

Abstract
Experimental evolution is a powerful tool to study genetic trajectories to antibiotic resistance under selection. A confounding factor is that outcomes may be heavily influenced by the choice of experimental parameters. For practical purposes (minimizing culture volumes), most experimental evolution studies with bacteria use transmission bottleneck sizes of 5 × 106 cfu. We currently have a poor understanding of how the choice of transmission bottleneck size affects the accumulation of deleterious versus high-fitness mutations when resistance requires multiple mutations, and how this relates outcome to clinical resistance. We addressed this using experimental evolution of resistance to ciprofloxacin in Escherichia coli. Populations were passaged with three different transmission bottlenecks, including single cell (to maximize genetic drift) and bottlenecks spanning the reciprocal of the frequency of drug target mutations (108 and 1010). The 1010 bottlenecks selected overwhelmingly mutations in drug target genes, and the resulting genotypes corresponded closely to those found in resistant clinical isolates. In contrast, both the 108 and single-cell bottlenecks selected mutations in three different gene classes: 1) drug targets, 2) efflux pump repressors, and 3) transcription-translation genes, including many mutations with low fitness. Accordingly, bottlenecks smaller than the average nucleotide substitution rate significantly altered the experimental outcome away from genotypes observed in resistant clinical isolates. These data could be applied in designing experimental evolution studies to increase their predictive power and to explore the interplay between different environmental conditions, where transmission bottlenecks might vary, and resulting evolutionary trajectories.
Keywords: experimental evolution, mutation supply, ciprofloxacin, Escherichia coli, bottleneck size, antibiotic resistance
Introduction
When facing a lethal selection, a population of organisms is dependent on the presence of preexisting individuals that unlike their ancestors have features that enable their survival and can rescue the population from extinction (Bell 2013). Such genetic variation within a population has relevance to the development of antibiotic resistance in bacteria where selection pressure can vary in magnitude across environments, and where high-level resistance may require the occurrence of multiple genetic alterations. The significance of population variation providing raw material for selection is illustrated by the evolution of resistance to ciprofloxacin (CIP) in Escherichia coli. Most resistant clinical resistant isolates carry several mutations altering different drug target genes, in particular, gyrA and parC (Hooper et al. 1987; Heisig 1996; Komp Lindgren et al. 2003; Huseby et al. 2017). With the exception of naturally transformable species such as Streptococcus pneumoniae (Ferrandiz et al. 2000; de la Campa et al. 2004) and one interesting example from E. coli where multiple chromosomal resistance mutations were acquired by horizontal genetic transfer (Tchesnokova et al. 2019), the evidence from clinical isolates supports a process involving the successive occurrence and selection of individual mutations, building up stepwise to confer a high-level resistance phenotype (Huseby et al. 2017). Such a process of change by the successive accumulation of mutations will at each step generate a fraction of individuals with a reduced susceptibility to the activity of the selecting drug. However, as beneficial mutations are typically rare events, the bacterial population should be of a size that can generate the required mutants at a frequency that reduces the risk of extinction. The population size necessary to enhance survival under strong selection will thus depend on the rate at which protective mutations appear in growing cells, which in turn depends on the mutation rate and the size of the mutational target. Mutational target size can be defined as the sum of all mutation rates that can generate a particular selected phenotype. If the phenotype requires a specific nucleotide substitution in one gene the target size for mutations will be very small, typically on the order of 10−10 per generation (Andersson et al. 2011; Huseby et al. 2017; Praski Alzrigat et al. 2017). In contrast, if the selection can be satisfied by a gene inactivation, or by many different substitution mutations, including in different genes, the target size for selectable mutations could be several orders of magnitude larger (Praski Alzrigat et al. 2017).
Experimental evolution allows for controlled testing of how the interplay between selection pressure, mutation rate, and population bottleneck size affect an evolutionary trajectory. A common experimental approach is to evolve a population of bacteria, by serial passage, for increased resistance to an antibiotic. In this process, a population of bacteria are grown to a known size (typically determined by culture volume), then a fraction of the population is transferred to a new culture vessel to seed the next growth cycle. The number of individuals in the fraction chosen for transfer creates a population transfer bottleneck, and the size of this bottleneck population will influence the probability that any favorable mutant that has arisen during the preceding growth cycle will be transferred to the next growth cycle. For example, we recently showed that one of the successive mutational steps in the evolution of high-level resistance to CIP in E. coli involves a choice between a high-frequency/low-fitness mutation and a low-frequency/high-fitness mutation (Huseby et al. 2017). At this important step, the supply of available mutations, largely determined by the transfer bottleneck, determines the future trajectory of evolution. Transfer bottlenecks are clinically relevant and occur frequently as part of the natural lifestyle of infectious bacteria. For example, transfer bottlenecks occur during the process of host-to-host transmission and can also occur within the host when bacteria encounter immune responses, physical barriers, or nutritional and spatial limitations (Abel et al. 2015). In general, the size of the transfer bottleneck, and the likelihood that the transferred population contains variant individuals with phenotypes that can survive the next selection, will impact the trajectory of evolution because the genotype(s) transferred create a founder effect on the evolving population (Abel et al. 2015).
The fluoroquinolone CIP is an important antibiotic that is used in therapy for a wide range of indications, including urinary tract infections caused by E. coli (Guay 2008; Chen et al. 2012; Castro et al. 2013). CIP targets Type II DNA topoisomerases (DNA gyrase and topoisomerase IV), essential bacterial enzymes that actively change the local density of chromosome supercoiling to facilitate efficient transcription, chromosome replication, and chromosome segregation (Drlica and Malik 2003; Hooper and Jacoby 2016). Fluoroquinolones exert their bactericidal activity by binding to the enzyme–DNA complex, after the enzyme has made a double-strand break in the DNA, and preventing the re-ligation reaction and generating a potentially lethal chromosome break (Chen et al. 1996; Malik et al. 2006). Wild-type E. coli is highly susceptible to CIP, with a minimal inhibitory concentration (MIC) typically in the range 0.008–0.03 mg/l (EUCAST 2019). The breakpoint defining clinical resistance to CIP in E. coli is 0.5 mg/l (EUCAST 2018), and multiple genetic alterations are required to raise the CIP MIC of a susceptible strain above this clinical breakpoint (Heisig and Tschorny 1994; Piddock 1999; Hooper 2001; Komp Lindgren et al. 2003; Marcusson et al. 2009). CIP resistance in clinical isolates of E. coli almost always involves mutations affecting both of the drug targets (DNA gyrase and topoisomerase IV), and it frequently also involves mutations that cause the upregulation of the AcrAB-TolC drug efflux system (Huseby et al. 2017). In addition, clinical isolates may carry one or more horizontally acquired resistance genes that contribute to resistance (Nazir et al. 2011), either by protecting the drug target (Qnr proteins), modifying the drug (aac(6′)-1b-cr, crpP) or increasing drug efflux (qepA, oqxAB) (Jacoby et al. 2003, 2014; Perichon et al. 2007; Yamane et al. 2007; Chavez-Jacobo et al. 2019).
Although the principle that transfer bottlenecks can impact evolution is well established, we currently have a poor understanding of how this might impact experimental studies of antibiotic resistance evolution where the aim is often to increase understanding of resistance evolution in clinical isolates. To address this, we experimentally evolved CIP resistance in E. coli using different transfer bottleneck sizes and examined how changing this parameter influenced the types of mutations that were accumulated. Both individual isolates and populations of bacteria were analyzed by whole-genome sequencing at different stages throughout the evolution. Our findings could help to improve the design of experimental studies into the evolutionary trajectories occurring in clinical and natural environments.
Results and Discussion
Experimental Evolution with Different Bottlenecks
Independent lineages (Materials and Methods) of antibiotic-susceptible E. coli K-12 strain MG1655 were evolved by cycles of growth with passaging at successively higher concentrations of CIP up to 32 mg/l as described in the Materials and Methods section. Experiments were made using three different population transfer bottlenecks referred to as small, medium, and large. The small bottleneck experiment was initiated by streaking 20 single colonies of MG1655 on agar without antibiotic and incubating at 37 °C for 24 h. From each streak, one colony was picked and streaked to obtain single colonies on agar containing CIP at the first concentration (0.016 mg/l). Thereafter, one single colony from each lineage was picked each day (choosing the largest colony if there was variation in size) and streaked onto the next, higher CIP concentration (0.023, 0.032, 0.048 mg/l, etc). Because each colony originates from an individual bacterium, this method of transfer effectively creates a single cell bottleneck at each successive step. One lineage went extinct after the 9th step, at 0.256 mg/l, whereas the remaining 19 lineages survived until the 17th step (4 mg/l). Thereafter, lineages successively went extinct until by the end-point 23rd step (32 mg/l) there was only one surviving lineage (supplementary fig. S1, Supplementary Material online). In the later transfer steps (from 4 mg/l), the incubation period was extended to 48–72 h because many colonies were slow growing. Our expectation was that the small bottleneck size would maximize the effects of genetic drift and permit the accumulation of a wide range of mutations that reduced susceptibility to CIP, even mutations that were somewhat deleterious to growth fitness.
In the medium bottleneck experiment, ten independent liquid culture lineages were initiated from single colonies. The population size at the end of each growth cycle was 6 × 109 cfu, and ∼3 × 108 cfu were transferred to initiate each successive growth cycle. In the large bottleneck experiment, five independent liquid culture lineages initiated from single colonies. In these lineages, ∼3 × 1010 cfu were transferred at each successive step, and the population size at the end of each growth cycle was 6 × 1011 cfu. Details of the liquid culture evolutions are given in the Materials and Methods section. The sizes of the medium and large transfer bottlenecks were chosen because they straddle the mutation rate (∼10−9 per generation) previously measured as the sum total of target mutations that reduce susceptibility to CIP (Huseby et al. 2017). The expectation was that larger transfer bottlenecks would increase the probability of rare high-fitness mutants being transferred at each step and having the opportunity to increase in relative frequency. All of the independent lineages of the medium and large bottleneck evolution experiments were successfully evolved up to a selective concentration of 32 mg/l CIP without any extinctions. Samples and clones (Materials and Methods) from each lineage and selection step were stored for future analysis.
Whole-Genome Sequence Analysis of Single Cell Bottleneck Evolution
From the single cell bottleneck experiment, ten lineages that evolved successfully to grow on agar at 6 mg/l CIP or greater were subjected to whole-genome sequence analysis. From each of the ten lineages, we sequenced the genomes of strains isolated at six different concentrations, from 0.048 mg/l (step 4) up to the highest concentration on which they grew (supplementary table S1, Supplementary Material online). There was a successive accumulation of mutations in each lineage as the strains were selected for growth on ever higher drug concentrations. The data revealed a number of unusual features, in particular, a very small number of target mutations relative to what is normally observed in resistant clinical isolates. All ten lineages acquired a single amino acid substitution mutation in the QRDR region of gyrA, but only one lineage acquired a mutation in parC. In addition, in most lineages, the mutations acquired in gyrA were, with the exception of lineages 9 and 12 (D87G, S83L, respectively), not the gyrA mutations that are commonly found in resistant clinical isolates (Huseby et al. 2017). All ten lineages acquired mutations in or immediately upstream of acrR (an important negative regulator of the AcrAB-TolC efflux pump) but in 6/10 cases the initial mutational event in these lineages was an amplification of the acrRAB region which later collapsed after acquisition of a point mutation in acrR. A large number of mutations were also acquired in genes that play central roles in the transcription and/or translation machinery. For example, all ten lineages acquired at least one mutation affecting tRNA genes and/or tRNA synthetase genes (supplementary table S1, Supplementary Material online). Mutations in various tRNA synthetases were recently shown to reduce susceptibility to CIP by causing an upregulation of multiple drug efflux systems (Garoff et al. 2018). Of course, the number of possible low-level resistance mutations is much higher than the number of target mutations. We conclude from these data that that the small transfer bottleneck facilitated the acquisition of mutations in a wide variety of different genes that cumulatively acted to reduce susceptibility to CIP.
Deep Sequence Analysis of Medium and Large Bottleneck Populations
To determine the identity of mutations and the order in which they were acquired during the evolution experiments with medium and large transfer bottlenecks, we performed deep sequencing on samples taken from these evolving populations after different selection steps. From the medium bottleneck experiment, we sequenced the populations from 9/10 lineages after every second selection step, from CIP 0.024 mg/l up to the end-point concentration 32 mg/l (lineage 2 was discarded because of suspected cross-contamination). From the large bottleneck experiment, we sequenced the population of 5/5 lineages after six different selection steps, from CIP 0.048 mg/l up to the end-point concentration 32 mg/l. The deep sequencing approach (∼100-fold coverage for each nucleotide) allowed the observation of a multiplicity of alleles present within each culture at each time point. A complete list of mutations meeting threshold criteria (Materials and Methods) in each lineage and drug concentration is shown in supplementary tables S2 and S3, Supplementary Material online.
In each lineage of both medium and large bottleneck populations, a common feature was that the first mutational event detected was the presence of alleles of gyrA with one of these alleles usually reaching fixation by cycle 8 (0.19 mg/l CIP). However, the alleles of gyrA differed significantly between the medium and large bottleneck populations. In the former, the gyrA D87G allele went to fixation in 8/9 lineages with gyrA D87Y in the remaining lineage (supplementary table S2, Supplementary Material online). In contrast, in the large bottleneck populations, the S83L allele of gyrA went to fixation in all 5/5 lineages as frequently observed in resistant clinical isolates (Huseby et al. 2017).
In medium bottleneck lineages, the second major event detected was the appearance of many different mutations affecting one or more of the efflux regulator genes. These typically appeared in cycles 8–10 (0.19–0.38 mg/l CIP), and reached fixation (or a high frequency) by cycles 14–16 (1.5–3 mg/l CIP). In 7/9 lineages of the same populations, the third wave of mutations selected, reaching high frequencies by cycle 14 or later, were mutations affecting genes involved in transcription and translation (e.g., rpoB, thrV, and mnmA). In several lineages, mutations in genes affecting transcription and/or translation reached fixation (rpoB lineage 1, 3, and 9; rpoN lineage 4; mnmA lineage 7; and thrV lineage 5 and 8). A fourth wave of mutations selected in the medium bottleneck populations affected drug target genes (gyrA, gyrB, parC, and parE) frequently reaching fixation by cycle 20 (12 mg/l CIP). There was a remarkable consistency in the dynamics of evolutionary succession between the independently evolved medium bottleneck lineages (fig. 1) with the selection of mutations in genes affecting transcription and/or translation being an integral part of the evolving mutant genotype (Materials and Methods). The effect on MIC of new mutations that decrease susceptibility by a small factor can be dramatic in a lineage that already carries a mutation conferring high-level resistance (Baquero and Martinez 2017).
Fig. 1.

Muller plots of medium bottleneck evolution. The Muller plots depict the evolutionary trajectory of medium bottleneck lineages 4 (A) and 7 (B). Frequencies and linkage of mutations were determined from population sequencing of individual selection steps (supplementary table S2, Supplementary Material online) and final clones (supplementary table S4, Supplementary Material online). The colors indicate the class into which mutations were sorted (target, efflux, or transcription/translation). A hatched pattern overlaying a color indicates the inferred presence (from sequencing data, linkage, or MIC) of a class of mutations in the population, but without any individual mutation rising above the 5% threshold for identification. Solid black lines indicate lineages which survived and were identified in the final culture, whereas dotted white lines indicate lineages that went extinct during the selection. For the lineages that became extinct during selection, where the linkage of mutations for constructing the figure was inferred, the data in supplementary tables S2 and S4, Supplementary Material online, should be considered authoritative.
In large bottleneck populations, although alleles of gyrA appeared and went to fixation first, the second mutational events reaching high frequencies and often going to fixation were typically mutations in parC. This was followed by a third mutational event also reaching high frequencies involving the selection of a second mutation in gyrA (supplementary table S3, Supplementary Material online). Thus, the populations evolved through the large bottleneck acquired a set of mutations in succession that closely mimicked the succession of mutations found in resistant clinical isolates, affecting gyrA residues S83, D87, and parC residue S80. This genotype profile is associated with selection for mutations that reduce CIP susceptibility with minimal negative effects on relative growth fitness (Huseby et al. 2017). Two examples of large bottleneck evolution are shown in figure 2. It is interesting to note that the ancestors of the mutants that dominated individual large bottleneck populations at the end-point of the experiment (32 mg/l CIP) were frequently present as relatively small subpopulations at earlier stages in the evolution (fig. 2). This suggests that part of the importance of large population bottlenecks might be that it enhances the probability of survival of genetic variants that can reveal favorable phenotypes subsequent to their initial occurrence. Lineage 5 acquired a mutation in mutL (encoding an enzyme in the DNA replication mismatch repair pathway) and as a result had a greatly increased mutation rate and acquired mutations in a very wide variety of genes unrelated to the selection (supplementary table S3, Supplementary Material online) making it atypical. With the exception of lineage 5, none of the lineages evolved with a large population bottleneck acquired any mutations affecting genes involved in transcription or translation, probably because such mutations are associated with relatively high-fitness costs (Pietsch et al. 2017; Garoff et al. 2018).
Fig. 2.

Muller plots of large bottleneck evolution. The Muller plots depict the evolutionary trajectory of large bottleneck lineages 3 (A) and 4 (B). Frequencies and linkage of mutations were determined from population sequencing of individual selection steps (supplementary table S3, Supplementary Material online) and end-point clones (supplementary table S4, Supplementary Material online). The colors indicates the class into which mutations were sorted. Solid black lines indicate lineages which survived and were identified in the final culture, whereas dotted white lines indicate lineages that went extinct during the selection. In some cases, for the lineages that became extinct during selection, the linkage of mutations for constructing the figure was inferred from the data in supplementary tables S3 and S4, Supplementary Material online, which should be considered authoritative.
A limitation of the population sequencing approach is that it does not provide definitive information on genetic linkage within strains. To address this gap, we choose individual clones from the end-point culture of each medium and large bottleneck evolution experiment and performed whole-genome sequence on them.
End-Point Clone Analysis and Genetic Linkage
Individual clones from the end-point cultures of nine medium bottleneck and five large bottleneck experiments were whole-genome sequenced. The data (supplementary table S4, Supplementary Material online) show that the large bottleneck clones each carry four mutations associated with reducing susceptibility to CIP (we exclude the lineage 5 clone from the analysis because it is a mutL mutator carrying >50 mutations). These four clones each carry 3–4 target mutations in gyrA, parC, and/or parE and 0–1 efflux mutations in marR or emrA (fig. 3). There are two minor points to explain about these data. One is that the additional 1–2 mutations that appear in these clones, in arcA, trkH, and treB (supplementary table S4, Supplementary Material online), are associated with media adaptation and have nothing directly to do with susceptibility to CIP (Knoppel et al. 2018). The second point is that three of the four clones carry a mutation predicted to upregulate CIP efflux (in marR or emrA), whereas efflux mutations did not appear as a major feature in the deep sequencing of the populations from which they originated (fig. 2). An explanation for this apparent discrepancy between population and clone sequencing is that there is a great diversity of efflux-related mutations present in the large bottleneck populations, but no individual mutant allele exceeds the 5% threshold for inclusion in the population allele lists. This hypothesis was tested in large bottleneck population lineage 1 by sequencing ten individual clones from the final selection step (32 mg/l). The sequencing confirmed the presence of two genotypes in the population with 2/10 clones having four target mutations, whereas 8/10 clones had three target mutations and one mutation affecting an efflux regulatory gene marR (supplementary table S5, Supplementary Material online). In each of the eight clones, the efflux regulator mutation was a different allele indicative of a great diversity of marR alleles. To further probe this phenomenon, 96 clones were pooled from the same population, and amplicons were sequenced from gyrA, parC, marR, acrR, and soxR (supplementary table S6, Supplementary Material online). The amplicon sequencing showed that 100% of the population carried two mutations in gyrA and at least one in parC. Approximately 35% of the population (identified by the distinguishing mutations gyrA D87Y and parC S80I) corresponded to the strains carrying four target mutations (supplementary table S5, Supplementary Material online). Approximately 47% of the population contained identifiable efflux regulator gene mutations, with 43 different marR mutations and a single acrR mutation being identified (supplementary table S6, Supplementary Material online). No individual efflux regulator mutation was present at a frequency >2.3%, indicating that each mutant allele likely represented 1 or 2 clones in the total of 96 pooled clones, further reinforcing the great diversity of efflux regulator alleles present in the population. The amplicon sequencing results likely represent an underestimate of the total diversity of efflux regulatory mutations present.
Fig. 3.
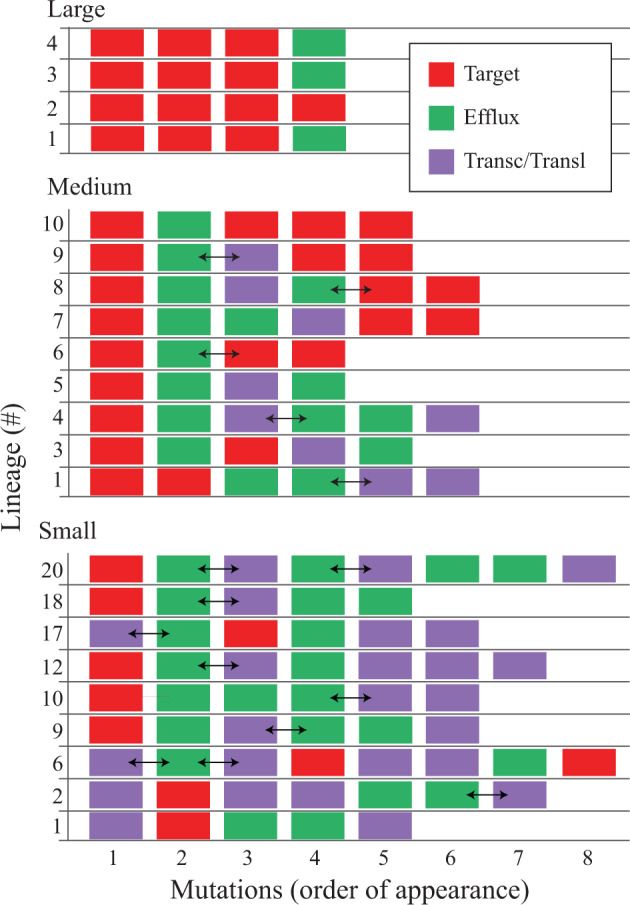
Mutation profile as a function of bottleneck size. Mutations classified into three groups (target, efflux, and transcription/translation) are shown in order of appearance as a function of evolution through large, medium, or small transfer bottlenecks. Horizontal double-headed arrows indicate mutations for which it is not possible to definitively state their order of appearance.
The mutational profile of the nine clones isolated from the medium bottleneck evolutions differs from that of the large bottleneck clones (supplementary table S4, Supplementary Material online). A significant difference is that the initial acquisition of a target mutation (gyrA) is almost always followed by the acquisition of an efflux-related mutation (acrR, marR, mdtK, and soxR). The differences continue with mutations in transcriptional/translational genes being acquired in 7/9 clones over the following three steps (fig. 3). By the end-point (32 mg/l CIP), 4/9 clones carry only 1 or 2 target mutations (the remaining 5 carry 3–4 target mutations), in contrast to the large bottleneck clones where all 5 clones sequenced carried 3–4 target mutations.
The single cell bottleneck clones also show a pattern where the first mutational step is dominated by target mutations (fig. 3). However, the succeeding mutational steps, 2–8, are dominated by the acquisition of mutations affecting transcription and translation genes (21/49 mutations) and drug efflux pumps (23/49 mutations). In a few cases, more than one mutation appeared at the same sequencing step (supplementary table S1, Supplementary Material online), and we cannot definitively state their order of appearance (indicated by a horizontal double-headed arrow in the figure).
The three different transfer bottlenecks resulted in two general differences in the observed evolutionary outcome. The number of resistance-associated mutations per clone decreased as bottleneck size increased probably reflecting the differential impact of different mutations on MIC, with large effect mutations dominating in the larger transfer populations. In addition, the predictability of the evolutionary trajectory increased as bottleneck size increased probably reflecting the impact of different mutations on the relative fitness of the mutants, with a great diversity of lower-fitness mutations dominating the smaller transfer populations. To test these conclusions, we measured MIC and relative fitness for individual clones from each evolution experiment.
Phenotypes of Clones from Different Evolution Regimes
The phenotypes of whole-genome-sequenced end-point clones, representing the different evolved lineages (ten small, nine medium, and five large bottleneck), were analyzed by measuring MIC to CIP and growth rate in Luria Broth (LB) relative to the parental strain MG1655 (supplementary table S7, Supplementary Material online). There were striking differences in phenotype associated with the different selection regimes (fig. 4). Each of the end-point clones from the large and medium bottleneck lineages had an MIC that was 3–4-fold higher than the concentration at which it was selected. In contrast, the clones evolved through single cell bottlenecks had MICs that did not exceed the MIC at which they were selected. The exponential growth rates of end-point clones, relative to the parental strain MG1655, were substantially higher in the five clones from large bottleneck populations (average relative growth rate 0.97), whereas clones from the medium and small bottleneck populations had much lower average relative growth rates of 0.50 and 0.35, respectively (fig. 4). The difference in relative growth rate between the medium and small bottleneck clones is significant (P = 0.02). These data show that differences in the transfer bottleneck during experimental evolution can significantly influence both the MIC and growth phenotypes of the evolved strains.
Fig. 4.
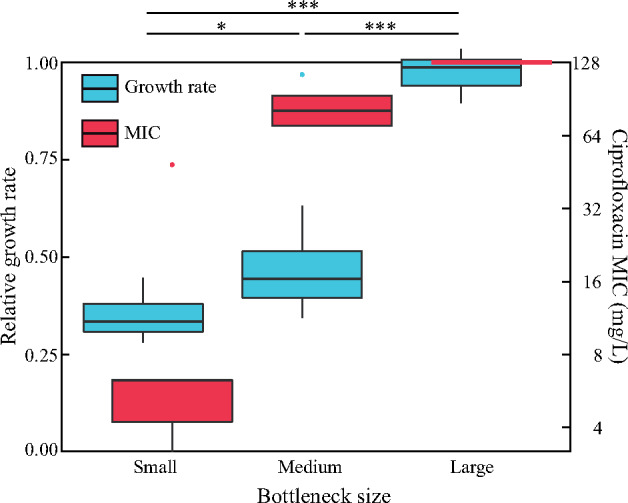
Phenotypic differences as a function of transfer bottleneck. Boxplot displaying the range of MICs (red) to CIP and relative growth rates (blue) of clones from the evolution experiments with different transfer bottlenecks. Growth rates are relative to the parental Escherichia coli MG1655 growing in LB. Single dots indicate outliers. Significant differences in relative growth rate between clones from different transfer bottleneck experiments are indicated by horizontal lines with associated P values (*significant at 0.05 and ***significant at 0.001).
Summary and Conclusions
Here, we have shown that the size of the transfer bottleneck size in experimental evolution can have profound effects on the set of genes in which mutations are selected, on the trajectory of evolution, and on the final phenotypes of the evolved strains. We propose that the critical issue is the size of the transfer bottleneck relative to the rate of mutations that affect the selected phenotype. For the selection of CIP resistance in E. coli, the critical mutation rate is the cumulative sum of mutations in drug target genes that reduce susceptibility. This rate, at ∼10−9, is several orders of magnitude lower that the cumulative rate of nontarget mutations, for example, those directly or indirectly affecting drug efflux (Garoff et al. 2018; Huseby et al. 2017). Most experimental evolution studies with bacteria use a transmission bottleneck size of ∼5 × 106 cfu (Lenski et al. 1991; Knopp and Andersson 2015; Knoppel et al. 2016, 2017; Vogwill et al. 2016; Roemhild et al. 2018; Lamrabet et al. 2019). This design is chosen for a very practical experimental purpose, mainly to minimize culture volume, which makes handling and experimental replication easier. However, bottlenecking affects the likelihood of selecting particular classes of resistance mutations, and because selected mutations can affect the potential for adaptation, it can influence mutational pathways and mutation supply rates in subsequent selection steps (Hall et al. 2010; Abel et al. 2015; Huseby et al. 2017). Narrow transmission bottlenecks reduce the supply of genetic variants to the next generation. This has the effect of reducing the power of natural selection for high-fitness individuals and instead increases genetic drift, making it more probable that deleterious mutants can exert a significant founder effect on the next generation (Heilbron et al. 2014; Vogwill et al. 2014).
To test the significance of this practical choice for the outcome of experimental evolution to CIP resistance, we tested three different transfer bottlenecks: one extremely small bottleneck (single cell) that is expected to maximize genetic drift and two bottlenecks (3 × 108 and 3 × 1010 cfu) that straddled the reciprocal of the cumulative drug target mutation rate. Note that the smaller of these transfer bottlenecks is still 100-fold larger than the typical experimental transfer bottleneck.
The large 1010 bottlenecks selected overwhelmingly mutations in drug target genes, and the resulting genotypes corresponded closely to those found in resistant clinical isolates. In contrast, both the medium 108 and small single cell bottlenecks selected mutations in three different gene classes: 1) drug targets, 2) efflux pump repressors, and 3) transcription-translation genes, including many mutations with low fitness (Pietsch et al. 2017; Garoff et al. 2018). Accordingly, bottlenecks smaller than the average nucleotide substitution rate significantly altered the experimental outcome away from genotypes observed in resistant clinical isolates (fig. 5). In the context of choosing an appropriate experimental design relevant to bacteria during a human infection, we note that there is a wide range of population sizes associated with different infections (as there is also in different natural environments). Some infecting populations can be very large, with up to 1010 CFU/bladder in urinary tract infections, but others, for example, in the lung, might be divided in small microecological compartments (Hughes and Andersson 2017).
Fig. 5.
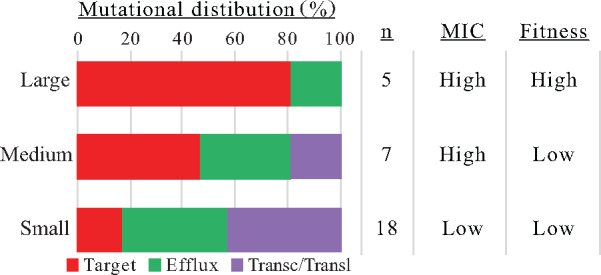
Overview of the genetic and phenotypic consequences of experimental evolution with different transfer bottlenecks. The distribution of mutations and between different functional/genetic classes. “n” is the total number of mutations in end-point clones (supplementary tables S1, S4, and S5, Supplementary Material online). MIC for CIP and relative fitness of end-point clones (see supplementary table S7, Supplementary Material online).
An interesting feature observed during the deep sequencing of populations undergoing this selection with the large bottleneck was that the genotypes dominating the populations in the final stages of the evolution were often minor subpopulations present at frequencies of <5% prior to their expansion later in the evolution (fig. 2 and supplementary table S3, Supplementary Material online). This is in contrast to the medium bottleneck evolution where clones that were ancestral to the strains dominating at the final step were typically also present at high frequencies earlier in the evolution (fig. 1 and supplementary table S2, Supplementary Material online). This implies that larger populations and larger transfer bottlenecks increase the effects of clonal interference, as more genetic variants are present that can potentially influence the selection coefficient for any given beneficial mutation, increasing the number of generations it might take to reach fixation in a population (Hall et al. 2010). An earlier study investigating E. coli and its resistance development to rifampicin has also shown that certain mutations, in terms of fitness, can be more beneficial at lower antibiotic concentrations but lose their advantage at higher concentrations, showing that the fitness effect of a mutation depends on the specific environment (Lindsey et al. 2013). This could also explain the expansion of minority subpopulations observed in our experiment with the large bottleneck, suggesting that the relative competitiveness of particular resistance genotypes may vary significantly as both the antibiotic concentration and the clonal population structure changes.
These data should be considered and applied practically in designing future experimental evolution studies to increase their predictive power and to explore the interplay between different environmental conditions, where transmission bottlenecks might vary, and resulting evolutionary trajectories.
Materials and Methods
Bacterial Strains and Growth Conditions
Bacterial strains are isogenic with E. coli MG1655. Selected mutants are referred in the text. Bacteria were grown in LB and on Luria Agar (LA, LB supplemented with 1.5% agar) where indicated in the text, with incubation at 37 °C. Throughout the article, the following definitions are applied. Genotype: the list of mutations that distinguish a mutant from an isogenic parent strain. Lineage: a temporal series of bacteria connected by a continuous line of descent from ancestor to descendant. Clone: a population of bacteria (usually a colony on agar) derived from a single founder cell.
Antibiotics and Susceptibility Testing
CIP was purchased from Sigma-Aldrich (Stockholm, Sweden) and was dissolved in 0.1 M HCl at 1 mg/ml before use. CIP was added to liquid and solid media for selections at the concentrations stated in the text. MICs were determined following CLSI and EUCAST guidelines by broth microdilution in Mueller Hinton II agar (Becton Dickinson & Company, France) with incubation for 18–20 h at 37 °C. Results were read after 45 h for slow-growing strains.
Evolution of Resistance to CIP
Independent lineages of the wild-type E. coli K12 strain MG1655 were initiated from single colonies grown on LA. Evolution of resistance to CIP was carried out with three different transfer bottleneck sizes, large, medium, and small: ≈3 × 1010 cfu, ≈3 × 108 cfu, and single colony, respectively. The concentration of CIP was increased stepwise during the evolution experiments (0, 0.016, 0.023, 0.032, 0.048, 0.064, 0.094, 0.128, 0.19, 0.25, 0.38, 0.5, 0.75, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0, 12.0, 16.0, 24.0, and 32 mg/l). For the large bottleneck experiment, five individual colonies were inoculated into 1-l LB, grown with shaking overnight at 37 °C, then 10 ml (1:100 dilution, transfer bottleneck ≈3 × 1010 cfu) was transferred to 1-l LB + CIP at 0.016 mg/l and incubated for ≈12 h. After ≈12 h, 10 ml was transferred to the same antibiotic concentration as previously, to allow for at least ten generations of growth at each antibiotic concentration. After the second incubation for ≈12 h, 10 ml was transferred to the next antibiotic concentration. For the medium bottleneck experiment, individual colonies were inoculated into 2-ml LB, grown with shaking overnight at 37 °C, then 100 μl was transferred to LB + CIP at 0.016 mg/l and incubated overnight at 37 °C (1:20 dilution, transfer bottleneck ≈3 × 108 cfu). Each 24 h, 100 μl was transferred into 2-ml LB containing CIP at 1.5-fold higher concentration than the current growth cycle. Cultures from each growth cycle were stored for future deep sequencing analysis at −80 °C. Finally, the single cell bottleneck evolution experiment was made by growing colonies on solid agar containing the same increasing concentrations of CIP as used in the liquid selection experiments. Accordingly, multiple individual colonies of wild-type, grown overnight on LA at 37 °C, were picked and streaked onto LA containing 0.016 mg/l CIP and incubated overnight at 37 °C. Typically several to many colonies grew along each streak on the drug-containing medium. From each streak, the largest colony was picked and restreaked onto agar with the same drug concentration and incubated overnight to complete one selection cycle (at higher drug concentrations, the incubation period was extended up to 72 h to facilitate slow-growing mutants). After incubation and growth, a single colony from each streak was picked and restreaked onto agar containing the next drug concentration. After each growth cycle step, each strain was stocked at −80 °C for future reference.
Growth Rate Measurements
Four biological replicates of liquid overnight cultures were diluted 1:1,000 in LB-media. Aliquots/well (300 μl) were pipetted into a Honeycomb 2 100-well plate (Oy Growth Curves AB Ltd, Finland), and growth measured using a Bioscreen C (Oy Growth Curves AB Ltd) with readings at 600 nm every 5 min for 18 h, 37 °C, continuous shaking. Negative controls were included to correct OD600 values by subtraction. The natural logarithm of each OD600 values was plotted against time, and the linear regression slope for ten subsequent OD600 readings calculated. Doubling time (min) was calculated by dividing ln(2) with the value of the maximal slope.
Whole-Genome Sequencing
For sequencing individual clones, genomic DNA was prepared using the MasterPure DNA Purification Kit (Epicentre, Illumina Inc, Madison, WI). Final DNA was resuspended in elution buffer. Genomic DNA concentrations were measured in a Qubit 2.0 Fluorometer (Invitrogen via ThermoFisher Scientific). DNA was diluted to 0.2 ng/ml in water (Sigma-Aldrich) and the samples were prepared for whole-genome sequencing according to Nextera XT DNA Library Preparation Guide (Illumina Inc). After the polymerase chain reaction cleanup-step, samples were validated for DNA fragment size distribution using the Agilent High Sensitivity D1000 ScreenTape System (Agilent Technologies, Santa Clara, CA). Sequencing was performed using a MiSeq desktop sequencer, according to the manufacturer’s instructions (Illumina Inc). The sequencing data were aligned and analyzed in CLC Genomics Workbench version 8.0.3 (CLCbio, Qiagen, Denmark).
Population sequencing was based on genomic DNA prepared from mixed (i.e., nonclonal) population cultures from intermediate steps in the experimental evolution corresponding to the CIP concentrations indicated in supplementary tables S2 and S3, Supplementary Material online. Three replicates of 100 μl were used from each frozen culture for genomic DNA extraction. Genomic DNA from populations and end-point clones was extracted using the MasterPureTM DNA Purification Kit (Epicentre, Madison, WI), according to the instructions of the manufacturer. Genomic libraries were prepared using Nextera XT DNA Library Preparation Kit and Nextera XT Indexes (Illumina). Whole-genome sequencing was performed with Illumina sequencing technology using the MiSeq platform.
Amplicon sequencing and primer design was done according to the guidelines of Illumina 16S Metagenomic Sequencing Library Preparation protocol. Amplicons of efflux regulatory genes (marR, acrR, and soxR) included regions upstream of the coding sequence to identify potential mutations of promotors or other regulatory sites. Sequencing data were aligned and analyzed using CLC Genomic Workbench v8 (CLCbio). For each culture, the three replicates were assembled and analyzed together. For identifying mutations of interest, the Basic Variant Detection tool was used with settings of minimum variant count of three reads, a minimum frequency of 5%, and a required forward/reverse read ratio >0. Exceptions to these thresholds were made for mutations in genes with known effects of reduced susceptibility to CIP (drug targets gyrA, gyrB, parC, and parE and drug efflux systems marR, acrR, and soxR), where only a minimum variant count of three reads was applied. Mutations only affecting prophages, repetitive regions, noncoding/nonpromoter regions, or mutations that were already fixed (i.e., at 100% frequency of reads) in populations isolated from 0.023 μg/ml CIP were disregarded.
Acknowledgments
This research was supported by grants to Diarmaid Hughes from Vetenskapsrådet (the Swedish Research Council) (2017-03593) and from the Scandinavian Society for Antimicrobial Chemotherapy (SLS-693211, SLS-876451). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Supplementary Material
References
- Abel S, Abel zur Wiesch P, Davis BM, Waldor MK.. 2015. Analysis of bottlenecks in experimental models of infection. PLoS Pathog. 11(6):e1004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Hughes D, Roth JR.. 2011. The origin of mutants under selection: interactions of mutation, growth and selection In: Böck A, Curtiss R III, Kaper JB, Karp PD, Neidhardt FC, Nyström T, Slauch JM, Squires CL, Ussery D, editors. EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. Washington (DC: ): ASM Press. [DOI] [PubMed] [Google Scholar]
- Baquero F, Martinez JL.. 2017. Interventions on metabolism: making antibiotic-susceptible bacteria. mBio 8(6):e01950–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G. 2013. Evolutionary rescue and the limits of adaptation. Philos Trans R Soc B 368(1610):20120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro W, Navarro M, Biot C.. 2013. Medicinal potential of ciprofloxacin and its derivatives. Future Med Chem. 5(1):81–96. [DOI] [PubMed] [Google Scholar]
- Chavez-Jacobo VM, Hernandez-Ramirez KC, Silva-Sanchez J, Garza-Ramos U, Barrios-Camacho H, Ortiz-Alvarado R, Cervantes C, Meza-Carmen V, Ramirez-Diaz MI.. 2019. Prevalence of the crpP gene conferring decreased ciprofloxacin susceptibility in enterobacterial clinical isolates from Mexican hospitals. J Antimicrob Chemother. 74:1253–1259. [DOI] [PubMed] [Google Scholar]
- Chen CR, Malik M, Snyder M, Drlica K.. 1996. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol. 258(4):627–637. [DOI] [PubMed] [Google Scholar]
- Chen YH, Ko WC, Hsueh PR.. 2012. The role of fluoroquinolones in the management of urinary tract infections in areas with high rates of fluoroquinolone-resistant uropathogens. Eur J Clin Microbiol Infect Dis. 31(8):1699–1704. [DOI] [PubMed] [Google Scholar]
- de la Campa AG, Balsalobre L, Ardanuy C, Fenoll A, Perez-Trallero E, Linares J, the Spanish Pneumococcal Infection Study Network G03/103. 2004. Fluoroquinolone resistance in penicillin-resistant Streptococcus pneumoniae clones. Emerg Infect Dis. 10(10):1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K, Malik M.. 2003. Fluoroquinolones: action and resistance. CTMC 3(3):249–282. [DOI] [PubMed] [Google Scholar]
- EUCAST. 2018. The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 8.1, valid from May 15, 2018. Available from: http://www.eucast.org/clinical_breakpoints/ Accessed January 25, 2020.
- EUCAST. 2019. MIC distributions and ECOFFs [Internet]. European Committee on Antimicrobial Susceptibility Testing. Available from: http://www.eucast.org/mic_distributions_and_ecoffs/ Accessed January 25, 2020.
- Ferrandiz MJ, Fenoll A, Linares J, De La Campa AG.. 2000. Horizontal transfer of parC and gyrA in fluoroquinolone-resistant clinical isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother. 44(4):840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff L, Huseby DL, Praski Alzrigat L, Hughes D.. 2018. Effect of aminoacyl-tRNA synthetase mutations on susceptibility to ciprofloxacin in Escherichia coli. J Antimicrob Chemother. 73(12):3285–3292. [DOI] [PubMed] [Google Scholar]
- Guay DR. 2008. Contemporary management of uncomplicated urinary tract infections. Drugs 68(9):1169–1205. [DOI] [PubMed] [Google Scholar]
- Hall AR, Griffiths VF, MacLean RC, Colegrave N.. 2010. Mutational neighbourhood and mutation supply rate constrain adaptation in Pseudomonas aeruginosa. Proc R Soc B 277(1681):643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilbron K, Toll-Riera M, Kojadinovic M, MacLean RC.. 2014. Fitness is strongly influenced by rare mutations of large effect in a microbial mutation accumulation experiment. Genetics 197(3):981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisig P. 1996. Genetic evidence for a role of parC mutations in development of high-level fluoroquinolone resistance in Escherichia coli. Antimicrob Agents Chemother. 40(4):879–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisig P, Tschorny R.. 1994. Characterization of fluoroquinolone-resistant mutants of Escherichia coli selected in vitro. Antimicrob Agents Chemother. 38(6):1284–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC. 2001. Emerging mechanisms of fluoroquinolone resistance. Emerg Infect Dis. 7(2):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC, Jacoby GA.. 2016. Topoisomerase inhibitors: fluoroquinolone mechanisms of action and resistance. Cold Spring Harb Perspect Med. 6(9):a025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper DC, Wolfson JS, Ng EY, Swartz MN.. 1987. Mechanisms of action of and resistance to ciprofloxacin. Am J Med. 82(4A):12–20. [PubMed] [Google Scholar]
- Hughes D, Andersson DI.. 2017. Evolutionary trajectories to antibiotic resistance. Annu Rev Microbiol. 71(1):579–596. [DOI] [PubMed] [Google Scholar]
- Huseby DL, Pietsch F, Brandis G, Garoff L, Tegehall A, Hughes D.. 2017. Mutation supply and relative fitness shape the genotypes of ciprofloxacin-resistant Escherichia coli. Mol Biol Evol. 34(5):1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby GA, Chow N, Waites KB.. 2003. Prevalence of plasmid-mediated quinolone resistance. Antimicrob Agents Chemother. 47(2):559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby GA, Strahilevitz J, Hooper DC.. 2014. Plasmid-mediated quinolone resistance. Microbiol Spectr. 2:PLAS-0006-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp M, Andersson DI.. 2015. Amelioration of the fitness costs of antibiotic resistance due to reduced outer membrane permeability by upregulation of alternative porins. Mol Biol Evol. 32(12):3252–3263. [DOI] [PubMed] [Google Scholar]
- Knoppel A, Knopp M, Albrecht LM, Lundin E, Lustig U, Nasvall J, Andersson DI.. 2018. Genetic adaptation to growth under laboratory conditions in Escherichia coli and Salmonella enterica. Front Microbiol. 9:756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoppel A, Nasvall J, Andersson DI.. 2016. Compensating the fitness costs of synonymous mutations. Mol Biol Evol. 33:1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoppel A, Nasvall J, Andersson DI.. 2017. Evolution of antibiotic resistance without antibiotic exposure. Antimicrob Agents Chemother. 61:e01495–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komp Lindgren P, Karlsson Å, Hughes D.. 2003. Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob Agents Chemother. 47(10):3222–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamrabet O, Martin M, Lenski RE, Schneider D.. 2019. Changes in intrinsic antibiotic susceptibility during a long-term evolution experiment with Escherichia coli. mBio 10(2):e00189–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, Tadler SC.. 1991. Long-term experimental evolution in Escherichia coli.1. Adaptation and divergence during 2,000 generations. Am Nat. 138(6):1315–1341. [Google Scholar]
- Lindsey HA, Gallie J, Taylor S, Kerr B.. 2013. Evolutionary rescue from extinction is contingent on a lower rate of environmental change. Nature 494(7438):463–467. [DOI] [PubMed] [Google Scholar]
- Malik M, Zhao X, Drlica K.. 2006. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol Microbiol. 61(3):810–825. [DOI] [PubMed] [Google Scholar]
- Marcusson LL, Frimodt-Moller N, Hughes D.. 2009. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog. 5(8):e1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazir H, Cao S, Hasan F, Hughes D.. 2011. Can phylogenetic type predict resistance development? J Antimicrob Chemother. 66(4):778–787. [DOI] [PubMed] [Google Scholar]
- Perichon B, Courvalin P, Galimand M.. 2007. Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob Agents Chemother. 51(7):2464–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddock LJ. 1999. Mechanisms of fluoroquinolone resistance: an update 1994–1998. Drugs 58(Suppl 2):11–18. [DOI] [PubMed] [Google Scholar]
- Pietsch F, Bergman JM, Brandis G, Marcusson LL, Zorzet A, Huseby DL, Hughes D.. 2017. Ciprofloxacin selects for RNA polymerase mutations with pleiotropic antibiotic resistance effects. J Antimicrob Chemother. 72(1):75–84. [DOI] [PubMed] [Google Scholar]
- Praski Alzrigat L, Huseby DL, Brandis G, Hughes D.. 2017. Fitness cost constrains the spectrum of marR mutations in ciprofloxacin-resistant Escherichia coli. J Antimicrob Chemother. 72(11):3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemhild R, Gokhale CS, Dirksen P, Blake C, Rosenstiel P, Traulsen A, Andersson DI, Schulenburg H.. 2018. Cellular hysteresis as a principle to maximize the efficacy of antibiotic therapy. Proc Natl Acad Sci U S A. 115(39):9767–9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchesnokova V, Radey M, Chattopadhyay S, Larson L, Weaver JL, Kisiela D, Sokurenko EV.. 2019. Pandemic fluoroquinolone resistant Escherichia coli clone ST1193 emerged via simultaneous homologous recombinations in 11 gene loci. Proc Natl Acad Sci U S A. 116(29):14740–14748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogwill T, Kojadinovic M, Furio V, MacLean RC.. 2014. Testing the role of genetic background in parallel evolution using the comparative experimental evolution of antibiotic resistance. Mol Biol Evol. 31(12):3314–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogwill T, Phillips RL, Gifford DR, MacLean RC.. 2016. Divergent evolution peaks under intermediate population bottlenecks during bacterial experimental evolution. Proc R Soc B 283(1835):20160749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Wachino J, Suzuki S, Kimura K, Shibata N, Kato H, Shibayama K, Konda T, Arakawa Y.. 2007. New plasmid-mediated fluoroquinolone efflux pump, QepA, found in an Escherichia coli clinical isolate. Antimicrob Agents Chemother. 51(9):3354–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.