

Abstract

An efficient method to access (E)-trisubstituted alkenes is reported via cobalt-catalyzed isomerization of 1,1-disubstituted alkenes using a phosphine-amido-oxazoline ligand. The reaction could also convert mono- and 1,2-disubstituted alkenes to (E)-internal alkenes with benzylic selectivity. This protocol is atom-economy and operationally simple and uses readily available starting materials with good functional tolerance. This catalytic system could be scaled up to gram scale smoothly with a catalyst loading of 0.1 mol %.
Introduction
Linear trisubstituted alkenes represent an important class of structural scaffolds in natural products1 and have wide applications in synthetic organic chemistry.2 Several classical methods, including Wittig olefination,3 metal-catalyzed cross-coupling,4 and olefin metathesis,5 have been developed for the construction of linear trisubstituted alkenes. However, generally, these approaches are minimally stereoselective or suffered from the production of stoichiometric amount of waste.6 In the view of green and sustainable chemistry, isomerization of alkenes with transition-metal catalysts is considered as an efficient strategy to access regio- and stereodefined alkenes with 100% atom-economy. During the past few decades, considerable effort has been made for developing catalysts for alkene isomerization.7 However, the method for alkene isomerization to access trisubstituted alkenes with high stereoselectivity has not been well explored. In 2009, RajanBabu and co-workers described a Pd-catalyzed isomerization of terminal alkenes into 2-alkenes, in which α-ethyl styrenes were tested to give trisubstituted alkenes with 2/1–19/1 E/Z selectivity (Scheme 1a).8 In 2012, the Grotjahn group disclosed an efficient ruthenium catalyst for E-selective alkene isomerization over one position; however, only 70–80% conversion was achieved in the case of α-ethyl styrenes (Scheme 1b).9 More recently, Zhao and Liu presented an enantioselective isomerization of homoallylic and bishomoallylic secondary alcohols with the rhodium catalyst. When α-ethyl styrene was subjected to the isomerization reaction, the corresponding (E)-trisubstituted alkene was yielded with stereoselectivity better than 10/1 (Scheme 1c).10 Hence, the development of efficient catalysts for the construction of stereodefined trisubstituted alkenes is still highly desirable.
Scheme 1. Stereoselective Isomerization of 1,1-Disubstituted Alkenes to (E)-Trisubstituted Alkenes.

In recent years, the earth-abundant transition metals including iron, cobalt, nickel, and copper have attracted a lot of interest owing to their low cost, environmentally benign nature, high natural abundance, and unique catalytic reactivity.11 The earth-abundant transition-metal catalysts have made notable progress in the field of alkene isomerization.12−21 In 2009, Oshima and Yorimitsu reported a catalytic isomerization of 1-alkenes to (E)-2-alkenes with a NHC–cobalt complex in the presence of a Grignard reagent.13 In 2014, Holland and co-workers developed a cobalt-catalyzed (Z)-selective isomerization of terminal alkenes with a bulky β-diketiminate ligand.14 Almost simultaneously, the Hilt group independently described a similar transformation using Ph2PH as the ligand.15 With a catalytic amount of Co–salen complex and organosilane, the Shenvi group reported a chemoselective isomerization of terminal alkenes over one position via a radical-based hydrogen atom transfer (HAT) mechanism.16 In 2016, Norton and co-workers found that Co(dmgBF2)2(THF)2 could generate a low concentration of H• donor under H2 pressure, which could enable isomerization and cycloisomerization of alkenes.17 In 2018, Liu and Jiao developed a regioselective isomerization of alkenes with a designed pyridine–amido–phosphine cobalt catalyst, affording the less hindered olefin products under high kinetic control.18 Employing a dual-visible-light-cobalt catalyst, König and co-workers accomplished a regiocontrollable isomerization of alkenes by switching the ligand.19 In 2019, the Schoenebeck group developed a reductant-free nickel-catalyzed double bond migration of alkenes via an intramolecular radical relocation approach.20 Very recently, our group reported a cobalt-catalyzed Z to E isomerization of alkenes with an amido–diphosphine ligand (PNP).21 However, to the best of our knowledge, there is no report on highly stereoselective isomerization of 1,1-disubstituted alkenes with an earth-abundant transition-metal catalyst.22 In this study, we present an efficient cobalt-catalyzed isomerization of 1,1-disubstituted alkenes to (E)-trisubstituted alkenes with a phosphine-amido-oxazoline (PAO) ligand.
Results and Discussion
In our initial experiments, we chose the isomerization of 1-(but-1-en-2-yl)-4-methoxybenzene (1a) as the model reaction (Table 1). When 1a was mixed with 2.5 mol % of CoCl2 and 7.5 mol % of NaBHEt3 in a solution of tetrahydrofuran (THF) at room temperature for 1 h, no isomerization product (2a) was formed (entry 1). Next, Xantphos and DPEphos were added as ligands in the reaction, giving 2a in 3 and 0% yields, respectively (entries 2–3). The use of the PNP ligand led to a slight increase in yield (entry 4, 8%) with good stereoselectivity (100/4 E/Z), while the bidentate amido–monophosphine ligand (PN) showed poor reactivity (entry 5). Then, we modified the PNP ligand by introducing an oxazoline ring to replace one of the PPh2 moieties. The PAO ligand could be easily synthesized from commercially available starting materials via three steps.21 To our delight, a significant increase in yield with excellent stereoselectivity was observed by using PAOdiMe as the ligand (entry 6, 60% yield, 100/1 E/Z). The difference observed between PNP and PAO ligands could be probably attributed to the smaller steric effect of the latter one. Solvent plays an important role in the reaction (entries 7–8). An excellent yield with a slight lower stereoselectivity was observed when using toluene (96%, 100/3 E/Z) or dioxane (96%, 100/4 E/Z) as the solvent. When the reaction time was increased to 12 h in toluene, no improvement of the yield and stereoselectivity was observed (entry 9). Interestingly, decreasing the reaction time to 30 min gave the desired product in the same yield with 100/1 E/Z selectivity (entry 10). Further decreasing the reaction time to 10 min led to a much lower yield (75%, entry 11).
Table 1. Optimization of Conditionsa.

| entry | ligand | solvent | Time | yield (%)b | E/Zc |
|---|---|---|---|---|---|
| 1 | THF | 1 h | 0 | ||
| 2 | Xantphos | THF | 1 h | 3 | |
| 3 | DPEphos | THF | 1 h | 0 | |
| 4 | PNP | THF | 1 h | 8 | 100/4 |
| 5 | PN | THF | 1 h | Trace | |
| 6 | PAO | THF | 1 h | 60 | 100/1 |
| 7 | PAO | toluene | 1 h | 96 | 100/3 |
| 8 | PAO | dioxane | 1 h | 96 | 100/4 |
| 9 | PAO | toluene | 12 h | 96 | 100/3 |
| 10 | PAO | toluene | 30 min | 96 | 100/1 |
| 11 | PAO | toluene | 10 min | 75 | 100/1 |

The reaction was conducted with 1a (0.5 mmol), CoCl2 (0.0125 mmol), ligand (0.015 mmol), and NaHBEt3 (0.038 mmol) in solvent (1 mL) at room temperature.
The yield of 2a was determined by 1H NMR analysis using mesitylene as an internal standard.
The E/Z ratio of 2a was determined by 1H NMR analysis.
With the optimal reaction conditions in hand, the substrate scope of 1,1-disubstituted alkenes is illustrated in Table 2. Under the standard conditions, α-ethyl styrene was converted to the isomerization product in 97% yield with 100/2 stereoselectivity (2b). Employing PAOdiMe or PAOMe as the ligand, various electron-donating and electron-withdrawing functional groups at the para-position of the phenyl ring, such as methyl, phenyl, fluoro, chloro, bromo, dimethyl aniline, silyl ether, thioether, ester, and trifluoromethyl, were well tolerated, giving the desired products in good yields with better than or equal to 100/2 stereoselectivity (2c–2l). The substituent at the meta-position of the phenyl ring has no influence on the reactivity and stereoselectivity (2m and 2n), whereas the reaction of styrene containing an ortho-substituent at the phenyl ring required a relatively longer time, giving the product with excellent stereoselectivity (2o, >100/1 E/Z). The styrenes bearing two or three substituents on the aryl ring resulted in similar yields and stereoselectivity (2p–2r), and the reaction of 1r was completed in only 3 min (TOF 736 h–1). The isomerization of α-alkyl styrenes proceeded smoothly to give the desired (E)-alkenes in 92–96% yields with up to 100/1 stereoselectivity (2s–2w), even if the alkyl chain was tethered with ester (2v) or free alcohol group (2w). Other aryl alkenes (2x–2z), such as 2-naphthalene, thiophene, and pyridine, were well tolerated to afford the desired products in excellent yields with up to >100/1 E/Z selectivity. Exocyclic alkenes were compatible with this catalytic system, producing the corresponding endocyclic olefins in quantitative conversion (2aa and 2ab). 1,1-Dialkyl alkene was also examined to give the corresponding trisubstituted olefin in 96% conversion (2ac); however, the catalytic reaction of methylenecyclooctane did not work.
Table 2. Substrate Scope of 1,1-Disubstituted Alkenesa.


The reaction was conducted with 1 (0.5 mmol), CoCl2 (0.0125 mmol), PAOdiMe (0.015 mmol), and NaHBEt3 (0.038 mmol) in toluene (1 mL) at room temperature for 30 min. The yields and E/Z ratios of 2 were determined by 1H NMR analysis.
Dioxane was used as a solvent.
CoCl2 (5 mol %), PAOMe (6 mol %), NaBHEt3 (7.5 mol %), and THF were used.
PAOMe was used as a ligand.
CoCl2 (5 mol %), PAOdiMe (6 mol %), NaBHEt3 (7.5 mol %), and dioxane were used.
The phenylpropenoids, including 2-propenylbenzenes and 1-propenylbenzenes, are an important class of naturally occurring compounds,23 which have wide applications in pharmaceutical chemistry,24 pesticide,25 and flavor and fragrance industry.26 Employing this cobalt catalytic system, estragole (3a), elemicin (3b), methyl eugenol (3c), and silyl-ethered eugenol (3d) could be efficiently converted to trans-anethole (4a), trans-isoelemicin (4b), trans-methyl isoeugenol (4c), and 4d, respectively, with 100/2 stereoselectivity under standard conditions in 1 min (Table 3, TOF = 2400 h–1). The reaction was compatible for double bond migration over two-carbon chain walk, producing benzylic olefin in quantitative conversion with 100/3 E-selectivity (4e). Internal alkene was also subjected to the reaction to give the corresponding (E)-β-alkyl styrene with high regio- and stereoselectivity (4f, 100/4 rr, 100/1 E/Z). Furthermore, this isomerization reaction was applicable for geometrical isomerization of alkene to produce the (E)-isomers with 100/2 selectivity using a mixture of (E)- and (Z)-alkenes as the starting material (3g).
Table 3. Substrate Scope of Mono- and 1,2-Disubstituted Alkenesa.


The reaction was conducted with 1 (0.5 mmol), CoCl2 (0.0125 mmol), PAOdiMe (0.015 mmol), and NaHBEt3 (0.038 mmol) in dioxane (1 mL) at room temperature.
The conversions and E/Z ratios were determined by 1H NMR analysis. Isolated yield in parenthesis.
The starting material was a mixture of 3f (59%) and 4f (41%).
To further showcase the synthetic utility, 2 g-scale reactions were performed with a lower catalyst loading (0.1 and 0.25 mol %), affording the desired product without compromising the stereoselectivity in 9227 and 97% isolated yields, respectively (eq 1). To gain insight into the catalytic cycle, deuterium-labeling and radical trap experiments were conducted under standard conditions (eq 1). The reaction of 1a was carried out in the presence of 1 equiv of deuterated 4-methoxy styrene (D-5). After 30 min, 79% proportion of the deuterium was incorporated at the C1 position of the isomerization product [D-(E)-2a], and the recovered D-5 displayed a decreased deuterium incorporation. The H/D scrambling experiment indicated that the catalytic reaction probably involved a cobalt-hydride catalysis or HAT mechanism. When the reaction of 1a was further performed with 9,10-dihydroanthracene and di-tert-butylhydroxytoluene, respectively (eq 4), no influence on the results was found, indicating that the radical-based HAT mechanistic pathway is unlikely.
 |
1 |
On the basis of the above results and previous reports,7d,21 a plausible catalytic cycle is proposed in Scheme 2. A cobalt-hydride species (A) was first formed by mixing CoCl2, the PAO ligand, and NaBHEt3. Following the coordination of 1,1-disubstituted alkene to the cobalt center, a double bond is inserted into the Co–H bond to generate a benzylic cobalt species (C). Subsequently, β-H elimination occurred to give the more stable (E)-trisubstituted alkene selectively.
Scheme 2. Proposed Catalytic Cycle.

Conclusions
In summary, we have developed an efficient cobalt-catalyzed isomerization of 1,1-disubstituted alkenes with the PAO ligand, producing the corresponding (E)-trisubstituted alkenes with good functional tolerance and high stereoselectivity (up to >100/1 E/Z). The reaction is also applicable for the isomerization of mono- and 1,2-disubstituted alkenes with benzylic selectivity (TOF 2400 h–1). This catalytic reaction could be scaled up to gram scale smoothly with a catalyst loading of 0.1 mol %. Preliminary mechanistic studies indicated that a cobalt-hydride species was involved in the catalytic reaction. Featuring a low-cost and environmentally benign cobalt catalyst, 100% atom-economy, mild reaction conditions, and readily available starting materials with good functional group compatibility, this is a practical method to access synthetically important (E)-alkenes.
Experimental Section
General Information
Toluene, THF, 1,4-dioxane, and dichloromethane (DCM) were taken from a solvent purification system (PS-400-5, Unilab Mbraun, Inc.). NaHBEt3 (1.0 M in THF) was purchased from Aldrich and used as received. CoCl2 (99%) was purchased from Strem and used as received. Oxydi-2,1-phenylene)bis(diphenylphosphine (DPEphos) (98%) and dimethylbisdiphenylphosphinoxanthene (Xantphos) (98%) were purchased from Energy Chemical and used as received. The other commercial available chemicals were used as received. Bis(2-(diphenylphosphanyl)phenyl)amine (PNP)28 and 2-(diphenylphosphanyl)-N-phenylaniline (PN)29 were prepared according to previously reported procedures, respectively. NMR spectra were recorded on a Bruker-400 instrument or Bruker-500 instrument. 1H NMR chemical shifts were referenced to tetramethylsilane signal (0 ppm), 13C NMR chemical shifts were referenced to the solvent resonance (77.00 ppm, CDCl3), and 19F NMR chemical shifts were referenced to the solvent resonance. The following abbreviations (or combinations thereof) were used to explain multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad, q = quadruplet. High-resolution mass spectrometry (HRMS) analysis was measured on a Bruker micrOTOF-Q II instrument (ESI) or a Waters GCT Premier instrument (EI-TOF).
Procedures for the Synthesis of Starting Materials
1-(4-Methoxyphenyl)propan-1-one (S1a), 1-(4-fluorophenyl)propan-1-one (S1e), 1-(4-chlorophenyl)propan-1-one (S1f), 1-(4-bromophenyl)propan-1-one (S1g), 1-(4-(trifluoromethyl)phenyl)propan-1-one (S1l), 1-(3-methoxyphenyl)propan-1-one (S1m), 1-(2-fluorophenyl)propan-1-one (S1o), 1-(benzo[1,3]dioxol-5-yl)butan-1-one (S1r), 1-phenylbutan-1-one (S1s), 1-phenylpentan-1-one (S1t), 1-phenylhexan-1-one (S1u), 1-(6-methoxynaphthalen-2-yl)propan-1-one (S1x), 1-(thiophen-2-yl)propan-1-one (S1y), 3,4-dihydronaphthalen-1(2H)-one (S1aa), 2,3-dihydro-1H-inden-1-one (S1ab), and 4-phenylbutan-2-one (S1ac) were purchased from Energy Chemical and used as received. 1-(4-((tert-Butyldimethylsilyl)oxy)phenyl)propan-1-one (S1i) was prepared according to previously reported procedures30 using 1-(4-hydroxyphenyl)propan-1-one and TBSCl as starting materials; 4-propionylphenyl acetate (S1k) was prepared according to previously reported procedures31 using 1-(4-hydroxyphenyl)propan-1-one as the starting material; and ethyl 5-(4-fluorophenyl)-5-oxopentanoate (S1v) was prepared according to the esterification reaction with ethanol using concentrated sulfuric acid as the catalyst.
All the other ketones were prepared by the following routes.
Route A: A 250 mL flame-dried Schlenk flask was cooled at room temperature under the argon atmosphere, charged with the corresponding benzaldehyde (50 mmol) and dry THF (40 mL), to which was added slowly EtMgBr (75 mmol, 1 M in THF, 1.5 equiv) at 0 °C, and then, the mixture was stirred at room temperature. After the aldehyde was fully consumed [monitored by thin-layer chromatography (TLC)], the reaction was quenched with saturated NH4Cl (25 mL) at 0 °C and extracted with ethyl acetate (30 mL × 3). The combined organic layers were washed with saturated brine, dried over Na2SO4, filtered, and concentrated by rotary evaporation to afford the crude alcohol.
A 250 mL round-bottomed flask was charged with the crude alcohol, DCM (100 mL), pyridinium chlorochromate (75 mmol, 1.5 equiv), and Celite (10 g). The mixture was stirred at room temperature until the alcohol was fully consumed (monitored by TLC). The resulted suspension was filtered through a pad of silica gel and washed with DCM. The filtrate was concentrated by rotary evaporation, and the residue was further purified by flash chromatography on silica gel to afford the desired ketone.
Route B: A 250 mL flame-dried Schlenk flask was cooled at room temperature under the argon atmosphere, charged with 4-(dimethylamino)benzonitrile (4.99 g, 34.12 mmol) and dry THF (50 mL). To the reaction mixture was added slowly EtMgBr (1 M in THF, 102.4 mL, 102.4 mmol) at 0 °C, followed by CuBr (0.45 g, 3.14 mmol) in one portion. The reaction was then heated at 60 °C for 4 h, cooled to 0 °C, and quenched cautiously with H2O (40 mL) and H2SO4 (1 M, 160 mL). The mixture was heated under reflux for 1 h, cooled to room temperature, and basified with NaOH (2 M) until pH = 9. The resulted solution was extracted with ethyl acetate (50 mL × 3), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was further purified by flash chromatography on silica gel to afford 1-(4-(dimethylamino)phenyl)propan-1-one (4.22 g, 23.83 mmol, 70%) as a pale yellow solid.
Propiophenone (S1b)32
Prepared according to route A. 1H NMR: (400.0 MHz, CDCl3): δ 8.00–7.95 (m, 2H), 7.59–7.53 (m, 1H), 7.49–7.43 (m, 2H), 3.01 (q, J = 7.2 Hz, 2H), 1.23 (t, J = 7.2 Hz, 3H).
1-([1,1′-Biphenyl]-4-yl)propan-1-one (S1d)32
Prepared according to route A. 1H NMR: (400.0 MHz, CDCl3): δ 8.10–7.99 (m, 2H), 7.75–7.57 (m, 4H), 7.52–7.35 (m, 3H), 3.04 (q, J = 7.2 Hz, 2H), 1.25 (t, J = 7.2 Hz, 3H).
1-(4-(Dimethylamino)phenyl)propan-1-one (S1h)32
Prepared according to route B. 1H NMR: (400.0 MHz, CDCl3): δ 7.91–7.85 (m, 2H), 6.67–6.62 (m, 2H), 3.05 (s, 6H), 2.91 (q, J = 7.4 Hz, 2H), 1.20 (t, J = 7.4 Hz, 3H).
1-(4-(Methylthio)phenyl)propan-1-one (S1j)33
Prepared according to route A. 1H NMR: (400.0 MHz, CDCl3): δ 7.90–7.86 (m, 2H), 7.26–7.24 (m, 2H), 2.96 (q, J = 7.2 Hz, 2H), 2.52 (s, 3H), 1.22 (t, J = 7.2 Hz, 3H).
1-(3,4-Dimethoxyphenyl)propan-1-one (S1p)34
Prepared according to route A. 1H NMR: (400.0 MHz, CDCl3): δ 7.65–7.50 (m, 2H), 6.95–6.85 (m, 1H), 3.94 (s, 6H), 2.97 (q, J = 7.2 Hz, 2H), 1.22 (t, J = 7.2 Hz, 3H).
1-(3,4,5-Trimethoxyphenyl)propan-1-one (S1q)35
Prepared according to route A. 1H NMR: (400.0 MHz, CDCl3): δ 7.23 (s, 2H), 3.92 (s, 9H), 2.99 (q, J = 7.2 Hz, 2H), 1.23 (t, J = 7.2 Hz, 3H).
1-(Pyridin-3-yl)propan-1-one (S1z)36
Prepared according to route A. 1H NMR: (400.0 MHz, CDCl3): δ 9.18 (S, 1H), 8.80–8.75 (m, 1H), 8.27–8.21 (m, 1H), 7.46–7.40 (m, 1H), 3.04 (q, J = 7.2 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H).
1-Allyl-4-methoxybenzene (3a), 5-allyl-1,2,3-trimethoxybenzene (3b), and 4-allyl-1,2-dimethoxybenzene (3c) were purchased from Energy Chemical and used as received. (4-Allyl-2-methoxyphenoxy)(tert-butyl)dimethylsilane (3d) was prepared according to a previously reported procedure using 4-allyl-2-methoxyphenol and TBSCl as starting materials.30
All the other alkenes were prepared according to the following procedure.
General Procedure for the Synthesis of Alkenes
A three-necked, round-bottomed flask (250 mL) was charged with Ph3PCH3Br (52 mmol, 1.3 equiv) and NaH (56 mmol, 1.4 equiv, 60% in oil) in dry THF (100 mL). The mixture was heated under reflux for 1.5 h and cooled to room temperature. Then, a solution of aldehyde (40 mmol in 20 mL of THF) was added dropwise to the mixture and heated to reflux. After the complete consumption of aldehyde (monitored by TLC), the reaction mixture was cooled to room temperature, quenched with saturated NH4Cl (1 mL), passed through a pad of silica gel, and washed with PE. The filtrate was concentrated by rotary evaporation, and the residue was further purified by flash chromatography or distillation to afford the desired alkene.
1-(But-1-en-2-yl)-4-methoxybenzene (1a)37
1H NMR: (400.0 MHz, CDCl3): δ 7.36 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 9.0 Hz, 2H), 5.21 (s, 1H), 4.98 (s, 1H), 3.81 (s, 3H), 2.49 (q, J = 7.2 Hz, 2H), 1.10 (t, J = 7.2 Hz, 3H).
But-1-en-2-ylbenzene (1b)38
1H NMR: (400.0 MHz, CDCl3): δ 7.45–7.38 (m, 2H), 7.36–7.29 (m, 2H), 7.28–7.23 (m, 1H), 5.27 (s, 1H), 5.06 (q, J = 1.4 Hz, 1H), 2.52 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H).
1-(But-1-en-2-yl)-4-methylbenzene (1c)37
1H NMR: (400.0 MHz, CDCl3): δ 7.36–7.29 (m, 2H), 7.18–7.11 (m, 2H), 5.25 (s, 1H), 5.02 (s, 1H), 2.50 (q, J = 7.4 Hz, 2H), 2.35 (s, 3H), 1.10 (t, J = 7.4 Hz, 3H).
4-(But-1-en-2-yl)-1,1′-biphenyl (1d)
White solid. mp 72–73 °C; 1H NMR: (400.0 MHz, CDCl3): δ 7.64–7.54 (m, 4H), 7.52–7.47 (m, 2H), 7.46–7.40 (m, 2H), 7.37–7.30 (m, 1H), 5.35 (s, 1H), 5.09 (q, J = 1.4 Hz, 1H), 2.55 (q, J = 7.6 Hz, 2H), 1.14 (t, J = 7.6 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 149.5, 140.8, 140.4, 140.1, 128.7, 127.2, 127.0, 126.9, 126.4, 111.0, 28.0, 13.0; HRMS (ESI): cald for [C16H16]: 209.1325 [M + H]+; found, 209.1329.
1-(But-1-en-2-yl)fluorobenzene (1e)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.42–7.30 (m, 2H), 7.05–6,94 (m, 2H), 5.22 (s, 1H), 5.04 (s, 1H), 2.53–2.40 (m, 2H), 1.13–1.03 (m, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 162.2 (d, J = 305.6 Hz), 149.0, 138 (d, J = 4.0 Hz), 127.5 (d, J = 9.4 Hz), 115.0 (d, J = 26.5 Hz), 110.9, 28.2, 12.8; 19F NMR: (470 MHz, CDCl3): δ −115.7; HRMS (EI): cald for [C10H11F]+ requires m/z, 150.0845; found m/z, 150.0846.
1-(But-1-en-2-yl)-4-chlorobenzene (1f)39
1H NMR: (400.0 MHz, CDCl3): δ 7.39–7.23 (m, 4H), 5.26 (s, 1H), 5.07 (s, 1H), 2.48 (q, J = 7.2 Hz, 2H), 1.09 (t, J = 7.2 Hz, 3H).
1-Bromo-4-(but-1-en-2-yl)benzene (1g)40
1H NMR: (400.0 MHz, CDCl3): δ 7.47–7.41 (m, 2H), 7.30–7.26 (m, 2H), 5.26 (s, 1H), 5.07 (s, 1H), 2.48 (q, J = 7.4 Hz, 2H), 1.09 (t, J = 7.4 Hz, 3H).
4-(But-1-en-2-yl)-N,N-dimethylaniline (1h)
Isolated as a mixture of 1h and 2h (4-(but-2-en-2-yl)-N,N-dimethylaniline) (1h/2h = 74/26). For 1h: 1H NMR: (400.0 MHz, CDCl3): δ 7.38–7.31 (m, 2H), 6.73–6.67 (m, 2H), 5.19 (s, 1H), 4.90 (q, J = 1.4 Hz, 1H), 2.95 (s, 6H), 2.49 (q, J = 7.4 Hz, 2H), 1.11 (t, J = 7.4 Hz, 3H). For 2h: 1H NMR: (400.0 MHz, CDCl3): δ 7.30–7.26 (m, 2H), 6.73–6.67 (m, 2H), 5.76 (q, J = 7.0 Hz, 1H), 2.93 (s, 6H), 2.02–1.98 (m, 3H), 1.77 (d, J = 7.0 Hz, 3H).
(4-(But-1-en-2-yl)phenoxy)(tert-butyl)dimethylsilane (1i)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.32–7.27 (m, 2H), 6.81–6.76 (m, 2H), 5.21 (s, 1H), 4.97 (s, 1H), 2.48 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H), 0.98 (s, 9H), 0.20 (s, 6H); 13C NMR: (125.5 MHz, CDCl3): δ 154.3, 137.1, 134.9, 126.3, 120.8, 119.6, 25.7, 18.2, 15.4, 14.2, −4.4; HRMS (ESI): calcd for [C16H26OSi], 263.1826 [M + H]+; found, 263.1834.
(4-(But-1-en-2-yl)phenyl)(methyl) Sulfane (1j)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.37–7.32 (m, 2H), 7.24–7.21 (m, 2H), 5.26 (s, 1H), 5.03 (q, J = 1.6 Hz, 1H), 2.49 (q, J = 7.4 Hz, 2H), 2.48 (s, 3H), 1.10 (t, J = 7.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 149.2, 138.2, 137.2, 126.4, 126.3, 110.5, 27.8, 15.8, 12.9; HRMS (ESI): calcd for [C11H14S], 179.0889 [M + H]+; found, 179.0881.
4-(But-1-en-2-yl)phenyl Acetate (1k)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.46–7.39 (m, 2H), 7.07–7.01 (m, 2H), 5.26 (s, 1H), 5.05 (q, J = 1.4 Hz, 1H), 2.49 (q, J = 7.6 Hz, 2H), 2.30 (s, 3H), 1.10 (t, J = 7.6 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 169.4, 149.8, 149.0, 139.1, 126.9, 121.1, 111.1, 27.9, 21.0, 12.8; HRMS (ESI): calcd for [C12H14O2], 191.1067 [M + H]+; found, 191.1065.
1-(But-1-en-2-yl)-4-(trifluoromethyl) Benzene (1l)39
1H NMR: (400.0 MHz, CDCl3): δ 7.58 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 5.33 (s, 1H), 5.16 (q, J = 1.3 Hz, 1H), 2.52 (q, J = 7.4 Hz, 2H), 1.11 (t, J = 7.4 Hz, 3H).
1-(But-1-en-2-yl)-3-methoxybenzene (1m)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.31–7.20 (m, 1H), 7.06–6.92 (m, 2H), 6.87–6.78 (m, 1H), 5.28 (s, 1H), 5.06 (s, 1H), 3.82 (s, 3H), 2.50 (q, J = 7.2 Hz, 2H), 1.10 (t, J = 7.2 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 159.5, 149.9, 143.1, 129.1, 118.5, 112.4, 112.0, 111.1, 55.1, 28.1, 12.9; HRMS (EI): calcd for [C11H14O]+ requires m/z, 162.1045; found m/z, 162.1045.
1-(But-1-en-2-yl)-3-chlorobenzene (1n)39
1H NMR: (400.0 MHz, CDCl3): δ 7.39 (s, 1H), 7.30–7.20 (m, 3H), 5.28 (s, 1H), 5.09 (s, 1H), 2.48 (q, J = 7.2 Hz, 2H), 1.10 (t, J = 7.2 Hz, 3H).
1-(But-1-en-2-yl)-2-fluorobenzene (1o)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.31–7.17 (m, 2H), 7.15–6.97 (m, 2H), 5.22 (s, 1H), 5.13 (s, 1H), 2.48 (q, J = 7.6 Hz, 2H), 1.05 (t, J = 7.6 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 159.8 (d, J = 307.2 Hz), 146.7, 130.2 (d, J = 18 Hz), 130.0 (d, J = 5.4 Hz), 128.6 (d, J = 10.2 Hz), 123.8 (d, J = 4.8 Hz), 115.6 (d, J = 28 Hz), 114.4 (d, J = 2.4 Hz), 29.3, 12.6; 19F NMR: (470 MHz, CDCl3): δ −115.5; HRMS (EI): calcd for [C10H11F]+ requires m/z, 150.0845; found m/z, 150.0844.
4-(But-1-en-2-yl)-1,2-dimethoxybenzene (1p)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.00–6.95 (m, 2H), 6.86–6.81 (m, 1H), 5.22 (s, 1H), 5.00 (q, J = 1.4 Hz, 1H), 3.90 (s, 3H), 3.89 (s, 3H), 2.49 (q, J = 7.4 Hz, 2H), 1.11 (t, J = 7.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 149.4, 148.6, 148.4, 134.3, 118.2, 110.8, 109.5, 109.4, 55.6, 27.9, 12.8; HRMS (EI): calcd for [C12H16O2]+ requires m/z, 192.1150; found m/z, 192.1150.
5-(But-1-en-2-yl)-1,2,3-trimethoxybenzene (1q)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.62 (s, 2H), 5.23 (s, 1H), 5.04 (q, J = 1.4 Hz, 1H), 3.88 (s, 6H), 3.86 (s, 3H), 2.49 (q, J = 7.4 Hz, 2H), 1.12 (t, J = 7.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 152.8, 150.0, 137.6, 137.4, 110.5, 103.5, 60.6, 56.0, 28.1, 12.8; HRMS (EI): calcd for [C13H18O3]+ requires m/z, 222.1256; found, m/z, 222.1253.
5-(Pent-1-en-2-yl)benzo[d][1,3]dioxole (1r)27
1H NMR: (400.0 MHz, CDCl3): δ 6.95–6.85 (m, 2H), 6.81–6.73 (m, 1H), 5.95 (s, 2H), 5.17 (s, 1H), 4.97 (s, 1H), 2.42 (t, J = 7.4 Hz, 2H), 1.51–1.40 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H).
Pent-1-en-2-ylbenzene (1s)27
1H NMR: (400.0 MHz, CDCl3): δ 7.44–7.37 (m, 2H), 7.35–7.29 (m, 2H), 7.29–7.22 (m, 1H), 5.28–5.25 (m, 1H), 5.06–5.03 (m, 1H), 2.48 (t, J = 7.6 Hz, 2H), 1.53–1.42 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H).
Hex-1-en-2-ylbenzene (1t)37
1H NMR: (400.0 MHz, CDCl3): δ 7.43–7.38 (m, 2H), 7.35–7.29 (m, 2H), 7.28–7.22 (m, 1H), 5.25 (s, 1H), 5.07–5.03 (m, 1H), 2.50 (t, J = 7.4 Hz, 2H), 1.47–1.40 (m, 2H), 1.39–1.30 (m, 2H), 0.89 (t, J = 7.2 Hz, 3H).
Hept-1-en-2-ylbenzene (1u)41
1H NMR: (400.0 MHz, CDCl3): δ 7.44–7.38 (m, 2H), 7.36–7.29 (m, 2H), 7.28–7.22 (m, 1H), 5.26 (s, 1H), 5.07–5.03 (m, 1H), 2.49 (t, J = 7.8 Hz, 2H), 1.50–1.40 (m, 2H), 1.36–1.25 (m, 4H), 0.87 (t, J = 7.2 Hz, 3H).
Ethyl 3-(4-Fluorophenyl)but-3-enoate (1v)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.40–7.33 (m, 2H), 7.05–6.97 (m, 2H), 5.25 (s, 1H), 5.07–5.03 (m, 1H), 4.12 (q, J = 7.2 Hz, 2H), 2.52 (t, J = 8.0 Hz, 2H), 2.32 (t, J = 7.4 Hz, 2H), 1.82–1.72 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 173.4, 162.2 (d, J = 305.6 Hz), 146.5, 136.8 (d, J = 4.0 Hz), 127.6 (d, J = 10.0 Hz), 115.1 (d, J = 26.4 Hz), 112.8, 60.2, 34.6, 34.5, 23.3, 14.2; 19F NMR: (470 MHz, CDCl3): δ −115.2; HRMS (ESI): calcd for [C14H17FO2], 237.1285 [M + H]+; found, 237.1283.
5-(4-Fluorophenyl)hex-5-en-1-ol (1w)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.40–7.32 (m, 2H), 7.05–6.96 (m, 2H), 5.22 (s, 1H), 5.05 (s, 1H), 3.64 (t, J = 6.4 Hz, 2H), 2.51 (t, J = 7.0 Hz, 2H), 1.65–1.45 (m, 4H), 1.25 (br, 1H); 13C NMR: (125.5 MHz, CDCl3): δ 162.2 (d, J = 243.8 Hz), 147.3, 137.2 (d, J = 3.6 Hz), 127.6 (d, J = 8.1 Hz), 115.0 (d, J = 20.5 Hz), 112.2, 62.5, 35.1, 32.2, 24.2; 19F NMR: (470 MHz, CDCl3): δ −115.4; HRMS (EI): cald for [C12H15FO]+ requires m/z, 194.1107; found m/z, 194.1105.
2-(But-1-en-2-yl)-6-methoxynaphthalene (1x)
White solid. mp 74–75 °C; 1H NMR: (400.0 MHz, CDCl3): δ 7.77 (s, 1H), 7.75–7.65 (m, 2H), 7.56 (dd, J = 8.6, 1.8 Hz, 1H), 7.16–7.09 (m, 2H), 5.39 (s, 1H), 5.14–5.10 (m, 1H), 3.91 (s, 3H), 2.62 (q, J = 7.4 Hz, 2H), 1.15 (t, J = 7.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 157.6, 149.7, 136.4, 133.9, 129.6, 128.8, 126.6, 125.1, 124.3, 118.8, 110.7, 105.5, 55.2, 28.0, 13.1; HRMS (ESI): cald for [C15H16O], 213.1274 [M + H]+; found, 213.1272.
2-(But-1-en-2-yl)thiophene (1y)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.15 (d, J = 5.2 Hz, 1H), 7.06–7.01 (m, 1H), 7.00–6.94 (m, 1H), 5.39 (s, 1H), 4.96 (s, 1H), 2.50 (q, J = 7.4 Hz, 2H), 1.18 (t, J = 7.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 145.5, 143.2, 127.2, 123.9, 123.0, 109.5, 28.2, 12.9; HRMS (EI): cald for [C8H10S]+ requires m/z, 138.0503; found m/z, 138.0504.
3-(But-1-en-2-yl)pyridine (1z)
Colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 8.69–8.66 (m, 1H), 8.52–8.48 (m, 1H), 7.71–7.66 (m, 1H), 7.27–7.23 (m, 1H), 5.32 (s, 1H), 5.18 (s, 1H), 2.52 (q, J = 7.2 Hz, 2H), 1.11 (t, J = 7.2 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 148.3, 147.4, 147.0, 136.8, 133.1, 123.0, 112.6, 27.7, 12.6; HRMS (ESI) calcd for [C9H11N], 133.0891 [M + H]+; found, 134.0963.
1-Methylene-1,2,3,4-tetrahydronaphthalene (1aa)42
1H NMR: (400.0 MHz, CDCl3): δ 7.68–7.60 (m, 1H), 7.20–7.05 (m, 3H), 5.47 (s, 1H), 4.95 (s, 1H), 2.84 (t, J = 6.2 Hz, 2H), 2.54 (t, J = 6.2 Hz, 2H), 1.92–1.83 (m, 2H).
1-Methylene-2,3-dihydro-1H-indene (1ab)
Isolated as a mixture of 1ab and 2ab (3-methyl-1H-indene) (1ab/2ab = 70/30). For 1ab27: 1H NMR: (400.0 MHz, CDCl3): δ 7.52–7.47 (m, 1H), 7.27–7.24 (m, 1H), 7.23–7.15 (m, 2H), 5.47–5.42 (m, 1H), 5.05–5.01 (m, 1H), 2.98 (t, J = 7.6 Hz, 2H), 2.83–2.76 (m, 2H). For 2ab8: 1H NMR: (400.0 MHz, CDCl3): δ 7.45 (d, J = 7.2 Hz, 1H), 7.36–7.28 (m, 2H), 7.20–7.15 (m, 1 H), 6.22–6.17 (m, 1H), 3.33–3.29 (m, 2H), 2.17 (q, J = 1.8 Hz, 3H).
(3-Methylbut-3-en-1-yl)benzene (1ac)27
1H NMR: (399.9 MHz, CDCl3): δ 7.32–7.24 (m, 2H), 7.22–7.15 (m, 3H), 4.74 (s, 1H), 4.71 (s, 1H), 2.76 (t, J = 7.8 Hz, 2H), 2.32 (t, J = 7.8 Hz, 2H), 1.77 (s, 3H).
(4-Allyl-2-methoxyphenoxy)(tert-butyl)dimethylsilane (3d)19
1H NMR: (400.0 MHz, CDCl3): δ 6.76 (d, J = 8.0 Hz, 1H), 6.68–6.66 (m, 1H), 6.63 (d, J = 8.0 Hz, 1H), 6.02–5.90 (m, 1H), 5.10–5.02 (m, 2H), 3.78 (s, 3H), 3.32 (d, J = 6.7 Hz, 2H), 0.99 (s, 9H), 0.14 (s, 6H).
But-3-en-1-ylbenzene (3e)19
1H NMR: (400.0 MHz, CDCl3): δ 7.32–7.24 (m, 2H), 7.22–7.15 (m, 3H), 5.92–5.80 (m, 1H), 5.09–4.95 (m, 2H), 2.71 (t, J = 8.2 Hz, 2H), 2.37 (q, J = 7.8 Hz, 2H).
Pent-2-en-1-ylbenzene (3f)
Isolated as a mixture of 3f and 4f (pent-1-en-1-ylbenzene) (3f/4f = 59/41). For 3f:431H NMR: (400.0 MHz, CDCl3): δ 7.32–7.25 (m, 2H), 7.22–7.15 (m, 3H), 5.58–5.49 (m, 2H), 3.42–3.30 (m, 2H), 2.21–2.00 (m, 2H), 1.05–0.92 (m, 3H). For 4f:431H NMR: (400.0 MHz, CDCl3): δ 7.37–7.32 (m, 2H), 7.31–7.25 (m, 2H), 7.21–7.15 (m, 1H), 6.38 (d, J = 16.0 Hz, 1H), 6.28–6.17 (m, 1H), 2.23–2.00 (m, 2H), 1.57–1.42 (m, 2H), 0.96 (t, 3H).
1-Methoxy-4-(prop-1-en-1-yl)benzene (3h, E/Z = 17/83)
For (E)-isomer:211H NMR: (400.0 MHz, CDCl3): δ 7.24 (d, J = 8.8 Hz, 2H), 6.86–6.81 (m, 2H) 6.41–6.30 (m, 1H), 6.14–6.04 (m, 1H), 3.79 (s, 3H), 1.84 (dd, J = 6.4, 1.4 Hz, 3H); for (Z)-isomer:211H NMR: (400.0 MHz, CDCl3): δ 7.23 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.42–6.30 (m, 1H), 5.75–5.65 (m, 1H), 3.81 (s, 3H), 1.89 (dd, J = 7.2, 1.8 Hz, 3H).
1-Methoxy-4-(vinyl-2,2-d2)benzene (D-5)
Prepared according to the general procedure of Wittig reaction using Ph3PCD3Br (prepared according to a previously reported method,44 81% D) and 4-methoxylbenzaldehyde as the starting materials, colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.38–7.32 (m, 2H), 6.89–6.83 (m, 2H), 6.71–6.62 (m, 1H), 5.64–5.56 (m, 0.19H), 5.15–5.08 (m, 0.19H), 3.81 (s, 3H); 2DNMR (400 MHz, CDCl3): δ 5.72.-5.60 (m, 0.81D), 5.25–5.10 (m, 0.81D).
Procedures for the Preparation of PAO Ligands
Synthesis of the PAOdiMe Ligand
![]()
2-(Diphenylphosphanyl)aniline (S1)
Prepared
according to a previously reported procedure:45 a flame-dried Schlenk tube (250 mL) was evacuated and refilled three
times with argon, charged with CuI (0.3861 g, 2 mmol), dry toluene
(80 mL), diphenylphosphines (7.65 mL, 44 mmol), and N,N′-dimethylethylenediamine (DMEDA) (1.54
mL, 14 mmol). The colorless solution was stirred for 10 min, to which
were added 2-iodoaniline (8.77 g, 40 mmol) and Cs2CO3 (26.10 g, 80 mmol) in one portion. The Schlenk tube was sealed
and stirred at 110 °C until the complete consumption of 2-iodoaniline
(monitored by TLC). The resulting suspension was cooled to room temperature,
passed through a pad of silica gel, and washed with EA. The filtrate
was concentrated under reduced pressure and purified by flash chromatography
on silica gel with PE/EA as the eluent to give 6.40 g (23.1 mmol,
58% yield) of S1 as a pale yellow solid. 1H NMR: (400.0 MHz, CDCl3): δ 7.37–7.28 (m,
10H), 7.20–7.13 (m, 1H), 6.79–6.72 (m, 1H), 6.71–6.64
(m, 2H), 4.09 (br, 2H).![]()
2-((2-(Diphenylphosphanyl)phenyl)amino)benzonitrile (S2)
A round-bottomed flask (250 mL) was evacuated
and refilled
three times with argon, charged with S1, KOtBu (8.55 g, 76.2 mmol), and dry DMSO (76 mL). The mixture was stirred
at room temperature for 10 min and cooled to −10 °C, fluorobenzonitrile
was added, and the mixture was stirred again at room temperature until
the complete consumption of S1 (monitored by TLC). The
resulted suspension was quenched with water (50 mL) and extracted
with ethyl acetate (50 mL × 3). The combined organic layers were
washed with brine, dried over Na2SO4, filtered,
and concentrated in vacuo. Recrystallization with ethanol afforded
8.20 g (21.7 mmol, 57% yield) of S2 as a pale yellow
solid. mp 109–110 °C; 1H NMR: (400.0 MHz, CDCl3): δ 7.40–7.30 (m, 14H), 7.11–7.04 (m,
1H), 6.96–6.90 (m, 2H), 6.79–6.70 (m, 1H), 6.42 (br,
1H); 13C NMR: (125.5 MHz, CDCl3): δ 147.1,
142.9 (d, J = 19.2 Hz), 135.0 (d, J = 9.0 Hz), 134.1 (d, J = 2.6 Hz), 134.0, 133.8,
133.5, 132.9, 131.9 (d, J = 10.9 Hz), 129.8, 129.2,
128.8 (d, J = 7.2 Hz), 124.9, 122.8, 119.2, 117.0,
114.5, 98.8; 31P NMR: (162 MHz, CDCl3): δ
−17.7; HRMS (ESI): cald for [C25H20N2P]+, 379.1359 [M + H]+; found, 379.1361.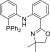
2-(4,4-Dimethyl-4,5-dihydrooxazol-2-yl)-N-(2-(diphenylphosphanyl)phenyl)aniline (PAOdime)
To a flame-dried flask (100 mL) were added amino-alcohol (0.95 g, 10.62 mmol), toluene (16 mL), S2 (3.35 g, 8.85 mmol), and Zn(OTf)2 (0.3217 g, 0.86 mmol) in sequence. The mixture was then heated under reflux for 4 days. The resulted suspension was cooled to room temperature, passed through a pad of silica gel, and washed with EA. The filtrate was concentrated under reduced pressure and purified by flash chromatography on silica gel to give 0.89 g (2.0 mmol, 31% yield) of PAOdiMe as a white solid. mp 125–126 °C; 1H NMR: (400.0 MHz, CDCl3): δ 10.37 (br, 1H), 7.69 (dd, J = 7.9, 1.5 Hz, 1H), 7.53–7.47 (m, 1H), 7.36–7.22 (m, 11H), 7.20–7.12 (m, 1H), 7.10–7.05 (m, 1H), 7.01 (t, J = 7.4 Hz, 1H), 6.89–6.83 (m, 1H), 6.68 (t, J = 7.5 Hz, 1H), 3.89 (s, 2H), 1.05 (s, 6H); 13C NMR: (125.5 MHz, CDCl3): δ 161.6, 146.1, 145.2, 145.1, 137.3 (d, J = 11.8 Hz), 134.4, 133.8, 133.7, 132.0 (d, J = 11.8 Hz), 131.7 (d, J = 10.9 Hz), 131.4, 129.5 (d, J = 5.4 Hz), 128.4 (d, J = 7.2 Hz), 124.1, 116.8, 113.6, 110.8, 77.4, 67.6, 28.1; 31P NMR: (162 MHz, CDCl3): δ −16.7; HRMS (ESI): calcd for [C29H28N2OP]+, 451.1934 [M + H]+; found, 451.1947.
Synthesis of the PAOMe Ligand
![]()
2-((2-Iodophenyl)amino)benzonitrile (S3)
A round-bottomed flask (250 mL) was evacuated
and refilled with argon,
charged with 2-iodoaniline (13.14 g, 60 mmol), KOtBu (13.47 g, 120 mmol), and dry DMSO (120 mL). The mixture was stirred
at room temperature for 10 min and cooled to −10 °C, and
fluorobenzonitrile was added. Then, the reaction mixture was stirred
at room temperature until the complete consumption of 2-iodoaniline
(monitored by TLC). The resulted suspension was quenced with water
(50 mL) and extracted with ethyl acetate (50 mL × 3). The combined
organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Recrystallization with
ethanol afforded 14.00 g (43.8 mmol, 73% yield) of S3 as a pale yellow solid. mp 97–98 °C; 1H NMR:
(400.0 MHz, CDCl3): δ 7.90–7.85 (m, 1H), 7.57–7.52
(m, 1H), 7.43–7.36 (m, 1H), 7.35–7.29 (m, 2H), 7.12–7.06
(m, 1H), 6.96–6.89 (m, 1H), 6.88–6.81 (m, 1H), 6.39
(br, 1H); 13C NMR: (125.5 MHz, CDCl3): δ
146.4, 141.1, 140.0, 133.8, 133.2, 129.2, 125.6, 121.4, 120.3, 117.2,
115.2, 100.0, 93.5; HRMS (ESI): calcd for [C13H10IN2]+, 320.9883 [M + H]+; found,
320.9883.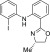
(S)-2-Iodo-N-(2-(4-methyl-4,5-dihydrooxazol-2-yl)phenyl)aniline (S4)
To a round-bottomed flask (250 mL) were
added amino-alcohol (2.42 g, 32.2 mmol), toluene (48 mL), S3 (8.60 g, 26.87 mmol), and Zn(OTf)2 (0.98 g, 2.69 mmol)
in sequence. The mixture was then heated under reflux for 4 days,
cooled to room temperature, passed through a pad of silica gel, and
washed with EA. The filtrate was concentrated under reduced pressure
and purified by flash chromatography on silica gel to give 6.30 g
(16.7 mmol, 62% yield) of S4 as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 10.59 (br, 1H),
7.93 (dd, J = 7.8, 1.4 Hz, 1H), 7.86 (dd, J = 7.8, 1.4 Hz, 1H), 7.51 (dd, J = 8.2,
1.4 Hz, 1H), 7.36–7.27 (m, 2H), 7.23 (dd, J = 8.2, 1.4 Hz, 1H), 6.88–6.78 (m, 2H), 4.58–4.46 (m,
2H), 3.99–3.89 (m, 1H), 1.47–1.41 (m, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 163.2, 144.9, 143.3,
140.0, 131.7, 129.9, 128.6, 124.4, 121.9, 117.7, 113.7, 111.3, 94.1,
72.5, 62.1, 21.5; HRMS (EI): calcd for [C16H15IN2O]+ requires m/z, 378.0229; found m/z, 378.0231.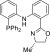
(S)-2-(Diphenylphosphanyl)-N-(2-(4-methyl-4,5-dihydrooxazol-2-yl)phenyl)aniline (PAOMe)
A round-bottomed flask (100 mL) was evacuated and refilled three times with argon, charged with CuI (0.1580 g, 0.84 mmol), diphenylphosphines (3.2 mL, 18.4 mmol), DMEDA (0.64 mL, 5.8 mmol), and anhydrous toluene (34 mL). The colorless solution was stirred for 10 min, to which were added S4 (6.30 g, 16.7 mmol) and Cs2CO3 (11.00 g, 33.4 mmol). The Schlenk tube was sealed and stirred at 110 °C for the complete consumption of S4 (monitored by TLC). The resulted suspension was cooled to room temperature, diluted with water, and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel with PE/EA to provide 4.60 g (10.5 mmol, 64% yield) of PAOMe as a white solid. mp 133–134 °C; 1H NMR: (400.0 MHz, CDCl3): δ 10.27 (br, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.52–7.46 (m, 1H), 7.35–7.25 (m, 11H), 7.16 (t, J = 7.8 Hz, 1H), 7.07 (d, J = 8.0 Hz, 1H), 7.01 (t, J = 7.2 Hz, 1H), 6.88–6.82 (m, 1H), 6.68 (t, J = 7.4 Hz, 1H), 4.29–4.23 (m, 1H), 4.05–3.94 (m, 1H), 3.76–3.70 (m, 1H), 0.97 (d, J = 6.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 163.0, 146.1, 144.9, 144.7, 137.14, 137.08, 137.05, 137.0, 134.2, 134.0, 133.93, 133.88, 133.8, 131.5, 129.6, 129.4, 128.6, 128.44, 128.42, 128.37, 124.2, 124.1, 116.8, 113.6, 110.6, 72.2, 61.8, 21.3; 31P NMR: (162 MHz, CDCl3): δ −16.4; HRMS (ESI): calcd for [C28H26N2OP]+, 437.1777 [M + H]+, found, 437.1780.
General Procedure for the Cobalt-Catalyzed Isomerization of Alkenes
To a 10 mL flame-dried Schlenk flask cooled under argon were added CoCl2 (0.0125 mmol), PAOdiMe (0.015 mmol), solvent (1 mL), and 1,1-disubstituted alkene (0.50 mmol) in sequence. The mixture was stirred at room temperature for 5 min, to which was added NaBHEt3 (38 μL, 0.038 mmol) dropwise. After stirring at room temperature for 30 min, the conversion of the starting material was determined by 1H NMR analysis using mesitylene as an internal standard and purified by flash column chromatography to give the isomerization product.
(E)-1-(But-2-en-2-yl)-4-methoxybenzene (2a)46
Prepared according to the general procedure using 0.0813 g (0.50 mmol) of 1a, 0.0016 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (96%) of 1a was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0803 g (0.49 mmol, 99% yield, E/Z = 100/1) of 2a as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.33–7.28 (m, 2H), 6.87–6.82 (m, 2H), 5.78 (qq, J = 6.8, 1.4 Hz, 1H), 3.80 (s, 3H), 2.02–1.98 (m, 3H), 1.78 (dq, J = 6.8, 1.0 Hz, 3H).
(E)-But-2-en-2-ylbenzene (2b)22
Prepared according to the general procedure using 0.0656 g (0.50 mmol) of 1b, 0.0017 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (97%) of 1b was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0573 g (0.43 mmol, 87% yield, E/Z = 100/2) of 2b as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.39–7.34 (m, 2H), 7.33–7.27 (m, 2H), 7.23–7.18 (m, 1H), 5.86 (qq, J = 6.8, 1.4 Hz, 1H), 2.05–2.00 (m, 3H), 1.80 (dq, J = 6.8, 1.0 Hz, 3H).
(E)-1-(But-2-en-2-yl)-4-methylbenzene (2c)22
Prepared according to the general procedure using 0.0748 g (0.51 mmol) of 1c, 0.0018 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (96%) of 1c was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0696 g (0.48 mmol, 93% yield, E/Z = 100/1) of 2c as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.27 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 5.83 (qq, J = 7.0, 1.2 Hz, 1H), 2.33 (s, 3H), 2.03–1.99 (m, 3H), 1.78 (dq, J = 7.0, 1.0 Hz, 3H).
(E)-4-(But-2-en-2-yl)-1,1′-biphenyl (2d)
Prepared according to the general procedure using 0.1054 g (0.50 mmol) of 1d, 0.0016 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (96%) of 1d was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1045 g (0.50 mmol, 99% yield, E/Z = 100/1) of 2d as a white solid. mp 99–100 °C; 1H NMR: (400.0 MHz, CDCl3): δ 7.63–7.57 (m, 2H), 7.57–7.52 (m, 2H), 7.47–7.39 (m, 4H), 7.36–7.30 (m, 1H), 5.94 (q, J = 7.0 Hz, 1H), 2.06 (s, 3H), 1.82 (d, J = 7.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 142.9, 140.9, 139.2, 135.0, 128.7, 127.1, 126.9, 126.8, 125.8, 122.6, 15.4, 14.4; HRMS (ESI): calcd for [C16H17]+, 209.1325 [M + H]+; found, 209.1318.
(E)-1-(But-2-en-2-yl)-4-fluorobenzene (2e)
Prepared according to the general procedure using 0.0735 g (0.49 mmol) of 1e, 0.0016 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (97%) of 1e was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0660 g (0.44 mmol, 90% yield, E/Z = 100/1) of 2e as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.34–7.28 (m, 2H), 7.01–6.94 (m, 2H), 5.79 (q, J = 6.8 Hz, 1H), 2.02–1.98 (m, 3H), 1.78 (dq, J = 6.8, 1.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 161.7 (d, J = 304.1 Hz), 140.1, 134.5, 126.9 (d, J = 9.4 Hz), 122.3, 114.8 (d, J = 26.6 Hz), 15.6, 14.3; 19F NMR: (470 MHz, CDCl3): δ −117.0; HRMS (EI): calcd for [C10H11F]+ requires m/z, 150.0845; found m/z, 150.0846.
(E)-1-(But-2-en-2-yl)-4-chlorobenzene (2f)8
Prepared according to the general procedure using 0.0838 g (0.50 mmol) of 1f, 0.0016 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (97%) of 1f was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0789 g (0.47 mmol, 94% yield, E/Z = 100/1) of 2f as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.31–7.23 (m, 4H), 5.85 (q, J = 6.8 Hz, 1H), 2.00 (s, 3H), 1.79 (d, J = 6.8 Hz, 3H).
(E)-1-Bromo-4-(but-2-en-2-yl)benzene (2g)47
Prepared according to the general procedure using 0.1077 g (0.50 mmol) of 1g, 0.0017 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 30 min, the conversion (93%) of 1g was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1042 g (0.49 mmol, 97% yield, E/Z = 100/1) of 2g as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.44–7.37 (m, 2H), 7.27–7.19 (m, 2H), 5.85 (q, J = 7.0 Hz, 1H), 1.99 (s, 3H), 1.78 (d, J = 7.0 Hz, 3H).
(E)-4-(But-2-en-2-yl)-N,N-dimethylaniline (2h)
Prepared according to the general procedure using 0.0846 g (0.50 mmol) of 1h (26% of 2h), 0.0018 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 15 min, the conversion (95%) of 1h was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0782 g (0.46 mmol, 92% yield, E/Z = 100/3) of 2h as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.30–7.26 (m, 2H), 6.72–6.67 (m, 2H), 5.75 (qq, J = 6.8, 1.2 Hz, 1H), 2.93 (s, 6H), 2.01–1.98 (m, 3H), 1.77 (dq, J = 6.8, 1.2 Hz, 3H); 13C NMR: (125.8 MHz, CDCl3): δ 149.4, 135.0, 132.5, 126.1, 119.1, 112.5, 40.6, 15.3, 14.2; HRMS (EI): calcd for [C12H17N]+ requires m/z, 175.1361; found m/z, 175.1360.
(E)-(4-(But-2-en-2-yl)phenoxy)(tert-butyl)dimethylsilane (2i)
Prepared according to the general procedure using 0.1316 g (0.50 mmol) of 1i, 0.0019 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 30 min, the conversion (95%) of 1i was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1309 g (0.50 mmol, 99% yield, E/Z = 100/2) of 2i as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.25–7.20 (m, 2H), 6.80–6.74 (m, 2H), 5.78 (q, J = 6.8 Hz, 1H), 1.99 (s, 3H), 1.77 (d, J = 6.8 Hz, 3H), 0.98 (s, 9H), 0.19 (s, 6H); 13C NMR: (125.5 MHz, CDCl3): δ 154.3, 137.1, 134.9, 126.3, 120.8, 119.6, 25.7, 18.2, 15.4, 14.2, −4.4. HRMS (ESI): calcd for [C16H26OSi], 263.1826 [M + H]+; found, 263.1825.
(E)-(4-(But-2-en-2-yl)phenyl)(methyl)sulfane (2j)
Prepared according to the general procedure using 0.0909 g (0.51 mmol) of 1j, 0.0019 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (97%) of 1j was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0858 g (0.48 mmol, 94% yield, E/Z = 100/2) of 2j as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.32–7.27 (m, 2H), 7.23–7.18 (m, 2H), 5.85 (q, J = 6.8 Hz, 1H), 2.47 (s, 3H), 2.00 (s, 3H), 1.79 (d, J = 6.8 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 140.9, 136.0, 134.7, 126.6, 125.8, 122.0, 16.0, 15.2, 14.3; HRMS (ESI): calcd for [C11H14S], 179.0889 [M + H]+; found, 179.0883.
(E)-4-(But-2-en-2-yl)phenyl Acetate (2k)
Prepared according to the general procedure using 0.0966 g (0.50 mmol) of 1k, 0.0032 g (0.025 mmol) of CoCl2, 0.0131 g (0.030 mmol) of PAOMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 h, the conversion (90%) of 1k was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0824 g (0.43 mmol, 85% yield, E/Z = 100/1) of 2k as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.40–7.32 (m, 2H), 7.28–7.23 (m, 2H), 5.89–5.78 (m, 1H), 2.29 (s, 3H), 2.01 (s, 3H), 1.83–1.75 (m, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 169.5, 149.1, 141.7, 134.6, 126.4, 122.6, 121.0, 21.0, 15.4, 14.2; HRMS (ESI): calcd for [C12H14O2]: 191.1067 [M + H]+; found, 191.1060.
(E)-1-(But-2-en-2-yl)-4-(trifluoromethyl)benzene (2l)
Prepared according to the general procedure using 0.1029 g (0.51 mmol) of 1l, 0.0017 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (98%) of 1l was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0914 g (0.46 mmol, 89% yield, E/Z = 100/1) of 2l as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.54 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.2 Hz, 2H), 5.94 (q, J = 7.0 Hz, 1H), 2.06–2.00 (m, 3H), 1.82 (dq, J = 7.0, 1.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 147.6, 134.6, 128.5 (q, J = 31.6 Hz), 125.8, 125.1 (q, J = 3.6 Hz), 124.7, 124.4 (q, J = 270.0 Hz), 15.3, 14.3; 19F NMR: (470 MHz, CDCl3): δ −62.4; HRMS (EI): calcd for [C11H11F3]+ requires m/z, 200.0813; found m/z, 200.0813.
(E)-1-(But-2-en-2-yl)-3-methoxybenzene (2m)
Prepared according to the general procedure using 0.0819 g (0.50 mmol) of 1m, 0.0018 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (97%) of 1m was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0754 g (0.46 mmol, 92% yield, E/Z = 100/1) of 2m as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.22 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 7.8 Hz, 1H), 6.93–6.89 (m, 1H), 6.79–6.74 (m, 1H), 5.87 (q, J = 6.8 Hz, 1H), 3.81 (s, 3H), 2.03–2.00 (m, 3H), 1.79 (d, J = 6.8 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 159.4, 145.6, 135.4, 129.0, 122.6, 118.1, 111.6, 111.4, 55.1, 15.5, 14.2; HRMS (EI): calcd for [C11H14O]+ requires m/z, 162.1045; found m/z, 162.1043.
(E)-1-(But-2-en-2-yl)-3-chlorobenzene (2n)22
Prepared according to the general procedure using 0.0847 g (0.51 mmol) of 1n, 0.0017 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (96%) of 1n was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0844 g (0.51 mmol, 99% yield, E/Z = 98/2) of 2n as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.36–7.32 (m, 1H), 7.24–7.15 (m, 3H), 5.88 (q, J = 6.8 Hz, 1H), 2.00 (s, 3H), 1.80 (dq, J = 6.8, 1.0 Hz, 3H).
(E)-1-(But-2-en-2-yl)-2-fluorobenzene (2o)
Prepared according to the general procedure using 0.0751 g (0.50 mmol) of 1o, 0.0017 g (0.0125 mmol) of CoCl2, 0.0066 g (0.015 mmol) of PAOMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 2 h, the conversion (96%) of 1o was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0679 g (0.45 mmol, 90% yield, E/Z = 100/0) of 2o as a colorless oil. H NMR: (400.0 MHz, CDCl3): δ 7.23–7.14 (m, 2H), 7.10–6.95 (m, 2H), 5.67 (q, J = 7.0 Hz, 1H), 2.03–1.98 (m, 3H), 1.78 (dq, J = 7.0, 1.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 159.9 (d, J = 244.8 Hz), 132.9 (d, J = 14.5 Hz), 132.1, 129.7 (d, J = 4.5 Hz), 127.9 (d, J = 8.1 Hz), 125.7 (d, J = 1.75 Hz), 123.8 (d, J = 3.6 Hz), 115.6 (d, J = 22.5 Hz), 16.6, 14.0; 19F NMR: (470 MHz, CDCl3): δ −115.7; HRMS (EI): calcd for [C10H11F]+ requires m/z, 150.0845; found m/z, 150.0846.
(E)-4-(But-2-en-2-yl)-1,2-dimethoxybenzene (2p)
Prepared according to the general procedure using 0.0990 g (0.51 mmol) of 1p, 0.0019 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (93%) of 1p was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0942 g (0.49 mmol, 95% yield, E/Z = 100/1) of 2p as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.95–6.89 (m, 2H), 6.84–6.79 (m, 1H), 5.80 (q, J = 6.9 Hz, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 2.01 (s, 3H), 1.79 (d, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 148.6, 147.9, 137.1, 135.0, 121.0, 117.7, 111.0, 109.2, 55.8, 55.7, 15.5, 14.1; HRMS (EI): calcd for [C12H16O2]+ requires m/z, 192.1150; found m/z, 192.1149.
(E)-5-(But-2-en-2-yl)-1,2,3-trimethoxybenzene (2q)
Prepared according to the general procedure using 0.1099 g (0.50 mmol) of 1q, 0.0019 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 15 min, the conversion (97%) of 1q was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1064 g (0.48 mmol, 97% yield, E/Z = 100/1) of 2q as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.58 (s, 2H), 5.81 (q, J = 7.0 Hz, 1H), 3.88 (s, 6H), 3.84 (s, 3H), 2.02 (s, 3H), 1.79 (d, J = 7.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 152.8, 140.1, 135.5, 126.8, 122.2, 103.1, 60.8, 56.0, 15.7, 14.2; HRMS (EI): calcd for [C13H18O3]+ requires m/z, 222.1256, found m/z, 222.1258.
(E)-5-(Pent-2-en-2-yl)benzo[d][1,3]dioxole (2r)
Prepared according to the general procedure using 0.0954 g (0.50 mmol) of 1r, 0.0017 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 3 min, the conversion (92%) of 1r was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0915 g (0.93 mmol, 93% yield, E/Z = 100/2) of 2r as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.91–6.88 (m, 1H), 6.87–6.82 (m, 1H), 6.77–6.72 (m, 1H), 5.93 (s, 2H), 5.67 (tq, J = 7.4, 1.2 Hz, 2H), 2.23–2.13 (m, 2H), 2.00–1.96 (m, 3H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 147.5, 146.1, 138.4, 133.5, 129.2, 118.8, 107.8, 106.2, 100.8, 21.9, 15.8, 14.0; HRMS (ESI): calcd for [C12H14O2], 191.1067 [M + H]+; found, 191.1070.
(E)-Pent-2-en-2-ylbenzene (2s)48
Prepared according to the general procedure using 0.0745 g (0.50 mmol) of 1s 0.0019 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (94%) of 1s was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0664 g (0.45 mmol, 89% yield, E/Z = 100/3) of 2s as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.41–7.35 (m, 2H), 7.33–7.26 (m, 2H), 7.23–7.16 (m, 1H), 5.77 (tq, J = 7.2, 1.4 Hz, 1H), 7.26–7.15 (m, 2H), 2.05–2.00 (m, 3H), 1.06 (t, J = 7.2 Hz, 3H).
(E)-Hex-2-en-2-ylbenzene (2t)49
Prepared according to the general procedure using 0.0808 g (0.50 mmol) of 1t, 0.0019 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 10 min, the conversion (96%) of 1t was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0781 g (0.49 mmol, 97% yield, E/Z = 100/1) of 2t as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.41–7.35 (m, 2H), 7.33–7.26 (m, 2H), 7.23–7.17 (m, 1H), 5.79 (tq, J = 7.2, 1.2 Hz, 1H), 2.18 (q, J = 7.2 Hz, 2H), 2.03 (s, 3H), 1.53–1.42 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H).
(E)-Hept-2-en-2-ylbenzene (2u)50
Prepared according to the general procedure using 0.0873 g (0.50 mmol) of 1u, 0.0019 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (93%) of 1u was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0855 g (0.49 mmol, 98% yield, E/Z = 100/1) of 2u as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.41–7.35 (m, 2H), 7.30 (t, J = 7.2 Hz, 2H), 7.21 (t, J = 7.2 Hz, 1H), 5.78 (tq, J = 7.4, 1.4 Hz, 1H), 2.20 (q, J = 7.4 Hz, 2H), 2.03 (s, 3H), 1.49–1.30 (m, 4H), 0.93 (t, J = 7.2 Hz, 3H).
Ethyl (E)-4-(4-Fluorophenyl)pent-3-enoate (2v)
Prepared according to the general procedure using 0.1204 g (0.54 mmol) of 1v, 0.0016 g (0.0125 mmol) of CoCl2, 0.0065 g (0.015 mmol) of PAOMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 2 h, the conversion (92%) of 1v was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1039 g (0.47 mmol, 86% yield, E/Z = 100/3) of 2v as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.35–7.28 (m, 2H), 7.02–6.94 (m, 2H), 5.67 (t, J = 7.2 Hz, 1H), 4.14 (q, J = 7.2 Hz, 2H), 2.56–2.40 (m, 4H), 2.03 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 173.1, 161.8 (d, J = 304.8 Hz), 139.6 (d, J = 4.0 Hz), 135.1, 127.1 (d, J = 10.1 Hz), 125.9, 114.8 (d, J = 26.6 Hz), 60.3, 34.1, 24.2, 15.9, 14.2; 19F NMR: (470 MHz, CDCl3): δ −116.4; HRMS (ESI): calcd for [C14H17FO2], 237.1285 [M + H]+; found, 237.1288.
(E)-4-(4-Fluorophenyl)pent-3-en-1-ol (2w)
Prepared according to the general procedure using 0.0958 g (0.53 mmol) of 1w, 0.0018 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 h, the conversion (95%) of 1w was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0932 g (0.52 mmol, 97% yield, E/Z = 100/2) of 2w as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.41–7.34 (m, 2H), 7.07–7.00 (m, 2H), 5.77 (t, 1H), 3.80–3.70 (m, 2H), 2.34 (q, J = 7.5 Hz, 2H), 2.07 (s, 3H), 1.83–1.73 (m, 2H), 1.35 (s, 1H); 13C NMR: (125.5 MHz, CDCl3): δ 161.8 (d, J = 243.8 Hz), 139.9 (d, J = 3.6 Hz), 134.4, 127.5, 127.0 (d, J = 8.1 Hz), 114.8 (d, J = 20.8 Hz), 62.4, 32.5, 25.0, 15.8; 19F NMR: (470 MHz, CDCl3): δ −116.6; HRMS (EI): calcd for [C12H15FO]+ requires m/z, 194.1107; found m/z, 194.1106.
(E)-2-(But-2-en-2-yl)-6-methoxynaphthalene (2x)
Prepared according to the general procedure using 0.1066 g (0.50 mmol) of 1x, 0.0019 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (96%) of 1x was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1061 g (0.50 mmol, 99% yield, E/Z = 100/0) of 2x as a white solid, mp 87–88 °C. 1H NMR: (400.0 MHz, CDCl3): δ 7.73–7.69 (m, 2H), 7.68–7.64 (m, 1H), 7.57–7.52 (m, 1H), 7.15–7.08 (m, 2H), 5.99 (q, J = 6.8 Hz, 1H), 3.91 (s, 3H), 2.14–2.10 (m, 3H), 1.85 (dq, J = 6.8, 1.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 157.3, 139.0, 135.2, 133.3, 129.5, 128.9, 126.4, 124.7, 123.6, 122.2, 118.7, 105.5, 55.2, 15.4, 14.4; HRMS (ESI): calcd for [C15H16O], 213.1274 [M + H]+; found, 213.1292.
(E)-2-(But-2-en-2-yl)thiophene (2y)
Prepared according to the general procedure using 0.0702 g (0.50 mmol) of 1y, 0.0016 g (0.0125 mmol) of CoCl2, 0.0065 g (0.015 mmol) of PAOMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 h, the conversion (96%) of 1y was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0576 g (0.41 mmol, 82% yield, E/Z = 100/2) of 2y as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.10–7.05 (m, 1H), 6.97–6.90 (m, 2H), 6.01 (q, J = 7.0 Hz, 1H), 2.03 (s, 3H), 1.78 (d, J = 7.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 148.1, 129.7, 127.1, 122.6, 121.7, 121.5, 15.4, 13.9; HRMS (EI): calcd for [C8H10S]+ requires m/z, 138.0503; found m/z, 138.0503.
(E)-3-(But-2-en-2-yl)pyridine (2z)
Prepared according to the general procedure using 0.0663 g (0.50 mmol) of 1z, 0.0033 g (0.025 mmol) of CoCl2, 0.0133 g (0.03 mmol) of PAOdiMe, 75 μL (1 M in THF, 0.075 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 h, the conversion (98%) of 1z was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.00573 g (0.43 mmol, 86% yield, E/Z = 100/2) of 2z as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 8.65–8.60 (m, 1H), 8.47–8.40 (m, 1H), 7.66–7.61 (m, 1H), 7.24–7.19 (m, 1H), 5.90 (qq, J = 7.0, 1.4 Hz, 1H), 2.06–2.02 (m, 3H), 1.82 (dq, J = 7.0, 1.0 Hz, 3H); 13C NMR: (125.5 MHz, CDCl3): δ 147.5, 147.1, 139.2, 132.64, 132.61, 124.3, 122.9, 15.1, 14.2; HRMS (ESI): calcd for [C9H11N], 133.0891 [M + H]+; found, 134.0963.
4-Methyl-1,2-dihydronaphthalene (2aa)22
Prepared according to the general procedure using 0.0705 g (0.50 mmol) of 1aa, 0.0016 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 30 min, the conversion (>99%) of 1aa was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0683 g (0.48 mmol, 97% yield) of 2aa as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.23–7.18 (m, 2H), 7.16–7.10 (m, 2H), 5.88–5.82 (m, 1H), 2.76 (t, J = 8.3 Hz, 2H), 2.29–2.20 (m, 2H), 2.07–2.03 (m, 3H).
3-Methyl-1H-indene (2ab)22
Prepared according to the general procedure using 0.0673 g (0.52 mmol) of 1ab (30% of 2ab), 0.0018 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of toluene. After 0.5 h, the conversion (>99%) of 1ab was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0662 g (0.51 mmol, 98% yield) of 2ab as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.51–7.41 (m, 1H), 7.40–7.15 (m, 3H), 6.26–6.15 (m, 1H), 3.37–3.27 (m, 2H), 2.23–2.14 (m, 3H).
(3-Methylbut-2-en-1-yl)benzene (2ac)51
Prepared according to the general procedure using 0.0733 g (0.50 mmol) of 1ac, 0.0034 g (0.025 mmol) of CoCl2, 0.0135 g (0.03 mmol) of PAOdiMe, 75 μL (1 M in THF, 0.075 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 6 h, the conversion (96%) of 1ac was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0696 g (0.47 mmol, 95% yield) of 2ac as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.30–7.25 (m, 2H), 7.18–7.14 (m, 3H), 5.37–5.29 (m, 1H), 3.34 (d, J = 7.4 Hz, 2H), 1.74 (s, 3H), 1.72 (s, 3H).
(E)-1-Methoxy-4-(prop-1-en-1-yl)benzene (4a)21
Method 1: prepared according to the general procedure using 0.0723 g (0.50 mmol) of 3a, 0.0018 g (0.0125 mmol) of CoCl2, 0.0069 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 min, the conversion (>99%) of 3a was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0653 g (0.45 mmol, 90% yield, E/Z = 100/2) of 4a as a colorless oil.
Method 2: prepared according to the general procedure using 0.0756 g (0.50 mmol) of 3g, 0.0019 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 min, the conversion (>99%) of 3g was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0736 g (0.49 mmol, 97% yield, E/Z = 100/2) of 4a as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.28–7.22 (m, 2H), 6.86–6.80 (m, 2H), 6.34 (dq, J = 15.8, 1.4 Hz, 1H), 6.15–6.03 (m, 1H), 3.79 (s, 3H), 1.85 (dd, J = 6.6, 1.6 Hz, 3H).
(E)-1,2,3-Trimethoxy-5-(prop-1-en-1-yl)benzene (4b)21
Prepared according to the general procedure using 0.0902 g (0.50 mmol) of 3b, 0.0018 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 min, the conversion (>99%) of 3b was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0894 g (0.50 mmol, 99% yield, E/Z = 100/2) of 4b as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.56 (s, 2H), 6.33 (dq, J = 15.8, 1.6 Hz, 1H), 6.21–6.10 (m, 1H), 3.87 (s, 6H), 3.84 (s, 3H), 1.88 (dd, J = 6.6, 1.6 Hz, 3H).
(E)-1,2-Dimethoxy-4-(prop-1-en-1-yl)benzene (4c)21
Prepared according to the general procedure using 0.0902 g (0.50 mmol) of 3c, 0.0018 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 min, the conversion (>99%) of 3c was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0894 g (0.49 mmol, 99% yield, E/Z = 100/2) of 4c as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.92–6.88 (m, 1H), 6.88–6.83 (m, 1H), 6.82–6.77 (m, 1H), 6.34 (d, J = 15.6 Hz, 1H), 6.16–6.05 (m, 1H), 3.89 (s, 3H), 3.87 (s, 3H), 1.87 (dd, J = 6.8, 1.4 Hz, 3H).
(E)-tert-Butyl(2-methoxy-4-(prop-1-en-1-yl)phenoxy)dimethylsilane (4d)19
Prepared according to the general procedure using 0.1409 g (0.50 mmol) of 3d, 0.0016 g (0.0125 mmol) of CoCl2, 0.0070 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 1 min, the conversion (>99%) of 3d was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.1358 g (0.48 mmol, 96% yield, E/Z = 100/2) of 4d as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 6.90 (s, 1H), 6.86–6.79 (m, 2H), 6.38 (d, J = 15.8 Hz, 1H), 6.21–6.09 (m, 1H), 3.86 (s, 3H), 1.91 (d, J = 6.6 Hz, 3H), 1.04 (s, 9H), 0.19 (s, 6H).
(E)-But-1-en-1-ylbenzene (4e)19
Prepared according to the general procedure using 0.0723 g (0.50 mmol) of 3e, 0.0019 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 10 min, the conversion (>99%) of 3e was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0574 g (0.43 mmol, 87% yield, E/Z = 100/3) of 4e as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.37–7.32 (m, 2H), 7.31–7.26 (m, 2H), 7.21–7.15 (m, 1H), 6.38 (d, J = 15.8 Hz, 1H), 6.31–6.22 (m, 1H), 2.27–2.18 (m, 2H), 1.09 (t, J = 7.4 Hz, 3H).
(E)-Pent-1-en-1-ylbenzene (4f)19
Prepared according to the general procedure using 0.0723 g (0.50 mmol) of 3f (39/61 E/Z) (41% of 4f), 0.0016 g (0.0125 mmol) of CoCl2, 0.0068 g (0.015 mmol) of PAOdiMe, 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3, and 1 mL of 1,4-dioxane. After 10 min, the conversion (>99%) of 3f was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give 0.0662 g (0.45 mmol, 90% yield, E/Z = 100/1) of 4f as a colorless oil. 1H NMR: (400.0 MHz, CDCl3): δ 7.37–7.32 (m, 2H), 7.31–7.26 (m, 2H), 7.22–7.15 (m, 1H), 6.38 (d, J = 15.6 Hz, 1H), 6.27–6.18 (m, 1H), 2.19 (q, J = 7.2 Hz, 2H), 1.55–1.44 (m, 2H), 0.95 (t, J = 7.4 Hz, 3H).
Procedures for Gram-Scale Reaction of 1r (eq 1)
To a 10 mL flame-dried Schlenk flask cooled under argon were added 0.0021 g (0.015 mmol) of CoCl2, 0.0083 g (0.018 mmol) of PAOdiMe, toluene (1.5 mL), and 2.8566 g (15 mmol) of 1r in sequence. The mixture was stirred at room temperature for 10 min, and 45 μL (1 M in THF, 0.045 mmol) of NaBHEt3 was added. Then, the mixture was stirred at room temperature for 1 h. After stirring at room temperature for 1 h, the resulted mixture was purified by flash column chromatography using PE as the eluent to give the crude product. The remaining 1r was removed by hydroboration27 to give 2.6188 g (13.8 mmol, 92% yield, E/Z = 100/2) of 2r as a colorless oil.
Procedures for Gram-Scale Reaction of 3a (eq 2)
To a 10 mL flame-dried Schlenk flask cooled under argon were added 0.0035 g (0.025 mmol) of CoCl2, 0.0138 g (0.03 mmol) of PAOdiMe, dioxane (1 mL), and 1.4803 g (10 mmol) of 3a in sequence. The mixture was stirred at room temperature for 5 min, and 75 μL (1 M in THF, 0.075 mmol) of NaBHEt3 was added dropwise. After 10 min, the crude reaction was quenched by PE. The mixture was filtered through a pad of silica gel and washed with PE. The filtrate was concentrated to afford 1.4311 g (9.7 mmol, 97% yield, E/Z = 100/2) of 4a as a colorless oil.
Deuterium-Labeling Reactions (eq 3)
To a 10 mL flame-dried Schlenk flask cooled under argon were added 0.0806 g (0.50 mmol) of 1a, 0.0697 g of D-5, 0.0019 g (0.0125 mmol) of CoCl2, 1 mL of toluene, and 0.0068 g (0.015 mmol) of PAOdiMe in sequence. The mixture was stirred at room temperature for 5 min, and 38 μL (1 M in THF, 0.038 mmol) of NaBHEt3 was added dropwise. After 30 min, the conversion (96%) of 1a was determined by 1H NMR analysis using mesitylene as an internal standard. The crude reaction mixture was purified by flash column chromatography using PE as the eluent to give a mixture of 2a and 5 as a colorless oil. For D-2a (E/Z = 100/1): 1H NMR: (400.0 MHz, CDCl3): δ 7.37–7.28 (m, 2H), 6.89–6.81 (m, 2H), 5.78 (q, J = 6.8 Hz, 1H), 3.80 (s, 3H), 2.02–1.96 (m, 2.21H), 1.78 (d, J = 6.8 Hz, 3H); 2D NMR (400 MHz, CDCl3): δ 2.02–1.96 (m, 0.79D).
Acknowledgments
Financial support was provided by NSFC (nos. 21801191, 21572163, and 21873074), Wenzhou Science & Technology Bureau (G20180016), College Students Innovation and Entrepreneurship Training Program (no. JWSC2018079), and Laboratory open project (no. JW19SK65) of Wenzhou University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00951.
Tolerance of reducible functional groups; copies of 1H NMR spectra of all the synthesized compounds; and copies of 1H, 13C, 19F, and 31P NMR spectra of all the new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Bhatt U.; Christmann M.; Quitschalle M.; Claus E.; Kalesse M. The First Total Synthesis of (+)-Ratjadone. J. Org. Chem. 2001, 66, 1885–1893. 10.1021/jo005768g. [DOI] [PubMed] [Google Scholar]; b Williams D. R.; Ihle D. C.; Plummer S. V. Total Synthesis of (-)-Ratjadone. Org. Lett. 2001, 3, 1383–1386. 10.1021/ol015753k. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Shang G.; Li W.; Zhang X.. Catalytic Asymmetric Synthesis; Ojima I., Ed.; Wiley: Weinheim, 2010; pp 344–436. [Google Scholar]; b Alexakis A.; Krause N.; Woodward S.. Copper-Catalyzed Asymmetric Synthesis; Alexakis A., Krause N., Woodward S., Eds.; Wiley: Weinheim, 2014; pp 33–68. [Google Scholar]; c Baslé O.; Denicourt-Nowicki A.; Crévisy C.; Mauduit M.. Copper-Catalyzed Asymmetric Synthesis; Alexakis A., Krause N., Woodward S., Eds.; Wiley: Weinheim, 2014; pp 85–119. [Google Scholar]; d Zhu Y.; Wang Q.; Cornwall R. G.; Shi Y. Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem. Rev. 2014, 114, 8199–8256. 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Vedejs E.; Peterson M. J. Stereochemistry and Mechanism in the Wittig Reaction. Top. Stereochem. 1994, 21, 1–157. 10.1002/9780470147306.ch1. [DOI] [Google Scholar]; b Maryanoff B. E.; Reitz A. B. The Wittig Olefination Reaction and Modifications Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and Selected Synthetic Aspects. Chem. Rev. 1989, 89, 863–927. 10.1021/cr00094a007. [DOI] [Google Scholar]
- For selected reviews, see:; a Reiser O. Palladium-Catalyzed Coupling Reactions for the Stereoselective Synthesis of Tri- and Tetrasubstituted Alkenes. Angew. Chem., Int. Ed. 2006, 45, 2838–2840. 10.1002/anie.200600025. [DOI] [PubMed] [Google Scholar]; b Negishi E.-I.; Huang Z.; Wang G.; Mohan S.; Wang C.; Hattori H. Recent Advances in Efficient and Selective Synthesis of Di-, Tri-, and Tetrasubstituted Alkenes via Pd-Catalyzed Alkenylation-Carbonyl Olefination Synergy. Acc. Chem. Res. 2008, 41, 1474–1485. 10.1021/ar800038e. [DOI] [PubMed] [Google Scholar]; c Negishi E.-i.; Wang G.; Rao H.; Xu Z. Alkyne Elementometalation-Pd-Catalyzed Cross-Coupling. Toward Synthesis of All Conceivable Types of Acyclic Alkenes in High Yields, Efficiently, Selectively, Economically, and Safely: “Green” Way. J. Org. Chem. 2010, 75, 3151–3182. 10.1021/jo1003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews, see:; a Handbook of Metathesis; Grubbs R. H., Wenzel A. G., O’Leary D. J., Khosravi E., Eds.; Wiley: Weinheim, 2015. [Google Scholar]; b Hoveyda A. H.; Zhugralin A. R. The remarkable metal-catalysed olefin metathesis reaction. Nature 2007, 450, 243–251. 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]; c Hoveyda A. H. Evolution of Catalytic Stereoselective Olefin Metathesis: From Ancillary Transformation to Purveyor of Stereochemical Identity. J. Org. Chem. 2014, 79, 4763–4792. 10.1021/jo500467z. [DOI] [PMC free article] [PubMed] [Google Scholar]; . For selected examples, see:; d Chatterjee A. K.; Grubbs R. H. Synthesis of Trisubstituted Alkenes via Olefin Cross-Metathesis. Org. Lett. 1999, 1, 1751–1753. 10.1021/ol991023p. [DOI] [PubMed] [Google Scholar]; e Chatterjee A. K.; Sanders D. P.; Grubbs R. H. Synthesis of Symmetrical Trisubstituted Olefins by Cross Metathesis. Org. Lett. 2002, 4, 1939–1942. 10.1021/ol0259793. [DOI] [PubMed] [Google Scholar]; f Morrill C.; Funk T. W.; Grubbs R. H. Synthesis of tri-substituted vinyl boronates via ruthenium-catalyzed olefin cross-metathesis. Tetrahedron Lett. 2004, 45, 7733–7736. 10.1016/j.tetlet.2004.08.069. [DOI] [Google Scholar]; g Koh M. J.; Nguyen T. T.; Zhang H.; Schrock R. R.; Hoveyda A. H. Direct synthesis of Z-alkenyl halides through catalytic cross-metathesis. Nature 2016, 531, 459–465. 10.1038/nature17396. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Nguyen T. T.; Koh M. J.; Shen X.; Romiti F.; Schrock R. R.; Hoveyda A. H. Kinetically controlled E-selective catalytic olefin metathesis. Science 2016, 352, 569–575. 10.1126/science.aaf4622. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Nguyen T. T.; Koh M. J.; Mann T. J.; Schrock R. R.; Hoveyda A. H. Synthesis of E- and Z-trisubstituted alkenes by catalytic cross-metathesis. Nature 2017, 552, 347–354. 10.1038/nature25002. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Mu Y.; Nguyen T. T.; Koh M. J.; Schrock R. R.; Hoveyda A. H. E- and Z-, di- and tri-substituted alkenyl nitriles through catalytic cross-metathesis. Nat. Chem. 2019, 11, 478–487. 10.1038/s41557-019-0233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissen M.; Lenoir D. Mass Efficiency of Alkene Syntheses with Tri- and Tetrasubstituted Double Bonds. ACS Sustainable Chem. Eng. 2017, 5, 10459–10473. 10.1021/acssuschemeng.7b02479. [DOI] [Google Scholar]
- For selected reviews, see:; a Hilt G. Double Bond Isomerisation and Migration-New Playgrounds for Transition Metal-Catalysis. ChemCatChem 2014, 6, 2484–2485. 10.1002/cctc.201402341. [DOI] [Google Scholar]; b Larionov E.; Li H.; Mazet C. Well-defined transition metal hydrides in catalytic isomerizations. Chem. Commun. 2014, 50, 9816–9826. 10.1039/c4cc02399d. [DOI] [PubMed] [Google Scholar]; c Hassam M.; Taher A.; Arnott G. E.; Green I. R.; van Otterlo W. A. L. Isomerization of Allylbenzenes. Chem. Rev. 2015, 115, 5462–5569. 10.1021/acs.chemrev.5b00052. [DOI] [PubMed] [Google Scholar]; d Vasseur A.; Bruffaerts J.; Marek I. Remote Functionalization through Alkene Isomerization. Nat. Chem. 2016, 8, 209–219. 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]; e Molloy J. J.; Morack T.; Gilmour R. Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy. Angew. Chem., Int. Ed. 2019, 58, 13654–13664. 10.1002/anie.201906124. [DOI] [PubMed] [Google Scholar]
- Lim H. J.; Smith C. R.; RajanBabu T. V. Facile Pd(II)- and Ni(II)-Catalyzed Isomerization of Terminal Alkenes into 2-Alkenes. J. Org. Chem. 2009, 74, 4565–4572. 10.1021/jo900180p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen C. R.; Grotjahn D. B. Stereoselective Alkene Isomerization over One Position. J. Am. Chem. Soc. 2012, 134, 10357–10360. 10.1021/ja3036477. [DOI] [PubMed] [Google Scholar]
- Huang R.-Z.; Lau K. K.; Li Z.; Liu T.-L.; Zhao Y. Rhodium-Catalyzed Enantioconvergent Isomerization of Homoallylic and Bishomoallylic Secondary Alcohols. J. Am. Chem. Soc. 2018, 140, 14647–14654. 10.1021/jacs.8b07007. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Bullck M.Catalysis without Precious Metals; Wiley-VCH: Weinheim, 2011. [Google Scholar]; b Chirik P.; Morris R. Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res. 2015, 48, 2495. 10.1021/acs.accounts.5b00385. [DOI] [PubMed] [Google Scholar]; c Chen J.; Lu Z. Asymmetric hydrofunctionalization of minimally functionalized alkenes via earth abundant transition metal catalysis. Org. Chem. Front. 2018, 5, 260–272. 10.1039/c7qo00613f. [DOI] [Google Scholar]; d Chen J.; Guo J.; Lu Z. Recent Advances in Hydrometallation of Alkenes and Alkynes via the First Row Transition Metal Catalysis. Chin. J. Chem. 2018, 36, 1075–1109. 10.1002/cjoc.201800314. [DOI] [Google Scholar]; e Wen H.; Liu G.; Huang Z. Recent advances in tridentate iron and cobalt complexes for alkene and alkyne hydrofunctionalizations. Coord. Chem. Rev. 2019, 386, 138–153. 10.1016/j.ccr.2019.01.024. [DOI] [Google Scholar]; f Ai W.; Zhong R.; Liu X.; Liu Q. Hydride Transfer Reactions Catalyzed by Cobalt Complexes. Chem. Rev. 2019, 119, 2876–2953. 10.1021/acs.chemrev.8b00404. [DOI] [PubMed] [Google Scholar]; . For selected examples, see:; g Baccalini A.; Vergura S.; Dolui P.; Maiti S.; Dutta S.; Maity S.; Khan F. F.; Lahiri G. K.; Zanoni G.; Maiti D. Cobalt-Catalyzed C(sp2)–H Allylation of Biphenyl Amines with Unbiased Terminal Olefins. Org. Lett. 2019, 21, 8842–8846. 10.1021/acs.orglett.9b03484. [DOI] [PubMed] [Google Scholar]; h Maity S.; Dolui P.; Kancherla R.; Maiti D. Introducing unactivated acyclic internal aliphatic olefins into a cobalt catalyzed allylic selective dehydrogenative Heck reaction. Chem. Sci. 2017, 8, 5181–5185. 10.1039/c7sc01204g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Liu X.; Li B. Base-Metal-Catalyzed Olefin Isomerization Reactions. Synthesis 2019, 51, 1293–1310. 10.1055/s-0037-1612014. [DOI] [Google Scholar]
- Kobayashi T.; Yorimitsu H.; Oshima K. Cobalt-Catalyzed Isomerization of 1-Alkenes to (E)-2-Alkenes with Dimethylphenylsilylmethylmagnesium Chloride and Its Application to the Stereoselective Synthesis of (E)-Alkenylsilanes. Chem.—Asian J. 2009, 4, 1078–1083. 10.1002/asia.200900111. [DOI] [PubMed] [Google Scholar]
- Chen C.; Dugan T. R.; Brennessel W. W.; Weix D. J.; Holland P. L. Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes. J. Am. Chem. Soc. 2014, 136, 945–955. 10.1021/ja408238n. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Nödling A. R.; Hilt G. An Alternative Mechanism for the Cobalt-Catalyzed Isomerization of Terminal Alkenes to (Z)-2-Alkenes. Angew. Chem., Int. Ed. 2015, 54, 801–804. 10.1002/anie.201409902. [DOI] [PubMed] [Google Scholar]
- Crossley S. W. M.; Barabé F.; Shenvi R. A. Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc. 2014, 136, 16788–16791. 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Kuo J. L.; Han A.; Abuyuan J. M.; Young L. C.; Norton J. R.; Palmer J. H. Radical Isomerization and Cycloisomerization Initiated by H• Transfer. J. Am. Chem. Soc. 2016, 138, 7698–7704. 10.1021/jacs.6b03509. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zhang W.; Wang Y.; Zhang Z.-X.; Jiao L.; Liu Q. Cobalt-Catalyzed Regioselective Olefin Isomerization Under Kinetic Control. J. Am. Chem. Soc. 2018, 140, 6873–6882. 10.1021/jacs.8b01815. [DOI] [PubMed] [Google Scholar]
- Meng Q. Y.; Schirmer T. E.; Katou K.; König B. Controllable Isomerization of Alkenes by Dual Visible-Light-Cobalt Catalysis. Angew. Chem., Int. Ed. 2019, 58, 5723–5728. 10.1002/anie.201900849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapat A.; Sperger T.; Guven S.; Schoenebeck F. E-Olefins through intramolecular radical relocation. Science 2019, 363, 391–396. 10.1126/science.aav1610. [DOI] [PubMed] [Google Scholar]
- Liu H.; Xu M.; Cai C.; Chen J.; Gu Y.; Xia Y. Cobalt-Catalyzed Z to E Isomerization of Alkenes: An Approach to (E)-β-Substituted Styrenes. Org. Lett. 2020, 22, 1193–1198. 10.1021/acs.orglett.0c00072. [DOI] [PubMed] [Google Scholar]
- During the time we were submitting the manuscript on our independent work, a similar work was reported byZhao J.; Cheng B.; Chen C.; Lu Z. Cobalt-Catalyzed Migrational Isomerization of Styrenes. Org. Lett. 2020, 22, 837–841. 10.1021/acs.orglett.9b04305. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Dewick P. M.Medicinal Natural Products-A Biosynthetic Approach, 3rd ed.; Wiley: Chippenham, 2009; pp 156–159. [Google Scholar]; b Petersen M.; Hans J.; Matern U.. Annual Plant Reviews, 2nd ed.; Wink M., Ed.; Wiley-Blackwell: Oxford, 2010; Vol. 40; pp 182–257. [Google Scholar]; c Boulogne I.; Petit P.; Ozier-Lafontaine H.; Desfontaines L.; Loranger-Merciris G. Insecticidal and antifungal chemicals produced by plants: a review. Environ. Chem. Lett. 2012, 10, 325–347. 10.1007/s10311-012-0359-1. [DOI] [Google Scholar]
- For selected reviews, see:; a Korkina L. G. Phenylpropanoids as Naturally Occurring Antioxidants: From Plant Defense to Human Health. Cell. Mol. Biol. 2007, 53, 15–25. [PubMed] [Google Scholar]; b Korkina L.; Kostyuk V.; De Luca C.; Pastore S. Plant Phenylpropanoids as Emerging Anti-Inflammatory Agents. Mini-Rev. Med. Chem. 2011, 11, 823–835. 10.2174/138955711796575489. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Popławski J.; Lozowicka B.; Dubis A.; Lachowska B.; Winiecki Z.; Nawrot J. Feeding-Deterrent Activity of α-Asarone Isomers against Some Stored Coleoptera. Pest Manage. Sci. 2000, 56, 560–564. . [DOI] [Google Scholar]; b Tewary D. K.; Bhardwaj A.; Sharma A.; Sinha A. K.; Shanker A. Bioactivity and Structure-Activity Relationship of Natural Methoxylated Phenylpropenes and Their Derivatives against Aphis craccivora Koch (Hemiptera: Aphididae). J. Pestic. Sci. 2006, 79, 209–214. 10.1007/s10340-006-0135-8. [DOI] [Google Scholar]; c Bhardwaj A.; Kumar Tewary D.; Kumar R.; Kumar V.; Kumar Sinha A.; Shanker A. Larvicidal and Structure-Activity Studies of Natural Phenylpropanoids and Their Semisynthetic Derivatives against the Tobacco Armyworm Spodoptera litura (Fab.) (Lepidoptera: Noctuidae). Chem. Biodiversity 2010, 7, 168–177. 10.1002/cbdv.200800345. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Buchbauer G.Handbook of Essential oils. Science, Technology and Applications; Baser K. H. C., Buchbauer G., Eds.; CRC Press: London, 2010; pp 235–280. [Google Scholar]; b Chapuis C.; Jacoby D. Catalysis in the Preparation of Fragrances and Flavours. Appl. Catal., A 2001, 221, 93–117. 10.1016/s0926-860x(01)00798-0. [DOI] [Google Scholar]
- The remained starting material was removed by hydroboration according to a previously reported procedure:Chen J.; Xi T.; Ren X.; Cheng B.; Guo J.; Lu Z. Asymmetric cobalt catalysts for hydroboration of 1,1-disubstituted alkenes. Org. Chem. Front. 2014, 1, 1306–1309. 10.1039/c4qo00295d. [DOI] [Google Scholar]
- Liang L.-C.; Chien P.-S.; Lin J.-M.; Huang M.-H.; Huang Y.-L.; Liao J.-H. Amido Pincer Complexes of Nickel(II): Synthesis, Structure, and Reactivity. Organometallics 2006, 25, 1399–1411. 10.1021/om050943a. [DOI] [Google Scholar]
- Zhu X.; Li W.; Luo X.; Deng G.; Liang Y.; Liu J. A catalyst-free and additive-free method for the synthesis of benzothiazolethiones from o-iodoanilines, DMSO and potassium sulfide. Green Chem. 2018, 20, 1970–1974. 10.1039/c8gc00477c. [DOI] [Google Scholar]
- Ikawa T.; Hattori K.; Sajiki H.; Hirota K. Solvent-modulated Pd/C-catalyzed deprotection of silyl ethers and chemoselective hydrogenation. Tetrahedron 2004, 60, 6901–6911. 10.1016/j.tet.2004.05.098. [DOI] [Google Scholar]
- Noji M.; Ohno T.; Fuji K.; Futaba N.; Tajima H.; Ishii K. Secondary Benzylation Using Benzyl Alcohols Catalyzed by Lanthanoid, Scandium, and Hafnium Triflate. J. Org. Chem. 2003, 68, 9340–9347. 10.1021/jo034255h. [DOI] [PubMed] [Google Scholar]
- Cosner C. C.; Cabrera P. J.; Byrd K. M.; Thomas A. M. A.; Helquist P. Selective Oxidation of Benzylic and Allylic Alcohols Using Mn(OAc)3/Catalytic 2,3-Dichloro-5,6-dicyano-1,4-benzoquinon. Org. Lett. 2011, 13, 2071–2073. 10.1021/ol200441g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamilselvan P.; Basavaraju Y. B.; Murugesan R.; Sampathkumar E. Cobalt(II) acetylacetonate catalyzed Friedel-Crafts acylation of anisole, thioanisole, and toluene. Catal. Commun. 2008, 10, 300–303. 10.1016/j.catcom.2008.09.025. [DOI] [Google Scholar]
- Park C. H.; Kim K. H.; Lee I. K.; Lee S. Y.; Choi S. U.; Lee J. H.; Lee K. R. Phenolic Constituents of Acorus gramineus. Arch. Pharmacal Res. 2011, 34, 1289–1296. 10.1007/s12272-011-0808-6. [DOI] [PubMed] [Google Scholar]
- Liu M.; Hyder Z.; Sun Y.; Tang W.; Xu L.; Xiao J. Efficient synthesis of alkyl aryl ketones & ketals via palladium-catalyzed regioselective arylation of vinyl ethers. Org. Biomol. Chem. 2010, 8, 2012–2015. 10.1039/c001004a. [DOI] [PubMed] [Google Scholar]
- Ito M.; Kitahara S.; Ikariya T. Cp*Ru(PN) Complex-Catalyzed Isomerization of Allylic Alcohols and Its Application to the Asymmetric Synthesis of Muscone. J. Am. Chem. Soc. 2005, 127, 6172–6173. 10.1021/ja050770g. [DOI] [PubMed] [Google Scholar]
- Chen J.; Chen C.; Ji C.; Lu Z. Cobalt-Catalyzed Asymmetric Hydrogenation of 1,1-Diarylethenes. Org. Lett. 2016, 18, 1594–1597. 10.1021/acs.orglett.6b00453. [DOI] [PubMed] [Google Scholar]
- Eisch J. J.; Adeosun A. A. Novel Alkylidenating Agents via the α Lithiation of Monoalkyl Group 4 Metal Derivatives: Methylidene-Metal Complexes and Their Active Lithiated Precursors. Eur. J. Org. Chem. 2005, 993–997. 10.1002/ejoc.200400808. [DOI] [Google Scholar]
- Zheng H.-X.; Qu J.-P.; Kang Y.-B. One-pot one-base conditions for two shots: organocatalyzed tandem isomerization-olefination of allylic alcohols. Org. Chem. Front. 2018, 5, 2349–2352. 10.1039/c8qo00505b. [DOI] [Google Scholar]
- McIntyre S.; Hörmann E.; Menges F.; Smidt S. P.; Pfaltz A. Iridium-Catalyzed Enantioselective Hydrogenation of Terminal Alkenes. Adv. Synth. Catal. 2005, 347, 282–288. 10.1002/adsc.200404256. [DOI] [Google Scholar]
- Zeng H.; Hua R. Palladium-Catalyzed Hydrophenylation of Alkynes with Sodium Tetraphenylborate under Mild Conditions. J. Org. Chem. 2008, 73, 558–562. 10.1021/jo7020554. [DOI] [PubMed] [Google Scholar]
- Phan D. H. T.; Kou K. G. M.; Dong V. M. Enantioselective Desymmetrization of Cyclopropenes by Hydroacylation. J. Am. Chem. Soc. 2010, 132, 16354–16355. 10.1021/ja107738a. [DOI] [PubMed] [Google Scholar]
- Deyris P.-A.; Cañeque T.; Wang Y.; Retailleau P.; Bigi F.; Maggi R.; Maestri G.; Malacria M. Catalytic Semireduction of Internal Alkynes with All-Metal Aromatic Complexes. ChemCatChem 2015, 7, 3266–3269. 10.1002/cctc.201500729. [DOI] [Google Scholar]
- Nguyen J.; Chong A.; Lalic G. Nickel-catalyzed anti-Markovnikov hydroarylation of alkenes. Chem. Sci. 2019, 10, 3231–3236. 10.1039/c8sc05445b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman D.; Jiang L.; Buchwald S. L. Copper-Catalyzed C-P Bond Construction via Direct Coupling of Secondary Phosphines and Phosphites with Aryl and Vinyl Halides. Org. Lett. 2003, 5, 2315–2318. 10.1021/ol0346640. [DOI] [PubMed] [Google Scholar]
- Lippincott D. J.; Trejo-Soto P. J.; Gallou F.; Lipshutz B. H. Copper-Catalyzed Oxidative Cleavage of Electron-Rich Olefins in Water at Room Temperature. Org. Lett. 2018, 20, 5094–5097. 10.1021/acs.orglett.8b01883. [DOI] [PubMed] [Google Scholar]
- Phelan J. P.; Lang S. B.; Compton J. S.; Kelly C. B.; Dykstra R.; Gutierrez O.; Molander G. A. Redox-Neutral Photocatalytic Cyclopropanation via Radical/Polar Crossover. J. Am. Chem. Soc. 2018, 140, 8037–8047. 10.1021/jacs.8b05243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W.; Dai X.-J.; Wang H.; Li C.; Yang X.; Li C.-J. Ruthenium(II)-catalyzed olefination via carbonyl reductive cross-coupling. Chem. Sci. 2017, 8, 8193–8197. 10.1039/c7sc04207h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.; Kumar R.; Rehbein J.; Wirth T. Enantioselective Oxidative Rearrangements with Chiral Hypervalent Iodine Reagents. Chem.—Eur. J. 2016, 22, 4030–4035. 10.1002/chem.201504844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.; Michael A. K.; McIntosh M. L.; Koren-Selfridge L.; Scott J. P.; Clark T. B. Stereoselective Formation of Trisubstituted Vinyl Boronate Esters by the Acid-Mediated Elimination of α-Hydroxyboronate Esters. J. Org. Chem. 2014, 79, 7199–7204. 10.1021/jo500773t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenebeck F.; Murphy J. A.; Zhou S.-Z.; Uenoyama Y.; Miclo Y.; Tuttle T. Reductive Cleavage of Sulfones and Sulfonamides by a Neutral Organic Super-Electron-Donor (S.E.D.) Reagent. J. Am. Chem. Soc. 2007, 129, 13368–13369. 10.1021/ja074417h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.