

Supplemental Digital Content is available in the text.
Keywords: aneurysm, angiotensin II, elastin, interleukin-6, signal transduction
Objective:
Excessive prostaglandin E2 production is a hallmark of abdominal aortic aneurysm (AAA). Enhanced expression of prostaglandin E2 receptor EP4 (prostaglandin E receptor 4) in vascular smooth muscle cells (VSMCs) has been demonstrated in human AAAs. Although moderate expression of EP4 contributes to vascular homeostasis, the roles of excessive EP4 in vascular pathology remain uncertain. We aimed to investigate whether EP4 overexpression in VSMCs exacerbates AAAs.
Approach and Results:
We constructed mice with EP4 overexpressed selectively in VSMCs under an SM22α promoter (EP4-Tg). Most EP4-Tg mice died within 2 weeks of Ang II (angiotensin II) infusion due to AAA, while nontransgenic mice given Ang II displayed no overt phenotype. EP4-Tg developed much larger AAAs than nontransgenic mice after periaortic CaCl2 application. In contrast, EP4fl/+;SM22-Cre;ApoE−/− and EP4fl/+;SM22-Cre mice, which are EP4 heterozygous knockout in VSMCs, rarely exhibited AAA after Ang II or CaCl2 treatment, respectively. In Ang II–infused EP4-Tg aorta, Ly6Chi inflammatory monocyte/macrophage infiltration and MMP-9 (matrix metalloprotease-9) activation were enhanced. An unbiased analysis revealed that EP4 stimulation positively regulated the genes binding cytokine receptors in VSMCs, in which IL (interleukin)-6 was the most strongly upregulated. In VSMCs of EP4-Tg and human AAAs, EP4 stimulation caused marked IL-6 production via TAK1 (transforming growth factor-β–activated kinase 1), NF-κB (nuclear factor-kappa B), JNK (c-Jun N-terminal kinase), and p38. Inhibition of IL-6 prevented Ang II–induced AAA formation in EP4-Tg. In addition, EP4 stimulation decreased elastin/collagen cross-linking protein LOX (lysyl oxidase) in both human and mouse VSMCs.
Conclusions:
Dysregulated EP4 overexpression in VSMCs promotes inflammatory monocyte/macrophage infiltration and attenuates elastin/collagen fiber formation, leading to AAA exacerbation.
Highlights.
Excessive prostaglandin E2–EP4 (prostaglandin E receptor 4) signaling in vascular smooth muscle cells caused abdominal aortic aneurysm in mice.
EP4 stimulation increased IL (interleukin)-6 production via PKA (protein kinase A)–TAK1 (transforming growth factor-β–activated kinase 1)–NF-κB (nuclear factor-kappa B)/JNK (c-Jun N-terminal kinase)/p38 pathways in vascular smooth muscle cells isolated from mouse aorta and human abdominal aortic aneurysms.
EP4-induced IL-6 in vascular smooth muscle cells promoted Ly6Chi monocyte/macrophage infiltration and MMP-9 (matrix metalloprotease-9) activation in the mouse aorta.
EP4 stimulation decreased lysyl oxidase protein expression in vascular smooth muscle cells isolated from mouse aorta and human abdominal aortic aneurysms.
Chronic inflammation is a pathological feature of diverse cardiovascular diseases. Prostaglandin E2 (PGE2)—a ubiquitous metabolite of arachidonic acid—is produced inducibly in inflamed tissues by COX-2 (cyclooxygenase-2) activity. PGE2, by binding to its receptors, EP1, EP2, EP3, and EP4 (prostaglandin E receptor 1, 2, 3, and 4) plays pleiotropic roles depending on environmental cues and exerts both pro- and anti-inflammatory effects to maintain homeostasis.1 The COX-2–PGE2 axis has been recognized to play a primary role in inflammatory conditions, but the mechanisms underlying its complexity of action in tissues composed of multiple cell types are still elusive.
Dysregulation of PGE2-mediated inflammatory responses may cause progressive vascular diseases, such as abdominal aortic aneurysm (AAA). Patient-derived tissues show high COX-2 and PGE2 expression,2,3 and mouse studies have shown that COX-2–PGE2 signaling inhibition or genetic depletion attenuated the progression of these diseases.4,5 Among PGE2 receptors, EP4 is gaining attention as an inducible proinflammatory factor and thus as a therapeutic target for some types of chronic inflammation, for example, rheumatoid arthritis.6 It has been demonstrated that EP4 expression was upregulated in human AAA tissues and was correlated with degeneration of elastic fibers,7,8 which is a fundamental cause of AAA. In addition, a recent study has reported that EP4 expression in human AAA was correlated with smoking habit,9 which is a strong risk factor for AAA.
It has been demonstrated that AAA formation induced by Ang II (angiotensin II) or periaortic application of CaCl2 was attenuated in EP4 heterozygous knockout mice.7 Pharmacological inhibition of EP4 signaling also inhibited AAA mouse models.10,11 On the other hand, when EP4 was completely depleted in vascular smooth muscle cells (VSMCs), Ang II administration exacerbated AAA formation.12 Similarly, homozygous deletion of EP4 on bone marrow–derived cells enhanced Ang II–induced AAA.13 These conflicting reports suggest that an adequate level of PGE2-EP4 signaling is required for VSMC homeostasis and to enable the protective effects of immune cells, but excessive EP4 signaling induces proinflammatory and destructive reactions in the aortic wall.
Based on previous reports of EP4 upregulation in VSMCs in human aneurysmal tissues7,14 and the inhibitory role of EP4 on elastic fiber formation,15 we hypothesized that excessive EP4 signaling in VSMCs plays an important role in AAA progression. In the present study, we generated EP4-transgenic mice, in which EP4 is overexpressed selectively in VSMCs, and tested the hypothesis using multiple AAA animal models, including Ang II infusion and periaortic CaCl2 application, as well as human AAA tissues.
Methods
The authors declare that all supporting data are available within the article and its Data Supplement.
Genetically Modified Mice
SM22-Cre mice (Stock Tg [Tagln-cre] 1Her/J) and ApoE (apolipoprotein E) knockout (ApoE−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). EP4-transgenic constructs were generated by Unitech (Chiba, Japan). The transgenic construct is composed of a CAG promoter, 2 loxP sites flanking the coding region of EGFP (extended green fluorescent protein), and human PTGER4 (EP4) cDNA (NM_000958.2), as shown in Figure IA in the Data Supplement. The transgenic construct was injected into C57BL/6J mouse zygotes, and 2 mice positive for the transgene were obtained (founders). The 2 founders, named line A and line B, were mated with wild-type C57BL/6J mice to obtain the F1 generation. EGFP expression driven by the CAG promoter was used to determine the presence of the transgene, which was further confirmed by polymerase chain reaction. Each line of mice was kept as a heterogeneous transgenic line and bred to SM22-Cre mice to generate EP4-Tg mice in which EP4 was selectively overexpressed in VSMCs.
We also generated a mouse line of EP4 heterozygous knockout specifically in VSMCs (EP4fl/+;SM22-Cre) by crossing mice in which the exon 2 of the EP4 gene was flanked by 2 loxP sites (EP4fl/fl)16 with the SM22-Cre mice. In addition, we generated a line that globally lacks ApoE and has EP4 heterozygous knockout in VSMCs (EP4fl/+;SM22-Cre;ApoE−/−). After EP4fl/fl and SM22-Cre mice were each crossed with ApoE−/−, we obtained EP4fl/fl;ApoE−/− and SM22-Cre;ApoE−/−, respectively. To generate EP4fl/+;SM22-Cre;ApoE−/− and EP4fl/+;ApoE−/−, EP4fl/fl;ApoE−/− mice were crossed with SM22-Cre;ApoE−/−. These mice were maintained with a C57BL/6 N genetic background through >10 iterations of backcrossing. Male mice at 12 to 16 weeks of age were used for experiments. We used male mice in this study because the high incidence of human AAA and Ang II–induced experimental AAA in male has been shown.17
Ang II Infusion and Treatment
Mice were infused with Ang II (Sigma-Aldrich, St. Louis, MO) subcutaneously via an ALZET mini-osmotic pump (DURECT, Cupertino, CA) at 1.0 or 3.0 μg/kg per min. Mice were anesthetized using Avertin (0.25 g/kg, intraperitoneal) before pump implantation. The day of pump implantation was considered to be day 0, and infusion was sustained until day 28 at maximum. EP4 antagonist (ONO-AE3-208) was diluted in 0.5% methylcellulose and orally administered at 0.1 mg/kg twice per day. Vehicle administration was used as a control. Anti–IL-6 receptor antibody (MR16-1) was diluted in PBS and administered via intraperitoneal injection at 10 mg/kg once every 2 days. The same amount of rat IgG was administered as a control. AE3-208 or MR16-1 was administrated starting 1 day before Ang II infusion. At sample collection, mice were euthanized with pentobarbital (13 mg, intraperitoneal). ONO-AE3-208 and MR16-1 were kindly provided by Ono Pharmaceutical Company (Osaka, Japan) and Chugai Pharmaceutical Company (Tokyo, Japan), respectively.
Flow Cytometry
Single-cell suspensions were obtained from mouse abdominal aorta by enzyme dissociation.18 Abdominal aorta between the diaphragm and bifurcation was freed of connective fat tissues. Aorta with adventitia was cut into 1-mm square pieces and treated for 60 minutes with the following enzyme solution: 125 U/mL of collagenase type XI (Sigma), 60 U/mL of type 1-s hyaluronidase (Sigma), 60 U/mL of type II DNase I (Sigma), 450 U/mL of type I collagenase (Worthington, Lakewood, NJ), 100 μmol/L CaCl2, and 0.1% BSA fraction V (Wako Pure Chemical Industries, Osaka, Japan) in PBS. The resulting single-cell suspension was treated with anti-CD16/32 and labeled with fluorescent dye-conjugated antibodies: BV421-CD45.2 (clone; 104), BV510-CD11b (clone; M1/70), PE-Ly6C (clone; HK1.4), PE-Cy7-F4/80 (clone; BM8), and APC-Cy7-Ly6G (clone; 1A8). Dead cells were labeled with 7AAD (7-amino-actinomycin D). All antibodies used in this assay were purchased from BioLegend (San Diego, CA). Flow cytometric analysis and sorting were performed using a FACSAria (BD Biosciences), and data were analyzed using FlowJo software (Flowjo, LLC, Ashland, OR).
Statistics
Data are presented as the mean±SEM of >3 independent experiments. Statistical comparison between 2 groups was performed using the Mann-Whitney U test. Statistical comparison among multiple groups was performed using the Kruskal-Wallis test, followed by Fisher least significant difference post hoc test, the Mann-Whitney U test. A P of <0.05 was considered significant. Time to death in the in vivo AAA model was compared by the log-rank test.
Study Approval
All experiments using animals and human samples were approved by the Institutional Review Board at Yokohama City University (F-A-16-011 and B130307001) and conformed to the principles outlined in the Declaration of Helsinki. All animal experiments were performed according to the National Institutes of Health guidelines (Guide for the Care and Use of Laboratory Animals). Human AAA specimens were obtained only after written informed consent had been obtained.
Results
Generation of VSMC-Selective EP4-Overexpressed Mice (EP4-Tg)
To investigate the role of excessive EP4 signaling in VSMCs, we constructed EP4-Tg mice, in which human EP4 is selectively overexpressed in VSMCs under the SM22α promoter (Figure IA and IB in the Data Supplement). Expression of endogenous (mouse) and human EP4 mRNA in the aorta was 7.0-fold higher in EP4-Tg mice (line A) than in littermate nontransgenic mice (non-Tg; Figure IC in the Data Supplement). We confirmed Cre/loxP site-specific recombination by lower GFP intensity in VSMCs of EP4-Tg mice (Figure ID in the Data Supplement). EP4 agonist (ONO-AE1-329)–induced cyclic AMP production was 8.9× greater in aortic VSMCs of EP4-Tg than in non-Tg VSMCs (Figure IE in the Data Supplement), suggesting that overexpressed EP4 is functionally active. This EP4 overexpression did not affect VSMC viability (Figure IF in the Data Supplement). There were no differences in the expressions of other PGE2 receptor subtypes, namely, EP1, EP2, and EP3, between EP4-Tg and non-Tg (Figure IG through II in the Data Supplement). Body weights, cardiac functions, and serum lipid profiles were similar between EP4-Tg and non-Tg mice (Table I in the Data Supplement).
In a different line of EP4-Tg (line B), EP4 was moderately but significantly overexpressed in VSMCs (Figure IIA and IIB in the Data Supplement). EP4 agonist–induced cyclic AMP production was 2.2× greater in aortic VSMCs of EP4-Tg than in non-Tg VSMCs (Figure IIC in the Data Supplement). There was no difference in the expression levels of EP1, EP2, and EP3 mRNAs between line B EP4-Tg and non-Tg (Figure IID through IIF in the Data Supplement). There was no difference in body weights, cardiac functions, and serum lipid profiles between EP4-Tg and non-Tg mice (Table II in the Data Supplement).
Ang II Infusion Induced Dissecting AAA and Rupture in EP4-Tg
Ang II is known as a potent inducer of PGE2-producing COX-2 expression in the aorta.4 There was no difference in blood pressure at baseline between EP4-Tg and non-Tg mice, and blood pressure was similarly increased by Ang II infusion in both EP4-Tg and non-Tg mice (Figure IIIA in the Data Supplement). However, a difference in aortic diameter, with EP4-Tg mice having larger diameters, appeared around day 7 of Ang II infusion and became prominent at day 14 (Figure 1A through 1D). Elastic fiber destruction preceded aortic diameter expansion and was detectable at day 4 of Ang II infusion (Figure 1B and 1D). Most EP4-Tg mice died of AAA rupture within 14 days after the start of Ang II infusion (1.0 μg/kg per min), and all Ang II–treated EP4-Tg mice died within 28 days (Figure 1E). In contrast, all non-Tg mice survived for all 28 days (Figure 1E). Oral administration of EP4 antagonist attenuated Ang II–induced AAA formation in EP4-Tg and improved the survival rate of EP4-Tg mice infused with Ang II (Figure IV in the Data Supplement).
Figure 1.
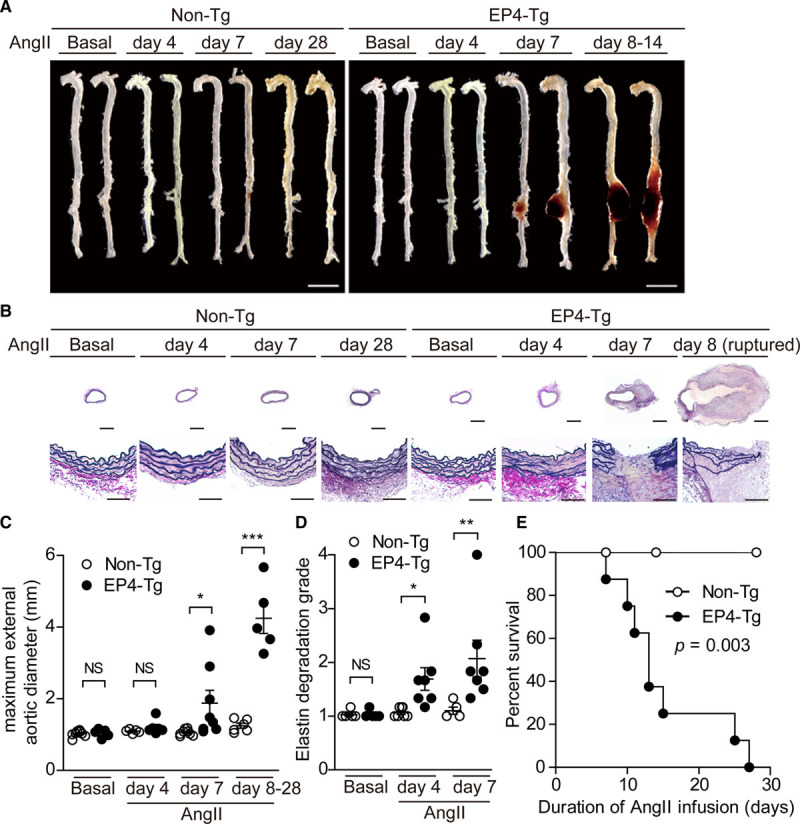
EP4 (prostaglandin E receptor 4) overexpression in vascular smooth muscle cells promoted dissecting abdominal aortic aneurysm and rupture after Ang II (angiotensin II) infusion. A, Representative images of aortas of nontransgenic (non-Tg) and EP4-Tg before and after Ang II infusion. Scale bars=5 mm. B, Elastica van Gieson–stained sections of the abdominal aortas of non-Tg and EP4-Tg mice after Ang II infusion. Lower stained sections (scale bars=20 μm) are magnifications of the upper stained sections (scale bars=500 μm). C and D, Maximum aortic diameter and elastin degradation grade of the aortas of non-Tg and EP4-Tg mice infused with Ang II. n=5 to 8. E, Survival rates of non-Tg and EP4-Tg mice after Ang II infusion. n=8 to 10. NS indicates not significant. *P<0.05, **P<0.01, ***P<0.001.
Like line A EP4-Tg, line B EP4-Tg mice exhibited increased aortic diameter and elastic fiber disruption after 28 days of Ang II infusion (3.0 μg/kg per min), but littermate line B non-Tg mice did not (Figure V in the Data Supplement). These data suggest that EP4 signaling in VSMCs contributes to elastic fiber disruption, leading to aortic rupture or dissection.
Ang II–Induced AAA Formation Was Inhibited in VSMC-Selective EP4 Heterozygous Knockout Mice
To further investigate the role of EP4 in VSMCs on AAA formation, we used VSMC-selective EP4 heterozygous knockout mice with ApoE deficiency (EP4fl/+;SM22-Cre;ApoE−/−), in which the expression level of EP4 mRNA was 47% lower compared with EP4fl/+;ApoE−/− mice (Table III in the Data Supplement). Body weights, cardiac functions, and serum lipid profiles were similar between these 2 lines (Table III in the Data Supplement). Aortic diameter and elastic fiber formation at baseline were not different between EP4fl/+;SM22-Cre;ApoE−/− and EP4fl/+;ApoE−/− mice (Figure 2).
Figure 2.
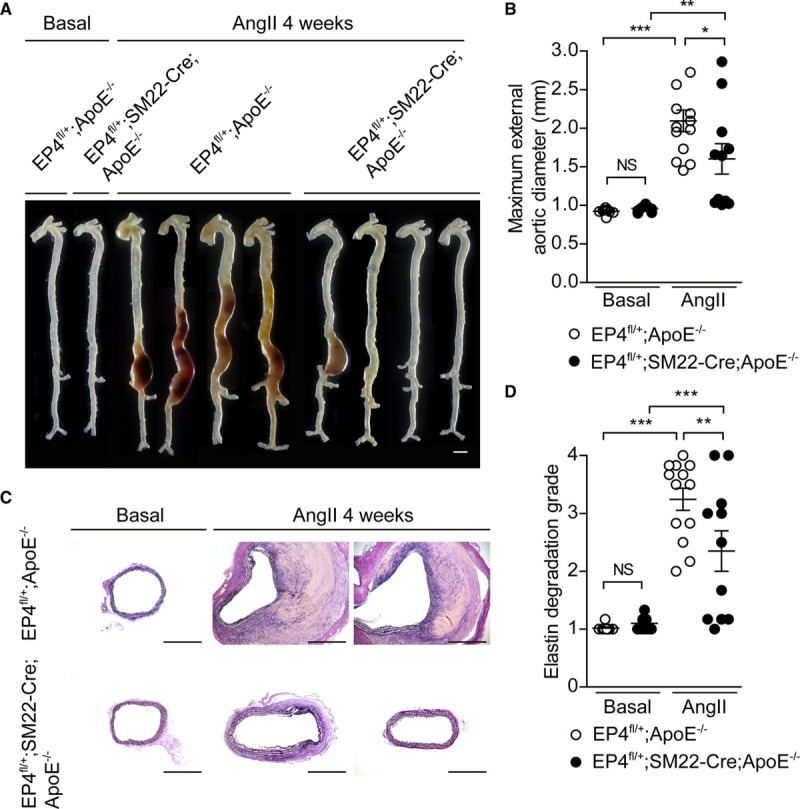
EP4 (prostaglandin E receptor 4) heterozygous knockout in vascular smooth muscle cells inhibited abdominal aortic aneurysm and rupture after Ang II (angiotensin II) infusion. A, Representative images of aorta for EP4fl/+;ApoE−/− and EP4fl/+;SM22-Cre;ApoE−/− mice before and after Ang II infusion. Scale bars=1 mm. B and D, Maximum aortic diameter and elastin degradation grade of the aorta in EP4fl/+;ApoE−/− and EP4fl/+;SM22-Cre;ApoE−/− mice infused with Ang II. n=9 to 13. C, Elastica van Gieson–stained sections of the abdominal aorta for EP4fl/+;ApoE−/− and EP4fl/+;SM22-Cre;ApoE−/− mice after Ang II infusion. Scale bars=500 μm. ApoE−/− indicates apolipoprotein E knockout; and NS, not significant. *P<0.05, **P<0.01, ***P<0.001.
Although Ang II–induced increase in blood pressure was higher in EP4fl/+;SM22-Cre;ApoE−/− than in EP4fl/+;ApoE−/− mice (Figure IIIB in the Data Supplement), Ang II infusion promoted AAAs in EP4fl/+;ApoE−/− mice, while reduced EP4 expression in VSMCs (EP4fl/+;SM22-Cre;ApoE−/−) inhibited Ang II–induced AAA formation (Figure 2A and 2B). In accordance with inhibited aortic expansion, elastic fiber degradation was attenuated in EP4fl/+;SM22-Cre;ApoE−/− (Figure 2C and 2D).
Periaortic CaCl2 Application–Induced AAA Formation Was Accelerated in EP4-Tg and Inhibited in VSMC-Selective EP4 Heterozygous Knockout Mice
We examined the role of EP4 in VSMCs using a CaCl2-induced mouse model of AAA as well.19 At 14 days after periaortic CaCl2 application, EP4-Tg mice exhibited enhanced dilatation of abdominal aorta and elastin disruption compared with non-Tg (Figure VIA through VID in the Data Supplement).
We then investigated the effect of reduced expression of EP4 using VSMC-selective EP4 heterozygous knockout mice without dyslipidemia background, that is, EP4fl/+;SM22-Cre, which have a 43% lower expression level of EP4 mRNA compared with EP4+/+;SM22-Cre mice (Table IV in the Data Supplement). Cardiac functions were similar between these two lines (Table IV in the Data Supplement). Aortic diameter and elastic fiber formation at baseline were not different between EP4fl/+;SM22-Cre and EP4+/+;SM22-Cre mice (Figure VIE through VIG in the Data Supplement). Periaortic CaCl2 application over 6 weeks induced aortic diameter expansion with elastic fiber disruption in EP4+/+;SM22-Cre mice, which were significantly attenuated in EP4fl/+;SM22-Cre (Figure VIE through VIH in the Data Supplement).
Immune Infiltrates Were Increased in EP4-Tg Aorta Treated With Ang II
To analyze the infiltration of immune cells into EP4-Tg mouse aorta under Ang II stimulation, we performed fluorescence-activated cell sorting analysis of the abdominal aorta (Figure 3A). Under baseline conditions in both EP4-Tg and non-Tg mice, CD45.2+/CD11b+ myeloid cells within the aorta were negative for both Ly6G (a marker of neutrophils) and Ly6C (a marker of inflammatory monocytes/macrophages) and positive for F4/80 (a marker of macrophages). Thus these myeloid cells were considered to be resident macrophages,20 and no obvious proinflammatory immune infiltrates were present in the aorta (Figure 3A) before Ang II infusion.
Figure 3.
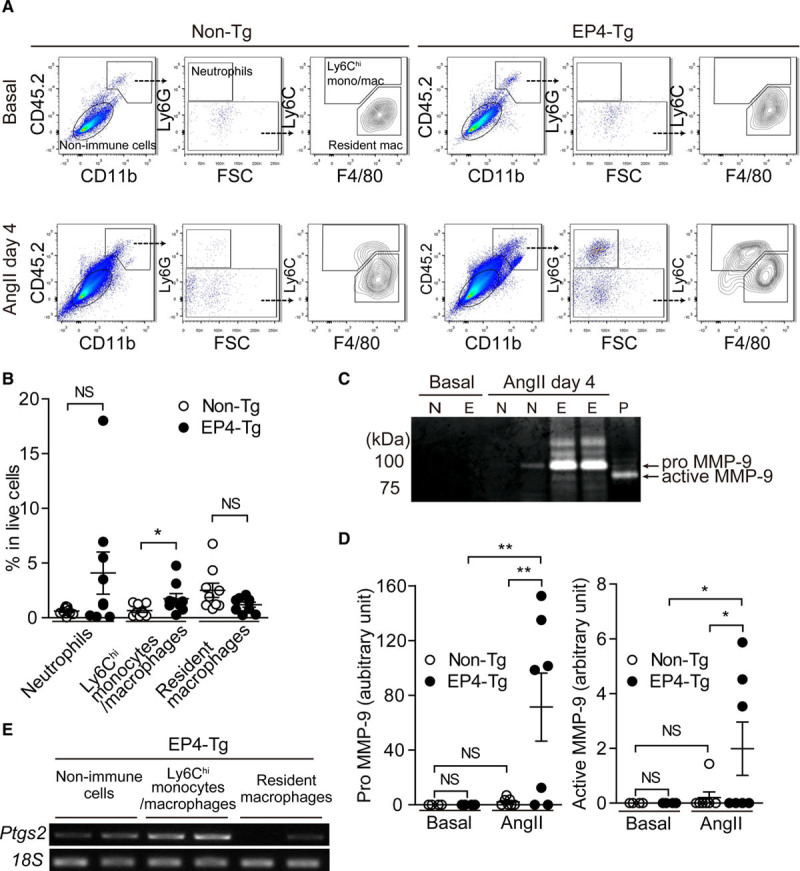
Inflammatory monocyte/macrophage invasion and MMP (matrix metalloprotease)-9 activity were enhanced in EP4 (prostaglandin E receptor 4)-Tg aorta at initial stage of abdominal aortic aneurysm formation. A, Representative fluorescence-activated cell sorting (FACS) plots of cell suspensions obtained from the abdominal aorta before and after Ang II (angiotensin II) infusion. B, Percentage of each cell fraction from FACS analysis. n=9. C and D, Representative image of gelatin zymography of cell suspensions obtained from the abdominal aorta for nontransgenic (non-Tg; N) and EP4-Tg (E) before and after Ang II infusion. Protein expression of pro-MMP-9 and activity of MMP from gelatin zymography. P: recombinant MMP-9 as a positive control. n=6 to 7. E, Semiquantitative polymerase chain reaction of cell fraction from FACS analysis. CD indicates cluster of differentiation; FSC, forward scatter; and NS, not significant. *P<0.05, **P<0.01.
Four days after Ang II infusion, we observed a marked increase in CD45.2+/CD11b+ myeloid cells in the EP4-Tg mouse aorta and a moderate increase in myeloid cells in the non-Tg mouse aorta. Among these cells, the number of Ly6Chi monocytes/macrophages was significantly higher in the EP4-Tg aorta than in the non-Tg aorta (Figure 3A and 3B). It is known that Ly6Chi cells represent the inflammatory features within the multiple roles of monocytes.21 The number of neutrophils tended to be higher in the EP4-Tg aorta than in the non-Tg aorta, but this difference was not significant (Figure 3B).
MMP-9 (matrix metalloproteinase-9) derived from immune infiltrates plays important roles in the progression of AAA.22 Aortic tissues of EP4-Tg mice infused with Ang II for 4 days exhibited marked increases in both pro- and active MMP-9 compared with baseline conditions, whereas active MMP-9 was not detected in the aorta of non-Tg mice infused with Ang II (Figure 3C and 3D).
To examine the cell types that produce PGE2, we examined COX-2 mRNA expression of nonimmune cells, infiltrated Ly6Chi monocytes/macrophages, and resident macrophages in EP4-Tg AAA induced by Ang II infusion for 4 days. Infiltrated Ly6Chi monocytes/macrophages highly expressed COX-2 mRNA and nonimmune cells, that is, VSMCs, moderately expressed COX-2, but resident macrophages had faint COX-2 mRNA expression (Figure 3E). These data suggest that EP4 signaling in VSMCs promoted infiltration of Ly6Chi monocytes/macrophages and increased PGE2 production and MMP-9 activity in the aorta.
EP4 Signaling Promoted IL-6 Production in VSMCs via TAK1–NF-κB/JNK/p38 Pathways
Based on our fluorescence-activated cell sorting analysis, we suspected that VSMC-derived factors may affect the observed increase in immune infiltration. We then performed a microarray analysis of EP4-Tg aortic VSMCs treated with PGE2 (accession number: GSE146758). Although immunofluorescence staining in EP4-Tg infused with Ang II and the previous study in human AAA8 indicated that infiltrated monocytes/macrophages produced IL (interleukin)-6 (Figure VIIA in the Data Supplement), a gene set enrichment analysis of microarray data revealed that PGE2-EP4 signaling upregulated the gene set related to gene ontology cytokine receptor binding in EP4-Tg VSMCs (Table V in the Data Supplement; Figure VIIB in the Data Supplement). In this gene set, the expression level of Il6 (IL-6)—the most dramatically upregulated gene—was 46.6-fold higher in EP4-Tg VSMCs treated with PGE2 than in untreated EP4-Tg VSMCs (Table VI in the Data Supplement).
In accordance with the microarray data, real-time reverse transcription polymerase chain reaction demonstrated that an EP4 agonist (ONO-AE1-329) or PGE2 stimulation significantly increased Il6 mRNA expression within 3 hours (Figure 4A) and that IL-6 protein secretion was increased 72-fold 6 hours after EP4 stimulation in EP4-Tg aortic VSMCs (Figure 4B).
Figure 4.
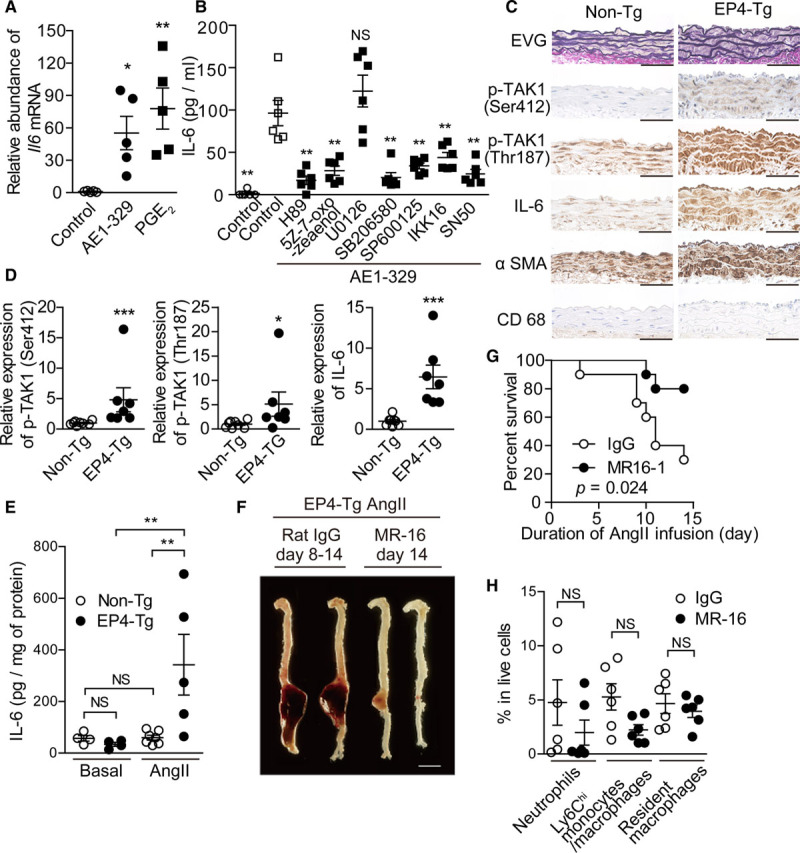
EP4 (prostaglandin E receptor 4)-PKA (protein kinase A)-TAK1 (transforming growth factor-β–activated kinase 1) and NF-κB (nuclear factor-kappa B) pathway increased IL (interleukin)-6 production and IL-6 inhibiton attenuated Ang II (angiotensin II)–induced abdominal aortic aneurysm. A, Expression of Il6 mRNA in EP4-Tg aortic vascular smooth muscle cells (VSMCs) stimulated with EP4 agonist and prostaglandin E2 (PGE2). n=5. B, IL-6 protein expression was measured by ELISA in EP4-Tg aortic VSMCs stimulated with the indicated drugs. n=6. C, Elastica van Gieson–stained (EVG) and immunohistochemically stained sections of the abdominal aortas of nontransgenic (non-Tg) and EP4-Tg mice after Ang II infusion. Scale bars=50 μm. D, Semiquantitative analysis of C. n=7 to 8. E, IL-6 protein expression was measured by ELISA in abdominal aorta from EP4-Tg before and after Ang II infusion. n=4 to 7. F, Representative images of aorta from Ang II–infused EP4-Tg treated with MR16-1. Scale bars=5 mm. G, Survival rates of Ang II–infused EP4-Tg treated with MR16-1 or control rat IgG. H, The number and proportion of cells in each gate from fluorescence-activated cell sorting analysis of abdominal aorta from Ang II–infused EP4-Tg at day 4. n=6. NS indicates not significant; and αSMA, alpha smooth muscle actin. *P<0.05, **P<0.01, ***P<0.001.
We then investigated the downstream signaling pathways of EP4 regarding IL-6 upregulation and found that EP4-induced IL-6 upregulation was attenuated by inhibition of PKA (protein kinase A). TAK1 (transforming growth factor-β–activated kinase 1) was reported to be a downstream molecule of PKA. PKA-induced phosphorylation of Ser-412 causes TAK1 activation and subsequent IL-6 upregulation.23,24 Thr-187 located at the TAK1 kinase domain is also required for TAK1 activation and cytokine production.24,25 Upon TAK1 activation, TAK1 phosphorylates IKK (IκB kinase), as well as JNK (c-Jun NH2-terminal kinase), and p38. Activated IKK further phosphorylates IκB proteins, leading to IκB protein degradation and thus activation of NF-κB (nuclear factor-kappa B)–dependent gene transcription.26,27 We used multiple inhibitors and found that TAK1, NF-κB, p38, and JNK, but not ERK1/2 (extracellular signal–regulated kinase 1/2), were involved in EP4-induced IL-6 production in VSMCs (Figure 4B).
EP4 agonist stimulation significantly increased the phosphorylation of TAK1 Ser-412 and Thr-187, JNK, and p38 and decreased IκBα in a time-dependent manner in EP4-Tg VSMCs (Figure VIIC through VIIH in the Data Supplement). In accordance with these in vitro data, immunohistochemical analysis revealed that aortic tissues isolated from EP4-Tg infused with Ang II exhibited increased TAK1 phosphorylation (Ser-412 and Thr-187) and elevated IL-6 proteins in VSMCs (Figure 4C and 4D). In addition, phosphorylated TAK1 (Ser-412 and Thr-187), JNK, p38, and IKKα/β proteins were present in AAA of EP4-Tg VSMCs (Figure VIIIA in the Data Supplement). These data suggested that EP4-PKA signaling increases IL-6 secretion via TAK1–NF-κB/JNK/p38 pathways.
Involvement of EP4-Induced IL-6 in Ang II–Induced AAA
Quantitative analysis using ELISA confirmed the significant upregulation of IL-6 protein levels in the abdominal aortas of EP4-Tg mice infused with Ang II but not in non-Tg mice (Figure 4E). We then examined the effect of IL-6 in AAA formation in EP4-Tg. Intraperitoneal injection of anti–IL-6R antibody (MR16-1) did not affect blood pressure, at least during the first 4 days of Ang II infusion (Figure IIIC in the Data Supplement), but inhibited AAA formation and significantly improved the survival rate of EP4-Tg infused with Ang II (Figure 4F and 4G). Fluorescence-activated cell sorting analysis revealed that the Ang II–induced increase in Ly6Chi monocytes/macrophages tended to be inhibited by MR16-1 (Figure 4H), although this difference did not reach significance (P=0.06; Figure 4H).
In addition to IL-6, PGE2 stimulation also increased Cxcl1 (C-X-C motif chemokine ligand 1) mRNA 9.7-fold in EP4-Tg aortic VSMCs (Table VI in the Data Supplement; Figure IXA in the Data Supplement). A ligand of CXCR2 (C-X-C motif chemokine receptor 2) CXCL1 plays a role in aortic inflammation and as a chemoattractant for neutrophils,28 and CXCL1 proteins were highly expressed in the abdominal aorta of EP4-Tg infused with Ang II compared with that of non-Tg (Figure IXB in the Data Supplement). However, Ang II–induced AAA-related death in EP4-Tg mice was not inhibited by CXCR2 antagonist administration (Figure IXC in the Data Supplement). These data suggested that EP4-induced IL-6 secretion plays roles in aortic inflammation and AAA formation to at least some degree.
EP4 Stimulation Decreased Protein Expression of LOX in VSMCs
LOX (lysyl oxidase) catalyzes the oxidative deamination of elastin and collagen monomers. By producing trimers, it contributes to the integrity and stabilization of vessel wall.29 Loss-of-function mutation in LOX causes impairment of elastin layer genesis and aortic aneurysm in mice and humans.30–32 Since it has been demonstrated that EP4 signaling reduced LOX protein expression via lysosomal degradation in fetal VSMCs,15 we examined whether EP4 signaling affected LOX expression in EP4-Tg VSMCs. Expression level of LOX mRNA was not changed by treatment with an EP4 agonist or PGE2 (Figure 5A), although an EP4 agonist decreased LOX protein expression in the supernatant of EP4-Tg VSMCs; the latter effect was attenuated by the lysosomal inhibitor bafilomycin (Figure 5B and 5C).
Figure 5.
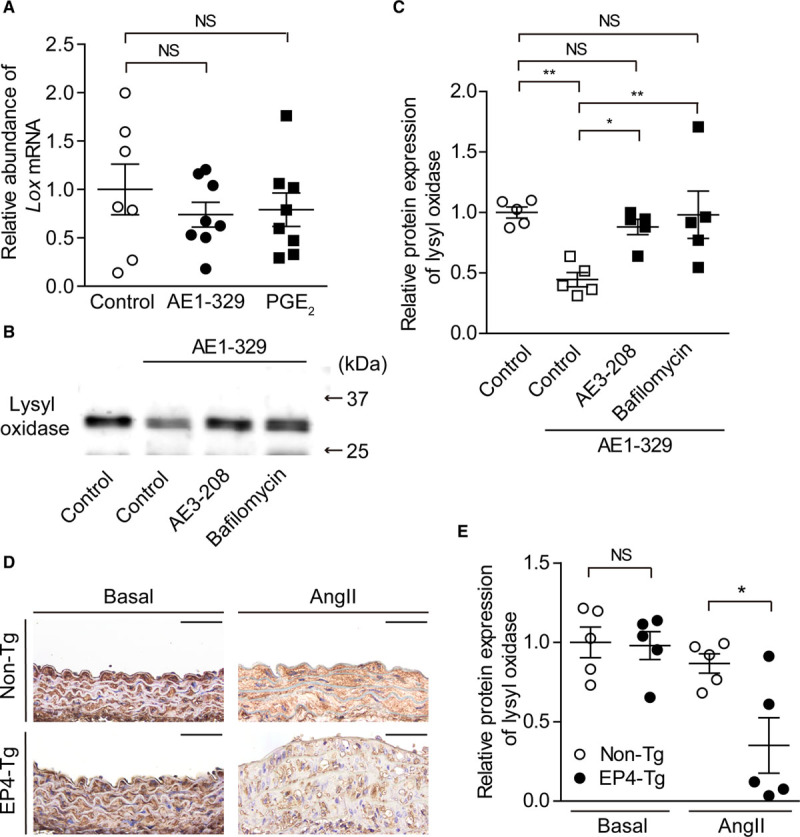
EP4 (prostaglandin E receptor 4) stimulation decreased lysyl oxidase expression via lysosomal degradation. A, Expression of Lox (lysyl oxidase) mRNA level in EP4-Tg vascular smooth muscle cells (VSMCs) stimulated with EP4 agonist or prostaglandin E2 (PGE2). n=7 to 8. B and C, Representative blot and relative protein expression of lysyl oxidase for supernatant of EP4-Tg VSMCs stimulated with EP4 agonist±EP4 antagonist or bafilomycin. n=5. D, Images of immunohistochemistry for lysyl oxidase of the abdominal aortas at baseline conditions and after Ang II (angiotensin II) administration (nontransgenic [non-Tg], day28; EP4-Tg, days 7–13). The aorta of EP4-Tg infused with Ang II was ruptured. Scale bars=50 µm. E, Semiquantitative analysis of D. n=5. NS indicates not significant. *P<0.05, **P<0.01.
Immunohistochemistry demonstrated that LOX protein expression was similar in both EP4-Tg and non-Tg VSMCs in vivo at basal conditions. After Ang II infusion, however, LOX protein expression was significantly lower in EP4-Tg VSMCs than in non-Tg, which was in accordance with the degree of elastic fiber formation (Figure 5D and 5E).
EP4-Induced IL-6 Production and LOX Downregulation in Human AAA
Based on these findings in mice, we investigated the roles of EP4 in human AAA specimens. In accordance with our in vitro mouse data, EP4 stimulation markedly increased IL-6 protein expression, and this upregulation was inhibited by inhibitors of PKA, TAK1, JNK, p38, or NF-κB in human VSMCs isolated from AAA tissues (Figure 6A). LOX proteins were significantly decreased by EP4 stimulation as well (Figure 6B and 6C). In human AAA tissues, immunohistochemical analysis revealed enhanced protein expressions of IL-6 and phosphorylated TAK1 (Ser-412 and Thr-187) in the tunica media (Figure 6D and 6E). In accordance with the results of EP4-Tg AAA, phosphorylated TAK1 (Ser-412 and Thr-187), JNK, p38, and IKKα/β proteins were present in human AAA VSMCs (Figure VIIIB in the Data Supplement).
Figure 6.
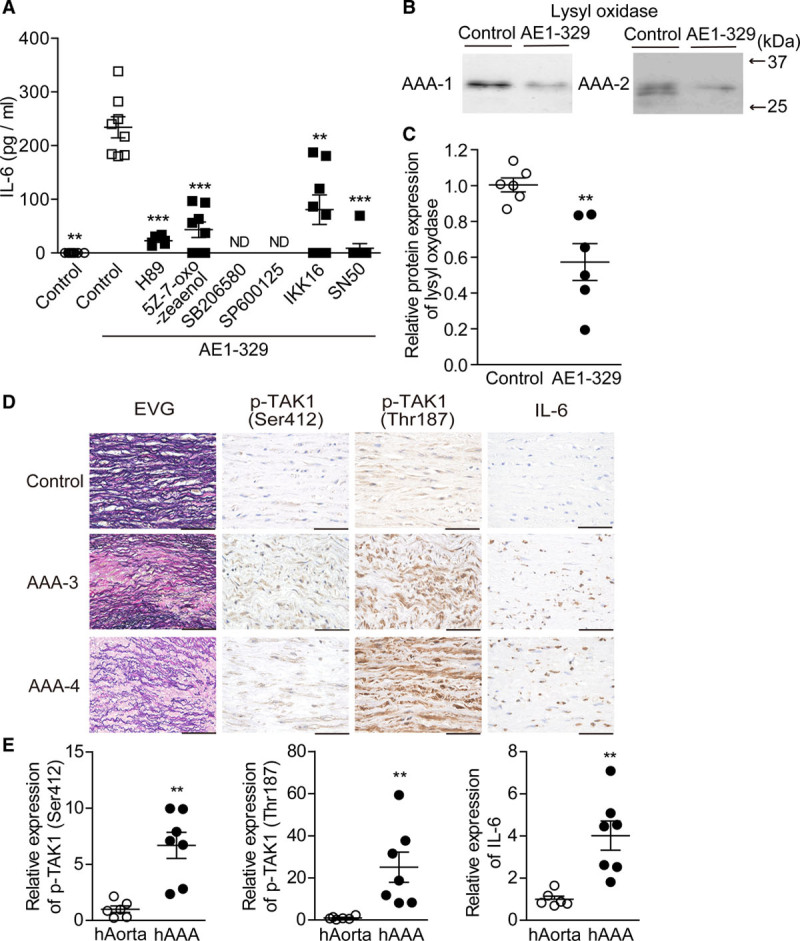
EP4 (prostaglandin E receptor 4) pathway induced IL (interleukin)-6 production and lysyl oxidase inhibition in human abdominal aortic aneurysm (AAA) vascular smooth muscle cells (VSMCs) and AAA tissues. A, IL-6 protein expression was measured by ELISA in human AAA (hAAA) VSMCs stimulated with the indicated drugs. n=5 to 8. B and C, Representative image and expression level of lysyl oxidase protein in supernatant of hAAA VSMCs stimulated with EP4 agonist. D, Elastica van Gieson–stained (EVG) and immunohistochemically stained sections of tissue from aorta and AAA. Scale bars=50 μm. E, Semiquantitative analysis of D in control aorta (hAorta) and hAAA. n=6 to 7. ND indicates not detected; and TAK1, transforming growth factor-β–activated kinase 1. *P<0.05, **P<0.01, ***P<0.001.
Discussion
It is well recognized that chronic inflammation underlies AAA pathology, and the involvement of immune cells in this condition is well documented. In addition, the concept that tissue constituent cells are actively involved in inflammatory responses has recently emerged. VSMCs are major vascular tissue constituent cells that contribute to vascular homeostasis and pathological remodeling. A study using VSMC-conditional gene modification demonstrated that VSMC phenotypic switch plays an important role in the progression of atherosclerosis.33 Selective depletion of genes such as LRP1 in VSMCs also causes aneurysmal remodeling.34,35 Although these studies clearly demonstrate the involvement of the VSMC phenotype per se in arterial diseases, the molecular mechanisms amplifying the inflammation through interactions of immune cells have not been thoroughly explained to date.
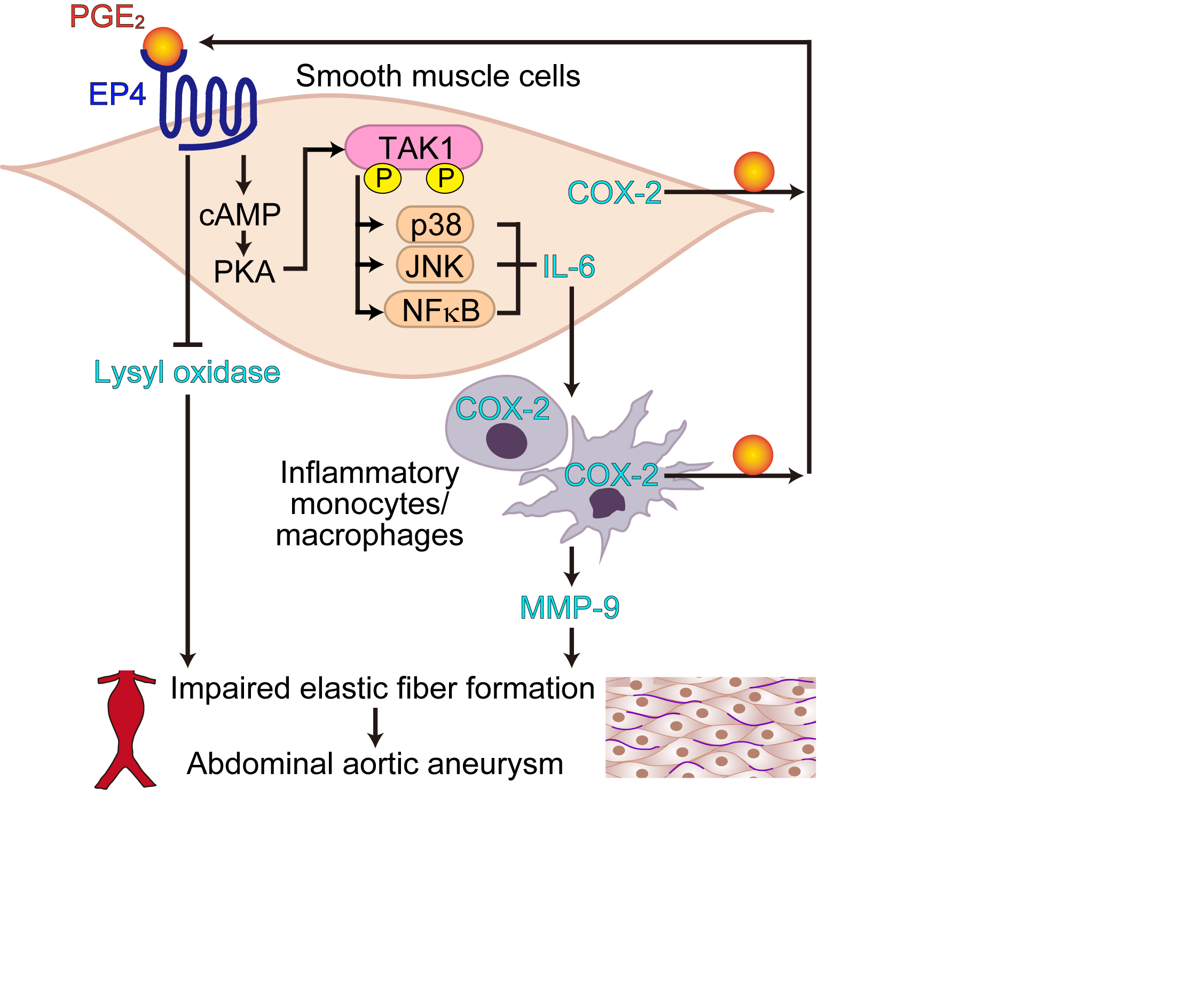
Here, we show that PGE2-EP4 signaling in VSMCs plays a critical role in the exacerbation of vascular inflammation during the interactions between immune cells. The present study suggests that excessive EP4 signaling in VSMCs promotes infiltration and activation of monocytes/macrophages in the aortic wall, in which, at least in part, VSMC-derived IL-6 is involved.
Ang II infusion into nonhyperlipidemic mice is known to promote inflammatory monocyte accumulation.36 In accordance with this finding, we observed that Ang II infusion slightly increased Ly6Chi monocytes/macrophages in aortic tissues of non-Tg mice (Figure 3A). It is known that periaortic CaCl2 application induces the infiltration of monocytes/macrophages into the media and adventitia.19 In the present study, EP4 overexpression in VSMCs per se did not enhance inflammation of the aorta and aneurysmal changes, and our preliminary data showed that EP4 agonist administration did not induce inflammatory responses and AAA/aortic dissection in EP4-Tg mice (data not shown). Hence, EP4-induced amplification of aortic inflammation was suggested to occur based on initial vascular injury followed by recruitment of inflammatory monocytes/macrophages. In addition to promoting immune infiltrates, Ang II is a potent inducer of COX-2 transcription,37 and it has been shown that Ang II infusion upregulates COX-2 expression even in the wild-type mouse aorta as early as 3 days after initiating infusion.4 An increase in PGE2 induced by Ang II could also be involved in the initiation of EP4-mediated enhancement of vascular inflammation. Our data demonstrated that infiltrated monocytes/macrophages and VSMCs expressed COX-2 mRNAs after Ang II infusion, suggesting that infiltration of monocytes/macrophages and Ang II administration further enhanced EP4 signaling in the vascular walls via PGE2 production.
It has been reported that Ang II causes aortic dissection in some types of mice without dyslipidemia.38,39 EP4-Tg infused with Ang II exhibited aortic aneurysm only in the abdominal aorta but not in the thoracic aorta. In EP4-Tg mice 4 days after Ang II infusion, a focal dissection in the suprarenal region accompanied with elastic lamella breaks and medial accumulation of macrophages were observed. Subsequently, luminal dilation and thrombus formation became evident as shown in Figure 1B. These processes are similar to that of Ang II–induced AAA in hyperlipidemic mice.40 Although the early stage of Ang II–induced AAA in EP4-Tg demonstrated a focal dissection in the tunica media, the typical aortic dissection induced by Ang II as reported in the literature38,39 was not observed in EP4-Tg mice.
In this study, we performed an unbiased analysis using PGE2-treated VSMCs of EP4-Tg. Gene set enrichment analysis using all gene sets belonging to the gene ontology molecular function revealed that 301 gene sets significantly positively correlated to EP4 signaling (false discovery rate, <0.25), in which 71 gene sets contain >100 genes in each gene set (Table V in the Data Supplement). We focused a gene set related to cytokine receptor binding. Among these genes, the most strongly upregulated gene was Il6. Our findings regarding anti–IL-6 receptor antibody suggest that EP4-mediated IL-6 upregulation is involved in enhancement of immune infiltrates and AAA formation in EP4-Tg mice. Because it has not been reported that IL-6 exhibits chemotactic activity, VSMC-derived IL-6 seems to activate Ang II–induced Ly6Chi monocytes/macrophages, resulting in further recruitment of inflammatory immune cells. In addition, infiltrated macrophages/monocytes were demonstrated to produce IL-6 via EP4 in human AAA8 and EP4-Tg mice, suggesting that immune infiltrates also contribute to IL-6–mediated inflammatory reaction.
It has been reported that Ang II–induced activated monocytes/macrophages promoted immune infiltration through macrophage-derived MCP-1 (monocyte chemoattractant protein-1).41 Chemoattractants such as MCP-1 may mediate the enhancement of immune infiltrates in the EP4-Tg aortic wall. Although our data indicate that IL-6 plays roles in aortic inflammation as an EP4-mediated proinflammatory cascade after 4 days of Ang II administration, we also observed that blockade of IL-6 receptor did not totally protect EP4-mediated AAA over longer periods (15 days). In addition to IL-6 production, multiple EP4-mediated VSMC-derived factors as shown in Table VI in the Data Supplement may also contribute to AAA formation. Further study is required to fully understand the cytokine/chemokine network involved in AAA exacerbation.
The present study demonstrated EP4 as an upstream receptor that stimulates the TAK1–NF-κB/JNK/p38 signaling pathways. Because of its multimodality, TAK1 is gaining attention as a key regulator of inflammatory diseases and cell survival.26 There is only one published investigation, however, on the role of TAK1 in VSMCs, namely, a study on neointima formation.42 To the best of our knowledge, less reports have shown the involvement of TAK1 in aneurysmal formation. TAK1 mediates various inflammatory stimuli,26 including the IL-1R (IL-1 receptor) and TLR (Toll-like receptor) signaling pathways, via phosphorylation of multiple sites such as Thr-187 in the TAK1 kinase domain in an autophosphorylated manner.25 In addition, recent reports have demonstrated that PKA catalytic subunit-α phosphorylates Ser-412 located at the C terminus outside of TAK1 kinase domain, which in turn leads to IL-6 production.23,24 Our data demonstrated that EP4-mediated PKA activation promoted the phosphorylation of both TAK1 Thr-187 and Ser-412. EP4-mediated cyclic AMP-PKA signaling may induce cross talk with these immune cell–derived IL-1R/TLR signaling pathways via modulating multiple TAK1 phosphorylation sites and amplifying inflammatory reactions such as IL-6 production in vivo.
LOX activity impairment causes aortic immature fiber formation in aortic elastin and collagen and aortic aneurysm in both in vivo mouse models and humans.30,32 The present study suggests that the EP4 signaling pathway is involved in the attenuation of cross-linking of elastin/collagen fibers during the development of AAA via a decrease in LOX protein expression. Collagen and elastic fibers are responsible for vascular stiffness and elasticity, which prevents the rupture of elastic arteries when exposed to high blood pressure.43 During AAA progression, LOX would play a protective role in maintaining vascular physical properties. The present study suggests the mechanism by which LOX protein expression is decreased in AAAs, although we currently do not know how this LOX downregulation affects the regeneration of extracellular matrix fiber formation in AAA in vivo. Our previous study demonstrated that EP4-PLC (phospholipase C) signaling promoted lysosomal degradation of LOX in the fetal artery.15 In accordance with those findings, the data in the present study suggest that LOX protein expression is regulated by degradation rather than transcriptional regulation. Further study is required to clarify the EP4 downstream signaling pathways that are involved in LOX protein expression in AAA.
AAA is a relatively common progressive lethal disease, but there is currently no proven medical therapy. In mouse models, the genetic depletion of COX-2 inhibits AAA formation.4 Accumulating evidence has demonstrated that inhibiting PGE2 production with nonsteroidal anti-inflammatory drugs or COX inhibitors attenuates AAA in mouse models.44–46 In humans, the progression of AAA was mitigated in patients taking COX-inhibiting nonsteroidal anti-inflammatory drugs.47 Based on this emerging evidence, inducible PGE2 may play a primary role in AAA progression, and inhibition of the PGE2-mediated signaling cascade appears to be a therapeutic strategy. However, nonsteroidal anti-inflammatory drugs and COX-2 inhibitors are not suitable for long-term administration because they have serious side effects. Hyperlipidemia and renin-angiotensin activation are known to be risk factors for AAA progression,48 but AAA expansion was not associated with hypercholesterolemia in clinical studies.49 The efficacy of renin-angiotensin system–inhibiting drugs also remains controversial.50
In our EP4-Tg models, we confirmed Cre/loxP site-specific recombination in VSMCs but could not confirm EP4 protein expression, which is the limitation of this study. These is no available anti-EP4 antibody that is selective enough for mouse and human EP4; however, line A and line B exhibited 7.0- and 3.7-fold increases, respectively, in EP4 mRNA expression, which are similar in magnitude to the overexpression range of EP4 in human AAA (a 3.2-fold increase).7 The degree of IL-6 production and elastic fiber destruction induced by Ang II or CaCl2 administration in the aorta was high in line A and mild in line B, suggesting that these responses are EP4 dependent. To inhibit excessive EP4 signaling, we administered a low dose of EP4 antagonist and found that AAA formation in EP4-Tg was attenuated. The experiments with EP4 heterozygous deletion mice in the present study also suggest that attenuation of excessive EP4 signaling inhibits AAA formation. In addition, previous reports have demonstrated that low-dose treatment with EP4 antagonist had inhibitory and therapeutic effects on AAA progression in mouse models.7,11
In contrast to the effects of excessive EP4 signaling, EP4 signaling at normal or baseline levels plays critical roles in vascular development and physiology.51,52 Global and VSMC-specific EP4-null mice died within 72 hours after birth due to patent ductus arteriosus.51–53 In accordance with these reports, we generated EP4fl/fl;SM22-Cre (EP4 null in VSMCs) using the EP4fl/fl mice,16 which was different from the abovementioned EP4fl/fl mice52 and found that all of these mice also died within 48 hours after birth due to the same phenotype (data not shown). Gruzdev et al52 have further demonstrated that EP4 plays certain roles in the development of contractility in the vessels. Another recent study has demonstrated that the VSMC-specific homologous deletion of EP4 promoted MCP-1 production and Ang II–induced AAA,12 although these VSMC-selective EP4-null mice did not display patent ductus arteriosus and lethal phenotypes. Anti-inflammatory roles of baseline expression level of EP4 has also been reported in endothelial cells.54 Hao et al54 demonstrated that total deletion of EP4 in endothelial cells exacerbated vascular neointima formation by attenuation of endothelial cell proliferation and promotion of endothelium-leukocyte adhesion and that an EP4 agonist suppressed neointima formation after vascular injury.
These studies suggest that a moderate or baseline expression level of EP4 is important for the maintenance of vascular homeostasis, while dysregulated EP4 signaling exacerbates vascular inflammation and attenuates arterial wall stiffness in AAAs. Inhibition of excessive EP4 signaling may be a therapeutic strategy for AAA.
Acknowledgments
We are grateful to Dr Matthew D. Breyer (Vanderbilt University) for providing EP4fl/fl mice. We also thank Yuka Sawada and Fumiko Kato for technical assistance.
Sources of Funding
This work was supported by JSPS (Japan Society for the Promotion of Science; U. Yokoyama: JP17K19403 and JP16H05358; Y. Ishikawa: JP19H036567 and JP18KT0073), AMED (Y. Ishikawa: JP19ek0210117), and Kitsuen Research Foundation (Y. Ishikawa: 1971000014) and partially supported by AMED (Japan Agency for Medical Research and Development; Y. Ishikawa: JP19ek0109240 and JP19lm0203087), JSPS (U. Yokoyama: JP20H03650), and Miyata Cardiac Research Promotion Foundation (U. Yokoyama).
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- AAA
- abdominal aortic aneurysm
- Ang II
- angiotensin II
- ApoE
- apolipoprotein E
- ApoE−/−
- apolipoprotein E knockout
- COX-2
- cyclooxygenase-2
- Cxcl1
- C-X-C motif chemokine ligand 1
- ERK1/2
- extracellular signal–regulated kinase 1/2
- IKK
- IκB kinase
- IL-1R
- IL-1 receptor
- IL-6
- interleukin-6
- JNK
- c-Jun N-terminal kinase
- LOX
- lysyl oxidase
- MCP-1
- monocyte chemoattractant protein-1
- MMP-9
- matrix metalloproteinase-9
- NF-κB
- nuclear factor-kappa B
- PGE2
- prostaglandin E2
- PKA
- protein kinase A
- PLC
- phospholipase C
- TAK1
- transforming growth factor-β–activated kinase 1
- TLR
- Toll-like receptor
- VSMC
- vascular smooth muscle cell
For Sources of Funding and Disclosures, see page 1572.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.314297.
References
- 1.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 2.Holmes DR, Wester W, Thompson RW, Reilly JM. Prostaglandin E2 synthesis and cyclooxygenase expression in abdominal aortic aneurysms. J Vasc Surg. 1997;25:810–815. doi: 10.1016/s0741-5214(97)70210-6. doi: 10.1016/s0741-5214(97)70210-6. [DOI] [PubMed] [Google Scholar]
- 3.Cipollone F, Prontera C, Pini B, Marini M, Fazia M, De Cesare D, Iezzi A, Ucchino S, Boccoli G, Saba V, et al. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E(2)-dependent plaque instability. Circulation. 2001;104:921–927. doi: 10.1161/hc3401.093152. doi: 10.1161/hc3401.093152. [DOI] [PubMed] [Google Scholar]
- 4.Gitlin JM, Trivedi DB, Langenbach R, Loftin CD. Genetic deficiency of cyclooxygenase-2 attenuates abdominal aortic aneurysm formation in mice. Cardiovasc Res. 2007;73:227–236. doi: 10.1016/j.cardiores.2006.10.015. doi: 10.1016/j.cardiores.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 5.Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Puré E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 6.Sakata D, Yao C, Narumiya S. Prostaglandin E2, an immunoactivator. J Pharmacol Sci. 2010;112:1–5. doi: 10.1254/jphs.09r03cp. doi: 10.1254/jphs.09r03cp. [DOI] [PubMed] [Google Scholar]
- 7.Yokoyama U, Ishiwata R, Jin MH, Kato Y, Suzuki O, Jin H, Ichikawa Y, Kumagaya S, Katayama Y, Fujita T, et al. Inhibition of EP4 signaling attenuates aortic aneurysm formation. PLoS One. 2012;7:e36724. doi: 10.1371/journal.pone.0036724. doi: 10.1371/journal.pone.0036724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayston T, Ramessur S, Reise J, Jones KG, Powell JT. Prostaglandin E2 receptors in abdominal aortic aneurysm and human aortic smooth muscle cells. J Vasc Surg. 2003;38:354–359. doi: 10.1016/s0741-5214(03)00339-2. doi: 10.1016/s0741-5214(03)00339-2. [DOI] [PubMed] [Google Scholar]
- 9.Dilmé JF, Solà-Villà D, Bellmunt S, Romero JM, Escudero JR, Camacho M, Vila L. Active smoking increases microsomal PGE2-synthase-1/PGE-receptor-4 axis in human abdominal aortic aneurysms. Mediators Inflamm. 2014;2014:316150. doi: 10.1155/2014/316150. doi: 10.1155/2014/316150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao RY, St Amand T, Li X, Yoon SH, Wang CP, Song H, Maruyama T, Brown PM, Zelt DT, Funk CD. Prostaglandin receptor EP4 in abdominal aortic aneurysms. Am J Pathol. 2012;181:313–321. doi: 10.1016/j.ajpath.2012.03.016. doi: 10.1016/j.ajpath.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Mamun A, Yokoyama U, Saito J, Ito S, Hiromi T, Umemura M, Fujita T, Yasuda S, Minami T, Goda M, et al. A selective antagonist of prostaglandin E receptor subtype 4 attenuates abdominal aortic aneurysm. Physiol Rep. 2018;6:e13878. doi: 10.14814/phy2.13878. doi: 10.14814/phy2.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu H, Du S, Fang B, Li C, Jia X, Zheng S, Wang S, Li Q, Su W, Wang N, et al. VSMC-specific EP4 deletion exacerbates angiotensin II-induced aortic dissection by increasing vascular inflammation and blood pressure. Proc Natl Acad Sci U S A. 2019;116:8457–8462. doi: 10.1073/pnas.1902119116. doi: 10.1073/pnas.1902119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang EH, Shvartz E, Shimizu K, Rocha VZ, Zheng C, Fukuda D, Shi GP, Sukhova G, Libby P. Deletion of EP4 on bone marrow-derived cells enhances inflammation and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2011;31:261–269. doi: 10.1161/ATVBAHA.110.216580. doi: 10.1161/ATVBAHA.110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akagi S, Nakamura K, Yokoyama U, Kasahara S, Sarashina T, Ejiri K, Ito H. Enhanced ep4 expression in a pulmonary artery aneurysm with dissection in a patient with pulmonary arterial hypertension. Circ Cardiovasc Imaging. 2017;10:e005839. doi: 10.1161/CIRCIMAGING.116.005839. doi: 10.1161/CIRCIMAGING.116.005839. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama U, Minamisawa S, Shioda A, Ishiwata R, Jin MH, Masuda M, Asou T, Sugimoto Y, Aoki H, Nakamura T, et al. Prostaglandin E2 inhibits elastogenesis in the ductus arteriosus via EP4 signaling. Circulation. 2014;129:487–496. doi: 10.1161/CIRCULATIONAHA.113.004726. doi: 10.1161/CIRCULATIONAHA.113.004726. [DOI] [PubMed] [Google Scholar]
- 16.Schneider A, Guan Y, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, Langenbach R, Breyer RM, Breyer MD. Generation of a conditional allele of the mouse prostaglandin EP4 receptor. Genesis. 2004;40:7–14. doi: 10.1002/gene.20048. doi: 10.1002/gene.20048. [DOI] [PubMed] [Google Scholar]
- 17.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from ATVB council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. doi: 10.1161/ATVBAHA.117.309524. doi: 10.1161/ATVBAHA.117.309524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gjurich BN, Taghavie-Moghadam PL, Galkina EV. Flow cytometric analysis of immune cells within murine aorta. Methods Mol Biol. 2015;1339:161–175. doi: 10.1007/978-1-4939-2929-0_11. doi: 10.1007/978-1-4939-2929-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Krishna S, Golledge J. The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis. 2013;226:29–39. doi: 10.1016/j.atherosclerosis.2012.09.010. doi: 10.1016/j.atherosclerosis.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M, Shikatani EA, El-Maklizi M, Williams JW, Robins L, et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol. 2016;17:159–168. doi: 10.1038/ni.3343. doi: 10.1038/ni.3343. [DOI] [PubMed] [Google Scholar]
- 21.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 22.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi Y, Mizoguchi T, Take I, Kurihara S, Udagawa N, Takahashi N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J Biol Chem. 2005;280:11395–11403. doi: 10.1074/jbc.M411189200. doi: 10.1074/jbc.M411189200. [DOI] [PubMed] [Google Scholar]
- 24.Ouyang C, Nie L, Gu M, Wu A, Han X, Wang X, Shao J, Xia Z. Transforming growth factor (TGF)-β-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit α (PKACα) and X-linked protein kinase (PRKX). J Biol Chem. 2014;289:24226–24237. doi: 10.1074/jbc.M114.559963. doi: 10.1074/jbc.M114.559963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–7368. doi: 10.1074/jbc.M407537200. doi: 10.1074/jbc.M407537200. [DOI] [PubMed] [Google Scholar]
- 26.Dai L, Aye Thu C, Liu XY, Xi J, Cheung PC. TAK1, more than just innate immunity. IUBMB Life. 2012;64:825–834. doi: 10.1002/iub.1078. doi: 10.1002/iub.1078. [DOI] [PubMed] [Google Scholar]
- 27.Ajibade AA, Wang HY, Wang RF. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013;34:307–316. doi: 10.1016/j.it.2013.03.007. doi: 10.1016/j.it.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 28.Anzai A, Shimoda M, Endo J, Kohno T, Katsumata Y, Matsuhashi T, Yamamoto T, Ito K, Yan X, Shirakawa K, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116:612–623. doi: 10.1161/CIRCRESAHA.116.304918. doi: 10.1161/CIRCRESAHA.116.304918. [DOI] [PubMed] [Google Scholar]
- 29.Csiszar K. Lysyl oxidases: a novel multifunctional amine oxidase family. Prog Nucleic Acid Res Mol Biol. 2001;70:1–32. doi: 10.1016/s0079-6603(01)70012-8. doi: 10.1016/s0079-6603(01)70012-8. [DOI] [PubMed] [Google Scholar]
- 30.Mäki JM, Räsänen J, Tikkanen H, Sormunen R, Mäkikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 31.Remus EW, O’Donnell RE, Jr, Rafferty K, Weiss D, Joseph G, Csiszar K, Fong SF, Taylor WR. The role of lysyl oxidase family members in the stabilization of abdominal aortic aneurysms. Am J Physiol Heart Circ Physiol. 2012;303:H1067–H1075. doi: 10.1152/ajpheart.00217.2012. doi: 10.1152/ajpheart.00217.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee VS, Halabi CM, Hoffman EP, Carmichael N, Leshchiner I, Lian CG, Bierhals AJ, Vuzman D, Mecham RP, Frank NY, et al. Brigham Genomic Medicine. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci U S A. 2016;113:8759–8764. doi: 10.1073/pnas.1601442113. doi: 10.1073/pnas.1601442113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 35.Davis FM, Rateri DL, Balakrishnan A, Howatt DA, Strickland DK, Muratoglu SC, Haggerty CM, Fornwalt BK, Cassis LA, Daugherty A. Smooth muscle cell deletion of low-density lipoprotein receptor-related protein 1 augments angiotensin II-induced superior mesenteric arterial and ascending aortic aneurysms. Arterioscler Thromb Vasc Biol. 2015;35:155–162. doi: 10.1161/ATVBAHA.114.304683. doi: 10.1161/ATVBAHA.114.304683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kossmann S, Hu H, Steven S, Schönfelder T, Fraccarollo D, Mikhed Y, Brähler M, Knorr M, Brandt M, Karbach SH, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550. doi: 10.1074/jbc.M114.604231. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derbyshire ZE, Halfter UM, Heimark RL, Sy TH, Vaillancourt RR. Angiotensin II stimulated transcription of cyclooxygenase II is regulated by a novel kinase cascade involving Pyk2, MEKK4 and annexin II. Mol Cell Biochem. 2005;271:77–90. doi: 10.1007/s11010-005-5386-9. doi: 10.1007/s11010-005-5386-9. [DOI] [PubMed] [Google Scholar]
- 38.LeMaire SA, Zhang L, Luo W, Ren P, Azares AR, Wang Y, Zhang C, Coselli JS, Shen YH. Effect of ciprofloxacin on susceptibility to aortic dissection and rupture in mice. JAMA Surg. 2018;153:e181804. doi: 10.1001/jamasurg.2018.1804. doi: 10.1001/jamasurg.2018.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fry JL, Shiraishi Y, Turcotte R, Yu X, Gao YZ, Akiki R, Bachschmid M, Zhang Y, Morgan KG, Cohen RA, et al. Vascular smooth muscle sirtuin-1 protects against aortic dissection during angiotensin II-induced hypertension. J Am Heart Assoc. 2015;4:e002384. doi: 10.1161/JAHA.115.002384. doi: 10.1161/JAHA.115.002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 41.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, 3rd, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song Z, Zhu X, Jin R, Wang C, Yan J, Zheng Q, Nanda A, Granger DN, Li G. Roles of the kinase TAK1 in CD40-mediated effects on vascular oxidative stress and neointima formation after vascular injury. PLoS One. 2014;9:e101671. doi: 10.1371/journal.pone.0101671. doi: 10.1371/journal.pone.0101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89:957–989. doi: 10.1152/physrev.00041.2008. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmes DR, Petrinec D, Wester W, Thompson RW, Reilly JM. Indomethacin prevents elastase-induced abdominal aortic aneurysms in the rat. J Surg Res. 1996;63:305–309. doi: 10.1006/jsre.1996.0265. doi: 10.1006/jsre.1996.0265. [DOI] [PubMed] [Google Scholar]
- 45.Miralles M, Wester W, Sicard GA, Thompson R, Reilly JM. Indomethacin inhibits expansion of experimental aortic aneurysms via inhibition of the cox2 isoform of cyclooxygenase. J Vasc Surg. 1999;29:884–892. doi: 10.1016/s0741-5214(99)70216-8. discussion 892. doi: 10.1016/s0741-5214(99)70216-8. [DOI] [PubMed] [Google Scholar]
- 46.King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006;26:1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 47.Walton LJ, Franklin IJ, Bayston T, Brown LC, Greenhalgh RM, Taylor GW, Powell JT. Inhibition of prostaglandin E2 synthesis in abdominal aortic aneurysms: implications for smooth muscle cell viability, inflammatory processes, and the expansion of abdominal aortic aneurysms. Circulation. 1999;100:48–54. doi: 10.1161/01.cir.100.1.48. doi: 10.1161/01.cir.100.1.48. [DOI] [PubMed] [Google Scholar]
- 48.Erbel R, Aboyans V, Boileau C, Bossone E, Di Bartolomeo R, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, et al. Grupa Robocza Europejskiego Towarzystwa Kardiologicznego (ESC) do spraw rozpoznawania i leczenia chorób aorty. [2014 ESC guidelines on the diagnosis and treatment of aortic diseases]. Kardiol Pol. 2014;72:1169–1252. doi: 10.5603/KP.2014.0225. doi: 10.5603/KP.2014.0225. [DOI] [PubMed] [Google Scholar]
- 49.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883–1889. doi: 10.1161/CIRCULATIONAHA.107.735274. doi: 10.1161/CIRCULATIONAHA.107.735274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malekzadeh S, Fraga-Silva RA, Trachet B, Montecucco F, Mach F, Stergiopulos N. Role of the renin-angiotensin system on abdominal aortic aneurysms. Eur J Clin Invest. 2013;43:1328–1338. doi: 10.1111/eci.12173. doi: 10.1111/eci.12173. [DOI] [PubMed] [Google Scholar]
- 51.Yokoyama U, Minamisawa S, Quan H, Ghatak S, Akaike T, Segi-Nishida E, Iwasaki S, Iwamoto M, Misra S, Tamura K, et al. Chronic activation of the prostaglandin receptor EP4 promotes hyaluronan-mediated neointimal formation in the ductus arteriosus. J Clin Invest. 2006;116:3026–3034. doi: 10.1172/JCI28639. doi: 10.1172/JCI28639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gruzdev A, Nguyen M, Kovarova M, Koller BH. PGE2 through the EP4 receptor controls smooth muscle gene expression patterns in the ductus arteriosus critical for remodeling at birth. Prostaglandins Other Lipid Mediat. 2012;97:109–119. doi: 10.1016/j.prostaglandins.2012.02.001. doi: 10.1016/j.prostaglandins.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, Malouf NN, Koller BH. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. doi: 10.1038/36342. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- 54.Hao H, Hu S, Wan Q, Xu C, Chen H, Zhu L, Xu Z, Meng J, Breyer RM, Li N, et al. Protective role of mPGES-1 (microsomal prostaglandin E synthase-1)-derived PGE2 (prostaglandin E2) and the endothelial EP4 (prostaglandin E receptor) in vascular responses to injury. Arterioscler Thromb Vasc Biol. 2018;38:1115–1124. doi: 10.1161/ATVBAHA.118.310713. doi: 10.1161/ATVBAHA.118.310713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.