

Graphical abstract

Keywords: Coronavirus, COVID-19, SARS-CoV-2, SARS-CoV-2 main protease, α-ketoamide, molecular docking, molecular dynamics simulations
Highlights
-
•
The SARS-CoV-2 main protease (Mpro) is an important target against COVID-19.
-
•
Peptidomimetic α-ketoamides represent prototypical inhibitors of the SARS-CoV-2 Mpro.
-
•
We confirmed the high affinity of the α-ketoamide 13b inhibitor to SARS-CoV-2 Mpro
-
•
MD simulations highlight α-ketoamide 13b stability in Mpro compared to amoxicillin
-
•
The α-ketoamide compound class could be pursued as potential antivirals against COVID-19
Abstract
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) causes an illness known as COVID-19, which has been declared a global pandemic with over 2 million confirmed cases and 137,000 deaths in 185 countries and regions at the time of writing (16 April 2020), over a quarter of these cases being in the United States. In the absence of a vaccine, or an approved effective therapeutic, there is an intense interest in repositioning available drugs or designing small molecule antivirals. In this context, in silico modelling has proven to be an invaluable tool. An important target is the SARS-CoV-2 main protease (Mpro), involved in processing translated viral proteins. Peptidomimetic α-ketoamides represent prototypical inhibitors of Mpro. A recent attempt at designing a compound with enhanced pharmacokinetic properties has resulted in the synthesis and evaluation of the α-ketoamide 13b analogue. Here, we performed molecular docking and molecular dynamics simulations to further characterize the interaction of α-ketoamide 13b with the active site of the SARS-CoV-2 Mpro. We included the widely used antibiotic, amoxicillin, for comparison. Our findings indicate that α-ketoamide 13b binds more tightly (predicted GlideScore = -8.7 and -9.2 kcal/mol for protomers A and B, respectively), to the protease active site compared to amoxicillin (-5.0 and -4.8 kcal/mol). Further, molecular dynamics simulations highlight the stability of the interaction of the α-ketoamide 13b ligand with the SARS-CoV-2 Mpro (ΔG = -25.2 and -22.3 kcal/mol for protomers A and B). In contrast, amoxicillin interacts unfavourably with the protease (ΔG = +32.8 kcal/mol for protomer A), with unbinding events observed in several independent simulations. Overall, our findings are consistent with those previously observed, and highlight the need to further explore the α-ketoamides as potential antivirals for this ongoing COVID-19 pandemic.
1. Introduction
At the end of 2019 on December 31st, a cluster of patients with pneumonia of unknown cause in the city of Wuhan, Hubei province of China were reported to the World Health Organization by national authorities in China (World Health Organization, 2020). A novel coronavirus was isolated and designated severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), causing coronavirus disease 2019 (COVID-19). As of April 16, 2020, this ongoing global health emergency has resulted in over 2,000,000 confirmed cases in 185 countries and regions, with more than 25% of confirmed cases in the United States (Dong et al., 2020). The global mortality rate has been estimated to be 5.7%, with higher mortality occurring among the elderly (Baud et al., 2020). The majority of deaths have occurred among adults aged greater than 60 years and those with serious underlying health conditions, with the highest fatality in those aged greater than 85 years ranging from 10% to 27% in the United States (CDC COVID-19 Response Team, 2020; Novel Coronavirus Pneumonia Emergency Response Epidemiology Team, 2020). Differences in disease prevalence are affected by sex, with data indicating that there is a higher prevalence of COVID-19 among men (Cai, 2020; Wang et al., 2020). The majority of early cases were linked to exposure to the Huanan Seafood Wholesale Market, potentially through zoonotic transmission (Li et al., 2020). Human-to-human transmission of SARS-CoV-2 was subsequently found to occur, with an attack rate within families of 83% suggestive of its high transmissibility (JF-W et al., 2020; Yuen et al., 2020).
The current outbreak of SARS-CoV-2 follows that of recent outbreaks of severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 and the Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012 (Munster et al., 2020). These coronaviruses are both zoonotic pathogens, with bats serving as the primary reservoir (de Wit et al., 2016). Masked palm civets were the intermediate reservoir for SARS-CoV, and dromedary camels for MERS-COV, where zoonotic transmission to humans subsequently occurred (de Wit et al., 2016). While SARS-CoV-2 appears to have lower fatality rates than SARS-CoV (9.5%) and MERS-CoV (34.4%), it has a greater ability to spread (Munster et al., 2020; Rajgor et al., 2020). Like SARS-CoV, the pathogenesis of SARS-CoV-2 involves the binding of its spike protein to angiotensin converting enzyme-2 (ACE2) in the host (Hoffmann et al., 2020; Walls et al., 2020). When cleavage occurs between the S1 and S2 subunits, the spike protein becomes activated for membrane fusion for entry into the host cell (Hoffmann et al., 2020; Walls et al., 2020). ACE2 is expressed on numerous tissues in the nasopharynx and intestinal epithelia, particularly in type II alveolar cells in the lung (Uhal et al., 2011; Mossel et al., 2008; Xu et al., 2020). Following entry of the virus into the host cells, viral RNA attaches to the host ribosome for translation of large polyproteins that are processed via proteolysis into components for new virions (Hilgenfeld, 2014; Morse et al., 2020). Along with the papain-like protease, the coronavirus main protease (Mpro) is responsible for this proteolysis (Hilgenfeld, 2014). Encoded by open reading frame 1 (ORF1) of the genome as non-structural protein 5 (Nsp5), Mpro cleaves at 11 sites in the polyproteins (Hilgenfeld, 2014).
To date, there is an absence of a vaccine and a lack of effective antiviral therapeutics against SARS-CoV-2. Therefore, there is an intense interest in identifying compounds that may interact with key viral molecular targets. Due to their functional importance and high degree of conservation among coronaviruses, Mpros have become an important target in the design of anti-coronaviral drugs (Hilgenfeld, 2014; Xue et al., 2008). The structure of the SARS-CoV-2 Mpro was initially solved by Jin et al. in late January of this year (Jin et al., 2020), accelerating the search for drugs that may act as lead compounds. Following the 2002 SARS outbreak, work by Hilgenfeld at al. aimed at designing compounds with broad-spectrum anti-coronaviral activity, focussing on main proteases (Hilgenfeld, 2014; Anand et al., 2003). Previously, they found that peptidomimetic α-ketoamides were potential candidates for broad-spectrum inhibitors of coronavirus and enterovirus replication (Zhang et al., 2020a). Most recently, work aimed at improving the biological properties to produce an inhibitor specific for the SARS-CoV-2 Mpro resulted in the potential antiviral agent α-ketoamide 13b (Zhang et al., 2020b). It was found that compound 13b demonstrates binds to the substrate-binding cleft and exhibits antiviral activity in vitro, inhibiting the SARS-CoV-2 Mpro with IC50 = 0.67 ± 0.18 μM (Zhang et al., 2020b).
Here, our aim was to further investigate the interaction of the α-ketoamide 13b with the SARS-CoV-2 Mpro in silico. To highlight the importance of molecular dynamics simulations, we compared the properties of α-ketoamide 13b, with one of the most widely prescribed antibiotics, amoxicillin. Amoxicillin was chosen for comparison for two reasons: 1) although it does not possess antiviral properties, it remains a mainstay as a frontline therapy for viral infections, including COVID-19, presumably to protect from opportunistic secondary bacterial infections, and 2) our initial screening indicates that it binds to the active site of the SARS-CoV-2 Mpro akin to α-ketoamide 13b, albeit with lower affinity.
2. Methods
2.1. Docking to the active site of the SARS-CoV-2 Mpro
Preparation of systems and docking calculations were carried out using the Schrodinger Suite (Schrödinger, 2018) molecular modelling package (version 2018-1) using default parameters unless otherwise specified. The PDBePISA (Proteins, Interfaces, Structures and Assemblies) server (Krissinel and Henrick, 2007) was used to assemble a homodimer complex of the SARS-CoV-2 Mpro (PDB ID: 6LU7). Crystallographic water molecules were removed. The homodimer protein complex was prepared using the Protein Preparation Wizard (Halgren, 2009). This was used to assign bond orders, add hydrogens, create zero-order bonds to metals, and create disulphide bonds. Hydrogen bonds were assigned and optimised, followed by restrained energy minimization. Ligand structures were pre-processed using LigPrep (Sastry et al., 2013), for the generation of ionization and stereoisomer variants of input molecules to obtain structures with optimised geometry.
Two receptor grids of 20 × 20 x 20 Å in size were generated around the active site of the protease, centroid to residues GLY-23, THR-24, GLY-143, HIS-163, THR-190, and ALA-191 on each chain. Ligands were docked to each chain separately. Docking was carried out using the Quantum Mechanics-Polarized Ligand Docking (QPLD) workflow (Cho et al., 2005) of Schrodinger. Initial docking was performed using the extra precision (XP) scoring function of Glide (Friesner et al., 2006). Partial charges on ligand atoms were then calculated using quantum mechanical methods using the ‘accurate’ setting in Jaguar (Bochevarov et al., 2013). Ligands were re-docked using the calculated charges with XP docking mode of Glide, and the final pose was selected based on GlideScore (Table 1 ).
Table 1.
Binding affinity calculated using molecular docking to the active site with QPLD in kcal/mol.
| Protomer A | Protomer B | Structure | |
|---|---|---|---|
| α-ketoamide 13b | −8.7 | −9.2 | 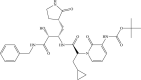 |
| Amoxicillin | −5.0 | −4.8 | 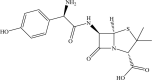 |
2.2. Molecular dynamics (MD) simulations
Classical MD simulations were performed using GROMACS 2018.2 software (Berendsen et al., 1995; Abraham et al., 2015) with the CHARMM27 force field (Bjelkmar et al., 2010; Vanommeslaeghe et al., 2010). Ligand topology was generated using SwissParam (Zoete et al., 2011). Protein-ligand complexes were solved using TIP3P water (Jorgensen et al., 1983) in a dodecahedral box with a minimum of 2.0 nm distance between any protein atom to the closest box edge. Sodium ions were added to the solvated system to neutralise the charge. Energy minimisation was performed using a steepest-descent gradient method for a maximum of 50,000 steps. Each complex was then restrained using an isothermal-isochloric (NVT) ensemble and isothermal-isobaric ensemble (NPT) for 100 ps. Temperature was maintained at 310 K with a modified Berendsen thermostat (Berendsen et al., 1984), and pressure at 1.0 bar with the Parrinello-Rahman barostat (Parrinello and Rahman, 1980). Bond lengths were constrained using the LINCS algorithm (Hess et al., 1997), with long-range electrostatic forces calculated using the particle-mesh Ewald scheme (PME) (Darden et al., 1993) (grid spacing 0.16 nm). Cutoff ratios of 1.2 nm for Coulomb and van der Waals potentials were used for the calculation of short-range nonbonded interactions. Simulations were carried out for 100 ns with a time-step of 2 fs in triplicate, with random generation of velocities according to a Maxwell distribution.
Visual Molecular Dynamics 1.9.3 (Humphrey et al., 1996) was utilised for analysis and visualisation of trajectories. Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA) was utilised for the quantification of free energy calculations (Baker et al., 2001). This was performed using the g_mmpbsa tool (Kumari et al., 2014). MM-PBSA calculations were performed on 1 ns segments of the triplicate stabilised trajectories (Hou et al., 2011). Energy contributions from electrostatic, van der Waals, and polar solvation terms were calculated using the adaptive Poisson-Boltzmann Solver (APBS) (Robert et al., 2012). Grid spacing was set to 0.05 nm, and values of 80 and 2 were used for solvent dielectric constant and solute dielectric constant, respectively. Solvent-accessible surface area (SASA) was used to approximate the non-polar energy contribution, with the probe radius set to 0.14 nm. Entropic energy terms were excluded from the calculations.
3. Results and Discussion
3.1. The α-ketoamide 13b ligand binds with relatively high affinity to the active site of the SARS-CoV-2 Mpro
Docking was performed using the QPLD workflow of Schrodinger to obtain a rigorous estimate of ligand binding affinities to the active site. The receptor grid was centred around the active site residues for a comprehensive search of binding poses using partial atomic charges calculated using quantum mechanical methods. The α-ketoamide 13b ligand bound to the protease with a GlideScore of -8.8 kcal/mol to the monomer, compared to -5.2 kcal/mol for amoxicillin. For binding to the dimer structure of the protease (Table 2 ), α-ketoamide 13b bound with strong affinities of -8.7 and -9.2 kcal/mol to protomers A and B, respectively. Amoxicillin bound with slightly weaker affinities that were similar between binding to both protomers, with glide energy values of -5.0 and -4.8 kcal/mol to protomers A and B, respectively. Hydrogen bonds are shown to be formed with GLU-166 in both protomers (Fig. 1 ). Ligand interaction diagrams reveal α-ketoamide 13b is docked in a position similar to that described by the crystal structure solved by Zhang et al., with the cyclopropyl ring of α-ketoamide 13b facing residues HID-41 and MET-49 in the hydrophobic S2 subsite (Fig. 1) (Zhang et al., 2020b). For both protomers, a hydrogen bond is shown to form with GLU-166, which forms part of the S1 substrate binding site. The docked α-ketoamide 13b forms a hydrogen bond with PHE-140 in protomer A, and ASN-142 in protomer B, which are both also residues located within the S1 substrate binding subsite.
Table 2.
Average energy contribution in kcal/mol of ligands bound to SARS-CoV-2 Mpro in kcal/mol.
| Type of energy | α-ketoamide 13b (Protomer A) | α-ketoamide 13b (Protomer B) | Amoxicillin (Protomer A) |
|---|---|---|---|
| van der Waal | −45.2 | −47.9 | −27.3 |
| Electrostatic | −4.5 | −8.2 | 12.9 |
| Polar solvation | 37.8 | 38.8 | 50.7 |
| SASA energy | −5.0 | −5.1 | −3.5 |
| Binding energy | -25.2 | -22.3 | 32.8 |
Fig. 1.

Ligand interaction diagrams following molecular docking of α-ketoamide 13b (A, B) and amoxicillin (C, D) to protomers A and B of SARS-CoV-2 Mpro. Pink arrows indicate formation of hydrogen bonds.
3.2. Ligand effects on overall structure of SARS-CoV-2 Mpro
Molecular dynamics simulations were performed on the protease dimer in its apo form and bound to α-ketoamide 13b and amoxicillin. Ligand-bound systems contain two compounds, with a single compound bound to the substrate binding site on each protomer, as determined by molecular docking described in the previous section. Systems were solvated in a water box and simulated in triplicate for 100 ns.
Root mean square deviation (RMSD) analysis indicates that the protein structure remains stable, and that structural rearrangements equilibrate after approximately 60 ns (Fig. 2 A). Subsequent analysis was performed on the last 40 ns of the trajectory, after protein complexes were stabilised. Compounds bound to the protease demonstrated a modestly higher average RMSD. The apo protease had an average RMSD of 0.24 nm, compared to 0.27 nm for α-ketoamide 13b and 0.26 nm for amoxicillin bound forms.
Fig. 2.

Stability of SARS-CoV-2 Mpro complex in the presence of α-ketoamide 13b and amoxicillin. Average root mean square deviation (RMSD) for protein fit to backbone (A) for 100 ns, and average root mean square fluctuation of whole protein (B) following stabilisation. Mpro apo form is shown in blue, α-ketoamide 13b bound in red, and amoxicillin bound is shown in green. (C) shows the RMSF values the apo form subtracted from ligand bound forms of the protein.
Root mean square fluctuation (RMSF) analysis measured the flexibility of the entire protein with respect to its average structure. For both apo and ligand-bound forms of the protease dimer, the largest fluctuations occurred at the C-terminal region of each protomer, as well as in four distinct areas: from residues THR-45 to ASN-51, at TYR-154, from VAL-186 to ALA-194, and at ARG-222 in both protomers (Fig. 2B). These regions lie within domain I, domain II, the connecting loop, and domain III of Mpro respectively (Jin et al., 2020). The remaining residues of the protomers remained relatively stable. When RMSF values of the apo protease were subtracted from the ligand bound forms, peaks persisted in the aforementioned regions, indicating more dramatic fluctuations in these residues when α-ketoamide 13b and amoxicillin are bound (Fig. 2C). Aside from C-terminal fluctuations, the greatest difference in RMSF between ligand and apo protease forms occurred between domain I residues THR-45 and ASN-51, and residues VAL-186 to ALA-194, particularly in protomer A. The fluctuating domain I residues are located within the substrate binding site of the protease. Residues of the connecting loop between domains I and II also show relatively high RMSF values, demonstrating decreased stability in this region.
3.3. The α-ketoamide 13b ligand binds with a strong free energy to SARS-CoV-2 Mpro
It is noted that from visual inspection, while α-ketoamide 13b remains strongly bound to the active site of the protease throughout the trajectory (Media S1), amoxicillin does not remain bound to the protease. Amoxicillin bound to protomer B (amoxicillin-B) of the protomer is seen to detach after approximately 40 ns (Media S2) and remains unbound in the solvent for the remainder of the trajectory. In another replicate, amoxicillin bound to protomer A (amoxicillin-A) detaches 13 ns into the trajectory and attaches to protomer B, where it interacts with the amoxicillin-B at 47 ns. At 70 ns, amoxicillin-A replaces amoxicillin-B in the active site of protomer B, while amoxicillin-B moves to bind to the apex of the protease. Fig. 3 depicts snapshots of frames along these trajectories. Thus, subsequent analysis using MM-PBSA for binding of amoxicillin are performed in duplicate on trajectories where amoxicillin-A remains bound to the protease. Analysis for α-ketoamide 13b is performed in triplicate.
Fig. 3.

Snapshots of 100 ns trajectory of α-ketoamide 13b and amoxicillin bound to SARS-CoV-2 Mpro. Protomer A and B are shown as blue and silver respectively. (A) α-ketoamide 13b bound to chain A and B are depicted in yellow and red respectively. (B) Amoxicillin bound to chain A at the start of the trajectory is shown in green, and (C) bound to chain B is purple. (A) and (B) depict respective final frames of the protein-ligand complex, while (C) depicts snapshots along the trajectory.
Molecular mechanics-Poisson Boltzmann surface area (MM-PBSA) calculations were performed to calculate binding free energy and to examine binding mechanisms. Table 2 indicates that van der Waals interactions are the predominant driving force for binding of α-ketoamide 13b to both protomers of the SARS-CoV-2 Mpro active site. Electrostatic and solvent-accessible surface area (SASA) energy had minor contributions to a favourable binding energy. α-ketoamide 13b bound to protomer A with a ΔG of -25.2 kcal/mol to, and -22.3 kcal/mol to protomer B of the protease, indicating that α-ketoamide 13b binds strongly to the protease. Amoxicillin, on the other hand, produced a positive ΔG of +32.8 kcal/mol. This indicates that amoxicillin interacts unfavourably with the protease, consistent with visual analysis of the trajectories where it detaches from the protein in some cases (Fig. 3, Media S2 and S3). While van der Waals forces were still the primary contributor for favourable binding energy, electrostatic interactions were strongly positive in contrast to α-ketoamide 13b binding. Polar solvation energy was also significantly more unfavourable compared to binding with α-ketoamide 13b. While MM-PBSA is a commonly used method for estimating free energy, it should be noted that entropy terms are excluded from calculations. While MM-PBSA methods have been used to rescore poses obtained through molecular docking, it should be noted that values produced should be treated as relative differences across a set of ligands, rather than absolute (Brown and Muchmore, 2009; Thompson et al., 2008). Nevertheless, the results presented here demonstrate that α-ketoamide 13b is able to bind strongly to the substrate binding site of the protease.
3.4. Key substrate binding residues contribute to favourable binding free energy to α-ketoamide 13b
Residue energy contributions are decomposed in Fig. 4 , where the average energy contributions are shown on a per-residue basis for the entire protease. For α-ketoamide 13b, energy contributions are largely confined to the protomer on which they are bound. Residue energy contributions are mostly favourable, shown as negative peaks below the x-axis (Fig. 5 ).
Fig. 4.

Average residue contributions to binding free energy of α-ketoamide 13b (A) and amoxicillin (B) to protease dimer. (A) shows residue contributions for both α-ketoamide 13b ligands bound to the dimer, with α-ketoamide 13b bound to protomer A in red, and α-ketoamide 13b bound to protomer B in light blue. For amoxicillin (B), residue contribution for the compound originally bound to protomer A is shown. Average residue contributions for α-ketoamide 13b were calculated based on triplicates, and for amoxicillin in duplicate.
Fig. 5.

Energy contribution of residues to binding of ligands to SARS-CoV-2 Mpro. (A) shows binding of α-ketoamide 13b to the substrate binding site of protomer A, (B) shows binding of α-ketoamide 13b to the substrate binding site of protomer B, and (C) depicts amoxicillin bound to protomer B at the final frame of a 100 ns trajectory. Residues are coloured according to their energy contribution in kcal/mol, with red indicative of a more favourable contribution.
The residue with the most negative energy contribution corresponding to the most favourable interaction is MET-49 on both protomers. This residue lies within the region where the large fluctuations in RMSF were observed in Fig. 2. A more favourable energy contribution was observed for this residue on protomer A on the dimer of -2.40 kcal/mol compared to -1.26 kcal/mol for protomer B. This is in line with the slightly stronger ΔG of for α-ketoamide 13b binding to protomer A compared to protomer B (Table 2). MET-49 is located in the S2 subsite of the substrate binding pocket, previously found to form a ‘lid’ in the closely related SARS-CoV Mpro S2 site facilitating hydrophobic interactions that enable inhibition of the enzyme with α-ketoamides (Zhang et al., 2020a). Another residue contributing favourably to binding to both protomers is CYS-145. The energy contribution was calculated to be -1.08 kcal/mol in protomer A, and -0.94 kcal/mol in protomer B. CYS-145 is conserved among viral proteases, and along with HIS-41 forms the catalytic dyad in the SARS-CoV-2 Mpro (Yang et al., 2003; Barretto et al., 2005). A nucleophilic attack occurs with the catalytic CYS-145 on the α-ketoamide 13b, while HIS-42 acts as a hydrogen acceptor (Zhang et al., 2020b).
A peak in energy contribution is only apparent for ASP-187 in α-ketoamide 13b binding to protomer A. This agrees with RMSF data where the residue fluctuations were more apparent in protomer A than B. A slightly unfavourable interaction with α-ketoamide 13b is observed, with an energy contribution of 0.34 kcal/mol for protomer A. The alkyl portion of the ASP-187 side chain forms part of the hydrophobic S2 subsite. Favourable energy contributions are seen in residues THR-25 and LEU-27 of protomer A that are not seen in protomer B, with contributions of -0.54 and -0.66 kcal/mol respectively. THR-25, along with THR-24 have previously been shown to interact with van der Waals forces with the benzyl group of α-ketoamide 13b in the S1’ site (Zhang et al., 2020b).
In protomer A, GLU-166 and MET-165 produce favourable energy contributions of -0.51 and -0.59 kcal/mol, and HIS-164 produces a large unfavourable energy of 0.34 kcal/mol. Interestingly, while a similar trend is observed in protomer B for MET-165 (-1.13 kcal/mol) and HIS-164 (0.85 kcal/mol), GLU-166 demonstrates a strong unfavourable energy contribution of 0.77 kcal/mol instead. This seems to be in line with work from Zhang et al., where they found that GLU-166 adopted an inactive conformation in protomer B (Zhang et al., 2020b). GLU-166 is a key residue essential for catalytic activity, interacting with NH2 terminal residues of each protomer at the dimer interface, shaping the S1 pocket of the substrate binding site (Zhang et al., 2020b; Anand et al., 2002).
When energy contribution from individual residues are decomposed for amoxicillin binding to protomer A of the protease, the range of energy is larger and greatly varied across the protein when compared to α-ketoamide 13b binding. While amoxicillin is bound to protomer A of the protease, energy contributions occur across both protomers, with extremes in both favourable and unfavourable directions. Larger energy contributions occur in the unfavourable direction above the x axis, contributing to the positive binding energy of the ligand. Along with the slightly larger fluctuations in RMSF values compare to α-ketoamide 13b (Fig. 3C), it may be that the structure of the protease is destabilised when amoxicillin is bound. Further analysis with more rigorous free energy prediction methods may be required to examine the effect of amoxicillin on the SARS-CoV-2 Mpro. Overall, our findings highlight the stability of the interaction of the α-ketoamide 13b ligand with the SARS-CoV-2 Mpro, confirming the need to further evaluate the antiviral properties of this compound.
Author Contributions Statement
TCK, KN, and AH conceptualized the aims and methodology and were involved in supervision. JL performed formal data analysis, was involved in data curation, and produced the first draft of the manuscript. CK performed formal data analysis and was involved in data curation. EP and KKD performed formal data analysis and validation. All authors contributed to editing and reviewing the manuscript.
Declaration of Competing Interest
Epigenomic Medicine Program (TCK) is supported financially by McCord Research (Iowa, USA), however there is no conflict of interest with respect to the SARS-CoV-2 main protease or α-ketoamide and amoxicillin described in the work. The remaining co-authors also have no conflicts of interest.
Acknowledgements
We would like to acknowledge intellectual and financial support by McCord Research (Iowa, USA). JL and KKD are supported by Australian Government Research Training Program Scholarships. We are indebted to Alfonso Perez Escuredo (crowdfightcovid) for enabling access to supercomputing facilities and to Matthew Gasperetti (Hypernet Labs; Galileo), for enabling cloud computing for this project. We thank the National Computing Infrastructure (NCI), and the Pawsey Supercomputing Centre in Australia (funded by the Australian Government). Further, we thank the Spartan High Performance Computing service (University of Melbourne), and the Partnership for Advanced Computing in Europe (PRACE) for awarding the access to Piz Daint, hosted at the Swiss National Supercomputing Centre (CSCS), Switzerland.
Footnotes
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.compbiolchem.2020.107292.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- World Health Organization . 2020. Novel Coronavirus (2019-nCoV): Situation Report, 1. [Google Scholar]
- Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. The Lancet Infectious Diseases. 2020 doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud D., Qi X., Nielsen-Saines K., Musso D., Pomar L., Favre G. Real estimates of mortality following COVID-19 infection. The Lancet Infectious Diseases. 2020 doi: 10.1016/S1473-3099(20)30195-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC COVID-19 Response Team Severe outcomes among patients with coronavirus disease 2019 (COVID-19)—United States, February 12–March 16, 2020. MMWR Morb Mortal Wkly Rep. 2020;69(12):343–346. doi: 10.15585/mmwr.mm6912e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novel Coronavirus Pneumonia Emergency Response Epidemiology Team The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Zhonghua liu xing bing xue za zhi= Zhonghua liuxingbingxue zazhi. 2020;41(2):145. [Google Scholar]
- Cai H. Sex difference and smoking predisposition in patients with COVID-19. The Lancet Respiratory Medicine. 2020;8(4):e20. doi: 10.1016/S2213-2600(20)30117-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. The Lancet. 2020;395(10223):470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. New England Journal of Medicine. 2020;382(13):1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JF-W Chan, Yuan S., Kok K.-H.-H., KK-W To, Chu H., Yang J. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. The Lancet. 2020;395(10223):514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen K.-S.-S., Ye Z.W., Fung S.-Y.-Y., Chan C.-P.-P., Jin D.-Y. SARS-CoV-2 and COVID-19: The most important research questions. Cell & Bioscience. 2020;10(1):40. doi: 10.1186/s13578-020-00404-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster V.J., Koopmans M., van Doremalen N., van Riel D., de Wit E. A Novel Coronavirus Emerging in China - Key Questions for Impact Assessment. The New England journal of medicine. 2020;382(8):692–694. doi: 10.1056/NEJMp2000929. [DOI] [PubMed] [Google Scholar]
- de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nature Reviews Microbiology. 2016;14(8):523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajgor D.D., Lee M.H., Archuleta S., Bagdasarian N., Quek S.C. The many estimates of the COVID-19 case fatality rate. The Lancet Infectious Diseases. 2020 doi: 10.1016/S1473-3099(20)30244-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020 doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.-J.-J., Tortorici M.A., Wall A., McGuire A.T., Structure Veesler D. Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020 doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhal B.D., Li X., Xue A., Gao X., Abdul-Hafez A. Regulation of alveolar epithelial cell survival by the ACE-2/angiotensin 1-7/Mas axis. Am J Physiol Lung Cell Mol Physiol. 2011;301(3):L269–L274. doi: 10.1152/ajplung.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel E.C., Wang J., Jeffers S., Edeen K.E., Wang S., Cosgrove G.P. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology. 2008;372(1):127–135. doi: 10.1016/j.virol.2007.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Zhong L., Deng J., Peng J., Dan H., Zeng X. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. International Journal of Oral Science. 2020;12(1):8. doi: 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. The FEBS Journal. 2014;281(18):4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse J.S., Lalonde T., Xu S., Liu W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem. 2020;21(5):730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J Virol. 2008;82(5):2515–2527. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science (New York, NY). 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis. and Activity Assessment. Journal of medicinal chemistry. 2020 doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science (New York, NY) 2020 doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger . 5 ed. LLC; New York: 2018. Maestro. 11. [Google Scholar]
- Krissinel E., Henrick K. Protein interfaces, surfaces and assemblies service PISA at European Bioinformatics Institute. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Halgren T.A. Identifying and characterizing binding sites and assessing druggability. Journal of chemical information and modeling. 2009;49(2):377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Cho A.E., Guallar V., Berne B.J., Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. Journal of computational chemistry. 2005;26(9):915–931. doi: 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of medicinal chemistry. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Bochevarov A.D., Harder E., Hughes T.F., Greenwood J.R., Braden D.A., Philipp D.M. Jaguar: A high‐performance quantum chemistry software program with strengths in life and materials sciences. International Journal of Quantum Chemistry. 2013;113(18):2110–2142. [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications. 1995;91(1):43–56. [Google Scholar]
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. [Google Scholar]
- Bjelkmar P., Larsson P., Cuendet M.A., Hess B., Lindahl E. Implementation of the CHARMM Force Field in GROMACS: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. Journal of Chemical Theory and Computation. 2010;6(2):459–466. doi: 10.1021/ct900549r. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of computational chemistry. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. Journal of computational chemistry. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics. 1983;79(2):926–935. [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., Gunsteren W.F.v., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics. 1984;81(8):3684–3690. [Google Scholar]
- Parrinello M., Rahman A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Physical Review Letters. 1980;45(14):1196–1199. [Google Scholar]
- Hess B., Bekker H., Berendsen H.J., Fraaije J.G. LINCS: a linear constraint solver for molecular simulations. Journal of computational chemistry. 1997;18(12):1463–1472. [Google Scholar]
- Darden T., York D., Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics. 1993;98(12):10089–10092. [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14(1) doi: 10.1016/0263-7855(96)00018-5. 33-8, 27-8. [DOI] [PubMed] [Google Scholar]
- Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proceedings of the National Academy of Sciences. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R., Lynn A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. Journal of Chemical Information and Modeling. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Hou T., Wang J., Li Y., Wang W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. Journal of chemical information and modeling. 2011;51(1):69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert K., Nathan A.B., McCammon J.A. iAPBS: a programming interface to the adaptive Poisson–Boltzmann solver. Computational Science & Discovery. 2012;5(1) doi: 10.1088/1749-4699/5/1/015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S.P., Muchmore S.W. Large-Scale Application of High-Throughput Molecular Mechanics with Poisson−Boltzmann Surface Area for Routine Physics-Based Scoring of Protein−Ligand Complexes. Journal of medicinal chemistry. 2009;52(10):3159–3165. doi: 10.1021/jm801444x. [DOI] [PubMed] [Google Scholar]
- Thompson D.C., Humblet C., Joseph-McCarthy D. Investigation of MM-PBSA rescoring of docking poses. J Chem Inf Model. 2008;48(5):1081–1091. doi: 10.1021/ci700470c. [DOI] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences. 2003;100(23):13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79(24):15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. The EMBO Journal. 2002;21(13):3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.