

Abstract
Acetaminophen (APAP) overdose-induced acute liver failure is an important clinical problem in the United States and the current antidote N-acetylcysteine, has a short early therapeutic window. Since most patients present late to the clinic, there is need for novel late-acting therapeutic options. Though the neuronal guidance cue netrin-1, has been shown to promote hepatic repair and regeneration during liver ischemia/reperfusion injury, its effect in APAP-induced hepatotoxicity is unknown. In the quest for a late-acting therapeutic intervention in APAP-induced liver injury, we examined the role of netrin-1 in a mouse model of APAP overdose. Male C57BL/6J mice were cotreated with exogenous netrin-1 or vehicle control, along with 300 mg/kg APAP and euthanized at 6, 12, and 24 h. Significant elevations in alanine aminotransferase indicative of liver injury were seen in control mice at 6 h and this was not affected by netrin-1 administration. Also, netrin-1 treatment did not influence mitochondrial translocation of phospho-JNK, or peroxynitrite formation indicating that there was no interference with APAP-induced injury processes. Interestingly however, netrin-1 administration attenuated liver injury at 24 h, as seen by alanine aminotransferase levels and histology, at which time significant elevations in the netrin-1 receptor, adenosine A2B receptor (A2BAR) as well as macrophage infiltration was evident. Removal of resident macrophages with clodronate liposomes or treatment with the A2BAR antagonist PSB1115 blocked the protective effects of netrin-1. Thus, our data indicate a previously unrecognized role for netrin-1 in attenuation of APAP hepatotoxicity by enhancing recovery and regeneration, which is mediated through the A2BAR and involves resident liver macrophages.
Keywords: acetaminophen, netrin-1, adenosine A2B, Kupffer cell, macrophage
Acetaminophen (APAP) is a widely used analgesic and antipyretic drug, which is safe at therapeutic doses, but causes severe hepatotoxicity, acute liver failure, and potentially death when taken as an overdose. The hepatotoxicity of APAP is initiated by formation of a reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI) during metabolism of the drug (Nelson, 1990). Although NAPQI can be detoxified by glutathione (GSH) when APAP is consumed at therapeutic doses, an overdose of APAP results in an excess of NAPQI, which depletes GSH and binds to cellular proteins (Cohen et al., 1997). Binding of NAPQI to mitochondrial proteins plays a critical role in the initiation of the injury (Qiu et al., 1998; Tirmenstein and Nelson, 1989; Xie et al., 2015), resulting in mitochondrial oxidant stress, c-Jun N-terminal kinase (JNK) activation in the cytosol and its translocation to the mitochondria. Mitochondrial JNK translocation ultimately activates the mitochondrial permeability transition pore opening, release of mitochondrial proteins to the cytosol, nuclear DNA fragmentation and hepatocyte necrosis (Bajt et al., 2006, 2008; Hanawa et al., 2008; Jaeschke and Bajt, 2006; Kon et al., 2004). Release of damage-associated molecular patterns (DAMPs) by dying hepatocytes stimulates an innate immune response with infiltration of neutrophils and macrophages into the liver, which facilitate removal of dead cells and induce a regenerative response to repair liver function (Jaeschke et al. 2012b; You et al., 2013).
The current treatment for APAP-induced liver injury in the clinic is infusion of N-acetylcysteine (NAC); however, in patients who deteriorate after developing acute liver failure, an orthotopic liver transplantation (OLT) is the only intervention available. Though NAC is the only clinically approved antidote for APAP hepatotoxicity, its therapeutic effectiveness is optimal when given within 8 h after APAP (Smilkstein et al., 1988). Unfortunately, most APAP-overdose patients present to the clinic relatively late, ie, during or at the peak of injury (Singer et al., 1995), missing the optimal therapeutic window for NAC. Limited efficacy of late NAC treatment increases the risk of developing acute liver failure, with OLT as the mainstay treatment with its associated with high cost and shortage of donor organs. Therefore, new therapeutic strategies are needed to treat patients with APAP overdose, especially those targeting the later phases of APAP hepatotoxicity (Jaeschke et al., 2020).
Netrin‐1, is a chemokine expressed in many tissues including the brain, lung, heart, liver, intestine, and kidney. It was shown to play a critical role in angiogenesis, cell migration, tissue morphogenesis, tumor progression, and regulation of inflammation (Fitamant et al., 2008; Hebrok and Reichardt, 2004; Nguyen and Cai, 2006; Rosenberger et al., 2009). Netrin-1 has been described as a survival factor, and showed protective effects in bone disease, acute kidney injury, acute lung injury, cardiac ischemia/reperfusion injury, and cancer (Bai et al., 2016; Maruyama et al., 2016; Mutz et al., 2010; Paradisi et al., 2013). A recent study in hepatic ischemia-reperfusion demonstrated a beneficial effect of netrin-1 against liver injury when administered 3 h after reperfusion, with netrin-1 modulating the inflammatory response and promoting liver regeneration (Schlegel et al., 2016). Since the APAP-induced innate immune response is a late event which facilitates repair and regeneration after APAP-induced hepatocyte necrosis, the efficacy of netrin-1 in protecting against APAP-induced liver injury was investigated. Our data indicate that netrin-1 provides a delayed protection against APAP hepatotoxicity, which is mediated through the adenosine A2B receptor (A2BAR), identifying a novel therapeutic target, which could provide significant benefits for late-presenting APAP-overdose patients.
MATERIALS AND METHODS
Animals
Eight-week-old male C57BL/6J mice with an average weight of 20–25 g were purchased from Jackson Laboratories (Stock No. 000664; Jackson Laboratories, Bar Harbor, Maine). All animals were kept in an environmentally controlled room with a 12-h light/dark cycle and free access to food and water. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center and followed the criteria of the National Research Council for the care and use of laboratory animals.
Experimental design
After an overnight fast, mice were i.p. injected with 300 mg/kg APAP (Sigma-Aldrich, St. Louis, Missouri) dissolved in warm saline. Just prior to APAP treatment, some mice were also treated with mouse recombinant netrin-1 (R&D Systems, Minneapolis, Minnesota) (1 µg/mouse) dissolved in 150 µl saline (with 0.2% BSA) i.v. as a cotreatment. All vehicle control mice received the same volume of saline (20 ml/kg, i.p. or 150 µl i.v.). The dose of 1 μg/mouse for netrin-1 was chosen based on earlier literature where this dose of netrin-1 was used for studies in the animal models of liver ischemia-reperfusion injury (Schlegel et al., 2016), inflammatory peritonitis (Mirakaj et al., 2011), and pulmonary inflammation (Mirakaj et al., 2010). The intravenous route was chosen for the administration of netrin-1 since it has been used earlier while evaluating effect of netrin-1 on liver pathophysiology (Schlegel et al., 2016) and to recapitulate systemic exposure. Mice were euthanized at 6, 12, or 24 h after APAP injection. Animals were refed in all experiments that lasted > 6 h. For experiments with clodronate liposomes, mice were injected with 100 µl clodronate liposomes or control liposomes (i.p.) (FormuMax Scientific, Sunnyvale, California). At 72 h after administration of liposomes, animals were treated with 300 mg/kg APAP (i.p.) dissolved in warm saline after overnight fast. Some mice were cotreated with mouse recombinant netrin-1 (i.v.) as described earlier and all mice were euthanized at 24 h after APAP. In some experiments, mice were cotreated with the A2BAR antagonist PSB1115 (Santa Cruz Biotechnology) (5 mg/kg, i.p.) dissolved in saline, followed by a second treatment with PSB1115 6 h after APAP, and euthanized at 24 h after APAP. PSB1115 is a widely used adenosine A2B antagonist, with doses up to 10 mg/kg administered i.p. (Eckle et al., 2008). Heparinized blood was collected and centrifuged at 14 000 × g for 3 min to collect plasma. Liver was resected and aliquots either snap frozen in liquid nitrogen, fixed in 4% paraformaldehyde for histology or embedded in OCT medium for cryosectioning.
Biochemical measurements
Plasma alanine aminotransferase (ALT) activities were measured via the Point Scientific ALT test (Point Scientific, Inc, Canton, Michigan) per the manufacturer’s instruction. Mouse plasma adenosine levels were measured using an adenosine assay kit (No. MET-5090, Cell Biolabs, Inc) according to the manufacturer’s instructions.
Histology and immunostaining
Tissue was fixed and embedded in paraffin. Hematoxylin and eosin (H&E) staining was performed for evaluation of the area of necrosis as described (Gujral et al., 2003). TUNEL staining was performed for evaluation of nuclear DNA damage per the manufacturer’s instructions (Roche Diagnostics, Basel, Switzerland). Nitrotyrosine staining was assessed by immunohistochemistry using an antinitrotyrosine antibody (Molecular Probes, Eugene, Oregon) with the DAKO LSAB peroxidase kit (K684; DAKO, Carpinteria, California). Neutrophil staining was carried out using naphthol AS-D chloroacetate esterase technique (Sigma), and the number of neutrophils present in sinusoidal spaces and extravasated into hepatic parenchyma was counted. Quantification was performed by randomly selecting 10 high power fields (×400) and counting positively stained cells consistent with neutrophil morphology. Staining for F4/80-positive macrophages was carried out using an F4/80 antibody from Cell signaling (F4/80 D2S9R XP Rabbit mAb No. 70076), while PCNA staining was performed by using an anti-PCNA antibody from Santa Cruz Biotechnology, (sc-7907). Immunofluorescence staining was performed using 6 µm cryosections made from OCT-embedded tissue. Sections were stained with augmenter of liver regeneration (ALR) (sc-365885, Santa Cruz Biotechnology, Dallas, Texas) or hepatocyte growth factor (HGF) (sc-374422, Santa Cruz Biotechnology) using the Mouse On Mouse fluorescent kit (FMK 2201, Vector Laboratories) according to the manufacturer’s instructions.
Western blotting
Snap frozen tissue was homogenized in a CHAPs containing protein buffer and total protein was measured using the BCA assay (Pierce Scientific, Waltham, Massachusetts). Gel electrophoresis was carried out on protein lysates from individual samples, which were then transferred to a nitrocellulose blot. Densitometry was performed to quantitatively assess differences using ImageJ software. In brief, densitometry was performed serially on blots and normalized to either the nonphosphorylated form (JNK) or the loading control, either porin or beta-actin. Antibodies to total JNK (catalog No. 9252), phospho-JNK (No. 9251), and β-actin (No. 4970) were purchased from Cell Signaling Technologies (Danvers, Massachusetts). The antibody against PCNA (sc-7907) was obtained from Santa Cruz Biotechnology. The antibody against netrin-1 (No. AF 1109) was obtained from R&D Systems. Horseradish peroxidase-coupled anti-mouse or anti-rabbit IgG was used as the secondary antibody. Proteins were visualized by enhanced chemiluminescence (Amersham Biosciences, Inc, Piscataway, New Jersey).
Real-time PCR analysis to quantify mRNA expression levels
cDNA was generated by reverse transcription of total RNA by M-MLV reverse transcriptase in the presence of random primers (Invitrogen, Carlsbad, California). Forward and reverse primers for the genes were designed using Primer Express software (Applied Biosystems, Foster City, California). After normalization of cDNA concentrations, the following primers were used: uncoordinated receptor 5B (UNC5B), 5′-GCT AAG TCC TGC CAC ATC CT-3′ (sense), 5′GGA CCT CCT TCA GTG CTA CA-3′ (antisense); A2B adenosine receptor (Adora2b), 5′GCA TTA CAG ACC CCC ACC AA-3′ (sense), 5′TTT ATA CCT GAG CGG GAC GC-3′ (antisense); DCC netrin-1 receptor (DCC), 5′-CAC CTC CTC TCA ACC CAA AC-3′ (sense), 5′GGG GTC AGT GGG ATC TGT TA-3′ (antisense), SYBR green PCR Master Mix (Bio-Rad, Hercules, California) was used for real-time PCR analysis. The relative differences in expression between groups were expressed using cycle time (Ct) values generated by the CFX384 instrument (Bio-Rad). All genes evaluated were first normalized to GAPDH and then expressed as a fold increase relative to control, which was arbitrarily set as 1.0. Calculations are made by assuming 1 cycle is equivalent to a 2-fold difference in copy number which is the 2^(−ddCt) formula.
Statistics
Data are expressed as means ± SE. Comparison between 2 groups was performed with Student’s t test. Comparisons between multiple groups were performed using 1-way ANOVA followed by Student-Neman-Keuls post hoc test for multiple groups. p < .05 was considered significant.
RESULTS
Exogenous Netrin-1 Attenuated APAP-induced Liver Injury
To assess the role of netrin-1 on APAP hepatotoxicity, we co-treated animals with mouse recombinant netrin-1 (1 µg/mouse) and 300 mg/kg APAP, followed by sacrifice at 0, 6, 12, or 24 h. APAP caused severe liver injury as indicated by the increase in plasma ALT levels peaking around 12 h (Figure 1A), as well as extensive centrilobular necrosis (Figure 1B). Although cotreatment with netrin-1 had no effect on APAP-induced injury at 6 or 12 h, it attenuated the increase in ALT activity by 50% at 24 h (Figure 1A) and significantly decreased the area of necrosis at 24 h (Figure 1B). Measurement of circulating netrin-1 levels at the 24-h time point indicated lower levels with APAP, which were restored to control levels after exogenous administration (Figure 1C). Examination of DNA fragmentation by TUNEL staining demonstrated significant increase by 6 h, which was again not influenced by netrin-1 treatment, whereas significant protection was evident in netrin-1-treated animals against the sustained DNA fragmentation seen at 24 h after APAP (Figure 1D). This suggests that netrin-1 has no influence on the early injury process after APAP, but interestingly has a delayed effect, showing significant protection only at 24 h after a 300 mg/kg dose of APAP.
Figure 1.

Exogenous netrin-1 attenuated acetaminophen (APAP)-induced liver injury. Fasted C57BL/6J mice were treated with either APAP (300 mg/kg) alone, APAP+netrin-1 (1 µg/mouse), or saline as controls after overnight fasting. Blood and liver tissues were obtained at 6, 12, and 24-h post-APAP. A, Plasma alanine aminotransferase activity. B, Hematoxylin and eosin staining of representative liver sections (×50 magnification). C, Plasma levels of netrin-1, 24 h after APAP or APAP+netrin-1 compared with controls. D, DNA fragmentation measured by the TUNEL assay (×50 magnification). Data represent means ± SE of n = 3–10 animals per group. *p < .05 (compared with control). #p < .05 (compared with APAP 24 h group). $p < .05 (compared with APAP group).
Netrin-1 Does Not Influence the Early Injury Process After APAP Overdose
Activation and translocation of JNK to the mitochondria and subsequent mitochondrial oxidant stress and formation of peroxynitrite are well-characterized hallmarks of APAP-induced hepatocyte injury (Ramachandran and Jaeschke, 2019). To confirm whether netrin-1 administration influences these mechanisms of liver injury, activation and mitochondrial translocation of JNK was investigated. As seen in Figure 2A robust JNK activation and mitochondrial translocation was evident in APAP-treated mice at 6 h after APAP and this was not altered after netrin-1 treatment. Formation of peroxynitrite was investigated by staining for nitrotyrosine and as seen in Figure 2B, significant increase was evident with APAP at 6 h, which was not affected by netrin-1 cotreatment. These data reiterate that netrin-1 does not influence the early cell signaling involved in APAP-induced cellular necrosis but provides delayed protection through an independent mechanism. Early depletion of hepatic glutathione is a hallmark of APAP-induced liver injury and modulation of its recovery could influence injury. Netrin-1 administration did not influence glutathione recovery at the 6 h time point, but enhanced total glutathione levels by 24 h compared with APAP alone (Figure 2C). However, the changes at this late time point without alterations at the earlier 6 h time point likely reflect enhanced recovery, since no protection against injury at 6 h was evident.
Figure 2.

Effect of netrin-1 on early injury processes and hepatic glutathione (GSH) levels after an acetaminophen (APAP) overdose. Fasted C57BL/6J mice were cotreated with 300 mg/kg APAP and netrin-1 (1 µg/mouse) or saline as control. Liver tissues were obtained at 6‐h post‐APAP. A, Total JNK and P-JNK were measured in cytosolic and mitochondrial liver fractions. B, Nitrotyrosine staining of representative liver sections (×50 magnification). C, Total GSH measured at both 6 and 24 h. Mice were refed at 6 h. Data represent means ± SE of n = 4–10 animals per group. *p < .05 (compared with controls), #p < .05 (compared with APAP alone).
APAP Overdose Induces an Upregulation of the A2BAR
Netrin-1 has been shown to act mainly through 3 receptors—UNC5B, the A2BAR, and the deleted in colorectal cancer (DCC) receptor (Ranganathan et al., 2014). We next determined the gene expression profiles of UNC5B, A2BAR, and DCC within hepatic tissue. Although netrin-1 co-treatment induced higher expression of A2B receptor expression at 6 h compared with APAP alone, both groups showed substantial elevations in receptor expression at 24 h (Figure 3A). Expression of the UNC5B receptor, however, did not show significant alterations along the time course, whereas DCC expression was not detectable in the liver under these conditions (data not shown). The unique early upregulation of the A2B receptor at 6 h in netrin-1-treated mice thus indicates that this could be mediating the protective effects of netrin-1 seen later at 24 h. Staining of liver sections demonstrate that this upregulation at 24 h seems to be predominantly seen in cells at the border of the necrotic area in both groups of animals (Figure 3B), indicating that this probably facilitates recovery and regeneration.
Figure 3.

Modulation of netrin-1 receptor expression and neutrophil infiltration after acetaminophen (APAP) overdose by exogenous netrin-1. Fasted mice were cotreated with 300 mg/kg APAP and netrin-1 (1 µg/mouse) or saline as control. Liver tissues were obtained at 6 and 24-h post-APAP. A, Adenosine A2B receptor mRNA was quantified by RT-PCR. B, Adenosine A2B receptor immunohistochemistry staining of representative liver sections (×50 magnification). The panels on the right show enlarged areas from the corresponding images. C, Neutrophil staining of representative liver sections (×50 magnification) showing APAP 6 (a) and 24 h (b) as well as APAP+netrin-1, 6 (c) and 24 h (d). D, Quantification of hepatic neutrophil recruitment based on neutrophil staining. Data represent means ± SE of n = 5–10 animals per group. *p < .05 (compared with controls). #p < .05 (compared with 6‐h post‐APAP). $p < .05 (compared with APAP-treated group).
Netrin-1 Attenuates Neutrophil Infiltration While Enhancing Macrophage Recruitment After APAP Overdose
Netrin-1 has been shown to be an immune mediator (Ranganathan et al., 2014) and acts as a potent inhibitor of migration of leukocytes, which express the netrin-1 receptor UNC5B (Ly et al., 2005). A recent study also demonstrated that netrin-1 reduces chemokinetic motility of human neutrophils, which is accompanied with reduced cell polarization and spreading (Hipoilito et al., 2018). APAP overdose mainly induces neutrophil infiltration at later stages after injury (Ramachandran and Jaeschke, 2019) and the effect of netrin-1 on this was investigated. As seen in Figure 3C(a), whereas no appreciable neutrophil infiltration was seen at 6‐h post‐APAP, a significant elevation in neutrophil numbers was observed at 24 h (Figure 3Cb). Although netrin-1 did not influence neutrophils at 6 h (Figure 3Cc), it significantly blunted neutrophil infiltration seen at 24 h (Figs. 3Cb, 3Cd, and 3D), suggesting that it does have immune modulatory effects in the liver in the context of APAP-induced liver injury. To further examine the role of immune cells in this process, liver macrophages were evaluated by F4/80 immunohistochemical staining in liver sections. As seen in Figure 4A, treatment with netrin-1 resulted in a significant increase in macrophage levels around the necrotic area by 24 h when compared with animals treated with APAP alone. The data so far indicated that netrin-1 induced an upregulation of A2BAR, as well as enhanced macrophage infiltration. To identify the cell type where A2BAR was upregulated, sequential liver sections were stained with A2BAR as well as F4/80 to identify macrophages. As seen in Figure 4B, staining with each of the probes illuminated distinct populations of cells, with A2BAR upregulation exclusively in hepatocytes and not on macrophages.
Figure 4.

Effect of netrin-1 on acetaminophen (APAP)-induced macrophage infiltration and liver regeneration. Fasted C57BL/6J mice were cotreated with 300 mg/kg APAP and netrin-1 (1 µg/mouse) or saline as control. Liver tissue was obtained at 6 and 24-h post-APAP. A, Immunohistochemistry staining for F4/80-positive macrophages (×50 magnification). Panels on the extreme right indicate enlargements of indicated areas from 24 h sections. B, Serial sections from 24 h APAP+netrin-1-treated mice probed for adenosine A2B or F4/80 as a marker of macrophages by immunohistochemistry (×50 magnification). Panels on the right are enlarged from areas marked on the left. C, Immunostaining for PCNA in liver sections obtained 24-h post-APAP or APAP+netrin-1 (×50 magnification).
Netrin-1 Treatment Augmented Liver Regeneration Induced by APAP Overdose
The effect of netrin-1 on liver regeneration after APAP overdose was then examined by staining liver sections for the regeneration marker PCNA. As seen in Figure 4C, APAP induces PCNA in cells around the area of necrosis at 24 h (Bajt et al., 2003); however, netrin-1 cotreatment resulted in significant increase in PCNA-positive cells in livers 24 h after APAP, again mainly in cells surrounding the necrotic area, confirming its role in upregulating regeneration and recovery in these cells.
Recruitment of Liver Macrophages After APAP-induced Injury Is Critical for Netrin-1-mediated Protection
To confirm the critical role of macrophages in late netrin-1-mediated protection against APAP-induced liver injury, experiments were repeated in mice treated with clodronate liposomes to remove resident macrophages, followed by administration of APAP and netrin-1. As seen in Figures 5A and 5C, whereas netrin-1 administration was protective against liver injury measured by ALT release, treatment with clodronate by itself resulted in an aggravation of APAP-mediated injury as reported previously (Ju et al., 2002), illustrating the importance of macrophages in recovery after injury. Interestingly, the protective effect of netrin-1 against APAP-induced liver injury was significantly blunted in the absence of liver macrophages, indicating that these cells were critical for netrin-1-mediated protection. This was also evident in liver histology, where the decreased areas of necrosis seen in APAP+netrin-1-treated animals were reversed in animals pretreated with clodronate (Figs. 5B and 5C). Though it has been reported that treatment with clodronate liposomes can increase circulating levels of ALT by delaying clearance in the absence of hepatocellular injury (Radi et al., 2011), the increase in area of necrosis in netrin-1+clodronate-treated animals compared with APAP+netrin-1 suggests that clodronate treatment blocked the protective effect of netrin-1.
Figure 5.

Removal of Kupffer cells attenuated protection by netrin-1. C57BL/6J mice were pretreated with clodronate liposomes or control liposomes (100 µl/mouse ip) 72 h before an overnight fast and treatment with 300 mg/kg acetaminophen (APAP) i.p. with or without netrin-1 (1 µg/mouse) i.v. Liver tissues were obtained 24 h after APAP treatment. A, Plasma alanine aminotransferase activity. B, Hematoxylin and eosin staining of representative liver sections (×50 magnification) and C, quantitation of areas of necrosis. Data represent mean ± SE of n = 5–10 animals per group. *p < .05 (compared with APAP alone). #p < .05 (compared with APAP+netrin-1).
Netrin-1-mediated Recruitment of Liver Macrophages Is Important in Recovery and Regeneration After APAP-induced Liver Injury
To further evaluate the effect of macrophage depletion on netrin-1 mediated upregulation of liver regeneration, liver sections from animals treated with APAP+netrin-1 either with, or without clodronate treatment were compared. As seen in Figure 6, clodronate treatment significantly diminished netrin-1 mediated upregulation of PCNA staining in cells surrounding the necrotic area, indicating that netrin-1-mediated macrophage recruitment was important for liver recovery and regeneration after APAP. To determine signaling mediators involved in induction of liver regeneration and recovery, expression of the ALR and HGF were examined in liver sections. Further confirmation of the role of netrin-1 mediated upregulation of macrophage infiltration in facilitating recovery was evident in the staining, where netrin-1 treatment upregulated both ALR and HGF staining in hepatocytes in the vicinity of the necrotic area, whereas clodronate treatment blunted this effect of netrin-1 (Figure 7).
Figure 6.
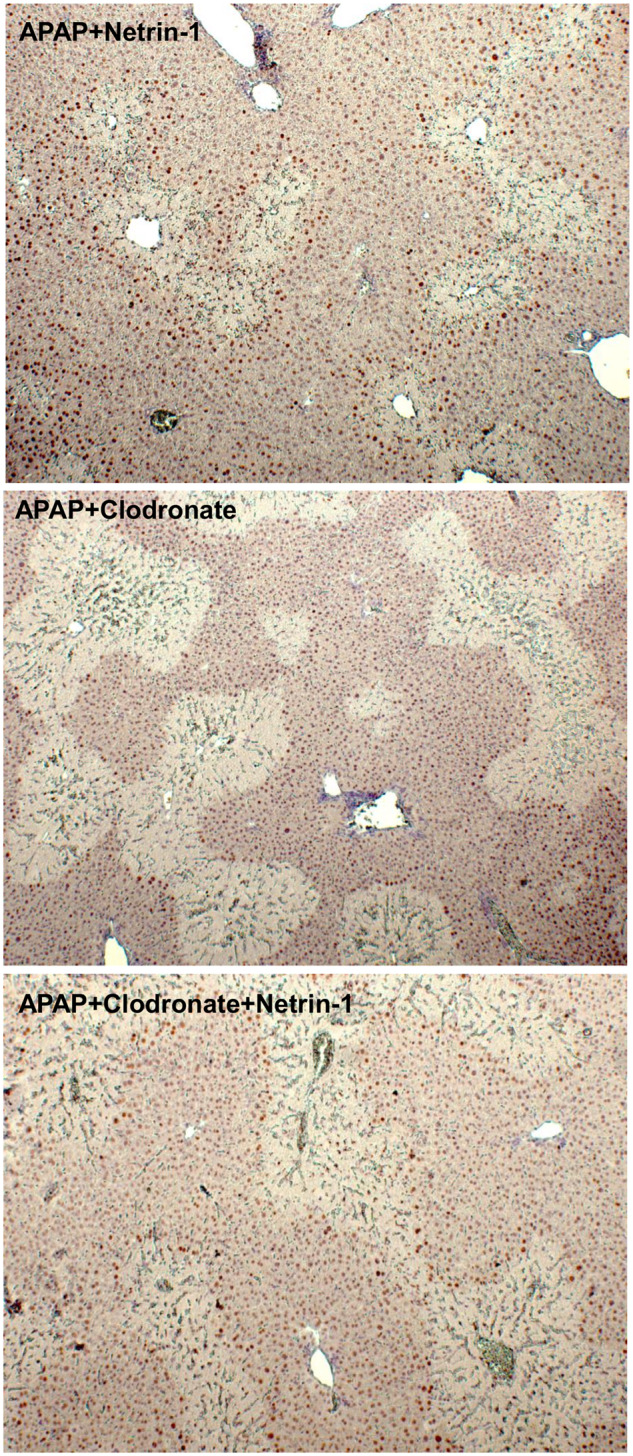
PCNA immunohistochemistry staining of liver sections (×50 magnification).
Figure 7.

Kupffer cell removal prevents netrin-1 induced increase in regenerative signals. C57BL/6J mice were treated with clodronate liposomes or control liposomes (100 µl/mouse ip) 72 h before an overnight fast and treatment with 300 mg/kg acetaminophen (APAP) i.p. with or without netrin-1 (1 µg/mouse) i.v. Liver tissues were obtained 24 h after APAP and sections probed for augmenter of liver regeneration (ALR) or hepatocyte growth factor (HGF) by immunofluorescence staining (×50 magnification) and inhibition by clodronate treatment quantified on the left.
Protective Effects of Netrin-1 Are Mediated Through the A2BAR
The data so far clearly showed that the delayed protection by netrin-1 against APAP hepatotoxicity was mediated by modulation of the innate immune response and enhancing infiltration of macrophages, which was essential for protection. But, how was netrin-1 influencing the immune response? The effect of netrin-1 administration on early elevation of the A2BAR indicates that this receptor could be playing a role, a notion supported by the fact that A2BAR has been shown to be involved in alternate macrophage activation to suppress inflammation and promote tissue restitution (Csoka et al., 2012). To investigate this, experiments were repeated with the A2BAR antagonist PSB1115. We hypothesized that if netrin-1 mediated its effects through the A2B receptor, blocking it would negate the protective effects of netrin-1. This seems to be the case, since treatment with PSB1115 removed the delayed protective effect of netrin-1 administration, as seen by the enhanced liver injury at 24 h as assessed by plasma ALT levels in animals treated with APAP along with netrin-1 and PSB1115 (Figure 8A). This was confirmed by histology (Figure 8B) and TUNEL staining, which also showed significant increase in DNA fragmentation in mice with PSB1115 when given along with netrin-1 and APAP (Figure 8C). Administration of PSB115 by itself to control mice did not cause any hepatotoxicity as assessed by liver histology and ALT levels (data not shown).
Figure 8.

The adenosine A2B receptor antagonist PSB1115 blocked protective effects of netrin-1. Fasted C57BL/6J mice were co-treated with 300 mg/kg acetaminophen (APAP) and netrin-1 (1 µg/mouse) or saline as control, with some mice also receiving PSB1115. Those mice received a second dose of PDB115 6 h after APAP, followed by sacrifice at 24 h and collection of blood and liver tissues. Plasma alanine aminotransferase activity (A) was measured and hematoxylin and eosin (B) and TUNEL (C) staining carried out on liver sections (×50 magnification). Data represent means ± SE of n = 5–10 animals per group. *p < .05 (compared with APAP alone). #p < .05 (compared with APAP+netrin-1).
Blocking the A2B Receptor Prevents Netrin-1-mediated Elevations in Macrophage Recruitment
To further confirm that the netrin-1-mediated effect on the infiltration of macrophages was mediated through the A2B receptor, this was investigated in liver sections. As seen in Figure 9, the enhanced recruitment of macrophages seen after netrin-1 treatment was almost completely blocked by administration of PSB1115. This confirms that the A2BAR is a key mediator of this delayed protection by netrin-1. Because adenosine is the canonical ligand for A2BAR, it is possible that netrin-1 was mediating its effects through induction of adenosine levels. However, measurement of circulating adenosine at 24 h after APAP or APAP+netrin-1 showed no change compared with control (data not shown), suggesting that netrin-1 effects were mediated independent of adenosine.
Figure 9.
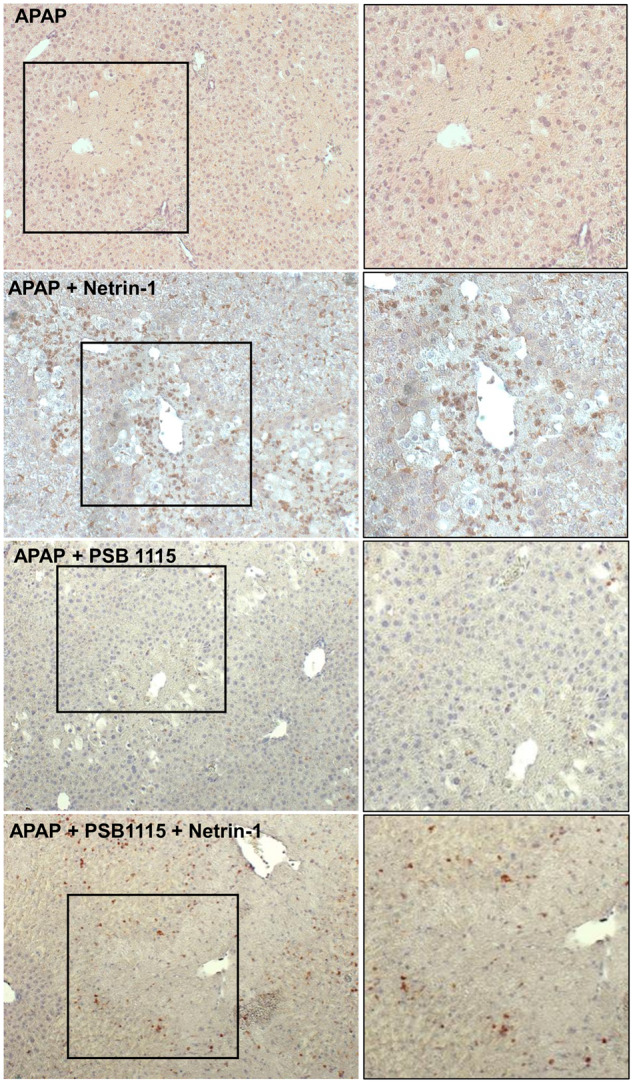
Blocking adenosine A2B receptor attenuated netrin-1-induced macrophage infiltration. Fasted mice were co-treated with 300 mg/kg acetaminophen (APAP) and netrin-1 (1 µg/mouse) or saline as control, with some mice also receiving PSB1115 at 0 and 6 h after APAP. Mice were sacrificed at 24 h and liver tissue sections used for immunohistochemistry staining of F4/80-positive macrophages (×50 magnification). Panels on the right represent an enlargement of the areas defined on the left.
DISCUSSION
Although netrin-1 was identified as a neuronal guidance cue directing neuronal axons to targets during development of the nervous system (Boyer and Gupton, 2018), netrin-1 is also widely expressed outside the nervous system. A recent study conducted by Schlegel et al. (2016) found that netrin-1 was expressed in the liver, with mRNA transcript and protein levels being significantly reduced after I/R injury. Exogenous netrin-1 administration was shown to dampen hepatic damage and activate generation of growth factors, which contributed to liver repair and regeneration (Schlegel et al., 2016). In APAP hepatotoxicity, we found that administration of exogenous netrin-1 had an interesting effect. No difference was noted with netrin-1 administration in early events of APAP-induced cell signaling such as mitochondrial oxidant stress and JNK activation and mitochondrial translocation, and netrin-1-treated animals had similar ALT release and areas of necrosis as the nontreated animals at 6‐h post‐APAP. However, by 24 h, significant protection against both ALT release and area of necrosis was notable. The fact that coadministration of netrin-1 did not influence APAP-induced injury but prevented progression of injury with protection only by 24 h suggests a unique pattern of delayed protection distinct from other interventions targeting reactive metabolite formation and oxidative stress such as NAC, Mito-TEMPO, or 4-methylpyrazole (Akakpo et al., 2018, 2019; Du et al., 2017; Saito et al., 2010).
Netrin-1 mediates its function predominantly through three receptors: DCC (Serafini et al., 1996), uncoordinated family member 5 (UNC5A–D or UNC5H1–4) (Hong et al., 1999), and A2BAR (Stein et al., 2001). Among the three, we only detected significant upregulation of A2BAR mRNA after APAP, which occurred earlier in the presence of netrin-1. Hence, our study was focused on the A2BAR since genetic studies have provided evidence for the role of this receptor in netrin-1-mediated protection during hepatic I/R injury (Schlegel et al., 2016). Our tissue immunohistochemistry data also showed a unique upregulation of A2BAR protein selectively in cells surrounding the necrotic areas after APAP-induced necrosis. The A2BAR has been implicated in modulation of the immune response (Fredholm et al., 2000), and the innate immune response after APAP is a late event initiated by 6–24 h after a 300 mg/kg dose (Jaeschke et al., 2012b). DAMP release from APAP-induced hepatocyte necrosis activate resident macrophages with formation of cytokines and chemokines, which, as mediators of inflammation can recruit inflammatory cells (neutrophils, monocytes) into the liver (Jaeschke et al., 2012a). Though the role of this immune response after APAP overdose was initially controversial, several studies have indicated that this is a beneficial response. It was demonstrated that a deficiency of intercellular adhesion molecule-1 (Cover et al., 2006), CD18 (Williams et al., 2010), or phox91 (James et al., 2003), all direct interventions against neutrophils, provided no protection against APAP-induced hepatotoxicity. Furthermore, pharmacological inhibitors of NADPH oxidase (Cover et al., 2006) were also ineffective, and neutrophils recruited into the liver after APAP overdose are not even primed or activated (Williams et al., 2010, 2014). These data strongly suggest that neutrophils do not contribute to the liver injury after an APAP overdose but arrive in response to hepatocyte necrosis to facilitate repair and regeneration (Yang et al., 2019). In addition to its effects on neurons, netrin-1 is also an immune mediator (Ranganathan et al., 2014) and was shown to reduce chemokinetic motility of human neutrophils (Hipoilito et al., 2018). Neutrophil function can also be modulated by adenosine in a concentration dependent manner. At low concentrations adenosine acts through the A1 and A3 adenosine receptor subtypes to promote neutrophil chemotaxis and phagocytosis (Barletta et al., 2012; Cronstein et al., 1990). However, at higher concentrations, adenosine acts at the A2A and A2B receptors to inhibit neutrophil trafficking and effector functions such as oxidative burst, inflammatory mediator production, and granule release (Barletta et al., 2012; Cronstein et al., 1990). Although our data indicate that circulating adenosine levels are not altered by APAP, engagement of A2BAR through netrin could still function to inhibit neutrophil trafficking. This could be one explanation for the decrease in neutrophil infiltration in netrin-1-treated mice. Another fact to be kept in mind is that the extent of immune cell infiltration is dependent on DAMP release from necrotic cells, and if netrin-1 delayed the rate of necrosis after the 6 h time point, infiltration of neutrophils at 24 h would also be reduced.
In contrast to neutrophils, cotreatment with netrin-1 significantly enhanced macrophage infiltration into the liver compared to untreated animals at 24 h, and these additional macrophages likely arise from bone marrow derived monocytes (Dambach et al., 2002; Holt et al., 2008; You et al., 2013). Several studies have focused on the effect of netrin-1 on macrophage migration (Mao et al., 2014; Ramkhelawon et al., 2014; Zhang et al., 2018), suggesting that netrin-1 promotes macrophage migration after organ damage. Studies on the role of macrophages in APAP hepatotoxicity indicate that they play an important role in liver recovery (Antoniades et al., 2012; Dambach et al., 2002; Holt et al., 2008; Merlin et al., 2016), and their enhancement by netrin-1, which also increased liver regeneration as measured by PCNA-positive cells, suggests a possible mechanism of the delayed protection exhibited by the molecule. This induction of liver regeneration corresponds to upregulation of ALR and HGF after netrin administration. This critical role of resident macrophages in netrin-1-mediated protection against APAP-induced liver injury was confirmed by the clodronate experiments, which remove resident liver macrophages (Ju et al., 2002) and clearly demonstrated that preventing macrophage infiltration attenuated netrin-1-mediated protection. They also indicate that macrophages were necessary for the netrin-1-mediated induction of proliferation, because PCNA staining as well as expression of HGF and ALR were also blunted in the presence of clodronate.
The persistence of circulating netrin-1 24 h after exogenous administration suggests that netrin-1 would be available to mediate its delayed effects by binding to its receptor A2BAR, and thus the confirmation of the role of A2BAR in netrin-1-mediated protection against APAP hepatotoxicity was important. Blocking of netrin-1’s effect on ALT release, area of necrosis and enhanced macrophage infiltration by the A2BAR receptor antagonist PSB1115 confirms that protective effects of netrin-1 in APAP hepatotoxicity were predominantly mediated through the A2BAR. However, since netrin-1 administration was carried out in parallel with APAP, and protection was observed 24 h later, systemic effects of netrin-1 due to interactions on extra-hepatic tissues cannot be ruled out. Despite this, these findings are very relevant clinically, because netrin-1 could then be substituted with an A2BAR agonist, which would have significant beneficial effect, especially in patients presenting after initiation of the injury phase. Since A2BAR-mediated protection seems to be independent of mechanisms causing early hepatocyte necrosis, the modulation of the subsequent innate immune response through the receptor would facilitate faster recovery and regeneration and hence even if a patient presents with injury already occurred, activation of the receptor would be protective.
In summary, the primary objective of this study was to evaluate the effect of exogenous netrin-1 on APAP hepatotoxicity and we demonstrate that co-treatment with netrin-1, while having no effect on the early APAP-induced injury processes, hastened subsequent recovery, significantly blunting liver injury by the later 24 h time point. Mechanistically, we identified activation of the A2BAR as a critical requirement for netrin-1-mediated protection against APAP-induced hepatotoxicity, which involves enhanced macrophage infiltration and activation of liver regeneration. The delayed protection by netrin-1 despite coadministration is unique, and the identification of A2BAR as a mediator of this delayed effect reveals novel therapeutic options targeting the receptor, which could function complementary to the current standard of care, NAC. This would especially benefit patients with ongoing injury who present later to the clinic after an APAP overdose, since the beneficial effect of A2BAR activation is independent of injury processes and activates the proregenerative innate immune response, which is activated late after APAP-induced injury. Thus, use of A2BAR agonists could have significant benefit in this group of patients.
FUNDING
National Institutes of Health (R01 DK102142 to H.J., P20 GM103549 to H.J., and P30 GM118247 to H.J.).
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Akakpo J. Y., Ramachandran A., Duan L., Schaich M. A., Jaeschke M. W., Freudenthal B. D., Ding W. X., Rumack B. H., Jaeschke H. (2019). Delayed treatment with 4-methylpyrazole protects against acetaminophen hepatotoxicity in mice by inhibition of c-Jun N-terminal kinase. Toxicol. Sci. 170, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akakpo J. Y., Ramachandran A., Kandel S. E., Ni H. M., Kumer S. C., Rumack B. H., Jaeschke H. (2018). 4-Methylpyrazole protects against acetaminophen hepatotoxicity in mice and in primary human hepatocytes. Hum. Exp. Toxicol. 37, 1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniades C. G., Quaglia A., Taams L. S., Mitry R. R., Hussain M., Abeles R., Possamai L. A., Bruce M., McPhail M., Starling C., et al. (2012). Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 56, 735–746. [DOI] [PubMed] [Google Scholar]
- Bai J., Hao J., Zhang X., Cui H., Han J., Cao N. (2016). Netrin-1 attenuates the progression of renal dysfunction by blocking endothelial-to-mesenchymal transition in the 5/6 nephrectomy rat model. BMC Nephrol. 17, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajt M. L., Cover C., Lemasters J. J., Jaeschke H. (2006). Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol. Sci. 94, 217–225. [DOI] [PubMed] [Google Scholar]
- Bajt M. L., Farhood A., Lemasters J. J., Jaeschke H. (2008). Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J. Pharmacol. Exp. Ther. 324, 8–14. [DOI] [PubMed] [Google Scholar]
- Bajt M. L., Knight T. R., Farhood A., Jaeschke H. (2003). Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J. Pharmacol. Exp. Ther. 307, 67–73. [DOI] [PubMed] [Google Scholar]
- Barletta K. E., Ley K., Mehrad B. (2012). Regulation of neutrophil function by adenosine. Arterioscler. Thromb. Vasc. Biol. 32, 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer N. P., Gupton S. L. (2018). Revisiting netrin-1: One who guides (axons). Front. Cell. Neurosci. 12, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. D., Pumford N. R., Khairallah E. A., Boekelheide K., Pohl L. R., Amouzadeh H. R., Hinson J. A. (1997). Selective protein covalent binding and target organ toxicity. Toxicol. Appl. Pharmacol. 143, 1–12. [DOI] [PubMed] [Google Scholar]
- Cover C., Liu J., Farhood A., Malle E., Waalkes M. P., Bajt M. L., Jaeschke H. (2006). Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 216, 98–107. [DOI] [PubMed] [Google Scholar]
- Cronstein B. N., Daguma L., Nichols D., Hutchison A. J., Williams M. (1990). The adenosine/neutrophil paradox resolved: Human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J. Clin. Invest. 85, 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csoka B., Selmeczy Z., Koscso B., Nemeth Z. H., Pacher P., Murray P. J., Kepka-Lenhart D., Morris S. M. Jr, Gause W. C., Leibovich S. J., et al. (2012). Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 26, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambach D. M., Watson L. M., Gray K. R., Durham S. K., Laskin D. L. (2002). Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology 35, 1093–1103. [DOI] [PubMed] [Google Scholar]
- Du K., Farhood A., Jaeschke H. (2017). Mitochondria-targeted antioxidant Mito-TEMPO protects against acetaminophen hepatotoxicity. Arch. Toxicol. 91, 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle T., Grenz A., Laucher S., Eltzschig H. K. (2008). A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J. Clin. Invest. 118, 3301–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitamant J., Guenebeaud C., Coissieux M. M., Guix C., Treilleux I., Scoazec J. Y., Bachelot T., Bernet A., Mehlen P. (2008). Netrin-1 expression confers a selective advantage for tumor cell survival in metastatic breast cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 4850–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm B. B., Arslan G., Halldner L., Kull B., Schulte G., Wasserman W. (2000). Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch. Pharmacol. 362, 364–374. [DOI] [PubMed] [Google Scholar]
- Gujral J. S., Farhood A., Bajt M. L., Jaeschke H. (2003). Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 38, 355–363. [DOI] [PubMed] [Google Scholar]
- Hanawa N., Shinohara M., Saberi B., Gaarde W. A., Han D., Kaplowitz N. (2008). Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 283, 13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebrok M., Reichardt L. F. (2004). Brain meets pancreas: Netrin, an axon guidance molecule, controls epithelial cell migration. Trends Cell Biol. 14, 153–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipoilito J., Peretz-Soroka H., Moya Torres A., Booy E., Yang K., Gupta M., Meier M., McKenna S., Koch M., Santos S., et al. (2018). The effect of netrin-1 on neutrophil and breast cancer cell migration and their migratory interaction. Biophys. J. 114, 325a. [Google Scholar]
- Holt M. P., Cheng L., Ju C. (2008). Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 84, 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K. S., Hinck L., Nishiyama M., Poo M. M., Tessier-Lavigne M., Stein E. (1999). A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 97, 927–941. [DOI] [PubMed] [Google Scholar]
- Jaeschke H., Akakpo J. Y., Umbaugh D. S., Ramachandran A. (2020). Novel therapeutic approaches against acetaminophen-induced liver injury and acute liver failure. Toxicol. Sci. 174, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H., Bajt M. L. (2006). Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 89, 31–41. [DOI] [PubMed] [Google Scholar]
- Jaeschke H., McGill M. R., Ramachandran A. (2012. a). Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 44, 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H., Williams C. D., Ramachandran A., Bajt M. L. (2012. b). Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 32, 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. P., McCullough S. S., Knight T. R., Jaeschke H., Hinson J. A. (2003). Acetaminophen toxicity in mice lacking NADPH oxidase activity: Role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic. Res. 37, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Ju C., Reilly T. P., Bourdi M., Radonovich M. F., Brady J. N., George J. W., Pohl L. R. (2002). Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem. Res. Toxicol. 15, 1504–1513. [DOI] [PubMed] [Google Scholar]
- Kon K., Kim J. S., Jaeschke H., Lemasters J. J. (2004). Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40, 1170–1179. [DOI] [PubMed] [Google Scholar]
- Ly N. P., Komatsuzaki K., Fraser I. P., Tseng A. A., Prodhan P., Moore K. J., Kinane T. B. (2005). Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 14729–14734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., Xing H., Mao A., Jiang H., Cheng L., Liu Y., Quan X., Li L. (2014). Netrin-1 attenuates cardiac ischemia reperfusion injury and generates alternatively activated macrophages. Inflammation 37, 573–580. [DOI] [PubMed] [Google Scholar]
- Maruyama K., Kawasaki T., Hamaguchi M., Hashimoto M., Furu M., Ito H., Fujii T., Takemura N., Karuppuchamy T., Kondo T., et al. (2016). Bone-protective functions of netrin 1 protein. J. Biol. Chem. 291, 23854–23868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin S., Bhargava K. K., Ranaldo G., Zanolini D., Palestro C. J., Santambrogio L., Prat M., Follenzi A., Gupta S. (2016). Kupffer cell transplantation in mice for elucidating monocyte/macrophage biology and for potential in cell or gene therapy. Am. J. Pathol. 186, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirakaj V., Gatidou D., Pötzsch C., König K., Rosenberger P. (2011). Netrin-1 signaling dampens inflammatory peritonitis. J. Immunol. 186, 549–555. [DOI] [PubMed] [Google Scholar]
- Mirakaj V., Thix C. A., Laucher S., Mielke C., Morote-Garcia J. C., Schmit M. A., Henes J., Unertl K. E., Köhler D., Rosenberger P. (2010). Netrin-1 dampens pulmonary inflammation during acute lung injury. Am. J. Respir. Crit. Care Med. 181, 815–824. [DOI] [PubMed] [Google Scholar]
- Mutz C., Mirakaj V., Vagts D. A., Westermann P., Waibler K., Konig K., Iber T., Noldge-Schomburg G., Rosenberger P. (2010). The neuronal guidance protein netrin-1 reduces alveolar inflammation in a porcine model of acute lung injury. Crit. Care 14, R189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S. D. (1990). Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis. 10, 267–278. [DOI] [PubMed] [Google Scholar]
- Nguyen A., Cai H. (2006). Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 6530–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradisi A., Creveaux M., Gibert B., Devailly G., Redoulez E., Neves D., Cleyssac E., Treilleux I., Klein C., Niederfellner G., et al. (2013). Combining chemotherapeutic agents and netrin-1 interference potentiates cancer cell death. EMBO Mol. Med. 5, 1821–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y. C., Benet L. Z., Burlingame A. L. (1998). Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J. Biol. Chem. 273, 17940–17953. [DOI] [PubMed] [Google Scholar]
- Radi Z. A., Koza-Taylor P. H., Bell R. R., Obert L. A., Runnels H. A., Beebe J. S., Lawton M. P., Sadis S. (2011). Increased serum enzyme levels associated with Kupffer cell reduction with no signs of hepatic or skeletal muscle injury. Am. J. Pathol. 179, 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A., Jaeschke H. (2019). Acetaminophen hepatotoxicity. Semin. Liver Dis. 39, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkhelawon B., Hennessy E. J., Menager M., Ray T. D., Sheedy F. J., Hutchison S., Wanschel A., Oldebeken S., Geoffrion M., Spiro W., et al. (2014). Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat. Med. 20, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan P., Mohamed R., Jayakumar C., Ramesh G. (2014). Guidance cue netrin-1 and the regulation of inflammation in acute and chronic kidney disease. Mediators Inflamm. 2014, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger P., Schwab J. M., Mirakaj V., Masekowsky E., Mager A., Morote-Garcia J. C., Unertl K., Eltzschig H. K. (2009). Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 10, 195–202. [DOI] [PubMed] [Google Scholar]
- Saito C., Zwingmann C., Jaeschke H. (2010). Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 51, 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel M., Kohler D., Korner A., Granja T., Straub A., Giera M., Mirakaj V. (2016). The neuroimmune guidance cue netrin-1 controls resolution programs and promotes liver regeneration. Hepatology 63, 1689–1705. [DOI] [PubMed] [Google Scholar]
- Serafini T., Colamarino S. A., Leonardo E. D., Wang H., Beddington R., Skarnes W. C., Tessier-Lavigne M. (1996). Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87, 1001–1014. [DOI] [PubMed] [Google Scholar]
- Singer A. J., Carracio T. R., Mofenson H. C. (1995). The temporal profile of increased transaminase levels in patients with acetaminophen-induced liver dysfunction. Ann. Emerg. Med. 26, 49–53. [DOI] [PubMed] [Google Scholar]
- Smilkstein M. J., Knapp G. L., Kulig K. W., Rumack B. H. (1988). Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N. Engl. J. Med. 319, 1557–1562. [DOI] [PubMed] [Google Scholar]
- Stein E., Zou Y. M., Poo M. M., Tessier-Lavigne M. (2001). Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science 291, 1976–1982. [DOI] [PubMed] [Google Scholar]
- Tirmenstein M. A., Nelson S. D. (1989). Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J. Biol. Chem. 264, 9814–9819. [PubMed] [Google Scholar]
- Williams C. D., Bajt M. L., Farhood A., Jaeschke H. (2010). Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int. 30, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. D., Bajt M. L., Sharpe M. R., McGill M. R., Farhood A., Jaeschke H. (2014). Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol. Appl. Pharmacol. 275, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., McGill M. R., Du K., Dorko K., Kumer S. C., Schmitt T. M., Ding W. X., Jaeschke H. (2015). Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol. Appl. Pharmacol. 289, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Tao Y., Wu Y., Zhao X., Ye W., Zhao D., Fu L., Tian C., Yang J., He F., et al. (2019). Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 10, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Q., Holt M., Yin H., Li G., Hu C. J., Ju C. (2013). Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem. Pharmacol. 86, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Y., Chen P., Di G. H., Qi X., Zhou Q. J., Gao H. (2018). Netrin-1 promotes diabetic corneal wound healing through molecular mechanisms mediated via the adenosine 2B receptor. Sci. Rep. 8, 5994. [DOI] [PMC free article] [PubMed] [Google Scholar]