

Abstract
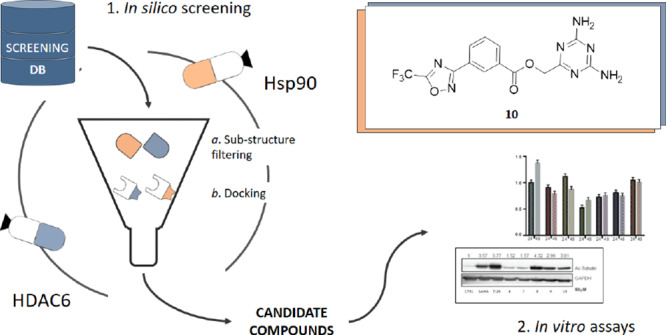
Histone deacetylase 6 (HDAC6) and heat shock protein 90 (Hsp90) are widely investigated anticancer drug targets. Importantly, several lines of evidence indicate that their regulation and activity are intimately linked, and that their combined inhibition may lead to impressive therapeutic benefits. In this study, we developed and applied an integrated computational strategy to design dual inhibitors of HDAC6 and Hsp90. Although the two targets share very little homology, an integrated ligand-based and structure-based virtual screening approach indicated a subset of compounds possessing the key structural requirements for binding at both targets. In vitro tests demonstrated that some of the selected candidates are able to selectively inhibit HDAC6 over HDAC1, to increase the acetylation levels of tubulin on cell assays and to reduce cell proliferation. The discovered compounds represent valuable starting points for further hit optimization.
Introduction
The molecular and genetic complexity of complex diseases suggests that targeting a single oncogenic pathway may not be sufficient to achieve durable remission in patients.1,2 As a consequence, drug discovery efforts have been mainly focused on targeting multiple signaling pathways, either via drug combinations or through the development of a single compound that is able to simultaneously hit multiple oncogenic targets. Indeed, the design of drugs acting on a single target has evolved into the concept of “polypharmacology” or “multitarget” drug discovery, which aims at developing a single drug that targets multiple biological systems simultaneously.2
Histone deacetylase (HDAC) and heat shock protein 90 (Hsp90) enzymes are widely investigated anticancer drug targets. The clinical relevance of HDAC inhibitors is testified by the presence of four drugs approved by the U.S. Food and Drug Administration, as well as by several drug candidates currently under clinical evaluation for a variety of cancers.3 An impressive number of Hsp90 inhibitors have been developed to date, but none of them has received clinical approval because of their low efficacy, toxicity, or de novo or acquired drug resistance. Experience from clinical trials suggests that the ability to reduce the dose without losing activity would be clinically beneficial. Importantly, owing to the observed beneficial synergistic effects, a number of clinical trials including Hsp90 inhibitors in combination with other anticancer drugs are currently ongoing.4 Importantly, Hsp90 and HDAC regulation and activity are intimately linked; their inter-dependence and participation in overlapping signaling networks in cancer cells making them ideal candidates for multi-targeting approaches.5−11 In cancer cells, Hsp90 refolds, stabilizes, and regulates the trafficking of many proteins responsible for uncontrolled proliferation and apoptotic resistance.12 Similarly, HDACs participate in the regulation of many oncogenic processes. Although HDAC inhibitors have demonstrated in vitro anticancer activity, clinical experience with single-agent HDAC inhibitors has been variable, suggesting that a combination of HDAC inhibitors with other agents is warranted.13,14
Interestingly, recent studies have identified Hsp90 as a substrate of histone deacetylase 6 (HDAC6).5−7 Although HDACs are mainly involved in chromatin remodeling and gene expression via direct modification of histones, HDAC6 is responsible for the deacetylation and activation of several cytosolic proteins, including Hsp90.5−7 Indeed, inactivation of HDAC6 leads to the accumulation of acetylated Hsp90, which is not able to form stable Hsp90–client complexes.15 In addition, HDAC6 specifically deacetylates α-tubulin and cortactin, two structural proteins involved in microtubule dynamics and actin network, which are mechanisms involved in cell division and migration.16 Therefore, the activity of HDAC6 heavily impacts on tumor cell invasion and metastasis. Interestingly, several combinations of class I HDAC or HDAC6 inhibitors with the first-in-class Hsp90 inhibitor 17-AAG synergistically induced the degradation of numerous Hsp90 client proteins in various cancer models.9,17−22 Moreover, impressive synergy has also been observed for the NVP-AUY922 and NVP-HSP990 Hsp90 inhibitors with the pan-HDAC inhibitor SAHA in multiple myeloma.23,24 Importantly, it was recently reported that administration of HDAC inhibitors was able to resensitize resistant cells toward the clinically relevant Hsp90 inhibitors 17-AAG and 17-DMAG, and that resistant cells were also identified as cross-resistant to structurally different Hsp90 inhibitors such as radicicol and other second-generation Hsp90 inhibitors.25 Dual inhibitors were recently reported.9,10 Altogether, these results strongly support the rationale that a dual HDAC6/Hsp90 inhibitor may represent a suitable therapeutic strategy to develop more effective anticancer drugs.
Based on the abovepresented evidence, we have developed and applied a tailored strategy to design small molecular weight dual inhibitors of Hsp90 and HDAC6 based on highly integrated pharmacophores.
Results and Discussion
The design of dual inhibitors of these targets is far from being trivial because Hsp90 and HDAC6 belong to different protein families and present a low sequence similarity (33%) and sequence identity (17%), as evaluated by the EMBOSS Water tool.26 Moreover, the two enzymes have different binding site architectures and interact with structurally different substrates in the cellular environment.27,28 Therefore, the design required the application of a specially devised in silico procedure that integrated different ligand- and structure-based approaches (Figure 1). Experimental details on the computational approaches are reported in the Experimental Methods section.
Figure 1.
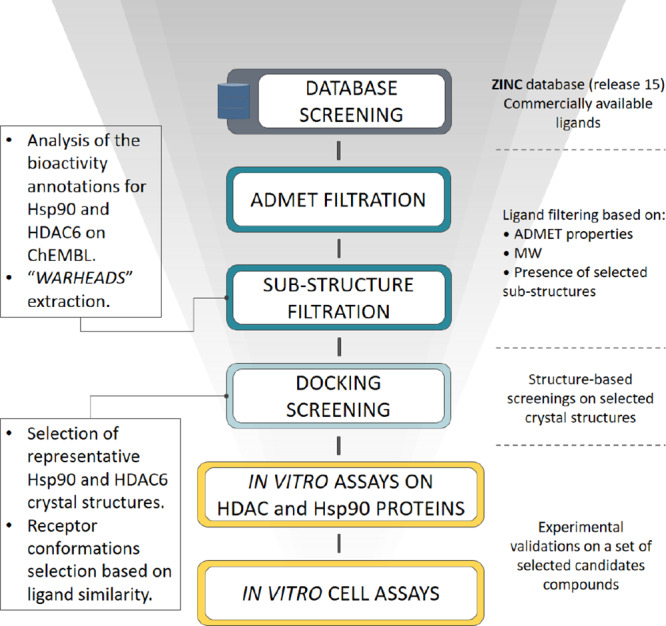
Workflow for the design of Hsp90/HDAC6 dual inhibitors. The devised workflow integrates different in silico approaches (i.e., data mining on publicly available databases to identify Hsp90 and HDAC6 warheads, ligand- and structure-based virtual screenings on commercial databases). The best candidates were tested by means of in vitro assays on purified Hsp90, HDAC6, and HDAC1 (to check for selectivity with respect to class I HDAC), as well as on cell lysates.
In particular, Hsp90 and HDAC6 inhibitors reported in ChEMBL29 were analyzed, as described in the Experimental Methods section to identify chemical “warheads,” that are able to establish favorable interactions with known enzyme hot spots, that is, Asp93 of Hsp90 and the catalytic zinc ion of HDAC6. These analyses, performed by using in-house developed tools based on OpenEye python toolkits,30 allowed for the identification of eight (HDAC6) and eleven (Hsp90) warheads (Figure 2), which were subsequently used to screen the commercially available compounds of the ZINC database31 (www.zinc.docking.org., accessed on February 20, 2018). Compounds in ZINC were first filtered to remove ligands with unfavorable ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, while preserving the molecular diversity. Then, qualified compounds were sequentially filtered for the presence of the identified Hsp90 and HDAC warheads, leading to a focused library of purchasable compounds possessing the warheads required to bind both targets.
Figure 2.

Hsp90 and HDAC6 warheads identified from the ligand-based analyses made on ChEMBL.
To evaluate which of the ligands in the filtered database presented good 3D complementary with the Hsp90 and HDAC6 binding sites, the compounds that passed the previous filtering steps were docked into selected Hsp90 and HDAC6 crystal structures, using Glide.32 Considering that the use of multiple crystallographic conformations helps accounting for potential binding site flexibility issues and may improve structure-based predictions,33,34 eleven Hsp90 and one HDAC6 conformation, which are representative of the conformational landscape of the two proteins, were selected from the Protein Data Bank (PDB)35 (see Table S1 in the Supporting Information). Moreover, Hsp90 crystal structures were prioritized according to the similarity of the screened ligands with the co-crystallized ones, as this approach has demonstrated to provide improved docking predictions.34,36 Visual inspection of the predicted poses and analysis of the docking scores led to the selection of 10 compounds (Table 1) to be tested in vitro. Interestingly, the best candidates included compounds bearing the 5-(trifluoromethyl)-1,2,4-oxadiazole chemical moiety previously reported in inhibitors selective for HDAC6.37 Compounds based on this scaffold were of particular interest to our aim, because the majority of the reported HDAC inhibitors are not isoform selective, and pan-inhibition of HDACs (e.g., by Vorinostat) is associated with side effects.38,39 To confirm the in vitro inhibitory activities of the selected candidate compounds, acetylation levels of tubulin (HDAC6 target) and of histone H3 (HDAC6 off-target) were evaluated in MCF7 cell-based assays (Figure 3, panel a).
Table 1. Inhibitory Activity of the Selected Candidate Compounds of HDAC6, HDAC1, and Hsp90.

Compounds that showed the percentage of inhibition higher than 60% were further investigated at lower concentrations.
Values were measured in triplicates. Percentages of inhibition are reported as mean ± SD.
Percentage of inhibition was evaluated at 1 μM.
Values were measured in singlicate. Hsp90 assays were performed only for compounds that significantly reduced the deacetylation levels of tubulin, that is, compounds 1, 4, 5, 8, 9, and 10.
N.D.—not determined.
N.A.—not active, when the compound showed a percentage of inhibition lower than 10%.
Figure 3.

HDAC in vitro assays. Panel (a): western blot analysis of acetylated histones and tubulin. Panel (b): MTT assay on MCF7 cells upon compound exposure.
After the induction of 24 h at a concentration of 50 μM, compounds 1, 4, 5, 8, 9, and 10 were confirmed to reduce the levels of ac-tubulin deacetylation, which depends on HDAC6, with no or weak effect on deacetylation of the H3K9/14ac histone, which depends on HDAC1 and other targets. These data corroborate the percentages of HDAC6 and HDAC1 enzyme inhibition reported in Table 1. Selected compounds were also used in MCF7 at a lower dose (5 μM), but in this case, the acetylation of tubulin was found to be negligible, suggesting that additional chemical modifications are necessary to ameliorate their chemical/biological properties (e.g., compound stability and cell permeability). Moreover, to evaluate whether, along with promising deacetylating/Hsp90 activities, selected compounds could impair cancer cell proliferation, MTT assay was performed in MCF7 cells (Figure 3, panel b). For 24 and 48 h of treatment, none of the compounds proved to be toxic, although 5 and 8 were shown to reduce breast cancer cell proliferation by about 50%. Altogether, these results suggest that the compounds are likely to exert in vitro antiproliferative effects through the simultaneous modulation of multiple targets. According to the predicted binding modes, the most active compound of the series (compound 10 in Table 1 and Figure 4, panel a) coordinates the zinc ion of the HDAC6 active site with the 5-(trifluoromethyl)-1,2,4-oxadiazole chemical moiety, adopting a conformation similar to that experimentally observed for other HDAC inhibitors based on this scaffold.40 The phenyl ring forms π–π stacking interactions with the Phe620 and Phe680 residues. Moreover, 2,4-diaminotriazine binds near His500, Pro501, and Leu749, extending toward a solvent-exposed area at the top of the HDAC6 cavity (residue numbering refers to 5EDU41). Interestingly, the latter chemical moiety was predicted to form a hydrogen bond network with residues Asp93, Wat2262, and Wat2137 of Hsp90 (Figure 4, panel b), as previously observed for other Hsp90 inhibitors bearing the 2,4-diaminotriazine moiety42 (residue numbering refers to 1UY643). Finally, in Hsp90, the phenyl ring of 10 was predicted to accommodate near residues Leu103, Leu107, and Phe138, while the 5-(trifluoromethyl)-1,2,4-oxadiazole ring extended toward a subpocket lined by residues Phe22, Gly108, Trp162, and Phe170. Although 10 did not show high activity toward Hsp90, the predicted binding modes provided useful structural hints for further hit optimization. Moreover, ligand similarity calculations on ChEMBL29 allowed for the identification of a significant degree of similarity between the selected compounds and known Hsp90 inhibitors (Table S2 in the Supporting Information), supporting that higher Hsp90 activity could be obtained with proper structural modifications of the identified scaffolds, which is currently ongoing.
Figure 4.
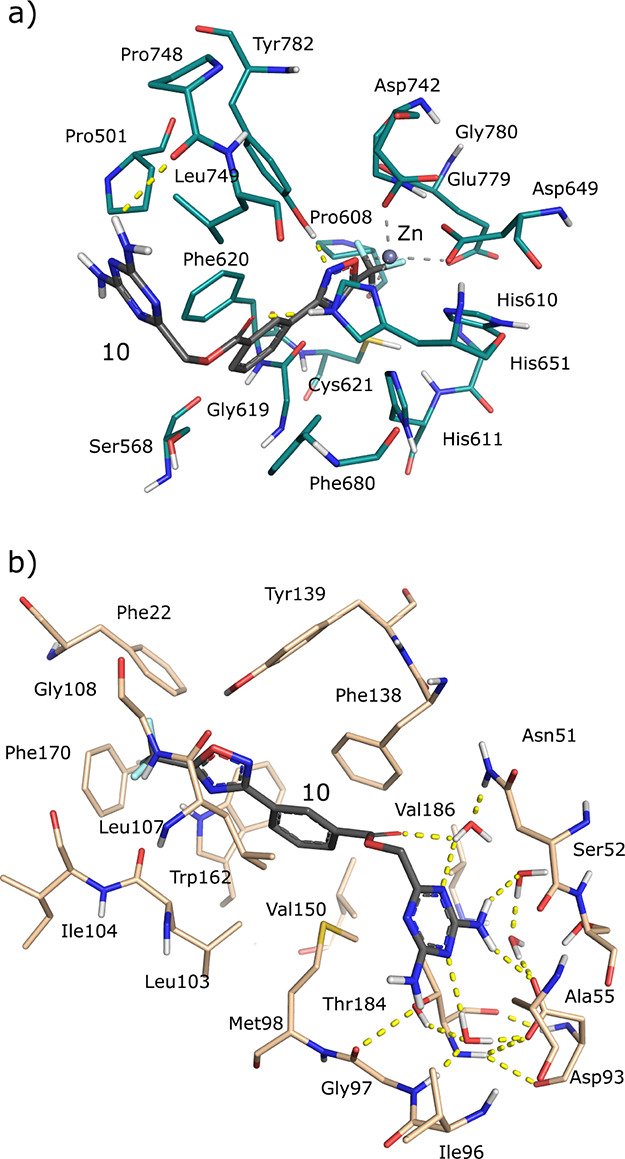
Predicted docking poses of 10 in HDAC6 and Hsp90 binding sites. In particular, panels (a,b) report the predicted binding modes of compound 10 in the 5EDU (HDAC6) and 1UY6 (Hsp90) crystal structures, respectively.
Conclusions
In conclusion, a careful analysis of HDAC6 and Hsp90 ligands reported in ChEMBL was first performed in order to identify proper warheads of these two targets. Then, ligand-based and structure-based screenings led to the selection of 10 compounds, which were evaluated for their activity on purified enzymes and cell lysates.
Notably, HDAC6 and Hsp90 belong to different families and possess very different binding sites, which makes the design of dual inhibitors particularly challenging. Although the identified compounds did not exhibit the desired balanced Hsp90/HDAC6 dual activity, some of them were able to selectively provide an increased level of acetylation of α-tubulin, while showing no effects on histone H3 acetylation. Moreover, two compounds also demonstrated the reduction of breast cancer cell proliferation, according to the in vitro assays. Interestingly, the identified scaffolds are small in size, which makes them valuable starting points for further structure-based optimization, which is currently ongoing.
Experimental Methods
In Silico Analyses
Identification of Hsp90 and HDAC6 Chemical “Warheads”
Hsp90 and HDAC6 inhibitors, whose activity annotations have been reported in terms of IC50, EC50, Ki, or Kd, were first collected from the ChEMBL database (accessed on February 20, 2018).29 Then, the activity records were filtered to retain only those derived by binding experiments on isolated proteins (assay confidence score equal to or higher than 8), leading to the identification of 1342 and 2753 unique compounds for Hsp90 and HDAC6, respectively. Afterward, the ligands were clustered by using the Canvas Similarity and Clustering utility available in Maestro.44,45 In particular, ligand similarity was assessed through 32-bit linear path-based chemical fingerprints, which encodes different atom types and bond orders and discriminates aromatic from nonaromatic groups. Afterward, hierarchical clustering was performed on the Tanimoto similarity matrix that resulted from ligand-based similarities, by using the “average” linkage method. A visual inspection of ligands in the clusters allowed for a final selection of compounds 11 and 8, from which representative substructures (herein referred to as “warheads”) interacting with the hotspots of Hsp90 (Asp93) and HDAC6 (Zn2+ ion) were, respectively, extracted (see Figure 2).
Selection of Representative Crystal Structures of Hsp90 and HDAC6
Crystal structures of human Hsp90 and HDAC6 (catalytic domain II) proteins were first downloaded from the PDB and split into their component chains (accessed on January 30, 2018) leading to 271 and 2 complexes, respectively. Then, the resulting complexes were preprocessed with the Protein Preparation Wizard utility available in Maestro (Schrödinger).44 In particular, atom types and bond connectivity issues were fixed, hydrogen atoms were added, and H-bond networks were optimized. Ions and water and solvent molecules were also removed from the complexes, except for water molecules establishing conserved H-bond networks with the ligand and the residues of the Hsp90 protein binding site.46 Afterward, the prepared crystal structures were aligned by using the protein structure alignment tool available in Maestro44 and manually clustered according to the spatial arrangements of the binding site residues, leading to a set of 11 and 1 clusters of structurally diverse Hsp90 and HDAC6 conformations, respectively. Finally, one ligand–protein complex was selected as a representative structure for each cluster (see Table S1). Redocking calculations using Glide32,47 were subsequently performed on the representative crystal complexes of Hsp90 and HDAC6 to validate the docking models. Ligands extracted from the representative complexes were prepared for the redocking calculations using the LigPrep utility available in the Schrödinger suite.48 In particular, every combination of ionization and tautomeric states potentially present at pH equal to 7 ± 2 were evaluated. Moreover, as HDAC6 contains a Zn ion in the active site, which is necessary for the catalytic activity of the protein, additional metal-binding states were also computed.
The prepared Hsp90 crystallographic ligands were finally redocked into the selected representative HDAC6 and Hsp90 enzymes. Default settings of the Standard Precision protocol were used to perform docking calculations, and receptor grids were centered at the centroids of the co-crystallized ligands. The ability of the models to reproduce the crystallographic poses of the ligands was evaluated in terms of rmsd, which always resulted to be lower than 2.0 Å (Table S1). Moreover, a visual inspection of the docking complexes was also carried out to better evaluate the protein–ligand interactions established in the predicted docking poses.
Virtual Screening
The ZINC31 database (release 15, containing ∼735 mL of purchasable compounds; accessed on February 20, 2018) was first downloaded and filtered using FILTER49,50 software of the OpenEye suite. In particular, ligands with molecular weights ≤200 and ≥650 and predicted poor ADMET properties were removed. Then, the screening compounds were sequentially filtered on the identified HDAC6 and Hsp90 chemical warheads (see Figure 2), which are important for binding to the two proteins. Substructure filtering was performed by using an in-house developed script implemented with OpenEye python toolkits.30 Afterward, compounds that passed the previous stages of filtering were prepared for docking calculations by using LigPrep with the same modalities described above. All combinations of stereoisomers were also generated for molecules with undefined stereocenters, at this stage of ligand preparation.
Docking calculations of the screening ligands were finally performed on Hsp90 and HDAC6 crystal structures, selected according to the similarity between the screened and co-crystallized ligands. In particular, HDAC6 and Hsp90 ligands were first extracted in their bound conformation from the prepared crystal structures and grouped according to the cluster membership of their parent protein conformations. Then, screening compounds were docked into the Hsp90 and HDAC6 representative crystal structures, whose cluster-based crystallographic ligands provided the highest similarity according to the ECFP4 and MACCS fingerprints.51,52 A visual inspection of the predicted binding modes and an analysis of the scores led to a selection of 10 candidate compounds to be tested in vitro on Hsp90- and HDAC6-purified proteins and on cell-based assays.
Ligand-based Similarity Calculations on ChEMBL
To evaluate whether the identified compounds present a high degree of structural similarity with respect to Hsp90 inhibitors reported in the literature, 2D similarity analyses were also carried out. In particular, Hsp90 inhibitors were first downloaded from the ChEMBL database and filtered to retain only those with activity annotations derived by binding experiments on isolated proteins (ChEMBL assay confidence score equal to or greater than 8), and expressed as IC50, EC50, KI, or Kd. Then, 2D similarity analyses were performed by using the MACCS and ECFP4 fingerprints’ implemented OpenEye python toolkits.30,51,52 These types of fingerprints were selected for the similarity estimations among those available, as they take into account different structural aspects of ligand similarity. The 2D fingerprint analyses revealed a notable degree of similarity between the identified molecules (i.e., 4, 5, 8, 9, and 10) and several Hsp90 ligands reported on ChEMBL (see Table S2). Interestingly, the majority of the identified similarity records resulted from the ECFP4fp-based calculations, with the evaluated scores being always higher than activity-relevant thresholds of similarity.53
Experimental Assays
Cell Line
The breast cancer cell line MCF7 (ATCC) was grown in DMEM medium (Euroclone, Milan, Italy) with 10% fetal bovine serum (Euroclone), 2 mM l-glutamine (Euroclone), and antibiotics (100 U/mL penicillin, 100 L g/mL streptomycin) (Euroclone). Cells were stimulated with drugs for indicated times at 5 and 50 μM.
Western Blot Analysis
To quantify the acetylation levels, we performed the total protein and histone extraction by evaluation of the acetylation effects of Ac-tubulin, as the target of HDAC6, and of histone H3K9,14ac, as the off-target.
Total Protein Extraction
After the induction with epidrugs at the indicated times and concentrations, MCF7 cells were then harvested, washed with phosphate-buffered saline (PBS) (Euroclone), and lysed for 15 min at 4 °C in a lysis extraction buffer with protease and phosphatase inhibitors (50 mM Tris–HCl of pH 8.0, 150 mM NaCl, 1% NP40, 10 mM sodium fluoride, 0.1 mM sodium orthovanadate, 40 mg/mL phenylmethylsulfonyl fluoride (PMSF), 20 g/mL aprotinin, 20 mg/mL leupeptin, 2 mg/mL antipain, 10 mM p-nitrophenyl phosphate, 10 mg/mL pepstatin A, and 20 nM okadaic acid). Cells were vortexed, then centrifuged at 13,000 rpm for 30 min at 4 °C. The protein concentration was determined by a colorimetric assay (Biorad, Italy). Cell extracts were 1:1 diluted in the 2× Laemmli sample buffer (0.217 M Tris–HCl with pH 8.0, 52.17% SDS, 17.4% glycerol, 0.026% bromophenol blue, and 8.7% β-mercaptoethanol), and then boiled for 3 min. Equal amounts of protein (50 μg) were run, separated by SDS–PAGE gel, transferred onto nitrocellulose membranes, and incubated with primary antibodies. Primary antibodies used were acetyl-tubulin (Sigma) and anti-GAPDH antibody (Santa Cruz Biotechnology) for loading control. The immune complexes were first detected with horseradish peroxidase-conjugated species-specific secondary antiserum (Bio-Rad Laboratories, Milan, Italy), and then by an enhanced chemiluminescence reaction (Bio-Rad Laboratories, Milan, Italy). Densitometric analysis of protein expression was performed by using the ImageJ image processing package.
Histone Extraction
After the induction with epidrugs at the indicated times and concentrations, MCF7 cells were harvested and washed twice with cold 1× PBS and lysed in Triton extraction buffer (TEB; PBS containing 0.5% Triton X 100 (v/v), 2 mM PMSF, and 0.02% (w/v) NaN3) for 10 min on ice, with gentle stirring. After centrifugation (2000 rpm at 4 °C for 10 min), the supernatant was removed, and the pellet was washed in half the volume of TEB and centrifuged as before. The pellet was overnight incubated in 0.2 N HCl at 4 °C on a rolling table. The samples were then centrifuged at 2000 rpm for 10 min at 4 °C; the concentration of protein in the supernatant was determined using a Bradford assay (Bio-Rad, CA, USA). For the detection of histone H3 acetylation, H3K9,14ac (Diagenode) was used. Histone H4 (Abcam) antibodies and Pounceau Red (Sigma) were used to normalize for equal loading. Semi-quantitative analysis was performed using ImageJ software.
Cell Viability Assay
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma-Aldrich, Milan, Italy) assay was used to determine the proliferation of MCF7 cells after treatment with epidrugs at indicated concentrations. 7 × 104 cells/well were plated in a 24-well plate and treated, in triplicates, with compounds at 50 and 5 μg for 24 and 48 hours of induction. After treatment, MTT solution was added for 3 h at 0.5 mg/mL, the purple formazan crystals were dissolved in dimethyl sulfoxide (Sigma-Aldrich, Milan, Italy), and the absorbance was read at a wavelength of 570 nm using a TECAN M-200 reader.
Fluorimetric HDAC Assays
HDAC1 and HDAC6 recombinant enzymes (10 ng per reaction; BPS Bioscience Catalog #: 50051 and 50006, respectively) were incubated in the HDAC buffer (50 mM Tris–HCl of pH 8.0, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2) with specific substrates (50 μM) and epidrugs, at concentration of 5 and 50 μM. As a HDAC1 substrate, the derivative of fluorescent tert-butyloxycarbonyl (Boc)-(Ac)-Lys-7-amino-4-methylcoumarin (AMC), called MAL, the benzyloxycarbonyl analogue Z-MAL was used. For HDAC6, the substrate was instead an (S)-[5-acetylamino-1-(2-oxo-4-trifluoromethyl-2H-chromen-7-ylcarbamoyl)pentyl]carbamic acid tert-butyl ester. Reactions were carried out for 1 h at 37 °C in black microplates (Corning Costar, cod: 266). Deacetylation sensitizes the substrate to the developer step [trypsin 6 mg/mL in trypsin buffer (50 mM Tris–HCl of pH 8.0 and 100 mM NaCl)], and it is directly proportional to the produced fluorophore. The fluorophore is excited with a 360 nm light, and the emitted light (460 nm) has been quantified with a TECAN Infinite M200 station. SAHA (5 μM) was used as the deacetylation control for HDAC1 assay and Tubastatin (5 μM) for HDAC6.
HSP90 Assay
Compounds that reduced the levels of ac-tubulin deacetylation, that is, 1, 4, 5, 8, 9, and 10 have also been tested in vitro to evaluate their Hsp90α inhibitory activity, in singlicate. The employed Hsp90α assay kit is designed for the identification of Hsp90α inhibitors using fluorescence polarization. The assay is a competitive binding assay, based on the binding of fluorescently labeled geldanamycin, an HSP90 inhibitor, to purified recombinant Hsp90α. The assay was carried out as suggested by the manufacturer (Hsp90α N-Terminal Domain Assay Kit, BPS Bioscience Catalog #50293). Epidrugs were used at the concentrations of 50 and 5 μM. 17-AAG (KOS953, APExBIO Catalog #: A4054) was used as a reference compound. Fluorescent polarizations of samples were performed at the intervals of 475–495 nm (excitation) and 518–538 nm (emission) in low-binding black microplates (Corning Costar, cod: 266).
Acknowledgments
We thank OpenEye Scientific Software, Inc., for the free academic license of the OpenEye Toolkit.
Glossary
Abbreviations
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- FDA
U.S. Food and Drug Administration
- HDAC
Histone deacetylase
- Hsp90
heat shock protein 90
- MCF7
Michigan Cancer Foundation-7
- PDB
protein data bank
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00559.
Table S1: HDAC6 and Hsp90 crystal structures employed in the study; Table S2: Hsp90 ChEMBL ligands that are structurally similar to the investigated compounds (PDF)
Author Contributions
L.P. and R.B. contributed equally to the study. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. L.P. performed the computational analyses; R.B. performed and supervised the biological assays; L.A. supervised the biological assays; and G.R. conceived and coordinated the study.
This work was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro [AIRC IG 23635]. Moreover, this research was also funded by Blueprint 282510; MIUR20152TE5PK; EPICHEMBIO CM1406; EPIGEN-MIUR-CNR; AIRC-17217; VALERE: Vanvitelli per la Ricerca; Campania Regional Government Technology Platform Lotta alle Patologie Oncologiche: iCURE; Campania Regional Government FASE2: IDEAL. MIUR, Proof of Concept POC01_00043.
The authors declare no competing financial interest.
Supplementary Material
References
- Iyengar R. Complex Diseases Require Complex Therapies. EMBO Rep. 2013, 14, 1039–1042. 10.1038/embor.2013.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Suraweera A.; O’Byrne K. J.; Richard D. J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Radiat. Oncol. 2018, 8, 92. 10.3389/fonc.2018.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- www.clinicaltrials.gov (accessed March 3, 2019).
- Kovacs J. J.; Murphy P. J. M.; Gaillard S.; Zhao X.; Wu J.-T.; Nicchitta C. V.; Yoshida M.; Toft D. O.; Pratt W. B.; Yao T.-P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607. 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Murphy P. J. M.; Morishima Y.; Kovacs J. J.; Yao T.-P.; Pratt W. B. Regulation of the Dynamics of Hsp90 Action on the Glucocorticoid Receptor by Acetylation/Deacetylation of the Chaperone. J. Biol. Chem. 2005, 280, 33792–33799. 10.1074/jbc.m506997200. [DOI] [PubMed] [Google Scholar]
- Kovacs J. J.; Cohen T. J.; Yao T.-P. Chaperoning Steroid Hormone Signaling via Reversible Acetylation. Nucl. Recept. Signal. 2005, 3, nrs.03004. 10.1621/nrs.03004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wang S.-Y.; Zhang X.-H.; Zhao M.; Hou C.-M.; Xu Y.-J.; Du Z.-Y.; Yu X.-D. FK228 Inhibits Hsp90 Chaperone Function in K562 Cells via Hyperacetylation of Hsp70. Biochem. Biophys. Res. Commun. 2007, 356, 998–1003. 10.1016/j.bbrc.2007.03.076. [DOI] [PubMed] [Google Scholar]
- Ojha R.; Huang H.-L.; HuangFu W.-C.; Wu Y.-W.; Nepali K.; Lai M.-J.; Su C.-J.; Sung T.-Y.; Chen Y.-L.; Pan S.-L.; et al. 1-Aroylindoline-Hydroxamic Acids as Anticancer Agents, Inhibitors of HSP90 and HDAC. Eur. J. Med. Chem. 2018, 150, 667–677. 10.1016/j.ejmech.2018.03.006. [DOI] [PubMed] [Google Scholar]
- Mehndiratta S.; Lin M.-H.; Wu Y.-W.; Chen C.-H.; Wu T.-Y.; Chuang K.-H.; Chao M.-W.; Chen Y.-Y.; Pan S.-L.; Chen M.-C.; et al. N-Alkyl-Hydroxybenzoyl Anilide Hydroxamates as Dual Inhibitors of HDAC and HSP90, Downregulating IFN-γ Induced PD-L1 Expression. Eur. J. Med. Chem. 2020, 185, 111725. 10.1016/j.ejmech.2019.111725. [DOI] [PubMed] [Google Scholar]
- Bali P.; Pranpat M.; Bradner J.; Balasis M.; Fiskus W.; Guo F.; Rocha K.; Kumaraswamy S.; Boyapalle S.; Atadja P.; et al. Inhibition of Histone Deacetylase 6 Acetylates and Disrupts the Chaperone Function of Heat Shock Protein 90: A Novel Basis for Antileukemia Activity of Histone Deacetylase Inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. 10.1074/jbc.c500186200. [DOI] [PubMed] [Google Scholar]
- Wu J.; Liu T.; Rios Z.; Mei Q.; Lin X.; Cao S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. 10.1016/j.tips.2016.11.009. [DOI] [PubMed] [Google Scholar]
- Zarour L.; Alumkal J. Emerging Therapies in Castrate-Resistant Prostate Cancer. Curr. Urol. Rep. 2010, 11, 152–158. 10.1007/s11934-010-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molife L. R.; Attard G.; Fong P. C.; Karavasilis V.; Reid A. H. M.; Patterson S.; Riggs C. E.; Higano C.; Stadler W. M.; McCulloch W.; et al. Phase II, Two-Stage, Single-Arm Trial of the Histone Deacetylase Inhibitor (HDACi) Romidepsin in Metastatic Castration-Resistant Prostate Cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- Aoyagi S.; Archer T. K. Modulating Molecular Chaperone Hsp90 Functions through Reversible Acetylation. Trends Cell Biol. 2005, 15, 565–567. 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Aldana-Masangkay G. I.; Sakamoto K. M. The Role of HDAC6 in Cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. 10.1155/2011/875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A.; Su L.; Campbell B.; Poulin N. M.; Nielsen T. O. Synergism of Heat Shock Protein 90 and Histone Deacetylase Inhibitors in Synovial Sarcoma. Sarcoma 2009, 2009, 794901. 10.1155/2009/794901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R.; Fiskus W.; Yang Y.; Lee P.; Joshi R.; Fernandez P.; Mandawat A.; Atadja P.; Bradner J. E.; Bhalla K. HDAC6 Inhibition Enhances 17-AAG--Mediated Abrogation of Hsp90 Chaperone Function in Human Leukemia Cells. Blood 2008, 112, 1886–1893. 10.1182/blood-2008-03-143644. [DOI] [PubMed] [Google Scholar]
- George P.; Bali P.; Annavarapu S.; Scuto A.; Fiskus W.; Guo F.; Sigua C.; Sondarva G.; Moscinski L.; Atadja P.; et al. Combination of the Histone Deacetylase Inhibitor LBH589 and the Hsp90 Inhibitor 17-AAG Is Highly Active against Human CML-BC Cells and AML Cells with Activating Mutation of FLT-3. Blood 2005, 105, 1768–1776. 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- Rahmani M.; Yu C.; Dai Y.; Reese E.; Ahmed W.; Dent P.; Grant S. . Coadministration of the Heat Shock Protein 90 Antagonist 17-Allylamino- 17-Demethoxygeldanamycin with Suberoylanilide Hydroxamic Acid or Sodium Butyrate Synergistically Induces Apoptosis in Human Leukemia Cells. Cancer Res. 2003, 63, 8420–8427. [PubMed] [Google Scholar]
- Rahmani M.; Reese E.; Dai Y.; Bauer C.; Kramer L. B.; Huang M.; Jove R.; Dent P.; Grant S. Cotreatment with Suberanoylanilide Hydroxamic Acid and 17-Allylamino 17-Demethoxygeldanamycin Synergistically Induces Apoptosis in Bcr-Abl+ Cells Sensitive and Resistant to STI571 (Imatinib Mesylate) in Association with down-Regulation of Bcr-Abl, Abrogation of Signal Transducer and Activator of Transcription 5 Activity, and Bax Conformational Change. Mol. Pharmacol. 2005, 67, 1166–1176. 10.1124/mol.104.007831. [DOI] [PubMed] [Google Scholar]
- Nimmanapalli R.; Fuino L.; Bali P.; Gasparetto M.; Glozak M.; Tao J.; Moscinski L.; Smith C.; Wu J.; Jove R.; et al. Histone Deacetylase Inhibitor LAQ824 Both Lowers Expression and Promotes Proteasomal Degradation of Bcr-Abl and Induces Apoptosis of Imatinib Mesylate-Sensitive or -Refractory Chronic Myelogenous Leukemia-Blast Crisis Cells. Cancer Res. 2003, 63, 5126–5135. [PubMed] [Google Scholar]
- Kaiser M.; Lamottke B.; Mieth M.; Jensen M. R.; Quadt C.; Garcia-Echeverria C.; Atadja P.; Heider U.; von Metzler I.; Türkmen S.; et al. Synergistic Action of the Novel HSP90 Inhibitor NVP-AUY922 with Histone Deacetylase Inhibitors, Melphalan, or Doxorubicin in Multiple Myeloma. Eur. J. Haematol. 2010, 84, 337–344. 10.1111/j.1600-0609.2009.01403.x. [DOI] [PubMed] [Google Scholar]
- Lamottke B.; Kaiser M.; Mieth M.; Heider U.; Gao Z.; Nikolova Z.; Jensen M. R.; Sterz J.; von Metzler I.; Sezer O. The Novel, Orally Bioavailable HSP90 Inhibitor NVP-HSP990 Induces Cell Cycle Arrest and Apoptosis in Multiple Myeloma Cells and Acts Synergistically with Melphalan by Increased Cleavage of Caspases. Eur. J. Haematol. 2012, 88, 406–415. 10.1111/j.1600-0609.2012.01764.x. [DOI] [PubMed] [Google Scholar]
- Chai R. C.; Vieusseux J. L.; Lang B. J.; Nguyen C. H.; Kouspou M. M.; Britt K. L.; Price J. T. Histone Deacetylase Activity Mediates Acquired Resistance towards Structurally Diverse HSP90 Inhibitors. Mol. Oncol. 2017, 11, 567–583. 10.1002/1878-0261.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F.; Park Y. M.; Lee J.; Buso N.; Gur T.; Madhusoodanan N.; Basutkar P.; Tivey A. R. N.; Potter S. C.; Finn R. D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf F. H.; Biebl M. M.; Buchner J. The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- Micelli C.; Rastelli G. Histone Deacetylases: Structural Determinants of Inhibitor Selectivity. Drug Discov. Today 2015, 20, 718–735. 10.1016/j.drudis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OpenEye Toolkits 2019. Oct 2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- Sterling T.; Irwin J. J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Caporuscio F.; Rastelli G. Exploiting Computationally Derived Out-of-the-Box Protein Conformations for Drug Design. Future Med. Chem. 2016, 8, 1887–1897. 10.4155/fmc-2016-0098. [DOI] [PubMed] [Google Scholar]
- Pinzi L.; Caporuscio F.; Rastelli G. Selection of protein conformations for structure-based polypharmacology studies. Drug Discov. Today 2018, 23, 1889–1896. 10.1016/j.drudis.2018.08.007. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzi L.; Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. 10.3390/ijms20184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.ebi.ac.uk/chembl/doc/inspect/CHEMBL3638656, (accessed Nov 29, 2019).
- Yoon S.; Eom G. H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam J. Med. Sci. 2016, 52, 1–11. 10.4068/cmj.2016.52.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P. A.; Xu W.-S. Histone Deacetylase Inhibitors: Potential in Cancer Therapy. J. Cell. Biochem. 2009, 107, 600–608. 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobera M.; Madauss K. P.; Pohlhaus D. T.; Wright Q. G.; Trocha M.; Schmidt D. R.; Baloglu E.; Trump R. P.; Head M. S.; Hofmann G. A.; et al. Selective Class IIa Histone Deacetylase Inhibition via a Nonchelating Zinc-Binding Group. Nat. Chem. Biol. 2013, 9, 319–325. 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- Hai Y.; Christianson D. W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth J. R.; Park C.; Petros A. M.; Kunzer A. R.; Wendt M. D.; Wang X.; Lynch C. L.; Mack J. C.; Swift K. M.; Judge R. A.; et al. Discovery and Design of Novel HSP90 Inhibitors Using Multiple Fragment-based Design Strategies. Chem. Biol. Drug Des. 2007, 70, 1–12. 10.1111/j.1747-0285.2007.00535.x. [DOI] [PubMed] [Google Scholar]
- Wright L.; Barril X.; Dymock B.; Sheridan L.; Surgenor A.; Beswick M.; Drysdale M.; Collier A.; Massey A.; Davies N.; et al. Structure-Activity Relationships in Purine-Based Inhibitor Binding to HSP90 Isoforms. Chem. Biol. 2004, 11, 775–785. 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2018-3: Maestro; Schrödinger, LLC: New York, NY, 2018.
- Schrödinger Release 2018-3: Canvas; Schrödinger, LLC: New York, NY, 2018.
- Sgobba M.; Rastelli G. Structure-Based and in Silico Design of Hsp90 Inhibitors. ChemMedChem 2009, 4, 1399–1409. 10.1002/cmdc.200900256. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release 2018-3: Glide; Schrödinger, LLC: New York, NY, 2018.
- Schrödinger Release 2018-3: LigPrep; Schrödinger, LLC: New York, NY, 2018.
- OMEGA 3.1.1.2; OpenEye Scientific Software: Santa Fe, NM. http://www.eyesopen.com.
- Hawkins P. C. D.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Data Dank and the Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Model. 2002, 42, 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Jasial S.; Hu Y.; Vogt M.; Bajorath J. Activity-Relevant Similarity Values for Fingerprints and Implications for Similarity Searching. F1000Research 2016, 5, 591. 10.12688/f1000research.8357.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.