staining. Samples were either expanded (for hydrogel samples) or refractive index matched with EC (for organic cleared samples) before imaging. A summary of the various reagents, buffers, quantities, and conditions for all data in this paper is listed in table S1.
Immunostaining procedures
All nontransfected samples covalently stained before gelation were processed in this sequence: carbohydrates, amines, antibodies, gelation, and lastly, DNA. For carbohydrate, amine, and DNA staining, see above section on General Covalent Staining Procedure for hydrogel samples and cleared tissues. For general immunostaining of cell samples before gelation, the sample was incubated first in block and permeabilization solution [BP, 0.3% (wt/v) BSA, 0.5% (wt/v) Triton X-100, and 1× PBS] for 30 min, followed by primary antibodies in BP for 90 min, and lastly, secondary antibodies in BP for 45 min. Samples were postfixed with 0.25% GA in 1× PBS for 5 min. For general immunostaining of tissue samples, the same procedure was followed but with different incubation conditions (see table S1 for details). After immunolabeling, tissue samples for hydrogel expansion were treated for 1 hour with freshly prepared 1 mM MA-NHS in 1× PBS.
All nontransfected cell samples covalently stained after gelation were proceeded in this sequence: antibodies, gelation, carbohydrates, and lastly, amine. For postgelation carbohydrate and amine staining, to immunostain before gelation, the procedure requires the use of secondary antibodies conjugated to biotin instead of a fluorescent dye because the mild oxidation used in carbohydrate staining step later may chemically bleach fluorophores on the secondary antibodies.
For the transfected cell sample in fig. S2 (R to V), immunostaining with anti-GFP Alexa Fluor 488 was performed after the postgelation covalent stains. The immunostain on the hydrogel sample was performed in BP for 15 hours at room temperature. Samples were either expanded (for hydrogel samples) or refractive index matched with EC (for organic cleared samples) before imaging. A summary of the various reagents, buffers, quantities, and conditions for all data in this paper is listed in table S1.
DNA fluorescence in situ hybridization
The DNA-FISH procedure was adapted from Beliveau et al. (35). FLARE-stained and MAP-expanded mouse kidney samples were first incubated in 0.5% Triton X-100 in 1× PBS for 2 hours. Samples were washed once in 1× PBS, then in 2× SSCT (0.1% Tween 20 in 2× SSC), and lastly, in hybridization buffer (50% formamide and 0.1% Tween 20 in 2× SSC) for 10 min each time. Samples were incubated in fresh hybridization buffer for 30 min at 60°C. The hybridization mixture (50% formamide, 10% dextran sulfate, 0.1% Tween 20, 3 mM sodium azide, 100 nM MaSat oligo probe, and 100 nM oligo reporter in 2× SSC) was preheated to 92.5°C for 5 min and then added to each sample at an approximate 2:1 volume ratio. Samples were denatured at 92.5°C for 10 min and hybridized at 37°C overnight. Samples were washed three times in 2× SSCT, first at 60°C, second at 37°C, and lastly at room temperature, 15 min each time. Samples were stored in 0.2× SSCT at 4°C for at least 1 hour or until needed for imaging (within a week). Before imaging, samples were fully expanded by replacing the sample buffer at least twice with water every 10 min at 4°C.
The MaSat oligo probe (5′-GGAATATGGC GAGAAAACTG AAAATCACGG AATGATACGG CGACCACCGA ACTGCTACAG-3′) contains 30 nt of the mouse major satellite repeat DNA sequence [obtained from Lehnertz et al. (36)] and 30 nt complimentary to the fluorophore conjugated oligo reporter (5′-/5ATTO647NN/CTGTAGCAGT TCGGTGGTCG CCGTATCATT-3′).
Organic solvent clearing
Stained tissue specimens were dehydrated using solutions of THF/H2O with ascending THF (v/v) concentrations (50, 80, and 100%) as follows. Stained 100-μm-thick tissue specimens were first incubated in 50% THF for 10 min, then in 80% THF twice for at least 15 min each, and lastly, in 100% THF twice for at least 15 min each. Stained 500- to 800-μm-thick tissue specimens were first incubated in 50% THF for 30 min, then in 80% THF twice for at least 1 hour each, and lastly, in 100% THF twice for at least 1 hour each. After THF dehydration, 100% dichloromethane (DCM) was then added and incubated until the specimen sank. DCM was removed, and 100% EC oil was added to the sample followed by incubation for 2 hours or more. For thicker specimens, EC was exchanged a few times to ensure good index matching.
Sample mounting
EC-cleared samples were transferred onto a rectangular no. 1.5 coverslip (24 mm by 50 mm; Fisher Scientific, #12544E) using a paintbrush. A piece of double-sided tape was attached to the surface of the coverslip along the each of the short edges of the coverslip, then a drop of EC was added on sample surface to prevent it from drying, and a second coverslip was pressed onto the first to form a sandwich structure held together by the double-sided tape. For the fully expanded hydrogel-embedded samples, the gel was adhered to a poly-l-lysine–coated coverslip and imaged immediately.
Fluorescence microscopy and imaging
All data, except for 1-mm EC-cleared mouse kidney and figs. S6 to S9, were acquired using a Leica SP5 inverted confocal point scanning microscope at the University of Washington Biology Imaging Facility. The objective lenses used on the confocal microscope were a HC Plan Apo CS 63× 1.2 numerical aperture (NA) water-immersion objective, a HC Plan APO CS 63× 1.4 NA oil-immersion objective, a HC Plan Apo CS 20× 0.7 NA air objective, and a HC Plan Apo CS 10× 0.4 NA air objective. A summary of the various imaging conditions used for each dataset can be found in table S1.
Figures S7 to S9 were acquired with a conventional wide-field epifluorescence inverted Nikon Ti-S microscope. The objective lenses used on the wide-field microscope were CFI Plan Apo VC 60× 1.2 NA (Nikon, Melville, NY, USA) water-immersion objective, CFI S Plan Fluor 20× 0.45 NA (Nikon) air objective, and CFI Plan Apo λ 4× 0.2 NA (Nikon) air objective. The illumination source was a four-channel light-emitting diode (LED4D251, Thorlabs, Newton, NJ, USA) that used a quad-band filter set (Chroma 89402). The detection unit used to capture the images was a ZWO-AS174MM-COOL CMOS camera (ZWO, Suzhou, China). When applicable, for single-channel observations, single-band emission filters (Chroma ET 525/50, ET 605/70, or ET700/75) were used.
Figure S6 was acquired with a homebuilt spinning disk confocal microscope using a Nikon CFI S Plan Fluor 20× 0.45 NA (Nikon) air objective lens. This microscope will be the subject of a future publication.
Three-dimensional (3D) volumetric imaging of the 1-mm EC-cleared mouse kidney was acquired using a recently published open-top light sheet microscope (26). Briefly, illumination light is coupled into the system by a single-mode fiber from a four-channel (405, 488, 561, and 638 nm) laser package (Skyra, Cobolt Lasers). The light is scanned using a galvanometer (6210H, Cambridge Technology) to create a digital light sheet with an NA of ~0.06. Fluorescence is collected by a multi-immersion objective (no. 54-10-12, Special Optics, distributed by Applied Scientific Instrumentation). This objective provides <1-μm in-plane resolution for clearing media with a usable refractive index range of 1.33 to 1.56. The collected fluorescence is focused onto a 2048 × 2048 pixel sCMOS camera (ORCA-Flash4.0 V2, Hamamatsu) by a tube lens, TL2 (TTL165, Thorlabs, f = 165 mm) which provides a sampling of ~0.45 μm per pixel at n = 1.56 which satisfies the Nyquist criterion. This results in a horizontal field of view of ~0.9 mm over the 2048 pixels of the camera. The vertical field of view is reduced to 256 pixels (113 μm) to closely match the depth of focus of the illumination light sheet (~110 μm). The 256 pixels are oriented parallel to the rolling shutter readout direction of the camera, which provides an exposure time of 1.25 ms and a frame rate of 800 Hz. 3D imaging is achieved using a combination of stage scanning and lateral/vertical tiling with a motorized XY stage and Z actuators (FTP-2050-XYZ, Applied Scientific Instrumentation).
Note added in proof: Recently published work further supports the general utility of NHS labeling strategy we describe here (37, 38).
Supplementary Material
Acknowledgments
We would like to thank the Biology Imaging Facility at the University of Washington for imaging assistance, R. D. Palmiter and H. Y. Kueh (University of Washington, Seattle, WA) for providing mice for this study, M. Davidson for providing mEmerald-Golgi-7 plasmids (Addgene), T. Rapoport for providing pAc-GFPC1-Sec61β plasmids (Addgene, Watertown, MA), and C. E. Alpers and R. O. L. Wong (University of Washington, Seattle, WA) for access to tissue-sectioning instruments. Funding: This work was supported by the University of Washington, NIDDK Diabetic Complications Consortium grants DK076169 and DK115255 (to J.C.V.), NIH grant numbers R01 MH115767 (to J.C.V.), R01 CA175391 (to J.T.C.L.), RO1 DK097598 (to S.J.S.), UH2 DK107343 (to S.J.S.), R01 AG046231 (to S.J.S.), K99 CA240681 (to A.K.G.), and DoD PCRP PC170176 (to J.T.C.L.) and by an NSF Graduate Research Fellowship DGE-1256082 (to T.J.C.). NW BioTrust, a core service for patient consenting, and NWBioSpecimen, a core service for procurement and annotation of research biospecimens, are supported by National Cancer Institute grant P30 CA015704 [G. Gilliland, principal investigator (PI)], Institute of Translational Health Sciences grant UL1 TR000423 (M. Disis, PI), the University of Washington School of Medicine and Department of Pathology, and Fred Hutchinson Cancer Research Center. Author contributions: C.M., M.Y.L., J.-R.J., A.R.H., M.A.W., A.K.G., J.W.P., S.J.S., J.T.C.L., and J.C.V. designed the experiments. C.M., M.Y.L., J.-R.J., A.R.H., M.A.W., T.J.C., L.S., A.K.G., and J.W.P. performed the experiments and analysis. C.M., M.Y.L., and J.C.V. wrote the paper, and all authors commented on the manuscript. J.C.V. supervised the project. Competing interests: J.T.C.L and A.K.G are co-founders and hold equity in Lightspeed Microscopy Inc., of which J.T.C.L. is also a board member. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding author.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba4542/DC1
REFERENCES AND NOTES
- 1.Richardson D. S., Lichtman J. W., Clarifying tissue clearing. Cell 162, 246–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahl S. J., Hell S. W., Jakobs S., Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 18, 685–701 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Sigal Y. M., Zhou R., Zhuang X., Visualizing and discovering cellular structures with super-resolution microscopy. Science 361, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tainaka K., Kuno A., Kubota S. I., Murakami T., Ueda H. R., Chemical principles in tissue clearing and staining protocols for whole-body cell profiling. Annu. Rev. Cell Dev. Biol. 32, 713–741 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Schermelleh L., Ferrand A., Huser T., Eggeling C., Sauer M., Biehlmaier O., Drummen G. P. C., Super-resolution microscopy demystified. Nat. Cell Biol. 21, 72–84 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Schnell U., Dijk F., Sjollema K. A., Giepmans B. N. G., Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 9, 152–158 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Bradbury A., Plückthun A., Reproducibility: Standardize antibodies used in research. Nature 518, 27–29 (2015). [DOI] [PubMed] [Google Scholar]
- 8.K. S. Suvarna, C. Layton, J. D. Bancroft, Theory and practice of histological techniques (Elsevier Churchill Livingston, Edinburgh, ed. 7, 2013). [Google Scholar]
- 9.Elfer K. N., Sholl A. B., Wang M., Tulman D. B., Mandava S. H., Lee B. R., Brown J. Q., DRAQ5 and Eosin (‘D & E’) as an analog to hematoxylin and eosin for rapid fluorescence histology of fresh tissues. PLOS ONE. 11, e0165530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ornstein L., Mautner W., Davis B. J., Tamura R., New horizons in fluorescence microscopy. J. Mt. Sinai. Hosp. N Y 24, 1066–1078 (1957). [PubMed] [Google Scholar]
- 11.Weinblatt F. M., Shannon Jr. W. A., Seligman A. M., A new fluorescent method for the demonstration of macromolecular aldehydes. Histochemistry 41, 353–359 (1975). [DOI] [PubMed] [Google Scholar]
- 12.G. Hermanson, Bioconjugate Techniques (Academic Press, ed.2, 2008). [Google Scholar]
- 13.Lomant A. J., Fairbanks G., Chemical probes of extended biological structures: Synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J. Mol. Biol. 104, 243–261 (1976). [DOI] [PubMed] [Google Scholar]
- 14.Bragg P. D., Hou C., Subunit composition, function, and spatial arrangement in the Ca2+− and Mg2+−activated adenosine triphosphatases of Escherichia coli and Salmonella typhimurium. Arch. Biochem. Biophys. 167, 311–321 (1975). [DOI] [PubMed] [Google Scholar]
- 15.Baskin J. M., Dehnert K. W., Laughlin S. T., Amacher S. L., Bertozzi C. R., Visualizing enveloping layer glycans during zebrafish early embryogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 10360–10365 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saitoh Y., Terada N., Saitoh S., Ohno N., Fujii Y., Ohno S., Three-dimensional reconstruction of living mouse liver tissues using cryotechniques with confocal laser scanning microscopy. J. Electron Microsc. (Tokyo) 59, 513–525 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Hill D. E., Fetterer R. H., Urban Jr. J. F., Biotin as a probe of the surface of Ascaris suum developmental stages. Mol. Biochem. Parasitol. 41, 45–52 (1990). [DOI] [PubMed] [Google Scholar]
- 18.Swirski F. K., Berger C. R., Figueiredo J.-L., Mempel T. R., von Andrian U. H., Pittet M. J., Weissleder R., A near-infrared cell tracker reagent for multiscopic in vivo imaging and quantification of leukocyte immune responses. PLOS ONE. 2, e1075 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen F., Tillberg P. W., Boyden E. S., Expansion microscopy. Science 347, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ku T., Swaney J., Park J.-Y., Albanese A., Murray E., Cho J. H., Park Y.-G., Mangena V., Chen J., Chung K., Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat. Biotechnol. 34, 973–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chozinski T. J., Halpern A. R., Okawa H., Kim H.-J., Tremel G. J., Wong R. O. L., Vaughan J. C., Expansion microscopy with conventional antibodies and fluorescent proteins. Nat. Methods 13, 485–488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renier N., Wu Z., Simon D. J., Yang J., Ariel P., Tessier-Lavigne M., iDISCO: A simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Klingberg A., Hasenberg A., Ludwig-Portugall I., Medyukhina A., Männ L., Brenzel A., Engel D. R., Figge M. T., Kurts C., Gunzer M., Fully automated evaluation of total glomerular number and capillary tuft size in nephritic kidneys using lightsheet microscopy. J. Am. Soc. Nephrol. 28, 452–459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mujumdar R. B., Ernst L. A., Mujumdar S. R., Lewis C. J., Waggoner A. S., Cyanine dye labeling reagents: Sulfoindocyanine succinimidyl esters. Bioconjug. Chem. 4, 105–111 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Murray E., Cho J. H., Goodwin D., Ku T., Swaney J., Kim S.-Y., Choi H., Park Y.-G., Park J.-Y., Hubbert A., McCue M., Vassallo S., Bakh N., Frosch M. P., Wedeen V. J., Seung H. S., Chung K., Simple, scalable proteomic imaging for high-dimensional profiling of intact systems. Cell 163, 1500–1514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glaser A. K., Reder N. P., Chen Y., Yin C., Wei L., Kang S., Barner L. A., Xie W., McCarty E. F., Mao C., Halpern A. R., Stoltzfus C. R., Daniels J. S., Gerner M. Y., Nicovich P. R., Vaughan J. C., True L. D., Liu J. T. C., Multi-immersion open-top light-sheet microscope for high-throughput imaging of cleared tissues. Nat. Commun. 10, 2781 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giacomelli M. G., Husvogt L., Vardeh H., Faulkner-Jones B. E., Hornegger J., Connolly J. L., Fujimoto J. G., Virtual hematoxylin and eosin transillumination microscopy using epi-fluorescence imaging. PLOS ONE. 11, e0159337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fagerberg L., Stadler C., Skogs M., Hjelmare M., Jonasson K., Wiking M., Åbergh A., Uhlén M., Lundberg E., Mapping the subcellular protein distribution in three human cell lines. J. Proteome Res. 10, 3766–3777 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Itzhak D. N., Tyanova S., Cox J., Borner G. H., Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5, e16950 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pahari S., Sun L., Alexov E., PKAD: A database of experimentally measured pKa values of ionizable groups in proteins. Database. 2019, baz024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y., Xie W., Glaser A. K., Reder N. P., Mao C., Dintzis S. M., Vaughan J. C., Liu J. T. C., Rapid pathology of lumpectomy margins with open-top light-sheet (OTLS) microscopy. Biomed. Opt. Express 10, 1257–1272 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie W., Chen Y., Wang Y., Wei L., Yin C., Glaser A. K., Fauver M. E., Seibel E. J., Dintzis S. M., Vaughan J. C., Reder N. P., Liu J. T. C., Microscopy with ultraviolet surface excitation for wide-area pathology of breast surgical margins. J. Biomed. Opt. 24, 1, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.E. D. Karagiannis, J. S. Kang, T. W. Shin, A. Emenari, S. Asano, L. Lin, E. K. Costa, IMAXT Grand Challenge Consortium, A. H. Marblestone, N. Kasthuri, E. S. Boyden, Expansion Microscopy of Lipid Membranes (Preprint, Bioengineering, 2019). [Google Scholar]
- 34.Unnersjö-Jess D., Scott L., Sevilla S. Z., Patrakka J., Blom H., Hjalmar B., Confocal super-resolution imaging of the glomeruluar filtration barrier enables by tissue expansion. Kidney Int. 93, 1008–1013 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Beliveau B. J., Joyce E. F., Apostolopoulos N., Yilmaz F., Fonseka C. Y., McCole R. B., Chang Y., Li J. B., Senaratne T. N., Williams B. R., Rouillard J.-M., Wu C.-T., Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. U.S.A. 109, 21301–21306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehnertz B., Ueda Y., Derijck A. A. H. A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A. H. F. M., Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13, 1192–1200 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Yu C.-C. J., Barry N. C., Wassie A. T., Sinha A., Bhattacharya A., Asano S., Zhang C., Chen F., Hobert O., Goodman M. B., Haspel G., Boyden E. S., Expansion microscopy of C. elegans. eLife 9, e46249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O. M’Saad, J. Bewersdorf, Light microscopy of proteins in their ultrastructural context. Cell Biol. [preprint] (2020); https://www.biorxiv.org/content/10.1101/2020.03.13.989756v1. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba4542/DC1