

Abstract

Peritoneal carcinomatosis (PC) has an extremely poor prognosis, which leads to a significantly decreased overall survival in patients with peritoneal implantation of cancer cells. Administration of sodium selenite by intraperitoneal injection is highly effective in inhibiting PC. Our previous study found that selenium nanoparticles (SeNPs) have higher redox activity and safety than sodium selenite. In the present study, we examined the therapeutic effect of SeNPs on PC and elucidated the potential mechanism. Our results revealed that intraperitoneal delivery of SeNPs to cancer cells in the peritoneal cavity of mice at a tolerable dose was beneficial for prolonging the survival time of mice, even better than the optimal dose of cisplatin. The underlying mechanism involved in SeNP-induced reactive oxygen species (ROS) production caused protein degradation and apoptotic response in cancer cells. Interestingly, N-acetyl-l-cysteine (NAC), recognized as a ROS scavenger, without reducing the efficacy of SeNPs, enhanced ROS production and cytotoxicity. The effect of NAC was associated with the following mechanisms: (1) the thiol groups in NAC can increase the biosynthesis of endogenous glutathione (GSH), thus increasing the production of SeNP-induced ROS and cytotoxicity and (2) redox cycling of SeNPs was directly driven by thiol groups in NAC to produce ROS. Moreover, NAC, without increasing the systematic toxicity of SeNPs, decreased SeNP-induced lethality in healthy mice. Overall, we demonstrated that SeNPs exert a potential cytotoxicity effect by inducing ROS production in cancer cells; NAC effectively heightens the property of SeNPs in vitro and in vivo.
Introduction
Peritoneal carcinomatosis (PC) is primarily induced through primary tumors occurring in organs confined to the peritoneal cavity, including ovarian, liver, stomach, pancreas, and colon.1 The treatment and prognosis of PC vary based on primary cancer. Although therapy with the intention to cure is offered to selected patients using cytoreductive surgery with chemotherapy, the prognosis remains poor for most of the patients.2,3 PC has the potential to disseminate and grow in the peritoneal cavity. It can also lead to tumor recurrence and the formation of malignant ascites or numerous small tumor nodules and various sizes of tumor masses, which are refractory to treatment and have been shown to significantly decrease overall survival in patients with peritoneal implantation of cancer cells.4 Intraperitoneal (ip) chemotherapy, which provides relatively higher and longer drug half-life of antineoplastic agents in the peritoneal cavity,5,6 is a promising approach for treating malignancies in the peritoneal cavity approved by the U.S. Food and Drug Administration.6
Selenium (Se) is an essential trace element with a series of health benefits for human health including anticancer properties. Certain Se compounds, such as selenite and methyl selenium, have a strong capacity of oxidizing thiols, thus leading to the formation of highly reactive and unstable metabolites, which undergo redox cycling with oxygen to form reactive oxygen species (ROS).7 Sodium selenite is known as one of the most redox-active Se compounds in inducing ROS production and a potent Se compound for inhibiting cancer cell proliferation for a long time.8,9 Administration of sodium selenite via ip injection is highly effective in inhibiting PC in mice bearing murine hepatocarcinoma 22 cells (H22 cells). The potential mechanisms involve the selective accumulation of Se in cancer cells in the form of selenium nanoparticles (SeNPs) and the abundant production of ROS.10 A previous study found that intraperitoneal administration of SeNPs is an effective and safe approach for preventing the proliferation of cancer cells in the peritoneal cavity.11 Our latest study found that SeNPs have higher redox activity than sodium selenite, especially under the circumstances of limited glutathione (GSH) and/or nicotinamide adenine dinucleotide phosphate (NADPH) levels, known as reducing equivalents in facilitating Se redox and biotransformation in vitro and in vivo.12 The anticancer activity and potential mechanisms of SeNPs have been intensively studied in certain cancer cell lines; nevertheless, the in vivo antitumor mechanisms need further investigation. Thus, this study revisited the therapeutic effect of SeNPs on PC in mice bearing H22 cells in the peritoneal cavity.
N-Acetyl-l-cysteine (NAC), recognized as a ROS scavenger, is commonly used as a tool for studying the mechanisms or explaining the consequences of oxidative stress in vitro and in vivo and as a therapeutic drug for antioxidant treatment in clinics.13,14 The redox chemistries of the group XVI elements, oxygen and sulfur (Figure 1), are due to the central role of NAC in biology.14 Nevertheless, a dual effect of NAC on selenite cytotoxicity has been revealed in HepG2 cells because NAC is a direct ROS scavenger and a precursor for the biosynthesis of GSH,13,15 which acts as either a well-recognized antioxidant to protect cell viability or a unique pro-oxidant for enhancing selenium-based ROS production and cytotoxicity.15 However, the interactions between NAC and SeNP-induced oxidative stress and apoptosis of cancer cells have not yet been reported. In the present study, we revealed the influences of NAC on the antineoplastic effects of SeNPs and elucidated the potential mechanism of action. Understanding the unique association between NAC and SeNP-induced oxidative stress and apoptosis of cancer cells may help to explain the controversy in the literature over the complex relationship between selenium and NAC and ultimately the anticancer properties of selenium.
Figure 1.
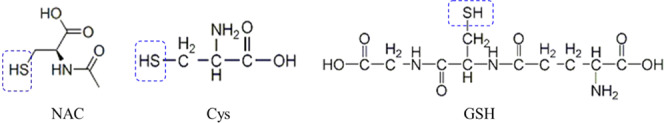
Chemical structures of NAC, Cys, and GSH.
Results and Discussion
Therapeutic Effect of SeNPs in Mice Bearing H22 Cells in the Peritoneal Cavity
Transmission electron microscopy (TEM) observation and dynamic light scattering (DLS) analysis showed that the mean size of SeNPs used in this study was 40 nm (Figure 2A,B). Since a previous study suggested that intraperitoneal administration of SeNPs is an effective and safe approach for preventing and inhibiting the proliferation of cancer cells in the peritoneal cavity,11 herein, we investigated the therapeutic effect of SeNPs in mice bearing H22 cells. First, we evaluated the effect of SeNPs on prolonging survival in mice bearing H22 cells. H22 model mice were ip injected with saline as control or SeNPs (3 mg Se/kg) once. Without therapy, cancer cells in the peritoneal cavity proliferated quickly as indicated by the abnormal body weight gain (Figures S1 and 2C) and the median survival time was only 8 days following inoculation with H22 cells (Figure 2D). On the contrary, SeNPs significantly suppressed the proliferation of cancer cells that was manifested by the decreased body weight in the first 3 days post SeNP treatment and the slower body weight gain in the next few days compared with the control (Figure 2C). Consequently, the median survival time increased to 24 days (Figure 2D) without obvious side effects. Thus, a higher dose of SeNPs (4 mg Se/kg) was used to explore its therapeutic potential; 9 mg/kg cisplatin was used in parallel for comparison.
Figure 2.

Therapeutic effects of SeNPs in mice bearing H22 cells in the peritoneal cavity. (A) TEM analysis of SeNPs. (B) Size distribution of SeNPs detected by DLS. Experiment 1. Survival of mice after treatment with SeNPs. Mice (n = 6/group) were ip injected with saline as control or SeNPs (3 mg Se/kg). (C) Body weight. (D) Survival time. Experiment 2. Survival of mice after treatment with SeNPs or cisplatin. Mice (n = 10) were ip injected with saline as control, SeNPs (4 mg Se/kg), or cisplatin (9 mg/kg). (E) Body weight. (F) Survival time. Data are presented as the mean ± standard error of the mean (SEM). **P < 0.01 and ***P < 0.001 compared to the control group; #P < 0.05, ##P < 0.01, and ###P < 0.001 compared to the cisplatin group.
Cisplatin at the dose of 9 mg/kg appeared as an optimal dose in this model. Evidence showed that a dose of 10 mg/kg triggered severe systemic toxicity, while a dose of 5 mg/kg was insufficient to effectively kill cancer cells,16 and a weekly ip injection of cisplatin at a dose of 8 mg/kg once, twice, or thrice presented a similar effect in the model.12 Thereby, to enhance the performance of cisplatin in prolonging survival without toxicological sequelae, 9 mg/kg cisplatin was chosen. Cisplatin was more efficient in suppressing body weight gain of mice than SeNPs (Figure 2E), but the medium survival time was only 12 days (Figure 2F), thus highlighting that the excessive body weight suppression was attributed to systemic toxicity rather than anticancer effect. Again, SeNPs significantly increased the median survival time of the highly malignant tumor model mice to 22 days (Figure 2F) at a highly tolerable dose of 4 mg Se/kg. Next, we studied the bioeffects in the early-stage post ip injection of SeNPs in mice bearing H22 cells for expounding the anticancer mechanism of SeNPs.
Rapid Apoptotic Response of H22 Cells Post SeNP Administration In Vivo
H22 model mice were ip injected with saline as control or SeNPs (4 mg Se/kg). Cancer cells in the peritoneal cavity quickly proliferated to approximately 100 million (Figure 3A) after 2 days of inoculation. SeNPs markedly suppressed cancer cell proliferation in a time-dependent manner (Figures 3A and S2). The morphological or apoptotic volume decrease of cancer cells was observed at 3 h after the injection (Figure 3B). At 24 h post treatment, SeNPs caused pronounced protein degradation (Figure 3C), indicating that cells suffered serious damage. Thus, we tested apoptotic response factors at 3 h post the injection. Apoptosis-associated proteins including caspase 9 and poly(ADP-ribose) polymerase (PARP) were downregulated, and cleaved PARP was upregulated (Figure 3D); prosurvival-associated proteins including AKT and NF-κBp65 were suppressed (Figure 3E); tissue injury and DNA repair-associated protein γ-H2AX was induced (Figure 3F); DNA repair-associated mRNA XPC was decreased (Figure 3G); and growth arrest and DNA damage-associated mRNAs including gadd45β and γ were increased (Figure 3G). These results suggested that cancer cells in the peritoneal cavity of mice suffered serious DNA damage and the apoptosis was already initiated at 3 h after SeNP treatment. The anticancer activities of SeNPs have been studied in several cancer cell lines; however, the mechanisms remain unclear and need further study. Therefore, next, we explored the potential mechanism of SeNP-induced H22 cell death.
Figure 3.

SeNP-triggered apoptotic responses of H22 cells in mice. Experiment 3. H22 model mice (n = 6/group) were ip injected with saline as a control, or SeNPs (4 mg Se/kg); mice in control and SeNP (3 h) groups were sacrificed at 3 h post the injection; mice in the SeNP (24 h) group were sacrificed at 24 h. (A) Viable cells. (B) Cellular morphology at 3 h post treatment. (C) Illustration of protein degradation at 24 h post treatment (n = 2/group). (D) Apoptosis-associated proteins. (E) Prosurvival-associated proteins. (F) γ-H2AX protein. (G) Tissue injury and DNA repair-associated mRNA levels. Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to control.
SeNP-Induced ROS Productions in H22 Cell Suspension and H22 Cell Lysate
SeNPs dose-dependently induced ROS production in the suspension of H22 cells (Figure 4A). But the increased ROS that was induced by SeNPs were effectively suppressed when CDNB (antagonist of GSH)17 was added to the suspension of H22 cells (Figure 4B). This is consistent with our previous study that GSH can dose-dependently stimulate redox and biotransformation of SeNPs to produce ROS in a pure enzyme system.11,18 NAC, recognized as a ROS scavenger,19,20 increased SeNP-induced ROS production in a dose-dependent manner (Figure 4C) but without suppressing the ROS levels as expected. To verify the results above obtained, we evaluated SeNP-related ROS production in the lysate of H22 cells. As expected, SeNPs dose-dependently increased ROS production (Figure 4D), the addition of CDNB dampened ROS levels (Figure 4E), and again, the addition of NAC enhanced ROS production (Figure 4F). These results reinforce the notion that intracellular GSH has an essential role in promoting SeNP biotransformation and ROS production, especially when GSH is the most abundant thiol-containing small molecule in cells.21 However, the cross talk between selenium and NAC in inducing ROS production has not been well elaborated. In the present study, we found that NAC participates in the redox and biotransformation of SeNP, which has not been reported hitherto. Thereby, we tested the influence of NAC on SeNP-induced cytotoxicity in vitro and in vivo.
Figure 4.

SeNP-induced ROS productions in H22 cell suspension and lysate. Experiments were carried out at 37 °C in H22 cell suspension. (A) Dose effect of SeNPs. (B) Effect of CDNB. (C) Effect of NAC. Experiments were carried out at 25 °C in the lysate of H22 cells. (D) Dose effect of SeNPs. (E) Effect of CDNB. (F) Influence of NAC. Experiments were carried out in cell suspension or cell lysate in the presence of 50 μM DCFH-DA. The cell suspension and cell lysate were preincubated with CDNB for 10 min at the indicated concentration and temperature, respectively. Data are presented as the mean of two replicates; the error bar represents the range. In some data points, the range was smaller than the symbol. The vehicle control has been subtracted from the treatments.
Effects of NAC on SeNP-Induced Cytotoxicity In Vitro and In Vivo
HepG2 cells were seeded in 96-well plates at a density of 50 000 cells per well for 24 h before experiment and were treated with SeNPs for another 24 h. SeNPs dose-dependently decreased viable cells at 24 h after treatment (Figure 5A), and the addition of NAC enhanced the cell-killing effect of SeNPs in a dose-dependent manner (Figure 5B). Similar results were also observed in the Tca8113 cell line (Figure 5C). Subsequently, we investigated the effects of NAC on SeNP-induced cytotoxicity in mice bearing H22 cells.
Figure 5.

Effects of NAC on SeNP-induced cancer cell proliferation inhibition in vitro. (A) Dose effect of SeNPs. (B) Influence of NAC on SeNP-induced cytotoxicity in HepG2 cells. (C) Influence of NAC on SeNP-induced cytotoxicity in Tca8113 cells. Data are presented as mean ± SEM (n = 6). **P < 0.01, ***P < 0.001 compared to control; #P < 0.05, ##P < 0.01, and ###P < 0.001 compared to SeNPs.
Mice were randomly divided into a SeNP-treated group and a SeNP-plus-NAC-treated group (n = 6/each). Mice in the SeNPs plus NAC group were ip injected with NAC (150 mg/kg) at 24 h after H22 cells were inoculated; all mice were ip injected with SeNPs (2 mg Se/kg) at 48 h postinoculation, and all mice were sacrificed after 1 h post-SeNP administration. Results showed that SeNPs plus NAC presented a higher inhibiting effect than SeNPs alone on H22 cells in the peritoneal cavity of mice (Figure 6A). Research has shown that GSH has a crucial role in cell defense mechanisms by acting as an antioxidant or conjugating with toxic electrophiles.22,23 The sensitivity of cancer cell lines to chemotherapeutic agents is inversely correlated with intracellular GSH levels.24−27 A positive correlation between the elevation of intracellular GSH levels and resistance to several chemotherapeutic agents (such as platinum or alkylating agents) has been established.25,28,29 Nevertheless, our previous study revealed that the key components in promoting Se biotransformation, including Grx-coupled GSH and Trx systems, were not compromised by treatment with sodium selenite or SeNPs when intracellular-pronounced apoptotic responses were initiated.10,12 SeNPs plus NAC treatment increased the most abundant thiol-containing small molecule GSH levels in cells (Figure 6B), showing that SeNPs can efficiently utilize GSH to generate excessive ROS and inhibit cancer cells when SeNPs were selectively accumulated in cancer cells post treatment.11,12 Selenium compounds, involving selenomethionine and selenite, can effectively reduce the multidrug resistance that is caused by cisplatin and the carboplatin-induced increase in intracellular GSH levels,30−32 thus prolonging the effectiveness of a repetitive platinum complex in human ovarian tumor xenograft treatment.31,33 Apoptotic shrinkage (Figures 3B and6C) and higher proliferation inhibition (Figure 6B,C,A) suggested that the increased GSH levels in H22 cells (Figure 6B), without inducing cell resistance to SeNPs, enhanced cytotoxicity (Figure 6A).
Figure 6.

Influence of NAC on the effects of SeNPs in vivo. Experiment 4. Mice in the SeNPs plus NAC group were ip injected with NAC (150 mg/kg) at 24 h post H22 cell inoculation, and then, all mice (n = 6) were ip injected with SeNPs (2 mg Se/kg) at 48 h post the inoculation. Mice were sacrificed at 1 h post SeNP administration. (A) Viable cells. (B) Intracellular GSH levels. (C) Cellular morphology. Experiment 5. Mice (n = 6/group) were ip injected with NAC at indicated doses at 24 h post H22 cell inoculation and were sacrificed at 24 h post NAC administration. (D) Intracellular GSH levels. Experiment 6. Mice in NAC and SeNPs plus NAC groups were ip injected with NAC; 24 h later, mice in SeNPs and SeNPs plus NAC groups were ip injected with SeNPs. (E) Survival time. #P < 0.05, ###P < 0.001 compared to SeNPs.
Another experiment was performed to investigate the dose effect of NAC on intracellular GSH levels. Mice were randomly divided into four groups (n = 6/each) and were ip injected with saline as control, or NAC (50, 150, or 300 mg/kg) at 24 h post H22 cell inoculation. Mice were sacrificed after 24 h post NAC administration. NAC (150 mg/kg) increased intracellular GSH levels by 37% without a significant statistical difference because of the large individual differences; 300 mg/kg NAC significantly increased intracellular GSH levels by 47% (Figure 6D), which is the key element to enhance SeNP-induced ROS production and cytotoxicity with the co-administration of SeNPs and NAC. Nevertheless, these results raised a concern whether NAC could increase the systemic toxicity of SeNPs, which was further explored through safety evaluation. Healthy mice were randomly divided into the following five groups (n = 8/each): NAC (250 mg/kg), SeNPs (5 or 6 mg Se/kg), and SeNPs (5 or 6 mg Se/kg) plus NAC (250 mg/kg). Mice in NAC and SeNPs plus NAC groups were ip injected with NAC (250 mg/kg); 24 h later, mice in SeNPs and SeNPs plus NAC groups were ip injected with SeNPs (5 or 6 mg Se/kg). Results showed that the administration of SeNPs (5 or 6 mg/kg) led to 25 or 50% survival in healthy animals (Figure 6E), while the co-administration of NAC (250 mg/kg) increased the survival to 87.5 or 100% (Figure 6E), indicating that NAC decreases systemic toxicity of SeNPs.
Redox Cycling of SeNPs Driven by GSH and/or NAC in Chemical Systems
Research showed that the basal GSH level in H22 cells was approximately 3.5 nmol per million cells, and the basal selenium level in H22 cells was approximately 8 pmol per million cells.11,34,35 Since intracellular selenium levels increased more than one hundred times in H22 cells of peritoneal cavity post therapeutic doses of selenium were delivered by ip injection,11,12 the molar ratio of GSH/Se in H22 cells from mice ranged from 1 to 4. SeNPs could be used as a superior Se species in such a low ratio of GSH/Se because SeNPs that present higher redox activity than selenite when reducing equivalents are limited.8,12 Yet, sufficient reducing equivalents could enhance ROS production especially when the molar ratio of GSH/Se is at the range of 25.7–257.8 Indeed, we found that both GSH and NAC dose-dependently induced ROS production in chemical systems at the ratio of 20 and 100 (Figure 7A,B). When the ratio of GSH/Se was set at 1, which is known as an inadequate reducing equivalent circumstance, NAC dose-dependently enhanced ROS production (Figure 7C); when the ratio of NAC/Se was set at 1, GSH also enhanced ROS production in a dose-dependent manner (Figure 7D). NAC as a precursor of GSH can facilitate intracellular GSH biosynthesis by increasing the supply of cysteine (Cys) sulfhydryl group,14,15 which is the core constituent of GSH and NAC (Figure 1). The thiol (−SH) in Cys, NAC, and GSH (Figure 1) can promote the biotransformation of selenium compounds with redox activity,7 but GSH was more efficient in promoting SeNP metabolism compared with NAC or Cys as indicated by the ROS production (Figures 7E and S3A) and ROS formation kinetics (Figures 7F and S3B).
Figure 7.

SeNP-induced ROS production in GSH and/or NAC systems. (A) Dose effect of GSH on SeNP-induced ROS production. (B) Dose effect of NAC on SeNP-induced ROS production. (C) Dose effects of NAC on SeNP-induced ROS production in the presence of GSH. (D) Dose effects of GSH on SeNP-induced ROS production in the presence of NAC. (E) Comparison between GSH and NAC in driving SeNP-induced ROS production. (F) ROS formation kinetics at the molar ratios (GSH/Se or NAC/Se) of 20 and 100. Experiments were carried out in 50 mM PBS (1 mM ethylenediamine tetraacetic acid disodium (EDTA-Na2), pH 7.5) at 37 °C in the presence or absence of 50 μM DCFH-DA. Data are presented as the mean of two replicates; the error bar represents the range. In some data points, the range was smaller than the symbol. The vehicle control has been subtracted from the treatments. *P < 0.05, **P < 0.01 compared to the GSH/SeNPs group.
In summary, the present study found that intraperitoneal delivery of SeNPs to cancer cells in the peritoneal cavity of mice at a tolerable dose was beneficial for prolonging survival time of mice, even better than the optimal dose of cisplatin. The underlying mechanism involved SeNP-induced abundant ROS production and pronounced apoptotic responses. The thiol groups in NAC increased the biosynthesis of endogenous GSH, thus promoting the biotransformation of SeNPs; moreover, the thiol groups in NAC directly participated in the redox cycling of SeNPs. Both aspects contributed to the increased ROS production and the enhanced cytotoxicity when intracellular reducing equivalents were limited by therapeutic doses of SeNPs that were delivered by ip injection. However, NAC, without increasing the systematic toxicity of SeNPs, decreased SeNP-induced lethality in healthy mice. Overall, the present study demonstrated that SeNPs have a potent cytotoxic effect by inducing ROS production and NAC can effectively heighten the property of SeNPs in vitro and in vivo.
There are several limitations in the present work: (1) compound containing free sulfhydryl groups, like cysteine, could be considered the precursor of GSH. Figure S3 shows that SeNPs can be driven by cysteine to produce ROS in a dose-dependent manner in a chemical system, indicating that cysteine may possess an effect similar to that of NAC. However, whether cysteine enhances the cytotoxicity by increasing selenium-induced ROS levels in mice bearing H22 cells remains unclear. (2) Zhong et al. indicated that superoxide dismutase (SOD) can protect against cytotoxicity of selenite by decreasing superoxide radicals induced by selenite in human prostate cancer cells;36 we also validated this conclusion with gallic acid (GA) as an antioxidant dose-dependently scavenged ROS induced by selenite, thus reducing ROS levels and cytotoxicity in TCA8113 oral cancer cells.10 However, the role of ROS in the enhanced cytotoxicity by SeNPs using antioxidants or overexpression of antioxidant enzymes has not been well illustrated in the present study, though SeNPs dose-dependently induce ROS production, thus killing cancer cells in a dose-dependent manner in mice bearing H22 cells.12
Materials and Methods
Chemicals and Drugs
Reduced glutathione (GSH), bovine serum albumin (BSA), 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB), cisplatin, sodium selenite, 1-chloro-2,4-dinitrobenzene (CDNB), and N-Acetyl-l-cysteine (NAC) were all obtained from Sigma (St. Louis, MO). Radioimmunoprecipitation assay (RIPA) reagent and bicinchoninic acid (BCA) protein assay kit were purchased from Beyotime Biotechnology (Shanghai, China). The primary antibodies against β-actin, protein kinase B (AKT), and nuclear transcription factor kappa-Bp65 (NF-κBp65) were acquired from Santa Cruz Biotechnology (Dallas, TX). The primary antibodies against phosphorylated histone 2AX (γ-H2AX), caspase 9, PARP, and antirabbit IgG, as well as antimouse IgG secondary antibodies, were all obtained from Cell Signaling Technology, Inc. (Boston, MA). ECL Plus reagent and poly(vinylidene difluoride) (PVDF) membrane were purchased from Bio-Rad Laboratories, Inc. (Hercules, California). Other chemicals were of the highest grade available.
Preparation of Elemental Se Nanoparticles and Characterization
SeNPs were prepared in a redox system of sodium selenite and GSH with BSA as a stabilizer.37−39 Transmission electron microscopy (TEM, HT7700, Hitachi, Tokyo, Japan) and dynamic light scattering (DLS, DelsaMax PRO, Beckman, Krefeld, Germany) were used to observe and analyze the average diameter of SeNPs.
Evaluation of Redox Cycling of SeNPs In Vitro
ROS levels were measured in the presence of 50 μM 2′,7′-dichlorofluorescin diacetate (DCFH-DA); fluorescence intensity of the DCFH-DA oxidation product was detected at an excitation wavelength of 488 nm and an emission wavelength of 525 nm in a microplate reader (Molecular Devices, Sunnyvale, CA). The reaction volume in all experiments was adjusted to 200 μL with 50 mM PBS (1 mM EDTA-Na2, pH 7.5) or cell lysates. Experiments using GSH or NAC and experiments using cell lysate were carried out at 37 and 25 °C, respectively.
Animals and H22 Model Mice
Male Kunming mice (20–22 g) and animal diet were all purchased from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China). Mice were housed at a controlled temperature of 22 ± 2 °C, relative humidity of 45 ± 10%, and 12 h light–dark cycles; they were provided with standard laboratory chow and tap water ad libitum. All animal studies (including the mice euthanasia procedure) were done in compliance with the regulations and guidelines of Jinan University (Guangzhou, China) institutional animal care and conducted according to the AAALAC and the IACUC guidelines.
H22 cells were obtained from Shanghai SLAC Laboratory Animal Co. Ltd., China, and propagated in the peritoneal cavity of mice. H22 cells were maintained in our laboratory. In brief, an ascitic fluid of 0.2 mL that contained 20 million viable cells was injected into the peritoneal cavity of mice, and the transplantation procedure was performed once a week. Forty-eight hours post H22 cells were inoculated, highly malignant H22 model mice were used for different experiments. The key parameters of animal experiments included the route of administration, experimental period, drug dose, and animal number, which are presented in the corresponding figure legends.
Cell Culture
Human hepatocellular carcinoma (HCC) cell line HepG2 was obtained from the Stem Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Human squamous cell carcinoma Tca8113 was obtained from the Key Laboratory of Oral Biomedicine of Shanghai Jiao Tong University, China. Both cell lines were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% (v/v) fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C under 95% air and 5.0% CO2. The medium for HepG2 cells additionally contained 2 mM l-glutamine.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
Cells were transferred to 96-well culture plates at a density of 50 000 cells per well for 24 h for full attachment before experiment. The attached cells were treated with SeNPs diluted in the full medium for 24 h, after which the medium was removed and 200 μL of fresh RPMI-1640 medium containing 100 mg of MTT was added to each well. After incubating for 4 h, the medium was replaced with 150 μL of dimethyl sulfoxide (DMSO), after which the absorbance at 490 nm was measured.
H22 Cell Collection
At the end of the animal experiment, H22 model mice were sacrificed by cervical dislocation; H22 cells suspended in the ascitic fluid were harvested and centrifuged at 400 g for 5 min. The cells were then washed twice with ice-cold saline and viable cells were counted in a hemocytometer using the trypan blue dye exclusion method.
GSH Detection
For the predominant intracellular nonprotein free thiol GSH assay, immediately after H22 cells (50 million/mL saline) were sonicated on ice for 2 min with a 3 s interval, a volume of cell lysis was removed and mixed with trichloroacetic acid (20%, w/v) to precipitate protein at a ratio of 10:1 in volume and then centrifuged at 12 000g and 4 °C for 5 min. Within 2 h after the centrifugation, the resulting supernatant was mixed with DTNB and read at 412 nm. GSH was presented as nmol/mg protein.40,41
Preparation of H22 Cell Lysate
H22 cells were mixed with lysis solution containing 0.1 M Tris–HCl (pH 8.0), 10 mM EDTA-Na2, and 0.05% (v/v) Triton X-100 at a ratio of 10 million cells/mL. The mixture was sonicated on ice for 2 min with a 3 s interval and then was centrifuged at 5000 rpm for 10 min at 4 °C. The supernatant was collected for the measurement of SeNP-induced ROS.
Intracellular ROS Measurement
H22 model mice were sacrificed by cervical dislocation, and H22 cells were collected by centrifugation (500g for 5 min at 4 °C) and washed with saline; the same procedure was performed twice. Finally, the cells were resuspended in 1640 serum-free medium for ROS detection by the aforementioned microplate reader using a 488 nm excitation wavelength and a 525 nm emission wavelength. Each sample was adjusted to 200 μL with 1640 serum-free medium, which contained 20 million/mL viable H22 cells, 50 μM DCFH-DA, and indicated concentrations of SeNPs, CDNB, or NAC. Experiments were carried out at 37 °C.
RNA Isolation and Analysis of mRNA Transcription Level by Real-Time Polymerase Chain Reaction (PCR)
Total RNA was extracted using TRIzol reagent (Takara Biotechnology) according to the manufacturer’s protocol. RNA samples with A260nm/A280nm between 1.8 and 2.2 were used for RT-PCR. The cDNA was prepared using 50 ng of total RNA, oligo dT primer, and PrimeScript RT Enzyme Mix (RT-for-PCR kit, Takara Biotechnology) according to the manufacturer’s instructions in a total volume of 20 μL. Real-time PCR was performed on a CFX System (Bio-Rad). ΔCT values were determined by normalization to RPs6. Fold change values were calculated using the 2–(ΔΔCT) method. The gene-specific primers are shown in Table 1.
Table 1. Primer Sequences for RT-PCR.
| genes | direction | sequences |
|---|---|---|
| XPC | forward | 5′-GACCAAGGCACTGATGAAGATG-3′ |
| reverse | 5′-AGACGGTGAGGTGGCAGAAT-3′ | |
| Gadd45β | forward | 5′-CAAGCGATCTGTCTTGCTCA-3′ |
| reverse | 5′-TAAAGCGCATGCTCCAGACT-3′ | |
| Gadd45γ | forward | 5′-AGTCCGCCAAAGTCCTGAATGT-3′ |
| reverse | 5′-GAACGCCTGAATCAACGTGAAA-3′ | |
| RPs6 | forward | 5′-ACTACTGTGCCTCGTCGGTTGG-3′ |
| reverse | 5′-TGCTTTGGTCCTGGGCTTCTTAC-3′ |
Western Blot Analysis
Total protein concentrations of H22 cells extracted with the RIPA reagent were determined by the BCA protein assay kit. Briefly, protein extracts were boiled with loading buffer at 95 °C for 10 min, then separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a PVDF membrane. After blocking with 5% nonfat dried milk in Tris-buffered saline with 0.05% Tween 20 (TBS-T) for 120 min at room temperature, the membrane was incubated with primary antibody diluted in TBS-T overnight at 4 °C according to the dilution ratio provided by the manufacturer. Then, the membrane was washed and incubated with a secondary antibody (2500–5000 dilution) for 60 min at room temperature and then washed three times with TBS-T for 30 min and one time with TBS for 10 min. Antibody bindings were detected using the ChemiDoc XRS + detection system (ECL, Bio-Rad). The Quantity One Image Analyzer software program (Bio-Rad) was used for densitometric analysis.
Statistical Analysis
Data are presented as means ± SEM. The significant differences between groups were examined by Student′s t-test or one-way analysis of variance (ANOVA) post hoc Tukey or Dunnett test, as appropriate. The log-rank test was used for survival comparison. Differences in body weight were examined by two-way ANOVA. All statistical analyses were performed using Prism (GraphPad Software, Inc., La Jolla, CA). A P-value of <0.05 was considered statistically significant.
Acknowledgments
This work was supported by Nano-modification and Application Research and development of polysaccharide from Macroalgae, Guangdong Medical University Marine Resources Public Service Platform Open Fund Project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01034.
Mice body weight and cancer cell number in peritoneal cavity of mice (Figure S1); time effect of SeNP-triggered apoptosis of H22 cells in peritoneal cavity of mice (Figure S2); comparison between GSH and Cys in driving SeNPs to produce ROS at a molar ratio of 20 and 80 (GSH/Se or Cys/Se) (Figure S3) (PDF)
Author Contributions
⊥ G.Z., R.D., and J.T. contributed equally to this work.
Author Contributions
G.Z., Y.W., H.P., and Z.W. conceived the study. G.Z. designed experiments. G.Z., R.D., J.T., L.Y., T.L., X.W., and Y.H. performed the experiments. G.Z. analyzed data and prepared the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Dakwar G. R.; Shariati M.; Willaert W.; Ceelen W.; De Smedt S. C.; Remaut K. Nanomedicine-based intraperitoneal therapy for the treatment of peritoneal carcinomatosis - Mission possible?. Adv. Drug Delivery Rev. 2017, 108, 13–24. 10.1016/j.addr.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Lambert L. A. Looking up: Recent advances in understanding and treating peritoneal carcinomatosis. Ca-Cancer J. Clin. 2015, 65, 283–298. 10.3322/caac.21277. [DOI] [PubMed] [Google Scholar]
- Almerie M. Q.; Gossedge G.; Wright K. E.; Jayne D. G. Treatment of peritoneal carcinomatosis with photodynamic therapy: Systematic review of current evidence. Photodiagn. Photodyn. Ther. 2017, 20, 276–286. 10.1016/j.pdpdt.2017.10.021. [DOI] [PubMed] [Google Scholar]
- Nissan A.; Stojadinovic A.; Garofalo A.; Esquivel J.; Piso P. Evidence-based medicine in the treatment of peritoneal carcinomatosis: Past, present, and future. J. Surg. Oncol. 2009, 100, 335–344. 10.1002/jso.21323. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Wang J.; Wientjes M. G.; Au J. L. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010, 6, 1625–1641. 10.2217/fon.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasovits C.; Clarke S. Pharmacokinetics and pharmacodynamics of intraperitoneal cancer chemotherapeutics. Clin. Pharmacokinet. 2012, 51, 203–224. 10.2165/11598890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Spallholz J. E. On the nature of selenium toxicity and carcinostatic activity. Free Radical Biol. Med. 1994, 17, 45–64. 10.1016/0891-5849(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Chen J. J.; Boylan L. M.; Wu C. K.; Spallholz J. E. Oxidation of glutathione and superoxide generation by inorganic and organic selenium compounds. Biofactors 2007, 31, 55–66. 10.1002/biof.5520310106. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Spallholz J. E. Generation of reactive oxygen species from the reaction of selenium compounds with thiols and mammary tumor cells. Biochem. Pharmacol. 1993, 45, 429–437. 10.1016/0006-2952(93)90080-G. [DOI] [PubMed] [Google Scholar]
- Wu X.; Zhao G.; He Y.; Wang W.; Yang C. S.; Zhang J. Pharmacological mechanisms of the anticancer action of sodium selenite against peritoneal cancer in mice. Pharmacol. Res. 2019, 147, 104360 10.1016/j.phrs.2019.104360. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sun K.; Tan Y.; Wu S.; Zhang J. Efficacy and safety of selenium nanoparticles administered intraperitoneally for the prevention of growth of cancer cells in the peritoneal cavity. Free Radical Biol. Med. 2014, 72, 1–10. 10.1016/j.freeradbiomed.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Zhao G.; Wu X.; Chen P.; Zhang L.; Yang C. S.; Zhang J. Selenium nanoparticles are more efficient than sodium selenite in producing reactive oxygen species and hyper-accumulation of selenium nanoparticles in cancer cells generates potent therapeutic effects. Free Radical Biol. Med. 2018, 126, 55–66. 10.1016/j.freeradbiomed.2018.07.017. [DOI] [PubMed] [Google Scholar]
- Dodd S.; Dean O.; Copolov D. L.; Malhi G. S.; Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin. Biol. Ther. 2008, 8, 1955–1962. 10.1517/14728220802517901. [DOI] [PubMed] [Google Scholar]
- Cotgreave I. A. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv. Pharmacol. 1996, 38, 205–227. 10.1016/s1054-3589(08)60985-0. [DOI] [PubMed] [Google Scholar]
- Shen H.; Yang C.; Liu J.; Ong C. Dual role of glutathione in selenite-induced oxidative stress and apoptosis in human hepatoma cells. Free Radical Biol. Med. 2000, 28, 1115–1124. 10.1016/S0891-5849(00)00206-9. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Peng D.; Lu H.; Liu Q. Attenuating the toxicity of cisplatin by using selenosulfate with reduced risk of selenium toxicity as compared with selenite. Toxicol. Appl. Pharmacol. 2008, 226, 251–259. 10.1016/j.taap.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Munro D.; Banh S.; Sotiri E.; Tamanna N.; Treberg J. R. The thioredoxin and glutathione-dependent H2O2 consumption pathways in muscle mitochondria: Involvement in H2O2 metabolism and consequence to H2O2 efflux assays. Free Radical Biol. Med. 2016, 96, 334–346. 10.1016/j.freeradbiomed.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chen P.; Zhao G.; Sun K.; Li D.; Wan X.; Zhang J. Inverse relationship between elemental selenium nanoparticle size and inhibition of cancer cell growth. Food Chem. Toxicol. 2015, 85, 71–77. 10.1016/j.fct.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Xie C.; Yi J.; Lu J.; Nie M.; Huang M.; Rong J.; Zhu Z.; Chen J.; Zhou X.; Li B.; Chen H.; Lu N.; Shu X. N-Acetylcysteine Reduces ROS-Mediated Oxidative DNA Damage and PI3K/Akt Pathway Activation Induced by Helicobacter pylori Infection. Oxid. Med. Cell. Longevity 2018, 2018, 1874985 10.1155/2018/1874985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasi M.; Wang M.; Chavan T. S.; Gaponenko V.; Hay N.; Gartel A. L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem J. 2013, 454, 201–208. 10.1042/BJ20130282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallis G. B.; Holmgren A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J. Biol. Chem. 1980, 255, 10261–10265. [PubMed] [Google Scholar]
- Davis W. Jr.; Ronai Z.; Tew K. D. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J. Pharmacol. Exp. Ther. 2001, 296, 1–6. [PubMed] [Google Scholar]
- Lu J.; Chew E. H.; Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12288–12293. 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andringa K. K.; Coleman M. C.; Aykin-Burns N.; Hitchler M. J.; Walsh S. A.; Domann F. E.; Spitz D. R. Inhibition of glutamate cysteine ligase activity sensitizes human breast cancer cells to the toxicity of 2-deoxy-D-glucose. Cancer Res. 2006, 66, 1605–1610. 10.1158/0008-5472.CAN-05-3462. [DOI] [PubMed] [Google Scholar]
- Dai J.; Weinberg R. S.; Waxman S.; Jing Y. Malignant cells can be sensitized to undergo growth inhibition and apoptosis by arsenic trioxide through modulation of the glutathione redox system. Blood 1999, 93, 268–277. 10.1182/blood.V93.1.268. [DOI] [PubMed] [Google Scholar]
- Meurette O.; Lefeuvre-Orfila L.; Rebillard A.; Lagadic-Gossmann D.; Dimanche-Boitrel M. T. Role of intracellular glutathione in cell sensitivity to the apoptosis induced by tumor necrosis factor {alpha}-related apoptosis-inducing ligand/anticancer drug combinations. Clin. Cancer Res. 2005, 11, 3075–3083. 10.1158/1078-0432.CCR-04-1764. [DOI] [PubMed] [Google Scholar]
- Wu X. X.; Ogawa O.; Kakehi Y. Enhancement of arsenic trioxide-induced apoptosis in renal cell carcinoma cells by L-buthionine sulfoximine. Int. J. Oncol. 2004, 24, 1489–1497. 10.3892/ijo.33.1.205. [DOI] [PubMed] [Google Scholar]
- Biroccio A.; Benassi B.; Fiorentino F.; Zupi G. Glutathione depletion induced by c-Myc downregulation triggers apoptosis on treatment with alkylating agents. Neoplasia 2004, 6, 195–206. 10.1593/neo.03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyano A.; Fernández C.; Sancho P.; de Blas E.; Aller P. Effect of glutathione depletion on antitumor drug toxicity (apoptosis and necrosis) in U-937 human promonocytic cells. The role of intracellular oxidation. J. Biol. Chem. 2001, 276, 47107–47115. 10.1074/jbc.M104516200. [DOI] [PubMed] [Google Scholar]
- Caffrey P. B.; Frenkel G. D. Selenium compounds prevent the induction of drug resistance by cisplatin in human ovarian tumor xenografts in vivo. Cancer Chemother. Pharmacol. 2000, 46, 74–8. 10.1007/s002800000127. [DOI] [PubMed] [Google Scholar]
- Caffrey P. B.; Frenkel G. D. Prevention of carboplatin-induced resistance in human ovarian tumor xenografts by selenite. Anticancer Res. 2013, 33, 4249–4254. [PubMed] [Google Scholar]
- Frenkel G. D.; Caffrey P. B. A prevention strategy for circumventing drug resistance in cancer chemotherapy. Curr. Pharm. Des. 2001, 7, 1595–1614. 10.2174/1381612013397186. [DOI] [PubMed] [Google Scholar]
- Caffrey P. B.; Frenkel G. D. Selenite enhances and prolongs the efficacy of cisplatin treatment of human ovarian tumor xenografts. In Vivo 2012, 6, 549–552. [PubMed] [Google Scholar]
- Ali-Osman F.; Stein D. E.; Renwick A. Glutathione content and glutathione-S-transferase expression in 1,3-bis(2-chloroethyl)-1-nitrosourea-resistant human malignant astrocytoma cell lines. Cancer Res. 1990, 50, 6976–6980. [PubMed] [Google Scholar]
- Barranco S. C.; Townsend C. M. Jr.; Weintraub B.; Beasley E. G.; MacLean K. K.; Shaeffer J.; Liu N. H.; Schellenberg K. Changes in glutathione content and resistance to anticancer agents in human stomach cancer cells induced by treatments with melphalan in vitro. Cancer Res. 1990, 50, 3614–3618. [PubMed] [Google Scholar]
- Zhao R.; Xiang N.; Domann F. E.; Zhong W. Expression of p53 enhances selenite-induced superoxide production and apoptosis in human prostate cancer cells. Cancer Res. 2006, 15, 2296–2304. 10.1158/0008-5472.CAN-05-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. S.; Gao X. Y.; Zhang L. D.; Bao Y. P. Biological effects of a nano red elemental selenium. Biofactors 2001, 15, 27–38. 10.1002/biof.5520150103. [DOI] [PubMed] [Google Scholar]
- Gao X. Y.; Zhang J. S.; Zhang L. D. Hollow sphere selenium nanoparticles: their in-vitro anti hydroxyl radical effect. Adv. Mater. 2002, 14, 290–293. . [DOI] [Google Scholar]
- Wang H.; Zhang J.; Yu H. Elemental selenium at nano size possesses lower toxicity without compromising the fundamental effect on selenoenzymes: comparison with selenomethionine in mice. Free Radical Biol. Med. 2007, 42, 1524–1533. 10.1016/j.freeradbiomed.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Ellman G. L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhang J.; Xu T. Cyclophosphamide-evoked heart failure involves pronounced co-suppression of cytoplasmic thioredoxin reductase activity and non-protein free thiol level. Eur. J. Heart Failure 2009, 11, 154–162. 10.1093/eurjhf/hfn012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.