

Abstract
Bacterial plasmids substantially contribute to the rapid spread of antibiotic resistance, which is a crisis in healthcare today. Coevolution of plasmids and their hosts promotes this spread of resistance by ameliorating the cost of plasmid carriage. However, our knowledge of plasmid–bacteria coevolution is solely based on studies done in well-mixed liquid cultures, even though biofilms represent the main way of bacterial life on Earth and are responsible for most infections. The spatial structure and the heterogeneity provided by biofilms are known to lead to increased genetic diversity as compared with well-mixed liquids. Therefore, we expect that growth in this complex environment could affect the evolutionary trajectories of plasmid–host dyads. We experimentally evolved Shewanella oneidensis MR-1 with plasmid pBP136Gm in biofilms and chemostats and sequenced the genomes of clones and populations. Biofilm populations not only maintained a higher diversity of mutations than chemostat populations but contained a few clones with markedly more persistent plasmids that evolved via multiple distinct trajectories. These included the acquisition of a putative toxin–antitoxin transposon by the plasmid and chromosomal mutations. Some of these genetic changes resulted in loss of plasmid transferability or decrease in plasmid cost. Growth in chemostats led to a higher proportion of variants with decreased plasmid persistence, a phenomenon not detected in biofilms. We suggest that the presence of more stable plasmid–host dyads in biofilms reflects higher genetic diversity and possibly unknown selection pressures. Overall, this study underscores the importance of the mode of growth in the evolution of antibiotic-resistant bacteria.
Keywords: plasmid, biofilm, experimental evolution, spatial structure, antibiotic resistance, horizontal gene transfer
Introduction
Plasmids are major agents shaping bacterial evolution through horizontal exchange of genetic information between bacteria (Ochman et al. 2000; Frost et al. 2005; Wiedenbeck and Cohan 2011). This exchange of adaptive traits is especially fostered by conjugative plasmids (Norman et al. 2009). Unfortunately, plasmids are increasingly shown to be vectors of the spread of resistance to so-called “last resort” antibiotics (Liu et al. 2016; McGann et al. 2016), resulting in antibiotic treatment failures (McCollister et al. 2016; Chen 2017). Indeed, plasmids increasingly make the headlines with alarming reports on their role in the spread of resistance to antimicrobials (World Health Organization 2014; White House 2015; European Centre for Disease Prevention and Control 2016). The development of strategies to abate the spread of antibiotic resistance may well depend on a better understanding of processes that promote the spread and persistence of resistance plasmids in bacteria.
Newly acquired resistance plasmids are not always stably maintained. Hereafter, we define “plasmid persistence” as the ability of a plasmid to maintain itself in a population in the absence of known selection for the plasmid. The improvement of such plasmid persistence over time is coined “plasmid stabilization.” The low persistence of plasmids in vitro is reflected in the rapid loss of plasmid-containing bacteria in serially transferred batch cultures (De Gelder et al. 2007). Selection for a plasmid-encoded trait such as antibiotic resistance has been shown to rapidly select for improved plasmid persistence. Such adaptive evolution is the result of genetic changes in the bacterial host, the plasmid, or both. The underlying mutations either ameliorate the plasmid fitness cost or decrease the plasmid loss rate during cell division (Sota et al. 2010; Hughes et al. 2012; San Millan et al. 2014; Harrison et al. 2015; Loftie-Eaton et al. 2016, 2017; Porse et al. 2016; Yano et al. 2016; Stalder et al. 2017). Some of these studies showed that plasmid cost reduction was linked directly or indirectly to chromosomally encoded helicases or transcriptional regulators, suggesting these proteins could be targets for future alternative drug therapies to reduce antibiotic-resistance spread. Thus, in the presence of antibiotics, bacteria and antibiotic resistance plasmids are known to rapidly adapt to each other, resulting in stabilization of the plasmid.
Unfortunately, almost all experimental evolution studies with plasmids have entirely been done using well-mixed liquid cultures that poorly mimic the habitat in which bacteria usually reside. In nature and the human body, bacteria typically grow in biofilms that are the source of many recalcitrant infections (Lewis 2001; Donlan 2012; Flemming and Wuertz 2019). Biofilms are populations or communities of microorganisms that grow on a surface in a matrix of extracellular polymeric substances (Hall-Stoodley et al. 2004; Lewandowski and Beyenal 2014). One intrinsic characteristic of biofilms is that they are spatially organized habitats in which individuals interact more frequently with neighbors than with more distant individuals. This spatial structure allows for habitat heterogeneity in which individuals at different locations experience different selection pressures. Both spatial structure and habitat heterogeneity are known to substantially impact evolution of microbial populations (Korona et al. 1994; Rainey and Travisano 1998; Kerr et al. 2002, 2006; Perfeito et al. 2008; Nahum et al. 2015; France et al. 2018, 2019). For example, by impeding global competition between cells, spatially structured environments protract selective sweeps of beneficial mutations and slow down the rate of adaptation (Perfeito et al. 2008; Nahum et al. 2015; France and Forney 2019). This promotes the coexistence of genetically distinct individuals, which would not coexist in a well-mixed environment, and results in the maintenance of genetic diversity allowing biofilm populations to harbor multiple distinct evolutionary outcomes (Boles et al. 2004; Ponciano et al. 2009; Eastman et al. 2011; Santos-Lopez et al. 2019). In addition, the intrinsic structure within biofilms generates environmental gradients of nutrients and electron acceptors creating habitat heterogeneity that drives local adaptation of subpopulations, further contributing to diversity. Extensive analysis of the mechanisms behind this diversification in biofilms was recently reviewed (Martin et al. 2016; Steenackers et al. 2016; France et al. 2018). The effect of biofilm growth on the evolutionary outcomes of bacteria that carry plasmids needs to be identified to gain insight into the spread and persistence of antibiotic resistance plasmids in natural or clinical settings.
Here, we addressed the role biofilms play in the evolution of a plasmid–host dyad that initially showed poor plasmid persistence. Because biofilms maintain genetic diversity, we hypothesized that biofilm growth allows for the emergence of some clones that show higher plasmid persistence than any clones evolved in well-mixed environments. To test our hypothesis, we conducted an experimental evolution study in biofilms and chemostats using Shewanella oneidensis MR-1 containing plasmid pBP136Gm. On an average, plasmid persistence in evolved biofilms clones was not higher than in the ancestor, but as expected, biofilm populations contained a small proportion of unique variants in which the plasmid was more persistent than in any clone evolved in chemostats. In contrast, chemostat growth led to variants that showed decreased plasmid persistence, something that was not detected in the biofilms. The various evolutionary and ecological processes that differ between biofilms and well-mixed systems, such as spatial structure and heterogeneity and selective pressures, are discussed. Our study shows that biofilm growth can lead to more persistent antibiotic-resistance plasmids that would probably be overlooked in the traditional liquid culture evolution experiments.
Results
To study how biofilm growth affects the evolutionary trajectories of a plasmid–host dyad, we evolved Shewanella oneidensis MR-1 with plasmid pBP136Gm for 28 days both as biofilms in four continuously fed flow cells (B1–4) and as well-mixed cultures in four chemostats (C1–4) (fig. 1A). This was done under antibiotic selection for the plasmid. The flow cells gradually produced a thick biofilm that covered the entire interior surface of the chamber (fig. 2). It was not possible to calculate the number of generations in the biofilms because the growth rates are known to vary throughout. Based on viable counts, the population densities in the biofilms and the chemostats were similar after 28 days: 1.60± 0.03×108 and 1.72±0.17×108 cfu ml−1, respectively (the total culture volume was 12 ml in both).
Fig. 1.

Diagram depicting the experimental design, sampling, and analyses of the Shewanella oneidensis evolution experiment. The isolated clones were named after their environment (B for biofilm and C for chemostat), and then numbered I1–5 (e.g., B2I3), as well as I6–35 for the first biofilm and chemostat, B1, and C1.
Fig. 2.
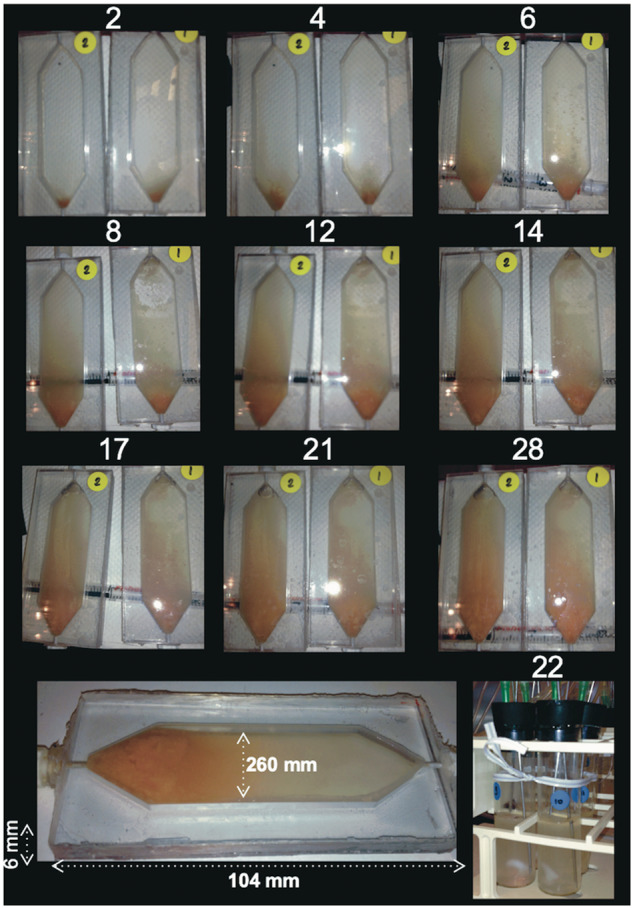
Photograph depicting biofilm growth over time in two replicate flow cells (time in days is indicated above each picture) and chemostats (bottom right). The bottom flow cell image shows the dimensions.
Evolutionary Outcomes of Plasmid Persistence
To test our hypothesis that evolution in biofilms allows the emergence of a few clones that show higher plasmid persistence as compared with evolution in well-mixed environments, we first measured plasmid persistence in evolved biofilm and chemostat clones (fig. 1C). These clones were randomly chosen from three of the four replicate populations (biofilms B1, B2, and B3 and chemostats C1, C2, and C3), and compared with the ancestor (fig. 3). They were named after their environment (B or C) followed by I (for isolated clone) and a clone number, for example, B2I3. Plasmid persistence was assayed by determining the fraction of cells with plasmids over time as the clones were passaged in serial batch cultures. This assay took the clones out of the environment they were evolved in (biofilm or chemostat), and placed them into a common environment (batch culture) to enable direct comparison of evolved plasmid persistence under similar conditions. By using this single “common garden” design, dependencies of evolved plasmid persistence on the local environment cannot be detected here. On an average, the persistence of plasmids in clones from biofilms was not significantly different from that of the ancestral plasmid–host dyad (fig. 3). However, when individual biofilm clones were compared with the ancestor, two stood out as showing a markedly higher plasmid persistence. Hereafter, we refer to these as “stabilized clones.” In contrast, the persistence of plasmids in bacteria evolved in chemostats was on an average significantly lower than in the ancestor, and no stabilized clones were observed in these populations. In fact, the opposite was true, as two chemostat clones showed significantly lower plasmid persistence than the ancestral dyad.
Fig. 3.
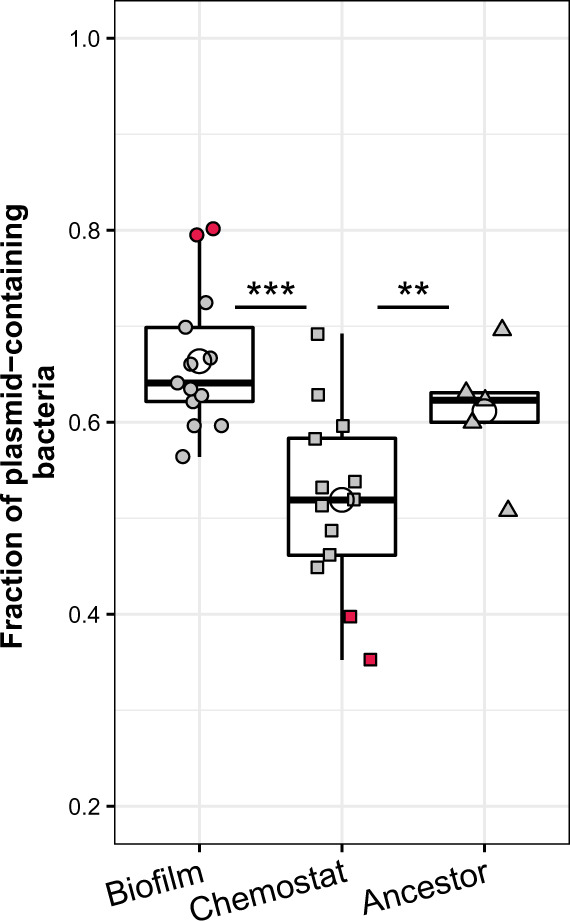
The persistence of plasmids in clones evolved in either biofilms or chemostats was compared with that in ancestral clones in 12-day plasmid persistence assays. For both biofilms and chemostats, we assayed the plasmid persistence of five clones from evolved populations 1 and 2, and three clones from population 3, compared with five ancestral clones (as two clones from biofilm B3 were lost, only three clones were analyzed for both biofilm B3 and chemostat C3, for consistency). The three filled symbols represent clones from biofilm (circle), chemostat (square), and ancestral (triangle) populations, with each point showing the average value from triplicate persistence assays for one clone. The open circles represent the overall average for each evolution environment and for the ancestor. The red filled symbols at the top or bottom denote clones in which plasmid persistence was markedly different from the ancestor (t-test P values; B1I3 < 0.01, B3I2 < 0.05, C3I1 < 0.05, and C3I5 < 0.01; tests were performed against the same sample size of randomly picked ancestor values). Boxes represent the median and the first and third quartiles, and the whiskers the lower and upper 1.5 interquartile range. No significant difference was observed between the biofilm and ancestral populations.
To further explore the presence of a low proportion of stabilized clones in biofilms, we sampled one biofilm (B1) and one chemostat (C1) population more deeply by determining plasmid persistence for 30 evolved clones from each population (figs. 1D and 4). As before, the average plasmid persistence in biofilm clones was not different from the ancestral dyad, but there were now three biofilm clones that stood out as showing higher plasmid persistence. Their final fraction of plasmid-bearing cells at the end of plasmid persistence assays exceeded 0.8 (vs. ∼0.65 in the ancestral population, fig. 4). We verified the persistence by repeating the plasmid persistence assay for these three clones in triplicate (data not shown). We then calculated the probability of observing a plasmid persisting at a fraction of 0.84 after 12 days (i.e., the lowest of the three high values in fig. 4) given the mean and SD of the plasmid persistence observed in the ancestral clones. This probability was 0.0047, thus less than 1 in 200, whereas we observed 3 clones out of 30 that showed a persistence ≥0.84. In contrast, none of the 30 clones evolved in the chemostat population showed plasmid persistence that exceeded that of the ancestor and three clones even showed a significantly lower plasmid persistence (fig. 4). These results suggest biofilms allow the emergence of a small proportion of clones that show clearly increased plasmid persistence. In contrast, no plasmid stabilization was observed among the chemostat clones, suggesting that mutations conferring higher plasmid persistence were not beneficial enough to sweep through the chemostat population in that time period. Thus, out of more than 40 evolved clones tested from each environment, 12% from biofilms, but none from chemostats, were found to much better retain their plasmid than the ancestor.
Fig. 4.
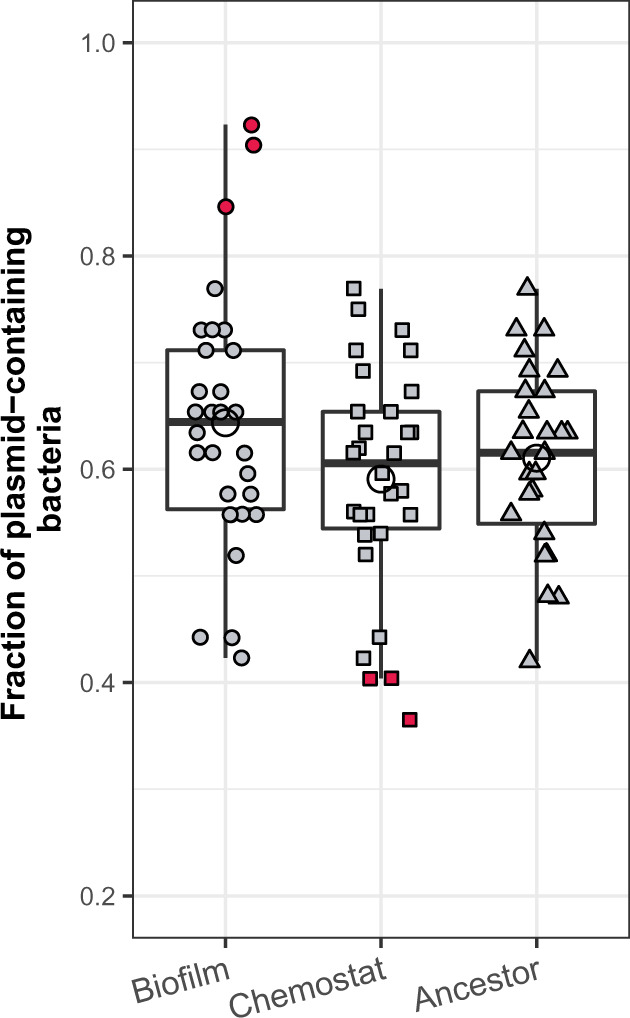
Plasmid persistence in 30 clones isolated from a single evolved biofilm population (B1), a single chemostat population (C1), and the ancestor. The three filled symbols depict clones from biofilm (circle), chemostat (square), and ancestral (triangle) populations, with each point representing a single measurement. The open circles represent the average for each environment. The red filled symbols at the top and bottom represent the clones for which plasmid persistence differed from the ancestor. Plasmid persistence in these clones was verified by repeating the assay in triplicate and the four values were compared with the same sample size of randomly picked ancestor values (t-test P values for the six clones were B1I22 < 0.1, B1I19 < 0.05, B1I25 < 0.01, C1I27 < 0.1, C1I25 < 0.01, C1I17 < 0.01). Boxes represent the median and the first and third quartiles, and the whiskers the lower and upper 1.5 interquartile range.
Evolutionary Pathways in Biofilms
Genotypes Suggest Different Evolutionary Trajectories in Biofilms
To determine if the five stabilized biofilm clones followed the same or different evolutionary trajectories, we compared their genome sequences (see red dots in figs. 3 and 4, and table 1). In addition, we sequenced three randomly chosen biofilm clones that showed no change in plasmid persistence (fig. 1E and table 1). Overall, there were as many as 7 unique genotypes among these 8 biofilm clones, and the mutations were in or near 20 distinct genes. The five stabilized biofilm clones had distinctly different genotypes, with three to six mutations per clone. Moreover, 18 of the 19 mutations were unique to each clone (table 1). Strikingly, none of these mutations was present in the three clones that did not show increased plasmid persistence. Only one of them, a mutation upstream of mxdA, was also found in a chemostat clone (see below, and tables 1 and 2).
Table 1.
Summary of the Mutations in the Isolated Clones of Shewanella oneidensis MR-1 (pBP136Gm) Evolved in Biofilms.
| Replicon | Gene | B1I3 | B1I19 | B1I22 | B1I25 | B3I2 | B1I1a | B1I2 | B1I5 | Position | Mutation | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome (NC 004347) | rpoA | x | 258578 | A>T (E272D) | RNA polymerase alpha subunit | |||||||
| pncC | x | 276015 | Tn6374b | Nicotinamide deamidase | ||||||||
| aaiD | x | 947021 | C>T (A212V) | Acyl homoserine lactone acylase | ||||||||
| SO1092 | xc | 1130985 | G>C (A105A) | Transcriptional regulator AraC family | ||||||||
| SO1278>/<SO1281 | x | 1332729 | 9 bp>G | Chemotaxis signal transduction system methyl accepting sensory transducer/hypothetical protein | ||||||||
| SO2045 | x | 2143919 | Deletion 6 bp | Cation efflux protein CDF family | ||||||||
| SO4822</ id="564">SO2290 | x | 2403573 | A>G | Hypothetical protein/rhodanese domain protein UPF0176 family | ||||||||
| tnpA | xc | 2405396 | A>C (T63T) | ISSod4 transposase | ||||||||
| wbqC | x | x | 3323205 | (T)7>6 | Putative glycine transferase in O-antigen biosynthesis cluster | |||||||
| x | 3323304 | tnpA b | ||||||||||
| SO3185 | x | 3323634 | (A)5>4 | Enzyme for biosynthesis of dTDP Qui4N | ||||||||
| rfbB | x | x | xd | 3326782 | C>A (R88L) | dTDP-glucose-4,6-dehydratase | ||||||
| fliP | x | 3358060 | deletion 1 bp | Flagellar export protein | ||||||||
| flgL | x | x | xd | 3377938 | +T | Flagellar hook-associated protein | ||||||
| cheR | x | 3389192 | tnpA b | Chemotaxis signal transduction system MCP methyltransferase | ||||||||
| x | 3389247 | G>A (A144V) | ||||||||||
| mxdA</<SO4181 | x | 4354008 | (A)6>7 | Diguanylate cyclase-like protein/RNA pseudouridylate synthase family protein | ||||||||
| SO4628 | x | 4824623 | 2 bp>AA | Membrane sulfatase HI1246 family | ||||||||
| SO4680 | x | 4879232 | tnpA b | CDP-glycerol glycerophosphotransferase family protein | ||||||||
| pBP136Gm | trbJ | x | 10326 | Tn6374b (TTTTT) | Mating pair formation and entry exclusion | |||||||
| trbO | xd | 14314 | Tn6374b (CAAGTGGG) | Transfer gene, unknown function | ||||||||
| klcA</>XF1597 | x | 40107 | Tn6374b (CTAAAT) | Possible anti-restriction system/hypothetical protein | ||||||||
| Total number of mutations | 3 | 6 | 3 | 4 | 4 | 2 | 2 | 3 | ||||
Gray shading highlights the evolved clones with no change in plasmid persistence.
The insertion of a transposable element at that position.
Silent mutation.
Detected in the marginal prediction.
Table 2.
Summary of the Mutations in the Isolated Clones of Shewanella oneidensis MR-1 (pBP136Gm) Evolved in Chemostats.
| Replicon | Gene | C1I17 | C1I25 | C3I1 | C3I5 | C1I27 | C1I16a | C1I26 | C1I12 | Position | Mutation | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome (NC 004347) | SO0569 | x | 590478 | C>T (G39D) | Diguanylate cyclase | |||||||
| cheC | xb | 590787 | C>T (R274H) | Chemotaxis signal transduction system | ||||||||
| SO1278>/<SO1281 | x | 1332264 | +ATGCCATCTCTG | Chemotaxis signal transduction system methyl accepting sensory transducer/hypothetical protein | ||||||||
| SO1699 | x | 1789463 | A>T (L127Q) | Transmembrane transcriptional regulator | ||||||||
| phoP | x | 2049619 | C>A (G59C) | Two component signal transduction system response regulator | ||||||||
| wbpA | xb | 3328917 | tnpA c | UDP-N-acetyl-d-glucosamine 6-dehydrogenase | ||||||||
| cheR | xb | xb | xb | 3388924 | tnpA c | Chemotaxis signal transduction system MCP methyltransferase | ||||||
| xb | 3389346 | G>A (A111V) | ||||||||||
| xb | xb | 3389372 | tnpA c | |||||||||
| metB | xb,d | xb,d | 4212022 | C>A (A137A) | Cystathionine gamma-synthase | |||||||
| MxdA</<SO4181 | xb | xb | 4354008 | (A)6>7 | Diguanylate cyclase-like protein/RNA pseudouridylate synthase family protein | |||||||
| x | x | xe | 4354022 | (A)5>6 | ||||||||
| pMR-1L | SOA0112; SOA0115 | x | 91763 | C>G (R1384G) | Putative lipoprotein | |||||||
| x | 91703 | G>T (D2775Y) | ||||||||||
| Total number of mutations | 4 | 2 | 3 | 3 | 2 | 3 | 2 | 2 | ||||
Gray shading highlights the evolved clones with no change in plasmid persistence.
Mutation detected by the whole-population sequencing approach.
The insertion of a transposable element at that position.
Silent mutation.
Detected in the marginal prediction.
In three of the five stabilized biofilm clones, plasmid pBP136Gm had acquired the transposon Tn6374 from a native plasmid. These were three independent transposition events. In one (B3I2), the transposon was inserted in a noncoding region 39 bp upstream of an operon composed of two putative proteins of unknown function (XF1597 and XF1596). In the two other clones (B1I25, B1I19), the transposon insertion disrupted two different plasmid transfer genes, trbJ and trbO, respectively. Only the latter disruption caused loss of the plasmid’s ability to transfer by conjugation (data not shown). Interestingly, such transposition events have been observed by us previously in the same strain, where they were shown to increase plasmid persistence (Stalder et al. 2017). The chromosomal mutations found in these three clones were not directly linked to mechanisms known to affect plasmid persistence. Instead, they were related to cell membrane composition and diffusion (SO4680), cell motility and chemotaxis (cheR and fliP), sulfur metabolism (SO2290 and SO4628), quorum sensing (aaiD), production of cyclic-di-GMP (mxdA), and the O-antigen synthesis pathway (wbqC) (table 1).
Of the remaining two stabilized clones, clone B1I22 had three chromosomal mutations in genes involved in cell membrane composition and diffusion (SO2045), chemotaxis (cheR), and an O-antigen synthesis pathway (SO3185). Finally, clone B1I3 also showed three chromosomal mutations, one of them in an intergenic region distant from its adjacent genes (>500 bp), and the other two in genes wbqC and rpoA. RpoA is the essential α subunit of the RNA polymerase. Interestingly, four of the five stabilized clones showed mutations in genes involved in the O-antigen synthesis pathway, which is part of the lipopolysaccharide and related to biofilm formation (Lau et al. 2009). We currently do not know which of these mutations is responsible for improved plasmid persistence, but the diversity of unique mutations in the five biofilm clones clearly indicates that each of them followed a different evolutionary trajectory.
As a few clones evolved in chemostats showed a lower plasmid persistence than the ancestor, a quite unusual observation, we analyzed the genome sequences of five of them (see red dots in figs. 3 and 4, and table 2) in addition to three clones with no change in plasmid persistence (table 2). Among these eight clones, there were five unique genotypes with mutations present in or near only ten genes. Seven of these eight clones, including those with no change in plasmid persistence, showed again at least one mutation in the chemotaxis signaling pathway che (cheR and cheC), and six clones had mutations within or upstream of genes involved in cyclic-di-GMP metabolism (SO0569 and mxdA). Cyclic-di-GMP has a prominent role in the switch between motile and biofilm forming cells. For example, in Pseudomonas aeruginosa, c-di-GMP controls the enzymatic activity of the methyltransferase CheR (Yan et al. 2018), and in Shewanella oneidensis MR-1, the (cyclic-di-GMP)-forming enzyme MxdA controls biofilm formation (Thormann et al. 2006). These data suggest that in the chemostats mutations linked to chemotaxis were beneficial.
Plasmid Stabilization through Multiple Mechanisms
Plasmid stabilization could result from mutations that affect one of at least three biological parameters: 1) the fitness cost of the plasmid, 2) the plasmid loss rate during cell division, and 3) horizontal plasmid transfer rate by conjugation (De Gelder et al. 2007). To gain insight into which of these parameters was affected by mutations in the five evolved stabilized biofilm clones, we estimated the cost of their plasmids (fig. 1F). This was done by measuring the fitness (W) of each evolved plasmid-bearing clone relative to its plasmid-free counterpart in competition experiments. These results were then compared with the relative fitness of the ancestral plasmid-bearing versus plasmid-free host (plasmid cost of the ancestral plasmid in the ancestral host was c = 1−W = 0.03; see fig. 5). The two evolved clones in which the transposon Tn6374 inserted in one of the plasmid’s transfer genes (B1I19, B1I25) showed a slightly but not significantly lower plasmid cost than the ancestor. To verify if the relative fitness (W) was significantly different from 1 meaning there is no plasmid cost, we changed the hypothesis of the t-test to W = 1. Interestingly, the plasmid cost in the clones B1I19 and B1I25 was no longer significantly different from zero, suggesting that the cost was ameliorated. Remarkably, the fitness of clone B1I25 tended to be even higher than that of its plasmid-free counterpart (W > 1 or c < 0) (t-test P value <0.1), suggesting the plasmid had become slightly beneficial. In contrast, the plasmid cost did not significantly change in clones B1I22 and B3I2, and became even larger in clone B1C3. Therefore, either the plasmid loss rate or horizontal transfer rate must have changed in these clones. For example, the three clones that acquired Tn6374 in pBP136Gm, including B3I2, may well have evolved a decreased plasmid loss rate. This transposon encodes a putative toxin–antitoxin that can inhibit the growth of plasmid-free bacteria generated during cell division, and a cointegrate resolution system that can help resolve plasmid multimers and thus improve proper plasmid segregation (Stalder et al. 2017). Our findings show that plasmid stabilization mechanisms were not the same among the clones, supporting the conclusion that the biofilm clones followed different evolutionary trajectories.
Fig. 5.
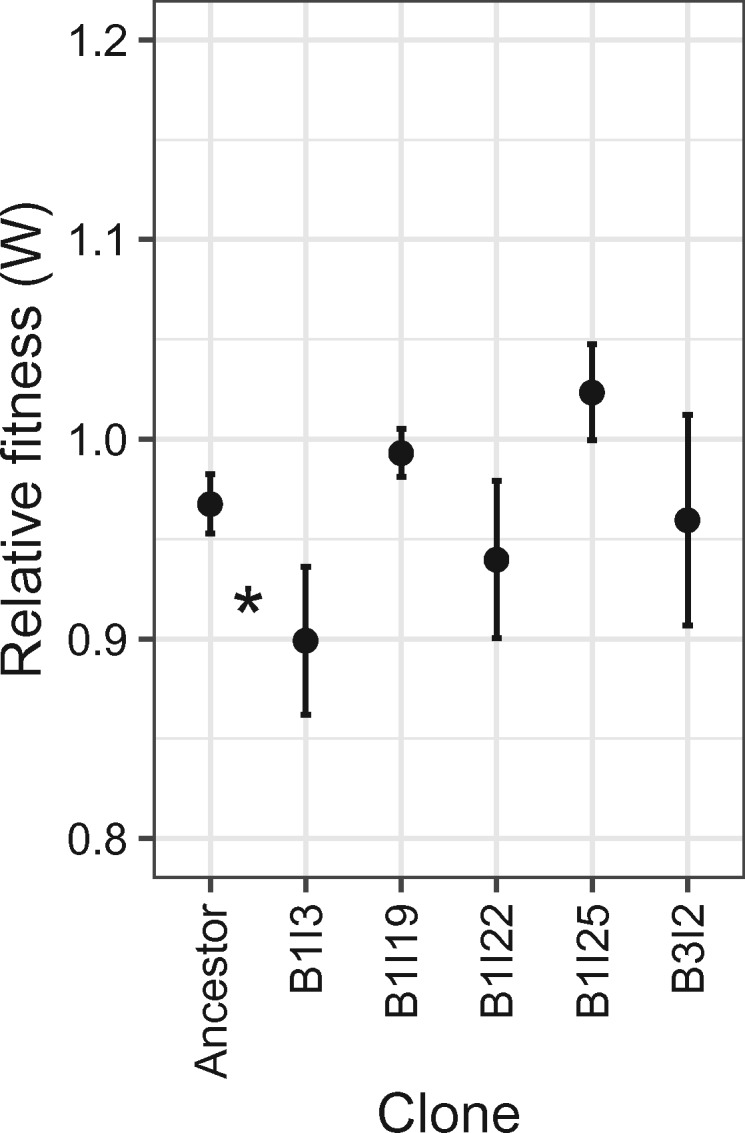
Fitness (W) of the ancestral plasmid–host dyad and five plasmid-bearing biofilm clones that showed the highest plasmid persistence, relative to their respective plasmid-free counterparts. Each point shows the average of three competition assays. As the plasmid cost equals 1−W, only clone B1I3 had a plasmid cost that had become statistically different from that of the ancestor (increased cost, P < 0.1). Plasmid-free counterparts of evolved hosts were obtained as one of the first segregants during the persistence assay protocol (>100 colonies were replica-plated on selective and nonselective media).
Whole-Genome Whole-Population Sequencing
Because we hypothesized that growth in biofilms would lead to greater genetic diversity, we also compared the frequency of genomic changes in all four evolved biofilm populations with those in the four evolved chemostat populations (fig. 1B). This was done using a whole-genome whole-population sequencing approach by sequencing DNA extracted from the evolved populations and from the ancestral strain. Henceforth, we refer to this approach as “whole-population sequencing.” A total of 45 unique mutations were detected in the four biofilm populations and only 22 in the four chemostat populations (fig. 6 and supplementary fig. S1, Supplementary Material online). On an average, the diversity of unique mutations measured by the Shannon diversity index (H) was higher in the biofilms than the chemostats (Hbiofilm = 2.24, HChemostat = 1.47, t-test P value <0.05; this index ranges from 0 for a population with only a single genotype to high values for populations that have many mutations, with each unique mutation found in a few reads). This supports the hypothesis that spatially structured biofilms maintain a higher diversity of mutations than well-mixed populations. We then tracked the frequencies of these genetic changes in the populations (i.e., equivalent to frequencies of new alleles). A frequency of 1 means that a mutation swept through the population, since every sequence read covering that locus had that genetic change. Values <1 represent the fraction of reads in which the new mutation was observed. On an average, the frequency of these mutations was significantly lower in the biofilm populations than in the chemostats, indicating proportionally fewer sweeps to fixation of beneficial mutations in biofilms (fig. 6). This is consistent with the finding that none of the mutations in the five biofilm clones was detected in any of the biofilms by whole-population sequencing, suggesting these five mutants were present at a rather low frequency. Indeed, average sequencing depth for the whole-population sequencing was only ∼100×, and each sequenced clone from biofilm B1 was present at only roughly 3% (1 out of 35 clones analyzed). It is thus plausible that their mutations were not observed among the whole-population sequence reads. This stands in contrast to the chemostat populations, where 50% of the mutations found in the sequenced clones were also detected by the whole-population sequencing approach, suggesting they were common as expected after a selective sweep (Table 2 and Figure S1). This population sequencing approach shows that bacterial evolution led to a higher diversity of detectable mutations in biofilms compared with chemostats, where a few mutations tended to sweep to fixation.
Fig. 6.
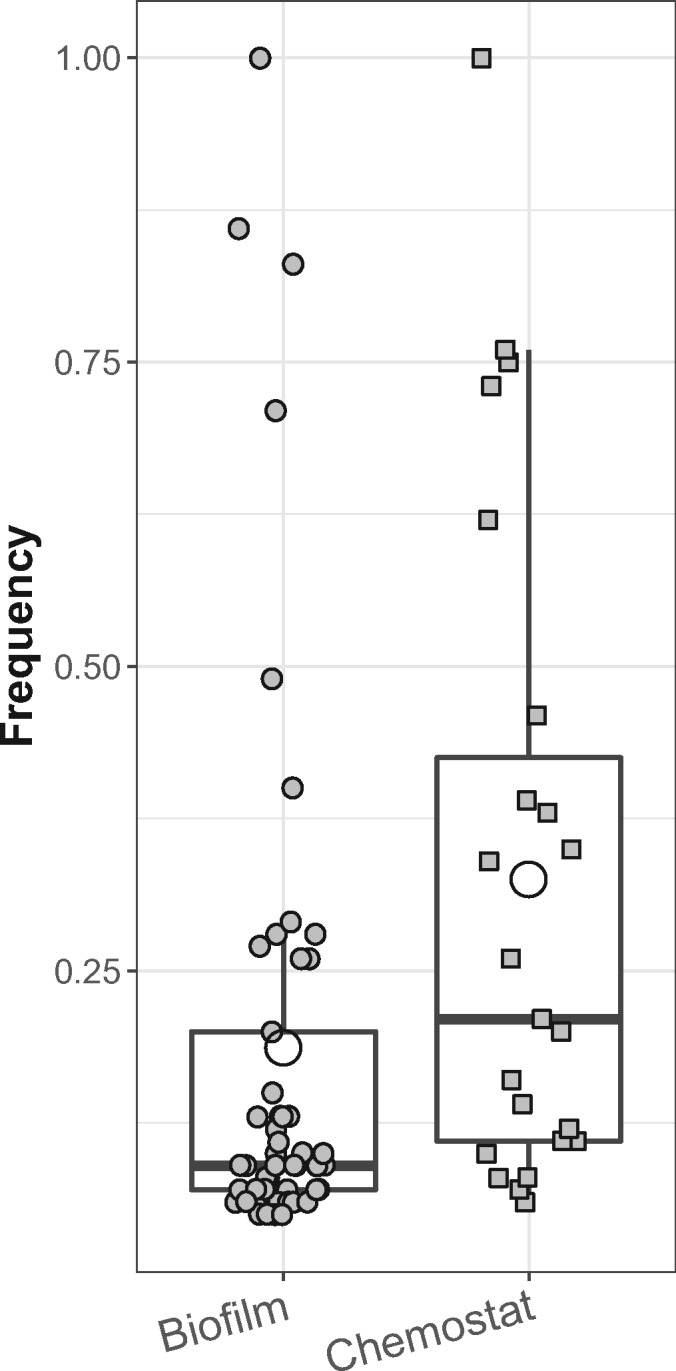
Frequency of mutations identified in biofilm and chemostat populations. Boxes represent the median (horizontal line) and the first and third quartiles, and the whiskers the lower and upper 1.5 interquartile range. The open circles represent the average frequency for each environment, which was lower for the biofilms than for the chemostats (P < 0.05).
Discussion
Very few studies have addressed the effect of biofilm growth on the coevolution of plasmids and their bacterial hosts (Ridenhour et al. 2017). Here, we postulated that the environment provided by biofilms affects the evolutionary outcomes of a plasmid-bacterium dyad by maintaining a higher diversity of genotypes, including clones with more persistent plasmids than in well-mixed environments. In line with other studies on bacterial evolution, we found a higher diversity of genotypes in bacterial biofilms than in mixed liquids, and most importantly, we showed that some of these evolved biofilm clones contained very persistent plasmids. Although the maintenance of a higher genetic diversity in biofilms can explain these findings, it seems that selection and pleiotropic effects also may have been at play.
We observed a higher diversity of mutations in populations that had evolved in biofilms compared with well-mixed chemostats. Such a result was expected as microbial evolution in biofilm populations is known to generate and preserve genetic diversity (Boles et al. 2004; Ponciano et al. 2009; Traverse et al. 2013). This observed diversification process in biofilms is also in line with other studies from our group, which showed higher diversity in plasmid persistence dynamics and corresponding genotypes after evolution in biofilms than in well-mixed liquid cultures for an Acinetobacter baumannii strain with a similar plasmid (Ridenhour et al. 2017; Metzger GA, Ridenhour BJ, France M, Forney LJ, Gliniewicz K, Millstein J, Settles ML, Stalder T, Top EM, personal communication). Diversification of biofilm populations also means there is a higher likelihood of discovery of mutants. Indeed, we showed here for the first time that in biofilms and not in chemostats a few genetically distinct individuals arose with a markedly more persistent plasmid than the ancestor or other clones of the same populations. Nevertheless, one could argue that if only genetic diversity was driving evolutionary outcomes this would translate in phenotypic diversity including both high- and low plasmid persistence phenotypes, as observed by (Ridenhour et al. 2017). Here, we did not detect less persistent variants in the biofilms. Hereafter, we discuss several scenarios that could explain such observations, each not being mutually exclusive but opening the way to further disentangling evolutionary trajectories of plasmid–host dyads in biofilms.
Several evolutionary and ecological processes have been proposed to explain patterns of diversification in biofilms, and are extensively reviewed in (Steenackers et al. 2016; France et al. 2018). For example, it has been suggested that populations evolving in biofilms may have a higher mutation rate (Ryder et al. 2012; Steenackers et al. 2016), which could increase the mutation supply of a biofilm compared with a chemostat population. In addition, local adaptation due to the environmental heterogeneity probably drives diversification of biofilm populations (Rainey and Travisano 1998). Greater heterogeneity in biofilms would thus lead to simultaneous evolutionary trajectories in multiple adaptive landscapes, leading to greater diversity. Furthermore, because biofilm populations are spatially structured, and can be divided into small subpopulations or microcolonies, genetic drift is likely to affect the exploration of the mutation landscape, and clonal interference is expected to be prolonged due to the protraction of selective sweeps (Wright 1932; Habets et al. 2006; Hallatschek et al. 2007; France and Forney 2019). Interestingly, the five clones showing increased plasmid persistence each had unique mutations and different underlying mechanisms of plasmid stabilization. Different mutations that promote plasmid persistence were thus competing in the same population, supporting the idea of clonal interference. Furthermore, none of these mutations swept to fixation. For the biofilm population from which 35 clones were analyzed, the frequency of each unique genotype with a higher plasmid persistence was <3% (1/35). Strikingly the mutations in these clones were not detected through whole-population sequencing, confirming that they were present at a rather low frequency. These results suggest that in biofilms there were a greater number of slower paced increases in new putative beneficial mutations to detectable frequencies, which led to a greater detectable standing diversity. It is precisely because these increases are not traditional selective sweeps that the diversity pattern in biofilms would appear as it does here. In some sense, the clonal interference in chemostats may have been shorter-lived as the very best mutation drove other good mutations to extinction (see below); whereas many different good mutations can coexist for longer in biofilms (Traverse et al. 2013)—that is, clonal interference is longer-lived in biofilms. Nevertheless, to fully understand the evolutionary dynamics of plasmid–bacteria dyads in biofilms, future studies should sample biofilm populations over time and space, and test the persistence of plasmids across a range of conditions, including those experienced by the evolving populations.
The diversity explanations above do not fully clarify why we did not observe individuals with less persistent plasmids in the biofilms, and why in chemostat we only detected clones with plasmid persistence similar or lower than in the ancestor. The former could be explained by positive selection on plasmid persistence and detection limits, but the latter requires an additional explanation. In the biofilms, one can argue that the presence of the antibiotic selected, however weakly, for individuals who better retained their resistance plasmid, thus slowly outcompeting less-stabilized clones. Even if these clones were present at just slightly lower frequency than the stabilized ones, we would not have found them by screening only 35 clones among ∼109 cfu per biofilm. Nevertheless, there are at least two more possible explanations for why in biofilms we found a few genetically distinct individuals with markedly higher plasmid persistence than in the ancestor, but not in chemostats. The first one is that plasmid-persistence-enhancing mutations could have had a higher selective value in biofilms than in chemostats, as supported by earlier findings that some plasmids can promote biofilm formation (Ghigo 2001; Madsen et al. 2012). Alternatively, other mutations beneficial to biofilm growth (not affecting plasmid persistence) may have had a relatively smaller selective value, in contrast to chemostat-selected mutations. Secondly, mutations beneficial to biofilm growth might have had unknown positive pleiotropic effects on plasmid persistence. The opposite observation that evolution in chemostats negatively affected the ability of the plasmid to persist (figs. 3 and 4) suggests antagonistic pleiotropy of mutations beneficial to chemostat growth. Indeed, the nature and high frequency of the mutations found in chemostat clones suggest that the motile–sessile transition pathway was under selection. Selection for wall growth formation in chemostats is well known (Hope et al. 2017), and may have resulted in a sweep of mutations linked to cell motility and attachment.
It should be noted that growth in a single biofilm resulted in more evolutionary solutions to plasmid stabilization than observed in our previous experimental evolution studies done in well-mixed serial batch cultures with a similar strain and plasmid (Sota et al. 2010; Hughes et al. 2012; Stalder et al. 2017). In those studies, mutations in the plasmid replication initiation protein TrfA1 were repeatedly observed, and insertion of the transposon Tn6374 into pBP136Km was seen in only one of six clones (Stalder et al. 2017) and never in its mini-replicon pMS0506 (Sota et al. 2010; Hughes et al. 2012). Here, transposition of Tn6374 was the only plasmid mutation observed in clones evolved in biofilms. Genome rearrangement and transposable elements have been suggested to play a critical role in the evolution of the S. oneidensis MR-1 genome (Romine et al. 2008), and more generally, in bacterial and plasmid evolution (Cohen 1976). By carrying out experimental evolution in biofilms instead of mixed liquids, we may thus have a better chance at observing the natural evolutionary trajectories of plasmid–host dyads.
This study illustrates how evolutionary dynamics in bacterial biofilms differ from those in well-mixed cultures. Given that 40–80% of cells on our planet live in biofilms or other forms of spatially structured communities (Flemming and Wuertz 2019), and that most of infections are caused by biofilms (Costerton et al. 1999; Hall-Stoodley et al. 2004; Lebeaux et al. 2014), it suggests that future studies on the evolution of clinically relevant antibiotic-resistance plasmids should include biofilms. Specifically, our findings show that growth in biofilms can result in the evolution of a small proportion of bacteria with more persistent self-transmissible antibiotic-resistance plasmids than expected from studies with well-mixed liquids. As many such resistance plasmids are ubiquitous in the clinic today (Mathers et al. 2015), this higher plasmid persistence could accelerate the spread of resistance among pathogens, which is of great medical concern.
Materials and Methods
Bacteria and Plasmid
The plasmid host in this experimental evolution study was S. oneidensis MR-1 ATCC 700550. The plasmid used was pBP136Gm, which was derived from pBP136, a cryptic IncP-1β plasmid isolated from the human pathogen Bordetella pertussis (Kamachi et al. 2006). The plasmid pBP136Gm was constructed by replacing the kanamycin-resistance gene cassette in pBP136Km (Kamachi et al. 2006) with the gentamycin (Gm)-resistance cassette using the lambda-Red recombinase expressed from pKD46 (Datsenko and Wanner 2000), and the PCR-amplified Gm-resistance gene (aacC1) cassette from pUC18-mini-Tn7T-LAC (Choi et al. 2005) using the primers set TraNup50aacC1 (5′-CAGGCCCGCATAAAAACGAAGCCCGGCGGTCGCCGGGCTTTTTTCTAGACCTTTGTCAACAGCAATGGATC-3′) and TraMdn50barA (5′-CAGCCCCCCCTCGGCGGGCCTCCCTCGCCAGAAATGGCGATGCTCTAGAAGTTCTGCTTTGCCTTCTCCAGCTTCT-3′). Briefly, the PCR-amplified Gm-resistance gene cassette containing fragments homologous to the flanking regions of the kanamycin-resistance gene cassette in pBP136Km was transformed into Escherichia coli EC100 containing the plasmids pBP136Km and pKD46 grown in 0.2% arabinose at 30 °C. Recombinant cells were selected on Gm and cured from pKD46 by growing the recombinant cells at 37 °C. Correct insertion of the Gm cassette was confirmed by PCR and Sanger sequencing. The ancestral plasmid–host dyad was constructed by electroporation of pBP136Gm into S. oneidensis MR-1, which had been preadapted for ∼100 generations in the culture medium (see below).
Culture Media and Conditions
All evolution and plasmid persistence assays were carried out in lactate mineral medium (Paulick et al. 2009) supplemented with 0.015 M sodium fumarate (LMF). Sodium fumarate was added as an alternative electron acceptor because oxygen can be limiting in our system. Experimental evolution was performed in LMF supplemented with 10 mg/l of Gm selecting for the retention of the plasmid pBP136Gm, and the plasmid persistence assays were done in the same medium without Gm. Dilution plating to obtain individual clones was done using LMF agar. All cultures were incubated at 30 °C.
Experimental Evolution
Evolution experiments were performed using an ancestral population that contained as little genetic diversity as possible. This was done by using an extinction-dilution procedure of the ancestral population (Ridenhour et al. 2017), wherein the highest dilution that showed growth was archived in glycerol stocks and used as inoculum for evolution experiments.
Shewanella oneidensis MR-1 (pBP136Gm) was evolved in parallel in four biofilms and four chemostat cultures for 28 days. We initiated the experiment by adjusting the optical density (OD600) of the ancestral culture to 0.01 using a spectrophotometer (BioMate 3, Thermo Fisher Scientific), and inoculated the flow cells and the chemostats with 12 ml of this cell suspension. The description of the setup of the flow cells is detailed in Ponciano et al. (2009) except that the flow cells were made of polycarbonate plastic and the 12-ml chambers were sealed in-between two slides using silicone adhesive. The chemostats were made of 21-ml glass tubes. After 4 h the flow of fresh medium was initiated (6 ml/h) and the chemostat culture volume was maintained at 12 ml. The flow cells, chemostats, media, and waste bottles were kept at 30 °C in an incubator, and media and waste bottles were changed approximately every 7 days. Flow cells and chemostats were checked daily for overgrowth into the tubing supplying the media and for leaks. Tubing and filters with substantial overgrowth were replaced as needed. To minimize biofilm growth on the chemostat walls, magnetic stir bars were used to keep the cells in suspension as much as possible. However, after 20 days, wall growth resulted in a thin visible biofilm layer on the chemostat walls. To avoid excessive wall growth, the chemostat populations were harvested on day 20, and cells were resuspended and transferred to new chemostats. This was repeated on day 26 as new wall growth became visible. The chemostat populations were grown for a total of 485 generations (28 days).
After 28 days, the biofilms were harvested by breaking the seal between the lid and body of a flow cell using a sterile scalpel blade and the cover was set aside. The cell mass was suspended in the overlying media and mixed by repeatedly pipetting up and down. The resulting suspensions were then transferred to 50-ml centrifuge tubes and vortexed for 1 min to disperse the cells. A comparable procedure was done for chemostat cultures (both planktonic cells and cells growing on the walls were harvested). Aliquots of these cell suspensions were used to determine total bacterial counts by spreading dilution series on LMF and LMF-Gm agar. In addition, 0.5- and 1-ml aliquots of biofilms and chemostats cultures, respectively, were harvested by centrifugation (10 min, 10,000×g). Cell pellets were stored at −20 °C for total DNA extraction. Another aliquot was archived in glycerol stocks at −70 °C.
Plasmid Persistence Assays
Individual clones were isolated from the glycerol stocks generated at the end of the evolution experiment. Clones from biofilm B4 and chemostat C4 were not analyzed; only whole-population sequencing analyses were performed on these fourth replicates. The persistence of plasmids in clonal populations was quantified as follows. Tubes containing 5 ml of LMF (without antibiotics) were inoculated with 4.9 µl of precultures which were grown overnight in LMF-Gm from archived clones (representing day 0 of the plasmid persistence assays). For the next 12 days, these cultures were serially transferred daily to fresh media without antibiotics selecting for the plasmid (4.9 µl into 5 ml, resulting in about ten doublings per day). The final overnight culture was serially diluted and aliquots were spread onto LMF agar plates. After overnight incubation, 52 single colonies were randomly selected and replica-plated onto LMF and LMF-Gm agar. The ratio of the number of colonies grown on LMF-Gm and LMF on day 12 was calculated and represents the fraction of plasmid-bearing bacteria in the population at the end of the serial transfer series.
Conjugative Plasmid Transfer Assays
Transmissibility of the plasmid by conjugation from the plasmid donors (D) to a plasmid-free recipient (R), resulting in transconjugants (T), was determined in filter matings. To distinguish R from D, R was a streptomycin (Sm)-resistant mutant of MR-1. Briefly, both D and R were grown from glycerol stocks in LB Miller broth using appropriate antibiotics for the selection of the plasmid-containing (Gm, 10 mg/l) and plasmid-free bacteria (Sm, 50 mg/l). After 24 h incubation, 1 ml of bacterial cultures were centrifuged (3 min, 8,000×g) and the pellets were rinsed by suspending in the same volume of phosphate-buffered saline pH 7.4 (PBS). Bacteria were harvested a second time by centrifugation and suspended in 0.1 volume of PBS, and 50 µl of D and R were mixed, harvested by centrifugation, and suspended in 50 µl of PBS. The mixture, as well as the same volumes of D and R separately, was placed on a 0.45-µm HTBP filter on a LB Miller agar plate, left to dry, and incubated. The filters were collected after 24 h, and for each the entire cell biomass was suspended in 2 ml of PBS. The population densities (CFU ml−1) of T, R, and D were determined by plate counting using selective LB Miller agar supplemented with the appropriate antibiotics.
Estimation of Plasmid Cost
Competition experiments were performed to estimate the plasmid cost. Each strain to be competed was inoculated from its glycerol stock into test tubes with 5 ml of LMF broth and grown for 16 h. Culture media were supplemented with Gm when appropriate. Cultures (1.5 ml) were harvested by centrifugation (10,000×g, 3 min) and the pellets were suspended in 1.5 ml of sterile PBS. Equal volumes of each competitor (100 μl) were mixed, and 4.9 µl was used to inoculate 5 ml of LMF broth. After 24 h of growth, 4.9 µl was transferred once again to 5 ml of LMF broth and incubated for 24 h. The number of CFU of the initial and final mixtures was determined by spread plating onto LMF agar. The fractions of plasmid-containing and -free bacteria were determined by replica plating 52 randomly selected colonies on LMF and LMF-Gm agar. The relative fitness (W) of the plasmid-containing relative to the plasmid-free bacteria was calculated according to (Lenski et al. 1991): Wp+/p− = ln(P+t2/P+t0)/ln(P−t2/P−t0), where P+t0 and P−t0 are the numbers of plasmid-containing and -free bacteria at the start of the 2-day assay, and P+t2 and P−t2 the final numbers. A plasmid cost of zero should result in a Wp+/p− of 1.
DNA Extractions and Sequence Analysis
For the whole-population sequencing approach, total genomic DNA was extracted from the biofilm and chemostat populations using the Power Biofilm DNA isolation kit (MoBio) on previously frozen cell pellets that were thawed on ice. The protocol was followed according to manufacturer’s instructions, except that during the lysis step the samples were vortexed by pulsing for 1 min instead of using the horizontal vortex adapter. For the genomic analyses of individual clones, total genomic DNA was extracted from 2 ml of an overnight culture using the GenEluteTM Bacterial Genomic DNA kit (Sigma-Aldrich, St. Louis, MO). The quality and the integrity of the gDNA were assessed on a 1% agarose gel and the concentrations were determined fluorometrically using Quant-iT PicoGreen dsDNA Reagent (ThermoFisher Scientific, Waltham, MA) with the TBS-380 Mini-Fluorometer (Turner BioSystems) (Molecular Devices, Sunnyvale, CA).
Full-genome sequencing was performed by the IBEST Genomics Resources Core using an Illumina MiSeq platform (Illumina, San Diego, CA) and 300-bp Paired-End Sample Preparation Kits (Illumina, San Diego, CA). Prior to analysis, the reads were preprocessed through the read-cleaning pipeline HTStream (https://github.com/ibest/HTStream; last accessed February 11, 2020) consisting of the following steps : 1) duplicate read pairs (possibly resulting from multicycle PCR reactions carried out as part of library preparation) were removed using the command super-deduper; 2) sequences were cleaned to remove sequencing adapters using the command adapter-trimmer; and 3) sequences were cleaned from low-quality bases using command q-window-trim, which is using a sliding window approach to remove low-quality ends of the reads.
Identification of the mutations was performed with the open source computational pipeline breseq 0.31.1 (http://barricklab.org/breseq; last accessed February 11, 2020) (Deatherage and Barrick 2014). For the whole-population sequencing analysis, the polymorphism mode (option -p) was turned on to the command and the flag -l was used to obtain coverage close to ∼100×. For each sample, the cleaned reads were mapped to the reference sequences of each replicon: 1) pBP136Gm, obtained in silico by introduction of the Gm cassette into the original pBP136 sequence (NC_008459), 2) the S. oneidensis MR-1 chromosome (NC_004347.2) and 3), the two putative native plasmids, here designated pSMR-1 and pLMR-1 (Stalder et al. 2017). In the whole-population sequencing approach, both the ancestral and the evolved populations were compared with the reference sequence. All SNPs and indels that were identified in genes present in both the ancestral and the evolved populations were removed, thus removing either mutations in the ancestral population relative to the reference sequence as well as possible standing variation in the ancestral population. For the clone analyses, mutations found in the sequenced ancestor were also subtracted, and marginal mutations were screened for consideration (mutations are “marginal” when evidence fails the established cutoffs, but there is still some support). In both analyses, all mutations resulting from the resolution of an N to a base were removed, and rearrangements not resolved by breseq (nonassigned junction evidence) were manually screened for the presence of mobile genetic elements (transposase, transposon, phages) integrated at a specific location. Since such structural variations can be difficult to detect in short-read sequencing data, future studies including long-read sequencing will probably give a better overview of the role of structural variation in bacterial genome evolution.
Statistics
Statistical tools were applied to the data set using R version 3.4.2. Pairwise comparisons were carried out using both the t-test and the nonparametric Mann–Whitney test, and when comparing more than two samples, we used the function aov() to fit an analysis of variance model. In cases where we combined the results from clones isolated from three distinct population (i.e., results in fig. 1), we nested the random factor “population” into the fixed factor “environment” (i.e., biofilm, chemostat, or ancestor), and considered the ancestor as one population. For all the models, we ran a post hoc analysis with Tukey’s test (all pairwise comparisons) or Dunnett’s test (when the means were compared with a control group), using the function glht() of the multcomp package. The P values are reported as *<0.1, **<0.05, and ***<0.01.
For the unbalanced design of the data in figure 1, we confirmed the analysis using a linear mixed-effect model approach using the nlme package. Using such a model, we nested the random factor “population” into the fixed factor “environment” and analyzed the variance tables for the model. To correct for the unbalanced design, we tested for a difference in the weighted marginal means (instead of the grand mean) using the option “type = ‘marginal’” in the function anova() or “type=‘c(‘III’)’” in the function Anova(). The corrected and uncorrected analyses of variance had almost same results (probably because the imbalance had a minor effect), and suggested that the means of the plasmid persistence were different between environments. We then performed a post hoc analysis on the model as described earlier.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation BEACON Center for the Study of Evolution in Action (Cooperative Agreement Number DBI-0939454), the National Institute of Allergy and Infectious Diseases (Grant No. R01 AI084918) of the National Institutes of Health (NIH), and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the NIH (Grant No. P20GM103408). The genome sequencing was carried out by the IBEST Genomic Resources Core, and made possible thanks to NIH NIGMS (Award No. P30 GM103324). We thank Ben Ridenhour for his advice on statistical analyses, Michael France for his advice on the chemostat and biofilm setup, and James Soderling and Alexandra Gal for technical assistance.
Author Contributions
E.M.T., L.J.F., and B.Kerr conceived the project. T.S. and E.M.T. wrote the article and L.J.F. and B.Kerr provided thorough revisions. T.S. led the project and did the genomic analyses. T.S. and B.K. ran the evolution experiment. B.C. performed the plasmid persistence assays and growth kinetics. J.L. performed additional persistence assays. S.D. significantly contributed to the competition and conjugation assays. H.Y. constructed pBP136Gm.
All sequencing data pertaining to this project have been made available at the National Center for Biotechnology Information (SRA accession number PRJNA531499). Other raw data are available upon request.
References
- Boles BR, Thoendel M, Singh PK.. 2004. Self-generated diversity produces “insurance effects” in biofilm communities. Proc Natl Acad Sci U S A. 101(47):16630–16635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. 2017. Notes from the field: pan-resistant New Delhi metallo-beta-lactamase-producing Klebsiella pneumoniae—Washoe County, Nevada, 2016. MMWR Morb Mortal Wkly Rep. 66(1):33. [DOI] [PMC free article] [PubMed]
- Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP.. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2(6):443–448. [DOI] [PubMed] [Google Scholar]
- Cohen SN. 1976. Transposable genetic elements and plasmid evolution. Nature 263(5580):731–738. [DOI] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP.. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284(5418):1318–1322. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL.. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 97(12):6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gelder L, Ponciano JM, Joyce P, Top EM.. 2007. Stability of a promiscuous plasmid in different hosts: no guarantee for a long-term relationship. Microbiology 153(2):452–463. [DOI] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE.. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 1151:165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlan RM. 2012. Biofilms: microbial life on surfaces. Emerg Infect Dis J. 8(9): 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman JM, Harmon LJ, LA H-J, Joyce P, Forney LJ.. 2011. The onion model, a simple neutral model for the evolution of diversity in bacterial biofilms. J Evol Biol. 24(11):2496–2504. [DOI] [PubMed] [Google Scholar]
- European Centre for Disease Prevention and Control. Last-line antibiotics are failing: options to address this urgent threat topatients and healthcare systems. Stockholm: ECDC; 2016. Available from: http://antybiotyki.edu.pl/edwa/pdf/AMR_Policy%20Briefing-final.pdf; last accessed February 11, 2020.
- Flemming H-C, Wuertz S.. 2019. Bacteria and archaea on Earth and their abundance in biofilms. Nat Rev Microbiol. 17(4):247–260. [DOI] [PubMed] [Google Scholar]
- France MT, Cornea A, Kehlet‐Delgado H, Forney LJ.. 2019. Spatial structure facilitates the accumulation and persistence of antibiotic resistant mutants in biofilms. Evol Appl. 12(3):498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- France MT, Forney LJ. 2019. The relationship between spatial structure and the maintenance of diversity in microbial populations. Am Nat. 193(4):503–513. [DOI] [PubMed] [Google Scholar]
- France MT, Ridenhour BJ, Forney LJ.. 2018. Effects of spatial structure and reduced growth rates on evolution in bacterial populations In: Rampelotto PH, editor. Molecular mechanisms of microbial evolution. Grand challenges in biology and biotechnology. Cham (Switzerland: ): Springer International Publishing; p. 175–197. [Google Scholar]
- Frost LS, Leplae R, Summers AO, Toussaint A.. 2005. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 3(9):722–732. [DOI] [PubMed] [Google Scholar]
- Ghigo JM. 2001. Natural conjugative plasmids induce bacterial biofilm development. Nature 412(6845):442–445. [DOI] [PubMed] [Google Scholar]
- Habets M, Rozen DE, Hoekstra RF, de Visser J.. 2006. The effect of population structure on the adaptive radiation of microbial populations evolving in spatially structured environments. Ecol Lett. 9(9):1041–1048. [DOI] [PubMed] [Google Scholar]
- Hallatschek O, Hersen P, Ramanathan S, Nelson DR.. 2007. Genetic drift at expanding frontiers promotes gene segregation. Proc Natl Acad Sci U S A. 104(50):19926–19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P.. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2(2):95–108. [DOI] [PubMed] [Google Scholar]
- Harrison E, Guymer D, Spiers AJ, Paterson S, Brockhurst MA.. 2015. Parallel compensatory evolution stabilizes plasmids across the parasitism-mutualism continuum. Curr Biol. 25(15):2034–2039. [DOI] [PubMed] [Google Scholar]
- Hope EA, Amorosi CJ, Miller AW, Dang K, Heil CS, Dunham MJ.. 2017. Experimental evolution reveals favored adaptive routes to cell aggregation in yeast. Genetics 206(2):1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JM, Lohman BK, Deckert GE, Nichols EP, Settles M, Abdo Z, Top EM.. 2012. The role of clonal interference in the evolutionary dynamics of plasmid-host adaptation. mBio 3(4):e00077–e00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamachi K, Sota M, Tamai Y, Nagata N, Konda T, Inoue T, Top EM, Arakawa Y.. 2006. Plasmid pBP136 from Bordetella pertussis represents an ancestral form of IncP-1beta plasmids without accessory mobile elements. Microbiology 152(12):3477–3484. [DOI] [PubMed] [Google Scholar]
- Kerr B, Neuhauser C, Bohannan BJM, Dean AM.. 2006. Local migration promotes competitive restraint in a host–pathogen “tragedy of the commons”. Nature 442(7098):75–78. [DOI] [PubMed] [Google Scholar]
- Kerr B, Riley MA, Feldman MW, Bohannan B.. 2002. Local dispersal promotes biodiversity in a real-life game of rock-paper-scissors. Nature 418(6894):171–174. [DOI] [PubMed] [Google Scholar]
- Korona R, Nakatsu CH, Forney LJ, Lenski RE.. 1994. Evidence for multiple adaptive peaks from populations of bacteria evolving in a structured habitat. Proc Natl Acad Sci U S A. 91(19):9037–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau PCY, Lindhout T, Beveridge TJ, Dutcher JR, Lam JS.. 2009. Differential lipopolysaccharide core capping leads to quantitative and correlated modifications of mechanical and structural properties in Pseudomonas aeruginosa biofilms. J Bacteriol. 191(21):6618–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaux D, Ghigo J-M, Beloin C.. 2014. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol Mol Biol Rev. 78(3):510–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE, Rose MR, Simpson SC, Tadler SC.. 1991. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am Nat. 138(6):1315–1341. [Google Scholar]
- Lewandowski Z, Beyenal H.. 2014. Fundamentals of biofilm research. 2nd ed Boca Raton (FL: ): CRC Press, Taylor & Francis Group. [Google Scholar]
- Lewis K. 2001. Riddle of biofilm resistance. Antimicrob Agents Chemother. 45(4):999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-Y, Wang Y, Walsh TR, Yi L-X, Zhang R, Spencer J, Doi Y, Tian G, Dong B, Huang X, et al. 2016. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 16(2):161–168. [DOI] [PubMed] [Google Scholar]
- Loftie-Eaton W, Bashford K, Quinn H, Dong K, Millstein J, Hunter S, Thomason MK, Merrikh H, Ponciano JM, Top EM.. 2017. Compensatory mutations improve general permissiveness to antibiotic resistance plasmids. Nat Ecol Evol. 1(9):1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftie-Eaton W, Yano H, Burleigh S, Simmons RS, Hughes JM, Rogers LM, Hunter SS, Settles ML, Forney LJ, Ponciano JM, et al. 2016. Evolutionary paths that expand plasmid host-range: implications for spread of antibiotic resistance. Mol Biol Evol. 33(4):885–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen JS, Burmølle M, Hansen LH, Sørensen SJ.. 2012. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Microbiol. 65(2):183–195. [DOI] [PubMed] [Google Scholar]
- Martin M, Hölscher T, Dragoš A, Cooper VS, Kovács ÁT.. 2016. Laboratory evolution of microbial interactions in bacterial biofilms. J Bacteriol. 198(19):2564–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers AJ, Peirano G, Pitout J.. 2015. The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clin Microbiol Rev. 28(3):565–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollister B, Kotter CV, Frank DN, Washburn T, Jobling MG.. 2016. Whole-genome sequencing identifies in vivo acquisition of a blaCTX-M-27-carrying IncFII transmissible plasmid as the cause of ceftriaxone treatment failure for an invasive Salmonella enterica serovar typhimurium infection. Antimicrob Agents Chemother. 60(12):7224–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann P, Snesrud E, Maybank R, Corey B, Ong AC, Clifford R, Hinkle M, Whitman T, Lesho E, Schaecher KE.. 2016. Escherichia coli harboring mcr-1 and blaCTX-M on a novel IncF plasmid: first report of mcr-1 in the United States. Antimicrob Agents Chemother. 60(7):4420–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahum JR, Godfrey-Smith P, Harding BN, Marcus JH, Carlson-Stevermer J, Kerr B.. 2015. A tortoise-hare pattern seen in adapting structured and unstructured populations suggests a rugged fitness landscape in bacteria. Proc Natl Acad Sci U S A. 112(24):7530–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman A, Hansen LH, Sørensen SJ.. 2009. Conjugative plasmids: vessels of the communal gene pool. Philos Trans R Soc B. 364(1527):2275–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA.. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405(6784):299–304. [DOI] [PubMed] [Google Scholar]
- Paulick A, Koerdt A, Lassak J, Huntley S, Wilms I, Narberhaus F, Thormann KM.. 2009. Two different stator systems drive a single polar flagellum in Shewanella oneidensis MR-1. Mol Microbiol. 71(4):836–850. [DOI] [PubMed] [Google Scholar]
- Perfeito L, Pereira MI, Campos PRA, Gordo I.. 2008. The effect of spatial structure on adaptation in Escherichia coli. Biol Lett. 4(1):57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponciano JM, La H-J, Joyce P, Forney LJ.. 2009. Evolution of diversity in spatially structured Escherichia coli populations. Appl Environ Microbiol. 75(19):6047–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porse A, Schønning K, Munck C, Sommer M.. 2016. Survival and evolution of a large multidrug resistance plasmid in new clinical bacterial hosts. Mol Biol Evol. 33(11):2860–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey PB, Travisano M.. 1998. Adaptive radiation in a heterogeneous environment. Nature 394(6688):69–72. [DOI] [PubMed] [Google Scholar]
- Ridenhour BJ, Metzger GA, France M, Gliniewicz K, Millstein J, Forney LJ, Top EM.. 2017. Persistence of antibiotic resistance plasmids in bacterial biofilms. Evol Appl. 10(6):640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine MF, Carlson TS, Norbeck AD, McCue LA, Lipton MS.. 2008. Identification of mobile elements and pseudogenes in the Shewanella oneidensis MR-1 genome. Appl Environ Microbiol. 74(10):3257–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder VJ, Chopra I, O’Neill AJ.. 2012. Increased mutability of staphylococci in biofilms as a consequence of oxidative stress. PLoS One 7(10):e47695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Millan A, Peña-Miller R, Toll-Riera M, Halbert ZV, McLean AR, Cooper BS, MacLean RC.. 2014. Positive selection and compensatory adaptation interact to stabilize non-transmissible plasmids. Nat Commun. 5:5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Lopez A, Marshall CW, Scribner MR, Snyder DJ, Cooper VS.. 2019. Evolutionary pathways to antibiotic resistance are dependent upon environmental structure and bacterial lifestyle. eLife 8:e47612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sota M, Yano H, Hughes JM, Daughdrill GW, Abdo Z, Forney LJ, Top EM.. 2010. Shifts in the host range of a promiscuous plasmid through parallel evolution of its replication initiation protein. ISME J. 4(12):1568–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder T, Rogers LM, Renfrow C, Yano H, Smith Z, Top EM.. 2017. Emerging patterns of plasmid-host coevolution that stabilize antibiotic resistance. Sci Rep. 7(1):4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenackers HP, Parijs I, Foster KR, Vanderleyden J.. 2016. Experimental evolution in biofilm populations. FEMS Microbiol Rev. 40(3):373–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thormann KM, Duttler S, Saville RM, Hyodo M, Shukla S, Hayakawa Y, Spormann AM.. 2006. Control of formation and cellular detachment from Shewanella oneidensis MR-1 biofilms by cyclic di-GMP. J Bacteriol. 188(7):2681–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse CC, Mayo-Smith LM, Poltak SR, Cooper VS.. 2013. Tangled bank of experimentally evolved Burkholderia biofilms reflects selection during chronic infections. Proc Natl Acad Sci U S A. 110(3):E250–E259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White House. 2015. Obama administration releases national action plan to combat antibiotic-resistant bacteria. whitehouse.gov [Internet]. Available from: https://www.whitehouse.gov/the-press-office/2015/03/27/fact-sheet-obama-administration-releases-national-action-plan-combat-ant. Last accessed February 11, 2020.
- Wiedenbeck J, Cohan FM.. 2011. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol Rev. 35(5):957–976. [DOI] [PubMed] [Google Scholar]
- World Health Organization. 2014. Antimicrobial resistance: global report on surveillance. Geneva (Switzerland: ): World Health Organization. [Google Scholar]
- Wright S. 1932. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc 6th Int Congr Genet. 1:356–366. [Google Scholar]
- Yan X-F, Xin L, Yen JT, Zeng Y, Jin S, Cheang QW, Fong R, Chiam K-H, Liang Z-X, Gao Y-G.. 2018. Structural analyses unravel the molecular mechanism of cyclic di-GMP regulation of bacterial chemotaxis via a PilZ adaptor protein. J Biol Chem. 293(1):100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Wegrzyn K, Loftie-Eaton W, Johnson J, Deckert GE, Rogers LM, Konieczny I, Top EM.. 2016. Evolved plasmid-host interactions reduce plasmid interference cost. Mol Microbiol. 101(5):743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.