

Abstract
Ferroptosis is a recently discovered form of iron-dependent regulated cell death (RCD) that occurs via peroxidation of phospholipids containing polyunsaturated fatty acid (PUFA) moieties. Activating this form of cell death is an emerging strategy in cancer treatment. Because multiple pathways and molecular species contribute to the ferroptotic process, predicting which tumors will be sensitive to ferroptosis is a challenge. We thus develop a mathematical model of several critical pathways to ferroptosis in order to perform a systems-level analysis of the process. We show that sensitivity to ferroptosis depends on the activity of multiple upstream cascades, including PUFA incorporation into the phospholipid membrane, and the balance between levels of pro-oxidant factors (reactive oxygen species, lipoxogynases) and antioxidant factors (GPX4). We perform a systems-level analysis of ferroptosis sensitivity as an outcome of five input variables (ACSL4, SCD1, ferroportin, transferrin receptor, and p53) and organize the resulting simulations into ‘high’ and ‘low’ ferroptosis sensitivity groups. We make a novel prediction corresponding to the combinatorial requirements of ferroptosis sensitivity to SCD1 and ACSL4 activity. To validate our prediction, we model the ferroptotic response of an ovarian cancer stem cell line following single- and double-knockdown of SCD1 and ACSL4. We find that the experimental outcomes are consistent with our simulated predictions. This work suggests that a systems-level approach is beneficial for understanding the complex combined effects of ferroptotic input, and in predicting cancer susceptibility to ferroptosis.
Keywords: Ferroptosis, Cancer biology, Discrete model, SCD1, ACSL4
1. Introduction
Ferroptosis is a recently discovered form of regulated cell death (RCD) that is triggered by iron-dependent lipid peroxidation (Stockwell et al., 2017). Ferroptosis is a member of a broader family of RCD pathways that also includes necroptosis, parthanatos, pyroptosis, apoptosis, and others (Stockwell et al., 2017; Vandenabeele et al., 2010; Conrad et al., 2016; Vanden Berghe et al., 2014; Galluzzi et al., 2018). Critical features of RCD processes include control by specific intrinsic cellular mechanisms, resulting in susceptibility of RCD to pharmacologic and genetic modulation. This distinguishes RCD from accidental death caused by severe chemical or mechanical insults (Vandenabeele et al., 2010; Conrad et al., 2016; Vanden Berghe et al., 2014). Different types of RCD serve complementary roles during homeostatic, developmental, and pathogenic processes. For example, apoptosis, perhaps the best-studied mechanism of RCD, is programmed to occur in normal physiological contexts such as development (Galluzzi et al., 2018). RCD can also serve as a form of intercellular communication: release of oxidized lipids resulting from a cell undergoing ferroptosis may serve to promote ferroptosis and other forms of cell death in nearby cells (Conrad et al., 2016). In addition, cellular internal state can drive a cell to undergo one vs. another form of RCD, as exemplified by the influence of autophagy, the process of regulated lysosomal degradation of cytoplasmic components, on the cellular decision to undergo RCD (Loos and Engelbrecht, 2009; Napoletano et al., 2019; Hou et al., 2016). Therefore, development of a systems-level understanding of ferroptosis becomes critical not only for understanding and predicting the process itself, but also for developing more comprehensive models of cell survival and death under a wide range of conditions.
Both intentional induction of ferroptosis and its prevention are the focus of current drug development efforts. Thus, ferroptosis inhibitors are being considered as agents for the treatment of neurodegenerative disease (Angeli et al., 2017; Wu et al., 2018); conversely, substantial effort is being directed at the discovery of ferroptosis-inducing agents to treat cancer (Stockwell et al., 2017; Shen et al., 2018). Further, recent work suggests that existing anti-cancer agents such as cisplatin and sorafinib trigger ferroptotic pathways (Guo et al., 2018; Lachaier et al., 2014a; Lachaier et al., 2014b). The finding that drug-tolerant “persister” cells are particularly vulnerable to ferroptosis inducers has further heightened interest in ferroptosis as a potential mechanism for treating drug resistant cancers. Ovarian cancer, a malignancy in which successful treatment is frequently thwarted by the emergence of drug resistance (Agarwal and Kaye, 2003; Norouzi-Barough et al., 2018), may be an ideal candidate for such an approach. Indeed, our laboratory has shown that ovarian cancer cells are exquisitely sensitive to ferroptosis inducers in vitro and in vivo (Basuli et al., 2017).
Sensitivity to ferroptosis is complex and depends on multiple interacting molecular components. Here, we use mathematical modeling to synthesize currently understood ferroptotic processes into a dynamic, predictive model to better understand how ferroptotic processes can be harnessed for anti-cancer therapy. Such a model can be used to predict the cellular phenotype(s) that are optimal for ferroptotic sensitivity, as well as the relative contributions of molecular species and submodules in the network to the ferroptotic response. Indeed, mathematical models of apoptosis have helped to answer a number of outstanding biological questions with respect to this form of regulated cell death (Schleich and Lavrik, 2013), as well as to develop optimized protocols for oncotherapy (Panetta and Fister, 2000; Konstorum et al., 2017). Models that explore the influence of autophagy on the cell decision process to undergo apoptosis (Tavassoly et al., 2015; Liu, 2017), and the cell decision process to survive or undergo apoptosis or necrotic cell death (Calzone et al., 2010), provide examples of how integration of different models for cell death and survival can be accomplished.
Kagan et al. (2017) recently developed a continuum model for ferroptosis with a focus on biochemical cascades occurring in the phosphatidylethanolamine (PE) class of phospholipids. While that model provides an excellent synthesis of specific processes involved in arachidonic acid metabolism, lipoxogynase activity, and GPX4 regulation, it excludes contributions of iron, lipid desaturases, and additional components of import to the lipid peroxidation processes involved in ferroptosis. Agmon et al. (2018) performed molecular dynamics simulations of membranes with compositions relevant to ferroptotic sensitivity, and showed how the biophysical properties of membranes are altered under ferroptotic-competent lipid compositions.
We developed our model to answer systems-level questions regarding ferroptosis sensitivity and its interdependence on the co-ordinated activities of several distinct molecular species that have been identified as central to the ferroptotic signaling pathway, with a focus on species that have been found important for the lipid peroxidation processes critically involved in ferroptosis (Dixon and Stockwell, 2014; Yang and Stockwell, 2016; Stockwell et al., 2017). We use a multistate discrete modeling approach to emphasize qualitative properties of signaling cascades relevant to ferroptosis. The discrete modeling approach allows us to investigate the relative importance of different drivers of ferroptosis using a wider range of data than would be available for a detailed kinetic model of the system. Discrete models can provide a mechanistic, systems-level, understanding of a process that can serve to better focus future detailed kinetic modeling. Discrete models have been used successfully to model a variety of important processes in cancer, including cell cycle progression driven by PI3K (Sizek et al., 2019), the topography of epithelial-mesenchymal transitions (Font-Clos et al., 2018), and transcription factor networks in small cell lung cancer (Sizek et al., 2019; Font-Clos et al., 2018; Udyavar et al., 2017).
Using this model, we connect signaling modules related to phosholipid precursor processing, phospholipid peroxidation, ROS signaling and generation, and GPX4 activity, and demonstrate how each contributes to ferroptosis sensitivity. We demonstrate how the model can be used to make novel predictions about the combined impact of multiple perturbations on the response to ferroptosis, and provide a proof-of-principle experimental validation of these predictions. We provide source-code for the model in (Konstorum, 2020).
2. Results
2.1. Model overview
Using the available literature, we developed a logic-based multistate model for the intracellular ferroptotic process, with a focus on pro- and anti-oxidant actors in lipid peroxidation (Fig. 1). All model components were chosen such that they were found to be central components in pro-and anti-oxidant processes in ferroptosis, and each individually was identified to play a promotional or inhibitory role in ferroptosis. The goal for model development was to investigate how the activity of these actors modifies ferroptotic sensitivity in a systems analysis. We provide an overview of how model components (underlined) were chosen, and refer the reader to Section 4 (Methods) for a more detailed description of individual component biology, evidence of importance for ferroptosis, and mechanism of model incorporation.
Fig. 1.
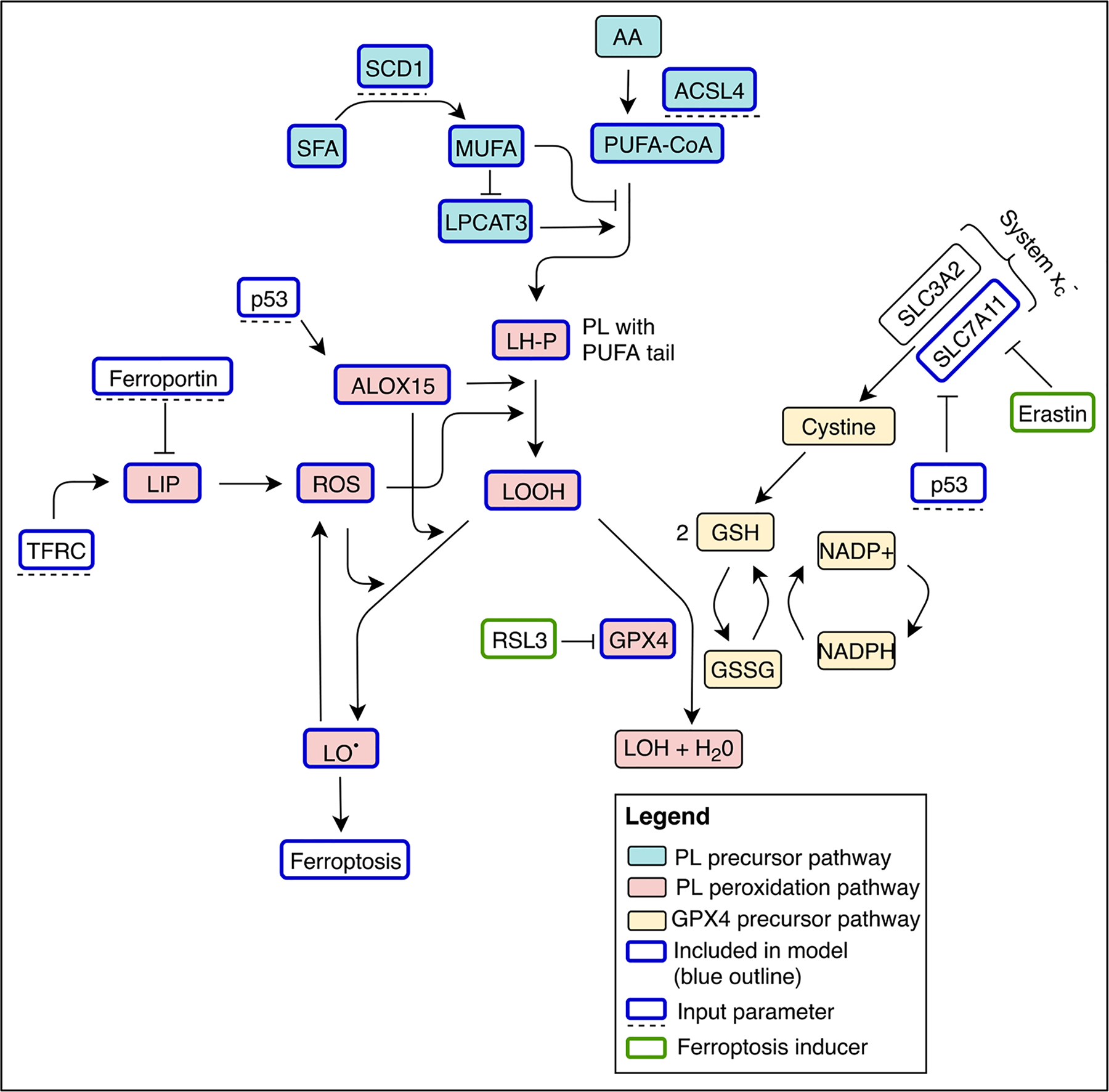
Diagrammatic overview of the multistate model for ferroptosis. Input parameters are model elements that do not change during the course of the simulation (blue outline; dashed line beneath), model variables on the other hand will change concentrations until reaching a steady state (blue outline). Molecular species that are implicitly included in the model but not simulated are also shown (black outline) as well as ferroptosis inducers (green outline). AA: arachidonic acid, LH-P: phospholipid conjugated to polyunsaturated fatty acid, LIP: labile iron pool, LOOH: lipid hydroperoxide, LO•: peroxide radical, LOH: redox-inert alcohol, MUFA: monounsaturated fatty acids, PL: phospholipid, PUFA-CoA: acetylated polyunsaturated fatty acids, ROS: reactive oxygen species, system xc−: cystine/glutamate transporter, SFA: saturated fatty acids.
Ferroptosis is characterized by iron-dependent lipid peroxidation that occurs in the presence of agents, such as erastin or RSL3, that inhibit counteractive antioxidant processes (Dixon et al., 2012). An antioxidant process found to be critical in protection from ferroptosis is mediated by the lipid peroxidase GPX4 (Yang et al., 2014). In order for ferroptosis to proceed, phospholipid molecules containing polyunsaturated fatty acids (LH-P) are generated from acetylated polyunsaturated fatty acids (PUFA-CoA) in a process that depends on the presence of the LPCAT3 enzyme (and relative absence of monounsaturated fatty acids, MUFAs, which can be produced by desaturation by SCD1 of saturated fatty acids (SFAs)). The PUFA-CoA moieties are produced from membrane polyunsaturated fatty acids (PUFAs) such as Arachidonic Acid (AA) by acyl-CoA synthetases such as ACSL4, an enzyme whose activity has been found to be critical for ferroptosis sensitivity (Doll et al., 2017; Dixon et al., 2015). The LH-P are oxidized to lipid hydroperoxides (LOOH) and eventually to lipid radicals (LO•) - which contribute to the initiation of ferroptosis - by reactive oxygen species (ROSs) and/or lipid peroxidating enzymes such as ALOX15, which act as a trigger for cell death (Feng and Stockwell, 2018). Presence of GPX4 can mitigate this process since it can reduce LOOH to LOH, thereby preventing its further oxidation. Iron, which promotes the formation of ROS through Fenton chemistry, is also required for ferroptosis (Dixon et al., 2012). Cellular levels of iron are dependent on several proteins, notably TFR1, an iron importer that is generally upregulated in cancer, and ferroportin (Fptn), an iron efflux pump that is downregulated in cancer (Torti and Torti, 2011; Torti and Torti, 2013; Torti et al., 2018). Additionally, p53 can act in two ways to promote ferroptosis: by increasing ALOX15 (Ou et al., 2016), and inhibiting SLC7A11, a component of the cystine/glutamate antiporter system xc− (cystine is a necessary precursor for GPX4 production).
The model is informed by results from multiple cell types and over 25 publications (see Methods for full model details). Each molecular species can take on a value of ‘low’ (0), ‘medium’ (1), or ‘high’ (2) activity/expression. Such a coarse-grained model can be used to understand qualitative characteristics of a system without detailed knowledge of biochemical rate parameters. Such models, which can specify functions using logical rule gates or transition tables, have been used successfully as described in the Introduction, as well in further studies such as to obtain novel biological understanding in cancer cell metabolism (Chifman et al., 2017), T cell receptor signaling (Saez-Rodriguez et al., 2007), innate immune response (Dimitrova et al., 2018), and a variety of other biochemical networks (Morris et al., 2010).
Initial conditions for the model are set as described in the Supplementary Information, Section S1 and Table S1. We note that simulation steady states do not depend on the initial conditions. The settings for the input parameters SCD, TFRC, Fptn, p53, and ACSL4 are all set to medium except for ACSL4, which is set to ‘high’. The latter condition is set since our simulations showed that a high ACSL4 is required to achieve a maximal ferroptotic response under any conditions of the other input variables. Other levels of ACSL4 and the other input variables are explored in Section 2.3.2. Further details on initial conditions for the model variables and input parameters are found in the Supplementary Information, Section S1. Full source-code for the model can be found in (Konstorum, 2020)
2.2. Response to ferroptosis inducers
We consider the steady state reached by the cells in our simulation with and without ferroptosis stimulation by erastin or RSL3, two small molecules specifically designed to induce ferroptosis that are widely used in the literature (Stockwell et al., 2017; Dixon et al., 2012) (Fig. 2). We observe that without ferroptosis induction, the cell has a high level of PUFA precursors for incorporation into phospholipids (PUFA-CoA), and a moderate incorporation of these moieties (LH-P), as well as moderate levels of hydroperoxides (LOOH). MUFA and LPCAT3 levels are moderate, as well as ROS. GPX4, SLC7A11, and ALOX15 are high. Importantly, although there are moderate levels of LOOH, there are low levels of peroxide radicals (LO•), which can lead to ferroptosis. One can consider such a cell primed for ferroptosis due to the presence of LOOH and oxidizers ALOX15 and ROS. The high levels of GPX4 prevent the cell from undergoing ferroptosis. Upon addition of erastin or RSL3, GPX4 levels are reduced, and we observe the appearance of moderate levels of ferroptosis via LO• (Fig. 2b). Note that erastin acts by inhibiting SLC7A11, which decreases availability of cysteine substrate for production of GSH, a co-factor required for GPX4 activity, whereas RSL3 inhibits GPX4 directly, hence SLC7A11 levels are still high in the RSL3+ cell. Thus the model accurately captures basic features of the ferroptotic response, an important first step towards its use for elucidating novel systems-level requirements for ferroptosis.
Fig. 2.
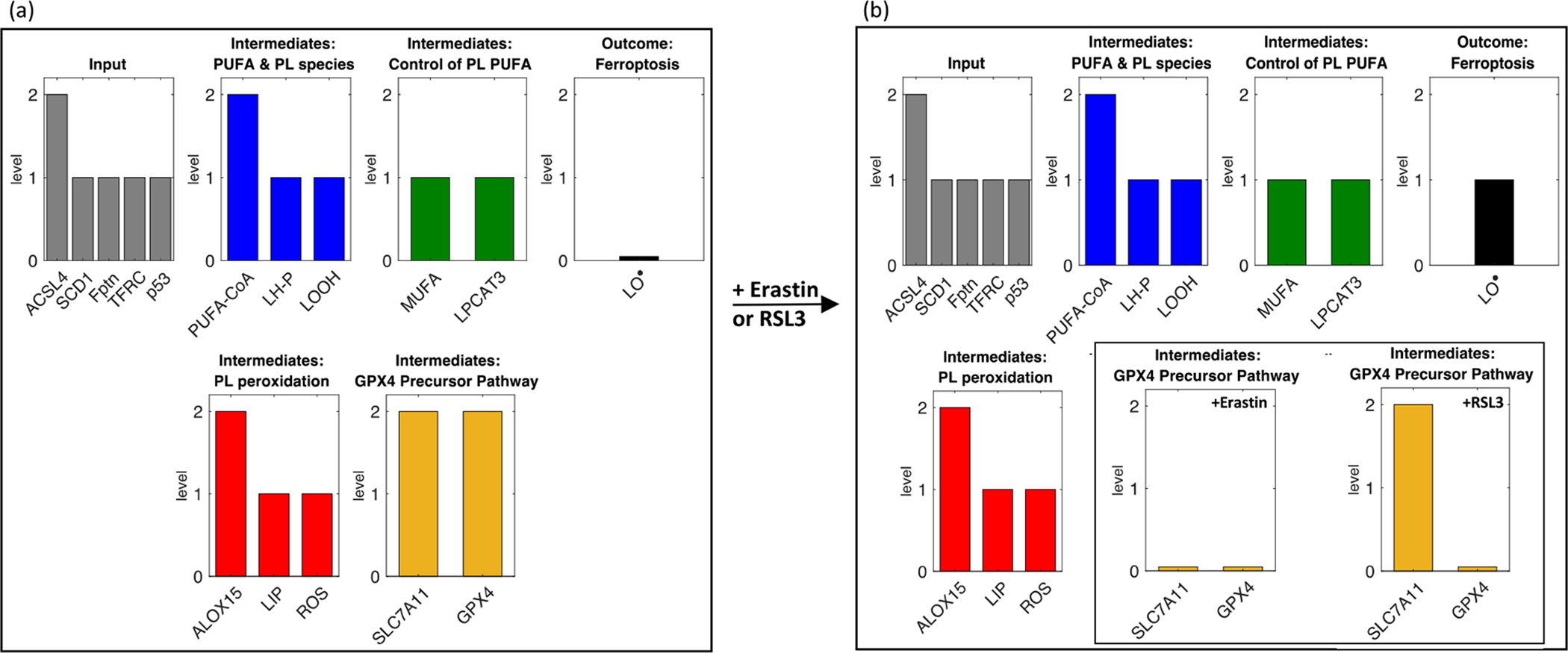
Addition of erastin or RSL3 in the baseline model stimulates a ferroptotic response. Steady state values of baseline model variables are shown (a) without and (b) with addition of the ferroptosis-inducers erastin or RSL3. Note that only SLC7A11 responds differently to erastin (no change) vs. RSL3 (lowered). Variables in gray (‘Input’) are external, and thus are set at time t = 0 and stay constant throughout the course of the simulations. The remainder of the variables are classified by types of molecular species or mode of action.
2.3. In silico system perturbation
2.3.1. Single perturbation: ACSL4
To test the model, we first performed a simple in silico perturbation that mimics an experiment with a known outcome. ACSL4 inhibition prevents ferroptosis (Doll et al., 2017). We use RSL3 as our ferroptosis-initiator in the model, following Doll et al. (2017). We observe that, compared to control baseline (Fig. 2a), in the ACSL4 knock-out model (Fig. 3a) there is low incorporation of PU-FAs into phospholipids, and thus less peroxidated phospholipids. Even with the addition of the GPX4 inhibitor RSL3 (Fig. 3b), we do not see an increase in peroxidated phospholipids because their precursors are not available - thus reproducing the findings of Doll et al. (2017).
Fig. 3.
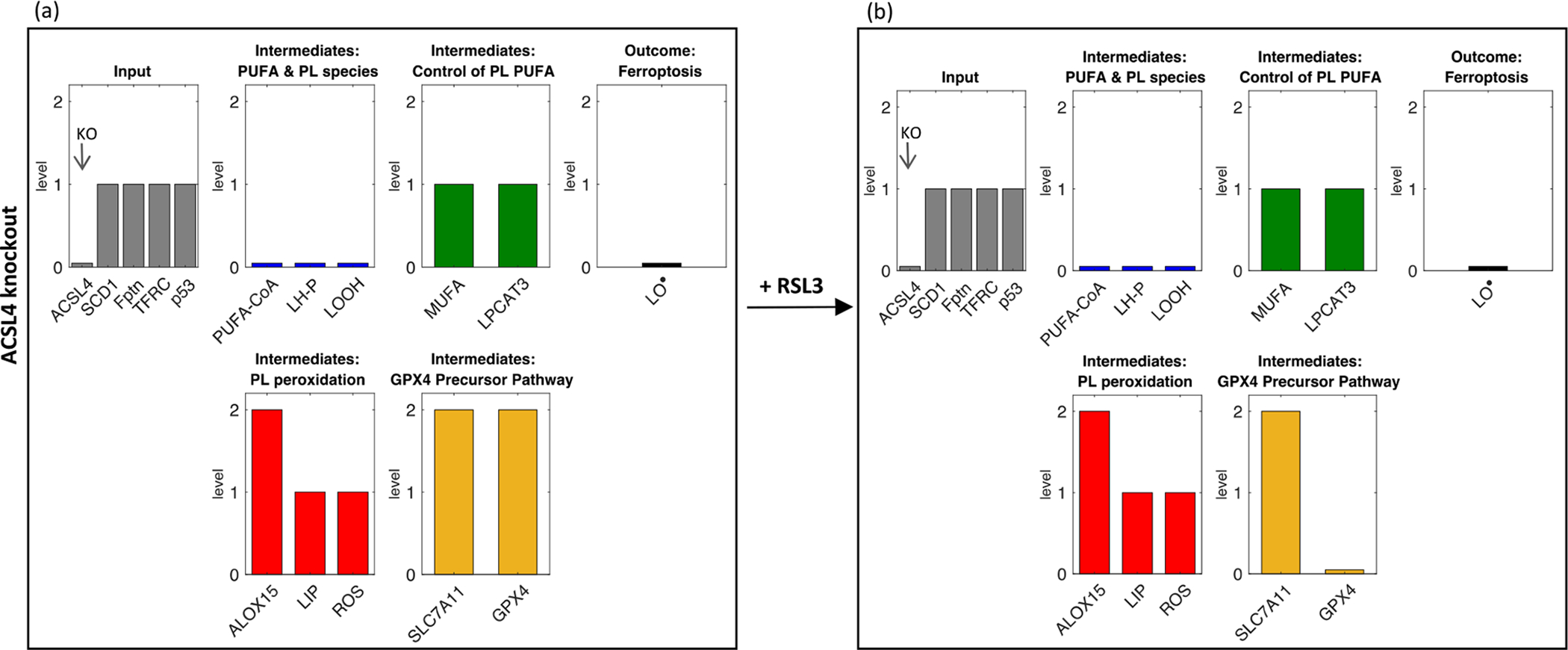
Effect of ACSL4 knock-out on model behavior (a) without and (b) with addition of RSL3.
2.3.2. Systems biology of ferroptosis
To more comprehensively understand how combinations of perturbations of system variables will impact ferroptotic response, we simulated the effect of all combinations of the input variables (35 = 243) on system behavior without or with addition of a ferroptotic agent (Fig. 4). We sort the perturbations by (1) decreasing LO• value in the erastin+ treatment group and (2) for each LO• value-subgroup in the erastin treatment group, increasing LO• value in the control treatment group. In this way, the patterns of the input variables that lead to the maximal ferroptotic response in each subgroup are easily identifiable. A comprehensive set of simulations is necessary to tease out the inter-related impact of the input variables on ferroptosis sensitivity, since the value of any one input variable is usually not, alone, predictive of ferroptotic response when all other input variables are allowed to vary (Supplementary Information, Section S2).
Fig. 4.
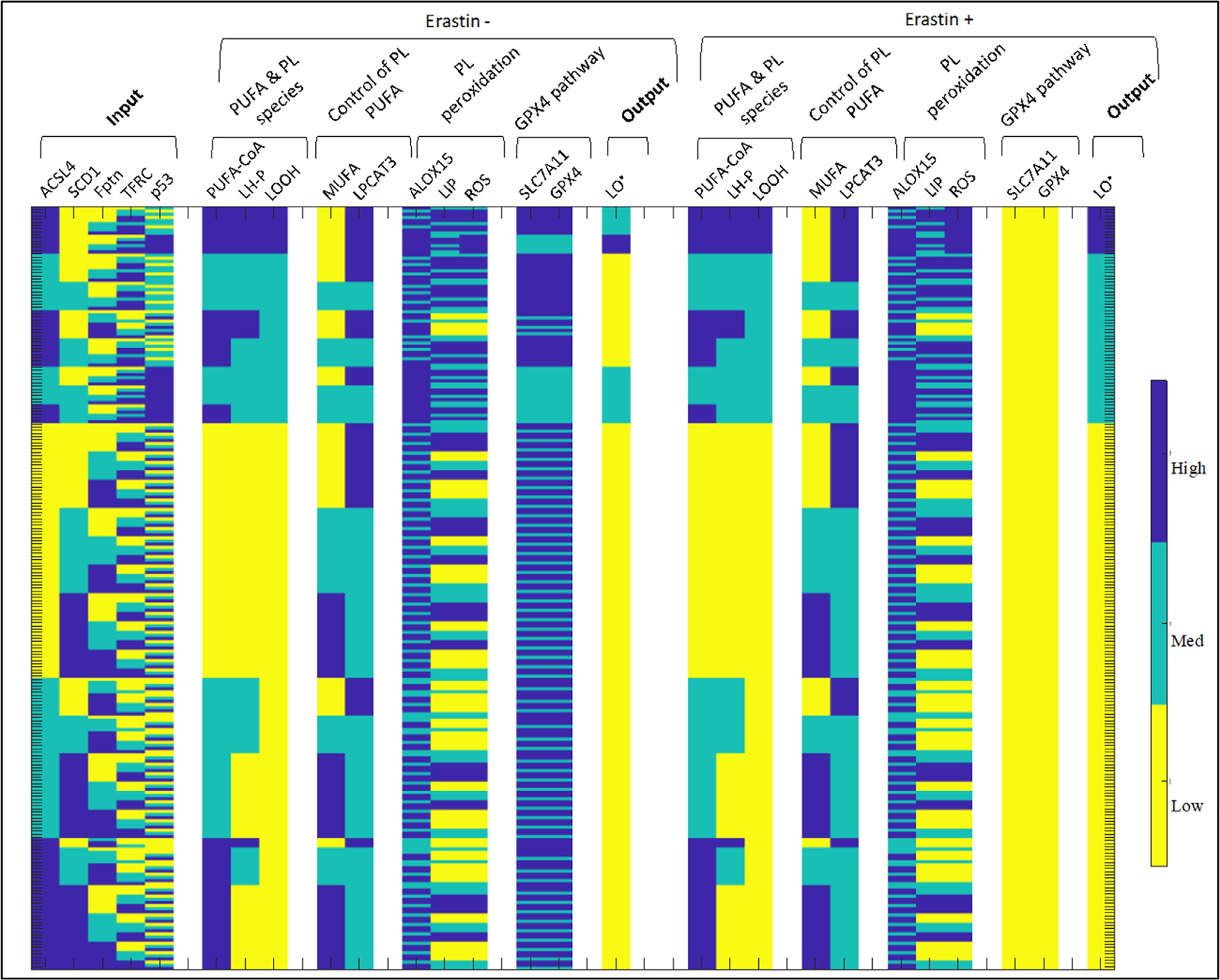
Ferroptosis model output with all combinations of input conditions, sorted by ferroptotic response.
We analyze the output of the respective high and low ferroptotic response subgroups, and include an analysis of the intermediate ferroptotic response in the Supplementary Information (Section S3). We conclude this subsection with a summary of the results in the form of a rule-based ‘algorithm’ for the cell that allows for a signature-based analysis of the input variables with respect to outcome in ferroptosis sensitivity.
2.3.2.1. High ferroptotic response.
The high ferroptotic response corresponds to ~6.17% of the input conditions, making it the most rare outcome. We note that for the cell to achieve a high ferroptotic response with erastin treatment, ACSL4 must be at its highest value, and SCD1 at its lowest value (Fig. 5). This combination yields maximal PUFA levels (due to high ACSL4) and maximal LH levels (due high ACSL4 and low SCD1/MUFA). While we observe high PUFA and LH in a small subset of low-ferroptotic response signatures (Fig. 6), only the high ferroptotic response signatures also have high LOOH, priming them for generating LO• in the absence of GPX4. The first peroxidation step is achieved by ROSs and/or ALOX15, and we see that ALOX15 and ROS levels are never low in the high ferroptotic signature cells. Additionally, for the cells to be sensitive to ferroptotic agents, p53 must be either at low or medium levels, otherwise SLC7A11, and hence GPX4 activity, is dampened, allowing for high LO• to be generated even in the control-treated cells. This may explain why results of p53 inhibition in the literature are varied: when p53 is lowered below maximal level, other factors contribute and may even predominate in determining ferroptosis sensitivity. This may be an interesting direction to pursue with respect to developing a more in-depth, kinetic model focusing on p53 in ferroptosis in order to further understand how this interplay impacts ferroptosis sensitivity.
Fig. 5.

Ferroptosis model output with all combinations of input conditions, sorted by ferroptotic response, displaying high ferroptosis after erastin treatment.
Fig. 6.
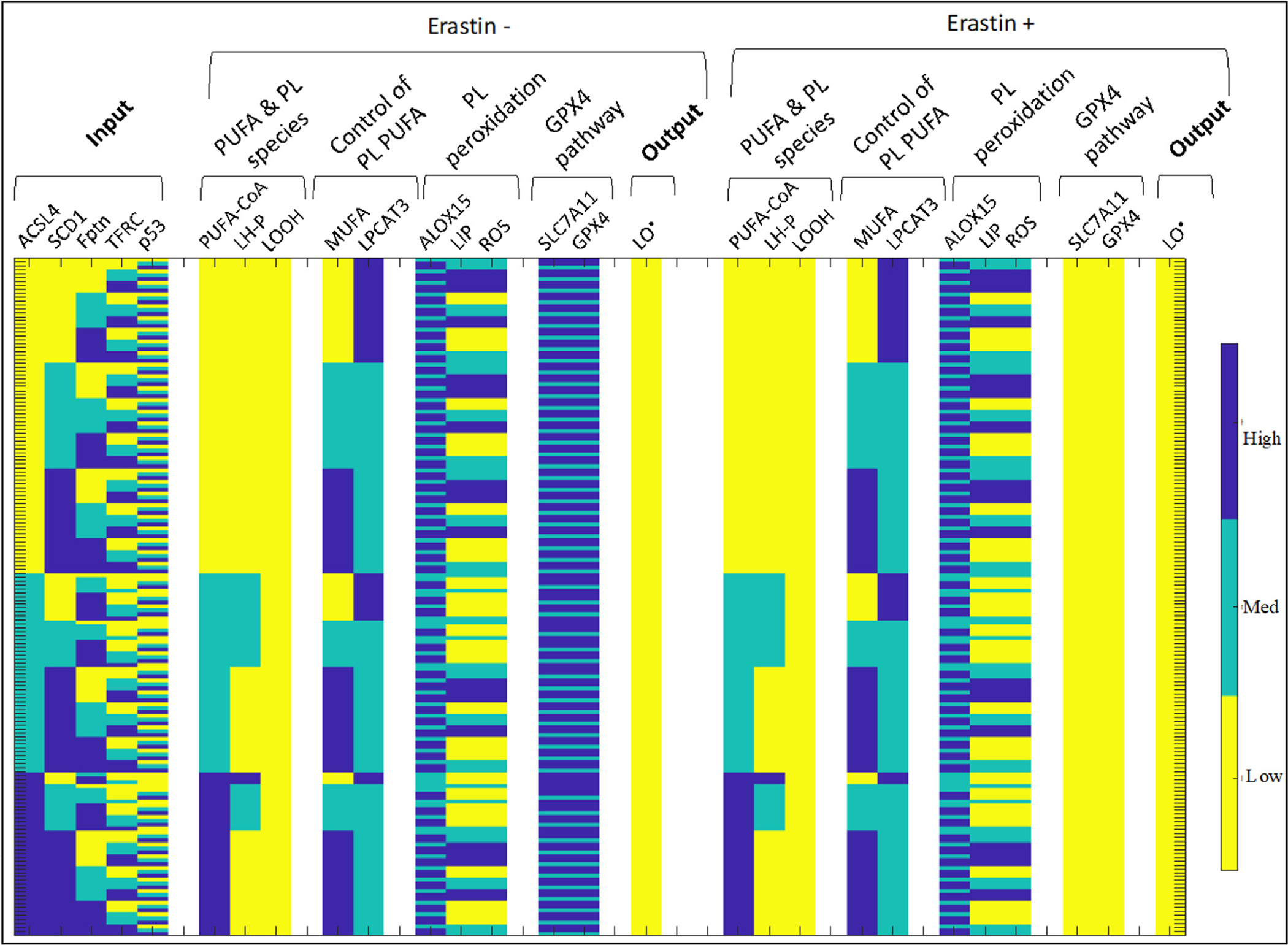
Ferroptosis model output with all combinations of input conditions, sorted by ferroptotic response, displaying low ferroptosis after erastin treatment.
2.3.2.2. Non-responsive ferroptosis cases.
We note that the no ferroptosis/non-responsive cases, which make up ~71.61% of the responses (Fig. 6), include a wide range of input variable values, but they all share one feature: there is no LOOH present in all the cases, whereas LOOH is at intermediate or high levels in the remainder of the simulations (Fig. 4). As can be observed in Fig. 1, LOOH provides the initial substrate for lipid peroxidation (another way to consider this is that LOOH is upstream to LO•, the peroxide radical that leads to ferroptosis). As can be observed from the simulations, and intuited from the diagram in Fig. 1, LOOH is thus necessary (but not sufficient) for cellular ferroptotic sensitivity. With no substrate for pro- or anti-oxidant activity, ferroptosis cannot be induced. As evidenced here, lack of LOOH can be induced by a large number of combinations of input conditions. Nevertheless, we do see certain conditions emerge that result in non-responsiveness: ACSL4, which catalyzes conversion of AA to PUFA-CoA, is necessary (but not sufficient) for production of LOOH. Therefore, all conditions with no ACSL4, regardless of the other input variables, are identified as non-responsive. Indeed, experiments in Section 2.4 bear out this observation for a subset of conditions.
2.3.2.3. A rule-based ‘algorithm’ for cellular sensitivity to ferroptosis.
We can summarize the results of the systems analysis using a rule-based approach for the input variable levels (Fig. 7). These rules can be deduced from Figs. 4–6, Methods (Section 4.2), and Figure S2. This approach allows for an analysis akin to a ‘gene-signature’ approach to determine ferroptosis sensitivity, as well as succinctly summarizes the various dependencies that the cell requires to initiate ferroptosis either with or without addition of a ferroptotic agent. For example, provided levels of input variables are available, one can predict whether a cell with high levels of PUFA-containing phospholipids (LOOH) will undergo spontaneous ferroptosis by observing the activity of p53 in the cell. Alternatively, a cell with a combination of input variables that results in moderate LOOH, but not high ROS levels, will not undergo spontaneous ferroptosis even under high levels of p53. Much of the intuition surrounding the rules in Fig. 7 can be found in the discussion contained within Sections 2.3.2.1 and 2.3.2.2. We consider this cellular ‘algorithm’ for ferroptosis sensitivity as a first approach towards considering what gene signatures may emerge as predictive for ferroptosis sensitivity in a translational setting.
Fig. 7.

A cellular algorithm for ferroptosis sensitivity. Provided known values of input variables (in blue), one can follow the algorithm to understand how a cell will ‘decide’ whether to undergo spontaneous or drug-induced ferroptosis. This algorithm provides a rule-based summary of the model steady state outcomes.
2.4. Predicting and testing the response of an ovarian cancer stem cell line to ferroptotic agents
We use our model to predict and then experimentally test the response of an ovarian cancer stem cell line to ferroptotic agents. FT-t cells are a model of high-grade serous ovarian cancer (HGSOC) stem cells that were derived from primary culture of fallopian tube stem cells (considered to be one of the major cells of origin for HGSOC (Kim et al., 2018)) via immortalization and transformation with h-TERT, SV40 large T antigen, and c-MYC (Yamamoto et al., 2016). When implanted in mice, FT-t cells form tumors that accurately mimic genetic features of HGSOC (Basuli et al., 2017; Yamamoto et al., 2016), rendering it a useful test case for how the model might predict the response of HGSOC to ferroptosis inducers. In particular, this cell line has low p53 (due to inactivation by SV40), high TFRC, and low ferroportin, thereby displaying a high iron retention phenotype (Basuli et al., 2017). We can therefore use our system to model this cell line by setting the input variables (p53, TFRC, ferroportin) = (low, high, low). We can then analyze the response of the simulated cell line to ferroptosis inducers as a function of the remaining two input variables ACSL4 and SCD1 (Fig. 8).
Fig. 8.
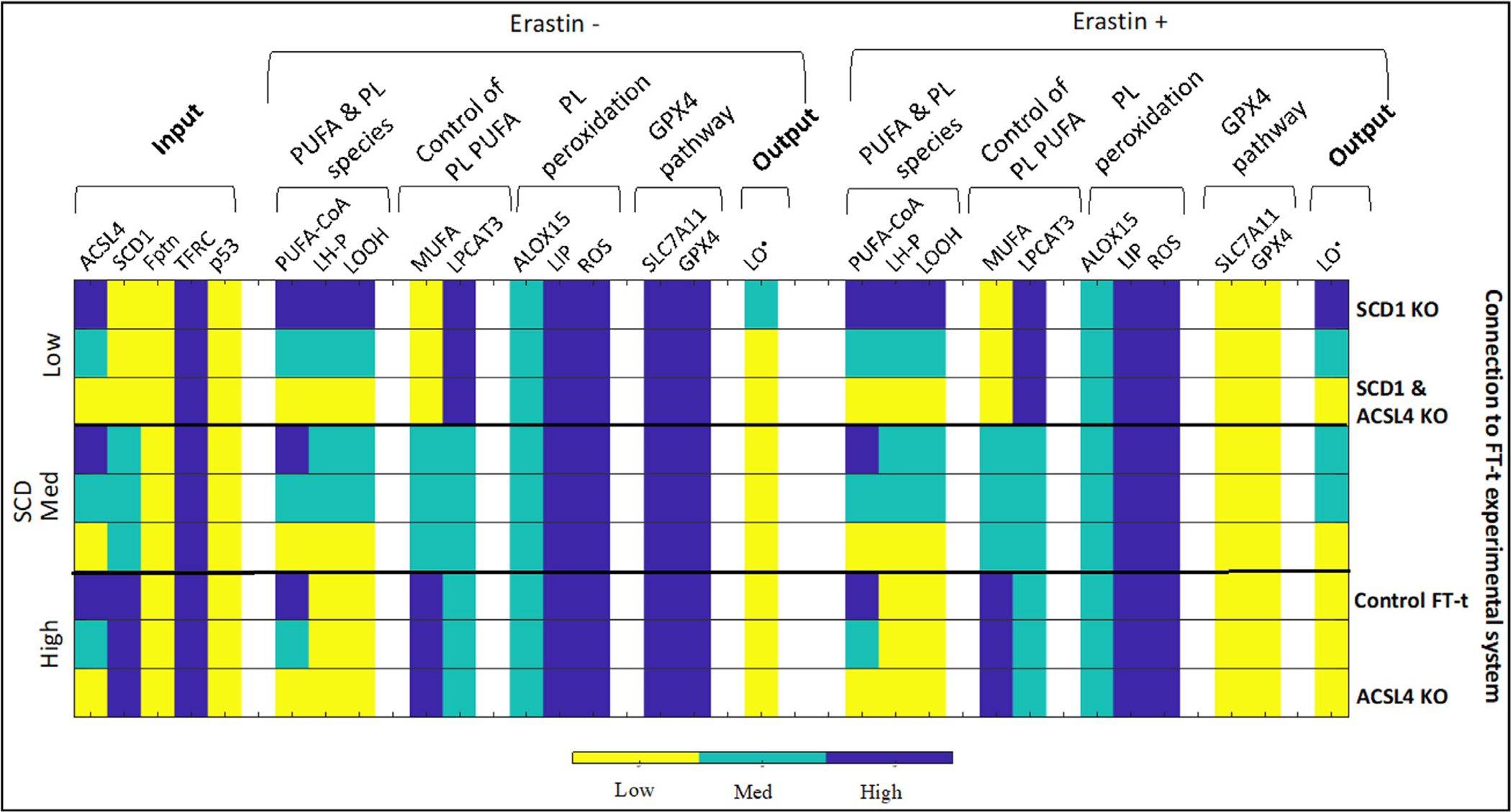
Modeling ferroptosis sensitivity in FT-t cells. Ovarian cancer stem cell line FT-t has low p53, high TFRC, and low Fptn. We consider the sensitivity of FT-t cells to erastin under varying SCD and ACSL4. The relationship between conditions experimentally tested and modeled is shown on the right-hand side. For example, the first row shows input conditions identical to the control FT-t row, except the input parameter SCD1 is set to low, which corresponds to a knock down of SCD1 in FT-t cells.
We observe that due to high TFRC and low Fptn, we have maximal LIP and ROS levels (while ROS can also be increased by feedback from LO• in lower iron conditions, an iron retention phenotype leads to maximal ROS in our model). Due to the low p53, we have intermediate ALOX15: while ALOX15 is boosted by p53 activity, lack of p53 does not completely abrogate its expression and function. Even though p53 activity is low, due to the high iron, these cells are primed to undergo ferroptosis in the right conditions.
If SCD1 activity is low, there are few MUFAs to inhibit LPCAT3, and directly inhibit PUFA incorporation into phospholipids. Therefore, LH-P levels are completely determined by ACSL4 activity, and LOOH is proportional to LH-P due to the high ROS. Thus, these cells will undergo ferroptosis upon inhibition of GPX4 activity if enough ACSL4 is present. If SCD1 activity is intermediate, there is partial inhibition of LPCAT3 and PUFA incorporation into phospholipids, evidenced by lower LH-P even at high ACSL4. Cells still respond to ferroptotic agents in the presence of ACSL4, but cannot maximize a ferroptotic response. Under conditions of high SCD1 activity, there is low LH-P due to MUFA-mediated inhibition of PUFA incorporation into phospholipids. Therefore, there is no substrate for ferroptosis to be initiated upon addition of ferroptotic agent.
Overall, the model predicts that in the genetic background of FT-t cells, SCD1 knock-out will increase sensitivity to ferroptosis, but a double-knockout of ACSL4 and SCD1 will prevent this SCD1-knockout induced increase (Fig. 8). To note, such a prediction is not possible using a static model, due to the complexity of interactions between the input parameters and model variables. For example, one can observe input parameter combinations where a low SCD1 and high ACSL4 background will not increase ferroptosis sensitivity (Fig. 6).
To test these predictions experimentally, we first ascertained the level of ACSL4 and SCD1 in FT-t cells in comparison to FT (primary fallopian tube stem cells) and FT-i (FT cells that have been immortalized but not transformed). We observed that both ACSL4 and SCD1 mRNA were elevated in FT-t in comparison to FT (Supplementary Figure 3), indicating that both of these genes are highly expressed in the model ovarian cancer stem cell line. Next, we knocked-down SCD1, ACSL4. or both genes using siRNA pools and assayed cell viability following treatment with the ferroptosis inducer RSL3. A knock down of SCD1 and/or ACSL4 corresponds to setting the respective input parameter to ‘low’ in the model with other parameters maintained as discussed previously (Fig. 8).
The results of this experiment are shown in Fig. 9. As seen in Fig. 9a, knockdown of SCD1 was successful, resulting in an approximately 80% decrease in levels of SCD1 mRNA. Similarly, knockdown of ACSL4 resulted in an 80% decrease in levels of ACSL4 when compared to a non-targeting control (Fig. 9b). Somewhat unexpectedly, knockdown of SCD1 slightly elevated ACSL4 mRNA (Fig. 9b) and conversely, knockdown of ACSL4 elevated levels of SCD1 mRNA (Fig. 9a) above that of controls. We currently have no explanation for this apparent crosstalk, which to our knowledge has not been previously reported and merits further investigation. Nevertheless, as expected, levels of both mRNAs remained low when both were knocked down together (Fig. 9a,b).
Fig. 9.
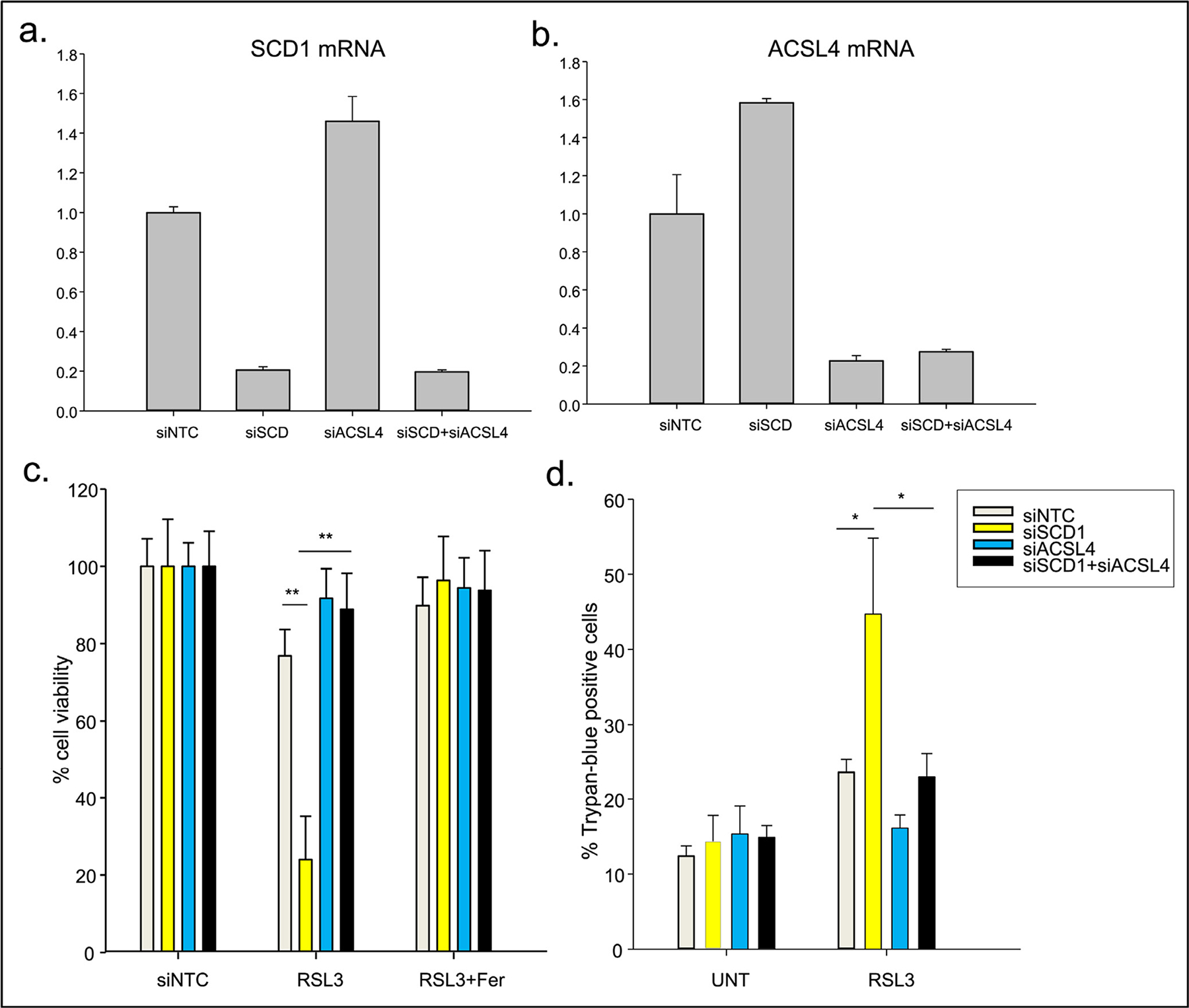
FT-t cells were transfected with siRNA targeted to SCD1 (siSCD1), ACSL4 (siACSL4), non-targeting siRNA (siNTC) or siRNA targeted to both SCD1 and ACSL4 (siSCD1 + siACSL4) for 48 h. a) Levels of SCD1 mRNA following knockdown of SCD1 and ACSL4 individually or together was Quantified by qRT-PCR. b) Levels of ACSL4 mRNA following knockdown of SCD1 and ACSL4 individually or together was quantified by qRT-PCR. c) Cells were treated with 0.5 to 2 μM RSL3 in the presence or absence of 2 μM ferrostatin-1 for 24 h to assess sensitivity to ferroptosis. Cell viability was measured using calcein-AM. **p<3.6E-29 (siNTC vs siSCD1 and siSCD1 vs siSCD1 + siACSL4) d) Cell death was determined using Trypan-blue exclusion. UNT, untreated controls. *p<2.4E-02 (siNTC vs siSCD1 and siSCD1 vs siSCD1 + siACSL4). Represented are means and standard deviations of three independent experiments, each performed using eight technical replicates per point.
The successful knockdown of SCD1 and ACSL4 enabled us to test the effect of both single and double knockdowns on sensitivity to ferroptosis. As shown in Fig. 9c, control cells were sensitive to the ferroptosis inducer RSL3, exhibiting an approximately 20% decrease in viability. Sensitivity was dramatically enhanced (to approximately 80%) when SCD1 was knocked down, consistent with model predictions and with a role of SCD1 as a cytoprotective protein (Tesfay et al., 2019). However, as predicted by the model, this enhanced cytotoxicity was blocked in cells in which ACSL4 was simultaneously reduced (Fig. 9c, compare yellow bar to black bar in the central panel). Concurrent treatment of cells with the ferroptosis inducer RSL3 and the ferroptosis inhibitor ferrostatin blocked cell death in all cases (Fig. 9c), demonstrating that effects on cell viability were attributable to the process of ferroptosis and not to another cell death mechanism. Further, staining with trypan blue, which directly stains dead cells (as opposed to the calcein staining shown in Fig. 9c, which stains living cells, with death being inferred from a reduction in viability) confirmed that knockdown of SCD1 only increases cell death in a genetic background in which ACSL4 has not been simultaneously reduced (Fig. 9d). Collectively, these data demonstrate that our systems-level model can accurately predict sensitivity to ferroptosis induction as an outcome of the activity of multiple components of the ferroptotic induction cascade.
3. Discussion
Ferroptosis is a recently-described mode of cell death that is distinct from other known cell death pathways. The discovery of ferroptosis has engendered wide-spread interest in the development of anti-cancer drugs that induce this mechanism of cell death (Stockwell et al., 2017). Although such drugs currently remain in a developmental stage, the complexity of the ferroptotic process has made it clear that the successful deployment of ferroptosis-inducing agents will ultimately require an understanding of how multiple parameters interact to dictate ferroptosis sensitivity or resistance in individual tumors.
In this work, we develop a dynamic multistate discrete model of the pathways underlying the ferroptotic process in order to achieve a systems-level understanding of cellular susceptibility to ferroptosis. We show that the model behaves in a manner that is consistent with experimental observations. The model has allowed us to explore global properties of ferroptosis susceptibility. Importantly, we show that a cell’s sensitivity to ferroptotic inducers is a product of several interacting pathways, including PUFA-CoA synthesis and incorporation into phospholipids, and the balance between pro- and anti-oxidant species.
More broadly, we show how different combinations of input variables (ACSL4, SCD1, Fptn, TFRC, and p53) alter ferroptosis sensitivity and obtain conclusions regarding the molecular profiles of these agents that can promote or inhibit sensitivity to ferroptosis induction (Figs. 4–8). For example, high ACSL4 and low SCD1 are necessary, but not sufficient, in our model to promote high ferroptosis sensitivity (Fig. 5). Conversely, a combination of parameters that minimizes production of LOOH - the first step in the lipid peroxidation pathway, will desensitize the cell to ferroptosis agents. As can be observed in Fig. 6, there are many combinations of input variables that will lead to this result, indicating that ferroptosis sensitivity is a systems-level process, and thus that predicting ferroptosis sensitivity may ultimately require a gene- or protein- signature approach in a patient setting. To this end, we use the model framework and simulation results to derive a cellular algorithm for ferroptosis sensitivity given known values of input parameters (Fig. 7).
By simultaneously modeling the expression of multiple genes, our model was able to expand and clarify observations in the literature. ACSL4 has previously been shown to enhance sensitivity to ferroptosis inducers (Doll et al., 2017). Indeed, when simulating ACSL4 knockdown, we observe that response to RSL3 is inhibited (Fig. 3). Nevertheless, our systems-level approach predicted that there exist conditions of SCD1 and other major players in the ferroptotic cascade that render a cell unresponsive to ferroptotic induction despite high levels of ACSL4 (Figs. 4–8).
We used an ovarian cancer stem cell model to test this prediction, since effective treatments for ovarian cancer remain limited, and ferroptosis inducers may ultimately offer a therapeutic option in this disease. The model we selected exhibits a gene expression pattern that is generally concordant with that observed in ovarian tumors. In particular, using conditions pertinent to FT-t ovarian cancer stem cells (Yamamoto et al., 2016) (setting Fptn, TFRC, and p53 to ‘low’, ‘high’, and ‘low’, respectively), the mathematical model predicted that under conditions of high SCD1, ACSL4 would be unable to exert this sensitizing effect (Fig. 8). This prediction was validated experimentally (Fig. 9). These results are consistent with recent observations implicating SCD1 as an inhibitor of ferroptosis (Tesfay et al., 2019), and the demonstration that its products (MUFAs) promote ferroptosis resistance (Magtanong et al., 2018); further, work from our laboratory has recently shown that prolonged inhibition of SCD1 can trigger cell death (Tesfay et al., 2019). Although this is somewhat surprising, given the ability of cells to take up MUFAs from exogenous sources (Kamphorst et al., 2013), a number of papers have similarly demonstrated that sustained inhibition of SCD1 induces cell death (Li et al., 2018; Leung and Kim, 2013; Mason et al., 2012; Minville-Walz et al., 2010; Hess et al., 2010). In the experiment shown in Fig. 9, we deliberately selected conditions under which the effects of SCD1 inhibition were modest, in order to focus on the combined influence of SCD1 and ACSL4 on the response of cells to ferroptosis. This experimental strategy successfully illustrated the interdependence of SCD1 and ACSL4 predicted by the model.
This example illustrates how intergenetic interactions can affect biological response in ways that are not inherently intuitive, and highlights the need for modeling to accurately capture the multiple variables that ultimately dictate biological response. Interpreted in the context of patient response to therapy, our results suggest that modeling approaches that incorporate the expression of multiple genes will ultimately be required to successfully tailor therapy to individual patients to realize “precision medicine”.
As our understanding of ferroptosis becomes more nuanced, it will become increasingly essential to use modeling to predict the combined contribution of multiple genes and pathways to ultimate cellular response, and to expand the model both within the core pathways of lipid peroxidation, and to additional pathways. Going forward, our model can serve as a framework for the addition of such new variables and pathways, which will ultimately improve the precision with which ferroptosis sensitivity can be predicted. For example, it has been suggested that iron concentration may impact the activity of iron-binding proteins involved in ferroptosis (e.g. ALOX15, SCD1) (Dixon and Stockwell, 2014; Konstorum et al., 2018), which may represent an additional layer of iron-mediated regulation of ferroptosis. Proteins that release or redistribute iron intracellularly (Torti and Torti, 2013; Torti et al., 2018; Chifman et al., 2014; Hower et al., 2009) may also impinge on ferroptosis. For example, HO-1, an enzyme that degrades heme and is induced as part of an antioxidant response mediated by Nrf2, contributes to ferroptosis in several different contexts (Fang et al., 2019; Hassannia et al., 2018; Kwon et al., 2015). Further, feedback loops that redistribute intracellular iron, such as the release of iron from degradation of ferritin in the authophagolysosome (Hou et al., 2016; Sui et al., 2019), also impact the ferroptotic cascade. Expansion or a focus of the model to intracellular iron distribution could yield rich insights into the contribution of intracellular iron distribution to ferroptosis. Additional contributors to ferroptosis that may ultimately factor into the model include the MAP kinase pathway, which plays a complex but as yet incompletely understood role in ferroptosis (Xie et al., 2016), as well as two recently-described modulators of ferroptosis, AMPK (Lee et al., 2020) and FSP1 (Doll et al., 2019; Bersuker et al., 2019). Finally, a more in-depth modeling study of the complex contribution of p53 to ferroptosis may help to resolve potentially conflicting experimental results, which have demonstrated both pro- and anti-ferroptotic effects of p53 (for examples, see (Magtanong et al., 2018; Jiang et al., 2015; Xie et al., 2017)). Our model indicates that low/medium levels of p53 are necessary, but not sufficient, to induce ferroptosis, suggesting that other factors may impinge on p53 to drive final cellular outcome. A detailed, kinetic study of wildtype as well as mutant p53 and their effects in different cellular contexts may be required to clarify the full range of roles that p53 can play in ferroptosis. Nevertheless, at present our model contains sufficient critical players and interactions to shed light on the processes regulating lipid peroxidation mechanisms leading to ferroptosis. If properly adjusted to cell line or patient data, the model could be used in the future to predict sensitivity to pro-ferroptotic therapy and/or to optimize combinatorial effects of ferroptotic agents with other anti-cancer agents.
4. Methods
4.1. A stochastic multistate discrete modeling framework
The mathematical model consists of five external inputs: ACSL4, SCD1, ferroportin (Fptn), TFRC, and p53 and eleven variables (MUFA, PUFAc, LPCAT3, LHP, ALOX15, ROS, SLC7A11, GPX4, LOOH, LOr, LIP), where PUFAc represents PUFA-CoA, LHP represents LH-P, and LOr represents the radical LO•. We represent the system with a state vector at discrete time t as
where xI ∈ {0, 1, 2} for all xi ∈ x. At each time step, the state vector is updated as follows,
where gi. (x(t)) = c(xi(t), fi(x(t*)).
For each variable xi, a transition table, fi, is specified (Fig. 10). The transition table takes as input all variables that act on xi, and outputs a corresponding value of xi for the next time step. We take x(t*) to represent a state of the system x such that a subset of xi ∈ x may have been updated. Then, gi imposes a continuity constraint, c, on the output fi(x(t*)) which limits the jump size to one level for each xi. The probability P that xi(t + 1) will update using gi is
For all i, we take pi = 0.5. An asynchronous update scheme is used at each time step, hence each variable is updated in a random order. The steady states of the system do not depend on the stochastic, continuous, or asynchronous update scheme in this framework (Murrugarra et al., 2012).
Fig. 10.
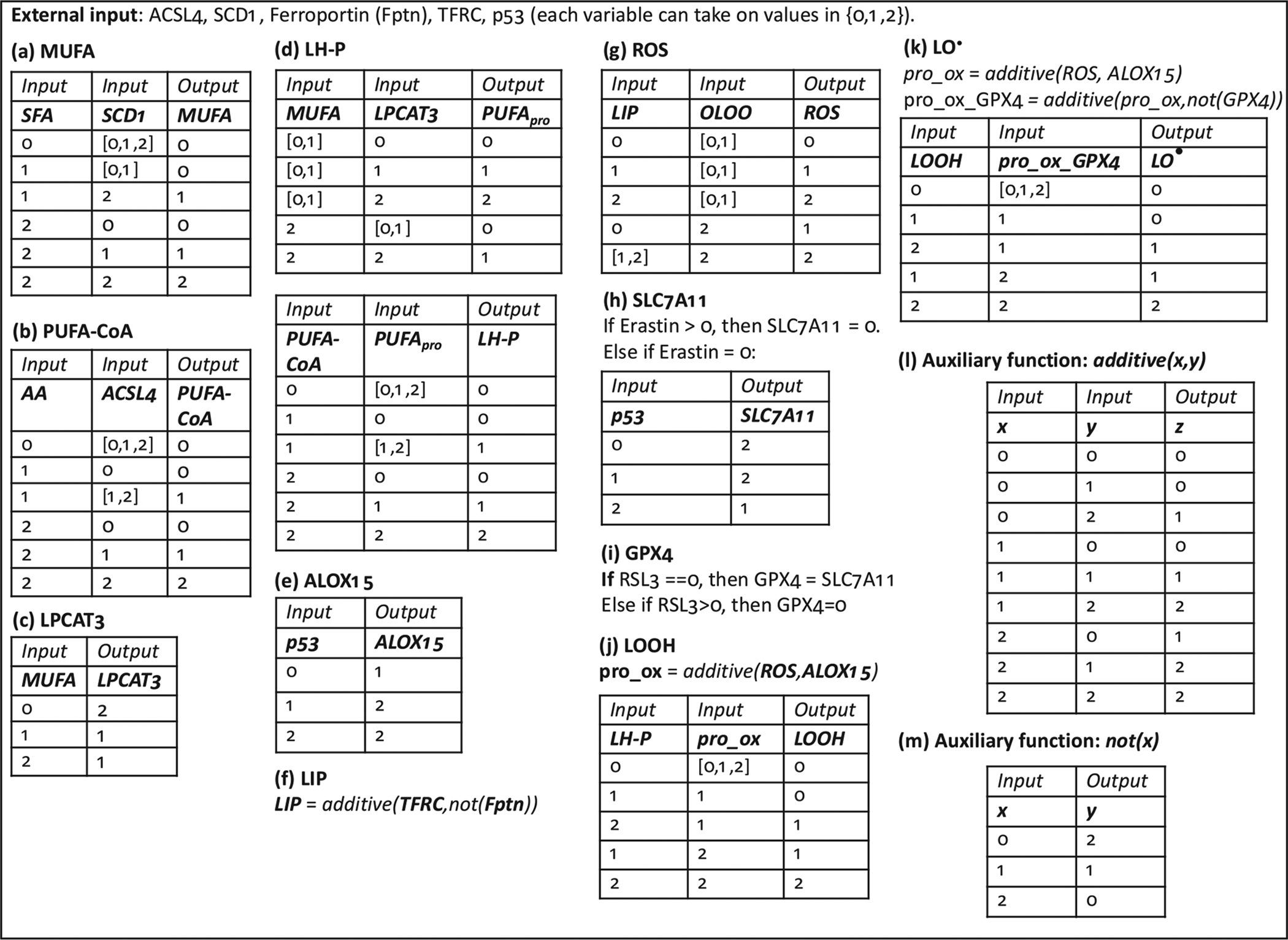
Transition tables for each modeled species in the multistate rule specification model of ferroptosis.
System behavior was simulated using Matlab R2017A. Choice of initial conditions of variables (aside from input variables, which are explored in Section 2.2.2) within a physiological range did not impact the steady states. Further details on the choice of initial conditions can be found in the Supplementary Information.
4.2. Transition tables
The transition tables for the multistate rule-specification model for ferroptosis are in Fig. 10. We shall now motivate each entry.
4.2.1. Monounsaturated fatty acids (MUFA)
Production of the most abundant MUFAs in mammalian cells, oleic and palmitoleic acid, depends on desaturation of the saturated fatty acids (SFAs) stearic acid and palmitic acid, respectively, catalyzed by stearoyl-CoA desaturase (SCD1) (Paton and Ntambi, 2009). In cancer cells, SCD1 is the main regulator of MUFA levels (Igal, 2011). Nevertheless, MUFAs can also be imported into cells directly, which can be modeled by increasing MUFA levels independently of SFA levels and SCD1 activity.
We assume that availability of SFAs is much greater than of MUFAs (Harayama et al., 2014), and thus when SFAs are converted to MUFAs, qualitatively, SFA levels are not lowered in a proportional manner.
4.2.2. Acetylated polyunsaturated fatty acids (PUFA-CoA)
Membrane polyunsaturated fatty acids (PUFAs) are direct targets for the ferroptotic cascade via their peroxidation (Skouta et al., 2014; Yang et al., 2016). In order for free fatty acids to be incorporated into phospholipid membranes, they must be converted to acyl-CoA moieties by acyl-CoA synthetases. ACSL4 is an acyl-CoA synthetase with a strong preference for arachidonic acid and other PUFAs (Kagan et al., 2017; Doll et al., 2017), and its activity has been found to be critical for sensitivity to ferroptosis inducers (Doll et al., 2017; Dixon et al., 2015). We model the effect of ACSL4 as follows: at moderate levels of AA, moderate and high ACSL4 result in moderate PUFA abundance, whereas at high AA, PUFA levels are directly related to ACSL4 levels.
4.2.3. LPCAT3
Lysophosphatidylcholine Acyltransferase 3 (LPCAT3) is involved in the glycerophospholipid remodeling pathway (Lands’ cycle) which allows for the fatty acid composition of phospholipids to be altered. LPCAT3 shows highest acyltransferase activity toward PUFAs (Shindou et al., 2009). Accordingly, it has been shown that LPCAT3 is essential for ferroptosis induced by inhibition of GPX4 (Dixon et al., 2015). It has been observed that in SCD1 knock-down conditions, when MUFA levels are reduced, LPCAT3 levels are increased (Ariyama et al., 2010). We thus model MUFAs as capable of partially inhibiting LPCAT3.
4.2.4. PUFA-containing phospholipids (LH-P)
Polyunsaturated fatty acids (PUFAs) are incorporated into phospholipids either during de novo biosynthesis (the Kennedy pathway) or maturation and remodeling (the Lands cycle). The acyltransferase LPCAT3 preferentially adds PUFAs to phospholipids via the Lands cycle (Shindou et al., 2009; Shindou and Shimizu, 2009; Hishikawa et al., 2008), and is critical for ferroptosis sensitivity, as discussed above. MUFAs can also inhibit PUFA incorporation into phospholipids via LPCAT3-independent mechanisms: Ariyama et al. (Ariyama et al., 2010) showed that knockdown of SCD1 (which would decrease MUFA levels) and LPCAT3 still resulted in an increase in phospholipid PUFA levels over control in HeLa cells.
4.2.5. ALOX15
ALOX15 is a non-heme iron-binding lipid peroxidating enzyme. It participates in reactions that lead to generation of lipid hydroperoxides (LOOH) from unsaturated lipids (LH) (this activity is termed its lipoxygenase activity), as well as generation of peroxyl radicals (LO•) (among others) from lipid hydroperoxides (termed its lipohydroperoxidase activity) (Ivanov et al., 2015; Girotti, 1998). The presence of ALOX15 has been found to be critical for ferroptosis sensitivity (Dixon et al., 2015; Shintoku et al., 2017), and it has recently been shown that p53 can modulate ferroptotic response by increasing ALOX15 expression via the upregulation of SAT1, which is proposed to act as a positive regulator of ALOX15 (Ou et al., 2016).
4.2.6. LIP
While intracellular iron regulation involves complex pathways of transcriptional and post-translational modifications (Torti and Torti, 2013), we consider a simple iron regulation model here, which only involves the iron import protein transferrin receptor 1 (TFRC) and export protein ferroportin, both which are found in the cellular plasma membrane. Loosely bound intracellular iron, termed the labile iron pool (LIP) will occur with high TFRC and low ferroportin, and minimal LIP will occur in the complementary conditions. Attesting to the importance of these pathways in the pathophysiology of iron regulation, alterations in levels of these proteins are sufficient to affect tumor growth in animal models (Basuli et al., 2017; Pinnix et al., 2010) and to predict survival of cancer patients (Basuli et al., 2017). Other regulators of LIP, such as the intracellular iron binding protein ferritin, or ferroportin inhibitor hepcidin, can be added to the model if considered important with respect to iron-mediated ferroptosis sensitivity of the cell.
4.2.7. ROS
The ability of iron to mediate production of hydroxyl free radicals via the Fenton reaction, which can then trigger peroxidation of LH-P to LOOH and LOOH to LO• implicates the labile iron pool as an important contributor to the reactive oxygen species (ROSs) that participate in formation of lipid peroxides. Further, LO• can trigger further free-radical mediated lipid peroxidation (Girotti, 1998). It has been demonstrated that the iron chelator deferoxamine (DFO) can inhibit ferroptosis (Dixon et al., 2012), while the strength of the feedback of LO• on the peroxidation process and sensitivity to ferroptosis has not yet been investigated. While iron can play a role in ferroptosis that is independent of its free-radical activity (Dixon and Stockwell, 2014), its contribution to the cellular ROS that is involved in lipid peroxidation is currently considered a significant contributor to ferroptosis sensitivity (Stockwell et al., 2017), and thus its effect on boosting ROS in the model is considered to be stronger than that of LO•.
4.2.8. SLC7A11
SLC7A11 is a component of the cystine/glutamate antiporter system xc−, and is the target of the pro-ferroptotic drug erastin (Dixon et al., 2012). Additionally, the tumor suppressor p53 has been shown to be a transcriptional regulator of SLC7A11 (Jiang et al., 2015). Therefore, we model erastin as an inhibitor of SLC7A11 independent of p53 activity, and p53 as a partial activator of SLC7A11 via transcriptional modulation.
4.2.9. GPX4
Glutathione peroxidase 4 (GPX4) is a lipid hydroperoxide reducer within biological membranes, and interference with its activity will promote ferroptosis: indeed, RSL3 acts as a ferroptosis inducer by direct inhibition of GPX4 (Yang et al., 2014). GPX4 requires GSH for its reducing activity, which in turn requires cystine, thereby explaining how erastin can initiate ferroptosis: by inhibiting cystine uptake via SLC7A11, it limits production of GSH and therefore the substrate for GPX4 activity (Stockwell et al., 2017; Yang and Stockwell, 2016). We model GPX4 activity to be inhibited by RSL3, independent of SLC7A11 activity, and in absence of RSL3, its activity is set proportional to SLC7A11.
4.2.10. LOOH and LO•
Lipid hydroperoxides (LOOH) and lipid radicals (LO•) are lipid peroxidation products that can be produced by ROS (Girotti, 1998) and lipoxygenases (e.g. ALOX15) (Ivanov et al., 2015) in a fashion independent of each others’ activity (Feng and Stockwell, 2018). The anti-oxidant activity of GPX4 is at the level of LOOH, which can be converted to LOH in the presence of GPX4 (Girotti, 1998). We thus model the generation of LOOH as a function of ROS and ALOX15 activity on the substrate LH-P. Generation of LO• is modeled in a similar fashion, where the substrate is now LOOH and GPX4 can mitigate the pro-oxidant effect of ROS and ALOX15. We consider each of these reactions to occur on a fraction of available substrate - therefore generation of LOOH does not deplete LH-P in a 1–1 fashion, but may be limited by limited availability of LH-P (and a similar consideration holds for generation of LO•). Since the presence of lipid radicals is considered to be the initiator of ferroptotic processes (Stockwell et al., 2017; Yang and Stockwell, 2016; Yang et al., 2016), we consider LO• as a proxy for ferroptosis in our model.
4.2.11. Auxiliary functions: additive(x,y) and not(x)
The additive(x,y) function represents the symmetric, additive effect of two variables (x,y) on a variable z, and can be represented with the following algorithm:
z = additive(x, y){
sumxy = x + y
if sumxy > 3 : z = 2
else if sumxy == 2 : z = 1
else : z = 0}
The second auxiliary function, not(x), represents the action of an inhibitor x on y.
4.3. In vitro experimental system
Cell culture.
FT, FT-i and FT-t cells (described in (Basuli et al., 2017; Yamamoto et al., 2016)) were cultured in DMEM (Gibco) containing 10% FBS (Gemini Bio-Products). Cells were passaged less than five times before use for experiments. All cells were maintained at 37°C in a humidified incubator at 5% CO2.
Real-time qRTPCR.
200 ng of RNA was reverse transcribed in a total volume of 50 μl using a reverse transcription reagents kit (Applied Biosystems). To make a standard curve, serial dilutions of RNA from one sample were added to the RT reaction. Aliquots (2 μl) of cDNA were added to a 18 μl reaction mixture containing 10 μl of 2× SYBR® Green PCR Master Mix (BioRad) and 400 nm primers. Absence of DNA contamination was confirmed by performing PCR from cDNA without reverse transcriptase. Primers used were:
human SCD1 forward: AAACCTGGCTTGCTGATG
human SCD1 reverse: GGGGGCTAATGTTCTTGTCA
human ACSL4 forward: CATCCCTGGAGCAGATACTCT
human ACSL4 reverse: TCACTTAGGATTTCCCTGGTCC
human β-Actin forward: TTG CCG ACA GGA TGC AGA AGG A
human β-Actin reverse: AGG TGG ACA GCG AGG CCA GGA T
Cell viability.
5000 FT-t cells were transfected with siRNA for 48 h. Afterwards, cells were treated with 0.5 μM of RSL3 (Selleckchem) in presence or absence of 2 μM ferrostatin-1 (Selleckchem) for 24 h. Cell viability was assessed using calcein-AM (Millipore), a fluorescent dye that stains living cells. Briefly, cells plated in 96 well black-walled plates (Corning) were incubated with 2 uM calcein-AM for 30 min at 37°C in a humidified incubator at 5% CO2. Fluorescence was measured in a fluorescent plate reader (BioTek, Synergy2) using 480/520 nm excitation/emission (Stoddart, 2011).
Trypan-blue exclusion assay.
1 × 106 FT-t cells were transfected with siRNA for 48 h. Afterwards, cells were treated with 0.5 to 2 μM of RSL3 (Selleckchem) for 24 hrs. Cells were counted following staining with 0.4% Trypan-blue (Invitrogen), a dye that stains dead cells but is excluded by living cells. The percentage of stained cells as a fraction of the total number of cells was quantified using a hemocytometer (Stoddart, 2011).
siRNA knock-down experiments.
All reagents were obtained from GE Dharmacon (Lafayette, CO, USA). 12 ng of ON-TARGETplus Human SCD1 siRNA (L-0 05,061–0 0–0010), ON-TARGETplus Human ACSL4 (2182) siRNA (L-0 09,364–0 0–0 005) and siNTC (cat#: d-001,810-10-05) were used for knockdown experiments. Transfections were performed according to the manufacturer’s recommendations using Dharmafect #1 transfection reagent (cat: T-2001). Knockdown efficiencies were confirmed at the time of harvest by RT-qPCR.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health, Postdoctoral Fellowship F32CA214030 (A.K.), Grant no. R01CA188025 (S.V.T), and Grant no. R01CA171101 (F.M.T.).
Footnotes
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jtbi.2020.110222.
References
- Agarwal R, Kaye SB, 2003. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev Cancer 3 (July(7)), 502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- Agmon E, Solon J, Bassereau P, Stockwell BR, 2018. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep 8 (March(1)), 5155. doi: 10.1038/s41598-018-23408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JPF, Shah R, Pratt DA, Conrad M, 2017. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol. Sci 38 (5), 489–498. doi: 10.1016/j.tips.2017.02.005, 05. [DOI] [PubMed] [Google Scholar]
- Ariyama H, Kono N, Matsuda S, Inoue T, Arai H, 2010. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem. 285 (July(29)), 22027–22035. doi: 10.1074/jbc.M110.126870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuli D, et al. , 2017. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 36 (29), 4089–4099. doi: 10.1038/onc.2017.11, 07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, et al. , 2019. The COQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575 (7784), 688–692. doi: 10.1038/s41586-019-1705-2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzone L, et al. , 2010. Mathematical modelling of cell-fate decision in response to death receptor engagement. PLoS Comput. Biol. 6 (March(3)), e1000702. doi: 10.1371/journal.pcbi.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman J, et al. , 2017. Activated oncogenic pathway modifies iron network in breast epithelial cells: a dynamic modeling perspective. PLoS Comput. Biol 13 (February(2)), e1005352. doi: 10.1371/journal.pcbi.1005352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifman J, Laubenbacher R, Torti SV, 2014. A systems biology approach to iron metabolism. Adv. Exp. Med. Biol. 844, 201–225. doi: 10.1007/978-1-4939-2095-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, et al. , 2015. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10 (July(7)), 1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Angeli JP, Vandenabeele P, Stockwell BR, 2016. Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 15 (5), 348–366. doi: 10.1038/nrd.2015.6, 05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova E, Caromile LA, Laubenbacher R, Shapiro LH, 2018. The innate immune response to ischemic injury: a multiscale modeling perspective. BMC Syst. Biol 12 (April(1)), 50. doi: 10.1186/s12918-018-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, et al. , 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149 (May(5)) 1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR, 2014. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10 (January(1)), 9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- Doll S, et al. , 2017. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13 (January(1)), 91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, et al. , 2019. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575 (7784), 693–698. doi: 10.1038/s41586-019-1707-0, 11. [DOI] [PubMed] [Google Scholar]
- Fang X, et al. , 2019. Ferroptosis as a target for protection against cardiomyopathy. In: Proceedings of the National Academy of Sciences of the United States of America, 116, pp. 2672–2680. doi: 10.1073/pnas.1821022116 February 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Stockwell BR, 2018. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. 16 (May(5)), e2006203. doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Font-Clos F, Zapperi S, La Porta CAM, 2018. Topography of epithelialmesenchymal plasticity. Proc. Natl. Acad. Sci. U S A 115 (23), 5902–5907. doi: 10.1073/pnas.1722609115, 06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, et al. , 2018. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ. 25 (3), 486–541. doi: 10.1038/s41418-017-0012-4, 03 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti AW, 1998. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 39 (August(8)), 1529–1542. [PubMed] [Google Scholar]
- Guo J, et al. , 2018. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res. Treat. 50 (April(2)), 445–460. doi: 10.4143/crt.2016.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T, et al. , 2014. Lysophospholipid acyltransferases mediate phosphatidylcholine diversification to achieve the physical properties required in vivo. Cell Metab. 20 (August(2)), 295–305. doi: 10.1016/j.cmet.2014.05.019. [DOI] [PubMed] [Google Scholar]
- Hassannia B, et al. , 2018. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 128 (8), 3341–3355. doi: 10.1172/JCI99032, August 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess D, Chisholm JW, Igal RA, 2010. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 5 (6), e11394. doi: 10.1371/journal.pone.0011394, June 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, Shimizu T, 2008. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl. Acad. Sci. U S A 105 (February(8)), 2830–2835. doi: 10.1073/pnas.0712245105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, et al. , 2016. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12 (8), 1425–1428. doi: 10.1080/15548627.2016.1187366, 08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hower V, et al. , 2009. A general map of iron metabolism and tissue-specific subnetworks. Mol. Biosyst. 5 (May(5)), 422–443. doi: 10.1039/b816714c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igal RA, 2011. Roles of stearoylcoa desaturase-1 in the regulation of cancer cell growth, survival and tumorigenesis. Cancers (Basel) 3 (2), 2462–2477. doi: 10.3390/cancers3022462, May 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I, Kuhn H, Heydeck D, 2015. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 573 (November(1)), 1–32. doi: 10.1016/j.gene.2015.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, et al. , 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520 (7545), 57–62. doi: 10.1038/nature14344, April 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, et al. , 2017. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13 (January(1)), 81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, et al. , 2013. Hypoxic and ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl. Acad. Sci. U S A 110 (May(22)), 8882–8887. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstorum A, Lynch ML, Torti SV, Torti FM, Laubenbacher RC, 2018. A systems biology approach to understanding the pathophysiology of high grade serous ovarian cancer: focus on iron and fatty acid metabolism. OMICS 22 (7). doi: 10.1089/omi.2018.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. , 2018. Cell origins of high-grade serous ovarian cancer 10.3390/cancers10110433. [DOI] [PMC free article] [PubMed]
- Konstorum A, 2020. Systems Biology of Ferroptosis: a Modeling Approach (source code), v1. Mendeley Data doi: 10.17632/sjkj7444xb.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstorum A , Vella AT, Adler AJ, Laubenbacher RC, 2017. Addressing current challenges in cancer immunotherapy with mathematical and computational modelling. J. R Soc. Interface 14 (131). doi: 10.1098/rsif.2017.0150, 06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MY, Park E, Lee SJ, Chung SW, 2015. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6 (27), 24393–24403. doi: 10.18632/oncotarget.5162, September 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaier E, et al. , 2014a. [Ferroptosis, a new form of cell death relevant to the medical treatment of cancer]. Med. Sci. (Paris) 30 (August-September(8–9)), 779–783. doi: 10.1051/medsci/20143008016. [DOI] [PubMed] [Google Scholar]
- Lachaier E, et al. , 2014b. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 34 (November(11)), 6417–6422. [PubMed] [Google Scholar]
- Lee H, et al. , 2020. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22 (February(2)), 225–234. doi: 10.1038/s41556-020-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JY, Kim WY, 2013. Stearoyl co-A desaturase 1 as a ccRCC therapeutic target: death by stress. Clin. Cancer Res. 19 (12), 3111–3113. doi: 10.1158/1078-0432. CCR-13–0800, June 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, et al. , 2018. Targeting stearoyl-CoA desaturase 1 to repress endometrial cancer progression. Oncotarget 9 (15). doi: 10.18632/oncotarget.24304, 12064–1.2078, February 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. , 2017. Quantitative assessment of cell fate decision between autophagy and apoptosis. Sci. Rep. 7 (1), 17605. doi: 10.1038/s41598-017-18001-w, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos B, Engelbrecht AM, 2009. Cell death: a dynamic response concept. Autophagy 5 (July(5)), 590–603. doi: 10.4161/auto.5.5.8479. [DOI] [PubMed] [Google Scholar]
- Magtanong L, et al. , 2018. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. doi: 10.1016/j.chembiol.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason P, et al. , 2012. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 7 (3), e33823. doi: 10.1371/journal.pone.0033823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minville-Walz M, et al. , 2010. Inhibition of stearoyl-CoA desaturase 1 expression induces CHOP-dependent cell death in human cancer cells. PLoS ONE 5 (12), e14363. doi: 10.1371/journal.pone.0014363, December 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MK, Saez-Rodriguez J, Sorger PK, Lauffenburger DA, 2010. Logic-based models for the analysis of cell signaling networks. Biochemistry 49 (April(15)), 3216–3224. doi: 10.1021/bi902202q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrugarra D, Veliz-Cuba A, Aguilar B, Arat S, Laubenbacher R, 2012. Modeling stochasticity and variability in gene regulatory networks. EURASIP J. Bioinform. Syst. Biol. (1) 5. doi: 10.1186/1687-4153-2012-5, Jun 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoletano F, Baron O, Vandenabeele P, Mollereau B, Fanto M, 2019. Intersections between regulated cell death and autophagy. Trends Cell Biol. 29 (4), 323–338. doi: 10.1016/j.tcb.2018.12.007, 04. [DOI] [PubMed] [Google Scholar]
- Norouzi-Barough L, Sarookhani MR, Sharifi M, Moghbelinejad S, Jangjoo S, Salehi R, 2018. Molecular mechanisms of drug resistance in ovarian cancer. J. Cell Physiol. 233 (June(6)), 4546–4562. doi: 10.1002/jcp.26289. [DOI] [PubMed] [Google Scholar]
- Ou Y, Wang SJ, Li D, Chu B, Gu W, 2016. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. U S A 113 (44), E6 806–E6 812. doi: 10.1073/pnas.1607152113, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panetta JC, Fister KR, 2000. Optimal control applied to cell-cycle-specific cancer chemotherapy. SIAM J. Appl. Math. 60, 1059–1072. doi: 10.1137/S0036139998338509. [DOI] [Google Scholar]
- Paton CM, Ntambi JM, 2009. Biochemical and physiological function of stearoyl- CoA desaturase. Am J. Physiol. Endocrinol. Metab 297 (July(1)), E28–E37. doi: 10.1152/ajpendo.90897.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnix ZK, et al. , 2010. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2 (August(43)), 43. doi: 10.1126/scisignal.3001127, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Rodriguez J, et al. , 2007. A logical model provides insights into t cell receptor signaling. PLoS Comput. Biol. 3 (August(8)), e163. doi: 10.1371/journal.pcbi.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleich K, Lavrik IN, 2013. Mathematical modeling of apoptosis. Cell Commun. Signal 11 (June(1)), 44. doi: 10.1186/1478-811X-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X, 2018. Emerging strategies of cancer therapy based on ferroptosis. Adv. Mater. 30 (March(12)), e1704007. doi: 10.1002/adma.201704007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou H, Hishikawa D, Harayama T, Yuki K, Shimizu T, 2009. Recent progress on acyl COA: lysophospholipid acyltransferase research. J. Lipid Res. 50, S46–S51. doi: 10.1194/jlr.R800035-JLR200, Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou H, Shimizu T, 2009. Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 284 (January(1)), 1–5. doi: 10.1074/jbc.R800046200. [DOI] [PubMed] [Google Scholar]
- Shintoku R, et al. , 2017. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 108 (November(11)), 2187–2194. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizek H, Hamel A, Deritei D, Campbell S, Ravasz Regan E, 2019. Boolean model of growth signaling, cell cycle and apoptosis predicts the molecular mechanism of aberrant cell cycle progression driven by hyperactive PI3K. PLoS Comput. Biol 15 (3), e1006402. doi: 10.1371/journal.pcbi.1006402, 03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R, et al. , 2014. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am Chem. Soc. 136 (March(12)), 4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, et al. , 2017. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171 (2), 273–285. doi: 10.1016/j.cell.2017.09.021, October 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart MJ, 2011. Cell viability assays: introduction In: Stoddart MJ, Totowa (Eds.), Mammalian Cell Viability: Methods and Protocols. NJ: Humana Press, pp. 1–6. [DOI] [PubMed] [Google Scholar]
- Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D, 2019. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell. Death Dis. 10 (5), 331. doi: 10.1038/s41419-019-1564-7, April 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti SV, Torti FM, 2011. Ironing out cancer. Cancer Res. 71 (March(5)), 1511–1514. doi: 10.1158/0008-5472.CAN-10-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti SV, Torti FM, 2013. Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13 (May(5)), 342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoly I, Parmar J, Shajahan-Haq AN, Clarke R, Baumann WT, Tyson JJ, Dynamic modeling of the interaction between autophagy and apoptosis in mammalian cells. CPT Pharm. Syst. Pharmacol. 4 (April(4)), 263–272. doi: 10.1002/psp4.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfay L, et al. , 2019. Steroyl-CoA desaturase 1 (SCD1) protects ovarian cancer cells from ferroptotic cell death. Cancer Res. doi: 10.1158/0008-5472.CAN-19-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM, 2018. Iron and cancer. Annu. Rev. Nutr. 38, 97–125. doi: 10.1146/annurev-nutr-082117-051732, August 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udyavar AR, et al. , 2017. Novel hybrid phenotype revealed in small cell lung cancer by a transcription factor network model that can explain tumor heterogeneity. Cancer Res. 77 (5), 1063–1074. doi: 10.1158/0008-5472.CAN-16-1467, 03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P, 2014. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15 (February(2)), 135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Bio.l 11 (October(10)), 700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Wu JR, Tuo QZ, Lei P, 2018. Ferroptosis, a recent defined form of critical cell death in neurological disorders. J Mol. Neurosci. 66 (October(2)), 197–206. doi: 10.1007/s12031-018-1155-6. [DOI] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U S A 113 (34), E4966–E4975. doi: 10.1073/pnas.1603244113, 08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR, 2016. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26 (March(3)), 165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, et al. , 2014. Regulation of ferroptotic cancer cell death by GPX4. Cell 156 (January(1–2)), 317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, et al. , 2016. Ferroptosis: process and function. Cell Death Differ. 23 (March(3)), 369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, et al. , 2017. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20 (7), 1692–1704. doi: 10.1016/j.celrep.2017.07.055, 08. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, et al. , 2016. In vitro and in vivo correlates of physiological and neoplastic human fallopian tube stem cells. J. Pathol. 238 (March(4)), 519–530. doi: 10.1002/path.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.