

Abstract
Early detection and effective treatment of severe COVID-19 patients remain major challenges. Here, we performed proteomic and metabolomic profiling of sera from 46 COVID-19 and 53 control individuals. We then trained a machine learning model using proteomic and metabolomic measurements from a training cohort of 18 non-severe and 13 severe patients. The model was validated using 10 independent patients, 7 of which were correctly classified. Targeted proteomics and metabolomics assays were employed to further validate this molecular classifier in a second test cohort of 19 COVID-19 patients, leading to 16 correct assignments. We identified molecular changes in the sera of COVID-19 patients compared to other groups implicating dysregulation of macrophage, platelet degranulation, complement system pathways, and massive metabolic suppression. This study revealed characteristic protein and metabolite changes in the sera of severe COVID-19 patients, which might be used in selection of potential blood biomarkers for severity evaluation.
Keywords: proteomics, metabolomics, COVID-19, serum, severity
Graphical Abstract
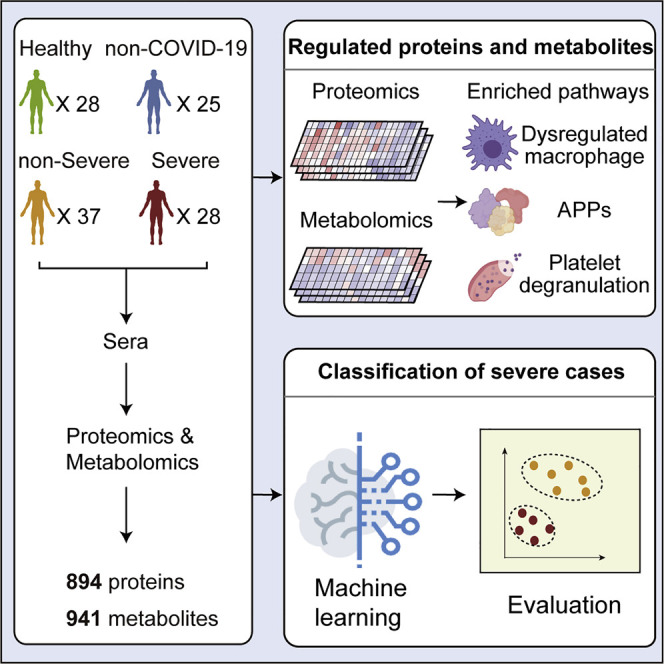
Proteomic and metabolomic analysis of COVID-19 sera identifies differentially expressed factors that correlate with disease severity and highlights dysregulation of multiple immune and metabolic components in clinically severe patients.
Introduction
Coronavirus disease 2019 (COVID-19) is an unprecedented global threat caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It is currently spreading around the world rapidly. The sudden outbreak and accelerated spreading of SARS-CoV-2 infection have caused substantial public concerns. Within about 3 months, over 2 million individuals worldwide have been infected, leading to over 150,000 deaths.
Most COVID-19 studies have focused on its epidemiological and clinical characteristics (Ghinai et al., 2020, Guan et al., 2020). About 80% of patients infected with SARS-CoV-2 displayed mild symptoms with good prognosis. They usually recover with, or even without, conventional medical treatment and therefore are classified as mild or moderate COVID-19 (Thevarajan et al., 2020). However, about 20% of patients suffer from respiratory distress and require immediate oxygen therapy or other inpatient interventions, including mechanical ventilation (Murthy et al., 2020, Wu and McGoogan, 2020). These patients, classified as clinically severe or critical life-threatening infections, are mainly diagnosed empirically based on a set of clinical characteristics, such as respiratory rate (≥30 times/min), mean oxygen saturation (≤93% in the resting state), or arterial blood oxygen partial pressure/oxygen concentration (≤300 mmHg). However, patients exhibiting these clinical manifestations have already progressed to a clinically severe phase and require immediate access to specialized intensive care; otherwise, they may die rapidly. Therefore, it is critical to develop new approaches to assess early which cases will likely become clinically severe. In addition, effective therapy for severe patients remains speculative, largely due to limited understanding of SARS-CoV-2 pathogenesis.
In this study, we hypothesized that SARS-CoV-2 induces characteristic molecular changes that can be detected in the sera of severe patients. These molecular changes may shed light on therapy development for COVID-19 patients. To test this hypothesis, we applied proteomic (Aebersold and Mann, 2016, Li et al., 2020) and metabolomic (Hou et al., 2020, Lee et al., 2019) technologies to analyze the proteome and metabolome of sera from COVID-19 patients and several control groups.
Results
Proteomic and Metabolomic Profiling of COVID-19 Sera
We procured a cohort of patients (Zheng et al., 2020) containing 28 severe COVID-19 patients. The detailed patient descriptions including the sampling date for each patient are shown in Figure 1 A, Table 1 , and Table S1. Controls with matched epidemiological features were included to identify severity-related molecular alterations. These controls were 28 healthy subjects, 25 non-COVID-19 patients (negative for the SARS-CoV-2 nucleic acid test) with similar clinical characteristics as COVID-19 patients, and 25 non-severe COVID-19 patients. A serum sample was obtained from each patient within a few days after hospital admission, with a few exceptions when samplings were performed at later disease stages. We analyzed 12 clinical measurements of the COVID-19 and non-COVID-19 patients (Figure S1 ), including white blood cell count, lymphocyte count, monocyte count, platelet count, C-reactive protein (CRP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), glutamyltransferase (GGT), total bilirubin (TBIL), direct bilirubin (DBIL), creatinine, and glucose. Compared to non-severe patients, severe patients showed significant suppression of lymphocyte count and monocyte count, as well as increase of CRP and AST (Figure S1).
Figure 1.

Summary of COVID-19 Patients and Machine Learning Design
(A) Summary of COVID-19 patients, including non-severe (n = 37) and severe (n = 28) patients with more details in Table S1. Patients labeled in red (y axis) indicate chronic infection of hepatitis B virus.
(B) Study design for machine-learning-based classifier development for severe COVID-19 patients. We first procured samples in a training cohort (C1) for proteomic and metabolomic analysis. The classifier was then validated in an independent test cohort (C2), followed by a second test cohort (C3).
Table 1.
Demographics and Baseline Characteristics of COVID-19 Patients
| Variables | Healthy Control (N = 28) | Non-COVID-19 (N = 25) | COVID-19 |
||
|---|---|---|---|---|---|
| Total (N = 65) | Non-severe (N = 37) | Severe (N = 28) | |||
| Sex - no. (%) | |||||
| Male | 21 (75.0) | 17 (68.0) | 41 (63.1) | 25 (67.6) | 16 (57.1) |
| Female | 7 (25.0) | 8 (32.0) | 24 (36.9) | 12 (32.4) | 12 (42.9) |
| Age - year | |||||
| Mean ± SD. | 44.4 ± 8.3 | 49.2 ± 14.0 | 48.2 ± 13.7 | 42.9 ± 12.5 | 55.1 ± 12.4 |
| Median (IQR) | 45.0 (38.0-51.0) | 53.0 (37.0-61.0) | 47.0 (37.0-57.5) | 43.0 (33.0-50.0) | 55.0 (47.0-64.8) |
| Range | 28.0-57.0 | 23.0-67.0 | 18.0-77.0 | 18.0-70.0 | 33.0-77.0 |
| BMI, kg/m2 | |||||
| Mean ± SD | 24.4 ± 2.7 | 23.5 ± 2.7 | 25.1 ± 3.0 | 24.5 ± 3.2 | 26.0 ± 2.4 |
| Median (IQR) | 24.2 (22.5-26.5) | 24.7 (20.9-25.8) | 24.9 (22.8-27.1) | 24.2 (21.8-26.8) | 26.2 (24.4-27.7) |
| Range | 19.9-32.9 | 19.1-27.4 | 18.9-31.3 | 18.9-30.7 | 22.2-31.3 |
| Smoke - no. (%) | 7 (10.8) | 2 (5.4) | 5 (17.9) | ||
| Alcohol - no. (%) | 6 (9.2) | 1 (2.7) | 5 (17.9) | ||
| Time from Onset to Admission, Days | |||||
| Mean ± SD. | 6.1 ± 4.4 | 4.6 ± 3.2 | 7.9 ± 5.1 | ||
| Median (IQR) | 4.0 (3.0-9.0) | 4.0 (2.5-5.5) | 8.5 (4.0-11.0) | ||
| Range | 1.0-24.0 | 1.0-15.0 | 1.0-24.0 | ||
| Time from Admission to Severe, Days | |||||
| Mean ± SD. | 2.4 ± 1.7 | ||||
| Median (IQR) | 2.0 (1.0-4.0) | ||||
| Range | 0.0-7.0 | ||||
| Symptoms - no. (%) | |||||
| Fever | 9 (36.0) | 49 (75.4) | 23 (62.2) | 26 (92.9) | |
| Cough | 11 (44.0) | 30 (46.2) | 14 (37.8) | 16 (57.1) | |
| Headache | 2 (8.0) | 9 (13.8) | 4 (10.8) | 5 (17.9) | |
| Fatigue | 2 (8.0) | 8 (12.3) | 4 (10.8) | 4 (14.3) | |
| Pharyngalgia | 0 (0.0) | 4 (6.2) | 3 (8.1) | 1 (3.6) | |
| Expectoration | 3 (12.0) | 13 (20.0) | 5 (13.5) | 8 (28.6) | |
| Diarrhea | 0 (0.0) | 6 (9.2) | 2 (5.4) | 4 (14.3) | |
| Chest tightness | 2 (8.0) | 2 (3.1) | 1 (2.7) | 1 (3.6) | |
| Chest CT - no. (%) | |||||
| Involvement of chest radiographs | 1 (4.0) | 63 (96.9) | 35 (94.6) | 28 (100.0) | |
| Comorbidity- no. (%) | |||||
| Hypertension | 10 (40.0) | 10 (15.4) | 3 (8.1) | 7 (25.0) | |
| Diabetes | 0 (0.0) | 10 (15.4) | 6 (16.2) | 4 (14.3) | |
| Respiratory system | 2 (8.0) | 4 (6.2) | 4 (10.8) | 0 (0.0) | |
| Other Endocrine system | 0 (0.0) | 4 (6.2) | 1 (2.7) | 3 (10.7) | |
| Chronic kidney disease | 0 (0.0) | 1 (1.5) | 0 (0.0) | 1 (3.6) | |
| Digestive system | 2 (8.0) | 6 (9.2) | 3 (8.1) | 3 (10.7) | |
| Oxygenation Index - mmHg | |||||
| Mean ± SD. | 394.4 ± 137.3 | 438.0 ± 130.0 | 336.7 ± 125.4 | ||
| Median (IQR) | 382.8 (307.2-459.5) | 447.6 (376.2-473.8) | 319.1 (259.2-382.4) | ||
| Range | 129.0-891.0 | 138.0-891.0 | 129.0-729.0 | ||
| Treatment - no. (%) | |||||
| Oxygen inhalation | 3 (12.0) | 40 (61.8) | 14 (37.8) | 26 (92.9) | |
| Antibiotics | 0 (0.0) | 22 (33.8) | 10 (27.0) | 12 (42.9) | |
| Antiviral drug | 25 (100) | 65 (100.0) | 37 (100.0) | 28 (100.0) | |
| Immunoglobulin | 0 (0.0) | 24 (36.9) | 0 (0.0) | 24 (85.7) | |
| Methylprednisolone | 0 (0.0) | 24 (36.9) | 1 (2.7) | 23 (82.1) | |
| Chinese medicine | 0 (0.0) | 65 (100.0) | 37 (100.0) | 28 (100.0) | |
no. (%), number; SD, standard deviation; IQR, interquartile range; CT, computed tomography.
Figure S1.

Twelve Clinical Parameters of COVID-19 Patients and Non-COVID-19 Patients, Related to Figure 1
Significance indicated by the asterisks (unpaired two-sided Welch’s t test. p value: ∗, < 0.05; ∗∗, < 0.01; ∗∗∗, < 0.001.)
We used stable isotope labeled proteomics strategy TMTpro (16plex) (Li et al., 2020) and ultra performance liquid chromatography/tandem mass spectrometry (UPLC-MS/MS) untargeted metabolomics approach to analyze the sera samples. Altogether, 894 proteins and 941 metabolites (including 36 drugs and their metabolites) were identified and quantified. Drugs are not used in the following analysis. The details of the peptides, proteins, and metabolites are shown in Table S2. For metabolomic analysis, both hydrophilic and hydrophobic molecules were analyzed using both positive and negative ionization to cover various endogenous biochemical classes. Our data were acquired with high degree of consistency and reproducibility. In quality control analysis, the median coefficient of variance (CV) values for proteomic and metabolomic data were 10% and 5%, respectively (Figure S2 A). Without molecule selection, the omics data of sera from SARS-CoV-2 infected patients were well resolved from healthy individuals, while the other groups showed certain degrees of separation (Figures S2B and S2C). We further checked the influence of age, sex, the time from disease onset to admission, and the time from sampling to diagnosis of severe cases, to the omics profiling in our training cohort (C1). As shown in the UMAP plots (Figures S2D and S2E), we did not observe substantial impact of these confounding factors to the omics data.
Figure S2.

Quality Control of Proteomic and Metabolomic Data, Related to Figure 1
(A) Coefficient of variation (CV) of the proteomic data is calculated by the proteins quantified in six quality control (QC) samples using the pooled samples from all samples in training cohort. CV of the metabolomic data is calculated by twelve QC samples using a set of isotopic internal spiked-in standards.
(B) Uniform Manifold Approximation and Projection (UMAP) of sera samples using 791 measured proteins in the training cohort.
(C) UMAP of sera samples using 847 metabolites excluding drugs.
(D) UMAP analysis of the COVID-19 patients using 791 measured proteins.
(E) UMAP analysis of the COVID-19 patients using 847 metabolites.
In D and E, patients labeled in red received serum test before they were diagnosed as severe. Inside the brackets are the sex, age, time from disease onset to admission and time from sampling to diagnose of severe case in sequence.
Identification of Severe Patients Using Machine Learning
We next investigated the possibility of identifying the severe cases from COVID-19 patients based on the molecular signatures of proteins and metabolites (Table S3). Five patients were excluded from this analysis because of failure to collect proteomic data. We built a random forest machine learning model based on proteomic and metabolomic data from 18 non-severe and 13 severe patients (Figure 1B), leading to prioritization of 29 important variables including 22 proteins and 7 metabolites (Figures 2A and 2B). This model reached an area under curve (AUC) of 0.957 in the training set (Figure 2C). One non-severe patient, XG3, was incorrectly classified as severe, possibly because this 70-year-old male patient was the oldest individual in this cohort (Figure 1A).
Figure 2.

Separation of Severe and Non-severe COVID-19 Patients by Machine Learning of Proteomic and Metabolomic Features
(A) Top 22 proteins and 7 metabolites prioritized by random forest analysis ranked by the mean decrease in accuracy.
(B) Network of prioritized proteins appeared in the classifier. Red and green nodes indicate upregulated and downregulated molecules, respectively. White nodes represent molecules not detected in our dataset.
(C) Receiver operating characteristic (ROC) of the random forest model in the training cohort (C1).
(D) Performance of the model in the test cohort (C2) of 10 COVID-19 patients.
(E) Performance of the model in the test cohort (C3) containing 19 COVID-19 patients. Patients labeled in red received serum test before they were diagnosed as severe.
We then tested the model on an independent cohort of 10 patients (Figure 2D; Table S4). All severe patients were correctly identified, except one patient, XG45, a 62-year-old male with the longest pre-admission treatment in this cohort. He had received traditional Chinese medicines for more than 20 days before admission. The extended administration of traditional Chinese medicines might have been a confounding factor for the model. An incorrectly classified non-severe patient was XG22, a 43-year-old male. This patient had chronic hepatitis B virus (HBV) infection, diabetes, and the longest hospitalization (>50 days) among all non-severe patients. His infection and treatment history might be confounding factors for our model. For reasons unclear, XG25, a 43-year-old male non-severe case, was incorrectly classified as severe. We also generated classifiers containing 29 molecules randomly selected from the omics profiles for 1,000 times, and these classifiers using random molecules had relatively low rates of classification correctness. The accuracies are from 48% to 49% within 95% confidence interval, which are significantly lower than our classifier using the 29 selected proteins and metabolites (p value < 2.2e−16 with one-sample t test).
To further validate this classifier, we developed targeted mass spectrometric assays for the 22 proteins and 7 metabolites (Figure 2A) and analyzed these 29 molecules in a second test cohort containing 19 COVID-19 patients (Figure 1; Table 1; Tables S1 and S5). Applying the classifier model using the 29 measured molecules to this new cohort led to correct assignment of 16 patients (Figure 2E). Severe patient X2-13 was incorrectly classified as non-severe, possibly because he was receiving methylprednisolone therapy before sampling. The methylprednisolone treatment might have suppressed his immune responses and distorted the classification. Another severe patient who was incorrectly classified as non-severe was X2-18. This is a 68-year-old female who had received mitral and aortic valve replacement and had a long warfarin treatment history. The only non-severe patient who was incorrectly classified as a severe patient was X2-22, a 66-year-old female who had hypertension and diabetes. On the day of blood sampling, her blood glucose level reached 27.8 mmol/L, which might have influenced the result of classification using our model.
Proteomic and Metabolomic Changes in Severe COVID-19 Sera
We found that 105 proteins were differentially expressed in the sera of COVID-19 patients but not the non-COVID-19 patients (Figures S3 and S4 ). After correlating their expression with clinical disease severity (Figure S5 ), 93 proteins showed specific modulation in severe patients. Pathway analyses and network enrichment analyses of the 93 differentially expressed proteins showed that 50 of these proteins belong to three major pathways (Figure S6 ), namely activation of the complement system, macrophage function, and platelet degranulation (Figure 3 A). It was found that 373 metabolites were significantly changed in COVID-19 patients (Figure S4B), whereas the change of 204 metabolites was correlated with disease severity as evaluated by mFuzz (Figure S5). Correspondingly, 80 significantly changed metabolites were also involved in the three biological processes revealed in the proteomic analysis. We summarized the key dysregulated molecules in Figure 5 and discussed them in the following sections.
Figure S3.

Differentially Expressed Proteins and Metabolites in Different Patient Groups in the Training Cohort, Related to Figure 4 and 5
(A-D) Volcano plots compare four pairs of patient groups as indicated in the plot. Proteins with log2 (fold-change) beyond 0.25 or below −0.25 with adjusted p value lower than 0.05 were considered as significantly differential expression. (E-H) Volcano plots for the metabolomics data. Number of significantly down- (blue) and up- (red) regulated proteins were shown on the top.
Figure S4.

Proteins and Metabolites Regulated in COVID-19 Patients but Not in Non-COVID-19 Patients, Related to Figure 4 and 5
Venn diagrams showing the overlaps between significantly regulated proteins (A) and metabolites (B) as identified in volcano plots. Proteins and metabolites labeled in red are the shortlisted molecules which differentially expressed in the COVID-19 patients but not in the non-COVID-19 patients.
Figure S5.

Identification of Specific Clusters of Proteins and Metabolites in COVID-19 Patients, Related to Figure 4 and 5
791 proteins (A) and 941 metabolites (B) were clustered using mFuzz into significant discrete clusters, respectively, to illustrate the relative expression changes of the proteomics and metabolomics data. The groups in proteomics and metabolomics data: 1: Healthy; 2: non-COVID-19; 3: non-Severe COVID-19; 4: Severe COVID-19.
Figure S6.

Pathway Analysis of 93 Differentially Expressed Proteins in COVID-19 Patients, Related to Figure 4 and 5
(A) The Gene Ontology (GO) processes enriched by Metascape.
(B) The GO terms enriched using the Cytoscape plug-in ClueGO.
(C) Ingenuine pathway analysis of most significantly relevant pathways with the predicted activation or inhibition state.
(D) Functional network analysis by GeNet identifies several communities.
Figure 3.

Dysregulated Proteins in COVID-19 Sera
(A) Heatmap of 50 selected proteins whose regulation concentrated on three enriched pathways.
(B) The expression level change (Z-scored original value) of six selected proteins with significant difference between non-severe and severe cases.
Asterisks indicate statistical significance based on unpaired two-sided Welch’s t test. p value: ∗, < 0.05; ∗∗, < 0.01; ∗∗∗, < 0.001.
Figure 5.

Key Proteins and Metabolites Characterized in Severe COVID-19 Patients in a Working Model
SARS-CoV-2 may target alveolar macrophages via ACE2 receptor, leading to an increase of secretion of cytokines including IL-6 and TNF-α, which subsequently induce the elevation of various APPs such as SAP, CRP, SAA1, SAA2, and C6, which are significantly upregulated in the severe group. Proteins involved in macrophage, lipid metabolism, and platelet degranulation were indicated with their corresponding expression levels in four patient groups.
Dysregulated Macrophage and Lipid Metabolism
Our data uncovered dysregulation of multiple apolipoproteins including APOA1, APOA2, APOH, APOL1, APOD, and APOM (Figure 3A). Most of them are associated with macrophage functions and were downregulated. Decrease of APOA1 in serum has been reported during the transition of COVID-19 patients from mild to severe illness (Nie et al., 2020). The APOM in sera of severe patients was downregulated compared with healthy and non-severe COVID-19 patients. Dysregulation of serum APOM has also been observed in patients with HBV infection (Gu et al., 2011).
Consistent with these proteomic findings, we also detected dysregulated metabolites involved in lipid metabolism. Accumulation of 11 steroid hormones in COVID-19 patients may contribute to macrophage modulation. Steroid hormones, including progesterone, androgens, and estrogens can promote the activity of macrophages, as well as other immune cells and non-immune cells (Cain and Cidlowski, 2017). We detected increased 21-hydroxypregnenolone, the essential intermediate for synthesizing corticosterone, suggesting that corticosterone biosynthesis may be a protective mechanism against SARS-CoV-2 infection.
We also found evidence of significant activation of the kynurenine pathway. Metabolites of kynurenate, kynurenine, and 8-methoxykynurenate were enriched in COVID-19 patients. Nicotinamide adenine dinucleotide (NAD+), the cofactor in many cellular redox reactions, can be synthesized from tryptophan by the kynurenine pathway and operates as a switch for macrophage effector responses (Minhas et al., 2019).
The macrophage process is closely related to lipid metabolism. Over 100 lipids were downregulated in severe patients. Our data showed decreased sphingolipids in both non-severe and severe COVID-19 patients (Figure 4 A). Sphingolipids and glycerophospholipids are important components of biomembranes, which mediate signal transduction and immune activation processes. Sphingolipids regulate diverse processes including growth regulation, cell migration, adhesion, apoptosis, senescence, and inflammatory responses (Hannun and Obeid, 2018). Sphingosine-1-phosphate has been reported to induce macrophage activation, inhibit macrophage apoptosis, and promote migration of macrophages to inflammatory sites (Weigert et al., 2009). Phagocytosis and platelet degranulation are coupled with changes in biomembrane lipid composition and fluidity and modulate the production of glycerophospholipids (Rouzer et al., 2007). In this study, we found continuous decrease of glycerophospholipids after SARS-CoV-2 infection. Glycerophospholipids and fatty acids such as arachidonic acid have been found significantly elevated in HCoV-229E-infected cells, and exogenous supplement of arachidonic acid significantly suppressed HCoV-229E and MERS-CoV replication (Yan et al., 2019). Our data suggest severe COVID-19 patients might benefit from this therapeutic strategy.
Figure 4.

Dysregulated Metabolites in COVID-19 Sera
(A) Heatmap of 80 regulated metabolites belonging to 10 major classes as indicated.
(B) The expression level change (Z-scored log 2-scaled original value) of eight selected regulated metabolites with significant difference between non-severe and severe cases. Asterisks indicate statistical significance as described in Figure 3.
Choline and its derivatives were downregulated in COVID-19 patients (Figure 4B), particularly in severe cases, while phosphocholine, the intermediate product for producing phosphatidylcholine (PC) was upregulated (Figure 4A). This was probably due to activated macrophage-mediated immunity (Sanchez-Lopez et al., 2019). Polarization of macrophages in response to pathogens requires increased absorption of choline for PC formation, thereby promoting cytokine secretion (Sanchez-Lopez et al., 2019). Other immunological functions may also contribute.
Activated Acute Phase Proteins and the Complement System
We detected ten activated acute phase proteins (APPs) among 20 proteins that are differentially expressed between non-severe and severe groups (Figure 2A). They are involved at the early stages of immune responses to virus infection. Among the most significantly upregulated proteins in the sera of the severe COVID-19 patients are APPs, including serum amyloid A-1 (SAA1), serum amyloid A-2 (SAA2), serum amyloid A-4 (SAA4), CRP, alpha-1-antichymotrypsin (SERPINA3), and serum amyloid P-component (SAP/APCS) (Figure 3B). Some of them, including SAA1, SAA2, and CRP, are known to be potential biomarkers for viral infections. Although CRP has been associated with COVID-19, the other proteins have not previously been reported in COVID-19 (Liang et al., 2020). SAA1 was reported to be elevated in severe SARS patients but was not specific to SARS-CoV (Pang et al., 2006). As a major contributor to acute phase response, complement system plays a crucial role in eliminating invading pathogens in the early stage of infection. Among those APPs, two proteins belong to the complement membrane attack complex, including complement 6 (C6) and complement factor B (CFB). Two other proteins, Properdin (CFP) and Carboxypeptidase N catalytic chain (CPN1), are regulators of complement system (Figure 3).
We also observed the accumulation of mannose and its derivatives in severe patients. In the complement system, binding of mannose to lectin leads to cleavage of C2 and C4, which then form a C3 convertase to promote complement activation (Ricklin et al., 2010).
Suppressed Platelet Degranulation in Severe COVID-19 Sera
Fifteen of 17 proteins involved in platelet degranulation were downregulated in SARS-CoV-2 infected patients, which may be associated with observed thrombocytopenia in this patient cohort (Zheng et al., 2020). Low platelet count is also reported to be associated with severe COVID-19 and mortality (Lippi et al., 2020). Two of the most intriguing proteins downregulated in severe patients are platelet-expressing chemokines pro-platelet basic protein (PPBP; also called macrophage-derived growth factor) and platelet factor 4 (PF4). PF4 was identified as a broad-spectrum HIV-1 inhibitor at the level of virus attachment and entry via interaction with the major viral envelope glycoprotein gp120 (Auerbach et al., 2012). In a proteomic investigation of sera in SARS patients, decreasing PF4 was found to be associated with poor prognosis (Poon et al., 2012), which is in consistent with our findings in COVID-19.
Most enterochromaffin cell-derived serotonins (5-hydroxytryptamine [5-HT]) are transported to platelets for storage and release (Baganz and Blakely, 2013). Serotonin level decreases as the severity of COVID-19 increases (Figure 4B) and so does the platelet count (Zheng et al., 2020). Compared with the healthy group, serotonin in non-severe and severe COVID-19 patients decreased by 2.07-fold (p = 1.86e-04) and 3.31-fold (p = 9.07e-07), respectively.
Massive Suppression of Amino Acid Metabolism in the Sera of COVID-19 Patients
Compared with the healthy controls, more than 100 metabolites, amino acids and their derivatives, in the sera of COVID-19 patients were significantly decreased, whereas their levels were either unchanged or even increased in the sera of non-COVID-19 patients. Enriched in these metabolites are 10 metabolites involved in arginine metabolism including glutamate, arginine, N-(l-arginino)-succinate, citrulline, ornithine, glutamine, 2-oxoglutarate, N-acetyl-L-glutamate, urea, and fumarate. In addition, some arginine derivatives such as argininate, asymmetric dimethylarginine, symmetric dimethylarginine, homoarginine, and N-acetylarginine were also significantly decreased in the sera of non-severe COVID-19 patients. This suggests hepatic dysfunction. Interferons have previously been shown to alter urea cycle activity during viral infection (Lercher et al., 2019), leading to regulation of multiple metabolites consistent with our data. It has been reported that arginine metabolism is suppressed in severe fever with thrombocytopenia syndrome caused by a SFTS bunyavirus (SFTSV) (Li et al., 2018b). Decreased arginine levels in SFTSV patients were associated with impaired anti-SFTSV functions of T cells.
Discussion
Classification of Severe COVID-19 Patients
Although COVID-19 can be diagnosed effectively by nucleic-acid-based methods at an early stage, it is equally critical to identify severe COVID-19 patients before the manifestation of severe symptoms to minimize mortality. In this study, we show that severe cases can be classified by molecular signatures of metabolites and proteins using a machine learning model based on the expression levels of 22 serum proteins and 7 metabolites (Figures 2A and 2B). We achieved an overall accuracy of 93.5% in the training set (C1). Classification of two patients did not match clinical diagnosis. One of them (patient XG3) is a non-severe individual who was the oldest patient in the non-severe group, reflecting the complexity of clinical cohort. In this training set (C1), five severe patients were correctly identified based on the analysis of their serum samples collected 1 to 4 days before they were clinically diagnosed as severe patients (Figure 1), suggesting that their serum protein and metabolite signatures at the sampling time may already point to further deterioration into severe state even when severe clinical symptoms have not started to appear yet.
The proteins and metabolites used in our classifier (Figure 2A) contain several known biomarkers for viral infections, such as SAA2, SAA1, and CRP, which have already been used empirically to monitor the severity of COVID-19. Our study suggests that more characteristic molecular changes at protein and metabolite levels may be used to build a diagnostic model for identification of severe cases. The classifier also included exceptionally high levels of other acute phase proteins, including SERPINA3, among others (Figures 2A and 2B). Our data suggest potential benefits of broader testing of these proteins in newly diagnosed cases to identify which COVID-19 patients are likely to progress to severe disease. The model contains molecules involved in hepatic damage. The elevation of glucose, glucuronate, bilirubin degradation product, and four bile acid derivatives, suggests suppressed hepatic detoxification (Rowland et al., 2013). Vascular cell adhesion protein 1 (VCAM-1), which helps to regulate transendothelial migration of leukocytes by stimulating production of reactive oxygen species (ROS), was upregulated in our data. As a potent antioxidant and inhibitor of VCAM-1-dependent cellular events (Keshavan et al., 2005), bilirubin was found to be downregulated in our metabolomic data.
Seven patients were correctly classified in the independent test cohort (C2) containing 10 patients. Two of three incorrectly classified cases might be explained by the patients’ complex comorbidity and medication history. We further validated this classifier using a second test cohort (C3), leading to 16 correct assignments out of 19 patients. The three incorrectly classified patients all had complex clinical history. Nevertheless, the false negative rate and false positive rate of C2 and C3 are 8%–33%, probably because of covariate imbalance and relatively small sample size, necessitating future validation studies in bigger cohorts with more rigorous study design.
Molecular Insights for the Pathogenesis of SARS-CoV-2 Infection
Our data shed light on the molecular changes reflected in COVID-19 sera, which could potentially yield critical diagnostic markers or therapeutic targets for managing severe COVID-19 patients (Figure 5 ). These molecular derangements may originate from binding of SARS-CoV-2 to alveolar macrophages via the ACE2 receptor (Hoffmann et al., 2020), resulting in release of interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) (Mehta et al., 2020) by macrophages (Gabay and Kushner, 1999). In response to elevated cytokines—especially IL-6, which triggers fever—various APPs are released by liver. Activation of APPs is accompanied by immunogenetics or organic damage (Gabay and Kushner, 1999). Our metabolomics data also provide plausible evidence for hepatic injury. In physiological condition, hormones or bilirubin binds to glucuronate, a derivative of glucose, for liver detoxification (Rowland et al., 2013). The elevation of glucose, glucuronate, bilirubin degradation product, and four bile acid derivatives in severe patients indicates potentially declined liver detoxification function (Figures 4 and 5). Our data also revealed upregulation of multiple APPs, including CRP and major attack complexes (MACs) in the severe sera. CRP can activate the complement system (Chirco and Potempa, 2018). This, on the one hand, leads to enhanced cytokine and chemokine production, potentially contributing to “cytokine storm”; and on the other hand, it overly recruits macrophages from the peripheral blood, which could result in acute lung injury (Chirco and Potempa, 2018, Narasaraju et al., 2011). Because about 50% of platelets are produced in the lung (Lefrançais et al., 2017), platelets may in turn respond to lung injury and activate macrophages by degranulation (Mantovani and Garlanda, 2013), which may further add to cytokine storm. A recent necropsy report revealed alveolar macrophage infiltration and activation in severe COVID-19 patients (Liao et al., 2020), supporting our findings.
Insights for COVID-19 Therapeutics
To date, few other therapies are proven effective for severe COVID-19 patients. Most patients receive standard supportive care and antiviral therapy (Wang et al., 2020). Corticosteroid treatment was effective in suppressing MERS-CoV and SARS-CoV (Arabi et al., 2018) but showed negligible effect on COVID-19 patients and may even have induced lung injury (Russell et al., 2020). The molecular changes revealed in this study in the COVID-19 sera might be useful for prioritizing therapeutic strategies for the severe patients.
Our proteomic data showed that proteins related to platelet degranulation were substantially downregulated in severe patients, a finding that was confirmed by low platelet counts (Zheng et al., 2020). The association between thrombocytopenia and viral infection has been observed in SARS-CoV (Zou et al., 2004), hepatitis C virus (HCV) (Assinger, 2014), and Dengue virus (Wilder-Smith et al., 2004). Thus, it might be useful to monitor changes in platelets during treatment.
Complement activation suppresses virus invasion and may lead to inflammatory syndromes (Barnum, 2017). Our data showed a general upregulation of complement system proteins, including MAC proteins such as C5, C6, and C8. Suppression of complement system has been reported as an effective immunotherapeutic in SARS-infected mouse model (Gralinski et al., 2018). C5a has been reported as highly expressed in severe SARS and MERS patients as well (Wang et al., 2015). Inhibition of C5a has been reported to alleviate viral infection-induced acute lung injury (Garcia et al., 2013, Jiang et al., 2018, Sun et al., 2015). Our data suggest that severe COVID-19 patients might benefit from suppression of complement system.
Our metabolomics results showed that more than 100 lipids including glycerophospholipid, sphingolipids, and fatty acids were downregulated in COVID-19 patient sera, probably because of damage to the liver, which is also reflected in aberrancy in bilirubin and bile acids. Glycerophospholipid, sphingolipids (one of the components of lipid rafts), and fatty acids have been reported to play an important role in the early development of enveloped viruses (Schoggins and Randall, 2013). Suppression of cholesterol synthesis by MβCD has been reported to be effective in inhibiting release of SARS-CoV particles in infected Vero E6 cells (Li et al., 2007). Drugs inhibiting lipid synthesis such as statin have been proposed to treat HCV (Heaton and Randall, 2011) and COVID-19 (Fedson et al., 2020). Our data suggest these potential therapeutics might be helpful in the treatment of severe COVID-19 patients.
Limitations of This Study and Outlook
SARS-CoV-2 is highly infectious, exerting huge pressure on the medical system worldwide. Upon COVID-19 outbreak, limited information of this pathogen was available, which restricted the collection of a large number of clinical specimens for this study mainly because of biosafety constraints. The median age of the severe patients is about 12 years older than the non-severe patients in our cohort (Table 1), so the impact of age on our data interpretation could not be precisely defined. The severe patients also exhibit slightly higher BMI and a higher proportion of comorbidities such as diabetes, which may influence the metabolomic profiles (Table 1). Samples from some severe patients were collected before or after the diagnosis of severe cases, although most of them were collected close to the diagnosis date. Nevertheless, sex, age, and variable hospitalization time and sampling time did not substantially distort the biological differences in the global proteomic and metabolomic profiles (Figures S2D and S2E). Although these confounding factors might be alleviated in future studies, we did identify multiple promising biomarker candidates (Figure 2).
The proteomic and metabolomic analysis in this study is not absolute quantification. If the model is to be applied in clinic, more rigorous quantification and extensive validation of these molecules using standard peptides and metabolites are required. Impact of drugs including traditional Chinese medicine to the proteomic/metabolomic profiles have to be evaluated, too. The sera samples were collected from different time points along the disease course, which could be potentially utilized to explore molecular dynamics during disease progression. However, the sample size is rather small. Future studies of sera from more time points are required for rigorous temporal analysis.
In conclusion, this study presents a systematic proteomic and metabolomic investigation of serum samples from multiple COVID-19 patient groups and control groups. We demonstrated the potential of identifying COVID-19 patients who may eventually become severe cases based on analysis of a panel of serum proteins and metabolites. Our data offer a landscape view of blood molecular changes induced by SARS-CoV-2 infection, which may provide useful diagnostic and therapeutic clues in the ongoing battle against the COVID-19 pandemic.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Serum samples from 65 COVID-19 patients and 25 non-COVID-19 patients and 28 healthy individuals | Taizhou Hospital | This paper(Table S1-Patient ID) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Triethylammonium bicarbonate buffer (TEAB) | Sigma-Aldrich | Cat # T7408 |
| Urea | Sigma-Aldrich | Cat # U1250 |
| Tris (2-carboxyethyl) phosphine (TCEP) | Adamas-beta | Cat # 61820E |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat # I6125 |
| Trypsin | Hualishi Tech | Cat # HLS TRY001C |
| Trifluoroacetic acid (TFA) | Thermo Fisher Scientific | Cat # 85183 |
| Water | Thermo Fisher Scientific | Cat # W6-4 |
| Acetonitrile | Thermo Fisher Scientific | Cat # A955-4 |
| Formic acid (FA) | Thermo Fisher Scientific | Cat # A117-50 |
| Ammonium hydroxide solution | Sigma-Aldrich | Cat # 221228 |
| Methanol | Sigma-Aldrich | Cat # 34860 |
| Critical Commercial Assays | ||
| TMTpro 16plex reagents | Thermo Fisher Scientific | Cat # A44520 |
| C-reactive protein (CRP) | Beckman Coulter | Cat # OSR6199 |
| Alanine aminotransferase (ALT) | Beckman Coulter | Cat # OSR6107 |
| Aspartate Aminotransferase (AST) | Beckman Coulter | Cat # OSR6209 |
| γ-glutamyltransferase (GGT) | Beckman Coulter | Cat # OSR6120 |
| Total bilirubin (TBIL) | Beckman Coulter | Cat # TB7074 |
| Direct bilirubin (DBIL) | Beckman Coulter | Cat # DB7084 |
| Creatinine | Beckman Coulter | Cat # OSR6178 |
| Glucose | Beckman Coulter | Cat # GL7210 |
| SARS-CoV-2 nucleic acid detection kit | Shanghai Zhijiang | Cat # Z-RR-0479-02025 Cat # Z-RR-0479-02-50 Cat # Z-RR-0479-02AT-50 |
| Stromatolyser-4DL (FFD-200A) | Sysmex | Cat # ZG900003(BN724234) |
| Stromatolyser-4DS (FFS-800A) | Sysmex | Cat # 984-1721-6(114010116) |
| Cellsheath (SE-90L) | Sysmex | Cat # ZG801001(83400324) |
| Cellpack (PK-30L) | Sysmex | Cat # ZG801000(88408711) |
| Cellclean (CL-50) | Sysmex | Cat # 834-0162-1 |
| Deposited Data | ||
| Mass spectrometry data | This paper | https://www.iprox.org/ (IPX0002106000 and IPX0002171000) |
| Data analysis codes | This paper | https://github.com/guomics-lab/CVDSBA |
| Software and Algorithms | ||
| Xcalibur | Thermo Fisher Scientific | Cat # OPTON-30965 |
| Proteome Discoverer Version 2.4.1.15 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| R version 3.5.2 | R Project | https://www.r-project.org |
| Skyline version 20.1.1.12 | MacLean et al., 2010 | https://skyline.ms/project/home/begin.view |
| Metascape | Zhou et al., 2019 | https://metascape.org/gp/index.html#/main/step1 |
| ClueGO 2.5.6 | Bindea et al., 2009 | https://cytoscape.org/ |
| Ingenuine pathway analysis | Krämer et al., 2014 | https://www.qiagen.com/cn/ |
| GeNet | Li et al., 2018a | http://apps.broadinstitute.org/genets# |
| Other | ||
| SOLAμ | Thermo Fisher Scientific | Cat # 62209-001 |
| ACQUITY UPLC Systems with 2D LC Technology | Waters Corporation | Cat # 186015001 |
| ACQUITY BEH C18 column, 2.1 × 100 mm, 1.7 μm | Waters Corporation | Cat # 186008316 |
| ACQUITY BEH Amide column, 2.1 × 100 mm, 1.7 μm | Waters Corporation | Cat # 186008315 |
Resource Availability
Lead Contact
Further information should be directed to and will be fulfilled by the Lead Contact Tiannan Guo (guotiannan@westlake.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The proteomics and metabolomics data are deposited in ProteomeXchange Consortium (https://www.iprox.org/). Project ID: IPX0002106000 and IPX0002171000. The project data analysis codes are deposited in GitHub (https://github.com/guomics-lab/CVDSBA).
Experimental Model and Subject Details
Patients and samples
Our team procured serum samples from 65 COVID-19 patients who visited Taizhou Hospital from January to March 2020. They were diagnosed as COVID-19 according to the Chinese Government Diagnosis and Treatment Guideline (Trial 5th version) (NHCPRC, 2020). For diagnosing COVID-19, nucleic acid from sputum or throat swab was extracted using nucleic acid extractor (Shanghai Zhijiang, China, EX3600) and virus nucleic acid extraction reagent (Shanghai Zhijiang, China, NO. P20200201) was used to extract nucleic acid. Fluorescence quantitative PCR (ABI7500) and SARS-CoV-2 nucleic acid detection kit (triple fluorescence PCR, Shanghai Zhijiang, China, NO. P20200203) were used for nucleic acid detection. This kit uses one step RT-PCR combined with Taqman technology to detect RdRp, E and N genes. Positive was concluded if RdRp gene was positive (Ct < 43), and one of E or N was positive (Ct < 43). Patients were also diagnosed as positive if two sequential tests of RdRp were positive while E and N were negative. According to the abovementioned guideline, COVID-19 patients are classified into four subgroups: 1) Mild: mild symptoms without pneumonia; 2) Typical: fever or respiratory tract symptoms with pneumonia; 3) Severe: fulfill any of the three criteria: respiratory distress, respiratory rate ≥ 30 times/min; means oxygen saturation ≤ 93% in resting state; arterial blood oxygen partial pressure/oxygen concentration ≤ 300 mmHg (1 mmHg = 0.133 kPa); 4) Critical: fulfill any of the three criteria: respiratory failure and require mechanical ventilation; shock incidence; admission to ICU with other organ failure. In this study, we included both severe and non-severe patients, with the latter composed of mild and typical COVID-19 patients. We also procured 25 non-COVID-19 patients with similar clinical characteristics including fever and/or cough as COVID-19 patients however negative in the nucleic acid test. Based on the Chinese Government Diagnosis and Treatment Guideline (Trial 5th version) (NHCPRC, 2020), patients are defined as suspected COVID-19 cases when they meet the following three clinical criteria: 1) fever or respiratory symptoms, 2) imaging manifestation of pneumonia, and 3) optional reduction of white blood cell or lymphocyte count at early stage. The patients only need to meet at least two of the above three criteria if they have been exposed to COVID-19 individuals. Most non-COVID-19 patients exhibited fever and cough. Causative analysis of the infection showed that four patients infected by herpes simplex virus, one patient infected by varicella-zoster virus, one by respiratory syncytial virus, one by klebsiella pneumoniae and acinetobacter baumanii, and one by enterococcus faecium. Some patients had other diseases including cancer, cerebral hemorrhage or lymphoma. No infection was detected in the other patients according to respiratory tract virus antigen test. We also collected serum samples from 28 healthy individuals as control. All the blood samples were drawn using serum separation tubes (Zhejiang GongDong Medical Technology Co., Ltd, China) between 6:00-6:30 am in the morning, stored for about 30 min in room temperature before sending the sample to the clinical chemistry laboratory using a biosafety transport box at 7 am. The laboratory obtained the sample about 7:30 am and centrifuged them at 1,500 g for 10 min. Then we collected the serum in new centrifuge tubes and immediately stored at −80°C. The samples from this study are from a clinical trial that our team initiated and registered in the Chinese Clinical Trial Registry with an ID of ChiCTR2000031365. This study has been approved by the Ethical/Institutional Review Board of Taizhou Hoapital and Westlake University. Contents from patients were waived by the boards.
Method Details
Proteome analysis
Serum samples were inactivated and sterilized at 56°C for 30 min, and processed as previously with some modifications. Five μL serum from each specimen was denatured in 50 μL buffer containing 8 M urea in 100 mM triethylammonium bicarbonate (TEAB) at 32°C for 30 min. The proteins were reduced with 10 mM tris (2-carboxyethyl) phosphine (TCEP) for 30 min at 32°C, then alkylated for 45 min with 40 mM iodoacetamide (IAA) in darkness at room temperature (25°C). The protein extracts were diluted with 200 μL 100 mM TEAB, and digested with double-step trypsinization (Hualishi Tech. Ltd, Beijing, China), each step with an enzyme-to-substrate ratio of 1:20, at 32°C for 60 min. The reaction was stopped by adding 30 μL 10% trifluoroacetic acid (TFA) in volume. Digested peptides were cleaned-up with SOLAμ (Thermo Fisher Scientific, San Jose, USA) following the manufacturer’s instructions, and labeled with TMTpro 16plex label reagents (Thermo Fisher Scientific, San Jose, USA) as described previously. The TMT samples were fractionated using a nanoflow DIONEX UltiMate 3000 RSLCnano System (Thermo Fisher Scientific, San Jose, USA) with an XBridge Peptide BEH C18 column (300 Å, 5 μm × 4.6 mm × 250 mm) (Waters, Milford, MA, USA)(Gao et al., 2020). The samples were separated using a gradient from 5% to 35% acetonitrile (ACN) in 10 mM ammonia (pH = 10.0) at a flow rate of 1 mL/min. Peptides were separated into 120 fractions, which were consolidated into 40 fractions. The fractions were subsequently dried and re-dissolved in 2% ACN/0.1% formic acid (FA). The re-dissolved peptides were analyzed by LC-MS/MS with the same LC system coupled to a Q Exactive HF-X hybrid Quadrupole-Orbitrap (Thermo Fisher Scientific, San Jose, USA) in data dependent acquisition (DDA) mode. For each acquisition, peptides were loaded onto a precolumn (3 μm, 100 Å, 20 mm∗75 μm i.d.) at a flow rate of 6 μL/min for 4 min and then analyzed using a 35 min LC gradient (from 5% to 28% buffer B) at a flow rate of 300 nL/min (analytical column, 1.9 μm, 120 Å, 150 mm∗75 μm i.d.). Buffer A was 2% ACN, 98% H2O containing 0.1% FA, and buffer B was 98% ACN in water containing 0.1% FA. All reagents were MS grade. The m/z range of MS1 was 350-1,800 with the resolution at 60,000 (at 200 m/z), AGC target of 3e6, and maximum ion injection time (max IT) of 50 ms. Top 15 precursors were selected for MS/MS experiment, with a resolution at 45,000 (at 200 m/z), AGC target of 2e5, and max IT of 120 ms. The isolation window of selected precursor was 0.7 m/z. The resultant mass spectrometric data were analyzed using Proteome Discoverer (Version 2.4.1.15, Thermo Fisher Scientific) using a protein database composed of the Homo sapiens fasta database downloaded from UniProtKB on 07 Jan 2020, containing 20412 reviewed protein sequences, and the SARS-CoV-2 virus fasta downloaded from NCBI (version NC_045512.2). Enzyme was set to trypsin with two missed cleavage tolerance. Static modifications were set to carbamidomethylation (+57.021464) of cysteine, TMTpro (+304.207145) of lysine residues and peptides’ N termini, and variable modifications were set to oxidation (+15.994915) of methionine and acetylation (+42.010565) of peptides’ N-termini. Precursor ion mass tolerance was set to 10 ppm, and product ion mass tolerance was set to 0.02 Da. The peptide-spectrum-match allowed 1% target false discovery rate (FDR) (strict) and 5% target FDR (relaxed). Normalization was performed against the total peptide amount. The other parameters followed the default setup. Different immunoglobulins as appeared in the fasta file are included, while other post-translational modifications and protein isoforms are not analyzed in this study, but they could be potentially analyzed in the future.
Quality control of proteome data
The quality of proteomic data was ensured at multiple levels. First, a mouse liver digest was used for instrument performance evaluation. We also run water samples (buffer A) as blanks every 4 injections to avoid carry-over. Serum samples of four patient groups from both training and test cohorts were randomly distributed in eight different batches. Every batch contains a pooled sample, i.e., a mixture of all peptide samples, as the control sample labeled by TMT pro-134N for aligning data from different batches and evaluation of quantitative accuracy. Six samples were injected in technical replicates.
Metabolome analysis
Ethanol was added to the serum samples and shaken vigorously to inactivate any potential viruses, then dried in a biosafety hood. The dried samples were further treated for metabolomics analysis. The metabolomic analysis was performed as described previously(Lee et al., 2019). Briefly, deactivated serum samples, 100 μL each, were extracted by adding 300 μL methanol extraction solution. The mixtures were shaken vigorously for 2 min. Proteins were denatured and precipitated by centrifugation. The supernatants contained metabolites of diverse chemical natures. To ensure the quantity and reliability of metabolite detection, four platforms were performed with non-target metabolomics. Each supernatant was divided into four fractions: two for analysis using two separate reverse-phase /ultra-performance liquid chromatography (RP/UPLC)-MS/MS methods with positive ion-mode electrospray ionization (ESI), one for analysis using RP/ UPLC-MS/MS with negative-ion mode ESI, and one for analysis using hydrophilic interaction liquid chromatography (HILIC)/UPLC-MS/MS with negative-ion mode ESI. Each fraction was dried under nitrogen gas to remove the organic solvent and later re-dissolved in four different reconstitution solvents compatible with each of the four UPLC-MS/MS methods.
All UPLC-MS/MS methods used ACQUITY 2D UPLC system (Waters, Milford, MA, USA) and Q Exactive HF hybrid Quadrupole-Orbitrap (Thermo Fisher Scientific, San Jose, USA) with HESI-II heated ESI source and Orbitrap mass analyzer. The mass spectrometer was operated at 35,000 mass resolution (at 200 m/z). In the first UPLC-MS/MS method, the QE was operated under positive electron spray ionization (ESI) coupled with a C18 column (UPLC BEH C18, 2.1 × 100 mm, 1.7 μm; Waters) was used in UPLC. The mobile solutions used in the gradient elution were water and methanol containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% FA; the gradient elution for methods using C18 columns was performed in a seven minutes run when the polar mobile phase was gradually increased from 5% to 95%. In the second method, the QE was still operated under ESI positive mode, and the UPLC used the same C18 column as in method one, but the mobile phase solutions were optimized for more hydrophobic compounds and contained methanol, acetonitrile, water, 0.05% PFPA, and 0.01% FA. The third method had the QE operated under negative ESI mode, and the UPLC method used a C18 column eluted with mobile solutions containing methanol and water in 6.5 mM ammonium bicarbonate at pH 8. The UPLC column used in the fourth method was HILIC column (UPLC BEH Amide, 2.1 × 150 mm, 1.7 μm; Waters), and the mobile solutions were consisted of water and acetonitrile with 10 mM ammonium formate at pH 10.8; gradient elution for this method is performed in a seven minutes run with the polar mobile phase decreased from 80% to 20%. The QE was operated under negative ESI mode. The QE mass spectrometer analysis was alternated between MS and data-dependent MS2 scans using dynamic exclusion. The scan range was 70-1,000 m/z. The MS capillary temperature was 350°C, sheath gas flow rate at 40, aux gas flow rate at 5 for both positive and negative methods.
After raw data pre-processing, peak finding/alignment, and peak annotation using in-house software, metabolites were identified by searching an in-house library containing more than 3,300 standards with library data entries generated from running purified compound standards through the experimental platforms. Identification of metabolites must meet three strict criteria: narrow window retention index (RI), accurate mass with variation less than 10 ppm and MS/MS spectra with high forward and reverse scores based on comparisons of the ions present in the experimental spectrum to the ions present in the library spectrum entries. Almost all isomers can be distinguished by these three criteria. All identified metabolites meet the level 1 requirements by the Chemical Analysis Working Group (CAWG) of the Metabolomics Standards Initiative (MSI) expect some asterisk labeled lipids which MS/MS spectral were in silico matched.
Quality control of metabolome analysis
Several types of quality control samples were included in the experiment: a pooled sample generated by taking a small volume of each experimental sample to serve as a technical replicate that was run multiple times throughout the experiment, extracted water samples served as blanks, and extracted commercial plasma samples for monitoring instrument variation. A mixture of internal standards was also spiked into every sample to aid chromatographic peak alignment and instrument stability monitoring. Instrument variability was determined by calculating the median relative SD (RSD) of all internal standards in each sample. The experimental process variability was determined by calculating the median RSD for all endogenous metabolites present in the pooled quality control samples.
Targeted protein analysis
Peptide samples were prepared in the same way as the previous proteomic section except no TMT labeling was performed. Eksigent NanoLC 400 System (Eksigent, Dublin, CA, USA) coupled with TripleTOF 6600 system (SCIEX, CA, USA) was applied for MRM-HR experiment. The peptide digests were separated at a 5 μL/min with a 10 min gradient (buffer B: 5%–10% for 1 min, 10%–40% for 6 min, 40%–80% for 0.1min, maintained 80% for 2.9 min, 80%–5% for 1 min) using an analytical column (3 μm, ChromXP C18CL, 120 Å, 150∗0.3 mm). IDA mode (rolling collision energy, +2 to +5 charge states with intensity criteria above 2,000,000 cps to guarantee all untargeted peptides will not be acquired) for time-scheduling was set up for 51 peptides including 10 iRT peptides (Escher et al., 2012) with a mass tolerance of 50 ppm. Accumulation time for TOF-MS scan (350-1250 m/z) and MS/MS scans (100-1500 m/z) was 250 ms and 50 ms, respectively. The data acquired by MRM-HR experiment were analyzed by Skyline (MacLean et al., 2010). The retention time was predicted by the iRT, and the isolation time window is 2 min. The mass analyzer for MS1 and MS/MS was set as “TOF” with the resolution power of 30,000.
Targeted metabolite analysis
For semiquantitative assay of the seven potential metabolite markers in the 19 COVID-19 patients in the test cohort 2, the sample preparation and analysis were carried out basically the same as detailed in the metabolomics assay. Briefly, each metabolite was analyzed using one of the 4 UPLC-MS/MS methods: reverse phase UPLC coupled with negative ESI-MS/MS, reverse phase UPLC coupled with positive ESI-MS/MS, reverse phase UPLC coupled with positive ESI-MS/MS for more hydrophobic metabolites, and HILIC UPLC coupled with negative ESI-MS/MS. The target metabolites were manually curated, and their peak areas were obtained using the Thermo Fisher Xcalibur 4.0 software.
Quantification and Statistical Analysis
Statistical analysis and machine learning
Metabolites and therapeutic compounds with over 80% missing ratios in a particular patient group were removed for the metabolomics dataset containing endogenous metabolites while full proteomics features were used for the subsequent statistical analysis. Missing values were imputed with the minimal value and zero in metabolomics and proteomics dataset respectively. Log2 fold-change (log2 FC) was calculated on the mean of the same patient group for each pair of comparing groups. Two-sided unpaired Welch’s t test was performed for each pair of comparing groups and adjusted p values were calculated using Benjamini & Hochberg correction. The statistical significantly changed proteins or metabolites were selected using the criteria of adjust p value less than 0.05 indicated and absolute log2 FC larger than 0.25. From the training cohort, we selected important protein and metabolite features with mean decrease accuracy larger than 3 using random forest. In the random forest analysis, a thousand trees were built using R package randomForest (version 4.6.14) with 10-fold cross validation, and this was repeated for 100 times. The normalized additive predicting probability was computed as the final predicting probability. The larger probability for the binary classification was adopted as the predictive label. For validation in the test cohort 2 (C3) generated by targeted proteomics and metabolomics, z-score normalization was applied before running the model validation. Those selected important features were used for the random forest analysis on the independent test cohort. We also ran the randomForest analysis with omics features after z-score normalization and got same classification results.
Pathway analysis
Four network pathway analysis tools were used for pathway analysis using 93 differentially expressed proteins (DEPs). The top Gene Ontology (GO) processes were enriched by Metascape web-based platform (Zhou et al., 2019). The GO terms is enriched using the Cytoscape plug-in ClueGO (Bindea et al., 2009). Ingenuine pathway analysis (Krämer et al., 2014) of the regulated proteins identifies most significantly relevant pathways with p value of determined based on right-tailed Fisher’s Exact Test with the overall activation or inhibition states of enriched pathways were predicted by z-score. Functional co-expression network analysis by GeNet(Li et al., 2018a) to represent statistical co-expressed protein modules.
Acknowledgments
This work is supported by grants from Tencent Foundation (2020), National Natural Science Foundation of China (81972492, 21904107, and 81672086), Zhejiang Provincial Natural Science Foundation for Distinguished Young Scholars (LR19C050001), and Hangzhou Agriculture and Society Advancement Program (20190101A04). We thank Drs. R. Aebersold, O.L. Kon, H. Yu, and D. Li and the Guomics team for invaluable comments to this study. We thank Westlake University Supercomputer Center for assistance in data storage and computation.
Author Contributions
T.G., Haixiao Chen, H.L., B.S., and Y. Zhu. designed and supervised the project. B.S., X.B., J.D., Y. Zhang, J.L., J.X., Z.H., B.C., J.W., H.Y., Y. Zheng, D.W., and J.Z. collected the samples and clinical data. X.Y., Y.S., F.Z., R.S., L.Q., W.G., W.L., S. Liang, Hao Chen, X.L., X.D., G.R., N.X., X.C., H.G., L.L., S.Li, Q.X., and T.L. conducted proteomic analysis. Data were interpreted and presented by all co-authors. C.Z., S.Q., Z. Kong, and Z. Kang conducted metabolomic analysis. T.G. wrote the manuscript with input from co-authors.
Declaration of Interests
The research group of T.G. is partly supported by Tencent, Thermo Fisher Scientific, SCIEX, and Pressure Biosciences Inc. C.Z., Z. Kong, Z. Kang, and S.Q. are employees of DIAN Diagnostics.
Published: May 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.05.032.
Supplemental Information
References
- Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Arabi Y.M., Mandourah Y., Al-Hameed F., Sindi A.A., Almekhlafi G.A., Hussein M.A., Jose J., Pinto R., Al-Omari A., Kharaba A., Saudi Critical Care Trial Group Corticosteroid Therapy for Critically Ill Patients with Middle East Respiratory Syndrome. Am. J. Respir. Crit. Care Med. 2018;197:757–767. doi: 10.1164/rccm.201706-1172OC. [DOI] [PubMed] [Google Scholar]
- Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front. Immunol. 2014;5:649. doi: 10.3389/fimmu.2014.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach D.J., Lin Y., Miao H., Cimbro R., Difiore M.J., Gianolini M.E., Furci L., Biswas P., Fauci A.S., Lusso P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. USA. 2012;109:9569–9574. doi: 10.1073/pnas.1207314109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baganz N.L., Blakely R.D. A dialogue between the immune system and brain, spoken in the language of serotonin. ACS Chem. Neurosci. 2013;4:48–63. doi: 10.1021/cn300186b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum S.R. Complement: A Primer for the Coming Therapeutic Revolution. Pharmacol. Ther. 2017;172:63–72. doi: 10.1016/j.pharmthera.2016.11.014. [DOI] [PubMed] [Google Scholar]
- Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.H., Pagès F., Trajanoski Z., Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain D.W., Cidlowski J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017;17:233–247. doi: 10.1038/nri.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirco K.R., Potempa L.A. C-Reactive Protein As a Mediator of Complement Activation and Inflammatory Signaling in Age-Related Macular Degeneration. Front. Immunol. 2018;9:539. doi: 10.3389/fimmu.2018.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M.J., Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedson D.S., Opal S.M., Rordam O.M. Hiding in Plain Sight: an Approach to Treating Patients with Severe COVID-19 Infection. MBio. 2020;11 doi: 10.1128/mBio.00398-20. e00398-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C., Kushner I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Gao H., Zhang F., Liang S., Zhang Q., Lyu M., Qian L., Liu W., Ge W., Chen C., Yi X. Accelerated Lysis and Proteolytic Digestion of Biopsy-Level Fresh-Frozen and FFPE Tissue Samples Using Pressure Cycling Technology. J. Proteome Res. 2020;19:1982–1990. doi: 10.1021/acs.jproteome.9b00790. [DOI] [PubMed] [Google Scholar]
- Garcia C.C., Weston-Davies W., Russo R.C., Tavares L.P., Rachid M.A., Alves-Filho J.C., Machado A.V., Ryffel B., Nunn M.A., Teixeira M.M. Complement C5 activation during influenza A infection in mice contributes to neutrophil recruitment and lung injury. PLoS ONE. 2013;8:e64443. doi: 10.1371/journal.pone.0064443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghinai I., McPherson T.D., Hunter J.C., Kirking H.L., Christiansen D., Joshi K., Rubin R., Morales-Estrada S., Black S.R., Pacilli M. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet. 2020;395:1137–1144. doi: 10.1016/S0140-6736(20)30607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralinski L.E., Sheahan T.P., Morrison T.E., Menachery V.D., Jensen K., Leist S.R., Whitmore A., Heise M.T., Baric R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. MBio. 2018;9 doi: 10.1128/mBio.01753-18. e01753–e01718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.-G., Zhu C.L., Cheng D.Z., Xie Y., Liu F., Zhou X. Enchanced levels of apolipoprotein M during HBV infection feedback suppresses HBV replication. Lipids Health Dis. 2011;10:154. doi: 10.1186/1476-511X-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., China Medical Treatment Expert Group for Covid-19 Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun Y.A., Obeid L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018;19:175–191. doi: 10.1038/nrm.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton N.S., Randall G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011;19:368–375. doi: 10.1016/j.tim.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.C., Yu H.C., Martin R., Cirulli E.T., Schenker-Ahmed N.M., Hicks M., Cohen I.V., Jönsson T.J., Heister R., Napier L. Precision medicine integrating whole-genome sequencing, comprehensive metabolomics, and advanced imaging. Proc. Natl. Acad. Sci. USA. 2020;117:3053–3062. doi: 10.1073/pnas.1909378117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Zhao G., Song N., Li P., Chen Y., Guo Y., Li J., Du L., Jiang S., Guo R. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 2018;7:77. doi: 10.1038/s41426-018-0063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavan P., Deem T.L., Schwemberger S.J., Babcock G.F., Cook-Mills J.M., Zucker S.D. Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J. Immunol. 2005;174:3709–3718. doi: 10.4049/jimmunol.174.6.3709. [DOI] [PubMed] [Google Scholar]
- Krämer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.H., Shannon C.P., Amenyogbe N., Bennike T.B., Diray-Arce J., Idoko O.T., Gill E.E., Ben-Othman R., Pomat W.S., van Haren S.D., EPIC Consortium Dynamic molecular changes during the first week of human life follow a robust developmental trajectory. Nat. Commun. 2019;10:1092. doi: 10.1038/s41467-019-08794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrançais E., Ortiz-Muñoz G., Caudrillier A., Mallavia B., Liu F., Sayah D.M., Thornton E.E., Headley M.B., David T., Coughlin S.R. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105–109. doi: 10.1038/nature21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher A., Bhattacharya A., Popa A.M., Caldera M., Schlapansky M.F., Baazim H., Agerer B., Gurtl B., Kosack L., Majek P. Type I Interferon Signaling Disrupts the Hepatic Urea Cycle and Alters Systemic Metabolism to Suppress T Cell Function. Immunity. 2019;51:1074–1087.e9. doi: 10.1016/j.immuni.2019.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.M., Li Y.G., Yamate M., Li S.M., Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007;9:96–102. doi: 10.1016/j.micinf.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Kim A., Rosenbluh J., Horn H., Greenfeld L., An D., Zimmer A., Liberzon A., Bistline J., Natoli T. GeNets: a unified web platform for network-based genomic analyses. Nat. Methods. 2018;15:543–546. doi: 10.1038/s41592-018-0039-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.K., Lu Q.B., Chen W.W., Xu W., Liu R., Zhang S.F., Du J., Li H., Yao K., Zhai D. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci. Transl. Med. 2018;10:eaat4162. doi: 10.1126/scitranslmed.aat4162. [DOI] [PubMed] [Google Scholar]
- Li J., Van Vranken J.G., Pontano Vaites L., Schweppe D.K., Huttlin E.L., Etienne C., Nandhikonda P., Viner R., Robitaille A.M., Thompson A.H. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods. 2020;17:399–404. doi: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T., Cai H., Chen Y., Chen Z., Fang Q., Han W., Hu S., Li J., Li T., Lu X. ALNAP; 2020. Handbook of COVID-19 Prevention and Treatment. [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Chen L., Li J., Wang X., Wang F. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv. 2020 doi: 10.1101/2020.1102.1123.20026690. [DOI] [Google Scholar]
- Lippi G., Plebani M., Henry B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta. 2020;506:145–148. doi: 10.1016/j.cca.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A., Garlanda C. Platelet-macrophage partnership in innate immunity and inflammation. Nat. Immunol. 2013;14:768–770. doi: 10.1038/ni.2666. [DOI] [PubMed] [Google Scholar]
- Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration, UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas P.S., Liu L., Moon P.K., Joshi A.U., Dove C., Mhatre S., Contrepois K., Wang Q., Lee B.A., Coronado M. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019;20:50–63. doi: 10.1038/s41590-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy S., Gomersall C.D., Fowler R.A. Care for Critically Ill Patients With COVID-19. JAMA. 2020 doi: 10.1001/jama.2020.3633. [DOI] [PubMed] [Google Scholar]
- Narasaraju T., Yang E., Samy R.P., Ng H.H., Poh W.P., Liew A.A., Phoon M.C., van Rooijen N., Chow V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011;179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHCPRC (National Health Commission of the PRC) 2020. Diagnosis and Treatment Protocol for COVID-19 (Trial Version 5)http://www.nhc.gov.cn/jkj/s3577/202002/a5d6f7b8c48c451c87dba14889b30147.shtml [Google Scholar]
- Nie S., Zhao X., Zhao K., Zhang Z., Zhang Z., Zhang Z. Metabolic disturbances and inflammatory dysfunction predict severity of coronavirus disease 2019 (COVID-19): a retrospective study. medRxiv. 2020 doi: 10.1101/2020.03.24.20042283. [DOI] [Google Scholar]
- Pang R.T., Poon T.C., Chan K.C., Lee N.L., Chiu R.W., Tong Y.K., Chim S.S., Sung J.J., Lo Y.M. Serum amyloid A is not useful in the diagnosis of severe acute respiratory syndrome. Clin. Chem. 2006;52:1202–1204. doi: 10.1373/clinchem.2006.068395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon T.C., Pang R.T., Chan K.C.A., Lee N.L., Chiu R.W., Tong Y.-K., Chim S.S., Ngai S.M., Sung J.J., Lo Y.M. Proteomic analysis reveals platelet factor 4 and beta-thromboglobulin as prognostic markers in severe acute respiratory syndrome. Electrophoresis. 2012;33:1894–1900. doi: 10.1002/elps.201200002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D., Hajishengallis G., Yang K., Lambris J.D. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C.A., Ivanova P.T., Byrne M.O., Brown H.A., Marnett L.J. Lipid profiling reveals glycerophospholipid remodeling in zymosan-stimulated macrophages. Biochemistry. 2007;46:6026–6042. doi: 10.1021/bi0621617. [DOI] [PubMed] [Google Scholar]
- Rowland A., Miners J.O., Mackenzie P.I. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 2013;45:1121–1132. doi: 10.1016/j.biocel.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Russell C.D., Millar J.E., Baillie J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. 2020;395:473–475. doi: 10.1016/S0140-6736(20)30317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Lopez E., Zhong Z., Stubelius A., Sweeney S.R., Booshehri L.M., Antonucci L., Liu-Bryan R., Lodi A., Terkeltaub R., Lacal J.C. Choline Uptake and Metabolism Modulate Macrophage IL-1beta and IL-18 Production. Cell Metab. 2019;29:1350–1362.e7. doi: 10.1016/j.cmet.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Randall G. Lipids in innate antiviral defense. Cell Host Microbe. 2013;14:379–385. doi: 10.1016/j.chom.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Zhao G., Liu C., Fan W., Zhou X., Zeng L., Guo Y., Kou Z., Yu H., Li J. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin. Infect. Dis. 2015;60:586–595. doi: 10.1093/cid/ciu887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevarajan I., Nguyen T.H.O., Koutsakos M., Druce J., Caly L., van de Sandt C.E., Jia X., Nicholson S., Catton M., Cowie B. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med. 2020;26:453–455. doi: 10.1038/s41591-020-0819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Xiao H., Guo R., Li Y., Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg. Microbes Infect. 2015;4:e28. doi: 10.1038/emi.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y. JAMA; China: 2020. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert A., Weis N., Brüne B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology. 2009;214:748–760. doi: 10.1016/j.imbio.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Wilder-Smith A., Earnest A., Paton N.I. Use of simple laboratory features to distinguish the early stage of severe acute respiratory syndrome from dengue fever. Clin. Infect. Dis. 2004;39:1818–1823. doi: 10.1086/426029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., McGoogan J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- Yan B., Chu H., Yang D., Sze K.-H., Lai P.-M., Yuan S., Shuai H., Wang Y., Kao R.Y.-T., Chan J.F.-W., Yuen K.Y. Characterization of the Lipidomic Profile of Human Coronavirus-Infected Cells: Implications for Lipid Metabolism Remodeling upon Coronavirus Replication. Viruses. 2019;11:73. doi: 10.3390/v11010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Zhang Y., Chi H., Chen S., Peng M., Luo L., Chen L., Li J., Shen B., Wang D. ). The hemocyte counts as a potential biomarker for predicting disease progression in COVID-19: a retrospective study. Clin Chem Lab Med. 2020 doi: 10.1515/cclm-2020-0377. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z., Yang Y., Chen J., Xin S., Zhang W., Zhou X., Mao Y., Hu L., Liu D., Chang B. Prognostic factors for severe acute respiratory syndrome: a clinical analysis of 165 cases. Clin. Infect. Dis. 2004;38:483–489. doi: 10.1086/380973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics and metabolomics data are deposited in ProteomeXchange Consortium (https://www.iprox.org/). Project ID: IPX0002106000 and IPX0002171000. The project data analysis codes are deposited in GitHub (https://github.com/guomics-lab/CVDSBA).