

Abstract
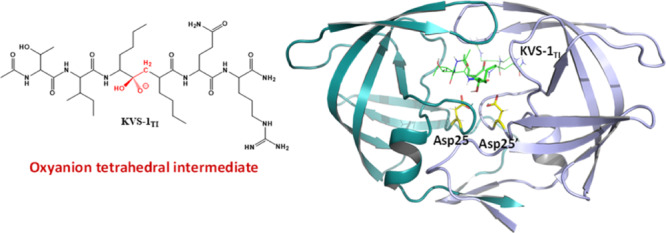
HIV-1 protease is indispensable for virus propagation and an important therapeutic target for antiviral inhibitors to treat AIDS. As such inhibitors are transition-state mimics, a detailed understanding of the enzyme mechanism is crucial for the development of better anti-HIV drugs. Here, we used room-temperature joint X-ray/neutron crystallography to directly visualize hydrogen atoms and map hydrogen bonding interactions in a protease complex with peptidomimetic inhibitor KVS-1 containing a reactive nonhydrolyzable ketomethylene isostere, which, upon reacting with the catalytic water molecule, is converted into a tetrahedral intermediate state, KVS-1TI. We unambiguously determined that the resulting tetrahedral intermediate is an oxyanion, rather than the gem-diol, and both catalytic aspartic acid residues are protonated. The oxyanion tetrahedral intermediate appears to be unstable, even though the negative charge on the oxyanion is delocalized through a strong n → π* hyperconjugative interaction into the nearby peptidic carbonyl group of the inhibitor. To better understand the influence of the ketomethylene isostere as a protease inhibitor, we have also examined the protease structure and binding affinity with keto-darunavir (keto-DRV), which similar to KVS-1 includes the ketomethylene isostere. We show that keto-DRV is a significantly less potent protease inhibitor than DRV. These findings shed light on the reaction mechanism of peptide hydrolysis catalyzed by HIV-1 protease and provide valuable insights into further improvements in the design of protease inhibitors.
Introduction
Enzymes ensure the existence of life by dramatically accelerating the rates of many chemical reactions that, if uncatalyzed, may have virtually insurmountable energy barriers. A mechanistic understanding of the remarkable catalytic efficiency of these biomacromolecular catalysts is crucial in designing drugs to battle many human diseases. Human immunodeficiency virus type 1 (HIV-1) that causes AIDS has been in the public eye for almost four decades. Drugs targeting virtually every enzyme of HIV-1 have been designed and further developed into clinical therapeutics,1,2 essentially transforming this deadly disease into a chronic condition for many patients. In this regard, inhibition of HIV-1 protease (PR) has played a crucial role in improving lives of many infected people.3,4 The design of protease inhibitors is considered as one of the greatest successes of structure-based drug design.5 Rapid accumulation of drug-resistant mutations and significant side effects, however, thwart the efficacy of protease inhibitors for prolonged use and necessitate the design of new drugs with better properties.3,5−7
HIV-1 PR catalyzes cleavage of peptide bonds at specific sites in the viral Gag and Gag-Pol polyproteins during the maturation stage of the HIV-1 replication cycle.8 The PR catalytic mechanism has been studied in depth since the first structures appeared in the literature.9−11 HIV-1 PR belongs to the class of aspartic proteases, and utilizes two co-located aspartic acid residues to mediate peptide bond hydrolysis through an acid-base catalytic mechanism.12 The active enzyme is a homodimer of two 99-amino acid subunits, each contributing a catalytic aspartate (Asp25 and Asp25′) to form the active site.13 Solution enzyme kinetics and NMR measurements have established important mechanistic details of the PR catalysis:14−19 (1) the two catalytic aspartates have very different pKa values of ∼3.5 ± 0.1 and ∼6.0 ± 0.5, indicating that the catalytic site is monoprotonated; (2) the lytic water activated by the catalytic Asp residues is required for the hydrolysis reaction; (3) a kinetically competent amide hydrate tetrahedral intermediate is formed along the reaction pathway, and (4) the tetrahedral intermediate collapses to the products in a rate-limiting step that involves proton transfer from a catalytic Asp to the nitrogen of the scissile C–N bond. However, atomic details of the PR catalytic mechanism are still being debated and proximity of multiple oxygen atoms in the catalytic site of PR indicates a possibility of multicenter (low-barrier) hydrogen bonds.16,20−22 Throughout the reaction pathway, three hydrogen (H) atoms within the catalytic site are key players in peptide hydrolysis—two Hs originate on the lytic water and one H on the Asp residues before a substrate binds (Figure 1). In the tetrahedral intermediate, these H atoms are distributed among six oxygen atoms and over a hundred different combinations (or isomers) can be envisioned. Importantly, the orientation of the lytic water and the exact positions of Hs on the carboxylic side chains of Asp25 and Asp25′ in the reactant (Michaelis) complex, and the tetrahedral intermediate and product structures are not known. Lack of this information has led to a multitude of proposals for the PR catalytic mechanism based on theoretical calculations, NMR measurements, and X-ray crystallography.
Figure 1.

Chemical diagrams of several possible HIV-1 PR catalytic site structures in the mechanism of peptide bond hydrolysis. H atoms involved in the reaction are colored red, possible hydrogen bond interactions are shown as dashed lines. Catalytic site of the substrate-free (and inhibitor-free) form of enzyme is drawn containing a low-barrier hydrogen bond formed between Oδ1 oxygen atoms of the Asp dyad and with H covalently bound to Asp25 Oδ1. Many other arrangements for the H atom positions in the substrate-free form, reactant and tetrahedral intermediate complexes are also possible.
In principle, the peptide bond hydrolysis reaction can proceed through a gem-diol tetrahedral intermediate, an oxyanion tetrahedral intermediate (Figure 1), or by a concerted mechanism with no stable intermediates along the reaction pathway.13 Several earlier molecular modeling studies focused on the PR catalytic mechanism that includes a gem-diol tetrahedral intermediate.23−26 Surprisingly, in these calculations the gem-diol tetrahedral intermediate was found to be either significantly more (−7 kcal/mol) or less (+12 kcal/mol) stable than the reactants, indicating that chosen models and/or levels of theory were inadequate. Other theoretical studies27,28 demonstrated that the reaction could also proceed through a high-energy oxyanion tetrahedral intermediate (+14–18 kcal/mol relative to reactants), in which both Asp25 and Asp25′ were protonated. In this case, proton tunneling for the protonation of the tetrahedral intermediate nitrogen by a catalytic Asp was proposed to accomplish the rate acceleration,16 in accordance with the inverse primary 15N kinetic isotope effect.19 More recent theoretical calculations considered many possible mechanisms and optimized structures of all 120 possible tetrahedral intermediates using density functional (DFT) and molecular orbital theories.29−34 For example, cluster QM calculations by Garrec et al.29 revealed a gem-diol tetrahedral intermediate having an energy of +3.4–4.8 kcal/mol relative to the reactants, whereas the only stable oxyanion tetrahedral intermediate was found to be greatly destabilized (+40 kcal/mol). On the other hand, QM/MM calculations of Krzeminska et al.31 resulted in a much more stable oxyanion tetrahedral intermediate (+8 kcal/mol) that could readily overcome a 2 kcal/mol energy barrier to convert into the gem-diol tetrahedral intermediate, which was more stable than the reactants by −3 kcal/mol. These calculations also demonstrated that a metastable zwitterion intermediate on the path from the oxyanion to the products could exist (Figure 1), whereas the concerted mechanism with a cyclic transition state would require overcoming a very large, 43.5 kcal/mol, energy barrier. In the most recent study, however, Lawal et al.34 revisited the concerted mechanism and showed that the pathway involving an acyclic transition state was feasible. Previous molecular modeling studies suggest that theoretical reaction energy barriers for the many modeled reaction pathways of the catalytic mechanism are in good agreement (based on the Eyring–Polanyi equation) with the corresponding experimental kcat values of ∼1–5 s–1 for substrate hydrolysis by PR14−19 at room temperature (RT), even though their specific atomic details are markedly different. Consequently, many different reaction pathways fit experimental measurements equally well, thus dissuading researchers from unequivocally determining the actual HIV-1 PR catalytic mechanism. In addition, based on the theoretically optimized gem-diol tetrahedral intermediate structures, one can hypothesize that a protease inhibitor containing the gem-diol chemical moiety with two hydroxyl groups should bind tighter to PR compared to the same inhibitor that has the commonly used hydroxyethylene isostere with one hydroxyl group, because the gem-diol moiety is capable of making an extra hydrogen bond with the catalytic Asp dyad. In the current study, we make an attempt to directly address these two crucial points pertaining to the PR catalysis and drug design.
X-ray crystallography has been a method of choice in structural biology to obtain accurate three-dimensional structures of biomacromolecules and to link structure to function. A limited number of HIV-1 PR crystal structures in complex with tetrahedral intermediates have been reported, which, nonetheless, indicated that such intermediates can be trapped in the crystal lattice.35−39 It was shown, initially, that nonhydrolyzable tetrahedral intermediates could be generated in the PR active site by a reaction with the lytic water attacking a pseudo-substrate where the amide nitrogen of the scissile peptide bond was replaced with a −CF2– or a keto group.35,36 Also, in certain cases, the reactive, amide hydrate, tetrahedral intermediates could be trapped and their structures solved.37−39 Nevertheless, positions of H atoms could not be resolved in these X-ray structures, leaving a significant gap in our understanding of the HIV-1 PR catalysis. The technique capable of accurately determining H atom positions and vizualizing hydrogen bonding in protein structures is neutron crystallography.40,41 It permits detection of H (and its heavy isotope deuterium, D) atom locations and, hence, direct determination of the protonation states. Moreover, cold neutrons with wavelengths of ∼2–5 Å used in neutron crystallographic experiments cause no radiation damage to protein crystals so that the diffraction data can be collected under near-physiological conditions (usually at RT), eliminating the need to freeze crystals and to use cryo-protectant chemicals. However, the neutron diffraction data are normally weaker and less complete than the X-ray data. Combination of the X-ray and neutron diffraction data in a joint X-ray/neutron (XN) refinement provides an increased data-to-parameter ratio to produce complete protein structures containing accurate positions of all atoms, where heavy atom positions are mainly defined by the X-ray data and D atom positions are defined by the neutron data.42
We present here novel atomic details pertinent to the hydrolysis reaction catalyzed by HIV-1 PR. We determined a RT joint XN structure of HIV-1 PR in complex with the nonhydrolyzable ketomethylene hexapeptide inhibitor KVS-1, which is converted in situ into a tetrahedral intermediate analogue (KVS-1TI)43,44 (Figures 2 and 3). We observed both catalytic Asp residues protonated on their Oδ1 oxygen atoms (labelled in Figure 3 and consistently referred to throughout the manuscript), and one protonated and one deprotonated oxygen on KVS-1TI. This rather unexpected result implies that the trapped tetrahedral intermediate moiety is an oxyanion, rather than the gem-diol. We support our experimental findings with cluster QM and QM/MM calculations. We also obtained a RT X-ray structure of HIV-1 PR in complex with keto-darunavir (keto-DRV), in which the hydroxyethylene isostere of the clinical inhibitor darunavir (DRV) was oxidized into the ketomethylene moiety. Similar to our joint XN structure, the ketomethylene of keto-DRV was converted to a tetrahedral intermediate analogue to give DRVTI bound in the PR active site cavity. The extent of inhibition of PR by keto-DRV was assessed using isothermal titration calorimetry (ITC).
Figure 2.

(A) Chemical diagram of the non-hydrolyzable hexapeptide KVS-1 and the resulting tetrahedral intermediate KVS-1TI. The reactive ketomethylene isostere in KVS-1 and the tetrahedral intermediate moiety in KVS-1TI are colored red. (B) Neutron structure of HIV-1 PR in complex with the oxyanion KVS-1TI (green sticks). The catalytic Asp25 and Asp25′ are shown as yellow sticks. D atoms on Asp25, Asp25′, and KVS-1TI are shown; nonexchangeable H atoms of KVS-1TI are omitted for clarity.
Figure 3.

Catalytic site in the HIV-1 PR/KVS-1TI complex. (A) 2FO-FC electron density map contoured at the 2.5 σ level. Oxygen atoms of the Asp25 and Asp25′ carboxylic groups are labeled as Oδ1 and Oδ2 (B) possible hydrogen bonding interactions between the catalytic Asp dyad and the oxygen atoms of the tetrahedral intermediate. O···O distances are given in Å. Only X-ray diffraction data were used to generate the figure; thus, H and D atoms are omitted to demonstrate that noncovalent interactions cannot be reliably interpreted when locations of H atoms are unknown.
Results
Neutrons Reveal Hydrated KVS-1 is a Tetrahedral Oxyanion Bound to HIV-1 PR
To obtain the complex with KVS-1TI we chose an HIV-1 PR triple mutant variant (PRTM), which contains three substitutions V32I, I47V, and V82I associated with drug resistance (Table S1), because of its demonstrated success to afford neutron diffraction quality crystals and because the resistance mutations do not introduce considerable distortions in ligand binding compared to the wild-type enzyme.45,46 We collected RT neutron crystallographic data to 2.2 Å resolution from a very small crystal of ∼0.15 mm3 in volume. The neutron data were refined jointly with a 1.85 Å resolution RT X-ray dataset to give the joint XN structure of the PRTM/KVS-1TI complex. The electron density maps calculated from the X-ray data (Figures 3A, and S1) clearly show that the central carbon atom is sp3 hybridized, confirming that the ketomethylene moiety of KVS-1 has reacted with a water molecule to give a formal tetrahedral intermediate with two oxygen atoms bound to the central carbon, in agreement with the previous low-temperature X-ray structures of HIV-1 PR in complex with this modified hexapeptide.43,44 The electron density for KVS-1TI is strong and well defined (Figure S1); thus, we saw no indication of the KVS-1TI static disorder in the PRTM/KVS-1TI complex, which was previously observed in many PR-ligand structures where the ligand can have two orientations related by a 180° rotation with similar occupancies because of the 2-fold symmetry of the PR dimer. Such static disorder has not been found in our joint XN structures of the deuterated HIV-1 PR.45,46 Examination of the O···O distances between the oxygen atoms of the tetrahedral intermediate moiety and the carboxylic groups of the Asp dyad reveals a possibility of six hydrogen bonds with the distances in the range of 2.7–3.1 Å (Figure 3B). Correct assignment of the hydrogen bonds, however, cannot be made based on the X-ray data, because H atom locations are not seen. Conversely, D atoms are visible within the catalytic site of the PRTM/KVS-1TI complex in the neutron scattering length density maps (also referred to as nuclear density maps) (Figures 4A and S2A); however, for simplicity, we use the standard chemical conventions such as protonation states and hydrogen bonds. There is a clear density identifying three D atoms—one each on Asp25, Asp25′, and a hydroxyl of the tetrahedral intermediate moiety (Figure 4A,B). Therefore, according to our joint XN structure, the carboxylic groups of the catalytic Asp dyad are both protonated on their Oδ1 atoms, and, thus, have neutral charges. However, within the tetrahedral intermediate moiety, one oxygen is protonated, whereas the other is not, hence having a −1 charge. This observation unequivocally leads us to conclude that the tetrahedral intermediate moiety is an oxyanion, rather than the gem-diol. Importantly, the Asp25 O-D bond is in the carboxylic group plane and is directed toward the protonated tetrahedral intermediate hydroxyl and the Asp25′ side chain, forming a bifurcated hydrogen bond (Figure 4C). However, the Asp25′ O-D bond is rotated away from both Asp25 and the protonated tetrahedral intermediate hydroxyl by almost 90° into a small hydrophobic cavity formed by the Thr26′-Gly27′-Ala28′ turn so that it makes no hydrogen bonding interactions (Figures 4B and S2B). The protonated tetrahedral intermediate hydroxyl group also makes a hydrogen bond with the Oδ2 atom of Asp25. This hydrogen bonding analysis demonstrates that only three out of six possible hydrogen bonds form within the tetrahedral intermediate moiety and the catalytic Asp dyad. The catalytic water molecule in the unliganded HIV-1 PR structure (PDB code 1LV1) lies within 0.7 Å from the protonated tetrahedral intermediate hydroxyl of KVS-1TI complexed to PRTM (Figure S3A). Based on these observations, we can now deduce that the protonated tetrahedral intermediate hydroxyl comes from the lytic water molecule and the oxyanion oxygen is from the ketomethylene carbonyl group of KVS-1. Furthermore, the D atom connected to Asp25 is the original proton present within the PR catalytic site before the substrate binds, whereas the D atoms on tetrahedral intermediate hydroxyl and Asp25′ come from the lytic water molecule.
Figure 4.

(A) The catalytic site of the HIV-1 PR/KVS-1TI complex. (B) Catalytic Asp25′, and residues Thr26′, Gly27′, and Ala28′ making a small hydrophobic pocket where D bonded to the Asp25′ carboxylic group is facing. (C) Hydrogen bonds (blue dashed lines with O···D distances in Å, and O-D···O angles in deg.) made between the catalytic Asp dyad and the oxygen atoms of the intermediate. The negatively charged oxygen atom of the oxyanion makes close 2.8–3.2 Å contacts with next carbonyl in the hexapeptide main chain. For panels A and B, 2FO-FC neutron scattering length density map at a 2.2 Å resolution is contoured at the 1.5 σ level; the FO-FC-omit difference neutron scattering length density map is the violet mesh contoured at the 3 σ level, indicating the locations of the three D atoms (dark gray spheres) involved in catalysis (other D atoms are light gray). H atoms of KVS-1TI are omitted for clarity.
KVS-1TI makes seven additional moderate hydrogen bonds with the main chain amide nitrogens of Asp29, Asp29′, Asp30′, and Gly48′, and with the main chain carbonyls of Gly48, Gly27′, and Gly48′ (Figure 5A), with D···O distances of 1.9–2.2 Å, and two weaker hydrogen bonds with Asp29′ and Asp30′ side chain carboxylates. It also interacts with the Gly48 main chain amide nitrogen through a water-mediated contact. The other water-mediated interaction connecting two carbonyl groups of KVS-1TI with the main chain amides of Ile50 and Ile50′ flap residues is rather peculiar (Figures 5B and S4). The flap D2O water molecule has such an orientation that it forms three hydrogen bonds instead of possible four. The flap D2O donates both D atoms to hydrogen bond with two carbonyls of KVS-1TI and accepts a main chain amide D to form a hydrogen bond with Ile50′. The main chain amide of Ile50 is also capable of participating in a hydrogen bond with the flap water, but the D···O distance is elongated (2.6 Å) and the amide makes an acute angle of 80° with the D2O plane. The geometry of this contact is, therefore, far outside the normal parameters for a hydrogen bond47 and this flap is anchored more weakly to the PR active site. Such an unexpected hydrogen bonding network around the flap water molecule has been observed previously in our joint XN structures of wild-type PR and PRTM in complex with clinical drugs amprenavir and DRV45,46 and agrees with the assessment based on NMR measurements44 that found one PR flap more dynamic than the other.
Figure 5.

(A) Hydrogen bonding and water-mediated interactions, shown as blue-dashed lines connecting O or N atoms with D atoms (O···D and N···D), between KVS-1TI and the active site residues in PRTM. The N–D···O contact with Gly27 main chain carbonyl is significantly distorted from the ideal hydrogen bond geometry, with the N–D vector being almost perpendicular to the carbonyl plane. (B) Water-mediated interactions with main chain amides of Ile50 and Ile50′. The flap water makes a hydrogen bond with the main chain amide of Ile50′, but not with Ile50 because the O···D distance of 2.6 Å is too long.
It is instructive to compare the binding of the hydrated KVS-1TI in the PRTM active site cavity to that of the actual amide hydrate tetrahedral intermediates published previously.37−39 Among the four amide hydrate tetrahedral intermediate structures determined at 100 K, three correspond to complexes with the wild type PR (PDB IDs 3B7V, 4FL8, and 5YRS), with two, 4FL8 and 5YRS, having the tetrahedral intermediate peptides disordered over two orientations related by a 180° flip, with 50% occupancy each, and 3B7V having the peptide in one orientation with 60% occupancy. The fourth structure (PDB ID 3B80) is of PR I54V mutant variant in which the tetrahedral intermediate has one orientation with 60% occupancy. Importantly, the electron density for the peptidic ligand is stronger in the 3B7V structure. Although all the complexes superimpose well, with the root mean square deviation (rmsd) on the main chain atoms of 0.2–0.4 Å, the tetrahedral intermediate moiety in the PRI54V-tetrahedral intermediate complex aligns best with that of our joint XN structure of PRTM/KVS-1TI (Figure S3B); however, its electron density is rather weak. All non-covalent O···O distances within the PRI54V catalytic site, except for one, are virtually the same compared to those in PRTM/KVS-1TI. In our XN structure, one O···O distance of 2.8 Å is longer than the equivalent one of 2.5 Å in the PRI54V mutant TI complex, and it corresponds to an interaction between the oxyanion oxygen and the Oδ2 of Asp25′. In the PRWT/tetrahedral intermediate structure 3B7V, this distance was found to be even shorter, at 2.3 Å, implying protonation of one of the oxygens and suggesting formation of a strong ionic hydrogen bond (Figure S3C). We would like to emphasize here that hydrogen atoms have not been observed in these low temperature X-ray structures, therefore, the accurate assignment of hydrogen bonds cannot be made.
Low Temperature Narrows the PRTM Substrate-Binding Channel
To find out if temperature has an effect on the structure of the PRTM/KVS-1TI complex, we obtained its X-ray structure at 100 K at 1.31 Å resolution. Overall, the room-temperature joint XN structure and the low-temperature X-ray structure superimpose well, with the rmsd on the main chain atoms of 0.3 Å and with the rmsd of 0.1 Å for all atoms of KVS-1TI. Closer examination of the PRTM substrate binding channel, however, reveals more significant differences in the relative positions of residues in the two structures. Many residues in the PRTM active site (substrate binding channel) have shifted by 0.3–0.4 Å toward KVS-1TI, reducing the distances across the substrate binding channel by at least 0.5–0.6 Å. The most dramatic shift is observed for the 80’s loops in PRTM, containing residues Thr80–Pro81–Ile82 and Thr80′–Pro81′–Ile82′ that are part of the active site (Figure S5). The distance between Pro81 and Pro81′ decreases by ∼1 Å in the low temperature structure, which is a significant change based on the estimated coordinate errors for the two structures of 0.05 Å (100 K X-ray structure) and 0.14 Å (room-temperature joint XN structure). Considering the shape of the PRTM substrate-binding channel as a cylinder and taking into account the shifts in residue positions between the room- and low-temperature structures, we estimate that at 100 K the active site volume has shrunk by ∼20–30 Å3 compared to its size at RT, which also leads to apparent shorter hydrophobic interactions between KVS-1TI and the side chains of the PRTM residues. Interestingly, the side chain conformations have mostly remained the same in the two structures unlike in the PRTM/APV structures reported previously,45 except for Ile82 whose two alternate conformations visible at low temperature are different from the single conformation observed in the RT joint XN structure.
Oxyanion Charge is Delocalized by Hyperconjugation, but KVS-1TI is Unstable
How is the formal negative charge of −1 on the oxyanion tetrahedral intermediate oxygen of KVS-1TI stabilized in the PRTM/KVS-1TI complex used to determine its structure? To understand how such stabilization can be achieved, we performed cluster QM calculations on a 194-atom model built using atomic coordinates from our joint XN structure (D atoms were replaced with H atoms) and performed NBO analysis of the model to estimate the strength of noncovalent interactions. The distance of 2.8–3.2 Å between the negatively charged oxygen of the tetrahedral intermediate and the carbonyl group of the next peptide bond (Figure 4C) suggests a possibility of charge delocalization by means of an n → π* charge transfer interaction, that is through-space hyperconjugation. Indeed, according to our NBO analysis, two such interactions of the charged tetrahedral intermediate oxygen lone pairs with an antibonding π* orbital of the carbonyl are present, with the energies of 1.92 and 0.67 kcal/mol, indicating a strong charge delocalization onto the carbonyl π system (Figure 6A,B). It is evident that the n → π* interactions occurring within the oxyanion tetrahedral intermediate should have a significant stabilization effect. These n → π* interactions are possibly enhanced by a strong repulsive electrostatic interaction with the Asp25′ carboxylic Oδ2 oxygen positioned 2.8 Å away from the negatively charged tetrahedral intermediate oxygen. The O···O repulsion, which is unfavorable for the oxyanion stability, however, may still dominate over the hypercojugation, as discussed below. We then shifted the H atom from Asp25′ carboxyl to the deprotonated oxygen of the tetrahedral intermediate to create a gem-diol, optimized its geometry, and performed NBO analysis. In our gem-diol tetrahedral intermediate Asp25′ Oδ2 now makes a hydrogen bond with the new hydroxyl group of the tetrahedral intermediate, instead of the repulsive interaction with the oxyanion. There is only one n → π* interaction of 1.52 kcal/mol with the carbonyl group of the next peptide bond in the gem-diol tetrahedral intermediate, in agreement with the previously computed gem-diol tetrahedral intermediate that had a different juxtaposition of H atoms.48 This interaction energy in our gem-diol tetrahedral intermediate is weaker, as expected, than in our oxyanion tetrahedral intermediate.
Figure 6.

Orbital interactions in the protease catalytic site. (A,B) Two n → π* interactions occur within KVS-1TI between two lone electron pairs of the negatively charged oxygen and an antibonding π* orbital of the nearby carbonyl group. (C) An n → σ* interaction between the protonated tetrahedral intermediate hydroxyl and Asp25′ hydroxyl, signifying a strong hydrogen bond formation.
To shed light on the Asp25′ Oδ1-H bond rotation away from the tetrahedral intermediate moiety hydroxyl of KVS-1TI, we examined the interaction of these two chemical groups. Our cluster QM calculation optimizes the H atom of the Asp25′ Oδ1-H to be in plane with the carboxylic group, resulting in a strong hydrogen bond with the protonated hydroxyl of the tetrahedral intermediate. The strength of this hydrogen bond can be measured by the charge transfer from a tetrahedral intermediate hydroxyl lone pair to the σ* orbital of the Asp25′ Oδ1-H bond (Figure 6C). NBO analysis reveals that this n → σ* interaction is 26 kcal/mol. By comparison, the n → σ* interactions of the two tetrahedral intermediate hydroxyl lone pairs with the Asp25 Oδ1-H bond are an order of magnitude weaker at 2.2 and 2.8 kcal/mol. When Asp25′ Oδ1-H was fixed in the geometry observed in the joint XN structure, the n → σ* interaction was reduced to just 0.1 kcal/mol, indicating that the charge transfer component of the hydrogen bond was essentially abolished.
We then optimized the geometry of the PRTM/KVS-1TI complex starting from the joint XN structure atomic coordinates using the QM/MM methodology. Unexpectedly, the strong hydrogen bond of Asp25 Oδ1-H with the protonated hydroxyl of the tetrahedral intermediate leads to a spontaneous proton transfer to the tetrahedral intermediate oxygen and the generated water molecule’s cleavage off the tetrahedral intermediate moiety. The tetrahedral intermediate carbon atom is rehybridized from sp3 to sp2 producing a ketone group. Therefore, both reactants, KVS-1 containing the initial ketomethylene isostere and the lytic water molecule, are regenerated during the QM/MM geometry optimization (Figure S6). This implies that the PRTM/KVS-1TI complex is metastable with the overall energy higher than that of the reactants.
Keto-DRV Binds to HIV-1 PRTM to Give a Tetrahedral Intermediate Complex
The clinical inhibitor DRV contains the hydroxyethylene isostere whose hydroxy group forms a hydrogen bond tightly with the catalytic Asp dyad.46 We oxidized this hydroxyl to give the ketomethylene isostere, as in KVS-1, thus producing keto-DRV (Figure 7A). Keto-DRV was co-crystallized with PRTM and a RT X-ray structure of the complex was obtained at 1.80 Å resolution. In the electron density map, we clearly observed that keto-DRV was converted to DRVTI bound to the catalytic Asp dyad, having the sp3-hybridized tetrahedral carbon connected to two oxygens (Figure 7B) similar to the structure and binding mode of KVS-1TI. One oxygen (O1 on Figure 7C) has an O···O contact of 2.4 Å with Asp25 Oδ2, which is somewhat shorter than that made by KVS-1TI. O1 of DRVTI is presumably protonated as in KVS-1TI and thus makes a strong hydrogen bond with Asp25, whereas all other distances to the Asp dyad carboxylic oxygens are very similar to those in the PRTM/KVS-1TI complex. As in PRTM/KVS-1TI, the second oxygen of DRVTI (O2 on Figure 7C) is 2.7 Å away from Asp25′ Oδ2 and, by analogy with the joint XN structure of PRTM/KVS-1TI is probably an oxyanion. We also superimposed our PRTM/DRVTI structure on the previously published neutron structure of the PRTM/DRV complex obtained at pH 6.46 The two structures aligned remarkably well, with the rmsd on main chain atoms of less than 0.1 Å. DRV and DRVTI essentially occupy identical positions in the PRTM active site channels (Figure S7). Their two matching oxygen atoms have virtually identical contacts with the Asp dyad. The only difference between the ligands is the presence of an extra oxygen in DRVTI. In PRTM/DRV, the drug’s hydroxy group participates in an unusual low-barrier hydrogen bond, with the Asp25′ D atom being equidistant to three oxygen atoms and donates its D to form a bifurcated hydrogen bond with Asp25 Oδ1 and Oδ2. We anticipate that the distribution of H atoms in PRTM/DRVTI would be identical to their locations in the joint XN structure of PRTM/KVS-1TI.
Figure 7.

(A) Chemical diagram of clinical inhibitor DRV, its oxidized analogue keto-DRV, and the resulting tetrahedral intermediate DRVTI. DRVTI is shown as an oxyanion assuming its protonation state is the same as for KVS-1TI. (B) Catalytic site of the PRTM/DRVTI complex showing the 2FO-FC electron density map contoured at the 2.0 σ level. (C) Possible hydrogen bonding interactions between the Asp dyad and the tetrahedral intermediate moiety of DRVTI are based on the O···O distances given in Å.
ITC Provides Evidence that Keto-DRV is Inferior to DRV
Because our QM/MM calculations on the PRTM/KVS-1TI complex demonstrated instability of the oxyanion tetrahedral intermediate, we reasoned that keto-DRV might be a weaker inhibitor of the HIV-1 PR than DRV, if the DRVTI we observed in the RT X-ray structure of PRTM/DRVTI is also an oxyanion. Therefore, we performed ITC and enzyme inhibition measurements using wild-type PR (PRWT), PRTM, and a clinical drug resistant variant PR20 with DRV and keto-DRV. From Table 1 and Figures 8 (and S8), it is apparent that the binding affinity of keto-DRV to the three PR variants is 2–4 orders of magnitude weaker than that of DRV, indicating that the formed PR/DRVTI complexes are markedly less stable than the corresponding PR/DRV complexes. Moreover, affinity of keto-DRV to PR20 could not be determined with ITC, which is in agreement with our failed attempts to grow crystals of this complex. We then determined how well keto-DRV inhibits PR-catalyzed hydrolysis of a substrate (Figures 8 and S9). The initial rates and activity plots of substrate hydrolysis by PRWT, PRTM, and PR20 clearly indicate that keto-DRV is a weaker inhibitor than DRV. To slow the enzyme activity below 20% of the initial values (i.e., when no inhibitor was present) requires 1:2 and 1:20 enzyme-to-keto-DRV molar ratio for PRWT and PRTM, respectively, whereas PR20 is not inhibited at all even at a 1:20 molar ratio. In contrast, DRV already fully inhibits PRTM at a 1:1 molar ratio at these concentrations.
Table 1. Comparison of the Binding Affinity of DRV and Keto-DRV to PRWT, PRTM, and PR20.
Not determined because of very weak binding (Kd ≫ 10 μM).
Figure 8.

Initial rates for the hydrolysis of the chromogenic substrate catalyzed by the mature PR variants (panels A–D) and binding isotherms for complex formation of keto-DRV with PRWT and PRTM and DRV with PRTM and PR20 (panels E–H).
Discussion
HIV-1 PR is an indispensable viral enzyme that drives the maturation stage of the HIV-1 lifecycle ensuring conversion of immature viral particles into infectious virions. Thus, PR has been considered a crucial target for the design and development of anti-HIV drugs, and its structure and function have been thoroughly studied.5,12 As a member of the aspartic protease family of enzymes, HIV-1 PR catalyzes peptide bond cleavage by utilizing a pair of co-located Asp residues, one of which is protonated, and a lytic water molecule that is symmetrically hydrogen bonded to the catalytic Asp dyad in the substrate-free (or inhibitor-free) form of the enzyme.13,16 The juxtaposition and interplay of the three H atoms present in the catalytic site of HIV-1 PR throughout the peptide bond hydrolysis reaction determines the actual chemical mechanism of amide bond hydrolysis. Although many reaction pathways have been considered using theoretical calculations,23,24 there is no experimental evidence on the exact location and movement of these H atoms within the PR catalytic site along the reaction coordinate because protons are normally not seen in X-ray structures. Consequently, an important unanswered mechanistic question remains: does the peptide bond hydrolysis reaction catalyzed by the HIV-1 PR proceed through a stable gem-diol intermediate, a metastable oxyanion intermediate, or possibly an oxyanion transition state? The latter case corresponds to a concerted mechanism with multiple possible pathways, each having no intermediates on the reaction potential energy surface.13,31,34
We approached this question by using neutron diffraction to probe the structure of a tetrahedral intermediate formed by the attack of the lytic water molecule on the carbonyl of the non-hydrolysable ketomethylene isostere of the hexapeptide KVS-1.43,44 Neutrons allowed us to directly visualize H atoms (observed as D atoms) in the resulting PRTM/KVS-1TI complex at RT and accurately determine their exact locations. We observed protonation of only one hydroxy group of the tetrahedral intermediate moiety, whereas the other appears to be deprotonated. Thus, the tetrahedral intermediate moiety is an oxyanion, having an OH group and a negatively charged O–, rather than a neutral gem-diol with two OH groups. Moreover, both Asp25 and Asp25′ were found to be protonated on the “inner” (Oδ1) atoms, so that the catalytic Asp dyad bears a neutral charge in the complex with a mimic of the anionic tetrahedral intermediate. The doubly protonated catalytic Asp dyad was previously considered in the HIV-1 PR catalytic mechanism calculations, with the reaction proceeding through a metastable oxyanion tetrahedral intermediate16,27,28,31 or an oxyanion transition state,34 where the two H atoms were either placed on both “outer” (Oδ2) Asp oxygens, or each positioned on the “inner” and “outer” oxygens. The two possible theoretical structures of the oxyanion tetrahedral intermediate with both Asp25 and Asp25′ protonated on the “inner” oxygens were optimized by Garrec et al.29 using cluster DFT calculations. Both structures were found to be unstable during geometry optimizations, in agreement with our QM/MM calculations on the joint XN structure of PRTM/KVS-1TI.
Interestingly, our results would agree with the concerted acyclic reaction mechanism proposed very recently by Lawal et al.,34 but would disagree with the NMR measurements of kinetic isotope effects done by Kipp et al.17 that are consistent with the formation of a gem-diol intermediate, which falls apart into products after a proton is transferred from Asp25′ to the amine of the scissile C–N bond in the rate-limiting step. Also, in the previous X-ray structures containing trapped amide hydrate tetrahedral intermediates, one of the two tetrahedral intermediate hydroxyls made very short interactions of <2.5 Å with Asp25′, indicative of strong hydrogen bonding and protonation of either of the oxygens, whereas in PRTM/KVS-1TI the corresponding distance is 2.8 Å and both oxygens are deprotonated. Hence, we note that one should not ignore a possibility that such ketomethylene isostere-containing compounds may not be proper analogues of the tetrahedral intermediate formed in the HIV-1 PR catalyzed hydrolysis of substrates, and caution has to be exercised when using them in mechanistic enzymatic studies.
It is of note that PRTM/KVS-1TI and PRTM/DRVTI complexes containing oxyanion tetrahedral intermediate mimics can be trapped in crystals, and that the PR/KVS-1TI structure could be previously studied in solution with NMR.43,44 Solution inhibition studies by Marinier et al.49 indicated that some peptides with the ketomethylene functionality showed a 10-fold increased inhibition of the PR as compared to those having the hydroxyethylene isostere. The fact that we were able to trap KVS-1TI as oxyanion tetrahedral intermediate within the PRTM/KVS-1TI crystal lattice correlates well with these solution inhibition studies, even though our QM/MM calculations indicate geometry-optimized PRTM/KVS-1TI reverts to reactants. In the case of keto-DRV, the resulting DRVTI is 2–4 orders of magnitude weaker inhibitor of PR than the unmodified clinical inhibitor DRV. Because the chemical modification of DRV to give keto-DRV only alters the central hydroxyl into a keto-group, the large differences in the binding affinity of DRV and keto-DRV (and by extension of DRVTI) may be because of this modification and/or because of the repulsion of the negatively charged oxyanion and Asp25′ carboxylic oxygen. This inference appears to be in excellent agreement with the conclusion of Sayer et al.50 stating that a major contribution to the binding affinities of PR inhibitors like DRV comes from specific hydrogen bonding interactions with the catalytic Asp dyad.
Based on our crystallographic and solution data, it is not unreasonable to suggest that if DRVTI were a gem-diol, it would have been a superior inhibitor to DRV because the second OH group of the tetrahedral intermediate moiety would create an additional hydrogen-bonding capability compared to DRV. However, based on our results of weaker PR inhibition by keto-DRV compared to DRV, we estimate that nonpeptidic inhibitors, such as DRV, containing a carbonyl functional group that is converted to the geminal oxyanion, would be inferior to those with a single hydroxyl interacting with the catalytic Asp dyad, as was also demonstrated in the early studies of HIV-1 PR inhibitors having a vicinal di-ketone isostere.35 Nevertheless, inhibitors containing two hydroxyl groups (or a hydroxyl and an amine group) in the vicinal configuration, potentially adding an extra hydrogen bond to the catalytic Asp residues, might be of interest for future drug design. Our results also highlight the importance of obtaining structural information at near physiological temperatures, especially for the drug-resistant protease complexes with clinical drugs, as was also documented by us previously.45
Conclusion
By using neutron diffraction, we visualized hydrogen atoms in an enzyme–substrate analogue complex. We demonstrate that the in situ generated tetrahedral hydrated form of the keto isostere of the KVS-1 peptide (KVS-1TI) bound to the HIV-1 PRTM is an oxyanion, with one oxygen atom protonated (OH) and the other deprotonated (O–). Asp25 and Asp25′ were found to be both protonated, and the Asp25′ carboxylic O–H bond is rotated away from Asp25 and the tetrahedral intermediate moiety into a hydrophobic pocket lined up by residues Thr26′-Gly27′-Ala28′. The trapped oxyanion tetrahedral intermediate is stabilized by the oxyanion negative charge delocalization into the π system of the adjacent carbonyl group through strong n → π* hyperconjugative interactions, even though it is unstable according to our QM/MM geometry optimizations. We also show that keto-DRV, similar to KVS-1, is capable of producing a tetrahedral intermediate DRVTI when bound to HIV-1 PR. However, keto-DRV turned out to be a much weaker inhibitor than DRV. Finally, our observations indicate that novel protease inhibitors may benefit from functionalities capable of making additional hydrogen bonds with the catalytic Asp dyad.
Materials and Methods
General Information
Protein purification supplies were purchased from GE Healthcare (Piscataway, New Jersey, USA). Crystallization reagents were purchased from Hampton Research (Aliso Viejo, California, USA). Synthesis of KVS-1 has been described previously.43,44 DRV was obtained through the NIH AIDS reagent program. Keto-DRV was custom synthesized from DRV by Nanosyn (Santa Clara, CA).
Protein Expression, Purification, and Crystallization
The HIV-1 protease (pseudo-wild type) construct bears the stabilizing substitution mutations Q7K, L33I, L63I, C67A, and C95A to restrict autoproteolysis and cysteine-thiol oxidation.51 The PRTM has additional substitutions V32I, I47V, and V82I associated with drug resistance. Expression and purification from inclusion bodies of wild-type PR, PRTM, and extremely drug-resistant clinical isolate PR20 (Table S1) using Luria–Bertani were performed in Escherichia coli (BL21-DE3) cells as described previously.52−54 To obtain deuterated PRTM, the minimal medium made with 99.8% D2O and hydrogenous glycerol as the sole carbon source was used, and the deuterated enzyme was isolated, purified, and refolded from inclusion bodies in H2O buffers using standard protocols.55 KVS-1 stock solution [40 mM in dimethyl sulfoxide (DMSO)] was mixed with 3.0 mg/mL HIV-1 PR in a molar ratio of 10:1 for crystallization of the complex. For neutron crystallography, crystals were grown in 200 μL drops made by mixing the sample and the reservoir solution (0.1 M MES, 0.9 M NaCl, and pH 6.0) at a 1:1 ratio in a sitting drop setup using a Hampton Research sandwich box setup. A neutron-diffraction quality crystal grew to ∼0.15 mm3 in volume and the labile H atoms in the crystal were allowed to exchange with D by the D2O vapor for several months before the neutron data collection. The crystal was mounted in a quartz capillary containing the reservoir solution made with 99.97% D2O for the neutron diffraction data. Smaller crystals grown under the same reservoir conditions were used for X-ray data collection. Keto-DRV (20 mM stock in DMSO) was mixed with 3.0 mg/mL PRTM in a molar ratio of 5:1. Crystals of the complex were grown in 300 μL drops made by mixing the sample and the reservoir solution (0.1 M MES, 1.0 M NaCl, pH 6.0 in H2O) at a 1:1 ratio in the 9-well glass plate/sandwich box sitting drop setup.
X-ray and Neutron Data Collection
RT X-ray crystallographic data for PRTM/KVS-1TI and PRTM/DRVTI crystals were collected on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator and Osmic VariMax optics. The diffraction images were obtained using an R-Axis IV++ image plate detector. Diffraction data were integrated and scaled using the HKL3000 software suite indicating no appreciable radiation damage.56 Low temperature 100 K crystallographic data for PRTM/KVS-1TI were collected on the 5.0.3 beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory, USA, and the diffraction data were integrated and scaled using the mosflm from CCP4 software suite.57 The preliminary neutron diffraction data at RT were collected on the IMAGINE58 instrument located at the High Flux Isotope Reactor (Oak Ridge National Laboratory). The full quasi-Laue neutron diffraction dataset to 2.2 Å resolution was collected at RT from a 0.15 mm3 PRTM/KVS-1TI crystal taken from the same crystallization drop that provided the crystal for room-temperature X-ray data on the LADI-III beamline at the Institut Laue-Langevin, Grenoble, France.59 Images were collected from three different crystal orientations. At each orientation, the crystal was held stationary at different φ settings for each 24 h exposure. The neutron data were processed using the Daresbury Laboratory LAUE suite program LAUEGEN modified to account for the cylindrical geometry of the detector.60,61 The program LSCALE62 was used to determine the wavelength–normalization curve using the intensities of symmetry-equivalent reflections measured at different wavelengths. No explicit absorption corrections were applied. These data were then merged in SCALA.63 The summary of experimental data statistics is given in Table S2.
Joint XN Refinement
The joint XN structure of the PRTM/KVS-1TI complex was determined using nCNS(42,64) and manipulated in Coot.65 After initial rigid-body refinement, several cycles of positional, atomic displacement parameter and occupancy refinement were performed. The structure was checked for the correctness of side-chain conformations, hydrogen bonding, and orientation of D2O water molecules, which were built based on the mFo-DFc difference neutron scattering length density maps. The 2mFo-DFc and mFo-DFc neutron scattering length density maps were then examined to determine the correct orientation of hydroxyl groups and protonation states of the enzyme residues. The protonation states of some disordered side chains could not be obtained directly and remained ambiguous. All water molecules were refined as D2O. Initially, water oxygen atoms were positioned according to their electron density peaks and then were shifted slightly in accordance with the neutron scattering length density maps. All H atom positions in PRTM and labile H positions in KVS-1TI were modeled as D because of ∼85% deuteration level of the enzyme, and then the occupancies of the D atoms were refined individually within the range of −0.56 to 1.00. Before depositing the neutron structure to the PDB, a script was run that converts a record for the coordinates of a D atom into two records corresponding to an H and a D partially occupying the same site, both with positive partial occupancies that add up to unity.
Low and RT X-ray Refinement
The low-temperature X-ray structure of the PRTM/KVS-1TI complex was solved by molecular replacement using the structure in PDB (PDB ID: 3DCR)43 as the search model in program Phaser, incorporated in PHENIX.66 The Ramachandran statistics for the structures reported here have residues in most favored regions 98.5–99.0% and residues in additional allowed regions 1.5–1.0%. The RT X-ray structure of PRTM/DRVTI complex was solved by molecular replacement using the PRTM/DRV structure (PDB ID: 5E5J)46 and refined using SHELX-97.67,68 Figures were generated using the PyMol molecular graphics software (Schrödinger LLC, v.2.2.0).
Cluster DFT and NBO Analysis
The cluster DFT model was constructed from the atomic positions extracted from our joint XN structure of the PRTM/KVS-1TI complex. The 194-atom model contained residues Asp25–Ala28, Asp25′–Ala28′, truncated Ile50 and Ile50′, the flap water, and truncated KVS-1TI where all the side chains were reduced to methyl groups. All D atoms were renamed to H. All termini were capped with methyl acetamido groups and the methyl Hs were placed in idealized geometries with the program GaussView 6 from Gaussian (Wallingford, CT). The cluster model geometry was optimized at the M06-2X/6-311+G(d,p) level of theory along with the integral equation formalism variant of the polarizable continuum model (IEFPCM) for water solvation by using Gaussian 16, Revision B software from Gaussian.69 During geometry optimizations of KVS-1TI model in the oxyanion and gem-diol forms, positions of C, N, and O atoms were kept in their joint XN structure coordinates, while positions of H atoms were fully optimized. The optimized structures were subjected to natural bonding orbital analysis to identify the electron donor–acceptor interactions by a second-order perturbative analysis within the integrated Gaussian 16/NBO3.0 program.70,71 Orbitals were depicted with GaussView 6.
QM/MM Calculations
The model was constructed using the atomic coordinates from the joint XN structure of the PRTM/KVS-1TI complex. 50 ps molecular dynamics simulations were first performed to equilibrate the model and the 11,141-atom model for QM/MM calculations was then constructed by retaining a 30 Å sphere of atoms centered around the hydroxyl oxygens of the KVS-1TI inhibitor. Detailed description of these calculations is given in the Supporting Information and in Table S3.
ITC and Enzyme Activity Measurements
Stock solutions of the clinical inhibitor DRV (100 mM in DMSO) and keto-DRV (25 mM in DMSO) were diluted in a range of 90–150 μM in 5 mM sodium acetate, pH 6, and then adjusted to a final concentration with 50 mM sodium acetate, pH 5. ITC titrations were performed in 50 mM sodium acetate buffer at 28 °C and pH 5 on an ITC200 microcalorimeter (Malvern Instruments Inc., Westborough, MA) using 9–15 μM (as dimer) PRWT, PRTM or PR20, and 90–150 μM inhibitor. The PR samples were dialyzed against the ITC buffer in the presence of 0.36–0.6% DMSO prior to the measurements to compensate for the DMSO present in the titrant when diluting DRV or keto-DRV stock solutions. For competitive inhibitors that bind at only one site, dissociation constants (Kd = 1/Ka) are equivalent to the inhibition constants measured by enzyme kinetics (Ki). Data were processed using the Origin ITC software. Enzyme inhibition assays were carried out in 50 mM sodium acetate buffer (pH 5) at a final concentration of 0.5 μM mature PR either in the absence or presence of DRV or keto-DRV and 380 μM of the chromogenic substrate (Lys-Ala-Arg-Val-Nle-[4-nitrophenylalanine]-Glu-Ala-Nle-NH2, California Peptide Research, Napa, CA) in a total volume of 120 μL at 28 °C.
PDB Depositions
Coordinates and structure factors have been deposited in the Protein Data Bank with the accession numbers: 6PTP for the RT joint XN structure of PRTM/KVS-1TI, 6KMP for the low-temperature X-ray structure of PRTM/KVS-1TI, and 6PU8 for RT X-ray structure of PRTM/DRVTI.
Acknowledgments
This research using IMAGINE beamline at the Oak Ridge National Laboratory’s (ORNL) High Flux Isotope Reactor (HFIR) was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. This research was supported by the Protein Crystallography Section, RB & HSD at Bhabha Atomic Research Centre (BARC), Mumbai, and by the Office of Biological and Environmental Research at Oak Ridge National Laboratory’s Center for Structural Molecular Biology (CSMB). We used facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy and National Facility for Structural Biology, Department of Atomic Energy (DAE, India). We thank Institut Laue-Langevin (ILL, Grenoble, France) for awarding neutron beamtime on neutron diffraction beamline LADI-III. The D2O used in this research was supplied by the United States Department of Energy Office of Science by the Isotope Program in the Office of Nuclear Physics. We thank beamline staff of LADI-III, and 5.0.3 beamline at BCSB, Advanced Light Source, Lawrence Berkeley National Laboratory, California, USA for their support. We also thank the Department of Science and Technology, France Embassy, New Delhi for travel support to ILL and Dr. R. Chidambaram (DAE), former Principal Scientific Advisor to the Government of India for discussions. A.D. was supported by BARC, DAE. A.K. was supported by DOE BES. We also acknowledge support from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health. This manuscript has been authored jointly by DAE, BARC, India and UT-Battelle LLC under DOE contract no. DE-AC05-00OR22725.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00835.
Details of the QM/MM calculations, sequence alignment of PRWT, PRTM, and PR20, crystallographic data collection and refinement statistics, distances (Å) from C(KVS-1) to O(Wcat) in QM/MM calculations, electron density of KVS-1TI, comparison of the joint XN structure with unliganded HIV-1 PR and PRI54V/TI, Figures 4A,B and 5B in crossed-eye stereo mode, comparison of the room- and low-temperature structures of the PRTM/KVS-1TI complex, catalytic site geometries from QM/MM calculations, comparison of PRTM/DRVTI and PRTM/DRV complexes, raw ITC plots, and the effect of DRV and keto-DRV on PR variant substrate hydrolysis (PDF)
Author Present Address
∇ TIFR Centre for Interdisciplinary Sciences, Tata Institute of Fundamental Research Hyderabad, Hyderabad 500107, India.
Author Contributions
A.D. designed research and planned experiments; A.D. and A.K. performed research; A.D. and M.P.B. collected neutron diffraction data; A.D., K.M., and A.K. collected X-ray diffraction data; A.D., A.K., K.M., M.K., M.P.B., and S.B.H.K. analyzed the data and results; A.K. and T.W. performed molecular modeling; J.M.L. collected and analyzed ITC and the enzyme kinetics data; and A.D., J.M.L., and A.Y.K. wrote the manuscript with input from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Engelman A.; Cherepanov P. The Structural Biology of HIV-1: Mechanistic and Therapeutic Insights. Nat. Rev. Microbiol. 2012, 10, 279–290. 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman T. L.; Buckheit R. W. Jr. The Continuing Evolution of HIV-1 Therapy: Identification and Development of Novel Antiretroviral Agents Targeting Viral and Cellular Targets. Mol. Biol. Int. 2012, 2012, 401965. 10.1155/2012/401965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensing A. M. J.; van Maarseveen N. M.; Nijhuis M. Fifteen Years of HIV Protease Inhibitors: Raising the Barrier to Resistance. Antiviral Res. 2010, 85, 59–74. 10.1016/j.antiviral.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Mitsuya H.; Maeda K.; Das D.; Ghosh A. K. Development of Protease Inhibitors and the Fight with Drug-Resistant HIV-1 Variants. Adv. Pharmacol. 2008, 56, 169–197. 10.1016/s1054-3589(07)56006-0. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Osswald H. L.; Prato G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbaiah M. A. M.; Meanwell N. A.; Kadow J. F. Design Strategies in the Prodrugs of HIV-1 Protease Inhibitors to Improve the Pharmaceutical Properties. Eur. J. Med. Chem. 2017, 139, 865–883. 10.1016/j.ejmech.2017.07.044. [DOI] [PubMed] [Google Scholar]
- Weber I.; Harrison RW Tackling the Problem of HIV Drug Resistance. Postepy Biochem. 2016, 62, 273–279. [PubMed] [Google Scholar]
- Beck Z.; Morris G.; Elder J. Defining HIV-1 Protease Substrate Selectivity. Curr. Drug Targets 2002, 2, 37–50. 10.2174/1568005024605837. [DOI] [PubMed] [Google Scholar]
- Navia M. A.; Fitzgerald P. M. D.; McKeever B. M.; Leu C.-T.; Heimbach J. C.; Herber W. K.; Sigal I. S.; Darke P. L.; Springer J. P. Three-Dimensional Structure of Aspartyl Protease from Human Immunodeficiency Virus HIV-1. Nature 1989, 337, 615–620. 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Miller M.; Jaskolski M.; Sathyanarayana B.; Baldwin E.; Weber I.; Selk L.; Clawson L.; Schneider J.; Kent S. Conserved Folding in Retroviral Proteases: Crystal Structure of a Synthetic HIV-1 Protease. Science 1989, 245, 616–621. 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- Lapatto R.; Blundell T.; Hemmings A.; Overington J.; Wilderspin A.; Wood S.; Merson J. R.; Whittle P. J.; Danley D. E.; Geoghegan K. F.; Hawrylik S. J.; Lee S. E.; Scheld K. G.; Hobart P. M. X-ray Analysis of HIV-1 Proteinase at 2.7 Å Resolution Confirms Structural Homology Among Retroviral Enzymes. Nature 1989, 342, 299–302. 10.1038/342299a0. [DOI] [PubMed] [Google Scholar]
- Aspartic Acid Proteases as Therapeutic Targets; Ghosh A. K., Ed.; Wiley-CVH Verlag GmbH & Co. KGaA, 2010; Vol. 45, p 615. [Google Scholar]
- Brik A.; Wong C.-H. HIV-1 Protease: Mechanism and Drug Discovery. Org. Biomol. Chem. 2003, 1, 5–14. 10.1039/b208248a. [DOI] [PubMed] [Google Scholar]
- Hyland L. J.; Tomaszek T. A. Jr.; Roberts G. D.; Carr S. A.; Magaard V. W.; Bryan H. L.; Fakhoury S. A.; Moore M. L.; Minnich M. D.; Culp J. S.; DesJarlais R. L.; Meek T. D. Human Immunodeficiency Virus-1 Protease: 1. Initial Velocity Studies and Kinetic Characterization of Reaction Intermediates by 18O Isotope Exchange. Biochemistry 1991, 30, 8441–8453. 10.1021/bi00098a023. [DOI] [PubMed] [Google Scholar]
- Hyland L. J.; Tomaszek T. A. Jr.; Meek T. D. Human Immunodeficiency Virus-1 Protease: 2. Use of pH Rate Studies and Solvent Kinetic Isotope Effects to Elucidate Details of Chemical Mechanism. Biochemistry 1991, 30, 8454–8463. 10.1021/bi00098a024. [DOI] [PubMed] [Google Scholar]
- Northrop D. B. Follow the Protons: A Low-Barrier Hydrogen Bond Unifies the Mechanisms of the Aspartic Proteases. Acc. Chem. Res. 2001, 34, 790–797. 10.1021/ar000184m. [DOI] [PubMed] [Google Scholar]
- Kipp D. R.; Hirschi J. S.; Wakata A.; Goldstein H.; Schramm V. L. Transition States of Native and Drug-Resistant HIV-1 Protease are the Same. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 6543–6548. 10.1073/pnas.1202808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbeev V. Y.; Kent S. B. H. Ionization State of the Catalytic Dyad Asp25/25’ in the HIV-1 Protease: NMR Studies of Site-Specifically 13C Labelled HIV-1 Protease Prepared by Total Chemical Synthesis. Org. Biomol. Chem. 2012, 10, 5887–5891. 10.1039/c2ob25569c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez E. J.; Angeles T. S.; Meek T. D. Use of Nitrogen-15 Kinetic Isotope Effects to Elucidate Details of the Chemical Mechanism of Human Immunodeficiency Virus 1 Protease. Biochemistry 1993, 32, 12380–12385. 10.1021/bi00097a015. [DOI] [PubMed] [Google Scholar]
- Torshin I. Y.; Harrison R. W.; Weber I. T. Close Pairs of Carboxylates: a Possibility of Multicenter Hydrogen Bonds in Proteins. Protein Eng. 2003, 16, 201–207. 10.1093/proeng/gzg027. [DOI] [PubMed] [Google Scholar]
- Piana S.; Carloni P. Conformational Flexibility of the Catalytic Asp Dyad in HIV-1 Protease: an Ab Initio Study on the Free Enzyme. Proteins: Struct., Funct., Genet. 2000, 39, 26–36. . [DOI] [PubMed] [Google Scholar]
- Porter M. A.; Molina P. A. The Low-Barrier Double-Well Potential of the Oδ1-H-Oδ1 Hydrogen Bond in Unbound HIV Protease: a QM/MM Characterization. J. Chem. Theory Comput. 2006, 2, 1675–1684. 10.1021/ct600200s. [DOI] [PubMed] [Google Scholar]
- Chatfield D. C.; P. Eurenius K.; Brooks B. R. HIV-1 Protease Cleavage Mechanism: a Theoretical Investigation Based on Classical MD Simulation and Reaction Path Calculations Using a Hybrid QM/MM Potential. J. Mol. Struct. 1998, 423, 79–92. 10.1016/s0166-1280(96)04875-0. [DOI] [Google Scholar]
- Okimoto N.; Tsukui T.; Hata M.; Hoshino T.; Tsuda M. Hydrolysis Mechanism of the Phenylalanine-Proline Peptide Bond Specific to HIV-1 Protease: Investigation by the Ab Initio Molecular Orbital Method. J. Am. Chem. Soc. 1999, 121, 7349–7354. 10.1021/ja9841106. [DOI] [Google Scholar]
- Piana S.; Carloni P.; Parrinello M. Role of Conformational Fluctuations in the Enzymatic Reaction of HIV-1 Protease. J. Mol. Biol. 2002, 319, 567–583. 10.1016/s0022-2836(02)00301-7. [DOI] [PubMed] [Google Scholar]
- Piana S.; Bucher D.; Carloni P.; Rothlisberger U. Reaction Mechanism of HIV-1 Protease by Hybrid Car-Parrinello/Classical MD Simulations. J. Phys. Chem. B 2004, 108, 11139–11149. 10.1021/jp037651c. [DOI] [Google Scholar]
- Trylska J.; Grochowski P.; McCammon J. A. The Role of Hydrogen Bonding in the Enzymatic Reaction Catalyzed by HIV-1 Protease. Protein Sci. 2004, 13, 513–528. 10.1110/ps.03372304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelic S.; Åqvist J. Catalysis and Linear Free Energy Relationships in Aspartic Proteases. Biochemistry 2006, 45, 7709–7723. 10.1021/bi060131y. [DOI] [PubMed] [Google Scholar]
- Garrec J.; Sautet P.; Fleurat-Lessard P. Understanding the HIV-1 Protease Reactivity with DFT: What Do We Gain from Recent Functionals?. J. Phys. Chem. B 2011, 115, 8545–8558. 10.1021/jp200565w. [DOI] [PubMed] [Google Scholar]
- Ribeiro A. J. M.; Santos-Martins D.; Russo N.; Ramos M. J.; Fernandes P. A. Enzymatic Flexibility and Reaction Rate: a QM/MM Study of HIV-1 Protease. ACS Catal. 2015, 5, 5617–5626. 10.1021/acscatal.5b00759. [DOI] [Google Scholar]
- Krzeminska A.; Moliner V.; Swiderek K. Dynamic and Electrostatic Effects on the Reaction Catalyzed by HIV-1 Protease. J. Am. Chem. Soc. 2016, 138, 16283–16298. 10.1021/jacs.6b06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker J. C. R.; Vincent M. A.; Popelier P. L. A. Using the Relative Energy Gradient Method with Interacting Quantum Atoms to Determine the Reaction Mechanism and Catalytic Effects in the Peptide Hydrolysis in HIV-1 Protease. Chem.—Eur. J. 2018, 24, 11200–11210. 10.1002/chem.201802035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadiq S. K.; Coveney P. V. Computing the Role of Near Attack Conformations in an Enzyme-Catalyzed Nucleophilic Bimolecular Reaction. J. Chem. Theory Comput. 2015, 11, 316–324. 10.1021/ct5008845. [DOI] [PubMed] [Google Scholar]
- Lawal M. M.; Sanusi Z. K.; Govender T.; Tolufashe G. F.; Maguire G. E. M.; Honarparvar B.; Kruger H. G. Unraveling the Concerted Catalytic Mechanism of the Human Immunodeficiency Virus Type 1 (HIV-1) Protease: a Hybrid QM/MM Study. Struct. Chem. 2019, 30, 409–417. 10.1007/s11224-018-1251-9. [DOI] [Google Scholar]
- Slee D. H.; Laslo K. L.; Elder J. H.; Ollmann I. R.; Gustchina A.; Kervinen J.; Zdanov A.; Wlodawer A.; Wong C.-H. Selectivity in the Inhibition of HIV and FIV Protease: Inhibitory and Mechanistic Studies of Pyrrolidine-Containing α-Keto Amide and Hydroxyethyleneamine Core Structures. J. Am. Chem. Soc. 1995, 117, 11867–11878. 10.1021/ja00153a008. [DOI] [Google Scholar]
- Silva A. M.; Cachau R. E.; Sham H. L.; Erickson J. W. Inhibition and Catalytic Mechanism of HIV-1 Aspartic Protease. J. Mol. Biol. 1996, 255, 321–340. 10.1006/jmbi.1996.0026. [DOI] [PubMed] [Google Scholar]
- Kovalevsky A. Y.; Chumanevich A. A.; Liu F.; Louis J. M.; Weber I. T. Caught in the Act: the 1.5 Å Resolution Crystal Structures of the HIV-1 Protease and the I54V Mutant Reveal a Tetrahedral Reaction Intermediate. Biochemistry 2007, 46, 14854–14864. 10.1021/bi700822g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A.; Mahale S.; Prashar V.; Bihani S.; Ferrer J.-L.; Hosur M. V. X-ray Snapshot of HIV-1 Protease in Action: Observation of Tetrahedral Intermediate and Short Ionic Hydrogen Bond SIHB with Catalytic Aspartate. J. Am. Chem. Soc. 2010, 132, 6366–6373. 10.1021/ja100002b. [DOI] [PubMed] [Google Scholar]
- Shen C.-H.; Tie Y.; Yu X.; Wang Y.-F.; Kovalevsky A. Y.; Harrison R. W.; Weber I. T. Capturing the Reaction Pathway in Near-Atomic-Resolution Crystal Structures of HIV-1 Protease. Biochemistry 2012, 51, 7726–7732. 10.1021/bi3008092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen E.; Chen J. C.-H.; Fisher S. Z. Neutron Crystallography for the Study of Hydrogen Bonds in Macromolecules. Molecules 2017, 22, 596. 10.3390/molecules22040596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkar R.; Bilheux H. Z.; Bordallo H.; Briber R.; Callaway D. J. E.; Cheng X.; Chu X.-Q.; Curtis J. E.; Dadmun M.; Fenimore P.; Fushman D.; Gabel F.; Gupta K.; Herberle F.; Heinrich F.; Hong L.; Katsaras J.; Kelman Z.; Kharlampieva E.; Kneller G. R.; Kovalevsky A.; Krueger S.; Langan P.; Lieberman R.; Liu Y.; Losche M.; Lyman E.; Mao Y.; Marino J.; Mattos C.; Meilleur F.; Moody P.; Nickels J. D.; O’Dell W. B.; O’Neill H.; Perez-Salas U.; Peters J.; Petridis L.; Sokolov A. P.; Stanley C.; Wagner N.; Weinrich M.; Weiss K.; Wymore T.; Zhang Y.; Smith J. C. Neutron Scattering in the Biological Sciences: Progress and Prospects. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74, 1129–1168. 10.1107/s2059798318017503. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Mustyakimov M.; Afonine P. V.; Langan P. Generalized X-ray and Neutron Crystallographic Analysis: More Accurate and Complete Structures for Biological Macromolecules. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 567–573. 10.1107/s0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbeev V. Y.; Mandal K.; Terechko V. A.; Kent S. B. H. Crystal Structure of Chemically Synthesized HIV-1 Protease and a Ketomethylene Isostere Inhibitor Based on the p2/NC Cleavage Site. Bioorg. Med. Chem. Lett. 2008, 18, 4554–4557. 10.1016/j.bmcl.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Torbeev V. Y.; Raghuraman H.; Hamelberg D.; Tonelli M.; Westler W. M.; Perozo E.; Kent S. B. H. Protein Conformational Dynamics in the Mechanism of HIV-1 Protease Catalysis. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 20982–20987. 10.1073/pnas.1111202108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlits O.; Keen D. A.; Blakeley M. P.; Louis J. M.; Weber I. T.; Kovalevsky A. Room Temperature Neutron Crystallography of Drug Resistant HIV-1 Protease Uncovers Limitations of X-ray Structure Analysis at 100K. J. Med. Chem. 2017, 60, 2018–2025. 10.1021/acs.jmedchem.6b01767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlits O.; Wymore T.; Das A.; Shen C.-H.; Parks J. M.; Smith J. C.; Weiss K. L.; Keen D. A.; Blakeley M. P.; Louis J. M.; Langan P.; Weber I. T.; Kovalevsky A. Long-Range Electrostatics-Induced Two-Proton Transfer Captured by Neutron Crystallography in an Enzyme Catalytic Site. Angew. Chem., Int. Ed. 2016, 55, 4924–4927. 10.1002/anie.201509989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner T. The hydrogen bond in the solid state. Angew. Chem., Int. Ed. 2002, 41, 49–76. . [DOI] [PubMed] [Google Scholar]
- Windsor I. W.; Gold B.; Raines R. T. An n→π* Interaction in the Bound Substrate of Aspartic Proteases Replicates the Oxyanion Hole. ACS Catal. 2019, 9, 1464–1471. 10.1021/acscatal.8b04142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinier A.; Toth M. V.; Houseman K.; Mueller R.; Marshall G. R. HIV-1 Protease Inhibitors: Ketomethylene Isosteres with Unusually High Affinity Compared with Hydroxyethylene Isostere Analogs. Bioorg. Med. Chem. 1994, 2, 919–925. 10.1016/s0968-0896(00)82041-6. [DOI] [PubMed] [Google Scholar]
- Sayer J. M.; Liu F.; Ishima R.; Weber I. T.; Louis J. M. Effect of the Active Site D25N Mutation on the Structure, Stability, and Ligand Binding of the Mature HIV-1 Protease. J. Biol. Chem. 2008, 283, 13459–13470. 10.1074/jbc.m708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis JM; Clore GM; Gronenborn AM Autoprocessing of HIV-1 Protease is Tightly Coupled to Protein Folding. Nat. Struct. Biol. 1999, 6, 868–75. 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- Wondrak E. M.; Louis J. M. Influence of Flanking Sequences on the Dimer Stability of Human Immunodeficiency Virus Type 1 Protease. Biochemistry 1996, 35, 12957–12962. 10.1021/bi960984y. [DOI] [PubMed] [Google Scholar]
- Mahalingam B.; Louis J. M.; Hung J.; Harrison R. W.; Weber I. T. Structural Implications of Drug-Resistant Mutants of HIV-1 Protease: High-Resolution Crystal Structures of the Mutant Protease/Substrate Analogue Complexes. Proteins: Struct., Funct., Genet. 2001, 43, 455–464. 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- Louis J. M.; Aniana A.; Weber I. T.; Sayer J. M. Inhibition of Autoprocessing of Natural Variants and Multidrug Resistant Mutant Precursors of HIV-1 Protease by Clinical Inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9072–9077. 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber I. T.; Waltman M. J.; Mustyakimov M.; Blakeley M. P.; Keen D. A.; Ghosh A. K.; Langan P.; Kovalevsky A. Y. Joint X-ray/Neutron Crystallographic Study of HIV-1 Protease with Clinical Inhibitor Amprenavir: Insights for Drug Design. J. Med. Chem. 2013, 56, 5631–5635. 10.1021/jm400684f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL3000: The Integration of Data, Reduction and Structure Solution – From Diffraction Images to an Initial Model in Minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 859–866. 10.1107/s0907444906019949. [DOI] [PubMed] [Google Scholar]
- Battye T. G. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. W. IMosflm: a New Graphical Interface for Diffraction-Image Processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 271–281. 10.1107/s0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilleur F.; Munshi P.; Robertson L.; Stoica A. D.; Crow L.; Kovalevsky A.; Koritsanszky T.; Chakoumakos B. C.; Blessing R.; Myles D. A. A. The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 2157–2160. 10.1107/s0907444913019604. [DOI] [PubMed] [Google Scholar]
- Blakeley M. P.; Teixeira S. C. M.; Petit-Haertlein I.; Hazemann I.; Mitschler A.; Haertlein M.; Howard E.; Podjarny A. D. Neutron Macromolecular Crystallography with LADI-III. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 1198–1205. 10.1107/s0907444910019797. [DOI] [PubMed] [Google Scholar]
- Campbell J. W. LAUEGEN, an X-Windows-Based Program for the Processing of Laue Diffraction Data. J. Appl. Crystallogr. 1995, 28, 228–236. 10.1107/s002188989400991x. [DOI] [Google Scholar]
- Campbell J. W.; Hao Q.; Harding M. M.; Nguti N. D.; Wilkinson C. LAUEGEN Version 6.0 and INTLDM. J. Appl. Crystallogr. 1998, 31, 496–502. 10.1107/s0021889897016683. [DOI] [Google Scholar]
- Arzt S.; Campbell J. W.; Harding M. M.; Hao Q.; Helliwell J. R. LSCALE - the New Normalization, Scaling and Absorption Correction Program in the Daresbury Laue Software Suite. J. Appl. Crystallogr. 1999, 32, 554–562. 10.1107/s0021889898015350. [DOI] [Google Scholar]
- Weiss M. S. Global Indicators of X-ray Data Quality. J. Appl. Crystallogr. 2001, 34, 130–135. 10.1107/s0021889800018227. [DOI] [Google Scholar]
- Mustyakimov M.; Langan P.. Copyright C-06, 104 Patch for CNS; nCNS an Open Source Distribution Patch for CNS for Macromolecular Structure Refinement; Los Alamos National Security: Los Alamos, NM, USA, 2007. [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/s0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/s0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
- Reed A. E.; Weinstock R. B.; Weinhold F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. 10.1063/1.449486. [DOI] [Google Scholar]
- Glendening E.; Reed A.; Carpenter J.; Weinhold F.. NBO Program, version 3.1; University of Wisconsin: Madison: 2001.
- King N. M.; Prabu-Jeyabalan M.; Nalivaika E. A.; Wigerinck P.; de Béthune M.-P.; Schiffer C. A. Structural and Thermodynamic Basis for the Binding of TMC114, a Next-Generation Human Immunodeficiency Virus Type 1 Protease Inhibitor. J. Virol. 2004, 78, 12012–12021. 10.1128/jvi.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.