

Abstract

NS3/4A protease of hepatitis C virus (HCV) plays an important role in viral RNA replication. A 1,4-diphenylbutanedicarboxylic acid derivative, namely, phyllanthin, extracted from the leaf of a herbal plant, Phyllanthus amarus, inhibits HCV NS3/4A protease and replication activities. However, the reduced aqueous solubility, high toxicity, and poor oral bioavailability are major impediments with phyllanthin. We herein present a design approach to generate phyllanthin congeners in order to potentiate inhibition activity against protease. The phyllanthin congeners were synthesized by chemical methods and subjected to systematic biological studies. One of the congeners, annotated as D8, is identified as a novel and potent inhibitor of the HCV-NS3/4Aprotease activity in vitro and the viral RNA replication in cell culture. Structural analysis using the computational-based docking approach demonstrated important noncovalent interactions between D8 and the catalytic residues of the viral protease. Furthermore, D8 was found to be significantly nontoxic in cell culture. More importantly, oral administration of D8 in BALB/c mice proved its better tolerability and bioavailability, as compared to native phyllanthin. Taken together, this study reveals a promising candidate for developing anti-HCV therapeutics to control HCV-induced liver diseases.
Introduction
With an estimated 171 million carriers, hepatitis C represents a public health burden globally.1 Hepatitis C virus (HCV) infection is a major cause of chronic liver diseases including hepatocellular carcinoma (HCC). HCV is categorized in the Hepacivirus genus of the Flaviviridae family. Its genome is a 9.6 kb long positive strand RNA encoding a single polyprotein of about 3010 amino acids.2 The cleavage of this polyprotein by cellular and viral proteases gives rise to 10 smaller proteins including three structural proteins (core, E1, and E2) and seven nonstructural proteins (P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B).3 The nonstructural proteins have critical functions in viral replication.4 Of these, NS3/4A assumes incredible significance as it is involved in many functions including suppression of the antiviral mechanism by evading host immune strategies. The NS3/4A protease has been shown to interfere with the pathogen recognition TLR3- and RIG-I/MDA5-mediated signaling pathways, by cleaving TRIF and MAVS signaling adaptors, respectively. This prevents the activation of type-I interferon stimulated genes (ISGs). For these reasons, the NS3/4A protease represents one of the most attractive targets for anti-HCV drug discovery.5−7
NS3 protease is a Zn(II)-dependent, chymotrypsin-like serine protease in which the catalytic triad (Ser139, His57, and Asp81) performs the enzymatic reaction.8,9 A majority of the current direct acting antivirals (DAAs) inhibit the catalytic activity of NS3 protease. However, a high replication rate and low fidelity of HCV RNA-dependent RNA-polymerase (RdRp) leads to the generation of heterogenous viral population and quasi-states leading to accumulation of drug-resistant mutations in NS3 protease and rendering the drugs less-effective.10−13 As a result, the search for classes of lead compounds assumes significance. In these efforts, we and others have reported successful attempts for the use various plant extracts and their derivatives to regulate the HCV infection.14−16 For example, punicalagin from pomegranate,16 curcumin from turmeric,17 silymarin from milk thistle18 exhibit considerable anti-HCV activity. In an earlier work, we demonstrated that the extract of the age-old folk plant, Phyllanthus amarus, successfully inhibited NS3 and NS5B-RdRp enzymes.19 Recently, we have also shown that corilagin, one of the active ellagitannins of the P. amarus extract, could inhibit HCV NS3 protease activity at sub-micromolar concentrations and NS5B-RdRp activity at higher concentrations.14
Phyllanthin is one of the active lignin, present in the leaf extracts of P. amarus. Earlier reports have demonstrated antioxidant, anticancer, antidiabetic, hepatoprotective and anti-inflammatory properties of phyllanthin.20−22 There are also reports which probed the role of phyllanthin in treating hepatitis B virus infection.23 In the present study, we report that phyllanthin, in its natural form, is capable of inhibiting NS3 protease activity. As phyllanthin is less water-soluble and has poor bioavailability, a rational structure-based design approach is exercised to identify phyllanthin structural congeners. The 1,4-diphenyl butane scaffold of phyllanthin with C-2, C-3 substitutions provides an opportunity to derive the structural congeners. The substitution pattern at C-2 and C-3 carbons was planned. The Stobbe condensation reaction of succinic ester with aromatic aldehydes was planned in order to synthesize the structural congeners. Following the synthesis of congeners, the compounds are evaluated for their NS3/4A protease inhibitory activity in vitro. Among these compounds, a dicarboxylic acid derivative, annotated as D8, has been observed to inhibit the NS3 protease inhibitory activity more effectively than phyllanthin. The aqueous solubility of D8 is the key reason for its better NS3 protease inhibition and bioavailability in vivo compared with parental phyllanthin. The systematic docking study enables evaluation of the mode of D8 binding to the active site of the protease. Furthermore, in vivo studies of D8 derivative proves its nontoxicity, bioavailability, and hence, safer use. Results of the computational, chemical and biological studies leading to identification of D8 as a potent HCV NS3 protease inhibitor are presented herein.
Results
One of the most promising approaches to an effective antiviral therapy is the development of small molecule inhibitors directed against the viral NS3 serine protease, which is an essential component required for the maturation of HCV polyprotein.24 Phyllanthin (Phy) is constituted with 1,4-diphenyl butane scaffold, with methoxymethyl and methoxy substituents in the butane and phenyl segments, respectively. Aqueous solubility of as isolated phyllanthin is 0.25 mg mL–1, necessitating further studies to be conducted in a mixture of organic-aqueous mixtures. The substituents around the scaffold provide an opportunity for modifications.
The importance of phyllanthin scaffold in binding to NS3 protease was assessed initially through a docking analysis. Similarities were observed between the aromatic rings and methoxy functional groups of matairesinol and phyllanthin structures. Matairesinol is a ligand bound to a dehydrogenase (PDB id: 2BGM),25 whose catalytic triad (Ser, Tyr and Lys) is similar to the catalytic triad residues of NS3 protease (Ser, Asp, and His). Our earlier studies have also demonstrated that the serine proteases, in general, are inhibited in the catalytic region. We have shown that at least 16 interactions could be correctly predicted in the docked NS3-telaprevir model with the aid of Autodock 4.2.16 Hence, to predict the binding mode of phyllanthin, it was docked into the active site of NS3 protease using Autodock 4.2. The interactions predicted by docking are shown in Figure 1, where hydrogen bonds between two of the three catalytic residues (Ser139 and His57) of NS3 protease and phyllanthin are shown. Other contacts between phyllanthin and NS3 are formed by Ala156, Lys136, and Val132. The interaction energy for this binding is calculated to be −3 kcal mol–1. Keeping in view that computational analysis showed the importance of hydrogen bonding and aromatic stacking interactions at the catalytic triad of phyllanthin–NS3 protease complex, the modifications warranted retaining the structural features intact for the above interactions.
Figure 1.
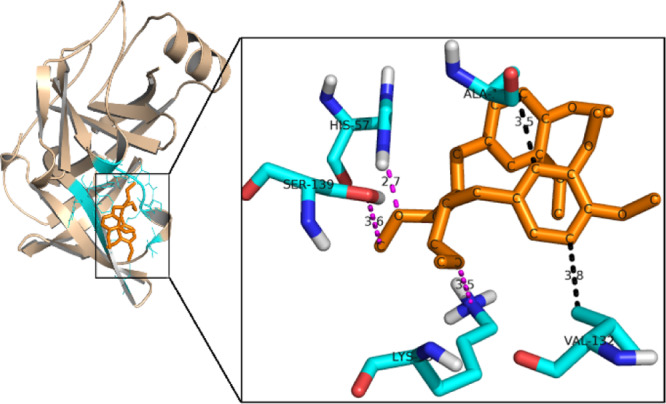
Molecular docking of the phyllanthin molecule with HCV NS3 protease. Color code: wheat: HCV NS3-protease; orange: phyllanthin; cyan: residues of the binding pocket which are involved in interactions. Zoom: view of the important interactions. Cyan: NS3 protease; pink: hydrogen bond interactions; black: hydrophobic interactions.
Chemical Synthesis
The chemical synthesis of phyllanthin structural congeners allowed variabilities in synthons undergoing the assembly to target molecules. Figure 2 shows the congener structures that are designed and synthesized in the present study.
Figure 2.
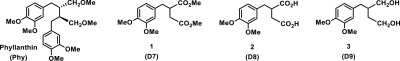
Molecular structures of phyllanthin (Phy) and congeners 1–3.
Naturally occurring phyllanthin (Phy) was extracted from P. amarus as reported previously.26 One of the early modifications planned was at C-2, C-3 carbons at the methoxymethylene side chain functionalities. A Stobbe condensation reaction of 3,4-dimethoxy benzaldehyde with diethyl succinate under basic condition formed the route to synthesize the congeners 1–3 (Scheme 1).
Scheme 1. Synthesis of Congeners 1–3.

3,4-Dimethoxy benzaldehyde was reacted with diethyl succinate, under a basic condition. Following work up, the reaction mixture was subjected to methyl ester formation upon treatment with SOCl2/MeOH. Purification of the reaction mixture afforded mono- and bis-substitution products.27 The desired monosubstitution product was isolated and subjected to Pd–C-mediated reduction to secure 1, in a moderate yield. In subsequent reactions, a base-mediated hydrolysis of ester functionalities afforded derivative 2,28−30 whereas an LAH-mediated reduction afforded bishydroxymethyl derivative 3. Both these derivatives were obtained in good to excellent yields. Physical characterizations are performed by NMR spectroscopy, mass spectrometry ascertained the structural homogeneities of derivatives 1–3 (D7–D9), and these derivatives are racemic.
Biochemical and Biological Evaluations
Effect of Phyllanthin and Its Synthetic Derivatives on HCV NS3/4A Protease Activity In Vitro
Phyllanthin (Phy) and three synthetic derivatives (Phy, D7–D9) were tested at first, in a fluorometric reaction-based NS3/4A protease activity assay. Among the synthetic derivatives tested in this assay, D8 was found to be the most effective inhibitor with an IC90 of 50 μM (Figure 3), and thus, subsequent studies were focused on this derivative.
Figure 3.

Effect of phyllanthin (Phy) and its derivatives (50 μM) on HCV NS3/4A protease activity. Inhibitions were compared with a standard control reaction with no test compound (Std) as 100%. Symbol * indicates a statistical significance with the P value (<0.05).
In order to investigate whether the inhibitory effect is dose-dependent, the above assay was performed at varying D8 concentrations, in the range from 10 to 100 μM. A dose-dependent NS3 protease inhibition activity was observed, and complete inhibition occurred at 75 μM of D8. Telaprevir, a standard NS3 protease inhibitor, was used as the positive control in this assay (Figure 4a).
Figure 4.

(a) Concentration-dependent inhibitory effect of D8 on NS3/4A protease activity used as a standard control. (b) Effect of D8 on cellular serine protease activity. Trypsin was incubated with the substrate in the presence of increasing concentrations of D8. Inhibitions were compared with a standard control reaction with no test compound (Std) as 100%. In this assay, the G. max soyabean inhibitor14 was used as a positive control. Symbol * indicates a statistically significant difference in terms of the P value (<0.05).
Structural Basis of NS3 Protease Inhibition by D8
The experimental studies showed several fold higher efficacy for D8 when compared to native phyllanthin. Hence, molecular docking was performed to further understand the interactions of D8 with NS3 protease enzyme (Figure 5). D8 showed better interaction profile than phyllanthin as observed by binding energy of −5.7 kcal mol–1 for D8 vis-à-vis the binding energy of −3 kcal mol–1 for phyllanthin. While D8 was also found to make interactions with Ser139 and His57 of the NS3 protease catalytic triad, it was observed that the presence of two carboxylic acid moieties further help D8 in making charged interactions with NH3+ of Lys136 from NS3 protease. These charged interactions contribute favorably to the binding energy. Phyllanthin, which lacks charged substituents, was unable to make these interactions and instead was found to be involved in the hydrogen bond with Lys136 (Figure 1). Furthermore, despite the presence of the aromatic ring in both phyllanthin and D8, it is in the D8/NS3 docked complex that the aromatic ring is properly stacked with His57 and contributes toward the aromatic stacking interactions (Figure 5). In the phyllanthin–NS3 docked complex, the aromatic ring is unable to stack properly with His57. Also, Ala156, which is one of the substrate-binding residues, participates in the hydrophobic interaction with D8. It is likely that D8 being smaller in size orients better with the catalytic residues as compared to phyllanthin, the fitting of which might be restricted because of the presence of other substituents. All these interactions contribute favorably to the interaction energy between D8 and NS3-protease, which can be considered as an indicator for stronger binding as compared to native phyllanthin. Taken together, although D8 is nearly half of the phyllanthin molecular structure, it can make better interactions with NS3 protease catalytic residues and thus better efficacy in the inhibition activities.
Figure 5.
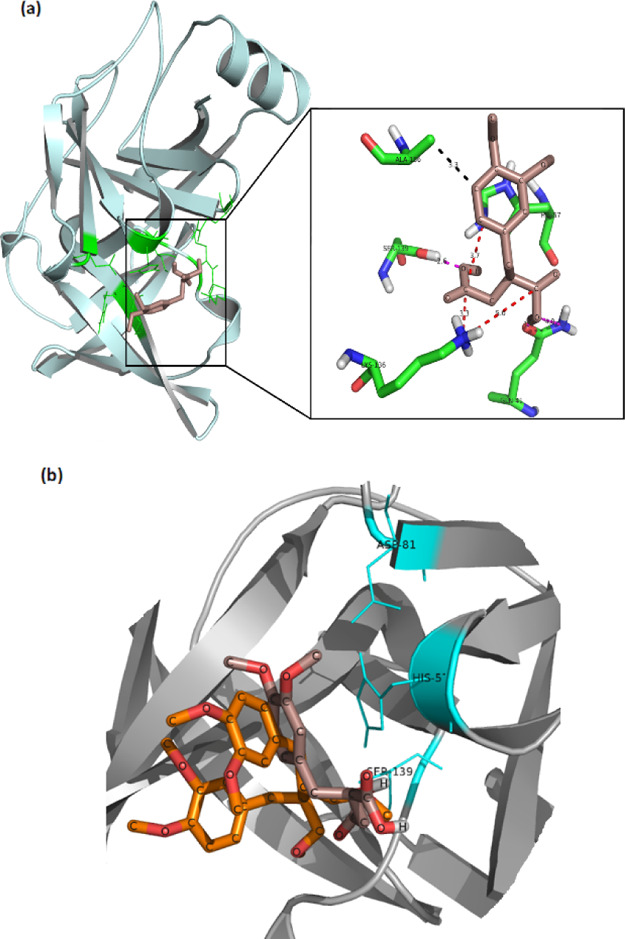
Molecular docking of D8 with HCV NS3 protease. (a) Left side shows the minimum energy binding of the D8 molecule to HCV NS3-protease (pale cyan) as obtained after docking. The D8 molecule is depicted in salmon color, and the residues of the binding pocket which are involved in interactions with it are shown in green. The right side shows the zoomed-in view of the important interactions. The NS3 protease residues are labeled and shown as sticks (green), and the interactions are shown as dotted lines. The pink color depicts hydrogen bond interactions, red depicts charged interactions, and black color depicts hydrophobic interactions. (b) Superposition of phyllanthin (orange) and D8 (salmon) in the binding pocket of NS3 protease (in gray). The catalytic residues are shown in cyan. The orientation of NS3 protease is different in A and B for representation purpose.
D8 is Specific to HCV NS3 Protease
The specificity of the D8 inhibition effect against HCV NS3 protease was conducted using another serine protease (cellular), namely, trypsin in a fluorometric assay. This assay involves cleavage of a casein substrate labeled with fluorescein isothiocyanate (FITC) by trypsin.31 The intensity of the fluorescence signal evolving from the cleaved FITC substrate in the presence of D8 is a quantitative measure of its inhibitory effect. Increasing concentrations of D8 were used in this assay. However, D8 did not show an inhibition, even at 250 μM. In contrast, an already known trypsin inhibitor from Glycine max soyabean,14 used here as a positive control, drastically reduced the trypsin activity. The assay suggested that D8 is a specific inhibitor of HCV NS3 serine protease (Figure 4b).
D8 is Considerably Nontoxic to Huh7.5 Cells Compared to Native Phyllanthin
As HCV replication in cell culture is limited to Huh7/Huh7.5 cells, toxic effect, if any, of D8 against these cells was assessed. The treatment of D8 up to 5 mM to Huh7.5 cells did not induce any toxicity (CC50 > 5 mM) in MTT assay, whereas more than 50% cell death was observed upon the treatment of phyllanthin at 5 mM. CC50 > 5 mM suggests D8 is significantly tolerable, as compared to native phyllanthin (Figure 6 and Table 1).
Figure 6.

MTT assay to compare the effect of D8 and phyllanthin (Phy) on cell viability. “C” represents control, namely, untreated Huh7.5 cells. Data represent mean ± SD from three independent experiments. Statistical significance is shown as *p < 0.05, **p < 0.01, ***p < 0.001.
Table 1. Effect of Phyllanthin and D8 on HCV RNA Replication Ex Vivo in Rep2a and Cell Viability Ex Vivoa.
| HCV RNA replication in Rep2a cell line | cell viability (MTT assay) | |
|---|---|---|
| compound | EC50 (μM) | CC50 (mM) |
| phyllanthin | 250 | 5.0 |
| D8 | 75 | >5.0 |
Note: EC50 = 50% of effective concentration; CC50: 50% of cellular cytotoxicity.
D8 Significantly Inhibits HCV Replication in Cell Culture
Following the successful assessment of toxicity of D8, the effect of D8 on HCV replication in the cell culture was performed. For this, Huh7 cells harboring HCV monocistronic replicon (Figure 7a) were incubated with increasing concentrations of D8. At 48 h post-treatment, HCV negative strand synthesis was quantified using RT-qPCR. Cells treated with D8 showed significant decrease in HCV RNA levels in a concentration-dependent manner. Also, this decrease was relatively more for D8, as compared to cells treated with phyllanthin (Figure 7b and Table 1), demonstrating better potency of D8 in inhibiting HCV replication.
Figure 7.

(a) Schematic representation of HCV monocistronic replicon; (b) HCV negative strand synthesis measured by RT-qPCR. “C” represents no treatment. Telaprevir was used as a standard positive control.
D8 Showed Higher Tolerance in BALB/C Mice Compared to Native Phyllanthin
In order to assess the safety of D8in vivo, acute and subacute toxicity analyses were performed. In acute toxicity study, BALB/c mice were orally fed with increasing doses (0–5000 mg/kg b. wt) of D8 and phyllanthin. The maximum acute tolerance dose of D8 was observed to be 4000 mg/kg/b. wt. At 5000 mg/kg b. wt, 40% death of mice was observed. However 100% death of mice was observed at 5250 mg/kg b. wt (50% lethal dose (LD50) of D8 lies between 5000 and 5250 mg/kg b. wt). The tolerance dose of D8 was ∼2.5 times higher than that of phyllanthin (1500 mg/kg/wt) and 100% death of mice observed at 2000 mg/kg b. wt. Suggesting that, the LD50 for phyllanthin was found to be between 1500 and 2000 mg/kg b. wt (Figure 8a–d). The body weights of mice from control groups, as well as acute tolerance doses of D8 via other modes of administrations (intravenous and intraperitoneal) in mice were also determined (data shown in the Supporting Information).
Figure 8.

Oral toxicity effect of phyllanthin and D8 in BALB/c mice. In acute toxicity studies, panels (a,b) show the maximum tolerance, whereas (c,d) show the induction of mortality at lethal doses of compounds. Sub-acute toxicity studies, figures (e,f) show no observed adverse effect of compounds after repeated administration (∼1/4th of the maximum tolerance dose) of compounds for 28 consecutive days. Note that sudden dropping lines to the x axis in figures c,d indicate death of the mice.
On the other hand, subacute toxicity study involved repeated administrations of D8 (1000 mg/kg b. wt/day/mouse), water (200 μL/day/mouse), PEG-400 (200 μL/day/mouse), and phyllanthin (500 mg/kg b. wt/day/mouse) for 28 consecutive days. All the treated mice groups did not exhibit any undesirable changes in general behavior and body weights compared to control groups (Figure 8e,f).
Treatment with D8 Does Not Alter the Histoarchitecture of the Mice Liver
As external signs of toxicity of D8 were not observed, histopathological analyses were performed next, so as to ensure no internal damage at the tissue level. Histology of the mice liver treated with D8 at maximum acute tolerance dose (4000 mg/kg b. wt p.o) showed normal histoarchitecture compared to the vehicle control (water). Similar observations were obtained for phyllanthin (1500 mg/kg b. wt p.o) and its vehicle control (PEG-400) groups [Figure 9a(i–iv)].
Figure 9.
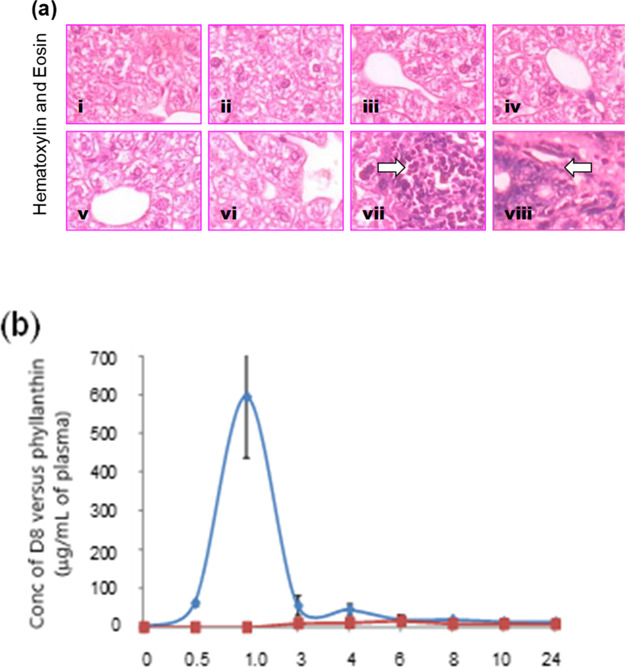
(a) Histopathological examination of the mice liver. A representative picture from the different groups (1–4) in the acute toxicity studies, (i) water control; (ii) D8 4000 mg/kg b. wt; (iii) PEG-400 (300 μL/mouse); (iv) phyllanthin (1500 mg/kg b. wt.). Similarly, the groups (5–8) in the subacute toxicity study, (v) water control; (vi) D8 (1000 mg/kg b. wt/day); (vii) PEG-400 (200 μL/day/mouse); (viii) phyllanthin (500 mg/kg b. wt/day). Arrow indicates multifocal aggregation of hematopoietic cells and liver necrosis. (b) Bioavailability of D8 (500 mg/kg b. wt p.o) and phyllanthin (1500 mg/kg b. wt p.o) in BALB/c mice. Values used to plot AUC are mean ± SD from the three independent experiments.
The liver sections of mice orally administrated with subacute dose of D8 (1000 mg/kg b. wt/day) for 28 days also showed normal histology. However, the histoarchitecture of mice livers obtained 28 days after repeated feeding of phyllanthin (500 mg/kg b. wt/day) showed tissue necrosis and multifocal aggregation of hematopoietic cells along with necrotic foci at some parts. It appears that the induction of liver necrosis could be due to repeated dosage of PEG-400, as similar features were observed in the mice group treated with PEG-400 alone [Figure 9a(v–vii)]. Taken together, these observations suggest that the aqueous solubility feature confers an advantage to D8 for its completely nontoxic behavior over its water-insoluble parental counterpart, namely, phyllanthin.
Orally Administered D8 is Systemically Bioavailable
It is important for a compound to be first absorbed into the blood in the process to reach a target tissue and exert its effects. As orally administered D8 did not show any toxic effect, it is pertinent to verify whether D8 is systemically available in blood and rule out the possibility of its unabsorbed elimination. Accordingly, mice were orally fed with 500 mg/kg b. wt of D8, and HPLC analyses were performed on plasma samples collected at different time points. Phyllanthin was also included in these assays for comparison. Results showed good systemic absorption of D8 with maximum bioavailability at 1 h (Tmax: 1 h). The half-life time (t1/2) of the compound and maximum concentration (Cmax) were observed as 2 h and ∼595 μg mL–1, respectively. In contrast, we could not observe any bioavailability (up to 24 h) in the mice group fed with phyllanthin at the same dose of 500 mg/kg b. wt p.o. A very low concentration of absorbed phyllanthin could only be observed when administered at a higher dose (1500 mg/kg b. wt) (Figure 9b). These observations proved better bioavailability of D8 over phyllanthin which might be due to its smaller size and increased aqueous solubility.
Discussion
The present study shows a structure-guided approach to identify potent derivatives inhibiting the activity of a serine-protease, namely, NS3/4A protease, a nonstructural protease having a critical role in the HCV viral replication. NS3/NS4A is a noncovalent, heterodimer complex formed by two HCV-encoded proteins, the N-terminal serine protease domain of NS3 (catalytic subunit) and the NS4A cofactor (activation subunit). This protein complex performs multiple important functions in HCV-infected cells, for example, NS3/4A is responsible for the proteolytic cleavages at the junctions of nonstructural proteins downstream of NS3 (NS3/NS4A, NS4A/NS4B, NS4B- NS5B-, and NS5A-NS5B) of the HCV polyprotein.32,33 Apart from this, it also directs proteolysis of its proline-rich adaptor protein, toll-IL-1 receptor domain-containing adaptor-inducing interferon-β (TRIF) and MAVS in the mitochondria, thus helps virus evade innate immunity by dislodging a pivotal antiviral protein from the mitochondria.34,35 Interestingly, the TRIF cleavage site has been described to have remarkable homology with the viral NS4B/5A substrate.34 Emphasis on this protease also arises as mutations in NS3 protease leads to multidrug resistance of the virus, abolishing its susceptibility to drugs, including direct acting antivirals.36,37 Our earlier study demonstrated that phyllanthin, a major constituent of the P. amarus extract, possesses a significant antiviral activity. NS3 protease was found to be the target enzyme to the phyllanthin inhibition activity. The aqueous solubility of phyllanthin was a major constraint, warranting a structure-based approach to identify most potent structures from a pool of structural congeners. A computational docking study of phyllanthin with the NS3 protease enzyme catalytic region revealed key ligand–receptor interactions, primarily that of aromatic stacking and hydrogen bonding interactions. Thus, structural modifications of the phyllanthin scaffold required retaining the moieties that promote these interactions.
Phyllanthin is a 1,4-diphenyl butane derivative possessing methyloxymethylene substituents at C-2 and C-3 carbons. Synthesis of congeners of this scaffold could be planned when the scaffold is also considered as benzylic substitutions at C-2 and C-3 carbons on a succinoyl synthon. Stobbe condensation of succinic acid esters with aromatic aldehydes as electrophilic acceptors is a suitable approach to synthesize congeners of phyllanthin. In effect, Stobbe condensation is used beneficially to synthesize the congeners or derivatives that possess aromatic and side chain functionalities to promote interactions with the receptor catalytic sites. The chemical synthesis also permits the side-chain functionality modifications at the alkyl segment. Furthermore, the synthesis afforded structures constituted with only one benzylic moiety in the succinoyl segment at C-2, without the presence of another benzylic moiety at C-3.
Having the congeners with varied functionality modifications, the efficacies of congeners to inhibit the NS3 protease activity was evaluated initially. Using the fluorometric assay, the emergence of fluorescence from the cleavage of the green fluorescent protein, namely, EGFP-NS5A/B-CBD, was utilized to identify the inhibitory potencies of the NS3 protease activity by synthetic congeners. Of the compounds studied in this assay, a derivative annotated as D8 was found to be the efficient inhibitor, surpassing the inhibition potency of native phyllanthin. Computational docking assessment of the interaction of D8 with NS3 protease indeed revealed a stronger binding energy in favor of D8, in comparison to native phyllanthin. This increased energy results from additional interactions between carboxylic acid moiety of D8 and proximal Lys residues at the catalytic triad of NS3 protease, a feature which is absent in the binding of phyllanthin with the same receptor. A series of subsequent experiments were undertaken with the D8 derivative, in efforts to establish the inhibition profile of this derivative.
A comparison to another protease, namely, trypsin, shows that inhibition of trypsin activity by D8 does not occur even at 250 μM, whereas the known trypsin inhibitor from G. max soyabean showed inhibition concentration IC50 at 1 μM. Thus, D8 retains a specificity as an inhibitor of NS3 protease, with IC50 < 10 μM, whereas native phyllanthin required an IC50 concentration of ∼50 μM.
The efficacy of D8 as the NS3 protease inhibitor warranted the assessment of the toxicity. When conducted in a thiazolium-based MTT assay on the Huh7.5 cell line, no perceptible cellular toxicity was observed with D8, even at 5 mM concentration, and phyllanthin exhibited 50% cell death at this concentration. When the same cell line harbored with HCV monocistronic replicon was treated with D8, a significant reduction in the HCV RNA replication is observed. The reduction in RNA replication is concentration-dependent and at 250 μM, the reduction in HCV negative strand synthesis was matching that of the inhibition by Telapravir. These in vitro assays established the inhibitory efficacy of D8, in comparison to native phyllanthin. Furthermore, augmented by higher aqueous solubility, assessments were taken up, so as to adjudge the validity of D8 as the potent inhibitor under the in vivo conditions. The in vivo toxicity experiments on Balb/c mice revealed a maximum acute tolerance of D8 on the mice at a concentration of 4000 mg/kg body weight of the mice. Repeated dosing D8 at 1000 mg/kg body mass also revealed significantly enhanced sub-acute toxicity of D8, in comparison to phyllanthin. These studies reveal that D8 possesses a significantly enhanced inhibition potency, outweighing the same exhibited by phyllanthin. Histopathological profiles of mice treated with D8, phyllanthin, and carrier control PEG400 also enabled to establish the complete preservation of histoarchitecture of liver sections on mice treated with D8 at the maximum acute tolerance concentration of 4000 mg/kg body mass, an effect which was not observed with phyllanthin and the carrier control, each of which exhibited loss in the histoarchitecture at much lesser concentrations.
Bioavailability of the inhibitors in mice blood circulation, when assessed through the HPLC technique on plasma samples withdrawn at varying time points, demonstrated a good bioavailability of D8 after 1 h, when orally fed with 500 mg/kg of body mass. After 2 h, plasma samples contained 595 μg mL–1. These results show a good systemic circulation of D8, and such a bioavailability was absent with native phyllanthin at such a concentration. The bioavailability of D8 may thus not be a limiting factor in advancing this new chemical entity.
The number of studies conducted on D8 as described above establishes that D8 is a newly discovered inhibitor of NS3 protease, which is one of the nonstructural proteins that regulate HCV replication and disease progression. A structure-based design effort, initiated from the phyllanthin core structure, has enabled us to discover this new inhibitor D8, presenting a formidable scope to develop further advanced studies toward a potent practical application of D8 to contain HCV infection.
Conclusions
The present study demonstrates a structure-based approach in order to identify structural congeners of phyllanthin as novel, potent inhibitor NS3 protease, a nonstructural protein involved with the HCV viral replication and disease progression. Facile synthesis by Stobbe condensation allowed construction of the congeners, initiated by the reaction of succinic ester with vanillin. The reaction led to mono- and disubstitution at the C-2 and C-3 carbons of succinic ester. The monosubstitution product of Stobbe condensation was taken through further modifications by either ester hydrolysis or ester reduction. Among three congeners, namely, a diester (D7), dicarboxylic acid (D8), and diol (D9), D8 exhibits a profound inhibition of the NS3/4A protease activity, at 75 μM concentration, for complete inhibition. A molecular docking study revealed that charged interactions were accrued further between carboxylic acid moieties of D8 and NH3+ side chain functionality of Lys136 residue located at the catalytic triad site of NS3 protease. Further studies leading to the assessment of the noncytotoxicity profile, inhibition of HCV replication, in vivo tolerance, and unalteration of the histoarchitecture of treated mice demonstrate that D8 surpasses phyllanthin in its inhibitory potency. The bioavailability of D8 further adds to the “drug-like” activity profile. The present study establishes that D8 is a new chemical entity, providing a greater scope to counter HCV viral infection.
Materials and Methods
General Methods
All reactions were carried out under a nitrogen atmosphere. Organic extracts were dried over anhydrous sodium sulphate. Solvents were removed in a rotary evaporator under reduced pressure. Silica gel (100–200 mesh size) was used for the column chromatography. Reactions were monitored using silica gel 60 F254-coated TLC plates. 1H NMR spectra were measured on an NMR instrument, operating at 400 MHz for 1H nucleus and 100 MHz for 13C nucleus, and the chemical shifts were calibrated with respect to tetramethylsilane or residual solvent peaks, as the internal standard. High-resolution mass spectra were recorded on an electrospray-QTOF instrument.
Plasmid Constructs and Reagents
Plasmids pYB-43 (single-chain HCV NS3/4A) and pYB-44 (EGFP-NS5A/B-CBD) encoding protease domain of NS3 and fusion substrate protein,36 respectively, were received as a kind gift from Dr. Itai Benhar, Tel Aviv University, Israel.
Cell Lines
The Huh7.5 cell line was received from Dr. C. M. Rice, Rockefeller University, USA. Huh7 cells harboring HCV monocistronic replicon genotype 2a (Rep2a)37 was received as a kind gift from Dr. Ralf Bartenschlager, Heidelberg University. All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/mL of penicillin (HiMedia), and 100 μg/mL of streptomycin sulphate (HiMedia). For Rep2a cell line maintenance, 25 μg per mL of Hygromycin B (Himedia) was added to the culture medium. All cell lines were maintained at 37 °C in 5% CO2.
Dimethyl 2-(3,4-dimethoxybenzyl)succinate (1) (D7)
NaH (0.9 g, 36 mmol) was added to a stirred solution of 3,4-dimethoxy benzaldehyde (3.0 g, 9.06 mmol) and diethyl succinate (3.18 g, 18.0 mmol) in PhMe (60 mL) at 0 °C. The reaction mixture was refluxed for 12 h, cooled, quenched with aq. HCl (5% v/v), extracted with ethyl acetate (4 × 20 mL), washed with water (3 × 10 mL), the organic layer was dried, solvents were evaporated in vacuo, and the residue was dried under high vacuum to afford the crude product (3.42 g, 72%). The residue (3.2 g, 12.0 mmol) in MeOH (60 mL) was treated with SOCl2 (2.60 mL, 30.0 mmol), the mixture was stirred at 0 °C for 6 h and at room temperature for 6 h. Solvents were evaporated in vacuo, the resulting product was dried under high vacuum and purified by column chromatography (SiO2) (pet. ether/EtOAc linear gradient) to afford a crude product as an amorphous powder (2.97 g, 84%). A suspension of this intermediate (2.7 g, 4.5 mmol) in MeOH (50 mL) and Pd–C (10%) (2.5 g) was stirred under a H2(g) atmosphere for 12 h, filtered through celite, washed with MeOH (30 mL), solvents evaporated in vacuo, and the resulting crude product purified by column chromatography (SiO2) (pet. ether/EtOAc linear gradient) to afford 1 as a gummy solid (1.56 g, 80%). 1H NMR (CDCl3): δ 6.70 (s, 1H, Ar), 6.60 (app. s, 2H, Ar), 3.75 (s, 3H, OMe), 3.73 (s, 3H, OMe), 3.56 (s, 3H, CO2Me), 3.53 (s, 3H, CO2Me), 3.01 (m, 1H, CH2), 2.90 (m, 1H, CH), 2.60 (m, 2H, CH2), 2.31 (app. dd, J = 12, 4 Hz, 1H, CH2); 13C NMR (CDCl3): δ 174.3 (C=O), 171.9 (C=O), 148.5 (Ar), 147.4 (Ar), 130.2 (Ar), 120.7 (Ar), 111.6 (Ar), 110.8 (Ar), 55.3 (OMe), 51.4 (CO2Me), 51.3 (CO2Me), 42.7 (CH), 36.9 (CH2), 34.4 (CH2). ESI-MS m/z [M + Na]+; calcd for C15H20O6Na, 319.1158; found, 319.1156.
2-(3,4-Dimethoxybenzyl)succinic Acid (2) (D8)
A mixture of D7 (1.4 g, 4.5 mmol) and LiOH.H2O (1.90 g, 45 mmol) in THF/water (5:1) (60 mL) was refluxed for 12 h, neutralized with Amberlite resin (H+ form), filtered, washed with Et2O (25 mL), the filtrate was dried, and solvents were evaporated in vacuo to afford 2 as an oil (1.18 g, 98%). 1H (CDCl3): δ 10.05 (br. S, 2H, OH), 6.78 (d, J = 8.0 Hz, 1H, Ar), 6.69 (d, J 8.0 Hz, 2H, Ar), 3.82 (s, 6H, OMe) 3.00 (m, 2H, CH2), 2.73 (m, 2H, CH2), 2.45 (m, 1H, CH); 13C (CDCl3): δ 180.3 (CO2H), 178.0 (CO2H), 149.0 (Ar), 147.9 (Ar), 130.1 (Ar), 121.2 (Ar), 112.0 (Ar), 111.3 (Ar), 55.88 (OMe), 55.84 (OMe), 42.9 (CH), 36.9 (CH2), 34.4 (CH2). ESI-MS m/z [M + Na]+; calcd for C13H16O6Na, 291.0845; found, 291.0847.
2-(3,4-Dimethoxybenzyl)butane-1,4-diol (3) (D9)
Derivative D7 (0.77 g, 1.29 mmol) in THF (20 mL) was added with LAH (0.14 g, 3.87 mmol), the reaction mixture was stirred for 12 h at room temperature, quenched with ice, solvents were evaporated in vacuo, dissolved in EtOAc (40 mL), washed with water (2 × 10 mL), dried, and evaporated in vacuo and purified by column chromatography (SiO2) (pet. ether/EtOAc linear gradient) to afford 3 as an amorphous solid (0.43 g, 80%). 1H NMR (CDCl3, 400 MHz): δ 6.70 (s, 1H, Ar), 6.62 (app. s, 2H, Ar), 3.90 (br, 2H, OH), 3.67 (s, 2H, OMe), 3.60–3.45 (m, 4H, OMe), 3.35–3.27 (m, 2H, CH2OH), 2.49 (m, 2H, CH2OH), 1.65 (m, 1H, CH), 1.50 (m, 4H, CH2); 13C NMR (CDCl3, 100 MHz): δ 148.6 (Ar), 147.1 (Ar), 132.8 (Ar), 121.0 (Ar), 112.2 (Ar), 111.0 (Ar), 65.2 (CH2OH), 60.7 (CH2OH), 55.7 (OMe), 41.1 (CH), 37.6 (CH2), 35.1 (CH2). ESI-MS m/z [M + Na]+; calcd for C13H20O4Na, 263.1259; found, 263.1277.
HCV NS3 Protease Assay
The effect of phyllanthin (Phy) and synthetic derivatives (D7–D9) at 50 μM on HCV NS3/4A protease activity was assessed using a high throughput fluorimetric assay as described earlier.38 Briefly, purified enzyme scNS3/4A (0.1 μM) was pre-incubated with a test compound (Phy, D7-D9), followed by the addition of purified fusion substrate protein (EGFP-NS5A/B-CBD, 0.5 μM). The reaction was incubated for another 1 h at 37 °C. The cleavage reaction was terminated by adding the cellulose slurry (40 μM) into the reaction mix. Then, reaction tube was centrifuged, and the supernatant was collected separately. Finally, the total amount of the fluorescence signal evolved from the cleaved EGFP substrate was quantified using a fluorimeter (Modulus TM Microplate Multimode Reader) using excitation filter 484 nm and emission filter 538 nm. Telaprevir (MedChem) was used as a positive control.
Cellular Serine Protease Assay
In order to demonstrate the specificity of D8, only to HCV NS3 serine protease but not to the cellular serine protease, a simple high throughput in vitro cellular serine protease (trypsin) assay was performed using a protease fluorescent detection kit (PF0100, Sigma-Aldrich). In this assay, cellular trypsin was pre-incubated with increasing concentrations of the test sample followed by the addition of the casein substrate protein (conjugated with fluoresce in isothiocyanate, FITC) and further incubation for 1 h at 37 °C. The reaction was terminated by adding the trichloroacetic acid (6.1 N), and the supernatants were collected by centrifugation. The amount of fluorescence signals evolved from the cleaved substrate was quantitated by fluorometry using an excitation filter of 485 nm and an emission filter of 538 nm. The trypsin inhibitor from G. max (Sigma-Aldrich) was used as the positive control.
Structural Analysis and Docking Protocol
Ligands similar to phyllanthin were searched by SMILES string (SMILES–the simplified molecular-input line-entry system: It describes the structure of chemical molecules) and molecular formula. Ligand bound protein structures were found using Ligand Expo and chemical component search in PDB.38 Instead of blind docking, the knowledge derived from the crystal structure of the protein–ligand complex (PDB id: 2BGM) was used to selectively dock phyllanthin around the catalytic region of NS3 (PDB id: 1DY8) using Autodock 4.2.39−42 Following docking parameters were used:
-
a
Polar hydrogens were added wherever missing.
-
b
Gasteiger charges were assigned.
-
c
Peptide bonds, amide bonds, guanidium bonds, and cyclic groups were treated as nonrotatable.
-
d
The grid was defined on the catalytic pocket and the protein was considered to be rigid.
-
e
Runs (100 GA) with medium evaluation because the number of rotatable bonds <10.
This directed docking was carried out upon observing the similarity between the functional groups of Matairesinol (PDB id: 2BGM) bound to similar catalytic triad residues of a dehydrogenase. The 3D structure of phyllanthin was obtained from PubChem.41 The best ranked model with lowest binding energy was analyzed using PLIP server42 and visualized using PyMOL (The PyMOL Molecular Graphics System Version 1.5.0.4).43 Similar docking protocol was used to dock D8 molecule with NS3 protease. Three-dimensional coordinates of D8 was saved using ChemDraw.
Cell Viability Assay
To investigate the cellular cytotoxicity, Huh7.5 cells were seeded in 96 well plates at a density of 2 × 104 cells in 100 μL of DMEM media containing 10% serum and incubated overnight at 37 °C. After 12 h, cells were treated with different concentrations of phyllanthin, and its synthetic derivative, D8, was treated. Cytotoxic effects of phyllanthin verses D8 on Huh7 cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma-Aldrich), as described earlier.44
HCV Replication Assay in Rep2a Cell Line
Huh-7 cells harboring the monocistronic replicon (7 × 104) were seeded in a 12 well plates containing DMEM (Sigma-Aldrich) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco) at 37 °C.37 In order to express the HCV subgenomic replicon system, 25 μg/mL of hygromycin B (Himedia) was used. After 12–16 h, the medium was changed, and the cells were treated with increasing concentrations of inhibitors (D8/phyllanthin) and further incubated to 48 h at 37 °C. Then, the total RNA was isolated using the TRIzol reagent, and HCV negative strand levels were quantified using analysis by reverse transcription quantitative PCR, as described earlier using HCV.14
Ethical Statement
Adult healthy BALB/c mice were obtained from the Institutional Centre Animal Facility. All study design was approved by the “Institutional Animal Ethics Committee”. Animals were maintained in accordance with the guidelines of the Indian Law on animal care and use.
Preparation of the Drugs for In Vivo Studies
The required concentrations of phyllanthin and D8 were dissolved in polyethylene glycol-400 (PEG-400) and water respectively. These solvents alone were used as vehicle controls.
Determination of Systemic Toxicity in Mice
For the safety assessment, the acute and sub-acute toxicity of phyllanthin and D8 was determined according to OECD guidelines (423 and 407).45,46 Hematoxylin and eosin stains were used to examine the histopathology of liver sections from treated and control groups of BALB/c mice. Body weight, food consumption, food and water intake, abnormal behavior, and adverse signs of toxicity were recorded throughout the study.
Histopathological Analysis
The formalin fixed mice liver tissues from both the control and treated groups were routinely processed in different alcoholic gradients and embedded in paraffin wax. Microtome sections (5 μm thickness) were cut, and permanent slides were prepared using haematoxylin and eosin stains (Sigma-Aldrich). The slides were examined under a light microscope and analyzed for any pathological signs.
Bioavailability of Phyllanthin and D8 in Mice
A total of 50 BALB/c mice (divided into 2 groups of 25 mice each) were used for this assay. All the animals selected for the study were kept under fasting overnight and were orally administered with phyllanthin and D8 separately to each group at 500 mg/kg b. wt/mouse. Blood was drawn by retro orbital puncture from three mice from each group at regular intervals between 0 and 24 h. Plasma was immediately separated by centrifuging blood for 2000 rpm for 20 min at 4 °C. Plasma thus obtained (500 μL) was mixed with the equal volume of 2% trifluoroacetic acid and subjected immediately to RP-HPLC using 0.02% acetonitrile and water as a mobile phase. The area under the curve (AUC) for plasma concentrations of phyllanthin and D8 was plotted, and pharmacokinetics parameters including maximum plasma concentration (Cmax), time to reach concentration maxima (Tmax), and half-life time (t1/2) were calculated.
Statistical Analyses
The results of all the experiments were expressed as the mean ± standard deviations (SD). The two-tailed unpaired “t” test was performed using GraphPad Prism 6.0 software to find out significance. The significant differences between the groups are represented by an asterisk (*p < 0.05, **p < 0.01, ***p < 0.001) compared with the control group (C).
Acknowledgments
We are thankful to Dr. Ralf Bartenschlager, Heidelberg University, Dr. Itai Benhar, Tel Aviv University, Israel, and Dr. Charles M. Rice Rockefeller University, USA, for sharing useful plasmid constructs and cell lines. We are also grateful to Central animal facility, Microtome, NMR, FACS, Analytical spectroscopy, and other central facilities of our Institute (funded by DST-FIST and IISc-DBT-partnership programme). We also thank Dr. Anuj Kumar for critical reading and editing of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00786.
1H and 13C NMR spectra of D7–D9 and in vivo toxicity of phyllanthin and D8 administered via different modes in BALB/c mice (PDF)
Author Present Address
⊥ Dept of Studies in Botany, V.S.K. University, Ballari 583105, India.
Author Contributions
The manuscript was written through contributions of all authors.
This work was supported by grant from the Department of Biotechnology (DBT) (BT/01/COE/06/02/10-IISc), Government of India, to S.D., N.S., and N.J. N.S., S.D., and N.J. also acknowledge JC Bose fellowship from DST for research support.
The authors declare no competing financial interest.
Notes
PDB ID codes: Matairesinol (PDB id: 2BGM); NS3 protease (PDB id: 1DY8); Crystal structure of protein–ligand complex (PDB id: 2BGM). Authors will release the atomic coordinates and experimental data upon article publication.
Supplementary Material
References
- Lee J.-Y.; Cortese M.; Haselmann U.; Tabata K.; Romero-Brey I.; Funaya C.; Schieber N. L.; Qiang Y.; Bartenschlager M.; Kallis S.; Ritter C.; Rohr K.; Schwab Y.; Ruggieri A.; Bartenschlager R. Spatiotemporal coupling of the hepatitis C virus replication cycle by creating a lipid droplet-proximal membranous replication compartment. Cell Rep. 2019, 27, 3602–3617. 10.1016/j.celrep.2019.05.063. [DOI] [PubMed] [Google Scholar]
- Simmonds P. The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 1–15. 10.1007/978-3-642-27340-7_1. [DOI] [PubMed] [Google Scholar]
- Penin F. o.; Dubuisson J.; Rey F. A.; Moradpour D.; Pawlotsky J.-M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Suzuki R.. Role of nonstructural proteins in HCV replication. Hepatitis C virus, Online: 29 October, 2016; Chapter 1, pp 129–148. [Google Scholar]
- McGivern D. R.; Masaki T.; Lovell W.; Hamlett C.; Saalau-Bethell S.; Graham B. Protease inhibitors block multiple functions of the NS3/4A protease-helicase during hepatitis C virus life cycle. J. Virol. 2015, 89, 5362–5370. 10.1128/jvi.03188-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatel-Chaix L.; Baril M.; Lamarre D. Hepatitis C virus NS3/4A protease inhibitors: A light at the end of the tunnel. Viruses 2010, 2, 1752–1765. 10.3390/v2081752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggakusuma B.; Brown R. J. P.; Banda D. H.; Todt D.; Vieyres G.; Steinmann E.; Pietschmann T. Hepacivirus NS3/4A proteases interfere with MAVS signaling in both their cognate animal hosts and humans: Implications for zoonotic transmission. J. Virol. 2016, 90, 10670–10681. 10.1128/jvi.01634-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stempniak M.; Hostomska Z.; Nodes B. R.; Hostomsky Z. The NS3 proteinase domain of hepatitis c virus is a zinc-containing enzyme. J. Virol. 1997, 71, 2881–2886. 10.1128/jvi.71.4.2881-2886.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco R.; Pessi A.; Steinkühler C. The hepatitis C virus NS3 proteinase: Structure and function of a zinc-containing serine proteinase. Antivir. Ther. 1998, 3, 99–109. [PubMed] [Google Scholar]
- Yi M.; Tong X.; Skelton A.; Chase R.; Chen T.; Prongay A.; Bogen S. L.; Saksena A. K.; Njoroge F. G.; Veselenak R. L.; Pyles R. B.; Bourne N.; Malcom B. A.; Lemon S. M. Mutations conferring resistance to SCH6, a novel hepatitis C virus NS3/4A protease inhibitor. Reduced RNA replication fitness and partial rescue by second-site mutations. J. Biol. Chem. 2006, 281, 8205. 10.1074/jbc.m510246200. [DOI] [PubMed] [Google Scholar]
- Howe A. Y. M.; Cheng H.; Johann S.; Mullen S.; Chunduru S. K.; Young D. C.; Bard J.; Chopra R.; Krishnamurthy G.; Mansour T.; O’Connell J. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 2008, 52, 3327–3338. 10.1128/aac.00238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Tuttle M.; Gao M.; Lemm J. A. Potency and resistance analysis of Hepatitis C virus NS5B polymerase Inhibitor BMS-791325 on all major genotypes. Antimicrob. Agents Chemother. 2014, 58, 7416–7423. 10.1128/aac.03851-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon J.; Beach N. M.; Moreno E.; Gallego I.; Pineiro D.; Martinez-Salas E.; Gregori J.; Quer J.; Esteban J. I.; Rice C. M.; Domingo E.; Perales C. Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J. Virol. 2014, 88, 12098–12111. 10.1128/jvi.01860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B. U.; Mullick R.; Kumar A.; Sharma G.; Bag P.; Roy C. L.; Sudha G.; Tandon H.; Dave P.; Shukla A.; Srinivasan P.; Nandhitha M.; Srinivasan N.; Das S. A natural small molecule inhibitor corilagin blocks HCV replication and modulates oxidative stress to reduce liver damage. Antiviral Res. 2018, 150, 47–59. 10.1016/j.antiviral.2017.12.004. [DOI] [PubMed] [Google Scholar]
- Jardim A. C. G.; Shimizu J. F.; Rahal P.; Harris M. Plant-derived antivirals against hepatitis C virus infection. Virol. J. 2018, 15, 34. 10.1186/s12985-018-0945-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B. U.; Mullick R.; Kumar A.; Sudha G.; Srinivasan N.; Das S. Small molecule inhibitors of HCV replication from pomegranate. Sci. Rep. 2015, 24, 5411. 10.1038/srep05411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.; Kim K. H.; Kim H. Y.; Cho H. K.; Sakamoto N.; Cheong J. Curcumin inhibits hepatitis C virus replication via suppressing the Akt-SREBP-1 pathway. FEBS Lett. 2010, 584, 707–712. 10.1016/j.febslet.2009.12.019. [DOI] [PubMed] [Google Scholar]
- Wagoner J.; Negash A.; Kane O. J.; Martinez L. E.; Nahmias Y.; Bourne N.; Owen D. M.; Grove J.; Brimacombe C.; McKeating J. A.; Pécheur E.-I.; Graf T. N.; Oberlies N. H.; Lohmann V.; Cao F.; Tavis J. E.; Polyak S. J. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 2010, 51, 1912–1921. 10.1002/hep.23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar Y. S.; Ray U.; Nandhitha M.; Perween A.; Raja Naika H.; Khanna N.; Das S. Inhibition of hepatitis C virus replication by herbal extract: Phyllanthus amarus as potent natural source. Virus Res. 2011, 158, 89–97. 10.1016/j.virusres.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Lee C.-Y.; Peng W.-H.; Cheng H.-Y.; Chen F.-N.; Lai M.-T.; Chiu T.-H. Hepatoprotective effect of Phyllanthus in Taiwan on acute liver damage induced by carbon tetrachloride. Am. J. Chin. Med. 2006, 34, 471–482. 10.1142/s0192415x06004004. [DOI] [PubMed] [Google Scholar]
- Ilangkovan M.; Jantan I.; Nasir S.; Bukhari A. Phyllanthin from Phyllanthus amarus inhibits cellular and humoral immune responses in Balb/C mice. Phytomedicine 2016, 23, 1441–1450. 10.1016/j.phymed.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Jagtap S.; Khare P.; Mangal P.; Kondepudi K. K.; Bishnoi M.; Bhutani K. K. Protective effects of phyllanthin, a lignan from Phyllanthus amarus, against progression of high fat diet induced metabolic disturbances in mice. RSC Adv. 2016, 63, 58343. 10.1039/c6ra10774e. [DOI] [Google Scholar]
- Ott M.; Thyagarajan S. P.; Gupta S. Phyllanthus amarus suppresses hepatitis B virus by interrupting interactions between HBV enhancer I and cellular transcription factors. Eur. J. Clin. Invest. 1997, 27, 908–915. 10.1046/j.1365-2362.1997.2020749.x. [DOI] [PubMed] [Google Scholar]
- Chanprapaph S.; Saparpakorn P.; Sangma C.; Niyomrattanakit P.; Hannongbua S.; Angsuthanasombat C.; Katzenmeier G. Competitive inhibition of the dengue virus NS3 serine protease by synthetic peptides representing polyprotein cleavage sites. Biochem. Biophys. Res. Commun. 2005, 330, 237–246. 10.1016/j.bbrc.2005.03.107. [DOI] [PubMed] [Google Scholar]
- Youn B.; Moinuddin S. G. A.; Davin L. B.; Lewis N. G.; Kang C. Crystal structures of apo-form and bina-ry/ternary complexes of podophyllum secoisolariciresinol dehydrogenase, an enzyme involved in formation of health-protecting and plant defense lignans. J. Biol. Chem. 2005, 280, 12917–12926. 10.1074/jbc.m413266200. [DOI] [PubMed] [Google Scholar]
- Krithika R.; Mohankumar R.; Verma R. J.; Shrivastav P. S.; Mohamad I. L.; Gunasekaran P.; Narasimhan S. Isolation, characterization and antioxidative effect of phyllanthin against CCl4-induced toxicity in HepG2 cell line. Chem. Biol. Interact. 2009, 181, 351–358. 10.1016/j.cbi.2009.06.014. [DOI] [PubMed] [Google Scholar]
- The bis-substitution product and its subsequent transformations were performed, to secure the congeners retained with the native 1,4-diphenyl butane scaffold of phyllanthin. The preliminary biological studies on these congeners showed that the inhibition activities were much weaker than phyllanthin, as a result, these bis-substitution products were not continued with further biological studies.
- Shao L.; Miyata S.; Muramatsu H.; Kawano H.; Ishii Y.; Saburi M.; Uchida Y. Asymmetric synthesis of (R)- and (S)-4-(substituted benzyl)dihydrofuran-2(3H)-ones: An application of the ruthenium-binap complex-catalysed asymmetric hydrogenation of alkylidene succinic acids. J. Chem. Soc., Perkin Trans. 1 1990, 1441–1445. 10.1039/p19900001441. [DOI] [Google Scholar]
- Barnier J.-P.; Blanco L.; Jampel G. E.; Rousseau G. Preparation of (R)-veratryl- and (R)-(3-methoxybenzyl)succinates. Tetrahedron 1989, 45, 5051–5058. 10.1016/s0040-4020(01)81084-x. [DOI] [Google Scholar]
- Pohmakotr M.; Soorukram D.; Tuchinda P.; Prabpai S.; Kongsaeree P.; Reutrakul V. Highly diastereoselective alkylation of vicinal dianions of chiralsuccinic acid derivatives: a new general strategy to (R)-β-arylmethyl-γ-butyrolactones. Tetrahedron Lett. 2004, 45, 4315–4318. 10.1016/j.tetlet.2004.04.008. [DOI] [Google Scholar]
- Twining S. S. Fluorescein isothiocyanate-labeled casein assay for proteolytic enzymes. Anal. Biochem. 1984, 143, 30–34. 10.1016/0003-2697(84)90553-0. [DOI] [PubMed] [Google Scholar]
- Failla C.; Tomei L.; De Francesco R. Both NS3 and NS4A are required for proteolytic processing of Hepatitis C virus nonstructural proteins. J. Virol. 1994, 68, 3753–3760. 10.1128/jvi.68.6.3753-3760.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou Y.-H.; Chang M.-F.; Wang Y.-M.; Hung T.-M.; Chang S. C. Differential requirements of NS4A for internal NS3 cleavage and polyprotein processing of Hepatitis C virus. J. Virol. 2007, 81, 7999–8008. 10.1128/jvi.00348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon J. C.; Ferreon A. C. M.; Li K.; Lemon S. M. Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J. Biol. Chem. 2005, 280, 20483–20492. 10.1074/jbc.m500422200. [DOI] [PubMed] [Google Scholar]
- Li X.-D.; Sun L.; Seth R. B.; Pineda G.; Chen Z. J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17717–17722. 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky Y.; Zemel R.; Bachmatov L.; Abramovich A.; Koren R.; Sathiyamoorthy P.; Golan-Goldhirsh A.; Tur-Kaspa R.; Benhar I. A novel high throughput screening assay for HCV NS3 serine protease inhibitors. J. Virol. Methods 2003, 107, 245–255. 10.1016/s0166-0934(02)00255-0. [DOI] [PubMed] [Google Scholar]
- Frese M.; Barth K.; Kaul A.; Lohmann V.; Schwärzle V.; Bartenschlager R. Hepatitis C virus RNA replication is resistant to tumour necrosis factor-α. J. Gen. Virol. 2003, 84, 1253–1259. 10.1099/vir.0.18997-0. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E.. The protein data bank. Structural Bioinformatics; Wiley, 2005; pp 181–198. [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDock tools 4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco S.; Rizzi M.; Volpari C.; Walsh M. A.; Narjes F.; Colarusso S.; De Francesco R.; Matassa V. G.; Sollazzo M. Inhibition of the Hepatitis C Virus NS3/4A Protease. J. Biol. Chem. 2000, 275, 7152–7157. 10.1074/jbc.275.10.7152. [DOI] [PubMed] [Google Scholar]
- Bolton E. E.; Chen J.; Kim S.; Han L.; He S.; Shi W.; Simonyan V.; Sun Y.; Thiessen P. A.; Wang J.; et al. PubChem3D: A new resource for scientists. J. Cheminf. 2011, 3, 32. 10.1186/1758-2946-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S.; Schreiber S.; Haupt V. J.; Adasme M. F.; Schroeder M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano W. L.The PyMOL Molecular Graphics System; Schrodinger, 2002. [Google Scholar]
- Ciofani G.; Danti S.; D’Alessandro D.; Moscato S.; Menciassi A. Assessing cytotoxicity of boron nitride nanotubes: Interference with the MTT assay. Biochem. Biophys. Res. Commun. 2010, 394, 405–411. 10.1016/j.bbrc.2010.03.035. [DOI] [PubMed] [Google Scholar]
- Organization for Economic Cooperation and Development . Test No. 423: Acute Oral toxicity–Acute Toxic Class Method. OECD Guidelines for the Testing of Chemicals: Paris, 2001. [Google Scholar]
- Organization for Economic Cooperation and Development . OECD Report of the Validation of the Updated Test Guideline 407: Repeat Dose 28 day Oral Toxicity Study in Laboratory Rats. Series on Testing and Assessment No 59, ENV/JM/MONO, 2006; p 26.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.