

Abstract
Protein kinase pathways are traditionally mapped by monitoring downstream phosphorylation. Meanwhile, the non-catalytic functions of protein kinases remain under-appreciated as critical components of kinase signaling. c-Src is a protein kinase known to have non-catalytic signaling function important in healthy and disease cell signaling. Large conformational changes in the regulatory domains regulate c-Src’s non-catalytic functions. Herein, we demonstrate that changes in the global conformation of c-Src can be monitored using a selective proteolysis methodology. Further, we use this methodology to investigate changes in the global conformation of several clinical and non-clinical mutations of c-Src. Significantly, we identify a novel activating mutation observed clinically, W121R, that can escape down-regulation mechanisms. Our methodology can be expanded to monitor the global conformation of other tyrosine kinases, including c-Abl, and represents an important tool toward the study of elucidating the non-catalytic functions of protein kinases.
Introduction
The 518 protein kinases (PKs) encoded by the human genome regulate important cellular processes such as cell growth and survival through post-translational phosphorylation [1–3]. All functional PKs consist of a highly conserved kinase domain (KD) responsible for the catalytic phosphorylation of protein substrates. Recently, specific non-catalytic functions of PKs have been identified, and these non-catalytic functions have been shown to play critical roles in cellular processes that are independent of kinase catalytic activity. These non-catalytic functions are commonly observed as protein-protein interactions (PPIs), and are often controlled through global conformational changes within the multi-domain protein kinase [4–8].
The tyrosine kinase c-Src, has been validated as an important target for many solid tumors via knockdown studies (e.g. siRNA) [9, 10]. Genomic knockdown leads to abrogation of both catalytic and non-catalytic functions of the targeted PK. In contrast, pharmacological inhibition using small molecule kinase inhibitors abrogates only the catalytic function of c-Src and has failed to recapitulate the effects of siRNA knockdown [11]. Together, these reports highlight the importance of non-catalytic functions in kinase signaling. The full signaling mechanism of c-Src includes physical protein-protein interaction (PPI) with other signaling proteins, including EGFR, FAK, and STAT3 [12–14]. These PPIs are governed by the global conformation of c-Src, which in turn is modulated by the phosphorylation state(s) of c-Src. Specifically, phosphorylation at Tyr-419 (human c-Src numbering, on the kinase activation loop), stabilizes an ‘open’ or ‘extended’ conformation in which the regulatory SH2 and SH3 domains are accessible for docking to signaling proteins [15]. Downregulation of c-Src, via phosphorylation at Tyr-530 (human c-Src numbering, C-terminal tail), stabilizes a ‘closed’ or ‘compact’ conformation, rendering the SH2 and SH3 domains inaccessible and ‘fully engaged’ to the kinase domain [16]. These large changes in quaternary structure enable c-Src to modulate non-catalytic functions.
Mutations within the kinase domain of PKs have been long understood to impact signaling by altering catalytic activity [17, 18]. However, data collected from next-generation sequencing (NGS) of patient-derived material, have uncovered uncharacterized mutations that exist outside the ATP-binding site of c-Src [19]. We hypothesize that mutations outside the kinase domain and ATP-binding site could modulate cell signaling via changes in global conformation.
Despite the growing appreciation for the non-catalytic functions of PKs, the difficulty in measuring protein conformation using biochemical approaches has hindered progress toward the mechanisms behind these important functions. Toward a potential selective proteolysis method to discern the global conformation of c-Src, MacAuley and Cooper reported in 1989 that c-Src is selectively proteolyzed by the bacterial protease thermolysin [20]. Furthermore, they reported that thermolysin cleavage of c-Src was decreased when Tyr-530 of c-Src is phosphorylated (which leads to the closed conformation) [20]. It was thus hypothesized that the thermolysin cleavage site is located on the linker between the SH2 domain and kinase domain (SH2-KD linker). We performed an analysis of publicly available crystal structures of 3-domain c-Src, which highlights significant differences in accessibility of the SH2-KD linker between the ‘open’ and ‘closed’ conformations (Figure 1). The SH2-KD linker is highly shielded by the SH3 domain and KD in the closed conformation, decreasing the accessibility of the linker to cleavage by thermolysin.
Figure 1. c-Src kinase in the closed (PDB: 2SRC) and the open (PDB: 1Y57) global conformations.
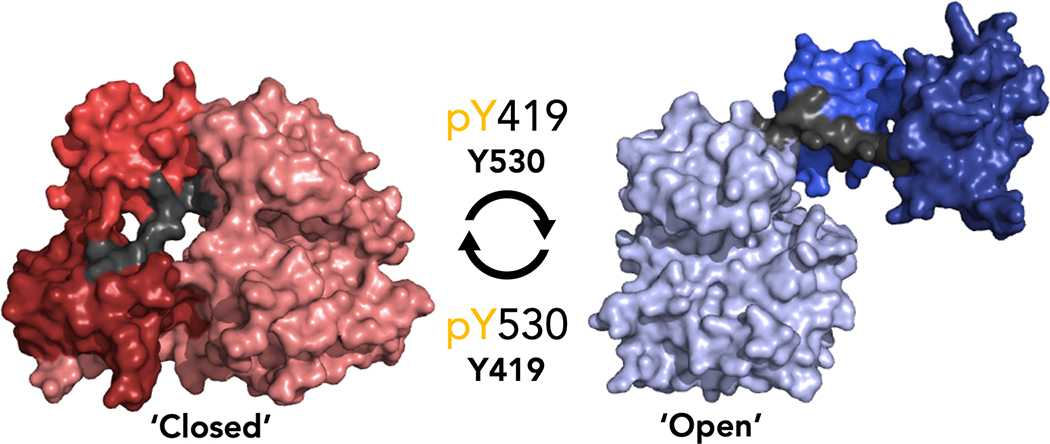
The linker connecting the kinase domain to the SH2 domain (colored grey) is shielded in the closed conformation (colored red) and accessible in the open conformation (colored blue).
On the basis of Cooper’s seminal report and our observations of publicly available structures, we adapted the selective proteolysis of c-Src by thermolysin to enable rapid identification of c-Src global conformation. Using this methodology, we analyzed and characterized clinical and non-clinical c-Src mutations. Significantly, our work has identified a novel gain-of-function mutation (W121R) that has not been characterized to date. Due to the robust nature of this assay, we provide a framework for its application to other tyrosine kinases. Our methodology has yielded a deeper understanding of the complete signaling mechanisms of c-Src, both catalytic and non-catalytic.
RESULTS AND DISCUSSION
Global conformational change of c-Src alters thermolysin cleavage rate
In 1989, MacAuley and Cooper reported that c-Src was selectively cleaved by thermolysin. Further, they reported that down-regulated c-Src (pY530) was less sensitive to proteolytic degradation [20]. From their SDS-PAGE analysis, they hypothesized that the cleavage site was within the SH2-KD linker, a linker that connects the kinase domain of c-Src to its regulatory domains. Recent crystallographic studies of c-Src show that the accessibility of the SH2-KD linker significantly changes between the active/open and inactive/closed conformations, and we thus hypothesized that the rate of thermolysin cleavage could be used to report on the global conformation of c-Src.
Incubation of c-Src with thermolysin and analysis via SDS-PAGE afforded two protein fragments, consistent with a single cleavage site. We characterized the half-life for cleavage with thermolysin to be 33 ± 2 min. We used mass spectrometry analysis to determine that the primary cleavage site was located on the SH2-KD linker, specifically between Gly-257 and Leu-258 (see Supplemental Information SIII). We hypothesized that thermolysin would cleave open-conformation c-Src more rapidly than the closed-conformation, due to the increased accessibility of Leu-258 (Supplemental Figure S1). As stabilization of these conformations is phospho-dependent, both Srcopen (pY419 c-SrcY530F) and Srcclosed (pY530 c-SrcY419F) 3-domain c-Src constructs were incubated with thermolysin, and the cleavage reaction was monitored over time (Figure 2). Consistent with our hypothesis, Srcclosed was cleaved at a significantly slower rate (t1/2 = 234 ± 26 min) than Srcopen (t1/2 = 12 ± 2 min). Given that several other protein kinases share the domain configuration of c-Src (SH3-SH2-KD), we hypothesized that this method could be expanded to homologous kinases. A sequence alignment of all kinases having an SH2-KD linker suggested that at least 9 additional kinases could be investigated using this method (Supplemental Figure S2). We confirmed the utility of this assay with one additional kinase, c-Abl (see supporting information SV).
Figure 2. Limited proteolysis using thermolysin can interrogate c-Src global conformation.

c-Src constructs (2 μM), or c-Src (2 μM) pretreated with conformation selective inhibitors (10 μM) were incubated with thermolysin (60 nM) and total c-Src levels were monitored over time. Open conformation c-Src is cut by thermolysin at a rate significantly faster than closed c-Src is cut.
Conformation-selective inhibitors alter the global conformation of c-Src
There has been recent excitement about the ability of ATP-competitive inhibitors to alter the global conformation of PKs, and these inhibitors have been termed ‘conformation-selective’ kinase inhibitors (CSKIs). CSKIs that have minor structural differences can promote large-scale conformational changes via long-range allosteric networks within a kinase [21–23]. More specifically, modulating the interaction between the inhibitor and the αC-helix of c-Src has been shown to stabilize the ‘open’ and ‘closed’ conformation [24]. Pushing the αC-helix using a steric element on the kinase inhibitor leads to a closed global kinase conformation, while hydrogen bonding to the αC-helix and keeping it more ‘inward’ leads to an open global kinase conformation [24]. In an effort to expand our limited proteolysis assay to study the impact of kinase inhibitors on the global conformation of c-Src, we incubated c-Src with CSKIs that we previously reported that are based on a dasatinib scaffold and bind the αC-helix-in (DAS-DFGO) or the αC-helix-out (DAS-CHO) conformations (see Supplemental Figure S4) [25].
Using our thermolysin assay, we found that DAS-DFGO bound to c-Src was degraded rapidly (t1/2 = 2 ± 0.1 min), while DAS-CHO bound to c-Src had a significantly slower cleavage rate (t1/2 = 80 ± 9 min) (Figure 2). From these data, we assign DAS-DFGO bound c-Src to be in an ‘open’ global conformation, while DAS-CHO bound c-Src is in the ‘closed’ conformation, consistent with crystal structures for these compounds published by our laboratory. The implications of these data on kinase inhibitor design are significant: compounds with similar or near identical binding potencies and selectivity profiles can have significantly different effects on global kinase conformation, which may impact non-catalytic function.
Reported mutation of c-Src alter the global kinase conformation
Several mutants of c-Src have been reported to interrogate the structure and function of c-Src. For example, K298M is frequently used to assess ‘kinase dead’ c-Src, often in comparison to c-Src knockdown [26]. In total, we surveyed 15 c-Src mutations using our limited proteolysis assay. c-Src mutants were expressed in E. coli and subsequently purified using adapted literature-reported protocols (see supplementary information for our modified protocols). To assign a global kinase conformation to each mutant, we utilized the thermolysin assay and obtained a half-life for cleavage for each mutant that could be compared to wild-type c-Src kinase (Figure 3). We utilized 5σ as the cutoff for a significant deviation from wild-type c-Src (5 * 2.4 min = 12 min).
Figure 3. c-Src mutations and their impact on the global kinase conformation.

All measurements were performed in triplicate and the error reported is the standard deviation.
We found that K298M and K298R, mutations that are frequently used to assess the impact of c-Src phosphorylation, both lead to significant, and opposite, changes in global kinase conformation [26, 27]. K298M (t1/2 = 11 ± 2 min) is significantly open compared to wild-type c-Src (t1/2 = 33 ± 2 min), while K298R (t1/2 = 92 ± 6 min) is significantly more closed. Given that c-Src has known non-catalytic roles, these findings are very important because the mutation is not impacting kinase activity alone. Rather, these mutations will impact both catalytic and non-catalytic functions. On this basis, we believe that K298 mutations are not acceptable choices for “kinase dead” c-Src. Toward identifying a mutation that inactivates c-Src while not altering its global conformation, we propose F408G. Phe-408 resides in the critical DFG-motif of c-Src and we observe no catalytic activity for F408G c-Src without altering the conformation of the kinase (F408G t1/2 = 31 ± 5 min; c-Src t1/2 = 33 ± 2 min).
Shokat and co-workers have developed a chemical genetic strategy that relies upon replacing the gatekeeper Thr of c-Src with a glycine (T341G) [28]. This mutation provides a ‘hole’ that a ‘bumped’ kinase inhibitor can interact with in a highly selective manner. We found that T341G is not silent to protein conformation and leads to a significant closure of the kinase (t1/2 = 71 ± 4 min).
Given that the T341G mutation led to changes in c-Src conformation, we next assessed the impact of gatekeeper mutations that are known to modulate the ability of many ATP-competitive ligands to bind c-Src. T341I and T341M have been reported as “drug-resistant” mutants of c-Src [29, 30]. We found that there was no significant change in the conformation with either T341I or T341M (t1/2 = 35 ± 1 and 43 ± 3 min, respectively). This suggests that the observed ability for these mutations to prevent inhibitor binding is unlikely to be due to conformational change of the kinase, but mainly from steric inclusion of the inhibitor.
Viral Src kinase (v-Src) is a viral homolog of the human c-Src kinase that leads to sarcoma formation in chickens [31]. Interestingly, v-Src and c-Src share extremely high sequence identity (98%) and while v-Src is constitutively active and transforming, c-Src’s activity is regulated and c-Src is not transforming. The sequence differences between c-Src and v-Src have been well characterized for their impact on catalytic activity and recognition by Csk, a negative regulator of c-Src. R98W and D120N (human kinase numbering) were identified as being crucial in enabling c-Src to escape down-regulation by Csk and yield a constitutively active kinase in S. pombe [32]. In our thermolysin proteolysis assay, we found that both R98W and D120N yielded a highly open kinase conformation (t1/2 = 12 ± 0.4 min and 11 ± 2 min, respectively). More recently, R98W and D120N mutations have been shown to modulate the protein-protein interaction between c-Src and Hsp90, however, no explanation for how these mutations impact Hsp90 binding was presented [33]. Given that global kinase conformation can modulate PPIs, this prior report is consistent with our findings that these mutations alter the conformation of c-Src kinase.
Seeliger and co-workers reported a detailed study of the kinase domain of c-Src’s allosteric regulatory network in 2015 [34]. Using molecular simulations, W263A and D407N were found to be important residues within the regulation of the kinase domain. Importantly, the Seeliger study was performed using only the kinase domain. Thus, we thought the thermolysin selective proteolysis assay could provide insight into the action of these two key residues on the global conformation of c-Src. Toward this goal, we expressed and purified the W263A and D407N mutants of c-Src and determined their half-life in our thermolysin assay. Consistent with these residues being important for the regulation of c-Src, we found that both W263A and D407N stabilized the open c-Src conformation (t1/2 = 3 ± 0.2 and 13 ± 2 min, respectively).
Unannotated clinical mutations alter the global conformation of c-Src
Given that we have shown a broad utility for our thermolysin selective proteolysis assay with a variety of previously reported c-Src mutations, we next wanted to apply our methodology to study clinical c-Src mutations. Single amino acid, nonsynonymous mutations in kinases have been reported to be transformative in a number of cancers including lung, breast, and colon. Next-generation sequencing of patient material has uncovered c-Src mutations that are distal to the ATP binding pocket and have not been previously characterized. To identify clinical mutations of c-Src, we utilized the COSMIC, NIH Center of Genomics, and the Cancer Cell Line Encyclopedia (CCLE) public databases [35–37]. Given how many non-clinical mutations we observed to alter the global conformation of c-Src, we hypothesized that clinical mutations of c-Src might also be perturbing c-Src’s conformation.
We expressed and purified a panel of 12 c-Src mutants that have been identified in patient materials (Figure 3). All but one of these clinical mutations have been previously uncharacterized. The lone characterized mutation, E527K, was recently reported as a dominant gain-of-function mutation that leads to thrombocytopenia, myelofibrosis, bleeding, and bone pathologies [38]. In our assay, we found that E527K stabilizes a closed conformation (t1/2 = 50 ± 10 min) relative to wild-type c-Src (t1/2 = 33 ± 2 min). Additionally, we found that E527K c-Src had reduced catalytic activity to that of wild-type c-Src in our biochemical assay (E527K c-Src Vmax = 28 ± 10 RFU/s; wild-type c-Src Vmax = 56 ± 6 RFU/s). While the changes we observe in our assays do not correspond to the gain-of-function reported for this mutation, it is possible that this mutation within the tail of c-Src prevents recognition by Csk, the cellular regulator of c-Src.
Four of the 11 previously uncharacterized clinical mutations were found to have no significant impact on kinase conformation (P171Q, D521N, I113F, and V140M). In contrast, four clinical mutations led to a closed global conformation of c-Src (Q529H, T341R, K298E, and P307R), while three mutations stabilized the open conformation (D407H, R163W, and W121R). We were particularly interested in mutations that stabilize the open conformation, which is generally associated with constitutive kinase activity. Indeed, we characterized the catalytic activity of W121R, the most open kinase mutant (t1/2 = 2.3 ± 0.2 min) and found it to be significantly more catalytically active (Vmax = 121 ± 21 RFU/s) than wild-type c-Src (Vmax = 56 ± 6 RFU/s).
We utilized published crystal structures to explore the potential mechanism by which W121R and R163W could stabilize a global conformation of c-Src. Both W121R and R163W mutations are located at an interface between a regulatory domain and the kinase domain (Figure 4). From the crystal structures, Arg-163 is located on the SH2 domain and in the closed kinase conformation, Arg-163 makes an electrostatic interaction with residue Asp-368 on the kinase domain. This salt bridge appears to aid in the stabilizing the closed c-Src conformation, and thus disruption of this interaction with R163W leads to an open kinase conformation. Trp-121 is located on the SH3 domain and in the closed conformation the sidechain of Trp-121 is bound within a hydrophobic pocket formed by the kinase domain. Thus, the W121R mutation disrupts this interaction and thus promotes an open kinase conformation.
Figure 4. Trp-121 and Arg-163 are key residues that stabilize the closed conformation of c-Src.

Trp-121 is located in the SH3 domain and binds within a hydrophobic pocket on kinase domain. Arg-163 is located on the SH3 domain and forms a salt-bridge with Asp-368 that stabilizes the closed conformation. Mutations to these residues destabilize the closed conformation: W121R prevents Trp-121 binding within a hydrophobic pocket of the kinase domain; R163W disrupts the salt bridge between Arg-163 and Asp-368.
The relationship of c-Src’s global conformation to its catalytic activity
On the basis of pY419 c-Src being open and highly active (and pY530 c-Src is closed and inactive), most literature reports assume that open c-Src is active and closed c-Src is inactive. Given that we have identified a number of mutations outside the kinase domain that stabilize either the open and closed kinase conformation, we are in a unique position to elucidate the relationship between c-Src’s conformation and its catalytic activity. Using a previously published activity assay utilizing a self-reporting synthetic peptide substrate, we determined the kinetic parameters for each c-Src mutant, including Vmax, KM,ATP, and KM,substrate (see Supplemental Information SVI, SVII) [39].
Consistent with the general literature assumption, all mutations that we identified that close c-Src cause decreased the catalytic activity of c-Src. Meanwhile, we identified mutations outside the ATP binding pocket that challenge the paradigm that open c-Src is active. W263A activates c-Src on the kinase-domain-only construct, but leads to inactivation of the three-domain c-Src construct compared to wild-type three-domain c-Src (Vmax W263A = 16 ± 2, Vmax WT = 56 ± 6). Meanwhile, this mutation leads to a highly open c-Src construct (t1/2 = 3.3 ± 0.2 min) compared to wild-type c-Src (t1/2 = 32.9 ± 2.4 min). Thus, W263 represents a mutation that opens the kinase, but allosterically reduces the catalytic activity of c-Src.
Closed c-Src resists phosphorylation by Csk
In the cell, c-Src activity is regulated by kinases, including Hck and Csk that phosphorylate c-Src at Tyr-419 and Tyr-530, respectively [40, 41]. Phosphorylation of Tyr-419 by Hck leads to activation, while phosphorylation of Tyr-530 by Csk leads to inactivation. Here, we wanted to determine whether the global conformation of c-Src can influence the activation or inactivation of c-Src by Hck and Csk, respectively.
To study the effect of conformation on activation, we monitored the rate of Hck phosphorylation of Tyr-419 of c-Src constructs stabilized in the open or closed conformation. To prevent autophosphorylation of c-Src (which occurs in vitro) while simultaneously locking c-Src into either the open or closed conformation, we utilized irreversible conformation-selective inhibitors developed by our laboratory [25, 42]. When ligated to c-Src, Das-DFGO-Irr stabilizes the open conformation (t1/2 = 14 ± 1 min) and Das-CHO-Irr ligated c-Src stabilizes the closed conformation (t1/2 = 272 ± 49 min). In addition to phosphorylating Tyr-419, we have observed that Hck can phosphorylate Tyr-530 in in vitro assays. Thus, to ensure we are only monitoring phosphorylation of Tyr-419, we ligated Das-DFGO-Irr and Das-CHO-Irr to the Y530F mutant of c-Src (which still leads to open (t1/2 = 18 ± 1 min) and closed (t1/2 = 204 ± 29 min) conformations, respectively). We purified the Y530F-Src–DFGO and Y530F-Src–CHO covalent complexes and incubated each with Hck and the reaction was initiated with ATP, while phosphorylation was monitored over time. We observed no change in the phosphorylation rates between the open or closed kinase complexes using either kinase domain-only or 3-domain Hck (Figure 5). These data suggest that the accessibility of Tyr-419 remains unchanged between the open and closed conformations of c-Src.
Figure 5. Irreversible conformation-selective analogs inhibitors impact inactivation by Csk.

c-Src kinase constructs with SrcY530F or SrcY419F (10 μM) were pretreated with irreversible conformation-selective inhibitors (100 μM) and purified. These constructs, Src-DFGO-Irr and Src-CHO-Irr, were incubated with 3-domain Hck or inactivating kinase Csk (60 nM) and initiated with 100 μM ATP. Phosphorylation at Tyr-419 and Tyr-530 were monitored over time. Reversible conformation-selective dasatinib analogs (PDB: 4YBJ and 4YC8) were overlaid with 3-domain c-Src (PDB: 2SRC and 1Y57).
An analogous experiment was performed to study the effect of global conformation on Csk phosphorylation at Tyr-530 using WT-Src (unlike Hck, we have found that Csk phosphorylates Tyr-530 with a high degree of specificity and does not phosphorylate Tyr-419). WT-Src–DFGO (open, t1/2 = 14 ± 1 min) and WT-Src–CHO (closed, t1/2 = 272 ± 49 min) complexes were incubated with Csk, the reaction was initiated with ATP, and phosphorylation of Tyr-530 was monitored over time. We observed that the open complex, WT-Src–DFGO, had a faster initial phosphorylation rate than the closed complex, Wt-Src–CHO. We hypothesize that the decrease in accessibility at Tyr-530 in the closed conformation is reflected in the decrease in phosphorylation rate by Csk. Importantly, these data are consistent with previously reported observations that closed kinase conformation leads to decreased phosphatase activity at pY530 by Maly and co-workers [24].
Mutations stabilizing the open conformation can prevent downregulation by Csk
It has been previously reported that R98W and D120N, two of the mutations found in v-Src and mapped onto c-Src, each escape down-regulation when c-Src is co-expressed with Csk in S. Pombe [32]. That is, when phosphorylated at Tyr-530 by Csk, R98W and D120N are reported to maintain catalytic activity (unlike wild-type c-Src). Using our assay, we found that R98W and D120N c-Src can individually stabilize the open kinase conformation (vide supra). We hypothesized that clinical mutations stabilizing the open conformation may also be able to escape down-regulation by Csk. To eliminate concern with auto-phosphorylation of c-Src during phosphorylation of Tyr-530 using Csk, we elected to use a pY530 c-Src mimetic to model down-regulated c-Src. The ‘SH2-engaged’ construct (SH2Eng) consists of a high affinity anionic sequence replacing the C-terminal tail, and mimics phosphorylation at Tyr-530 [43]. Analysis via the selective proteolysis and kinetic parameters verifies this construct as an acceptable pY530 c-Src mimetic that stabilizes the closed conformation (t1/2 = 356 ± 34 min).
Using the SH2Eng construct, we determined the effect of open conformation-stabilizing residues on kinase conformation (Figure 6). T341MSH2Eng was used as a negative control because previously found that the T341M mutation has a similar conformation to wild-type c-Src (T341M t1/2 = 43 ± 3 min, WT c-Src t1/2 = 33 ± 2 min). As expected, T341MSH2Eng adopts a closed kinase conformation (t1/2 = 283 ± 32 min). Consistent with previous reports, we observe that R98WSH2Eng c-Src adopts an open conformation relative to WTSH2Eng (R98WSH2Eng t1/2 = 38 ± 6, WTSH2Eng t1/2 = 283 ± 32). In addition, we found that R98WSH2Eng leads to a restoration of catalytic activity compared to WTSH2Eng (R98WSH2Eng Vmax = 70 ± 21 RFU/s, WTSH2Eng Vmax = 6.5 ±0.5 RFU/s).
Figure 6. W121R mutation resists canonical down-regulation.

Opening mutation were introduced onto the SrcSH2Eng constructs and their catalytic activity and global conformation were assessed. Error reported is the standard deviation of three independent measurements.
We next introduced the previously uncharacterized clinical mutation W121R onto this construct (W121RSH2Eng). Like R98W, W121RSH2Eng maintained an open conformation (t1/2 = 4 ± 0.1 min) and possessed high catalytic activity (Vmax = 118 ± 7 RFU/s). An alternative to our model (Figure 6) is that the kinase tail remains associated with the SH2 domain while the SH3 domain is disengaged from the linker. Both hypotheses are consistent with increased linker accessibility and faster thermolysin cleavage rate. The latter hypothesis is supported by Smithgall and Engen who have shown that activation of Hck (a Src family kinase) by HIV-1 Nef occurs via SH3 disengagement while the kinase tail remains bound to the SH2 domain of Hck [44]. Thus, further studies are required to elucidate the full mechanism for W121R’s ability to overcome Csk inactivation. Significantly, these data reveal that mutations which stabilize the open conformation have the potential not only to disrupt protein-protein interactions, but can also lead to a constitutively active kinase, as observed with W121R c-Src. Thus, our methodology has identified a novel gain-of-function clinical mutation for c-Src.
Conclusions
We used the bacterial protease thermolysin to develop a rapid and sensitive assay to detect changes in the global conformation of c-Src. This method relies upon the differences in accessibility of Gly-257 and Leu-258 residues located on the SH2-KD linker in the ‘open’ and ‘closed’ conformations. The analysis of a number of mutations reported in the literature to be involved in c-Src regulation showcase the methodology and provides new insight into how these mutations impact c-Src conformation. Significantly, we identified a previously uncharacterized clinical mutation of c-Src (W121R) that leads to a constitutively active, open-conformation kinase. We further found that W121R is immune to down-regulation by Csk. Our results both provide a novel means to survey protein kinase conformation, but also yield important new insight into the role of conformation in c-Src regulation. We anticipate that several of the mutations we identified and characterized can be useful for studying the non-catalytic activities of c-Src kinase. Finally, on the basis of our work, continued effort toward designing conformation-selective inhibitors that interact with a specific open/closed form of kinases is warranted.
Supplementary Material
Acknowledgements
We thank A. Mapp (U. Michigan) for contributive discussions. This work was funded by the National Institutes of Health Grant R01 GM125881 to M.B.S. A National Institutes of Health Chemistry—Biology Interface Training Grant (GM008597) supported, in part, F.E.K. A National Institutes of Health Cellular Biotechnology Training Grant (GM008353) supported, in part, T.K.J.
Footnotes
Dedication
We dedicate this work to Ronald T. Raines on the occasion of his 60th birthday.
References
- (1).Hunter T (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80, 225–236. [DOI] [PubMed] [Google Scholar]
- (2).Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S. (2002) The Protein Kinase Complement of the Human Genome. Science. 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- (3).Hunter T (1987) A Thousand and One Protein Kinases. Cell. 50, 823–829. [DOI] [PubMed] [Google Scholar]
- (4).Pawson T, Gish GD (1992) SH2 and SH3 Domains: from Structure to Function. Cell. 71, 359–362. [DOI] [PubMed] [Google Scholar]
- (5).Taylor SS, and Kornev AP (2011) Protein Kinases: Evolution of Dynamic Regulatory Proteins. Trends Biochem. Sci. 36, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Soderling TR (1990) Protein kinases. Regulation by Autoinhibitory Domains. J. Biol. Chem. 265, 1823–1826. [PubMed] [Google Scholar]
- (7).Huse M, and Kuriyan J (2002) The Conformational Plasticity of Protein Kinases. Cell. 109, 275–282. [DOI] [PubMed] [Google Scholar]
- (8).Kung JE, and Jura N (2016) Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure. 24, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gilani RA, Phadke S, Bao LW, Lachacz EJ, Dziubinski ML, Brandvold KR, Steffey ME, Kwarcinski FE, Graveel CR, Kidwell KM, and Merajver SD (2016) UM-164: a Potent c-Src/p38 Kinase Inhibitor with in Vivo Activity Against Triple-Negative Breast Cancer. Clin. Can. Re. 22, 5087–5096. [DOI] [PubMed] [Google Scholar]
- (10).Bjorge JD, Pang AS, Funnell M, Chen KY, Diaz R, Magliocco AM, and Fujita DJ (2011) Simultaneous siRNA Targeting of Src and Downstream Signaling Molecules Inhibit Tumor Formation and Metastasis of a Human Model Breast Cancer Cell Line. PLoS One. 6, e19309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Balzer EM, Whipple RA, Thompson K, Boggs AE, Slovic J, Cho EH, Matrone MA, Yoneda T, Mueller SC and Martin SS, 2010. c-Src differentially regulates the functions of microtentacles and invadopodia. Oncogene. 29, p.6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Maa MC, Leu TH, McCarley DJ, Schatzman RC, and Parsons SJ (1995) Potentiation of Epidermal Growth Factor Receptor-mediated Oncogenesis by c-Src: Implications for the Etiology of Multiple Human Cancers. Proc. Natl. Acad. Sci. U.S.A. 92, 6981–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Avizienyte E, and Frame MC (2005) Src and FAK Signalling Controls Adhesion Fate and the Epithelial-to-mesenchymal Transition. Curr. Opin. Cell Biol. 17, 542–547. [DOI] [PubMed] [Google Scholar]
- (14).Xi S, Zhang Q, Dyer KF, Lerner EC, Smithgall TE, Gooding WE, Kamens J, and Grandis JR (2003) Src Kinases Mediate STAT Growth Pathways in Squamous cell Carcinoma of the Head and Neck. J. Biol. Chem. 278, 31574–31583. [DOI] [PubMed] [Google Scholar]
- (15).Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, and Meyer T (2005) The Crystal Structure of a c-Src Complex in an active Conformation Suggests Possible steps in c-Src Activation. Structure. 13, 861–871. [DOI] [PubMed] [Google Scholar]
- (16).Young MA, Gonfloni S, Superti-Furga G, Roux B, and Kuriyan J (2001) Dynamic Coupling Between the SH2 and SH3 Domains of c-Src and Hck Underlies their Inactivation by C-terminal Tyrosine Phosphoryation. Cell. 105, 115–126. [DOI] [PubMed] [Google Scholar]
- (17).Blume-Jensen P, and Hunter T (2001). Oncogenic kinase signalling. Nature. 411, 355. [DOI] [PubMed] [Google Scholar]
- (18).Bikker JA, Brooijmans N, Wissner A, and Mansour TS (2009) Kinase domain mutations in cancer: implications for small molecule drug design strategies. J. Med. Chem. 52, 1493–1509. [DOI] [PubMed] [Google Scholar]
- (19).Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R and Yeatman TJ, 1999. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 21, p.187. [DOI] [PubMed] [Google Scholar]
- (20).MacAuley AL, and Cooper JA (1989) Structural Differences Between Repressed and Derepressed Forms of p60c-src. Mol. Cell. Biol. 9, 2648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Agius MP, and Soellner MB (2014). Modulating noncatalytic function with kinase inhibitors. Chem. Biol. 21, 569–571. [DOI] [PubMed] [Google Scholar]
- (22).Novotny CJ, Pollari S, Park JH, Lemmon MA, Shen W, and Shokat KM (2016) Overcoming Resistance to HER2 Inhibitors through State-specific Kinase Binding. Nat. Chem. Biol. 12, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Maly DJ, and Papa FR (2014) Druggable Sensors of the Unfolded Protein Response. Nat. Chem. Biol. 10, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Leonard SE, Register AC, Krishnamurty R, Brighty GJ, and Maly DJ (2014) Divergent Modulation of Src-family Kinase Regulatory Interactions with ATP-competitive Inhibitors. ACS Chem. Biol. 9, 1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kwarcinski FE, Brandvold KR, Phadke S, Beleh OM, Johnson TK, Meagher JL, Seeliger MA, Stuckey JA, and Soellner MB, (2016) Conformation-selective Analogues of Dasatinib Reveal Insight into Kinase Inhibitor Binding and Selectivity. ACS Chem. Biol. 11, 1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schwartzberg PL, Xing L, Hoffmann O, Lowell CA, Garrett L, Boyce BF, and Varmus HE (1997) Rescue of Osteoclast Function by Transgenic Expression of Kinase-deficient Src insrc−/− Mutant Mice. Genes Dev. 11, 2835–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, Burns M, Julian B, Peng XP, Hieronymus H and Maglathlin RL, 2008. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 27, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liu Y, Shah K, Yang F, Witucki L, and Shokat KM (1998) Engineering Src Family Protein Kinases with Unnatural Nucleotide Specificity. Chem. Biol. 5, 91–101. [DOI] [PubMed] [Google Scholar]
- (29).Blencke S, Zech B, Engkvist O, Greff Z, Őrfi L, Horváth Z, Kéri G, Ullrich A, and Daub H (2004) Characterization of a Conserved Structural Determinant Controlling Protein Kinase Sensitivity to Selective Inhibitors. Chem. Biol. 11, 691–701. [DOI] [PubMed] [Google Scholar]
- (30).Azam M, Seeliger MA, Gray NS, Kuriyan J, and Daley GQ (2008) Activation of Tyrosine Kinases by Mutation of the Gatekeeper Threonine. Nat. Struct. Mol. Biol. 15, 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Collett MS, and Erikson RL (1978). Protein kinase activity associated with the avian sarcoma virus src gene product. Proc. Natl. Acad. Sci. 75, 2021–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Superti-Furga G, Fumagalli S, Koegl M, Courtneidge SA, and Draetta G (1993). Csk inhibition of c-Src activity requires both the SH2 and SH3 domains of Src. EMBO J. 12, 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Luo Q, Boczek E, Wang Q, Buchner J, and Kaila VRI (2017). Hsp90 dependence of a kinase is determined by its conformational landscape. Sci. Reports. 7, 43996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Foda ZH, Shan Y, Kim ET, Shaw DE, and Seeliger MA (2015). A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat. Commun. 6, 5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L and Stefancsik R (2016). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, pp.D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, Iorio F, Sousa FG, Elloumi F, Aladjem MI, Thomas A, Sander C., Kohn KW, Benes CH, Garnett M, Reinhold WC, and Pommier Y (2018). CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience. 10, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cancer Cell Line Encyclopedia (CCLE). https://portals.broadinstitute.org/clle (accessed June 24, 2019).
- (38).Turro E, Greene D, Wijgaerts A, Thys C, Lentaigne C, Bariana TK, Westbury SK, Kelly AM, Selleslag D, Stephens JC and Papadia S 2016. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci. Transl. Med. 8, 328ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wang Q, Cahill SM, Blumenstein M and Lawrence DS, 2006. Self-reporting fluorescent substrates of protein tyrosine kinases. J. Am. Chem. Soc. 128, 1808–1809. [DOI] [PubMed] [Google Scholar]
- (40).Abram CL, and Courtneidge SA (2000) Src Family Tyrosine Kinases and Growth Factor Signaling. Exp. Cell Res. 254, 1–3. [DOI] [PubMed] [Google Scholar]
- (41).Okada M, Nada S, Yamanashi Y, Yamamoto T, and Nakagawa H (1991) CSK: a Protein-Tyrosine Kinase Involved in Regulation of Src Family Kinases. J. Biol. Chem. 266, 24249–24252. [PubMed] [Google Scholar]
- (42).Kwarcinski FE, Fox CC, Steffey ME and Soellner MB, 2012. Irreversible inhibitors of c-Src kinase that target a nonconserved cysteine. ACS Chem. Biol. 7, 1910–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lerner EC, and Smithgall TE. (2002) SH3-dependent Stimulation of Src-family Kinase Autophosphorylation Without Tail Release from the SH2 Domain in vivo. Nat. Struct. Mol. Biol. 9, 365–369. [DOI] [PubMed] [Google Scholar]
- (44).Wales TE, Hochrein JM, Morgan CR, Emert-Sedlak LA, Smithgall TE, and Engen JR Biochem. 54, 6382–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.