

Mycobacteria are major environmental microorganisms and cause many significant diseases, including tuberculosis. Mycobacteria make an unusual vitamin-like compound, F420, and use it to both persist during stress and resist antibiotic treatment. Understanding how mycobacteria make F420 is important, as this process can be targeted to create new drugs to combat infections like tuberculosis. In this study, we show that mycobacteria make F420 in a way that is different from other bacteria. We studied the molecular machinery that mycobacteria use to make F420, determining the chemical mechanism for this process and identifying a novel chemical intermediate. These findings also have clinical relevance, given that two new prodrugs for tuberculosis treatment are activated by F420.
KEYWORDS: cofactor biosynthesis, deazaflavin, F420, Mycobacterium, Mycobacterium smegmatis, structural biology
ABSTRACT
F420 is a low-potential redox cofactor used by diverse bacteria and archaea. In mycobacteria, this cofactor has multiple roles, including adaptation to redox stress, cell wall biosynthesis, and activation of the clinical antitubercular prodrugs pretomanid and delamanid. A recent biochemical study proposed a revised biosynthesis pathway for F420 in mycobacteria; it was suggested that phosphoenolpyruvate served as a metabolic precursor for this pathway, rather than 2-phospholactate as long proposed, but these findings were subsequently challenged. In this work, we combined metabolomic, genetic, and structural analyses to resolve these discrepancies and determine the basis of F420 biosynthesis in mycobacterial cells. We show that, in whole cells of Mycobacterium smegmatis, phosphoenolpyruvate rather than 2-phospholactate stimulates F420 biosynthesis. Analysis of F420 biosynthesis intermediates present in M. smegmatis cells harboring genetic deletions at each step of the biosynthetic pathway confirmed that phosphoenolpyruvate is then used to produce the novel precursor compound dehydro-F420-0. To determine the structural basis of dehydro-F420-0 production, we solved high-resolution crystal structures of the enzyme responsible (FbiA) in apo-, substrate-, and product-bound forms. These data show the essential role of a single divalent cation in coordinating the catalytic precomplex of this enzyme and demonstrate that dehydro-F420-0 synthesis occurs through a direct substrate transfer mechanism. Together, these findings resolve the biosynthetic pathway of F420 in mycobacteria and have significant implications for understanding the emergence of antitubercular prodrug resistance.
IMPORTANCE Mycobacteria are major environmental microorganisms and cause many significant diseases, including tuberculosis. Mycobacteria make an unusual vitamin-like compound, F420, and use it to both persist during stress and resist antibiotic treatment. Understanding how mycobacteria make F420 is important, as this process can be targeted to create new drugs to combat infections like tuberculosis. In this study, we show that mycobacteria make F420 in a way that is different from other bacteria. We studied the molecular machinery that mycobacteria use to make F420, determining the chemical mechanism for this process and identifying a novel chemical intermediate. These findings also have clinical relevance, given that two new prodrugs for tuberculosis treatment are activated by F420.
INTRODUCTION
Factor 420 (F420) is a deazaflavin cofactor that mediates diverse redox reactions in bacteria and archaea (1). Chemically, F420 consists of a redox-active deazaflavin headgroup (derived from the chromophore Fo) that is conjugated to a variable-length polyglutamate tail via a phosphoester linkage (2). While the Fo headgroup of F420 superficially resembles flavins (e.g., flavin adenine dinucleotide [FAD], flavin mononucleotide [FMN]), three chemical substitutions in the isoalloxazine ring give it distinct chemical properties more reminiscent of nicotinamides (e.g., NADH, NADPH) (1). These properties include a low standard potential (−350 mV) and obligate two-electron (hydride) transfer chemistry (3, 4). The electrochemical properties of F420 make it ideal to reduce a wide range of otherwise recalcitrant organic compounds (5–7). Diverse microorganisms are known to synthesize F420, but the compound is best characterized for its roles in methanogenesis in archaea, antibiotic biosynthesis in streptomycetes, and metabolic adaptation of mycobacteria (1, 8–11). In mycobacteria, F420 is involved in a plethora of processes: central carbon metabolism, cell wall synthesis, recovery from dormancy, resistance to oxidative stress, and inactivation of certain bactericidal agents (7, 12–14). In the human pathogen Mycobacterium tuberculosis, F420 is also critical for the reductive activation of the newly approved clinical antitubercular prodrugs pretomanid and delamanid (15–17).
Following the elucidation of the chemical structure of F420 in the 1970s, the F420 biosynthesis pathway in archaea was determined through a combination of in situ biochemistry and recombinant protein analysis (1, 2). Described briefly, the deazaflavin fluorophore Fo is synthesized through condensation of 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione and l-tyrosine by the S-adenosyl-l-methionine (SAM)-radical enzymes CofG and CofH (18). The putative enzyme CofB synthesizes 2-phospholactate (2PL), which links Fo to the glutamate tail of mature F420 (19). Subsequently, the nucleotide transferase CofC condenses 2PL with GTP to form the reactive intermediate l-lactyl-2-diphospho-5ʹ-guanosine (LPPG) (20). The phosphotransferase CofD then transfers 2PL from LPPG to Fo, leading to the formation of F420-0 (i.e., F420 with no glutamate tail) (21). Finally, the GTP-dependent glutamate ligase CofE adds a variable-length γ-linked glutamate tail to produce mature F420 (22, 23). With the exception of the putative lactate kinase CofB, the enzymes responsible for F420 biosynthesis in archaea have been identified and characterized to various extents (1). Crystal structures have been obtained for CofC, CofD, and CofE from methanogenic archaea, providing some insight into how these enzymes function, but questions surrounding their catalytic mechanisms remain unresolved (23–25). For example, the crystal structure of CofD from Methanosarcina mazei was solved in the presence of Fo and GDP; however, no divalent cation(s) required for catalysis was present in the structure, and the ribosyl tail group of Fo, which receives the 2PL moiety from LPPG was disordered, precluding an understanding of the catalytic mechanism of this step in F420 biosynthesis (21, 25).
It was assumed that the biosynthesis pathway for archaeal F420 was generic to all F420-producing organisms (1). However, recent studies have shown that the structure and biosynthesis of F420 vary among F420-producing organisms (24, 26). F420 produced by the proteobacterial fungal symbiont Paraburkholderia rhizoxinica was found to incorporate 3-phospho-d-glycerate (3PG) in the place of 2PL, producing a chemically distinct F420 (26). In parallel, analysis of purified F420 biosynthesis enzymes from mycobacteria indicated that the central glycolytic and gluconeogenic intermediate phosphoenolpyruvate (PEP), rather than 2PL, is a precursor for F420 biosynthesis (24). In contrast to P. rhizoxinica, in mycobacteria, mature F420 is chemically analogous to that produced by archaea (27). All mycobacterial species possess the four enzymes required for F420 biosynthesis. However, as these enzymes catalyze reactions distinct from their archaeal homologues, the following alternative nomenclature is applied compared to the archaeal enzymes: FbiD (homologous to CofC), FbiC (a single protein with domains homologous to CofG and CofH), FbiA (homologous to CofD), and FbiB (N-terminal domain homologous to CofE) (8). In addition to its CofE-like domain, FbiB possesses an FMN-binding C-terminal domain, and biochemical evidence suggests that it is responsible for the reduction of the moiety derived from PEP (24, 28).
However, several findings have cast doubt on whether the proposed revised biosynthesis pathway of F420 is physiologically relevant. The predicted use of PEP in F420 biosynthesis in mycobacteria would lead to the formation of the oxidized intermediate compound dehydro-F420-0 (DH-F420-0). The production of DH-F420-0 was detected in a coupled enzyme assay containing purified FbiD and FbiA with PEP supplied as the substrate but has yet to be detected in mycobacterial cells (24). The study also showed that CofC from Methanocaldococcus jannaschii utilized PEP rather than 2PL for F420 biosynthesis (24), leading the authors to conclude that PEP is the general precursor for F420 biosynthesis in microorganisms. However, these findings contradict previous analysis of CofC activity in M. jannaschii cell lysates (19, 21), as well as recent biochemical analysis, which shows that CofC preferentially utilizes 2PL for F420 biosynthesis (26). In turn, these findings cast doubt on whether PEP is truly the preferred substrate for mycobacterial F420 biosynthesis and whether DH-F420-0 is the physiological intermediate in this pathway.
In this work, we first resolved this ambiguity by analyzing the F420 biosynthetic pathway in Mycobacterium smegmatis in whole cells. We demonstrate that PEP, not 2PL, is the substrate for F420 biosynthesis in mycobacterial cells, suggesting that divergent biosynthesis pathways are utilized to generate F420 in different microbial species. Consistent with this result, we determine that DH-F420-0 is the physiological intermediate for F420 biosynthesis in mycobacteria and that it is present in high quantities in cells lacking FbiB and comes bound to FbiA purified from M. smegmatis. Furthermore, to elucidate the catalytic mechanism for the formation of the novel intermediate DH-F420-0, we determined the crystal structure of FbiA in the presence and absence of its substrate and product compounds. These data resolve long-standing questions about the catalytic mechanism of FbiA and CofD in F420 biosynthesis. Moreover, they provide a target for therapeutic intervention through the inhibition of F420 biosynthesis, as well as insight into potential mechanisms for the emergence of delamanid and pretomanid drug resistance through mutations in FbiA.
RESULTS
Phosphoenolpyruvate is the substrate for the biosynthesis of F420 in mycobacterial cells.
To determine whether PEP or 2PL is the substrate for F420 biosynthesis in mycobacteria (Fig. 1A), we spiked clarified cell lysates from M. smegmatis with GTP and either PEP or 2PL, and monitored the synthesis of new F420 species through fast-performance liquid chromatography (FPLC) coupled with fluorescence detection. In cell lysates spiked with PEP, a species corresponding to F420-0 in the F420 standard was present, which was absent from both the untreated and 2PL-spiked lysates (Fig. 1B). The formation of this F420-0-like species in PEP-spiked lysates corresponded to a decrease in Fo levels, suggesting that synthesis of DH-F420-0 from PEP is occurring (Fig. 1B). These data strongly suggest that PEP, not 2PL, is the precursor for F420 biosynthesis in M. smegmatis.
FIG 1.
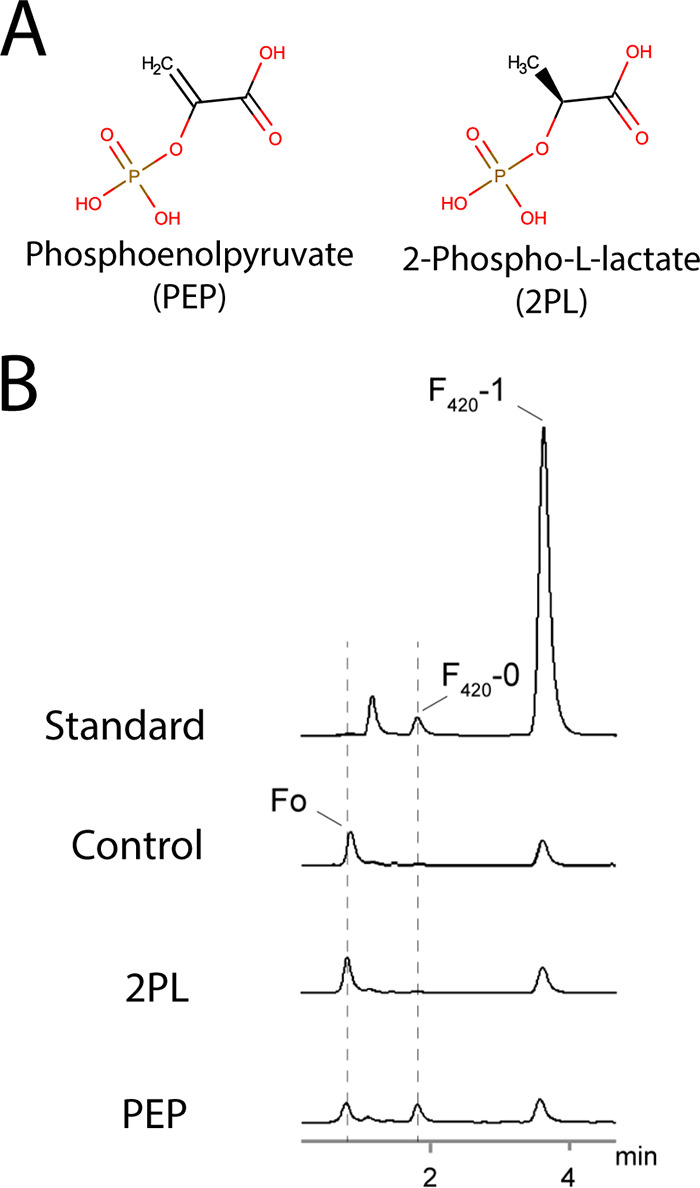
PEP, but not 2PL, stimulates DH-F420-0 synthesis in M. smegmatis cell lysates. (A) Two-dimensional (2D) structures of PEP and 2PL demonstrating the difference (double bond or single bond) in bonding between carbon 2 and 3. (B) Fluorescence emission detection chromatogram from HPLC of M. smegmatis lysates spiked with either 2PL or PEP or an unspiked control. Synthesis of a species with characteristic F420 fluorescence (excitation, 420 nm; emission, 480 nm) corresponding to F420-0 from the purified standard was detected only in the PEP-spiked lysate. The appearance of this F420-0-like species coincided with a decrease in the presence of Fo, suggesting that PEP is the precursor for F420 synthesis in M. smegmatis in cells. F420-1 in the standard corresponds to F420 with a single glutamate moiety.
While the lysate spiking experiment establishes that PEP is specifically utilized for F420 synthesis in M. smegmatis, the fluorescence detection method utilized does not chemically differentiate between F420-0 or DH-F420-0. As PEP is utilized, it would be expected that DH-F420-0 is produced. However, DH-F420-0 may be rapidly reduced to F420-0 rather than accumulating in the cell. To confirm the synthesis of the DH-F420-0 in M. smegmatis, we created isogenic deletions in the four F420 biosynthesis genes: fbiD, fbiC, fbiA, and fbiB (Fig. 2A). The genome sequences of these deletion strains were determined, confirming clean deletion with no secondary mutations present. We then detected the deazaflavin species present in clarified cell lysates from these strains using fluorescence-coupled high-performance liquid chromatography (HPLC) and liquid chromatography coupled to mass spectrometry (LC-MS). As expected, based on the proposed function of these enzymes, mature F420 was detected only in wild-type cell lysates and possessed a polyglutamate tail length of three to eight (Fig. 2B; see also Fig. S1 in the supplemental material). Fo was detected in the wild type and all mutants except the ΔfbiC mutant, consistent with the function of this enzyme in the synthesis of the Fo-deazaflavin moiety (Fig. 2A and C and Fig. S1). The proposed biosynthetic intermediate DH-F420-0 was detected only in cell lysates of the ΔfbiB strain (Fig. 2D and Fig. S1). No F420-0 was detected in wild-type or mutant strains.
FIG 2.

Mutagenic dissection of the F420 biosynthesis pathway in M. smegmatis reveals that DH-F420-0 is the biosynthetic intermediate in mycobacteria. (A) A schematic of the F420 biosynthesis pathway in M. smegmatis with PEP, rather than 2PL, utilized by FbiD to create the reaction intermediate EPPG. The enzymes responsible for catalytic steps are shown, along with the 2D structures of proposed pathway intermediates and mature F420. The yellow box highlights the reduction of DH-F420-0, proposed to be mediated by the C-terminal domain of FbiB using FMNH2. (B to D) LC-MS detection of mature F420 species (B), Fo (C), and DH-F420-0 (D) in M. smegmatis cell lysates of the wild type (Wt) and F420 biosynthesis pathway mutants confirming the proposed function of the F420 biosynthetic genes detecting the novel intermediate DH-F420-0 in whole cells. F420-X species in panel B correspond to different lengths of the polyglutamate chain where X = n tail length.
Fluorescence-coupled HPLC analysis of clarified cell lysates from M. smegmatis F420 biosynthesis pathway mutants. Fluorescence (excitation wavelength [Ex λ] of 420 nm; emission wavelength [Em λ] of 480 nm) trace for wild-type (A), ΔfbiC (B), ΔfbiA (C), ΔfbiD (D), and ΔfbiB (E) strains showing the formation of mature F420 species in the wild-type strain only and accumulation of DH-F420-0 in the ΔfbiB strain. LU, fluorescence intensity. For ΔfbiC, shown in panel B, mass spectrometry analysis confirmed that no Fo or F420 species were present, despite observed low-level fluorescence. Download FIG S1, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The presence of DH-F420-0 (and absence of detected F420-0) in whole cells demonstrates that it is the central physiological intermediate in mycobacterial F420 biosynthesis. This also lends support to the biochemical and cellular assays indicating that PEP, not 2PL, is the substrate for this pathway in mycobacteria. Furthermore, in addition to its role as the F420 glutamyl-ligase, structural and biochemical analysis suggests that FbiB is responsible for the reduction of DH-F420-0 (24, 28). The detection of DH-F420-0 only in the ΔfbiB strain demonstrates that this intermediate is rapidly turned over in the cell and supports the hypothesis that FbiB and not another enzyme performs this step in mycobacterial F420 biosynthesis.
FbiA copurifies with its product dehydro-F420-0.
In order to determine the catalytic mechanism for the synthesis of the novel intermediate DH-F420-0, we overexpressed and purified FbiA from M. smegmatis. Purified FbiA from M. smegmatis possessed a light yellow color, indicating copurification with a product or substrate molecule (Fig. S2A). The nature of this substrate was investigated using fluorescence spectroscopy, with purified FbiA found to have a broad absorbance peak at 400 nm and a corresponding emission peak at 470 nm (Fig. S2B), which is consistent with the presence of a deazaflavin with a protonated 8-OH group (16). We then utilized LC-MS to identify the deazaflavin species associated with FbiA and found that the major species was its product DH-F420-0 (Fig. S2C). In addition, significant quantities of mature F420 species were also associated with FbiA, suggesting that it also binds to mature F420 present in the cytoplasm (Fig. S2C).
FbiA recombinantly expressed in M. smegmatis copurifies with its product DH-F420-0. (A) Purified, concentrated (16 mg · ml−1) recombinant FbiA produced in M. smegmatis showing a characteristic yellow color associated with a bound F420 species. (B) The absorbance (purple) and fluorescence spectra (blue) of purified FbiA from panel A, which is characteristic of F420 species with a protonated deazaflavin 8-OH group. (C) LC-MS analysis of F420 species bound to purified FbiA, showing that DH-F420-0 is the predominantly bound species and the presence of some mature F420 species. (D) SEC-MALS analysis of purified FbiA shows that it has a molecular weight in solution consistent with a homodimeric species. Download FIG S2, TIF file, 2.3 MB (2.4MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The crystal structure of FbiA reveals an active site with open and closed states.
In order to resolve the catalytic mechanism of DH-F420-0 synthesis, purified FbiA was crystallized, and its structure was determined at 2.3 Å by X-ray crystallography (see Table S1 in the supplemental material). FbiA crystallized as a dimer mediated by the interaction of three α-helices and a β-sheet (Fig. 3A). This dimer is predicted to be stable by the protein-interaction prediction program PISA (29), and the molecular weight of FbiA determined by size exclusion chromatography coupled to multiangle light scattering (SEC-MALS) shows that it forms a dimer in solution (Table S2 and Fig. S2D). The structure of CofD from M. mazei, a homologous enzyme that instead utilizes LPPG derived from 2PL as its substrate, also crystallized as a dimer with an analogous interface to FbiA (25). Despite the copurification of FbiA with DH-F420-0, only weak electron density attributable to DH-F420-0 was observed in the catalytic site of molecule B (Mol. B) of the FbiA dimer (Fig. S3). To obtain the product-bound structure of FbiA, DH-F420-0 was purified from recombinant FbiA and soaked into existing crystals of FbiA. Using this procedure, electron density clearly attributable to the Fo and phosphate moieties of DH-F420-0 was observed in Mol. B of FbiA, allowing modeling of the product-bound structure (Fig. S3). Density for the carbonyl group of DH-F420-0 was less well resolved, suggesting that it exists in multiple conformations in product-bound FbiA (Fig. S3). Similarly, FbiA crystals were soaked with Fo and GDP, individually or in combination, and structures of substrate-bound FbiA were determined (Fig. S3 and Table S1).
FIG 3.
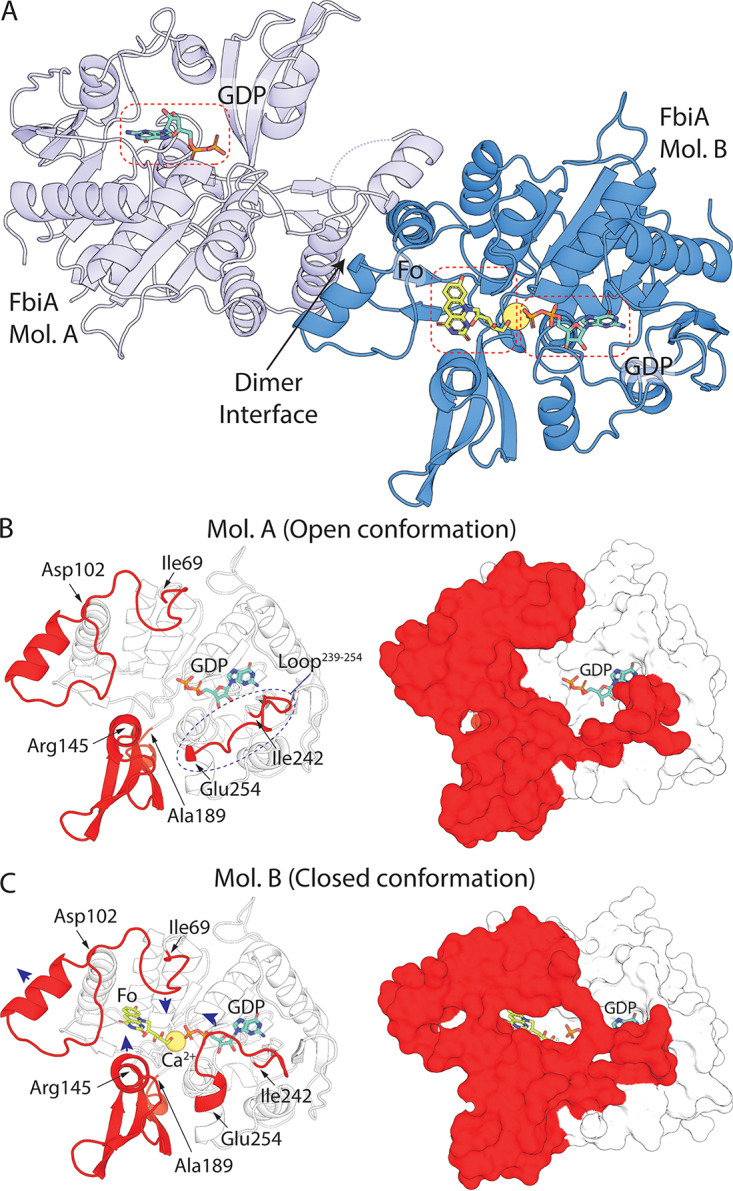
The crystal structure of FbiA captures the enzyme in open and closed states. (A) The crystal structure of FbiA from M. smegmatis in complex with Fo and GDP. FbiA is shown as a cartoon representation with molecule B (Mol. B) in sky blue and Mol. A in light blue. GDP and Fo are shown as stick representations, and Ca2+ is shown as a yellow sphere. (B) Mol. A from the FbiA structure exists in an open conformation. (Left) Mol. A as a cartoon with loops and subdomains which differ in conformation in Mol. B highlighted in red. (Right) Mol. A as a surface representation with mobile regions highlighted in red. (C) Mol. B of FbiA structure exists in a closed “catalytically ready” state. (Left) Mol. B displayed as in panel B, with the direction of movement of loops compared to Mol. A shown with blue arrows. (Right) Mol. B as in panel B, demonstrating how the mobile regions enclose the FbiA active site.
Electron density corresponding to FbiA substrates and products in cocrystal structures. The panel corresponds to the cocrystal structures indicated in the bottom right. A composite omit map is shown carved to visible molecules at a distance of 2 Å and contoured to 1 σ. Download FIG S3, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Crystallographic data collections and refinement statistics. Download Table S1, XLSX file, 0.01 MB (12.1KB, xlsx) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
FbiA dimer interface statistics reported by PISA (29). Download Table S2, XLSX file, 0.01 MB (10KB, xlsx) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of the Mol. A and Mol. B from the FbiA dimer reveals the active site of the enzyme in distinct open and closed conformations (Fig. 3). In Mol. A, the active site is locked in an open state due to participation of an extended loop (amino acids [aa] 239 to 254) in crystal packing (Fig. 3B). In this open state, FbiA has a lower apparent substrate affinity, with no density attributable to Fo or DH-F420-0 and only weak density for GDP observed in the respective cocrystal structures (Fig. S3). In contrast, in Mol. B, the extended loop (aa 239 to 254) is partially disordered in the non-GDP-bound structures and encloses GDP in the active site in GDP-bound structures (Fig. 3C). In Mol. B, additional conformational changes are observed in amino acids 69 to 102 and a subdomain composed of amino acids 145 to 189, creating the binding pocket for the deazaflavin moiety of Fo or DH-F420-0, which is not present in Mol. A (Fig. 3B and C). The conformational differences observed between Mol. A and Mol. B are consistent in the apo-, substrate-, and product-bound structures, demonstrating that they are not substrate induced and are likely representative of conformational differences of the enzyme in solution.
The crystal structures of FbiA in substrate- and product-bound forms provide mechanistic insight into dehydro-F420-0 synthesis.
The resolution of the structure of FbiA in the presence of its substrate and product compounds provides key insights into the catalytic mechanism of this unique phosphotransferase. It was previously established that FbiA and its archaeal homologue CofD require the presence of the divalent cation Mg2+ for activity (21, 24). However, it remained to be resolved whether Mg2+ is bound stably in the FbiA active site during catalysis and the mode of coordination of the ion(s). In the GDP-bound structures of FbiA, a single metal ion was present in the active site of Mol. B of FbiA (Fig. 4A; see also Fig. S4A and Fig. S5). Interestingly, no metal ion was observed in Mol. A, despite the presence of GDP, suggesting that its recruitment is conformation dependent (Fig. S2). As calcium acetate is present at high concentration (0.2 M) in the crystallization condition, and magnesium is absent, we modeled this ion as Ca2+. The activity of FbiA in the presence of Ca2+ has not been tested. Ca2+ and Mg2+ have been shown to be interchangeable in some phosphohydrolase enzymes, while for some Mg2+-dependent kinases, Ca2+ is a competitive inhibitor of activity (30, 31). Despite this, the coordination of Ca2+ is very similar to Mg2+, allowing for analysis of the enzyme active site with Ca2+ bound (31). In the GDP-only structure of FbiA, the Ca2+ ion is directly coordinated by aspartates 45 and 57, an oxygen atom of the β-phosphate of GDP, two H2O molecules, and a glycerol molecule (Fig. S4B). Aspartate 57 exhibits bidentate coordination of the Ca2+ ion, leading to a coordination number of seven with distorted octahedral geometry.
FIG 4.

Resolution of the structure of FbiA in the presence of Fo, GDP, and DH-F420-0 provides insight into its catalytic mechanism. (A) Fo and GDP in complex with Mol. B of FbiA in coordination with the catalytic Ca2+ ion. FbiA is shown as a sky blue cartoon, Fo and GDP as sticks, and Ca2+ as a sphere. (B) Stereoview of the catalytic center of the FbiA active site in complex with Fo and GDP, showing FbiA side chains involved in coordinating the catalytic metal ion and a coordinating H2O molecule. Bond distances of <3.2 Å are shown as yellow dashed lines, and the distance between the terminal OH of Fo and P of the β-phosphate of GDP is highlighted in blue. (C) DH-F420-0 in complex with FbiA, shown as in panel A. (D) Stereoview of the FbiA catalytic center with the reaction substrate EPPG model in place of GDP displayed in panel C, with the close proximity between the carboxylic acid group of EPPG and the terminal OH of Fo highlighted with a red dashed line. (E) Schematic showing the proposed catalytic mechanism for the formation of DH-F420-0 by FbiA.
The crystal structure of FbiA in complex with Fo and GDP. (A) GDP in complex with Mol. B of FbiA. FbiA is represented as a sky blue cartoon, bound GDP and glycerol molecules are shown as stick models, and bound Ca2+ ion is shown as a yellow sphere. (B) A stereoview of the catalytic center of the FbiA active site in complex with GDP and glycerol, showing FbiA side chains involved in coordinating the catalytic metal ion and a coordinating H2O molecule. Bond distances of <3.2 Å are shown as yellow dashed lines. (C) Fo in complex with Mol. B of FbiA is shown as a sky blue cartoon, and Fo is shown as a stick model. Download FIG S4, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The full interaction network of the FbiA active site in the presence of substrates (Fo and GDP) and product (DH-F420-0). Download FIG S5, TIF file, 2.0 MB (2.1MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In the GDP and Fo-bound structure, the coordination of Ca2+ is analogous to the GDP-only structure. However, the glycerol molecule and one of the H2O molecules observed in the GDP-only structure are displaced by the ribosyl chain of Fo, resulting in a coordination number of six with octahedral geometry (Fig. 4B and Fig. S4B). The terminal hydroxyl group of Fo is significantly closer to the Ca2+ ion (2.6 Å) and to the β-phosphate of GDP (2.8 Å from O and 3.0 Å from P) than the coordinating hydroxyl of glycerol, which is not within the bonding distance of GDP. These bond distances between the hydroxyl of Fo and GDP, as well as the central orientation of the hydroxyl of Fo toward the β-phosphate of GDP, place it in an ideal position to act as the acceptor substrate for the transfer of PEP catalyzed by FbiA (Fig. 4B). In the Fo- and DH-F420-0-bound structures, no density corresponding to a Ca2+ ion was observed (Fig. 4C and Fig. S4C). This suggests that binding of FbiA to its catalytic metal ion is contingent on complex formation with enolpyruvyl-diphospho-5ʹ-guanosine (EPPG) (Fig. 2A), which is substituted for GDP in our structures due to the instability of the F420 pathway intermediate (24). The ability of FbiA to bind Fo in the absence of GDP and Ca2+ suggests that substrate binding to FbiA is not sequential; however, recruitment of all three components is required for catalysis to proceed.
On the basis of these structural data and previous biochemical characterization of FbiA and CofD, we propose a catalytic mechanism for synthesis of the DH-F420-0 (Fig. 4E) (21, 25). Our structural data agree with previous work that suggests that CofD does not form a covalent intermediate as part of the reaction mechanism (21), but rather the reaction proceeds through direct nucleophilic attack of the β-phosphate of EPPG by the terminal hydroxyl of Fo. This leads to the formation of a pentavalent transition state between Fo and EPPG that is stabilized by the catalytic metal ion (Fig. 4E). In order for the hydroxyl group of Fo to perform nucleophilic attack, it needs to be activated through deprotonation. The carboxylic acid group of EPPG is a likely candidate for this activation, as it is the only acidic group in close proximity to the hydroxyl group of Fo when EPPG is modeled in the FbiA structure in place of GDP (Fig. 4D). Additionally, the activation of Fo by the carboxylic acid group of EPPG would provide FbiA with substrate specificity for EPPG over GDP and GTP. Following the formation of the pentavalent reaction intermediate, GMP would act as the leaving group, leading to the formation of the phosphodiester bond between PEP and Fo and the formation of DH-F420-0 (Fig. 4E).
DISCUSSION
Integrating these findings with other recent literature, it is now clear that the substrate for the initial stage of F420 tail biosynthesis differs between F420-producing organisms (19, 24, 26). We definitively show here that, in mycobacteria, PEP is the substrate for F420 biosynthesis, resolving the ambiguity in the literature (24, 26). In contrast, in the archaeal and proteobacterial species that have been analyzed, 2PL and 3PG, respectively, are preferentially utilized (19, 26). This divergent substrate utilization occurs despite the enzymes responsible for this stage of synthesis (FbiD/CofC and FbiA/CofD) sharing a common evolutionary history (8). This suggests that the substrate specificity of these enzymes has evolved in response to selection to maintain compatibility between the substrate used for F420 biosynthesis and what is available in the cellular metabolite pool. Both PEP and 3PG are intermediates in central metabolic pathways, including glycolysis, whereas 2PL is not thought to be present in significant quantities in most organisms (24, 26, 32). This makes PEP and 3PG compatible substrates for F420 biosynthesis in Mycobacterium spp. and P. rhizoxinica, respectively, with the specifics of cellular metabolism of each organism likely dictating which compound was selected for F420 biosynthesis. In contrast, in the archaeon Methanobacterium thermoautotrophicum, 2PL is present at micromolar concentrations (19). However, in archaeal species, it remains to be determined how 2PL is synthesized and whether this compound plays a wider role as a general metabolite beyond F420 biosynthesis.
Phylogenetic analysis of FbiD/CofC and FbiA/CofD suggests that these proteins were horizontally transferred between bacteria and archaea (8). Based on this analysis, it is curious that mycobacteria reduce DH-F420-0 produced via PEP to F420, rendering it chemically identical to that produced with 2PL. The redox properties of the deazaflavin group of DH-F420 and F420 are identical, and chemically the molecules are very similar, posing the question: why is reduction of DH-F420-0 is required? A plausible explanation is that actinobacteria originally utilized 2PL for F420 synthesis, with a switch to PEP occurring at a later stage in evolution. As a result, the F420-dependent enzymes present in mycobacteria evolved to recognize the nonplanar 2PL moiety of F420, requiring reduction of DH-F420 to maintain compatibility after the substrate switch. Previous structural and biochemical analysis suggests that the C-terminal domain of mycobacterial FbiB is responsible for the reduction of DH-F420-0 (24, 28). This domain is present in all mycobacterial species but is absent from FbiB/CofE in most other F420-producing organisms, including M. mazei and P. rhizoxinica, that produce F420 through pathways that do not require this reductive step (8, 26). This conclusion is supported by our cellular analysis of F420 biosynthesis in M. smegmatis, which shows that DH-F420-0 accumulates in the ΔfbiB strain (Fig. 2A and D).
The structural analysis of FbiA that we present in this work provides unprecedented insight into the catalytic mechanism for the novel phosphotransferase reaction employed at this step in F420 biosynthesis. The crystal structure of FbiA shows that this enzyme employs a flexible active site to capture Fo and EPPG, precisely positioning them for catalysis. Determination of the fully resolved FbiA substrate complex, in the presence of a single catalytic metal ion, provides a clear picture of the mechanism of catalysis of this enzyme. In this structure, the terminal hydroxyl group of Fo is ideally positioned for nucleophilic attack of the β-phosphate of EPPG, strongly suggesting that DH-F420-0 biosynthesis occurs through direct transfer of PEP to Fo, via a pentavalent phosphate intermediate that is stabilized by the catalytic metal ion. The positioning of the Fo terminal hydroxyl group in our structure in relation to EPPG is strikingly similar to that of the attacking ribose in the final step in DNA ligation by T4 ligase, recently resolved by X-ray crystallography (33). This is consistent with both reactions resulting in the formation of a phosphodiester bond through direct nucleophilic attack of a diphosphonucleoside intermediate. As no acidic side chains are present in proximity of the terminal hydroxyl of Fo in our structure of FbiA, it is likely that deprotonation of this group for nucleophilic attack is EPPG induced, possibly by the PEP carboxyl moiety. These data are also consistent with biochemical analysis of CofD from M. mazei, which did not detect the formation of a catalytic reaction intermediate during the synthesis of F420-0 (21).
The resolution of the F420 biosynthesis pathway also has implications for tuberculosis treatment. It has been proposed that F420 biosynthesis represents a promising target for the development of drugs for the treatment of M. tuberculosis, given the pleiotropic role of this cofactor and its absence from human cells. While nonessential for the growth of mycobacteria under optimal conditions, F420 has been shown to be important for persistence, recovery from dormancy, and antibiotic resistance, and hence is likely to contribute to M. tuberculosis infection (1). Given that FbiA mediates the key step in F420 biosynthesis, the structural insights into FbiA catalysis provide a basis for the development of inhibitory compounds targeting F420 biosynthesis (24). In addition, loss of function of FbiA causes resistance to the clinical nitroimidazole prodrugs delamanid and pretomanid, both of which are activated by the F420H2-dependent reductase Ddn (15, 17, 34). Hence, the structural and mechanistic insights provided here will enable prediction of which substitutions are likely to impair or inactivate FbiA, thus conferring resistance to these compounds.
MATERIALS AND METHODS
Creation of M. smegmatis F420 biosynthesis mutant strains.
M. smegmatis MSMEG_5126 was deleted in wild-type M. smegmatis mc2155 using a two-step allelic replacement strategy. Two 0.8-kb fragments containing sequences from the left and right flanking regions of the MSMEG_5126 (fbiC) gene were cloned as separate constructs and later combined to make the deletion construct. The left flanking fragments were amplified using ProofStart DNA polymerase (Qiagen) with primers MSMEG_5126left and MSMEG_5126leftrev, and the PCR product was subsequently cloned into the SacI/BamHI sites of pUC18, creating plasmid pUC-MSMEG_5126left (additional information on the primers in Table S3 in the supplemental material). A 0.8-kb fragment containing sequence from the right side of MSMEG_5126 was amplified using primers MSMEG_5126right and MSMEG_5126rightrev and cloned into the XbaI/BamHI sites of pUC18, creating plasmid pUC-MSMEG_5126right. The right flanking sequence was then excised from pUC-MSMEG_5126right using XbaI/SacI and subcloned into Xba/SacI-digested pUC-MSMEG_5126left, fusing the left and right flanking sequences to create plasmid pUC-ΔMSMEG_5126.
Oligonucleotide primers utilized in this study. Download Table S3, XLSX file, 0.01 MB (10.9KB, xlsx) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The 1.6-kb fused insert was then liberated using Xba/SacI and subcloned into Xba/SacI-digested pMSS vector (35), a suicide plasmid for M. smegmatis that contains streptomycin selection and sucrose counterselection markers. The resultant plasmid, pMSS:ΔMSMEG_5126, was sequenced and then electroporated into electrocompetent M. smegmatis mc2155 cells, using an ECM 630 electroporator (BTX), selecting for streptomycin-resistant colonies (30 μg/ml), which were then screened for sensitivity to 10% (wt/vol) sucrose. DNA from confirmed streptomycin-resistant, sucrose-sensitive colonies was PCR amplified using primer pair MSMEG_5126screen-F and MSMEG_5126KOright-R. The resultant PCR product was confirmed by DNA sequencing.
A confirmed single crossover (SCO) strain was grown for 3 days in the absence of antibiotic selection, serially diluted, and plated on LB plates containing 10% (wt/vol) sucrose to select for potential double crossover (DCO) strains (i.e., MSMEG_5126 deletion mutants). Genomic DNA was extracted from sucrose-resistant, streptomycin-sensitive clones, digested using ClaI/NcoI, and subjected to Southern blot analysis using an MSMEG_5126-specific probe to confirm deletion of the MSMEG_5126 gene. For Southern blotting, 2 μg of genomic DNA (gDNA) was digested with appropriate restriction enzymes (NEB) at 37°C for 16 h. Purified samples and digoxigenin (DIG)-labeled, HindIII-digested λ DNA markers were separated on a 1% agarose gel, followed by depurination, denaturation, neutralization, and capillary transfer onto a nylon membrane (Thermo Fisher). The membrane was then hybridized at 67°C with a gene-specific probe prepared by DIG labeling a 3.0-kb PCR product obtained using primers MSMEG_5126screen-R and MSMEG_5126screen-F. Once confirmed by Southern blotting, the genomes of mutant strains were sequenced at the Peter Doherty Institute for Infection and Immunity at the University of Melbourne, and mapped to the wild-type strain, confirming that the strains were otherwise isogenic.
MSMEG_1829 (fbiB), MSMEG_2392 (fbiD), and MSMEG_1830 (fbiA) deletion mutants were generated by using the same methods used for MSMEG_5126, using gene-specific primer combinations (Table S3). Individual deletion mutants of fbiA, fbiB, and fbiD were confirmed by Southern blotting following digestion with PvuII and then genome sequencing.
Purification of Fo, DH-F420-0, and F420.
Fo was purified from culture supernatants of M. smegmatis mc2155 overexpressing FbiC from M. tuberculosis cloned into the acetamide-inducible vector pMyNT. The cells were grown at 37°C in 7H9 medium to an optical density at 600 nm (OD600) of ∼3.0 before FbiC expression was induced by the addition of 0.2% acetamide. The cells were grown for an additional 72 h at 37°C with shaking, and supernatant was clarified by centrifugation at 10,000 × g for 20 min. The clarified supernatant was filtered (0.45 μm) and applied to a C18-silica column equilibrated in distilled H2O (dH2O). Bound Fo was eluted with 20% methanol in dH2O, and the solvent was removed by vacuum evaporation. Fo was resuspended in dH2O and centrifuged (20,000 × g for 20 min) to remove insoluble contaminants, before reapplication to a C18-silica column equilibrated in dH2O. Fo was again eluted with 20% methanol in dH2O, vacuum evaporated, and stored at –20°C for further analysis.
F420 was expressed and purified as previously described in M. smegmatis mc24517 overexpressing FbiA, FbiB, and FbiC in the expression vector pYUBDuet-FbiABC (36). The cells were grown in LB broth plus 0.05% Tween 80 at 37°C with shaking to an OD600 of ∼2.0 before the expression of the fbi genes was induced with 0.2% acetamide. The cells were grown for an additional 72 h before harvesting by centrifugation at 10,000 × g for 20 min. The cells were resuspended in 50 mM Tris (pH 7.5) at a ratio of 10 ml of buffer per 1 g of cells (wet weight) and lysed by autoclaving. The autoclaved cell suspension was clarified by centrifugation at 20,000 × g for 20 min. The clarified supernatant was applied to a High Q Anion Exchange Column (Bio-Rad) equilibrated in 50 mM Tris (pH 7.5). Bound species were eluted with a gradient of 0 to 100% of 50 mM Tris and 1 M NaCl (pH 7.5). Fractions containing F420 were identified via visible spectroscopy based on their distinctive absorbance peak at 420 nm. Fractions containing F420 were pooled and applied to a C18-silica column equilibrated in dH2O. F420 was eluted with 20% methanol in H2O, vacuum evaporated, and stored at –20°C for further analysis.
DH-F420-0 was extracted from purified FbiA expressed in M. smegmatis as described below. Purified concentrated FbiA (∼20 mg ml−1; prior to cleavage of the polyhistidine tag) was denatured in a buffer containing 50 mM Tris and 8 M urea (pH 7.0). This solution containing denatured FbiA and free DH-F420-0 was applied to a nickel-agarose column, with denatured FbiA binding to the column due to its hexahistidine (hexahis) tag and DH-F420-0 eluting in the flowthrough. The flowthrough containing DH-F420-0 was applied to a Superdex 30 10/300 column equilibrated in dH2O, and eluted fractions containing DH-F420-0 were identified based on their absorbance at 420 nM. DH-F420-0-containing fractions were pooled, vacuum evaporated, resuspended in 500 μl of dH2O, and reapplied to the Superdex 30 10/300 column equilibrated in 20% acetonitrile in dH2O. DH-F420-0-containing fractions were then pooled, vacuum evaporated, and stored at –20°C for further analysis.
FbiA expression and purification.
The DNA coding sequence corresponding to FbiA from M. smegmatis was amplified by PCR using the primers in Table S3, resulting in a DNA fragment with 5′ NcoI and 3′ HindIII sites, respectively. This fragment was cloned into pMyNT by restriction enzyme cloning using the aforementioned sites, yielding pMyNTFbiAMS, which expresses FbiA with a tobacco etch virus (TEV)-cleavable N-terminal hexahis tag. This vector was cloned and propagated in Escherichia coli DH5α in LB medium/agar with the addition of 200 μg ml−1 hygromycin B. Sequence-confirmed pMyNTFbiAMS was transformed into M. smegmatis mc2155 via electroporation, with successful transformants selected for in LB plus 0.05% Tween 80 (LBT) agar in the presence of 50 μg ml−1 hygromycin B. Colonies from this transformation were used to inoculate 50 ml of LBT medium plus 50 μg ml−1 hygromycin B, which was grown with shaking at 37°C until stationary phase (2 or 3 days). This starter culture was used to inoculate 5 liters of terrific broth plus 0.05% Tween 80 (TBT), giving a 1:100 dilution of the starter culture. The cells were grown with shaking at 37°C for 24 h until approximately mid-log phase, and protein production was induced through the addition of 0.2% acetamide. The cells were grown with shaking at 37°C for an additional 72 h before they were harvested via centrifugation at 5,000 × g for 20 min. Harvested cells were either lysed immediately or stored frozen at –20°C.
The cells were resuspended in Ni binding buffer (20 mM HEPES, 300 mM NaCl, 20 mM imidazole; pH 7.5) at a ratio of approximately 5 ml of buffer per 1 g of cells (wet weight). Lysozyme (1 mg ml−1), DNase (0.5 mg ml−1), and complete protease inhibitor tablets (Roche) were added, and cells were lysed with a cell disruptor (Constant Systems). The cell lysate was stored on ice and clarified by centrifugation at 4°C at 30,000 × g. Clarified lysate was passed through a column containing Ni2+ agarose resin equilibrated in Ni binding buffer. The column was washed with Ni binding buffer, and protein was eluted with a gradient of Ni gradient buffer (20 mM HEPES, 300 mM NaCl, 500 mM imidazole; pH 7.5). Fractions containing FbiA were identified based on absorbance at 280 nm and their yellow color due to F420 precursor copurification and pooled. Pooled fractions were applied to a Superdex S200 26/600 size exclusion chromatography (SEC) column, equilibrated with SEC buffer (20 mM HEPES, 150 mM NaCl; pH 7.5), and fractions containing FbiA were identified as described above and pooled. The hexahis tag was cleaved from purified FbiA through the addition of 0.5 mg of hexahistidine-tagged TEV protease (expressed and purified as described in reference 37) per mg of FbiA, plus 1 mM dithiothreitol (DTT). Digestion was performed at room temperature for ∼6 h before the sample was passed through a Ni2+ agarose column to remove TEV and the cleaved hexahis tag. The resulting flowthrough from this column was collected, concentrated to ∼15 mg ml−1, and snap-frozen at –80°C. Purified FbiA was light yellow in color due to copurification with F420 precursors, with a yield of 5 to 10 mg per liter of culture. The molecular weight of purified FbiA was determined by size exclusion coupled to multiangle laser light scattering (SEC-MALS), using a Superdex S200 Increase 10/300 column equilibrated in 200 mM NaCl and 50 mM Tris (pH 7.9), coupled to fast performance liquid chromatography (FPLC) (Shimadzu) with MALS detection (Wyatt Technology).
FbiA crystallization, ligand soaking, and structure solution.
Purified FbiA was screened for crystallization conditions using a sparse matrix approach, with approximately 600 individual conditions screened. Thin intergrown plate crystals of FbiA formed under a number of conditions, with a condition containing 0.1 M Tris (pH 8.0), 0.2 M Ca acetate and 20% polyethylene glycol 3350 (PEG 3350) chosen for optimization. Diffraction quality crystals were obtained by microseeding into 0.1 M Tris (pH 8.5), 0.2 M Ca acetate, and 16% PEG 3350 with or without 20% glycerol. Crystals grew as bunches of very thin plates and were slightly yellow in color. Crystals from conditions containing glycerol were looped and directly flash cooled to 100 K in liquid N2, providing “apo” crystals for data collection. Crystals from non-glycerol-containing wells were transferred into well solution with 20% glycerol and either Fo, GDP, and Fo and GDP or DH-F420-0. Crystals were incubated in this solution for 1 to 5 min before they were looped and flash cooled to 100 K in liquid N2.
Data were collected at the Australian Synchrotron, with crystals diffracting anisotropically to ∼2.2 to 3.0 Å, and processed using XDS and merged using Aimless from the CCP4 package (38, 39). The structure of FbiA was solved by molecular replacement using Phaser (40), with a search model derived from the structure of CofD from M. mazei (PDB identifier [ID] 3C3D) prepared based on the amino acid sequence for FbiA from M. smegmatis using sculptor from the Phenix package (40). Native and ligand-soaked structures of FbiA were built and refined using Coot and phenix.refine from the Phenix package (40, 41). Structural coordinates for Fo, DH-F420-0 and F420 were generated using the AceDrg program within the CCP4 suite (39, 42).
LC-MS detection of F420 and precursors.
Wild-type and Fbi mutant M. smegmatis mc2155 strains were grown in 20 ml of LBT medium until stationary phase (2 or 3 days) and harvested by centrifugation at 5,000 × g for 20 min. The cells were resuspended in 2 ml of dH2O and lysed by boiling for 5 min before clarification by centrifugation at 25,000 × g for 10 min. The soluble fraction was then decanted for mass spectrometry analysis. Samples were analyzed by hydrophilic interaction liquid chromatography (HILIC) coupled to high-resolution mass spectrometry (LC-MS) according to a previously published method (43). In brief, the chromatography utilized a guard (20 by 2.1 mm) in series with an analytical column (150 by 4.6 mm) (both ZIC-pHILIC; Merck). The column temperature was maintained at 25°C with a gradient elution of 20 mM ammonium carbonate (solution A) and acetonitrile (solution B) (linear gradient time−percent solution B as follows: 0 min−80%, 15 min−50%, 18 min−5%, 21 min−5%, 24 min−80%, 32 min−80%) on a Dionex RSLC3000 ultrahigh performance liquid chromatograph (UHPLC) (Thermo). The flow rate was maintained at 300 μl min−1. Samples were kept at 4°C in the autosampler, and 10 μl was injected for analysis. The mass spectrometric acquisition was performed at 35,000 resolution on a Q-Exactive Orbitrap MS (Thermo) operating in rapid switching positive (4 kV) and negative (−3.5 kV) mode electrospray ionization (capillary temperature, 300°C; sheath gas, 50; auxiliary [Aux] gas, 20; sweep gas, 2; probe temperature, 120°C). The resulting LC-MS data were processed by integrating the area below the extracted ion chromatographic peaks using TraceFinder 4.1 (Thermo Scientific). All species were detected in negative mode as the singly deprotonated anion (Fo and DH-F420-0) or in the case of the F420-n species the double deprotonated dianion.
2-Phospholactate synthesis.
Synthesis of 2-phospholactate was achieved in high-yield via one-step catalytic hydrogenation of phosphoenolpyruvate as previously described (44). Briefly, 26 mg of phosphoenolpyruvate monosodium salt and 12 mg palladium on carbon (Pd/C) (10% wet concentration) were transferred into a 25 ml round bottom flask and dissolved in 5 ml water. After fixing the flask with a hydrogen bag, the solution was stirred for 5 h at room temperature and 1 atm pressure. The solution was filtered, dried by rotary evaporation, and stored at –20°C. Successful production of a racemic mix of 2-phospholactate was verified by mass spectrometry and 1H NMR.
Stimulation of DH-F420-0 production in spiked M. smegmatis cell lysates and detection of F420 species by HPLC.
To detect F420 synthesis in spiked M. smegmatis lysates, 500-ml cultures were grown in LBT for 3 days at 37°C with shaking. The cells were harvested by centrifugation at 8,000 × g for 20 min at 4°C. The pellet was resuspended in 50 ml lysis buffer (50 mM morpholinepropanesulfonic acid [MOPS], 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM DTT, 5 mM MgCl2, 2.5 mg · ml−1 lysozyme, 2.5 mg DNase I). An M-110P Microfluidizer (Fluigent) pressure-lysis maintained at 4°C was used to lyse the cells. The lysate was centrifuged at 10,000 × g at 4°C for 20 min. One-milliliter aliquots of lysate were spiked with either 1 mM phosphate buffer (pH 7.0) plus GTP and 2PL or GTP and PEP. These spiked samples, along with a “no spike” control, were incubated at 4 h at 37°C. To terminate the reaction, the aliquots were heated at 95°C for 20 min and then centrifuged at 16,000 × g for 10 min. The supernatants were filtered through a 0.22-μm polyvinylidene difluoride (PVDF) filter and moved to analytical vials.
F420 biosynthetic intermediates present in the filtered M. smegmatis cell lysates were analyzed by separation and detection using an Agilent 1200 series HPLC system equipped with a fluorescence detector and a Poroshell 120 EC-C18 column (2.1 by 50 mm; 2.7 μm). The system was run at a flow rate of 0.3 ml min−1, and the samples were excited at 420 nm, and emission was detected at 480 nm. A gradient of two buffers were used: buffer A containing 20 mM ammonium phosphate and 10 mM tetrabutylammonium phosphate, pH 7.0, and buffer B, 100% acetonitrile. A gradient was run from 25% to 40% buffer B as follows: 0 to 1 min, 25%; 1 to 10 min, 25% to 35%; 10 to 13 min, 35%; 13 to 16 min, 35 to 40%; and 16 to 19 min, 40% to 25%.
Data availability.
The crystallographic coordinates and associated structure factors for the FbiA structures produced in this study are available in the Protein Data Bank (PDB) with the following accession codes: FbiA Apo, 6UVX; FbiA plus Fo, 6UW1; FbiA plus GDP, 6UW3; FbiA plus GDP plus Fo, 6UW5; FbiA plus DH-F420-0, 6UW7.
ACKNOWLEDGMENTS
This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. We thank the Monash Crystallisation Facility for their assistance with sample characterization, crystallographic screening, and optimization. pMyNT was a gift from Katherine Beckham, Annabel Parret, and Matthias Wilmanns at EMBL Hamburg. We thank Ghader Bashiri for helpful discussions, Matthew Belousoff for his assistance in the development of F420 derivative purification protocols, and Phillip Holt in the School of Chemistry, Monash University, for his assistance with fluorescence HPLC analysis.
This work was supported by an ARC DECRA Fellowship (DE170100310; awarded to C.G.), an NHMRC EL2 Fellowship (APP1178715; awarded to C.G.), an NHMRC New Investigator Grant (APP1142699; awarded to C.G.), an NHMRC grant (APP1139832; awarded to C.J.J., C.G., and G.M.C.), a Monash University Science-Medicine Seed Grant (awarded to C.G. and M.J.C.), Monash University Doctoral Scholarships (awarded to P.R.F.C. and D.L.G.), and an ARC Discovery Grant (DP180102463; awarded to R.L.C.).
C.G. and R.G. conceived and supervised the study. Different authors contributed to cellular spiking assays (B.N., C.G., C.J.J., A.C.W., and M.C.T.), knockout construction (C.G., R.B., R.L.C., P.K.C., G.M.C., J.G.O., M.C.T., L.K.H., G.T., and D.A.W.), LC-MS analysis (C.K.B., R.G., R.B.S., and C.G.), protein purification (R.G., C.G., B.N., P.R.F.C., and C.J.J.), and crystallographic analysis (R.G., P.R.F.C., D.L.G., C.G., T.I., and M.J.C.). R.G. and C.G. analyzed data and wrote the paper with input from all authors.
We declare that we have no competing financial interests.
REFERENCES
- 1.Greening C, Ahmed FH, Mohamed AE, Lee BM, Pandey G, Warden AC, Scott C, Oakeshott JG, Taylor MC, Jackson CJ. 2016. Physiology, biochemistry, and applications of F420-and Fo-dependent redox reactions. Microbiol Mol Biol Rev 80:451–493. doi: 10.1128/MMBR.00070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eirich LD, Vogels GD, Wolfe RS. 1978. Proposed structure for coenzyme F420 from Methanobacterium. Biochemistry 17:4583–4593. doi: 10.1021/bi00615a002. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson F, Walsh C. 1984. Properties of 7,8-didemethyl-8-hydroxy-5-deazaflavins relevant to redox coenzyme function in methanogen metabolism. Biochemistry 23:979–988. doi: 10.1021/bi00300a028. [DOI] [Google Scholar]
- 4.Edmondson DE, Barman B, Tollin G. 1972. Importance of the N-5 position in flavine coenzymes. Properties of free and protein-bound 5-deaza analogs. Biochemistry 11:1133–1138. doi: 10.1021/bi00757a003. [DOI] [PubMed] [Google Scholar]
- 5.Taylor MC, Jackson CJ, Tattersall DB, French N, Peat TS, Newman J, Briggs LJ, Lapalikar GV, Campbell PM, Scott C, Russell RJ, Oakeshott JG. 2010. Identification and characterization of two families of F420H2-dependent reductases from mycobacteria that catalyse aflatoxin degradation. Mol Microbiol 78:561–575. doi: 10.1111/j.1365-2958.2010.07356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W, Khullar A, Chou S, Sacramo A, Gerratana B. 2009. Biosynthesis of sibiromycin, a potent antitumor antibiotic. Appl Environ Microbiol 75:2869–2878. doi: 10.1128/AEM.02326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greening C, Jirapanjawat T, Afroze S, Ney B, Scott C, Pandey G, Lee BM, Russell RJ, Jackson CJ, Oakeshott JG, Taylor MC, Warden AC. 2017. Mycobacterial F420H2-dependent reductases promiscuously reduce diverse compounds through a common mechanism. Front Microbiol 8:1000. doi: 10.3389/fmicb.2017.01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ney B, Ahmed FH, Carere CR, Biswas A, Warden AC, Morales SE, Pandey G, Watt SJ, Oakeshott JG, Taylor MC, Stott MB, Jackson CJ, Greening C. 2017. The methanogenic redox cofactor F420 is widely synthesized by aerobic soil bacteria. ISME J 11:125–137. doi: 10.1038/ismej.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu W-T, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 10.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, Hooper SD, Pati A, Lykidis A, Spring S, Anderson IJ, D’haeseleer P, Zemla A, Singer M, Lapidus A, Nolan M, Copeland A, Han C, Chen F, Cheng J-F, Lucas S, Kerfeld C, Lang E, Gronow S, Chain P, Bruce D, Rubin EM, Kyrpides NC, Klenk H-P, Eisen JA. 2009. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spang A, Poehlein A, Offre P, Zumbrägel S, Haider S, Rychlik N, Nowka B, Schmeisser C, Lebedeva EV, Rattei T, Böhm C, Schmid M, Galushko A, Hatzenpichler R, Weinmaier T, Daniel R, Schleper C, Spieck E, Streit W, Wagner M. 2012. The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ Microbiol 14:3122–3145. doi: 10.1111/j.1462-2920.2012.02893.x. [DOI] [PubMed] [Google Scholar]
- 12.Gurumurthy M, Rao M, Mukherjee T, Rao SPS, Boshoff HI, Dick T, Barry CE, Manjunatha UH. 2013. A novel F420-dependent anti-oxidant mechanism protects M ycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol Microbiol 87:744–755. doi: 10.1111/mmi.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jirapanjawat T, Ney B, Taylor MC, Warden AC, Afroze S, Russell RJ, Lee BM, Jackson CJ, Oakeshott JG, Pandey G, Greening C. 2016. The redox cofactor F420 protects mycobacteria from diverse antimicrobial compounds and mediates a reductive detoxification system. Appl Environ Microbiol 82:6810–6818. doi: 10.1128/AEM.02500-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purwantini E, Mukhopadhyay B. 2013. Rv0132c of Mycobacterium tuberculosis encodes a coenzyme F420-dependent hydroxymycolic acid dehydrogenase. PLoS One 8:e81985. doi: 10.1371/journal.pone.0081985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cellitti SE, Shaffer J, Jones DH, Mukherjee T, Gurumurthy M, Bursulaya B, Boshoff HI, Choi I, Nayyar A, Lee YS, Cherian J, Niyomrattanakit P, Dick T, Manjunatha UH, Barry CE, Spraggon G, Geierstanger BH. 2012. Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 20:101–112. doi: 10.1016/j.str.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohamed A, Ahmed F, Arulmozhiraja S, Lin C, Taylor M, Krausz E, Jackson C, Coote M. 2016. Protonation state of F420H2 in the prodrug-activating deazaflavin dependent nitroreductase (Ddn) from Mycobacterium tuberculosis. Mol Biosyst 12:1110–1113. doi: 10.1039/c6mb00033a. [DOI] [PubMed] [Google Scholar]
- 17.Lee BM, Almeida DV, Afriat-Jurnou L, Aung HL, Forde BM, Hards K, Pidot SJ, Ahmed FH, Mohamed AE, Taylor MC. 2019. The evolution of nitroimidazole antibiotic resistance in Mycobacterium tuberculosis. bioRxiv doi: 10.1101/631127. [DOI] [PMC free article] [PubMed]
- 18.Graham DE, Xu H, White RH. 2003. Identification of the 7,8-didemethyl-8-hydroxy-5-deazariboflavin synthase required for coenzyme F420 biosynthesis. Arch Microbiol 180:455–464. doi: 10.1007/s00203-003-0614-8. [DOI] [PubMed] [Google Scholar]
- 19.Graupner M, White RH. 2001. Biosynthesis of the phosphodiester bond in coenzyme F420 in the methanoarchaea. Biochemistry 40:10859–10872. doi: 10.1021/bi0107703. [DOI] [PubMed] [Google Scholar]
- 20.Grochowski LL, Xu H, White RH. 2008. Identification and characterization of the 2-phospho-L-lactate guanylyltransferase involved in coenzyme F420 biosynthesis. Biochemistry 47:3033–3037. doi: 10.1021/bi702475t. [DOI] [PubMed] [Google Scholar]
- 21.Graupner M, Xu H, White RH. 2002. Characterization of the 2-phospho-L-lactate transferase enzyme involved in coenzyme F420 biosynthesis in Methanococcus jannaschii. Biochemistry 41:3754–3761. doi: 10.1021/bi011937v. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Graupner M, Xu H, White RH. 2003. CofE catalyzes the addition of two glutamates to F420-0 in F420 coenzyme biosynthesis in Methanococcus jannaschii. Biochemistry 42:9771–9778. doi: 10.1021/bi034779b. [DOI] [PubMed] [Google Scholar]
- 23.Nocek B, Evdokimova E, Proudfoot M, Kudritska M, Grochowski L, White R, Savchenko A, Yakunin A, Edwards A, Joachimiak A. 2007. Structure of an amide bond forming F420: γγ-glutamyl ligase from Archaeoglobus fulgidus−a member of a new family of non-ribosomal peptide synthases. J Mol Biol 372:456–469. doi: 10.1016/j.jmb.2007.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bashiri G, Antoney J, Jirgis ENM, Shah MV, Ney B, Copp J, Stuteley SM, Sreebhavan S, Palmer B, Middleditch M, Tokuriki N, Greening C, Scott C, Baker EN, Jackson CJ. 2019. A revised biosynthetic pathway for the cofactor F420 in prokaryotes. Nat Commun 10:1558. doi: 10.1038/s41467-019-09534-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forouhar F, Abashidze M, Xu H, Grochowski LL, Seetharaman J, Hussain M, Kuzin A, Chen Y, Zhou W, Xiao R, Acton TB, Montelione GT, Galinier A, White RH, Tong L. 2008. Molecular insights into the biosynthesis of the F420 coenzyme. J Biol Chem 283:11832–11840. doi: 10.1074/jbc.M710352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braga D, Last D, Hasan M, Guo H, Leichnitz D, Uzum Z, Richter I, Schalk F, Beemelmanns C, Hertweck C, Lackner G. 2019. Metabolic pathway rerouting in Paraburkholderia rhizoxinica evolved long-overlooked derivatives of coenzyme F420. ACS Chem Biol 14:2088–2094. doi: 10.1021/acschembio.9b00605. [DOI] [PubMed] [Google Scholar]
- 27.Bair TB, Isabelle DW, Daniels L. 2001. Structures of coenzyme F420 in Mycobacterium species. Arch Microbiol 176:37–43. doi: 10.1007/s002030100290. [DOI] [PubMed] [Google Scholar]
- 28.Bashiri G, Rehan AM, Sreebhavan S, Baker HM, Baker EN, Squire CJ. 2016. Elongation of the poly-γ-glutamate tail of F420 requires both domains of the F420: γ-glutamyl ligase (FbiB) of Mycobacterium tuberculosis. J Biol Chem 291:6882–6894. doi: 10.1074/jbc.M115.689026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 30.Grinter R, Roszak AW, Cogdell RJ, Milner JJ, Walker D. 2012. The crystal structure of the lipid II-degrading bacteriocin syringacin M suggests unexpected evolutionary relationships between colicin M-like bacteriocins. J Biol Chem 287:38876–38888. doi: 10.1074/jbc.M112.400150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knape MJ, Ahuja LG, Bertinetti D, Burghardt NC, Zimmermann B, Taylor SS, Herberg FW. 2015. Divalent metal ions Mg2+ and Ca2+ have distinct effects on protein kinase A activity and regulation. ACS Chem Biol 10:2303–2315. doi: 10.1021/acschembio.5b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fothergill-Gilmore LA, Michels PA. 1993. Evolution of glycolysis. Prog Biophys Mol Biol 59:105–235. doi: 10.1016/0079-6107(93)90001-z. [DOI] [PubMed] [Google Scholar]
- 33.Shi K, Bohl TE, Park J, Zasada A, Malik S, Banerjee S, Tran V, Li N, Yin Z, Kurniawan F, Orellana K, Aihara H. 2018. T4 DNA ligase structure reveals a prototypical ATP-dependent ligase with a unique mode of sliding clamp interaction. Nucleic Acids Res 46:10474–10488. doi: 10.1093/nar/gky776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haver HL, Chua A, Ghode P, Lakshminarayana SB, Singhal A, Mathema B, Wintjens R, Bifani P. 2015. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:5316–5323. doi: 10.1128/AAC.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cashmore TJ, Klatt S, Yamaryo-Botte Y, Brammananth R, Rainczuk AK, McConville MJ, Crellin PK, Coppel RL. 2017. Identification of a membrane protein required for lipomannan maturation and lipoarabinomannan synthesis in Corynebacterineae. J Biol Chem 292:4976–4986. doi: 10.1074/jbc.M116.772202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bashiri G, Rehan AM, Greenwood DR, Dickson JM, Baker EN. 2010. Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLoS One 5:e15803. doi: 10.1371/journal.pone.0015803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tropea JE, Cherry S, Waugh DS. 2009. Expression and purification of soluble His 6-tagged TEV protease, p 297–307. In Doyle SA. (ed), High throughput protein expression and purification: methods and protocols. Humana Press, Springer, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 38.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long F, Nicholls RA, Emsley P, Graǽulis S, Merkys A, Vaitkus A, Murshudov GN. 2017. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr D Struct Biol 73:112–122. doi: 10.1107/S2059798317000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoessel D, Nowell CJ, Jones AJ, Ferrins L, Ellis KM, Riley J, Rahmani R, Read KD, McConville MJ, Avery VM, Baell JB, Creek DJ. 2016. Metabolomics and lipidomics reveal perturbation of sphingolipid metabolism by a novel anti-trypanosomal 3-(oxazolo [4,5-b] pyridine-2-yl) anilide. Metabolomics 12:126. doi: 10.1007/s11306-016-1062-1. [DOI] [Google Scholar]
- 44.Shchepin RV, Coffey AM, Waddell KW, Chekmenev EY. 2012. PASADENA hyperpolarized 13C phospholactate. J Am Chem Soc 134:3957–3960. doi: 10.1021/ja210639c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fluorescence-coupled HPLC analysis of clarified cell lysates from M. smegmatis F420 biosynthesis pathway mutants. Fluorescence (excitation wavelength [Ex λ] of 420 nm; emission wavelength [Em λ] of 480 nm) trace for wild-type (A), ΔfbiC (B), ΔfbiA (C), ΔfbiD (D), and ΔfbiB (E) strains showing the formation of mature F420 species in the wild-type strain only and accumulation of DH-F420-0 in the ΔfbiB strain. LU, fluorescence intensity. For ΔfbiC, shown in panel B, mass spectrometry analysis confirmed that no Fo or F420 species were present, despite observed low-level fluorescence. Download FIG S1, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
FbiA recombinantly expressed in M. smegmatis copurifies with its product DH-F420-0. (A) Purified, concentrated (16 mg · ml−1) recombinant FbiA produced in M. smegmatis showing a characteristic yellow color associated with a bound F420 species. (B) The absorbance (purple) and fluorescence spectra (blue) of purified FbiA from panel A, which is characteristic of F420 species with a protonated deazaflavin 8-OH group. (C) LC-MS analysis of F420 species bound to purified FbiA, showing that DH-F420-0 is the predominantly bound species and the presence of some mature F420 species. (D) SEC-MALS analysis of purified FbiA shows that it has a molecular weight in solution consistent with a homodimeric species. Download FIG S2, TIF file, 2.3 MB (2.4MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Electron density corresponding to FbiA substrates and products in cocrystal structures. The panel corresponds to the cocrystal structures indicated in the bottom right. A composite omit map is shown carved to visible molecules at a distance of 2 Å and contoured to 1 σ. Download FIG S3, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Crystallographic data collections and refinement statistics. Download Table S1, XLSX file, 0.01 MB (12.1KB, xlsx) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
FbiA dimer interface statistics reported by PISA (29). Download Table S2, XLSX file, 0.01 MB (10KB, xlsx) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The crystal structure of FbiA in complex with Fo and GDP. (A) GDP in complex with Mol. B of FbiA. FbiA is represented as a sky blue cartoon, bound GDP and glycerol molecules are shown as stick models, and bound Ca2+ ion is shown as a yellow sphere. (B) A stereoview of the catalytic center of the FbiA active site in complex with GDP and glycerol, showing FbiA side chains involved in coordinating the catalytic metal ion and a coordinating H2O molecule. Bond distances of <3.2 Å are shown as yellow dashed lines. (C) Fo in complex with Mol. B of FbiA is shown as a sky blue cartoon, and Fo is shown as a stick model. Download FIG S4, TIF file, 2.7 MB (2.8MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The full interaction network of the FbiA active site in the presence of substrates (Fo and GDP) and product (DH-F420-0). Download FIG S5, TIF file, 2.0 MB (2.1MB, tif) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotide primers utilized in this study. Download Table S3, XLSX file, 0.01 MB (10.9KB, xlsx) .
Copyright © 2020 Grinter et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The crystallographic coordinates and associated structure factors for the FbiA structures produced in this study are available in the Protein Data Bank (PDB) with the following accession codes: FbiA Apo, 6UVX; FbiA plus Fo, 6UW1; FbiA plus GDP, 6UW3; FbiA plus GDP plus Fo, 6UW5; FbiA plus DH-F420-0, 6UW7.