

SUMMARY
Neurons and oligodendrocytes communicate to regulate oligodendrocyte development and ensure appropriate axonal myelination. Here, we show that Glycerophosphodiester phosphodiesterase 2 (GDE2) signaling underlies a neuronal pathway that promotes oligodendrocyte maturation through the release of soluble neuronally derived factors. Mice lacking global or neuronal GDE2 expression have reduced mature oligodendrocytes and myelin proteins but retain normal numbers of oligodendrocyte precursor cells (OPCs). Wild-type (WT) OPCs cultured in conditioned medium (CM) from Gde2-null (Gde2KO) neurons exhibit delayed maturation, recapitulating in vivo phenotypes. Gde2KO neurons show robust reduction in canonical Wnt signaling, and genetic activation of Wnt signaling in Gde2KO neurons rescues in vivo and in vitro oligodendrocyte maturation. Phosphacan, a known stimulant of oligodendrocyte maturation, is reduced in CM from Gde2KO neurons but is restored when Wnt signaling is activated. These studies identify GDE2 control of Wnt signaling as a neuronal pathway that signals to oligodendroglia to promote oligodendrocyte maturation.
Graphical Abstract
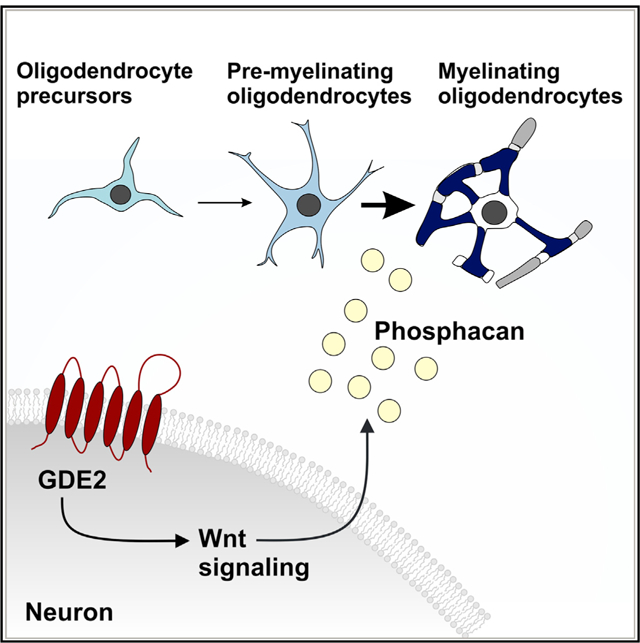
In Brief
Communication between neurons and oligodendroglial cells regulates oligodendrocyte development. Here, Choi et al. show that the six-transmembrane GPI-anchor-cleaving enzyme GDE2 stimulates canonical Wnt signaling in neurons to release soluble factors, such as phosphacan, to promote oligodendrocyte maturation.
INTRODUCTION
Oligodendrocytes (OLs) are important regulators of neural circuit function. OLs produce myelin, a lipid-rich extension of their plasma membrane that wraps axons and facilitates the fast, saltatory conduction of action potentials. In addition, OLs serve as a source of metabolic support for neurons that help promote neuronal health and survival (Nave, 2010). The remarkable match between the number of myelinating OLs and axons that require myelination (Davison and Peters, 1970) suggests that communication between axons and OL lineage cells is involved in coordinating OL proliferation, survival, and maturation. However, neuronal pathways that control the timing of OL maturation are not well understood.
OLs in the brain are generated from three major waves of OL precursor cell (OPC) production that originate first subcortically and then cortically (Kessaris et al., 2006). OPCs exhibit regional diversity in terms of their proliferative, migratory, and remyelination properties (Lentferink et al., 2018; Power et al., 2002; Spitzer et al., 2019). However, genetic ablation studies indicate that ventrally and dorsally derived OPC populations are functionally redundant (Kessaris et al., 2006); thus, the physiological basis of OPC diversity remains unclear. OPCs cultured in vitro can proliferate and differentiate into myelinating OLs in the absence of neurons (Barres et al., 1993); nevertheless, neurons in vivo appear to play important roles in coordinating multiple aspects of OL development. Nerve transection or silencing of neuronal activity shows profound loss of OPC proliferation, survival, and myelination (Barres and Raff, 1993; Ueda et al., 1999), and roles for experience, learning, and environmental factors are emerging as important contributors to myelination in development and in adulthood (Gibson et al., 2014; Makinodan et al., 2012; Mayoral and Chan, 2016).
What are the mechanisms by which neurons regulate OL differentiation and myelination? OPCs that make stable contact with axons differentiate into myelinating OLs, and this is mediated by surface-localized receptors and adhesion molecules that converge to stimulate activity of the non-receptor Srcfamily tyrosine kinase Fyn in OPCs (Umemori et al., 1994). Interestingly, many contact-mediated cues appear to inhibit OL differentiation, presumably to ensure the appropriate timing of axonal myelination during development. For example, polysialylated neuronal cell adhesion molecule (PSA-NCAM) inhibits OPC differentiation and is downregulated to coincide with myelination (Charles et al., 2000), as is the canonical Notch ligand Jagged, which is expressed on axons and binds the Notch receptor on OPCs to inhibit OL differentiation (Wang et al., 1998). The finding that OLs cultured with inert polystyrene fibers exhibit a size-dependent ensheathment of 0.4 μm fibers or more suggests that axonal caliber also contributes to OL myelination (Lee et al., 2012). Of note, both myelinated and unmyelinated axons range in diameter from 0.2 to 0.8 μm in vivo (Remahl and Hildebrand, 1982), suggesting the existence of repulsive and instructive axonal cues that integrate axonal caliber with OL developmental mechanisms. One such cue is likely to involve Akt-mTOR signaling, as activation of this pathway increases the caliber of normally unmyelinated cerebellar axons and expands OPC progenitors and production of myelinating OLs (Goebbels et al., 2016). Another major factor that influences OL proliferation, differentiation, and maturation is neuronal activity. Neuronal activity releases adenosine and glutamate, which regulates the proliferation and differentiation of OPCs into myelinating OLs (Stevens et al., 2002; Yuan et al., 1998). ATP released by electrically active neurons can stimulate astrocytes to produce leukemia inhibitory factor (LIF), which promotes OL differentiation (Ishibashi et al., 2006). Thus, contact-mediated signals, axon caliber, and neuronal activity are important for OL development. Other neuronally derived pathways that regulate OL differentiation and maturation are not well defined.
Glycerophosphodiester phosphodiesterase 2 (GDE2 or GDPD5) is a six-transmembrane protein that contains an external enzymatic domain that is homologous to bacterial glycerophosphodiester phosphodiesterases (GDPDs) (Rao and Sockanathan, 2005). GDE2 and its family members GDE3 and GDE6 are the only known enzymes in vertebrates that regulate the function of glycosylphosphatidylinositol (GPI)-anchored proteins on the plasma membrane through cleavage at the GPI-anchor (Park et al., 2013). During embryonic development, GDE2 regulates the timing of cortical and spinal motor neuron differentiation to promote late-born neuronal subtypes by downregulating Notch signaling (Rodriguez et al., 2012). In developing spinal motor neurons, GDE2 downregulates Notch activation by releasing the GPI-anchored Notch activator reversion-inducing cysteine-rich protein with Kazal motifs (RECK) from motor neuron surfaces (Park et al., 2013). GDE2 GPI-anchor cleavage activity is also implicated in promoting neuroblastoma differentiation, in this case through release of the heparan sulfate proteoglycan GPC6 (Matas-Rico et al., 2016). In addition, GDE2 is required for motor neuron survival, and genetic studies indicate that these functions are distinct from its role in embryonic development (Cave et al., 2017).
We show here that GDE2 functions in neurons to regulate the timing of OL development. GDE2 is required to maintain canonical Wnt signaling in neurons, and this pathway is responsible for the release of soluble factors such as phosphacan that promote OL maturation. These studies identify a neuronal mechanism that controls OL differentiation and maturation and reveals roles for soluble, neuronally derived factors in regulating the production of myelinating OLs.
RESULTS
Gde2 Is Primarily Expressed in Neurons in the Postnatal Brain
Fluorescence in situ hybridization (FISH) detects Gde2 transcripts in the hippocampus, thalamus, caudoputamen, cortex (CTX), and medial habenula at postnatal day 11 (P11) mouse brain (Figure 1A). Gde2 mRNA is initially expressed in deep cortical layers V and VI but expands to upper cortical layers at later stages (Figures 1B, 1C, and S1C). Western blot confirms GDE2 protein expression in postnatal cortices, with increasing GDE2 expression from P7 to P14 and continued expression at 1 and 2 months of age (Figures S1A–S1C). FISH combined with immunohistochemical detection of neuronal (NeuN) markers detects Gde2 transcript expression in neurons at P11 with ~98% of NeuN+ neurons expressing Gde2 mRNA (Figures 1B and 1D). Western blot of protein extracts from cultured cortical neurons confirms neuronal expression of GDE2 protein (Figure S1D). Gde2 transcripts are also detected in ~20% of Olig2+ oligodendroglial cells, but levels of Gde2 transcript expression in Olig2+ cells are markedly lower than in neurons (Figure 1B and 1D). Quantitative PCR (qPCR) from cultured OPCs isolated from P6 cortices reveal minimal Gde2 expression in proliferating OPCs; however, differentiated OLs express both Gde2 transcripts and GDE2 protein (Figures S1E and S1F). Thus, Gde2 is predominantly expressed in neurons during early postnatal development, with lower levels of expression in a subset of OLs.
Figure 1. Gde2 Is Expressed in Neurons and Oligodendrocytes (OLs) in Postnatal Brain.
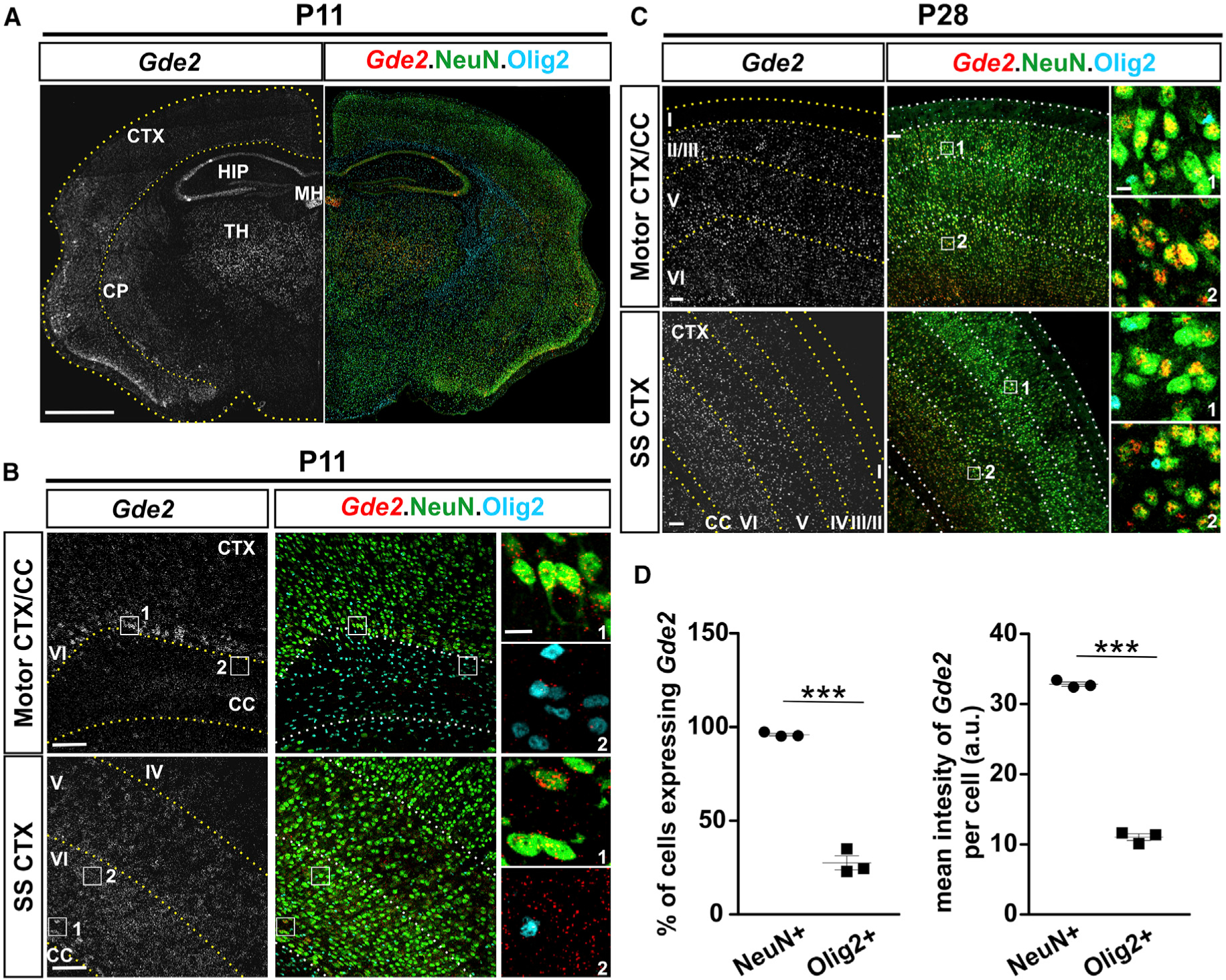
(A–C) FISH of Gde2 mRNA in coronal cortical sections in different brain areas and postnatal stages. CTX, cortex; SS, somatosensory; CC, corpus callosum; HIP, hippocampus; TH, thalamus; CP, caudoputamen; MH, medial habenula. Hatched lines mark cortical layers. Boxes 1 and 2 in (B) and (C) are magnified in the corresponding panels. (D) Graphs quantifying Gde2 mRNA expression. a.u., arbitrary units. ***p < 0.0001. n = 3 WT, n = 3 Gde2KO. Data are presented as mean ± SEM; two-tailed unpaired Student’s t test. Scale bars represent 1,000 μm (A), 100 μm (B and C), and 10 μm (insets, B and C).
GDE2 Ablation Delays OL Maturation
Mice genetically ablated for GDE2 (Gde2KO) show delayed production of deep-layer neurons and increased production of superficial cortical neurons during embryonic development (Rodriguez et al., 2012). We examined Gde2KO animals at P11–P15 when neuronal migration is complete and detected no discernible differences in cortical lamination, neuronal numbers, or morphology in Gde2KO animals compared with wild-type (WT) littermates, suggesting that early perturbations in neurogenesis have normalized by this time point (Figure S2A). The period of increased GDE2 expression in mouse CTX (P7–P14; Figure S1A) coincides with the period of OL differentiation and maturation (Trapp et al., 1997). Further, the spatiotemporal expression of GDE2 correlates with the pattern of cortical OL maturation and myelination, which initiates in deep cortical layers and extends to superficial laminae (Tomassy et al., 2014) (Figures 1A–1C). To determine if GDE2 regulates OL maturation, we examined OL development in Gde2KO animals at P7 and at P11, focusing specifically on the corpus callosum (CC) and adjacent motor and retrosplenial CTX. OPCs that are actively proliferating are identified by coexpression of the OL lineage determinants Olig2 and Sox10 and the proliferation marker Ki67 (Kuhlbrodt et al., 1998; Zhou et al., 2000) (Figures S2B and S2C). Quantification of Ki67/SOX10/Olig2+ OPCs showed equivalent numbers of proliferating OPCs in WT and Gde2KO CC and CTX at P7, suggesting that loss of GDE2 does not affect OPC production (Figure S2D). OPCs stop dividing and differentiate into premyelinating and myelinating OLs, which express CC1, and myelin basic protein (Mbp) transcripts (Bhat et al., 1996; Dugas et al., 2006) (Figure S2B). At P11, overall numbers of OL lineage cells (Olig2+) in CC and CTX were equivalent between Gde2KO animals and WT controls (Figure 2A and 2B). However, Gde2KO animals exhibited a 30% reduction of Olig2+ CC1+ cells and decreased Mbp expression in CC and CTX compared with WT (Figure 2A and 2B). Further, cells that expressed Mbp in Gde2KO animals had consistently less elaborations than their WT counterparts (Figure 2A). The number of CC1+ cells in Gde2KO cortices was reduced in rostral, medial, and caudal regions, indicating a requirement for GDE2 in CC1+ OL generation across the rostral-caudal axis (Figure S2G). Notably, no changes in the number of immature, newly differentiating OLs (TCF4+/CC1−) were found between WT and Gde2KO brain at these stages (Figures S2E and S2F). These observations suggest that GDE2 is not required for the generation or initiation of OPC differentiation but is instead required for OL maturation. In support of this notion, the number of mature myelinating OLs, identified by expression of aspartocylase (ASPA) protein (Madhavarao et al., 2004), was markedly reduced in P15 Gde2KO mice compared with WT littermates (Figures S3A and S3B). Further, western blot of P14 cortical extracts revealed robust reduction of myelin proteins MBP and myelin OL glycoprotein (MOG) (Solly et al., 1996) in Gde2KO condition but equivalent levels of Olig2 and platelet-derived growth factor receptor alpha (PDGFRα) (Figures 2C and 2D).
Figure 2. GDE2 Ablation Impairs OL Maturation.
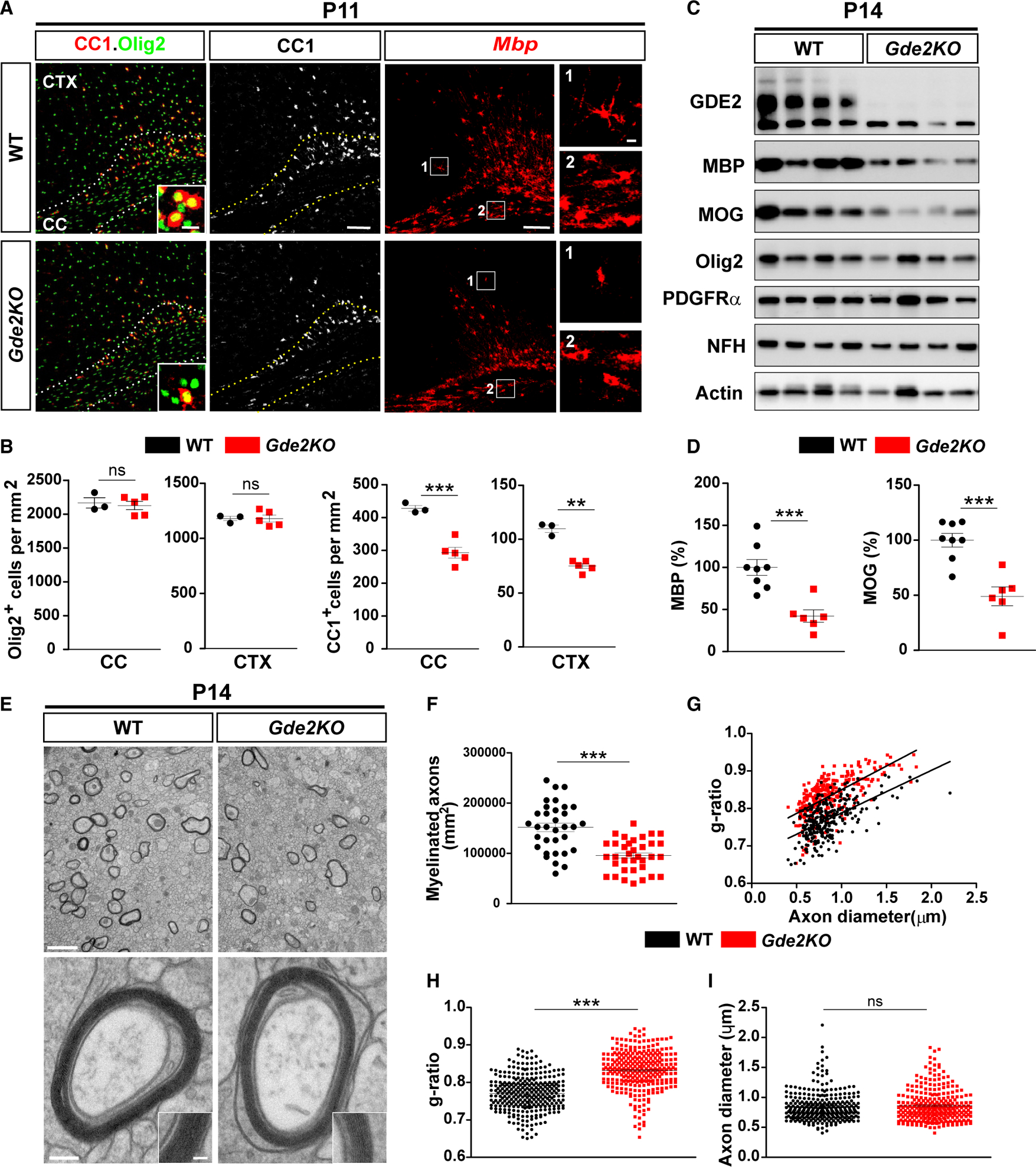
(A) Coronal sections of motor CTX and CC. Hatched lines mark the CC. Insets and boxed areas show high magnification in the corresponding panels. (B) Graphs quantifying Olig2+ and CC1+ cells in CC and CTX. Nonsignificant (ns), p > 0.05; ***p = 0.0007; **p = 0.0035. n = 3 WT, n = 5 Gde2KO. (C) Western blot of cortical extracts. Actin was used as a loading control (Olig2, p = 0.1745; PDGFRα, p = 0.5163). (D) Graphs quantifying western blots for MBP (***p = 0.0005) and MOG (***p = 0.0009). n = 8 WT, n = 6 Gde2KO. (E) TEM of CC. (F–I) Graphs quantifying myelinated axons (F) (***p < 0.0001, points represent individual regions of interest [ROIs]), g-ratios (G and H) (***p < 0.0001, points represent individual myelinated axons), and axon diameter (I) (ns, p = 0.5523). n = 3 WT, 3 Gde2KO. All graphs show mean ± SEM, two-tailed unpaired Student’s t test. Scale bars represent 100 μm (A) (insets, 5 μm), 2 μm (E, top), and 100 nm (E, bottom) (inset, 50 nm).
Electron microscopy (EM) of P14 cortices showed that Gde2KO animals had fewer numbers of myelinated axons compared with WT at P14 (Figures 2E and 2F). Notably, axons that were myelinated had increased g-ratio (ratio of axonal diameter to outer diameter) indicative of hypomyelination (Figures 2G and 2H). Axonal diameters between Gde2KO animals and WT littermates were comparable (Figure 2I), suggesting that the decrease in myelin thickness observed in Gde2KO animals is not a consequence of altered axon caliber. These collective observations suggest that GDE2 is required for promoting OL maturation during the peak period of developmental myelination in postnatal brain. By P28, numbers of ASPA+ myelinating OLs and levels of MBP and MOG proteins in Gde2KO animals had normalized to WT amounts, suggesting that the loss of GDE2 results in delayed OL maturation (Figures S3C–S3E). However, EM analysis of 10-week animals showed that in contrast to WT, there was a dramatic reduction in myelinated larger-diameter axons in Gde2KO animals, although the incidence and myelination of smaller-diameter axons were normal (Figure S3F). This observation suggests that the temporal control of OL maturation by GDE2 is necessary for appropriate axonal myelination.
Neuronal GDE2 Promotes OL Maturation
GDE2 is predominantly expressed in neurons, suggesting that GDE2 acts non-cell autonomously to regulate the timing of OL maturation. To test this hypothesis, we used Cre-lox genetics to ablate GDE2 function in neurons. Nex-Cre mice express Cre recombinase under the control of the endogenous promoter of the NEX transcription factor, which targets Cre expression in cortical excitatory pyramidal neurons and hippocampus, but not in proliferating neural progenitors, interneurons, OLs, or astrocytes (Goebbels et al., 2006). Thus, Gde2-lox/−;Nex-Cre mice (N-Gde2KO) will lack GDE2 expression and function in pyramidal neurons but retain GDE2 OL function. Western blot of P14 cortical extracts shows that GDE2 expression is reduced by 80% in N-Gde2KO condition compared with Gde2+/−; Nex-Cre controls (Ctrl) (Figures 3E and 3F). This confirms efficient ablation of GDE2 expression and supports our earlier observation that GDE2 expression is predominantly neuronal (Figure 1). N-Gde2KO showed no differences in the number of NG2/Olig2 cells, confirming that neuronal GDE2 does not influence OPC production (Figures 3A and 3B). However, N-Gde2KO animals showed a 15% reduction in Olig2+ CC1+ OLs in the CC and a more marked 30% reduction of Olig2+ CC1+ OLs in the CTX compared with controls (Figures 3C and 3D). In addition, western blot of P14 cortical extracts showed robust reduction of MBP and MOG in N-Gde2KO mice compared with control littermates (Figures 3E and 3F). Both genotypes showed equivalent levels of Olig2 and PDGFRα, which are expressed primarily in oligodendroglial cells and OPCs respectively, suggesting that overall numbers of oligodendroglia are not disrupted in N-Gde2KO animals (Figure 3E). Moreover, the amounts of axonal Neurofilament Heavy Chain (NFH) protein is similar between N-Gde2KO and control animals confirming earlier observations that cortical neuronal numbers and lamination are grossly intact in both cases (Figure 3E). Taken together, our observations in N-Gde2KO brain recapitulate the OL phenotypes of Gde2KO animals and provide genetic evidence that GDE2 neuronal function is required to promote OL maturation.
Figure 3. Neuronal GDE2 Promotes OL Maturation.
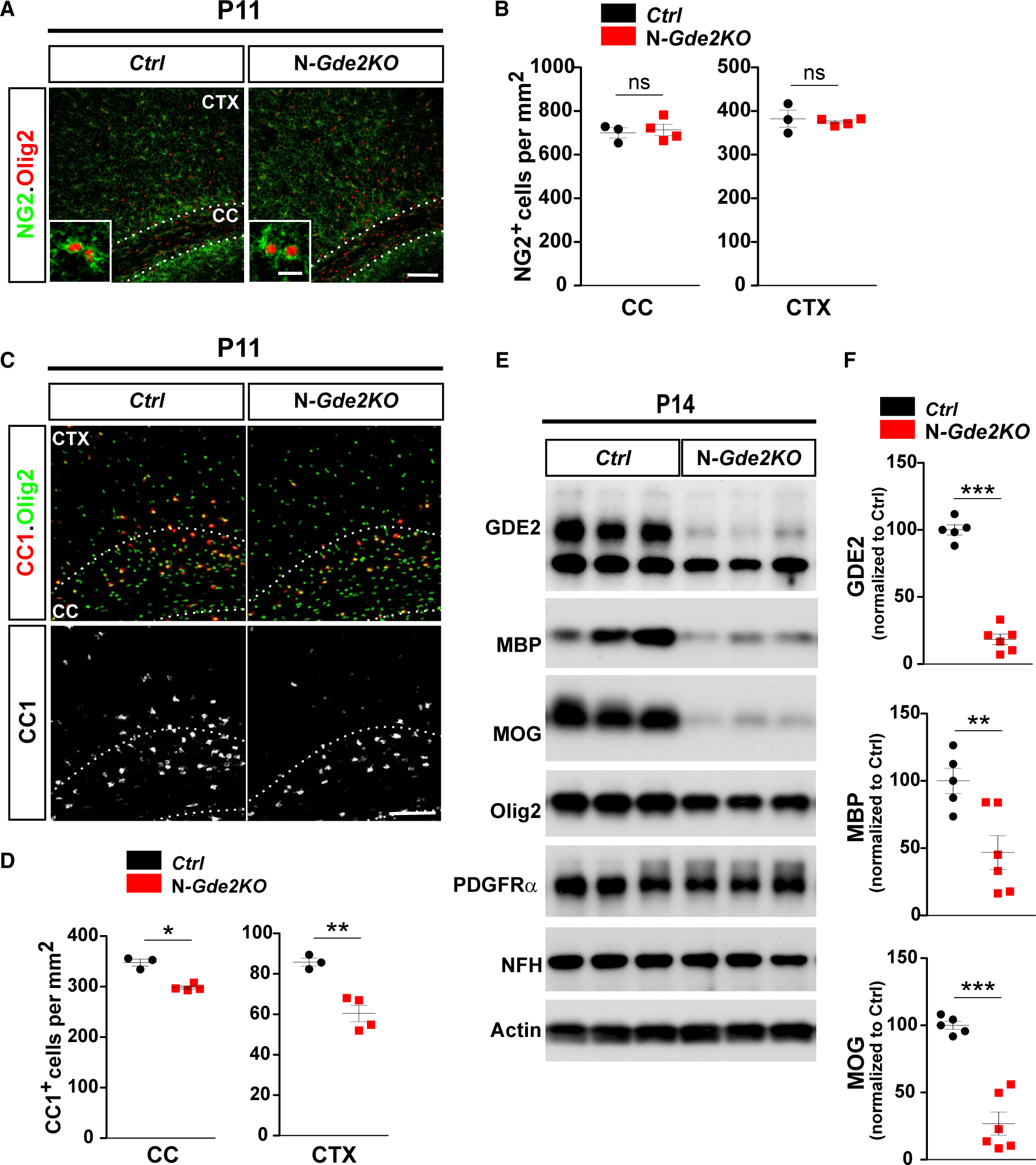
(A and C) Coronal sections of motor CTX and CC showing NG2 (A) and CC1 (C) expression with Olig2 . Hatched lines mark the CC. Ctrl: Gde2+/−;Nex-Cre N-Gde2KO: Gde2lox/−;Nex-Cre. (B and D) Graphs quantifying NG2+ OPCs and CC1+ cells in CC and CTX. (B) ns p > 0.05, (D) *p = 0.0227, **p = 0.0052. n = 3 Ctrl, 4 N-Gde2KO. (E) Western blot of cortical extracts. Actin was used as a loading control. Levels of Olig2 (p = 0.5804) and PDGFRα (p = 0.4708) are unchanged between genotypes. (F) Graphs quantifying western blots (GDE2, ***p < 0.0001; MBP, **p = 0.0091; MOG, ***p = 0.0002). n = 5 Ctrl, n = 6 N-Gde2KO. All graphs show mean ± SEM, two-tailed unpaired Student’s t test. Scale bars represent 100 μm (A and C), 10 μm (inset A).
Neuronal GDE2 Releases Factors to Promote OL Maturation
To define the mechanisms by which neuronal GDE2 enhances OL maturation, we co-cultured purified WT and Gde2KO neurons with WT OPCs. Cortical neurons were derived from embryonic day 16.5 (E16.5) embryos and cultured for 3 days in vitro (DIV3); at this stage, neurons are immature and are undergoing active axonal and dendritic growth similar to neurons in postnatal brain at the time of OL maturation. WT and Gde2KO neuronal cultures were equivalent and typically composed of 95% neurons (β-tubulin type III+) and ~2% astrocytes (GFAP+), with no Olig2+ oligodendroglia (Figure S4A). On DIV3, OPCs purified from P6 WT cortices were plated on WT and Gde2KO neurons in the absence of mitogenic factors and co-cultured for an additional 3 days (Figure 4A). Cultures were then fixed and examined for OL maturation. When compared with OPCs cocultured with WT neurons, OPCs cocultured with Gde2KO neurons showed a 33% reduction in the number of mature MBP+ OLs (Figure 4B), and the number of myelinated segments in 9-day co-cultures was markedly reduced (Figure 4B). Total numbers of Olig2+ OL lineage cells were equivalent between the two conditions (Figure S4B). These observations recapitulate our in vivo data indicating that GDE2 neuronal function is required for OL maturation.
Figure 4. GDE2 Releases Neuronally Derived Factors that Promote OL Maturation.
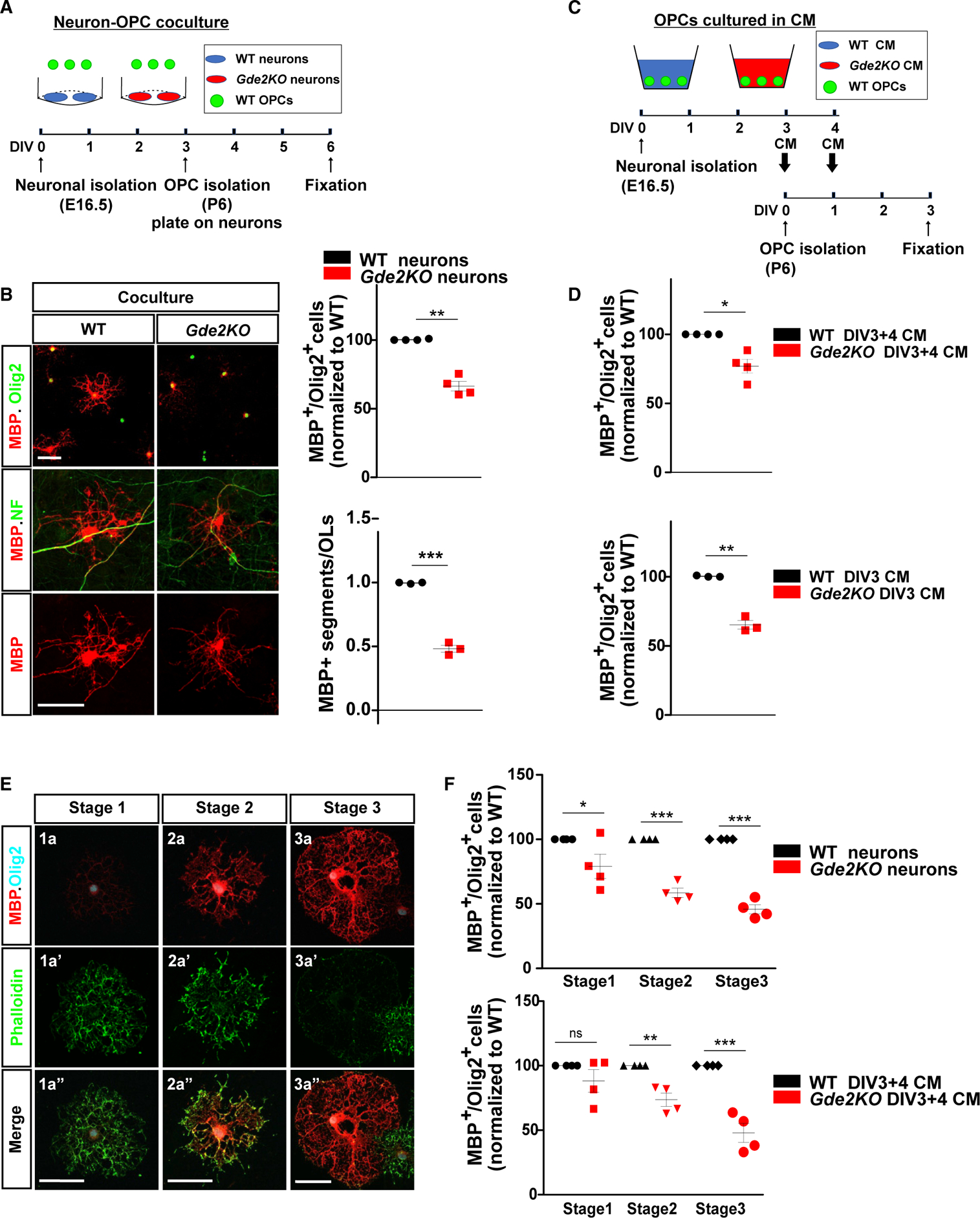
(A) Schematic of neuron-OPC co-culture. (B) WT OPCs co-cultured with WT or Gde2KO neurons. Graphs quantifying the percentage of MBP+ Olig2+ OLs (normalized to WT) (**p = 0.0019, n = 4 WT, n = 4 Gde2KO) and numbers of MBP+ segments (***p < 0.0001, n = 3 WT, 3 Gde2KO). (C) Schematic of OPCs cultured with neuronal conditioned medium (CM) (DIV3+4 CM). (D) Graphs quantifying percentage of MBP+ Olig2+ OLs (normalized to WT). DIV3+4 CM, *p = 0.0209, n = 4 WT, n = 4 Gde2KO CM; DIV3 CM, **p = 0.0078, n = 3 WT, n = 3 Gde2KO CM. (E) Representative images of the three stages of OL maturation in vitro. (F) Graphs quantifying percentage of MBP+ Olig2+ OLs. Top: co-culture two-way ANOVA ***p < 0.0001 (Bonferroni correction), *p < 0.05, ***p < 0.001; n = 4 WT, n = 4 Gde2KO. Bottom: WT OPCs cultured with CM, two-way ANOVA ***p < 0.0001 (Bonferroni correction); ns, p > 0.05; **p < 0.01; ***p < 0.001; n = 4 WT, n = 4 Gde2KO CM. All graphs show mean ± SEM (B and D), two-tailed unpaired Student’s t test. Scale bars represent 50 μm (B and E). See Table S1 for cell numbers.
We next treated freshly purified WT OPCs with conditioned medium (CM) collected from WT or Gde2KO neurons at DIV3 and DIV4 (Figure 4C). Specifically, WT OPCs were cultured for 1 day in DIV3 CM and on the next day cultured with CM collected between DIV3 and DIV4 for 2 days and then fixed and analyzed for OL maturation (Figure 4C). The total number of MBP+ OLs in cultures treated with Gde2KO CM was reduced by ~25% compared with WT CM (Figure 4D; Table S1). CM prepared from DIV3 WT and Gde2KO neurons alone recapitulated these changes in OL maturation (Figure 4D; Table S1). These observations suggest that neuronal GDE2 does not utilize contact-mediated signals to regulate OL maturation. Instead, GDE2 stimulates the release of soluble OL maturation factors, and these factors are released by DIV3 neurons.
OLs in vitro undergo stereotypic morphological changes, increase expression of myelin proteins, and shift from actin assembly to disassembly coincident with myelination (Zuchero et al., 2015). We defined three stages of OL maturation based on their morphology, MBP expression, and F-actin network visualized by phalloidin staining (Figure 4E; Zuchero et al., 2015). Differentiating Olig2+ OLs in vitro are arborized, with weak cell-body MBP expression and robust phalloidin labeling in the cell body and distal processes (stage 1, immature). Partially differentiated OLs show strong MBP expression and phalloidin labeling in distal processes, with occasional flattening of the myelin sheath in distal structures (stage 2, premyelinating), while more mature OLs show ring-like or lamellar morphology with increased MBP expression throughout the membrane sheath with near absence of the actin cytoskeleton (stage 3, myelinating). WT OPCs co-cultured with Gde2KO neurons after DIV3 showed a 40% and 50% reduction in the number of OLs at stages 2 and 3 of maturation and an ~25% reduction in the number of OLs at stage 1 (Figure 4F; Table S1). Similarly, WT OPCs grown in DIV3+4 CM showed a 25% and 50% reduction in the number of OLs at stages 2 and 3 of maturation when treated with Gde2KO CM but no change in the number of stage 1 OLs (Figure 4F; Table S1). These observations suggest that GDE2-dependent pathways in neurons release factors that promote OL maturation.
Neuronal activity is a central driver of OL maturation. We utilized optical recording of intracellular calcium via the calcium indicator Fluo-4 to monitor neuronal activity in cultured WT and Gde2KO neurons at DIV3 (Figure S4C), when cultured neurons utilize GDE2 pathways to release factors required for OL maturation. Analysis of calcium transients (ΔF/Fo) over a 3.5-min recording period showed no detectable calcium transients in DIV3 WT and Gde2KO neurons (Figure S4C). Treatment with the ionophore ionomycin, which ensures calcium internalization, results in robust signal confirming efficient Fluo-4 loading in neurons. These observations indicate that DIV3 WT and Gde2KO neurons are immature and largely inactive at this stage, suggesting that GDE2 regulation of OL maturation is unlikely to involve neuronal activity-dependent mechanisms. Stimulation of cultured neurons through addition of bicuculline did not alter GDE2 protein levels, supporting activity-independent roles for GDE2 in regulating OL maturation (Figure S4D).
GDE2 Maintains Canonical Wnt Signaling in Neurons
To gain insight into potential pathways that mediate GDE2 control of OL maturation, we performed bulk RNA sequencing (RNA-seq) of WT and Gde2KO nervous system tissue. 454 genes were differentially expressed in Gde2KO tissues compared with WT (Table S2), and Gene Ontology (GO) analysis using the STRING database (v.11) highlighted pathways associated with canonical Wnt signaling (Figures S5A and S5B). Known canonical Wnt target genes (https://web.stanford.edu/group/nusselab/cgi-bin/wnt/target_genes) were downregulated in the Gde2KO condition, implying that GDE2 normally potentiates Wnt pathway activation (Figure S5C). Wnt ligands bind their cognate receptors to ultimately stabilize and promote nuclear translocation of β-catenin (Janda et al., 2012). Nuclear, activated β-catenin (ABC) interacts with transcription factors to regulate expression of Wnt target genes, which include the transcription factor Lef1 (Hovanes et al., 2001; Shimogori et al., 2004). qPCR analysis showed a 32% reduction in Lef1 expression in cDNAs prepared from P10 Gde2KO cortical tissue compared with WT, and this decrease was recapitulated in Gde2KO cultured cortical neurons (Figure 5A). Further, levels of ABC detected by antibodies specific to β-catenin that is dephosphorylated on residues Ser37 or Thr41 (Liu et al., 2002) are decreased in Gde2KO DIV3 neuronal extracts compared to WT, while total levels of β-catenin are unchanged (Figure 5B). Immunohistochemical and biochemical analyses reveal that ABC levels in both nuclear and cytoplasmic compartments of Gde2KO DIV3 neurons are decreased (Figures 5C and 5D). These observations suggest that canonical Wnt signaling in neurons is reduced when neuronal GDE2 function is disrupted.
Figure 5. Canonical Wnt Signaling Is Reduced in Gde2KO Neurons and Oligodendroglia.
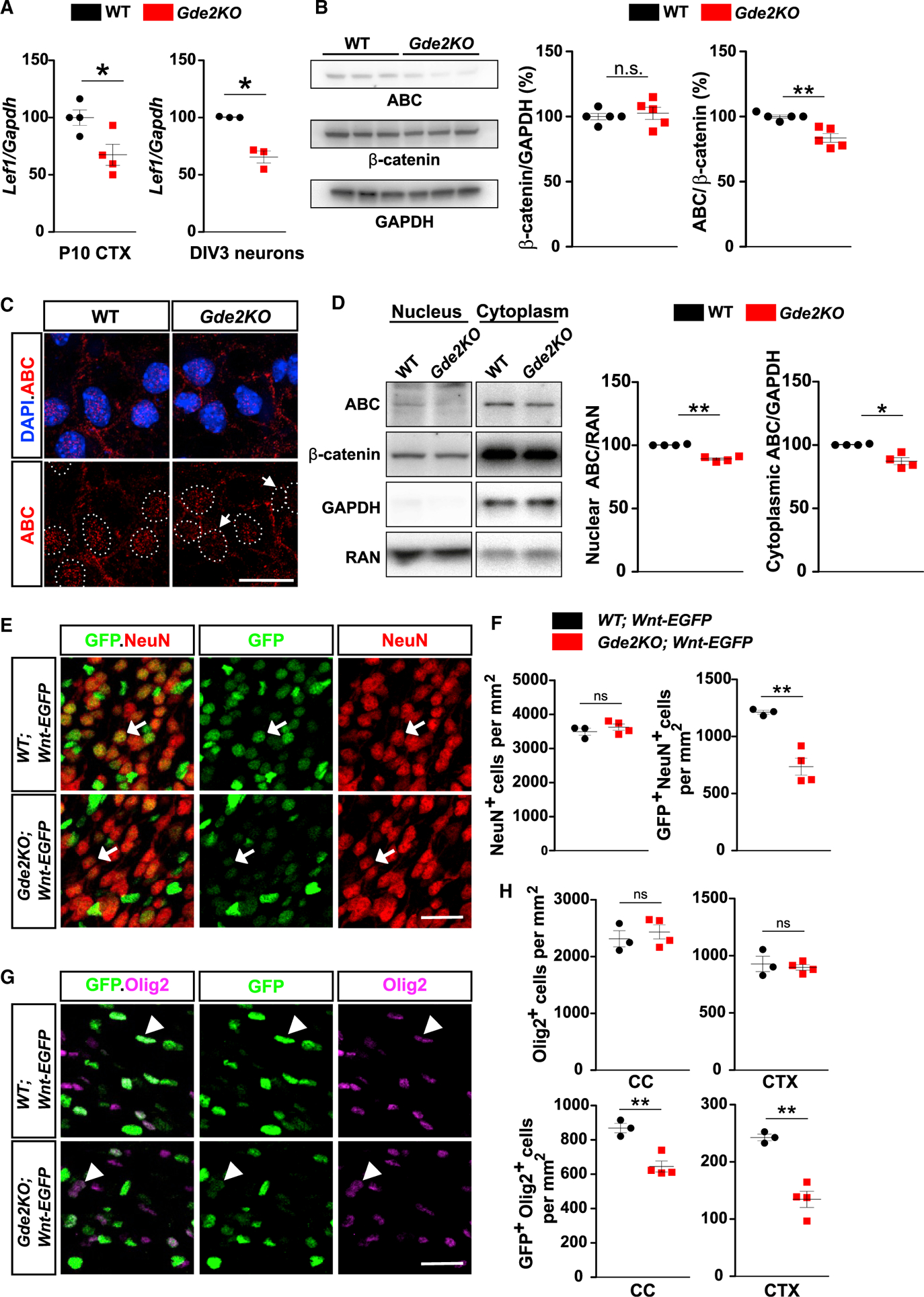
(A) Graphs quantifying qPCR of Lef1 transcripts normalized to Gapdh mRNAs. CTX, *p = 0.0231, n = 4 WT, n = 4 Gde2KO; DIV3 cortical neurons, *p = 0.0362, n = 3 WT, n = 3 Gde2KO. (B) Western blot of DIV3 cortical neurons with associated quantification. ns, p = 0.6465; **p = 0.0018; n = 5 WT, n = 5 Gde2KO. (C) Images of cultured cortical neurons. Arrows mark reduced ABC nuclear (hatched lines) staining. (D) Western blot of fractionated DIV3 cortical neuron extracts. Graphs quantifying ABC normalized to RAN and GAPDH, **p = 0.0019, *p = 0.0186, n = 4 WT, n = 4 Gde2KO. (E and G) Coronal sections of P11 CTX. (E) Arrows show differential GFP expression in neurons in WT and Gde2KO;Wnt-EGFP mice. (G) Arrowheads mark differential GFP expression in Olig2+ cells in WT and Gde2KO;Wnt-EGFP mice. (F) Graphs quantifying neurons and GFP+ neurons in WT and Gde2KO;Wnt-EGFP mice. ns, p = 0.3936; **p = 0.0078. H) Graphs quantifying Olig2+ and GFP+ Olig2+ cells in WT and Gde2KO;Wnt-EGFP mice. ns, p > 0.05, CC **p = 0.006, CTX **p = 0.0057. For (F) and (H), n = 3 WT;Wnt-EGFP, 4 Gde2KO;Wnt-EGFP. All graphs show mean ± SEM, two-tailed unpaired Student’s t test. Scale bars represent 5 μm (C) and 20 μm (E and G).
We next examined the temporal and spatial pattern of canonical Wnt pathway activation in vivo using a mouse reporter line that visualizes in situ Wnt pathway activation through expression of EGFP (Rosa26 Tcf./Lef-H2B-EGFP mice or Wnt-EGFP) (Cho et al., 2017). Analysis of Wnt-EGFP mice at P7 and P11 show robust nuclear EGFP expression in neurons and Olig2+ cells, coincident with the period of OL maturation (Figures S5D and S5E). In contrast, little to no neuronal GFP expression is detected at P28 when developmental myelination is almost complete. However, GFP continues to be expressed in ~20% of Olig2+ cells (Figures S5D and S5E). Thus, Wnt activation in WT neurons overlaps with GDE2 neuronal expression and the temporal profile of GDE2 requirement in OL maturation.
To determine if Wnt signaling is dependent on GDE2 expression, we introduced the Wnt-EGFP reporter into Gde2KO animals. Total numbers of GFP+ cells were reduced in Gde2-KO;Wnt-EGFP animals compared with WT controls, suggesting reduced Wnt pathway activation in the absence of GDE2 (Figures S5F and S5G). Gde2KO;Wnt-EGFP animals showed a marked 40% reduction in the number of EGFP-expressing neurons (NeuN+) compared with WT littermates (Figures 5E and 5F). This indicates that GDE2 is required to maintain canonical Wnt signaling in neurons at the time of OL maturation. P11 Gde2KO;Wnt-EGFP cortices also show 25% and 40% reduced EGFP expression in oligodendroglia (Olig2+ cells) in CC and CTX, respectively (Figures 5G and 5H). The numbers of neurons and oligodendroglia were equivalent in both Wnt-EGFP and Gde2KO;Wnt-EGFP animals (Figures 5E–5H). NG2 progenitors show robust GFP expression while immature (TCF4+ CC1−) and mature (TCF4− CC1+) OLs show minimal EGFP expression, suggesting that Wnt activity is restricted to OPCs (Figure S5H). Because GDE2 is expressed in OLs and not OPCs, this implies that GDE2-dependent activation of Wnt signaling in OPCs is non-cell autonomous. These observations indicate that GDE2 maintains canonical Wnt activity in neurons and OPCs at the time of OL maturation in the developing postnatal CTX.
Increasing Neuronal Wnt Activity in Gde2KO Mice Rescues OL Maturation
To test if the reduction in canonical Wnt activity is causal for OL maturation deficits in Gde2KOs, we increased Wnt signaling in Gde2KO animals by genetically stabilizing β-catenin in vivo. Ctnnb1flex3 mice harbor loxP sites flanking exon 3 of β-catenin (β-catex3), which contains phosphorylation sites for GSK3-β that target β-catenin for degradation (Harada et al., 1999). Cre-dependent excision of exon 3 prevents GSK3-β phosphorylation of β-catenin, thus stabilizing β-catenin and increasing canonical Wnt signaling. We first stabilized β-catenin in neurons of Gde2KO mice by generating Gde2KO;Nex-Cre;β-catex3 animals (Gde2KO;N-β-catex3). Western blots from P14 cortical extracts confirmed Cre-dependent removal of exon 3 in β-catenin protein in Gde2KO;N-β-catex3 animals, but not in WT;β-catex3 and Gde2-KO;β-catex3 controls (Figure S6A). All three genotypes had equivalent numbers of Olig2+ cells, suggesting that β-catenin stabilization in neurons had minimal effect on the number of OL lineage cells (Figure S6B). However, there was a substantial increase of Olig2+ Mbp+ mature OLs in Gde2KO;N-β-catex3 cortices compared to Gde2KO;β-catex3 controls that restored numbers of mature Olig2+ Mbp+ OLs to WT levels (Figures 6A and 6B). Gde2KO;N-β-catex3 cortices also showed recovery of CC1+ Olig2+ OLs to WT levels in CC with partial rescue in CTX (Figures 6C and 6D). These observations suggest that GDE2 mediates OL maturation through stimulation of canonical Wnt signaling in neurons.
Figure 6. Stabilizing β-catenin in Neurons Rescues Gde2KO OL Maturation.
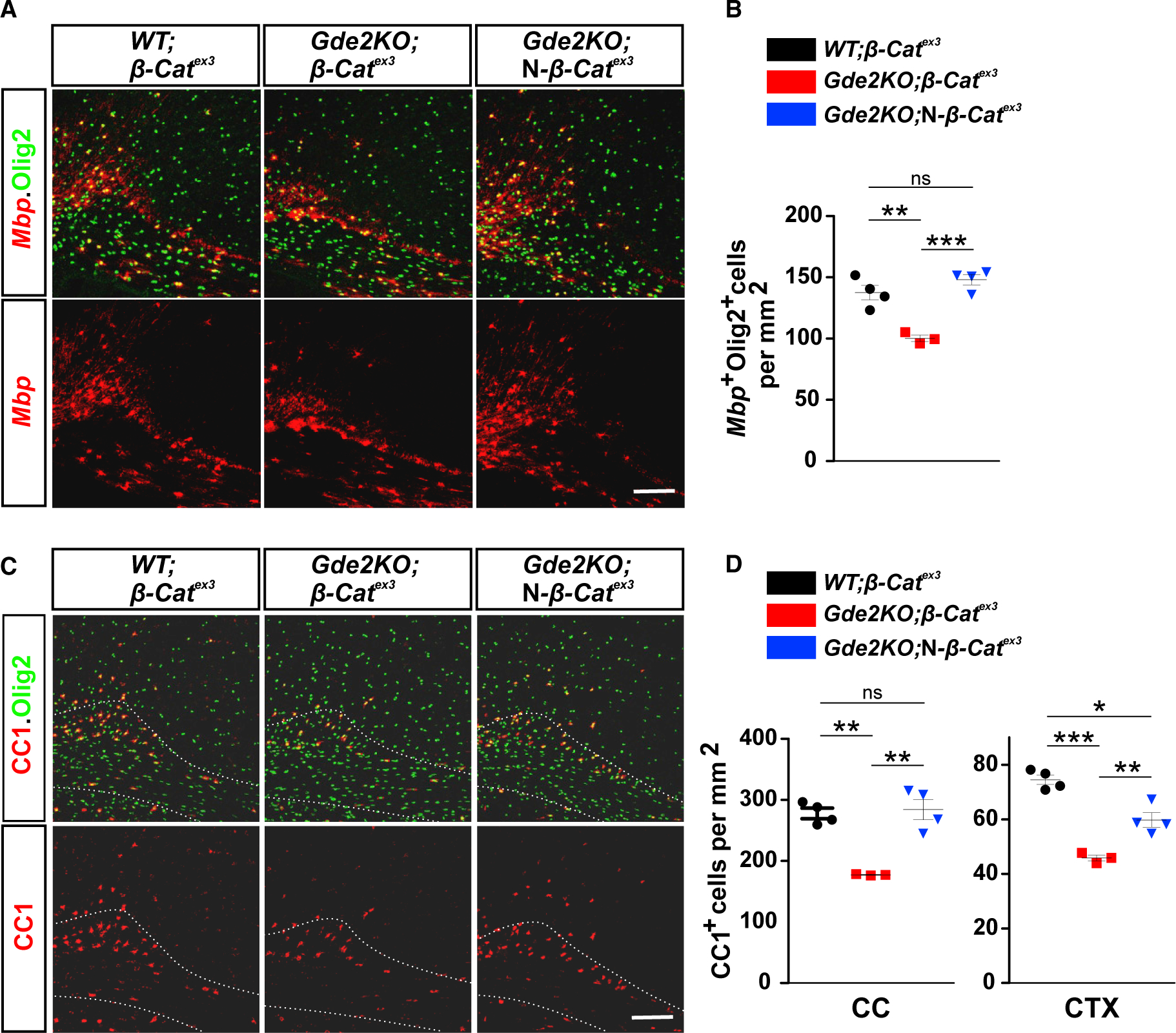
(A and C) Coronal sections of motor CTX and CC showing expression of Mbp (A) and CC1 (C) expression in oligodendroglia (Olig2+). Hatched lines mark the CC. (B) Graph quantifying Olig2+ cells expressing Mbp transcripts. **p = 0.0047, ***p = 0.0006, ns = 0.204. (D) Graph quantifying numbers of CC1+ OLs. In CC, **p = 0.0014 (WT;β-catex3 versus Gde2KO;β-catex3), **p = 0.0078 (Gde2KO;β-catex3 versus Gde2KO;N-β-catex3), ns p = 0.7592 (WT;β-catex3 versus Gde2KO;N-β-catex3). In CTX, ***p = 0.0006 (WT;β-catex3 versus Gde2KO;β-catex3), **p = 0.0098 (Gde2KO;β-catex3 versus Gde2KO;N-β-catex3), *p = 0.0156 (WT;β-catex3 versus Gde2KO;N-β-catex3). n = 4 WT;β-catex3, 3 Gde2KO;β-catex3, n = 4 Gde2KO;N-β-catex3. All graphs show mean ± SEM, two-tailed unpaired Student’s t test. Scale bars represent 100 μm (A and C).
Gde2KO animals also display reduced Wnt signaling in OPCs (Figures 5G and 5H). We stabilized β-catenin in OPCs by generating Gde2KO;β-catex3;PDGFαR-CreER animals (Gde2KO;O-β-catex3), which express Cre recombinase in OPCs in response to 4 hydroxytamoxifen (4-HT) (Kang et al., 2010). We administered 4-HT to Gde2KO;O-β-catex3 mice and WT;β-catex3and Gde2KO;β-catex3 controls at P7 and examined OL maturation at P11. β-catenin stabilization in OPCs in Gde2KO;O-β-catex3 mice resulted in reduced numbers of Olig2+ oligodendroglia in CC compared with controls, whereas Olig2+ cells in CTX were equivalent between genotypes (Figures S7A and S7B). OL maturation was further retarded in both CC and CTX in Gde2KO;O-β-catex3 CTX (Figures S7C–S7E). Because OL maturation phenotypes are not rescued in Gde2KO;O-β-catex3 animals, we conclude that GDE2 regulation of canonical Wnt signaling in OPCs does not promote OL maturation.
Neuronal Wnt Activity Releases OL Maturation Factors
To test if GDE2 stimulation of Wnt signaling in neurons is required for the release of OL maturation factors, we generated CM from neurons isolated from Gde2KO;β-catex3 and Gde2KO;N-β-catex3 animals that were cultured till DIV3 (Figure 7A). WT OPCs were treated for 3 days with CM and examined for OL maturation. Strikingly, CM from Gde2KO neurons with stabilized β-catenin (Gde2KO;N-β-catex3) showed an 60% increase in the number of MBP+ OLs compared with CM from Gde2KO neurons (Gde2KO;β-catex3), as well as a robust increase in MBP protein by western blot (Figures 7A and 7B; Table S1). Further, we observed a 60% increase in the number of stage 1, stage2, and stage3 MBP+ OLs in CM from Gde2KO;N-β-catex3 neurons compared with CM from Gde2KO;β-catex3 condition (Figure 7B; Table S1). Thus, stabilization of β-catenin in Gde2KO neurons is sufficient to release factors that stimulate OL maturation. This is consistent with the model that GDE2 stimulates canonical Wnt signaling in neurons and that this pathway potentiates the release of neuronally derived factors that promote OL maturation. The increase in stage 1 OLs in the presence of CM from Gde2KO neurons with stabilized β-catenin contrasts with our observation that the production of stage 1 OLs is not dependent on GDE2 neuronal CM (Figures 7B and 4F). We attribute this to the robust and continuous release of OL maturation factors when β-catenin is constitutively stabilized in neurons.
Figure 7. Stabilized β-catenin in Neurons Stimulates Release of OL Maturation Factors.
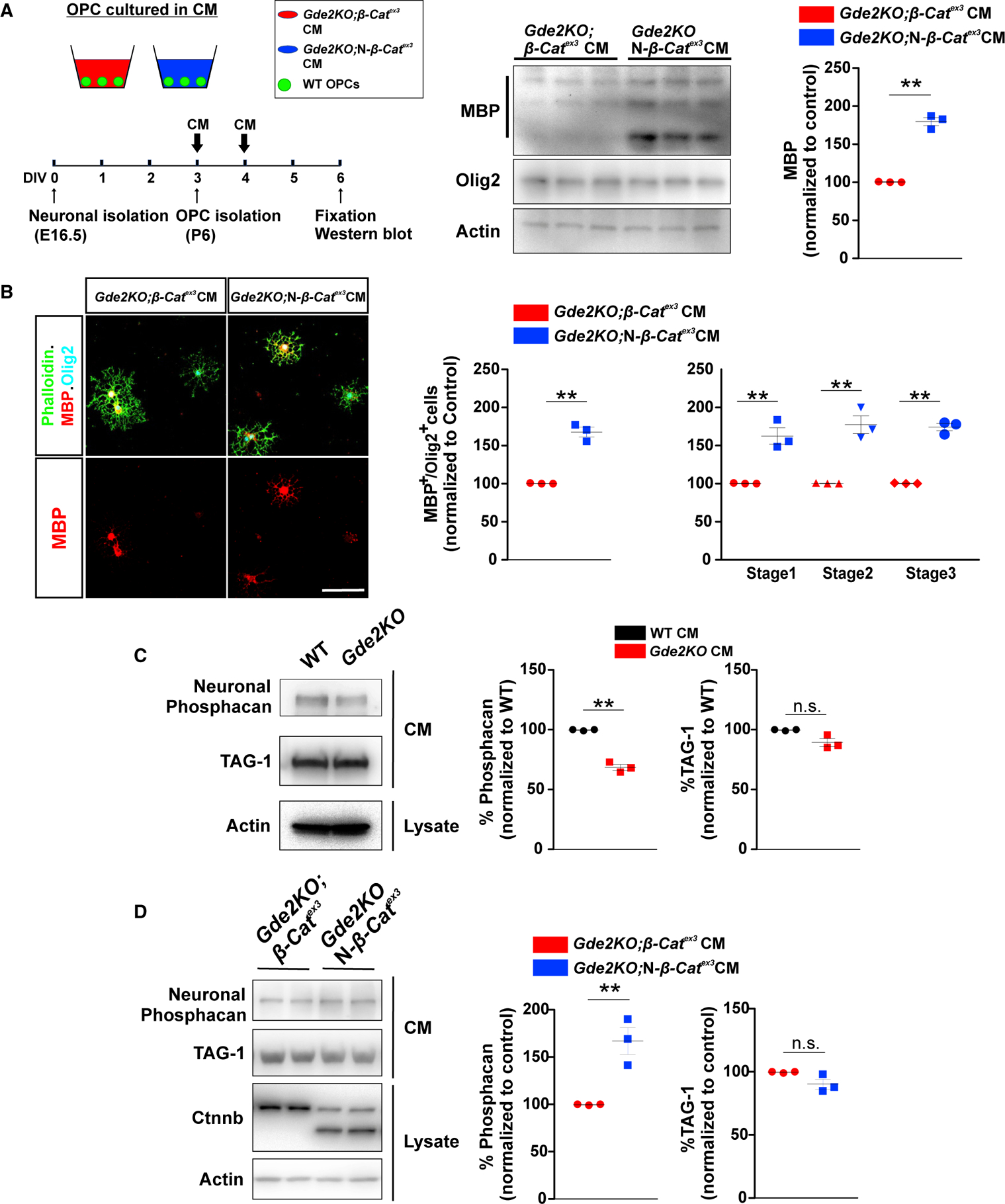
(A) Schematic of OPCs cultured with neuronal CM. Western blot of WT OPC lysates + CM and MBP quantification. **p = 0.0043, n = 3 Gde2KO;β-catex3, n = 3 Gde2KO;N-β-catex3CM. (B) Representative images of OPCs cultured in CM. Graphs quantifying the percentage of MBP+ Olig2+ cells. **p = 0.009. This increase spans all three stages of OL maturation: two-way ANOVA. ***p < 0.0001 (Bonferroni correction), **p < 0.001, n = 3 Gde2KO;β-catex3, n = 3 Gde2KO;N-β-catex3 CM. See Table S1 for cell numbers. (C) Western blot and protein quantification. **p = 0.0062; ns, p = 0.0864. n = 3 WT, n = 3 Gde2KO CM. (D) Western blot and protein quantification. **p = 0.009; ns, p = 0.0781. n = 3 Gde2KO;β-catex3, n = 3 Gde2KO;N-β-catex3CM. All graphs show mean ± SEM, two-tailed unpaired Student’s t test. Scale bar represents 20 μm (B).
Candidate Factors Released by GDE2/Wnt Signaling in Neurons
To identify OL maturation factors released by GDE2 neuronal function, we collected WT and Gde2KO CM and analyzed the protein content by mass spectrometry. We identified 149 proteins that were expressed at 40% or higher in Gde2KO CM compared to WT CM (Table S3). GDE2 releases GPI-anchored proteins from the plasma membrane; however, no GPI-anchored proteins were differentially expressed in WT and Gde2KO CM (Figure S8A). This is consistent with our model that GDE2 stimulates Wnt signaling in neurons, which drives the release of neuronal factors that promote OL maturation. We identified 11 secreted/extracellular matrix (ECM) associated proteins that were differentially expressed in WT and Gde2KO neuronal CM, with 10 proteins showing decreased expression in Gde2KO CM (Figure S8B). Of these 10 proteins, soluble receptor-type tyrosine-protein phosphatase zeta (RPTPzeta, or phosphacan) can promote OL maturation through interaction with contactin-1 in OPCs (Lamprianou et al., 2011). Phosphacan is expressed in neurons, astrocytes, and oligodendroglia (Cahoy et al., 2008; Dwyer et al., 2015); accordingly, released phosphacan is a promising candidate for mediating GDE2-dependent regulation of OL maturation. Neuronally derived phosphacan is distinguished from glial phosphacan by antibodies that recognize cell-type-specific O-mannosyl glycans (Dwyer et al., 2015). Western blots reveal that levels of neuronal phosphacan are reduced in Gde2KO CM compared to WT. In contrast, GPI-anchored protein TAG1 levels are equivalent between conditions (Figure 7C). These observations confirm the proteomic analysis of phosphacan and Tag1 expression in Gde2KO and WT CM (Figures S8A and S8B). To determine if reduced levels of phosphacan mediate the delay in OL maturation exerted by Gde2KO CM, we depleted phosphacan from WT CM using antibodies to neuronal phosphacan conjugated to protein L (Figure S8C). OPCs cultured with phosphacan depleted CM largely recapitulated the delay in OL maturation elicited by Gde2KO CM; specifically, the number of MBP+ cells was decreased with concomitant reductions in stage 1, stage 2, and stage 3 MBP+ OLs (Figures S8D and S8E; Table S1). The degree of OL maturation was not changed when WT CM incubated with protein L alone was used (Figure S8E; Table S1). Our genetic studies suggest that GDE2 stimulation of canonical Wnt signaling in neurons mediates OL maturation. We thus performed western blot analysis on CM prepared from Gde2KO;N-β-catex3 and Gde2-KO;β-catex3 neuronal cultures. Levels of soluble neuronally derived phosphacan were markedly increased in CM from Gde2KO;N-β-catex3 cortical cultures compared with control Gde2KO;β-catex3 CM (Figure 7D). These observations suggest that GDE2 stimulation of Wnt signaling in neurons releases soluble factors, such as phosphacan, that promote OL maturation.
DISCUSSION
Our studies reveal that GDE2 neuronal function promotes the maturation of premyelinating OLs in the developing CTX. During the period of developmental myelination, GDE2 is predominantly expressed in cortical neurons, where it is required to maintain canonical Wnt signaling. Activation of canonical Wnt pathways causes the release of soluble factors from neurons such as phosphacan that promote the production of premyelinating and myelinating OLs from OPCs (Figure S8F). These observations identify roles for neuronal GDE2 in regulating OL maturation and suggest that GDE2 regulation of Wnt signaling in neurons is part of the complex interplay between neurons and glia that coordinates axonal myelination during cortical development.
Neuronal GDE2 and OL Maturation
Known factors in neurons that regulate OL development include contact-dependent signals between axons and oligodendroglia, axon caliber, and neuronal activity (Charles et al., 2000; Gibson et al., 2014; Lee et al., 2012). Here, we show that GDE2 activity in neurons stimulates OL maturation through pathways that are apparently separate from these mechanisms. Our studies using WT and Gde2KO neuronal CM suggest that GDE2 regulates OL maturation through the release of soluble promaturation factors and not through contact-mediated pathways. Further, CM with promaturation activity was derived from DIV3 cultured neurons that are immature and lack obvious neuronal activity, as assayed by calcium imaging, thus ruling out possible contributions of activity-dependent mechanisms. In addition, measurements of axonal diameters indicate that axonal caliber is unaffected by disruption of GDE2 function. Our studies provide support for a mechanism driven by GDE2, whereby neurons release soluble factors that promote nearby OPCs to differentiate and initiate the myelination program. This idea is consistent with the expression of GDE2 in the developing CTX, which matches the pattern of OL maturation and myelination that initiates in deep layers and broadens to the superficial laminae (Tomassy et al., 2014). We note that GDE2 loss leads to a delay in OL maturation; by P28, the numbers of myelinating OLs and myelin-associated proteins in Gde2KO animals are equivalent to WT. This recovery is likely due to the robust compensatory mechanisms known to occur when OL development is disrupted. However, the increased incidence of unmyelinated large-diameter axons in Gde2KO adult animals suggests that the timing of OL maturation regulated by GDE2-dependent mechanisms is important to ensure appropriate axonal myelination. GDE2 is vertebrate specific (Nogusa et al., 2004). We speculate that GDE2 could constitute one of several regulatory pathways that have evolved in vertebrates to myelinate axons in order to facilitate complex neural circuit function.
GDE2 Regulation of Canonical Wnt Signaling and OL Maturation
Canonical Wnt signaling has established roles in cortical progenitor proliferation, differentiation, and neuronal migration (Bocchi et al., 2017; Chenn and Walsh, 2002; Munji et al., 2011). Our analysis of Wnt-EGFP reporter mice confirms Wnt signaling is active in neurons, OPCs, and OLs during the period of developmental myelination. By P28, when developmental myelination is almost complete, there is remarkably little Wnt activation in neurons, although subpopulations of oligodendroglial cells exhibit substantial reporter gene expression. We show here that Wnt activation in neurons and OPCs is dependent in part on GDE2 function. Activation of canonical Wnt signaling in OPCs in Gde2KO animals worsens OL maturation phenotypes and is consistent with previous studies showing that elevating Wnt signaling delays OL maturation and negatively regulates terminal OL differentiation (Fancy et al., 2009; Ye et al., 2009). In contrast, genetic stabilization of β-catenin in neurons of Gde2KO animals rescues their OL maturation phenotypes, indicating that GDE2-dependent maintenance of Wnt signaling in neurons is important for appropriate OL development. This observation identifies roles for neuronal canonical Wnt signaling in the cross-talk between neurons and oligodendroglia that coordinates the timing of OL maturation. While Wnt activation in neurons declines from P28, GDE2 continues to be expressed in neurons. This observation raises two main questions: how does GDE2 promote Wnt activation during developmental myelination, and how is this pathway switched off after P28? Greater insight into these questions will be gleaned from further investigation into the mechanisms by which GDE2 regulates canonical Wnt signaling. GDE2 is a membrane-bound enzyme that functions at the cell surface to regulate GPI-anchored protein function by cleavage of the GPI-anchor and release of the protein from the cell surface (Matas-Rico et al., 2016; Park et al., 2013). Accordingly, a plausible mechanism for GDE2 regulation of Wnt signaling is through regulation of GPI-anchored protein function. RECK and the heparan sulfate proteoglycans GPC6 and GPC4 are established substrates of GDE2 (Matas-Rico et al., 2016; Park et al., 2013). RECK can bind Wnt and can interact in a multiprotein complex with Gpr124 and Frizzled to stimulate Wnt signaling (Cho et al., 2017; Eubelen et al., 2018). GPC6 and GPC4 areknown to regulate Wnt pathway activation and can function as activators or inhibitors of Wnt signaling (Han et al., 2005; Lebensohn et al., 2016; Sakane et al., 2012). The known contributions of RECK, GPC6, and GPC4 in the regulation of Wnt signaling warrant further investigation into whether they mediate GDE2-dependent control of OL maturation.
Neuronal Wnt Signaling Releases OL Maturation Factors from Neurons
Our studies suggest that GDE2 function in neurons is required for the release of soluble factors that promote OL maturation. Strengthening the idea that GDE2 activation of canonical Wnt signaling mediates this release is that CM from Gde2KO neurons expressing stabilized β-catenin promotes OL maturation to an extent that is more potent than WT neuronal CM. Our proteomic analysis of WT and Gde2KO CM identified phosphacan within a cohort of secreted and ECM-associated proteins that were reduced in Gde2KO CM. Phosphacan is a splice variant of PTPRzeta encoded by the Ptprz1 gene. It contains the extracellular domain of the full-length membrane-bound isoform of PTPRzeta that consists of an N-terminal carbonic anhydrase like domain, threefibronectin type III repeats, and attachment sites for chondroitin sulfate proteoglycan (Maurel et al., 1994). Loss of PTPRzeta results in increased OPC proliferation and impaired OL differentiation, and studies of cultured OPCs indicate that phosphacan regulates the rate of OL maturation via interaction with contactin-1 (Harroch et al., 2002; Lamprianou et al., 2011). PTPRzeta is widely expressed during developmental myelination, but the relevant cellular source for phosphacan that regulates OL maturation has not been defined (Faissner et al., 2006). We find that Gde2KO CM has reduced levels of neuronal phosphacan and that phosphacan levels are restored in CM prepared from Gde2KO neurons with stabilized β-catenin. Further, WT neuronal CM depleted for phosphacan mimics OL maturation deficits observed when OPCs are incubated with Gde2KO CM. These observations support the model that phosphacan released by GDE2-dependent activation of Wnt signaling in neurons mediates OL maturation. How Wnt signaling promotes phosphacan release is not clear. Phosphacan transcripts are not altered in our bulk RNA-seq data from Gde2KO tissue, favoring models that involve secondary mechanisms that impact phosphacan protein production and secretion. We have focused here on phosphacan because it is exemplar of a factor with known activities in promoting OL maturation. Given that CM is a complex mixture of proteins and RNA species, it is possible that Wnt signaling pathways in neurons promotes the release of additional factors important for OL maturation.
In summary, we have identified GDE2 regulation of canonical Wnt signaling in neurons as a pathway that controls the rate of OL maturation through the release of soluble promaturation factors that include phosphacan. This study provides insight into the complex communication pathways between axons and oligodendroglia that collectively regulate developmental myelination.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Shanthini Sockanathan (ssockan1@jhmi.edu).
Materials Availability
Antibodies against mouse GDE2 will be provided freely with no restrictions by the Lead Contact upon request.
Data and Code Availability
The RNA-seq raw data generated during this study are publicly available at National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO). The accession number for the data reported in this paper is GSE147144.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD018080.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The following mouse lines were used in this study: Gde2KO (Sabharwal et al., 2011), Gde2flox (Sabharwal et al., 2011), PDGFRαCre-ER (Kang et al., 2010), Nex-Cre (Goebbels et al., 2006), Rosa26 Tcf./Lef H2B-EGFP (Cho et al., 2017), Ctnnb1flex3 (Harada et al., 1999). All mice were housed and handled according to the approved Institutional Animal Care and Use Committee (IACUC) protocol of the Johns Hopkins Medical Institution. Both males and females were used for analysis. The age of the animals analyzed are stated in the figures, figure legends and main text.
METHOD DETAILS
Tissue processing and immunohistochemistry
Mice were deeply anesthetized with Avertin solution (1.3% 2,2,2-Tribromorethanol (Fluka 90710) and 0.7% 2-methyl-2-butanol (Sigma 240486) in Phosphate Buffered Saline (PBS) at 0.02 ml/g body weight and perfused transcardially with 0.1 M Phosphate Buffer (PB) followed by fixation solution (4% paraformaldehyde in 0.1 M PB). The brains were postfixed in the fixation solution overnight at 4°C and transferred to 30% sucrose solution and stored at 4°C for more than 48 hr. The tissues were embedded in O.C.T. Compound (Tissue-Tek 62550–12), flash frozen, and coronally sectioned (50 μm for P7 brain tissues and 35 μm for the rest) with a cryostat (Thermo Fisher Scientific HM550). Immunofluorescence was performed on free-floating sections. Brain sections were boiled in sodium citrate buffer (10 mM sodium citrate with 0.05% Tween-20) at 95°C before blocking. Tissue sections were pre-incubated in blocking solution (1% normal goat serum, 0.3% Triton X-100 in PBS) for 2 hours at room temperature, then incubated in primary antibodies overnight at 4°C. The primary antibodies and secondary antibodies used are listed in the Key Resources Table. Sections were mounted onto slides with mounting reagent (Polysciences 18606). Images were acquired using confocal microscopy (Zeiss LSM700) with 10 or 20x objective. A total of 4–5 sections were assessed per mouse and 3–5 mice were analyzed per group. For all studies, a region of interest (ROI) was chosen based on the anatomical structures of CC and CTX based on DAPI staining, subsequent to quantification (see section on Quantification and Statistical Analyses).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Olig2 | Millipore | Cat# AB9610; RRID:AB_570666 |
| Guinea Pig anti-Olig2 | Ben Novitch, University of California Los Angeles | N/A |
| Mouse anti-NeuN | Millipore | Cat# MAB377; RRID:AB_2298772 |
| Rabbit anti-NeuN | Millipore | Cat# MABN140; RRID:AB_2571567 |
| Rabbit anti-TCF4/TCF7L2 | Cell Signaling Technology | Cat# 2569; RRID:AB_2199816 |
| Mouse anti-CC1 | Calbiochem | Cat# OP80; RRID:AB_2057371 |
| Rabbit anti-ASPA | Gene Tex | Cat# GTX113389; RRID:AB_2036283 |
| Rabbit anti-Ki67 | Abcam | Cat# ab15580; RRID:AB_443209 |
| Goat anti Sox10 | Santa Cruz | Cat# Sc-17342; RRID:AB_2195374 |
| Chicken anti-GFP | Aves Labs | Cat# GFP-1020; RRID:AB_10000240 |
| Rabbit anti-GFP | Life Technologies | Cat# A11122; RRID:AB_221569 |
| Sheep Anti-Digoxigenin, POD Conjugated | Roche | Cat# 11207733910; RRID:AB_514500 |
| Mouse anti-MBP | Covance | Cat# SMI-99P-100; RRID:AB_10120129 |
| Rat anti-MBP | Milipore | Cat# MAB386; RRID: AB_94975 |
| Mouse anti-beta-Tubulin III | Sigma-Aldrich | Cat# T8578; RRID:AB_1841228 |
| Rabbit anti-GFAP | Agilent | Cat# Z0334; RRID:AB_10013382 |
| Mouse anti-Active-β-Catenin (Anti-ABC) | Millipore | Cat# 05–665; RRID:AB_309887 |
| Mouse anti-Ran | BD Biosciences | Cat# 610341; RRID: AB_397731 |
| Rabbit anti-Cux1 | Proteintech Group | Cat# 11733–1-AP; RRID:AB_2086995 |
| Rat anti-Ctip2 | Abcam | Cat# ab18465; RRID:AB_2064130 |
| Mouse anti-MOG | Millipore | Cat# MAB5680; RRID:AB_1587278 |
| Rabbit anti-Neurofilament H | Millipore | Cat# AB1989; RRID:AB_11212727 |
| Rabbit anti-GDE2 | This study | N/A |
| Mouse anti-Actin | Millipore | Cat# MAB1501; RRID:AB_2223041 |
| Rabbit anti-PDGF receptor alpha | Cell Signaling Technology | Cat# 3174; RRID:AB_2162345 |
| Rabbit anti- β-Catenin | Cell Signaling Technology | Cat# 8480; RRID:AB_11127855 |
| Goat anti-Contactin-2/TAG1 | R and D Systems | Cat# AF4439; RRID:AB_2044647 |
| Mouse anti-Chondroitin Sulfate Proteoglycan (CAT-315) | Millipore | Cat# MAB1581; RRID:AB_94270 |
| Rabbit anti-GAPDH | Cell Signaling Technology | Cat# 8884; RRID:AB_11129865 |
| Goat anti-rabbit IgG (H+K), secondary antibody, FITC and Alexa 647 conjugates | Jackson ImmunoResearch Labs | Cat# 111-095-144; RRID:AB_2337978, Cat# 111-605-144; RRID:AB_2338078 |
| Goat anti-mouse IgG (H+K), secondary antibody, Cy3 and Alexa 488 conjugates | Jackson ImmunoResearch Labs | Cat# 115-165-146; RRID:AB_2338690, Cat# 115-545-166; RRID:AB_2338852 |
| Goat anti-guinea pig IgG (H+K), secondary antibody, Alexa 647 conjugate | Jackson ImmunoResearch Labs | Cat# 106-605-003; RRID:AB_2337446 |
| Donkey anti-goat IgG (H+L), secondary antibody, Cy3 conjugate | Jackson ImmunoResearch Labs | Cat# 705-165-147; RRID:AB_2307351 |
| Donkey anti-chicken IgY (H+L), secondary antibody, Cy3 conjugate | Jackson ImmunoResearch Labs | Cat# 703-165-155; RRID:AB_2340363 |
| Peroxidase Donkey anti-mouse IgG (H+L), secondary antibody | Jackson ImmunoResearch Labs | Cat# 715-035-150; RRID:AB_2340770 |
| Peroxidase Donkey anti-rabbit IgG (H+L), secondary antibody | Jackson ImmunoResearch Labs | Cat# 711-035-152; RRID:AB_10015282 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (Z)-4-Hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| Sunflower seed oil from Helianthus annuus | Sigma-Aldrich | Cat# S5007 |
| Neurobasal media | GIBCO | Cat# 21103–049 |
| DMEM/F12 media | GIBCO | Cat# 10565–018 |
| Sodium Pyruvate 100mM | GIBCO | Cat# 11360–070 |
| Glutamax | GIBCO | Cat# 35050–061 |
| N2B supplement | STEMCELL Technologies | Cat# 7156 |
| SM1 supplement | STEMCELL Technologies | Cat# 5711 |
| Penicillin-Streptomycin | GIBCO | Cat# 15140–122 |
| Poly-D-lysine hydrobromide | Sigma-Aldrich | Cat# P0899 |
| Laminin | Sigma-Aldrich | Cat# L2020 |
| PureCol Type I Bovine Collagen Solution | Advanced Biomatrix | Cat# 5005-B |
| Forskolin | Calbiochem | Cat# 344270 |
| CNTF | PeproTech | Cat# 450–5 |
| HEPES | GIBCO | Cat# 15630080 |
| N-Acetyl-Cysteine | Sigma-Aldrich | Cat# A8199 |
| DAPI | Invitrogen | Cat# R37606 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Fluo-4, AM | Thermo Fisher Scientific | Cat# F14201 |
| Ionomycin | Tocris | Cat# 1704 |
| Trizol | Thermo Fisher Scientific | Cat# 15596018 |
| Fast SYBR® Green Master Mix | Thermo Fisher Scientific | Cat# 4385612 |
| Alexa Fluor® 488 Phalloidin | Invitrogen | Cat# A12379 |
| Chondroitinase ABC | Sigma-Aldrich | Cat# C3667 |
| Protein L magnetic beads | Thermo Fisher Scientific | Cat# 88849 |
| Bicuculine | Tocris | Cat# 0131 |
| Critical Commercial Assays | ||
| Neural Tissue Dissociation Kit (P) | Miltenyi Biotec | Cat# 130-092-628 |
| Miltenyi Isolation Starting Kit | Miltenyi Biotec | Cat# 130-090-312 |
| Anti-O4 Microbeads | Miltenyi Biotec | Cat# 130-094-543 |
| SuperScript III | Thermo Fisher Scientific | Cat# 18080051 |
| TSA Plus Cy3 system | PerkinElmer | Cat# NEL744001KT |
| TruSeq® Stranded mRNA LT - Set A | Illumina | Cat# RS-122–2101 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| NE-PER Nuclear and cytoplasmic extraction reagents | Thermo Fisher Scientific | Cat# 78833 |
| Deposited Data | ||
| RNA-seq data | This study | NCBI’s GEO: GSE147144 |
| Mass spectrometry data | This study | ProteomeXchange: PXD018080 |
| Experimental Models: Cell Lines | ||
| Primary cortical oligodendrocyte progenitor cells | This study | N/A |
| Primary cortical neuronal cells | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Gde2KO | Sabharwal et al., 2011 | N/A |
| Mouse: Gde2flox | Sabharwal et al., 2011 | N/A |
| Mouse: PdgfrαCre-ER | Kang et al., 2010 | N/A |
| Mouse: Nex-Cre | Goebbels et al., 2006 | N/A |
| Mouse: Rosa26 Tcf./Lef H2B-EGFP | Cho etal., 2017 | N/A |
| Mouse: Ctnnb1flex3 | Harada et al., 1999 | N/A |
| Oligonucleotides | ||
| Gde2 PCR primers Forward: 5′ CAGAAGGGACCAAGCACTCA 3′ | Sabharwal et al., 2011 | N/A |
| Gde2 PCR primers: Reverse: 5′ CCCGTTGGTTGACATTCGTG 3′ | Sabharwal et al., 2011 | N/A |
| Gde2 in situ hybridization primers: Forward: 5′ CCTCAAGACCGACCCCTT 3′ | Allen brain atlas | http://mouse.brain-map.org/ |
| Gde2 in situ hybridization primers: Reverse: 5′ GGGGCATGATCCAGAGTG 3′ | Allen brain atlas | http://mouse.brain-map.org/ |
| Mbp in situ hybridization primers: Forward: 5′ GAGGCCTGGATGTGATGG 3′ | Allen brain atlas | http://mouse.brain-map.org/ |
| Mbp in situ hybridization primers: Reverse: 5′ GGGGAACAAGTCAGGGCT 3′ | Allen brain atlas | http://mouse.brain-map.org/ |
| Gde2 qPCR primers Forward: 5′ GGCTCCAGAACACACAGTGA 3′ | This study | N/A |
| Gde2 qPCR primers Reverse: 5′ CAGGACAGTCCAGTTGAGCA 3′ | This study | N/A |
| Lef1 qPCR primers: Forward: 5’′ ATGCACGTGAAGCCTCAACA 3′ | Elbazetal., 2016 | N/A |
| Lef1 qPCR primers: Reverse: 5′ AGCTGCACTCTCCTTTAGCG 3′ | Elbazetal., 2016 | N/A |
| Gapdh qPCR primers: Forward: 5′ CGTCCCGTAGACAAAATGGT 3′ | Lebrun-Julien et al., 2014 | N/A |
| Gapdh qPCR primers: Reverse: 5′ TTGATGGCAACAATCTCCAC 3′ | Lebrun-Julien et al., 2014 | N/A |
| Software and Algorithms | ||
| ImageJ/Fiji version 1.52d | National Institutes of Health | https://imagej.nih.gov/ij/index.html |
| Corel Draw X8 | Corel | http://www.corel.com/en/ |
| Adobe Photoshop CC 2017 | Adobe | https://www.adobe.com |
| GraphPad Prism 5 | GraphPad | https://www.graphpad.com/ |
| Imaris | BITPLANE | https://imaris.oxinst.com |
| FastQC | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Fqtrim | Johns Hopkins University | https://ccb.jhu.edu/software/fqtrim/ |
| Tophat2 v2.1.1 | Johns Hopkins University | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks v2.2.1 | Cole Trapnell, University of Washington | http://cole-trapnell-lab.github.io/cufflinks/ |
| Mouse UniProt protein database (released on May 2018) | Uniprot Consortium | https://www.uniprot.org/ |
| MaxQuant v1.5.5.1 | Cox and Mann, 2008 | https://www.biochem.mpg.de/5111795/maxquant |
4-HT preparation and administration
4-hydroxytamoxifen (4-HT, sigma H7904) was prepared as described previously (Badea et al., 2003). 0.2 mg of 4HT was injected to P7 pups intraperitoneally. Pups were sacrificed to collect tissue samples at P11.
Fluorescent in situ hybridization (FISH)
Embedded frozen tissues were sectioned at 16 mm. Sections were incubated in 3% hydrogen peroxide to block endogenous peroxidase activity and permeabilized with 0.3% Triton X-100 in PBS. Sections were acetylated in 0.3% acetic anhydride. After pre-hybridization, slides were hybridized with digoxigenin-labeled sense and antisense probes overnight at 65°C. Primers used to generate probes against Gde2 and Mbp are listed in the Key Resources Table. To couple FISH with immunohistochemistry, sections were blocked in blocking solution (PerkinElmer FP1020) and incubated with sheep anti-digoxigenin-POD, 1:500 (Roche 11207733910) and relevant primary antibodies overnight at 4°C. After incubation with secondary antibodies (1 hour, room temperature), fluorescent signals were developed with TSA Plus Cy3 system (PerkinElmer NEL744001KT) according to the manufacturer’s instructions. Images were acquired on a confocal microscope (Zeiss LSM700) with 20x objective and on Zeiss LSM800 with 10x objective for tiling (9×9).
Transmission Electron Microscopy (TEM)
Anesthetized mice were perfused intracardially with fixative containing 2% PFA (EM grade), 2% glutaraldehyde, 50 mM PO4, 5 mM MgCl2, in 50mM sodium cacodylate buffer, pH 7.4, for 30 min at a rate of 1 ml/min for P14 and 2min/ml for 10-week-old mice. Brains were post-fixed in the same fixative overnight at 4°C. CC and adjacent CTX containing the ROI were carefully dissected out from 1000 μm coronally sectioned brain slices. For TEM imaging and analysis of the CC, the sagittal surface near the midline of the CC was sectioned further. All tissues were serially dehydrated, embedded, and sectioned by the JHU SOM Microscope Facility as previously described (Baxi et al., 2015). Images were acquired with a Hitachi 7600 TEM. Images (under 9700x and 65000x magnification) were obtained from the CC below the CTX at random with the operator blinded. ImageJ (National Institutes of Health) software was used to measure the number of myelinated axons and g-ratio per unit area. For g-ratio analysis, the diameters of axons and outer myelinated axons were calculated from the surface area derived from the circumference of each. For g-ratio analysis of P11 samples, selection of myelinated axons was unbiased. Specifically, a grid was first created, and axons located at grid line intersections were selected for g-ratio analysis. 100 myelinated axons were counted from 10~13 ROIs (under 9700x magnification) per animal and three animals were used per condition. For myelin sheath analysis of 10 week old samples, more than 500 axons with diameter greater than 0.5 μm were counted from 6~9 ROIs (under 9700x magnification) per animal with three animals per condition.
Cell culture
Mouse primary cortical neuronal cultures were prepared from embryonic day 16.5 (E16.5) fetuses from timed-pregnant mice using Neural Tissue Dissociation Kits (Miltenyi Biotec 130-092-628) according to the manufacturer’s recommendations. Cortical preparations were plated at a density of 2.5 × 105 cells per cm2 on plastic wells (6-well plate) or glass coverslips coated with poly-D-lysine (0.1 mg/ml in 0.1 M Trizma buffer pH 8.5) containing 1% laminin (Sigma-Aldrich L2020) and 1% PureCol Type I Bovine Collagen Solution (Advanced Biomatrix 5005-B). Cells were initially cultured in neurobasal medium (GIBCO 21103–049) containing 5% fetal horse serum, 1% penicillin/streptomycin (GIBCO 15140122), 1% Glutamax-I (GIBCO 35050061), 1% sodium pyruvate (GIBCO 11360070), 30 mM Glucose, and 2% SM1 supplement (STEMCELL Technologies 5711). The next day on DIV1, the medium was replaced with Neurobasal medium with 1% penicillin/streptomycin, 1% Glutamax-I, 1% sodium pyruvate, 30 mM Glucose, 2% SM1 supplement, and 1% N2B (STEMCELL Technologies 7156). OPCs were obtained from P6 cortices of WT pups using Neural Tissue Dissociation and Isolation kits (Miltenyi Biotec 130-090-312) with magnetic beads (Miltenyi Biotec 130-094-543) to positively select O4+ cells according to the manufacturer’s recommendations. For neuron-OL cocultures, freshly isolated OPCs in coculture media (half DMEM:F12 and half Neural basal media containing 10 mM HEPES (GIBCO 15630080), 2% SM1 supplement, 1% N2B, 0.5% penicillin/streptomycin, 5 μg/mL N-Acetyl-Cysteine (Sigma A8199), 5 μM Forskolin (Calbiochem 344270), 10ng/mL CNTF (PeproTech 450–50) were added at a density of 30,000 cells on top of DIV3 neurons and cocultured for indicated time periods. Neuronally conditioned media (CM) were collected from neuronal cultures on DIV3 and spun at 3,000xg for 10 minutes at 4°C to remove cellular debris. Freshly isolated WT OPCs in DIV3 CM were plated at a density of 15,000 cells per cm2 on plastic wells (24-well plate) or glass coverslips coated with poly-D-lysine (0.1 mg/ml in distilled water) containing 1% laminin. One day after plating, cells were replenished with CM collected from neurons on DIV4 and cultured for another 2 days prior to further analyses. For depletion of phosphacan from CM, CAT-315 antibodies recognizing neuronal phosphacan were bound to protein L magnetic beads (Thermo Fisher Scientific 88849). Antibody-bound protein L magnetic beads were subsequently incubated with WT CM overnight at 4°C.
Immunocytochemistry
Cultured cells were fixed with 4% PFA solution for 10 minutes and permeabilized in PBS with 0.3% Tween-20 for 10 minutes followed by blocking with PBS containing 1% bovine serum albumin (BSA) and 0.15% Tween-20 for 1 hour at room temperature. The cells were incubated with primary antibodies diluted in PBS (1:500) overnight at 4°C. Primary antibodies used were as follows: Rat anti-MBP (Millipore MAB386), rabbit anti-Olig2 (Millipore AB9610), guinea pig anti-Olig2 (from B. Novitch), mouse anti-MBP (Covance SMI-99P-100), mouse anti-beta-Tubulin III (Sigma-Aldrich T8578), Rabbit anti-Neurofilament H (Millipore AB1989), rabbit anti-GFAP (Agilent Z0334), mouse anti-Active-β-catenin (Anti-ABC) (Millipore 05–665). After incubation with appropriate secondary antibodies (1 hour room temperature), cells were counterstained with DAPI (Invitrogen R37606). To visualize F-actin network, cells were stained with Alexa Fluor 488-phalloidin (Invitrogen A12379) during secondary antibody incubation. Cells were mounted on slides with mounting reagent and imaged using confocal microscope (Zeiss LSM700) with 20x objective and on epifluorescence microscope (Keyence BZ-X710) with 10x objective for tiling (3×3).
Calcium transient imaging and analysis
Cortical neurons were loaded with the synthetic calcium indicator Fluo-4, AM (Thermo Scientific F14201) on DIV3. 4 mM Fluo-4 stock solution in DMSO (Sigma D8418) was diluted in HEPES-buffered extracellular solution (143 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.2, osmolality 305–310 mOsm) to yield 2 μM working solution. At the end of image acquisition, 2 μM ionomycin (Tocris 1704) was added into each well as a positive control. Live recorded images of spontaneous calcium activity were acquired with epifluorescence microscope (Keyence BZ-X710) under 10X objective. Images were streamed at 3 Hz frame rate for 3.5 minutes. Each image frame was 680 × 480 pixels, which corresponded to 0.32 mm2 rectangular area. F0 (baseline) and F are the mean fluorescence intensities and fluorescence intensities at a given time in each ROI, respectively. A change in fluorescence (Δ F/F0) was considered as a Ca2+ rise if it was > 10%. For peak analysis, F0 for each ROI trace was manually adjusted to zero. Each data point represents mean value of Δ F/F0 from at least 11 recordings per group at a given time.
mRNA-sequencing analysis
RNA-seq studies were performed using 5 week WT and Gde2KO spinal cords. The ventral half of the lumbar spinal cord was freshly dissected from 3 WT and 3 Gde2KO animals, and poly-adenylated mRNA was extracted using a QIAGEN RNeasy Plus Mini Kit (QIAGEN, 74134). cDNA libraries were prepared using the Illumina TruSeq Stranded mRNA Library Prep Kit (Illumina, RS-122–2101). Paired-end reads, 50 bp in length, were generated on an Illumina HiSeq 2500 system yielding between 50–61 million reads per sample. To analyze the RNA-seq data, reads were quality checked and trimmed using the programs fastqc (Andrews, 2010) and fqtrim (Pertea, 2010). Reads were then mapped to the mouse genome mm10 using the spliced alignment program Tophat2 v2.1.1 (Kim et al., 2013) and assembled into transcripts using Cufflinks v2.2.1 (Trapnell et al., 2012). Mapping rates ranged from 91.7% to 95.2%, with 83%–88% representing exonic reads, indicating very high-quality sequences. Transcript assemblies across all samples were merged with Cuffcompare v.2.2.1, using GENCODE v.M5 (https://www.gencodegenes.org/) (Mudge and Harrow, 2015) as reference, to create a set of gene and transcript annotations that was later used in the differential analyses. Lastly, Cuffdiff v2.2.1 was run on each pairwise comparison to determine statistically significant differentially expressed genes (cutoffs: p-val ≤ 0.05).
Mass spectrometry analysis
Neuronal CM samples were subjected to SDS-PAGE, followed by in-gel trypsin digestion (Shevchenko et al., 2006). The extracted peptides were analyzed on an Orbitrap Fusion Lumos Tribrid Mass Spectrometer coupled with the UltiMate 3000 RSLCnano liquid chromatography system (Thermo Fisher Scientific). The peptides were loaded on Acclaim PepMap100 Nano-Trap Column (100 μm × 2 cm, Thermo Fisher Scientific). Peptides were resolved at 300-nl/min flow rate using a linear gradient of 10% to 35% solvent B (0.1% formic acid in 95% acetonitrile) over 95 minutes on an EASY-Spray column (50 cm × 75 μm ID, Thermo Fisher Scientific). MaxQuant (v1.5.5.1) software was used for quantitation and identification of proteins from the mass spectrometry data using mouse UniProt database (released on May 2018) with common contaminant proteins (Cox and Mann, 2008). Search parameters included, a) trypsin as a proteolytic enzyme with up to 2 missed cleavages; b) first search peptide mass error tolerance of 20 ppm and the main search peptide mass error tolerance of 4 ppm; c) fragment mass error tolerance of 20 ppm; d) carbamidomethylation of cysteine (+57.02146 Da) as a fixed modification: e) oxidation of methionine (+15.99492 Da) and protein acetyl (+42.01056 Da) on N terminus as dynamic modifications. Peptides and proteins were filtered at 1% false-discovery rate.
Immunoblotting
Samples were sonicated in lysis buffer (20 mM Tris-HCl pH 8.0, 130 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% NP-40, and 0.2% sodium deoxycholate) containing protease inhibitor cocktail (Sigma-Aldrich P8340), subjected to reducing SDS-PAGE, and transferred to PVDF membranes for immunoblotting. For detection of phosphacan in CM, CM samples were equilibrated at a final concentration of 3 mg/ml in chondroitinase buffer (50 mM Trizma, 60 mM sodium acetate, pH 8.0) and treated with chondroitinase ABC (0.25 U per 200 μg protein) from Proteus Vulgaris (Sigma-Aldrich, C3667) for 8 hours at 37°C. CM samples were then boiled in 1x gel loading buffer for electrophoresis and immunoblotting. Nuclear and cytosolic fractions were isolated by Nu-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific 78833) according to the manufacturer’s instructions. Transferred membranes were incubated with primary antibodies overnight at 4°C. Primary antibodies used were as follows: Rabbit anti-Olig2 (1:2,000 Millipore AB9610), Mouse anti-MBP (1:2,000 Covance SMI-99P-100), Mouse anti-MOG (1:3,000 Millipore MAB5680), Rabbit anti-Neurofilament H (1:10,000 Millipore AB1989), Rabbit anti-GDE2 (1:1000), Rabbit anti-PDGF receptor alpha (1:2,000 Cell Signaling Technology 3174), rabbit anti-Olig2 (Millipore AB9610), Mouse anti-ABC (1:1,000 Millipore 05–665), Ran (10,000 BD Biosciences 610341), Rabbit anti-β-catenin (1:2,000 Cell Signaling Technology 8480), Goat anti-Contactin-2/TAG1 (1:1000 R and D Systems AF4439), Mouse anti-Chondroitin Sulfate Proteoglycan (CAT-315) (1:5000 Millipore MAB1581), Rabbit anti-GAPDH (1:1,000 Cell Signaling Technology 8884), Mouse anti-Actin (1:10,000 Millipore MAB1501). After incubation with appropriate HRP-conjugated secondary antibodies (1 hour room temperature), membranes were developed by film or by using a digital imaging system (KwikQuant, Kindle Biosciences).
Quantitative real-time PCR
Total RNA from in vivo or in vitro samples was extracted using Trizol (Thermo Fisher Scientific 15596018) and reverse transcription was carried out using SuperScript III (Thermo Fisher Scientific 18080051) according to the manufacturer’s instructions. SYBR-green labeling (Thermo Fisher 4385612) was used for quantitative real-time PCR (Applied Biosystems). The Comparative CT (DDCt) method was used to determine the relative quantities of mRNA. The primers used are listed in the Key Resources Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
3D Image quantification was performed using semi-automated Imaris (Bitplane). For quantification, 4–5 sections per animal and 3–5 animals per group were used. Littermate controls were utilized throughout the study. Images were obtained from brain regions of CC and CTX corresponding to retrosplenial and motor areas. After 3D image reconstruction, each ROI (CC and CTX) was created using the contour function in Imaris. Using spot and surface detection functions, parameters were set for size and fluorescent signal intensity threshold to create an object representing a nucleus or a cell body. After manual validation of parameter settings, the entire z stack was subjected to automated quantification using Imaris. Then, false-positive and false negative spots and surfaces were manually corrected. Number of cells was normalized to surface area of each ROI. All studies were blinded to the investigator. For quantifying intensity of Gde2 transcript signal in FISH, mean signal intensity of Gde2 channel (TSA-cy3) within each created 3D object (Olig2+ or NeuN+ cells) was obtained using Imaris. Percent cells expressing Gde2 and mean intensity of Gde2 signal per cell were analyzed. For quantifying GFP+ nuclei in Rosa26 Tcf./Lef-H2B-EGFP mice, spots were designated as GFP+ when above a set threshold signal intensity. This threshold was applied across all analysis. The number of NeuN+ GFP+ and Olig2+ GFP+ cells in each ROI was analyzed using MATLAB modules in Imaris. For quantifying MBP+ OLs at Stage1-Stage3 in culture, 3×3 tiled images with 10x objective (3.99 mm2 area) were acquired from 2 wells per animal with at least three biological samples per experiment. The number of MBP+ cells for each stage was divided by the number of Olig2+ cells in each group and then normalized to control. GraphPad Prism 5 software was used to generate plots and to conduct statistical analysis. The mean ± SEM are shown. Statistical significance was determined by a two-tailed, unpaired Student’s t test, 1-way or 2-way ANOVA test and is shown by * p < 0.05, ** p < 0.01, *** p < 0.001. Power analysis (Sample Size Calculator, provided by UCSF Clinical & Translational Science Institute; https://www.sample-size.net/sample-size-means/) was conducted for all analyses. All samples sizes are sufficient to reach 80% power that detect estimated difference in means with a 5% significance level.
Supplementary Material
Highlights.
GDE2 is expressed in neurons and a subset of oligodendrocytes
Loss of neuronal GDE2 delays oligodendrocyte maturation and impairs myelination
GDE2 stimulates canonical Wnt signaling in neurons, which releases phosphacan
Neuronally derived phosphacan promotes oligodendrocyte maturation
ACKNOWLEDGMENTS
We thank C. Wladyka and Y. Li for technical assistance; Drs. D. Bergles, P. Calabresi, and S. Bouyain and the Sockanathan lab for discussions; L. Florea for bioinformatics; Drs. J. Nathans (Rosa26 Tcf/Lef H2B-EGFP), M. Taketo (Ctnnb1flex3/+), K. Nave (Nex:Cre), and D. Bergles (PDGFαR-CreER) for mice; and the Multiphoton Imaging Core of the Johns Hopkins P30 Center for Neuroscience Research (NS050274). B.-R.C. was supported by a Fulbright Graduate Study Award (Science and Engineering). This work was supported by the National Institutes of Health (grant R01NS046336 to S.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107540.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data, Available at. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Badea TC, Wang Y, and Nathans J (2003). A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J. Neurosci 23, 2314–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, and Raff MC (1993). Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361, 258–260. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, and Raff MC (1993). Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 118, 283–295. [DOI] [PubMed] [Google Scholar]
- Baxi EG, DeBruin J, Tosi DM, Grishkan IV, Smith MD, Kirby LA, Strasburger HJ, Fairchild AN, Calabresi PA, and Gocke AR (2015). Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. J. Neurosci 35, 8626–8639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, and Baraban JM (1996). Expression of the APC tumor suppressor protein in oligodendroglia. Glia 17, 169–174. [DOI] [PubMed] [Google Scholar]
- Bocchi R, Egervari K, Carol-Perdiguer L, Viale B, Quairiaux C, De Roo M, Boitard M, Oskouie S, Salmon P, and Kiss JZ (2017). Perturbed Wnt signaling leads to neuronal migration delay, altered interhemispheric connections and impaired social behavior. Nat. Commun 8, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave C, Park S, Rodriguez M, Nakamura M, Hoke A, Pletnikov M, and Sockanathan S (2017). GDE2 is essential for neuronal survival in the postnatal mammalian spinal cord. Mol. Neurodegener 12, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, and Lubetzki C (2000). Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc. Natl. Acad. Sci. USA 97, 7585–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, and Walsh CA (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369. [DOI] [PubMed] [Google Scholar]
- Cho C, Smallwood PM, and Nathans J (2017). Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron 95, 1056–1073.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Davison AN, and Peters A (1970). Myelination (Charles C. Thomas), p. 56. [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, and Barres BA (2006). Functional genomic analysis of oligodendrocyte differentiation. J. Neurosci 26, 10967–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer CA, Katoh T, Tiemeyer M, and Matthews RT (2015). Neurons and glia modify receptor protein-tyrosine phosphatase z (RPTPz)/phosphacan with cell-specific O-mannosyl glycans in the developing brain. J. Biol. Chem 290, 10256–10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz B, Traka M, Kunjamma RB, Dukala D, Brosius Lutz A, Anton ES, Barres BA, Soliven B, and Popko B (2016). Adenomatous polyposis coli regulates radial axonal sorting and myelination in the PNS. Development 143, 2356–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubelen M, Bostaille N, Cabochette P, Gauquier A, Tebabi P, Dumitru AC, Koehler M, Gut P, Alsteens D, Stainier DYR, et al. (2018). A molecular mechanism for Wnt ligand-specific signaling. Science 361, eaat1178. [DOI] [PubMed] [Google Scholar]
- Faissner A, Heck N, Dobbertin A, and Garwood J (2006). DSD-1-proteoglycan/phosphacan and receptor protein tyrosine phosphatase-beta isoforms during development and regeneration of neural tissues. Adv. Exp. Med. Biol 557, 25–53. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, and Rowitch DH (2009). Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 23, 1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave KA (2006). Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Wieser GL, Pieper A, Spitzer S, Weege B, Yan K, Edgar JM, Yagensky O, Wichert SP, Agarwal A, et al. (2016). A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat Neurosci. 20, 10–15. [DOI] [PubMed] [Google Scholar]
- Han C, Yan D, Belenkaya TY, and Lin X (2005). Drosophila glypicans Dally and Dally-like shape the extracellular Wingless morphogen gradient in the wing disc. Development 132, 667–679. [DOI] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, and Taketo MM (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, Buxbaum JD, and Schlessinger J (2002). A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat. Genet 32, 411–414. [DOI] [PubMed] [Google Scholar]
- Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe RF, and Waterman ML (2001). Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet 28, 53–57. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, and Fields RD (2006). Astrocytes promote myelination in response to electrical impulses. Neuron 49, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda CY, Waghray D, Levin AM, Thomas C, and Garcia KC (2012). Structural basis of Wnt recognition by Frizzled. Science 337, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, and Bergles DE (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, and Richardson WD (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci 9, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS, Lo M, Pak S, Mattis J, Lim BK, et al. (2013). Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature 496, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, and Wegner M (1998). Sox10, a novel transcriptional modulator in glial cells. J. Neurosci 18, 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprianou S, Chatzopoulou E, Thomas JL, Bouyain S, and Harroch S (2011). A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc. Natl. Acad. Sci. USA 108, 17498–17503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn AM, Dubey R, Neitzel LR, Tacchelly-Benites O, Yang E, Marceau CD, Davis EM, Patel BB, Bahrami-Nejad Z, Travaglini KJ, et al. (2016). Comparative genetic screens in human cells reveal new regulatory mechanisms in WNT signaling. eLife 5, e21459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Julien F, Bachmann L, Norrmén C, Trötzmüller M, Köfeler H, Rüegg MA, Hall MN, and Suter U (2014). Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J. Neurosci 34, 8432–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Leach MK, Redmond SA, Chong SY, Mellon SH, Tuck SJ, Feng ZQ, Corey JM, and Chan JR (2012). A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat. Methods 9, 917–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentferink DH, Jongsma JM, Werkman I, and Baron W (2018). Grey matter OPCs are less mature and less sensitive to IFNg than white matter OPCs: consequences for remyelination. Sci. Rep 8, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, and He X (2002). Control of beta-catenin phosphorylation/degradation by a dualkinase mechanism. Cell 108, 837–847. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, and Jacobowitz DM (2004). Immunohistochemical localization of aspartoacylase in the rat central nervous system. J. Comp. Neurol 472, 318–329. [DOI] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, and Corfas G (2012). A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matas-Rico E, van Veen M, Leyton-Puig D, van den Berg J, Koster J, Kedziora KM, Molenaar B, Weerts MJ, de Rink I, Medema RH, et al. (2016). Glycerophosphodiesterase GDE2 promotes neuroblastoma differentiation through glypican release and is a marker of clinical outcome. Cancer Cell 30, 548–562. [DOI] [PubMed] [Google Scholar]
- Maurel P, Rauch U, Flad M, Margolis RK, and Margolis RU (1994). Phosphacan, a chondroitin sulfate proteoglycan of brain that interacts with neurons and neural cell-adhesion molecules, is an extracellular variant of a receptor-type protein tyrosine phosphatase. Proc. Natl. Acad. Sci. USA 91, 2512–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayoral SR, and Chan JR (2016). The environment rules: spatiotemporal regulation of oligodendrocyte differentiation. Curr. Opin. Neurobiol 39, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge JM, and Harrow J (2015). Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm Genome 26, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, and Pleasure SJ (2011). Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J. Neurosci 31, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA (2010). Myelination and support of axonal integrity by glia. Nature 468, 244–252. [DOI] [PubMed] [Google Scholar]
- Nogusa Y, Fujioka Y, Komatsu R, Kato N, and Yanaka N (2004). Isolation and characterization of two serpentine membrane proteins containing glycerophosphodiester phosphodiesterase, GDE2 and GDE6. Gene 337, 173–179. [DOI] [PubMed] [Google Scholar]
- Park S, Lee C, Sabharwal P, Zhang M, Meyers CL, and Sockanathan S (2013). GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 339, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea G (2010). Fqtrim: trimming and filtering of next-gen reads. http://ccb.jhu.edu/software/fqtrim/.
- Power J, Mayer-Pröschel M, Smith J, and Noble M (2002). Oligodendrocyte precursor cells from different brain regions express divergent properties consistent with the differing time courses of myelination in these regions. Dev. Biol 245, 362–375. [DOI] [PubMed] [Google Scholar]
- Rao M, and Sockanathan S (2005). Transmembrane protein GDE2 induces motor neuron differentiation in vivo. Science 309, 2212–2215. [DOI] [PubMed] [Google Scholar]
- Remahl S, and Hildebrand C (1982). Changing relation between onset of myelination and axon diameter range in developing feline white matter. J. Neurol. Sci 54, 33–45. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Choi J, Park S, and Sockanathan S (2012). Gde2 regulates cortical neuronal identity by controlling the timing of cortical progenitor differentiation. Development 139, 3870–3879. [DOI] [PubMed] [Google Scholar]
- Sabharwal P, Lee C, Park S, Rao M, and Sockanathan S (2011). GDE2 regulates subtype-specific motor neuron generation through inhibition of Notch signaling. Neuron 71, 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane H, Yamamoto H, Matsumoto S, Sato A, and Kikuchi A (2012). Localization of glypican-4 in different membrane microdomains is involved in the regulation of Wnt signaling. J. Cell Sci 125, 449–460. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Shimogori T, VanSant J, Paik E, and Grove EA (2004). Members of the Wnt, Fz, and Frp gene families expressed in postnatal mouse cerebral cortex. J. Comp. Neurol 473, 496–510. [DOI] [PubMed] [Google Scholar]
- Solly SK, Thomas JL, Monge M, Demerens C, Lubetzki C, Gardinier MV, Matthieu JM, and Zalc B (1996). Myelin/oligodendrocyte glycoprotein (MOG) expression is associated with myelin deposition. Glia 18, 39–48. [DOI] [PubMed] [Google Scholar]
- Spitzer SO, Sitnikov S, Kamen Y, Evans KA, Kronenberg-Versteeg D, Dietmann S, de Faria O Jr., Agathou S, and Káradóttir RT (2019). Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron 101, 459–471.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, and Fields RD (2002). Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, and Arlotta P (2014). Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, and Macklin W (1997). Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J. Cell Biol 137, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Levine JM, Miller RH, and Trapp BD (1999). Rat optic nerve oligodendrocytes develop in the absence of viable retinal ganglion cell axons. J. Cell Biol 146, 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemori H, Sato S, Yagi T, Aizawa S, and Yamamoto T (1994). Initial events of myelination involve Fyn tyrosine kinase signalling. Nature 367, 572–576. [DOI] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, and Barres BA (1998). Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21, 63–75. [DOI] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, et al. (2009). HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci 12, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Eisen AM, McBain CJ, and Gallo V (1998). A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 125, 2901–2914. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang S, and Anderson DJ (2000). Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 25, 331–343. [DOI] [PubMed] [Google Scholar]
- Zuchero JB, Fu MM, Sloan SA, Ibrahim A, Olson A, Zaremba A, Dugas JC, Wienbar S, Caprariello AV, Kantor C, et al. (2015). CNS myelin wrapping is driven by actin disassembly. Dev. Cell 34, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq raw data generated during this study are publicly available at National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO). The accession number for the data reported in this paper is GSE147144.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD018080.