

Abstract
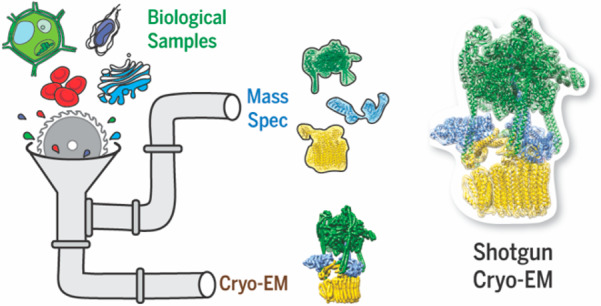
Rapid developments in cryogenic electron microscopy have opened new avenues to probe the structures of protein assemblies in their near native states. Recent studies have begun applying single -particle analysis to heterogeneous mixtures, revealing the potential of structural-omics approaches that combine the power of mass spectrometry and electron microscopy. Here we highlight advances and challenges in sample preparation, data processing, and molecular modeling for handling increasingly complex mixtures. Such advances will help structural-omics methods extend to cellular-level models of structural biology.
With the sequencing of thousands of genomes, large biological data sets (-omics data) have become pervasive in most fields of biology, including development,1,2 the classification of organisms,3,4 and disease,5−7 among many others. Disciplines embracing -omics strategies reach well beyond the central dogma of biology—genomics, transcriptomics, and proteomics—into such areas as metabolomics,8 epigenomics,9 pharmacogenomics,10 and interactomics.11 As with these other endeavors, structural biology has also expanded to embrace -omics approaches.
Major historic interactions of structural biology and -omics approaches have included, for example, electron tomography12 to provide cellular context and spatial information to complement proteomics and interactomics data,13−15 many efforts at proteome-scale modeling of three-dimensional (3D) structures and interactions,16−18 and the entire field of structural genomics.19−22 Structural genomics has employed techniques such as X-ray crystallography, NMR spectroscopy, and electron microscopy (EM) to solve structures of purified macromolecules in a high-throughput manner, targeting new protein folds and entire proteomes, which have been supplemented by molecular modeling and structure prediction to extend structural insights to new molecules.
The Potential of Shotgun Cryo-EM Methods
More recently, advances in single particle cryogenic electron microscopy (cryo-EM) have opened interesting new opportunities to connect -omics approaches and structural biology. In particular, cryo-EM boasts several important features: it requires only small amounts of sample, there is no requirement for crystal screening and optimization, and as a result, it is possible to capture several states of a macromolecular machine of interest. Cryo-EM is also capable of imaging a large field of individual macromolecular complexes in a single image. With the advent of direct electron detectors, ultrastable electron microscopes, automated data collection strategies,23 and real-time data processing,24 the “resolution revolution” in cryo-EM provides a definite route forward for increasing the throughput of structural biology.25 We can anticipate that structures from these methods, in combination with electron tomography, will produce information-rich cell atlases capturing high-resolution structures of the proteome and its spatial context that will synergize with other -omics approaches. Here we focus specifically on efforts to increase the applicability of single-particle cryo-EM to increasingly complex and heterogeneous samples, approaching cell lysates in complexity (as in shotgun cryo-EM), thus furthering the transformation of cryo-EM into a pipeline for structural-omics.
Mass spectrometry combined with electron microscopy has been shown to be well-suited for characterizing the architecture of protein complexes without purifying a specific target molecule, as demonstrated in yeast,16Desulfovibrio vulgaris,26 macrophage cytoplasm,27 the nuclear pore complex,28−30 and most recently Plasmodium falciparum.31 Protein–protein interactions identified through mass spectrometry in conjunction with advances in 3D structure determination have been used to investigate the architecture of multiple distinct protein complexes from mixtures such as fractionated cell lysate or even single cells.32−34 To date, such studies have largely been limited to the identification of protein complexes that were easily recognizable (e.g., the proteasome and ribosome) or of high enough resolution to identify the proteins by comparing contiguous stretches of highly resolved amino acids to a reference proteome.31 Currently, the field lacks robust and systematic computational pipelines for sorting, identifying, and performing molecular modeling of the myriad of structures that can potentially be solved from mixtures. The question remains: how can we break through these barriers?
Challenges in Sample Preparation of Heterogeneous Mixtures
In fact, even before the challenges of molecular modeling of mixtures of structures obtained from shotgun cryo-EM methods, several challenges exist for high-throughput cryo-EM data collection and processing of mixtures. Sample preparation is often a major bottleneck in structural studies. In our hands, finding suitable freezing conditions for heterogeneous mixtures has proven equally difficult as for a single purified sample,35 with the addition of several new challenges. Notably, in the case of cell extracts, the presence of dominating, highly abundant macromolecules can make screening difficult, especially when the sizes and shapes of other, less abundant proteins are unfamiliar. Although multiple orthogonal chromatographic separations might help simplify mixtures, we find that sample preparation with similar-sized macromolecules improves the chances of success. We have also found that different buffers in combination with different support substrates such as graphene oxide can produce an additional “purification” step, ultimately determining which complexes are present on the grid. Furthermore, many 3D reconstructions are built from large data sets containing hundreds of thousands of particles per complex. Scaling this to samples containing tens to hundreds of complexes, which may be present in different quantities, could prove challenging simply from a data collection perspective. It will also be important to incorporate improved denoising and particle picking algorithms to assist users in picking difficult to recognize particles with multiple shapes and sizes.36−38 Despite these challenges, several groups have already produced multiple structures to <5 Å resolution from fractionated lysates.31,32
While work on sample preparation methods for investigating fractionated or whole-cell lysates is ongoing, there already exist many approaches that can be used to reduce the complexity or target specific molecules from a mixture. Modified grid surfaces have been used for capturing proteins by His-tag,39,40 biotin,41 and antibody affinity.42 These approaches can alleviate the need for purification, target low-abundance proteins, help with orientation bias, and be readily integrated in combination with clonal sets such as the ASKA library.43 Other approaches include using microfluidic devices that can isolate and enrich target molecules.44 To date, many of these studies have been limited to identifying only a few symmetric molecules from a mixture, and scaling these approaches for high throughput has yet to be attempted.
Advances in Data Processing of Heterogeneous Mixtures
Apart from optimization of sample preparation and data collection, new data processing schemes will also need to be introduced. Currently, most cryo-EM data processing software operates under the assumption that samples contain one dominant structure that may contain conformational or subunit heterogeneity. In order to adapt such software for use on highly heterogeneous samples, we developed an auxiliary algorithm based on the principles of the projection-slice theorem to presort particles into homogeneous subsets prior to conventional 3D classification and therefore avoid the need to guess the number of underlying structures present in the data.35 A subsequent challenge will be to identify the resulting models, which can range from low to high resolution. Recently, the cryoID software package was introduced, which uses a unique approach to sequence by structure from highly resolved, contiguous amino acids in a 3D reconstruction.31 However, the challenges from sample preparation suggest that it is more likely that these studies will produce a number of low- to mid-resolution maps, and there still remains a significant challenge for identifying and modeling low- to mid-resolution reconstructions from a mixture when their identities are not known a priori.
Approaches for Docking Atomic Models into Low- to Mid-Resolution Reconstructions
Because of the likelihood that lower-abundance proteins in mixtures will only achieve low- to mid-resolution 3D reconstructions, if simply as a function of fewer particles, there will continue to be a need to better leverage other structural data. For this reason, an important focus remains improving approaches for fitting both predicted and currently available atomic structures into these lower-resolution 3D reconstructions (Figure 1). These range from user-intensive to computation-intensive approaches. Ideally, given the ambiguity of fitting numerous subunits into 3D reconstructions of unknown identity, one would prefer a quick, efficient, and computationally driven method. The challenge of fitting subunits into a 3D reconstruction becomes increasingly difficult for multi-subunit complexes and may be additionally complicated by considerations of symmetry. Techniques such as MBP and Fab labeling of individual subunits have been used to identify specific subunits within multi-subunit complexes.45,46 While this would prove cumbersome for identifying proteins in multiple complexes within a cell lysate, it may be useful for targeting a specific complex of interest.
Figure 1.

A structural-omics pipeline. A broad goal in the field is to develop a high-throughput structural-omics approach for reconstructing complexes from a heterogeneous mixture. For example, whole-cell lysates, organelle lysates, and heterogeneous mixtures might be analyzed by both cryo-EM and mass spectrometry. Cryo-EM produces multiple 3D reconstructions of protein complexes, while mass spectrometry provides identity and interaction information for the proteins present in the sample. To merge the two, even more efficient computational pipelines are needed to build or retrieve individual structures of proteins, organize them by interactions, assemble them into complexes, and match them to their 3D reconstructions obtained from a sample.
One commonly employed user-driven approach for fitting atomic structures into 3D reconstructions involves segmenting the maps either manually or using the Segger tool47 followed by rigid-body docking using Fit-in-Map into these segmented regions in UCSF Chimera.48,49 Scoring of this approach can be optimized using a flexible fitting tool50,51 such as MDFF,50 which applies forces proportional to the density gradient of the EM map, while conserving stereochemistry, to fit atomic structures into EM maps with resolutions as low as 15 Å. While these methods may work well if structural information is known a priori, any manual approach of rigid docking faces the possibility of getting caught in a local minimum, suffering from user bias, and requiring numerous user hours. Furthermore, fitting atomic models into complexes becomes extremely challenging when their identities are incompletely known.
The development of integrative methods allows for a more hands-off approach, eliminating some of these biases.52−54 These approaches combine data retrieved from various experiments such as yeast two-hybrid (Y2H) assays, mutagenesis, cross-linking, small-angle X-ray scattering, electron microscopy, and X-ray crystallography to build the multiprotein model.55,56 Such methodologies have been successful in building models for a number of multiprotein complexes such as the nuclear pore complex,57 16S rRNA complexed with methyltransferase A small subunit,56 and the BBSome.58 Recently, several models predicted by integrative modeling were validated against their experimentally determined high-resolution structures.52 The results showed that for all atom models the positions of subunit centers were within 5 Å of the true model, demonstrating the power of this approach.59−64 For those structures with resolution higher than 10 Å, not only can secondary structure elements be detected, but orientation and connectivity may also be predicted to validate the integrative models.65 While these methods are promising for building a single multiprotein assembly with abundant data, they are computationally intensive, and whether they will be equally applicable to mixtures of multiple complexes from structural-omics data remains untested. Methods that could simplify model building by further constraining possible orientations, interactions, or flexibility may help moving forward.
Approaches for Identifying Molecular Machines within Complex Mixtures
Because of the size and complexity of the data that describe extremely heterogeneous samples, corresponding mass spectrometry data become pivotal in identifying the proteins present, estimating their relative abundances, and identifying those that interact to form complexes in the sample. Previous studies have shown that machine learning combined with co-fractionation mass spectrometry can be used to detect proteins that interact to form complexes on the basis of their elution profiles from multiple separation techniques.66 These predicted complexes can be prioritized by relative abundance for modeling. Additionally, identification of previously solved structures could reduce the number of 3D reconstructions that need to be considered for subsequent modeling. Pipelines such as GEM-PRO could accomplish this by streamlining rapid searches of the Protein Data Bank by returning protein structures given a gene or protein sequence, while also evaluating the quality of the structures and preparing sequences for comparative modeling for those that do not have a known structure.67
Recently, improved shape-based searches for protein complexes have been developed to better accommodate the low- to mid-resolution EM data produced from tomography.68 Such shape-search tools might prove useful for searching 3D reconstructions in order to identify those known from prior structures. The 3D reconstructions that have been resolved and identified could then be used to revisit raw micrographs and pick specific particles with template matching approaches.69 The remaining 3D models would subsequently have to be built de novo on the basis of, e.g., protein identities from mass spectrometry performed on the same samples. Importantly, beyond the structures of proteins already solved and available in the Protein Data Bank,70 3D structural models have now been computationally generated by many research groups at the proteome scale, a success of the Protein Structure Initiative (such as those indexed by the Uniprot71 database), using techniques of comparative modeling,67,72 evolutionary couplings,73 or even ab initio(74) approaches.
Any structural modeling of native protein assemblies would most likely require prior knowledge of which specific protein–protein interactions were occurring75,76 as well as the stoichiometries of the interacting subunits. The latter, if unknown, might be obtainable using mass spectrometry.57,66,77−79 Other approaches to deciphering stoichiometry might include using volume constraints, where volumes of different numbers of individual subunits are compared to the volume of a 3D reconstruction. Cross-linking mass spectrometry, where large numbers of pairwise protein interactions may be identified, can help in elucidating protein interaction partners.80 Additionally, other pairwise restraints may be added, such as protein docking predictions, to reveal new assemblies.81−83 However, protein docking becomes significantly more complex with more than two proteins and no knowledge of interaction interfaces or order of assembly.
Moving toward Structural-Omics
Given knowledge of interacting subunits and their stoichiometries, the task becomes fitting them into the correct map in the correct assembly. The problem resembles a jigsaw puzzle, where subunits must fit into the molecular envelope while respecting mutual packing interfaces. In general, such packing problems are known to be NP-complete84 and cannot be solved computationally in polynomial time. Nonetheless, additional restraints can be brought to bear to reduce the search complexity. For example, like a puzzle, one might determine interacting interfaces among the subunits, either by docking18 or more approximate approaches, ideally algorithms that are rapid and partner-specific. In our own work, we have developed reduced representations of protein surfaces to help predict complementary interaction interfaces, which add a measure of robustness to minor structural deformations upon binding.85 Combinations of such packing restraints could then be employed to help pack and refine 3D protein structures to EM maps. In parallel, researchers have improved computational search algorithms for packing problems by using reduction or backtracking,86,87 and the potential exists to crowdsource the problem, employing the visual acuity of humans to manually fit subunits into 3D reconstructions.88
Structural-omics stands to benefit strongly from the cryo-EM resolution revolution, and in turn these approaches have the potential to greatly enhance our understanding of biology from a systems perspective. Toward this end, it is already clear that various low- to high-resolution complexes may be reconstructed from a cell lysate using single-particle electron microscopy. The development of new computational tools to efficiently sort and build atomic models into these low- to mid-resolution reconstructions or to solve the high-resolution structures from mixtures of increasing complexity will certainly help to further advance this field and put it on a path toward even richer structural cell atlases.
Acknowledgments
This work was supported in part by Welch Foundation Research Grants F-1938 (to D.W.T.) and F-1515 (to E.M.M.), Army Research Office Grant W911NF-15-1-0120 (to D.W.T.), a Robert J. Kleberg, Jr., and Helen C. Kleberg Foundation Medical Research Award (to D.W.T.), and grants from the National Institutes of Health (GM122480, DK110520, and HD085901) to E.M.M. C.L.M is an NSF Graduate Research Fellow supported by the National Science Foundation (2019238253). D.W.T is a CPRIT Scholar supported by the Cancer Prevention and Research Institute of Texas (RR160088) and an Army Young Investigator supported by the Army Research Office (W911NF-19-1-0021). The authors thank Angel Syrett for assistance with Figure 1.
The authors declare no competing financial interest.
References
- Wang Z.; Gerstein M.; Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Tan Y.; Cahan P. Understanding development and stem cells using single cell-based analyses of gene expression. Development 2017, 144, 17–32. 10.1242/dev.133058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce A. R.; Palsson B. Ø. The model organism as a system: integrating’omics’ data sets. Nat. Rev. Mol. Cell Biol. 2006, 7, 198. 10.1038/nrm1857. [DOI] [PubMed] [Google Scholar]
- Raupach M. J.; Amann R.; Wheeler Q. D.; Roos C. The application of “-omics” technologies for the classification and identification of animals. Org. Divers. Evol. 2016, 16, 1–12. 10.1007/s13127-015-0234-6. [DOI] [Google Scholar]
- Karczewski K. J.; Snyder M. P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299. 10.1038/nrg.2018.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y.; Seldin M.; Lusis A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. 10.1186/s13059-017-1215-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter S. S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479. 10.1038/s41581-018-0021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riekeberg E.; Powers R. New frontiers in metabolomics: from measurement to insight. F1000Research 2017, 6, 1148. 10.12688/f1000research.11495.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. A.; Baylin S. B. The epigenomics of cancer. Cell 2007, 128, 683–692. 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly A. K. Pharmacogenetics: a general review on progress to date. Br. Med. Bull. 2017, 124, 65–79. 10.1093/bmb/ldx035. [DOI] [PubMed] [Google Scholar]
- Luck K.; Sheynkman G. M.; Zhang I.; Vidal M. Proteome-scale human interactomics. Trends Biochem. Sci. 2017, 42, 342–354. 10.1016/j.tibs.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lučić V.; Förster F.; Baumeister W. Structural studies by electron tomography: from cells to molecules. Annu. Rev. Biochem. 2005, 74, 833–865. 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Güell M.; Van Noort V.; Yus E.; Chen W.-H.; Leigh-Bell J.; Michalodimitrakis K.; Yamada T.; Arumugam M.; Doerks T.; Kühner S.; et al. Transcriptome complexity in a genome-reduced bacterium. Science 2009, 326, 1268–1271. 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- Kühner S.; van Noort V.; Betts M. J.; Leo-Macias A.; Batisse C.; Rode M.; Yamada T.; Maier T.; Bader S.; Beltran-Alvarez P.; et al. Proteome organization in a genome-reduced bacterium. Science 2009, 326, 1235–1240. 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- Yus E.; Maier T.; Michalodimitrakis K.; van Noort V.; Yamada T.; Chen W.-H.; Wodke J. A.; Güell M.; Martínez S.; Bourgeois R.; et al. Impact of genome reduction on bacterial metabolism and its regulation. Science 2009, 326, 1263–1268. 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- Aloy P.; Böttcher B.; Ceulemans H.; Leutwein C.; Mellwig C.; Fischer S.; Gavin A.-C.; Bork P.; Superti-Furga G.; Serrano L. Structure-based assembly of protein complexes in yeast. Science 2004, 303, 2026–2029. 10.1126/science.1092645. [DOI] [PubMed] [Google Scholar]
- Baker D.; Sali A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Vakser I. A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. 10.1016/j.bpj.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-H. Shining a light on structural genomics. Nat. Struct. Biol. 1998, 5, 643. 10.1038/1334. [DOI] [PubMed] [Google Scholar]
- Skolnick J.; Fetrow J. S.; Kolinski A. Structural genomics and its importance for gene function analysis. Nat. Biotechnol. 2000, 18, 283. 10.1038/73723. [DOI] [PubMed] [Google Scholar]
- Stevens R. C.; Yokoyama S.; Wilson I. A. Global efforts in structural genomics. Science 2001, 294, 89–92. 10.1126/science.1066011. [DOI] [PubMed] [Google Scholar]
- Chandonia J.-M.; Brenner S. E. The impact of structural genomics: expectations and outcomes. Science 2006, 311, 347–351. 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- Li Y.; Cash J. N.; Tesmer J. J. G.; Cianfrocco M. A. High-throughput cryo-EM enabled by user-free preprocessing routines. bioRxiv 2019, 885541. 10.1101/2019.12.20.885541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegunov D.; Cramer P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 2019, 16, 1146–1152. 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt W. The resolution revolution. Science 2014, 343, 1443–1444. 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- Han B.-G.; Dong M.; Liu H.; Camp L.; Geller J.; Singer M.; Hazen T. C.; Choi M.; Witkowska H. E.; Ball D. A.; et al. Survey of large protein complexes in D. vulgaris reveals great structural diversity. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16580–16585. 10.1073/pnas.0813068106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maco B.; Ross I. L.; Landsberg M. J.; Mouradov D.; Saunders N. F.; Hankamer B.; Kobe B. Proteomic and electron microscopy survey of large assemblies in macrophage cytoplasm. Mol. Cell. Proteomics 2011, 10, M111.008763. 10.1074/mcp.M111.008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber F.; Dokudovskaya S.; Veenhoff L. M.; Zhang W.; Kipper J.; Devos D.; Suprapto A.; Karni-Schmidt O.; Williams R.; Chait B. T.; et al. The molecular architecture of the nuclear pore complex. Nature 2007, 450, 695–701. 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- Alber F.; Dokudovskaya S.; Veenhoff L. M.; Zhang W.; Kipper J.; Devos D.; Suprapto A.; Karni-Schmidt O.; Williams R.; Chait B. T.; et al. Determining the architectures of macromolecular assemblies. Nature 2007, 450, 683–694. 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- Beck M.; Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73. 10.1038/nrm.2016.147. [DOI] [PubMed] [Google Scholar]
- Ho C.-M.; Li X.; Lai M.; Terwilliger T. C.; Beck J. R.; Wohlschlegel J.; Goldberg D. E.; Fitzpatrick A. W. P.; Zhou Z. H. Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat. Methods 2020, 17, 79–85. 10.1038/s41592-019-0637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastritis P. L.; O’Reilly F. J.; Bock T.; Li Y.; Rogon M. Z.; Buczak K.; Romanov N.; Betts M. J.; Bui K. H.; Hagen W. J.; et al. Capturing protein communities by structural proteomics in a thermophilic eukaryote. Mol. Syst. Biol. 2017, 13, 936. 10.15252/msb.20167412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke E. J.; Mallam A. L.; Drew K.; Marcotte E. M.; Taylor D. W. Classification of single particles from human cell extract reveals distinct structures. Cell Rep. 2018, 24, 259–268. 10.1016/j.celrep.2018.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X.; Verbeke E. J.; Chang Y.; Dickinson D. J.; Taylor D. W. Electron microscopy snapshots of single particles from single cells. J. Biol. Chem. 2019, 294, 1602–1608. 10.1074/jbc.RA118.006686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke E. J.; Zhou Y.; Horton A. P.; Mallam A. L.; Taylor D. W.; Marcotte E. M. Separating distinct structures of multiple macromolecular assemblies from cryo-EM projections. J. Struct. Biol. 2020, 209, 107416. 10.1016/j.jsb.2019.107416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T.; Noble A. J.; Berger B. Topaz-Denoise: general deep denoising models for cryoEM. bioRxiv 2019, 838920. 10.1101/838920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T.; Merino F.; Stabrin M.; Moriya T.; Antoni C.; Apelbaum A.; Hagel P.; Sitsel O.; Raisch T.; Prumbaum D.; et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2019, 2, 218. 10.1038/s42003-019-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T.; Morin A.; Noble A. J.; Brasch J.; Shapiro L.; Berger B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Res. Comput. Mol. Biol. 2018, 10812, 245–247. [PMC free article] [PubMed] [Google Scholar]
- Kelly D. F.; Abeyrathne P. D.; Dukovski D.; Walz T. The Affinity Grid: a pre-fabricated EM grid for monolayer purification. J. Mol. Biol. 2008, 382, 423–433. 10.1016/j.jmb.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin C. J.; Wright K. J.; Bolton S. C.; Hyun S.-H.; Krynski K.; Grover M.; Yu G.; Guo F.; Kinzer-Ursem T. L.; Jiang W.; Thompson D. H. Selective capture of histidine-tagged proteins from cell lysates using TEM grids modified with NTA-graphene oxide. Sci. Rep. 2016, 6, 32500. 10.1038/srep32500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.-G.; Walton R. W.; Song A.; Hwu P.; Stubbs M. T.; Yannone S. M.; Arbeláez P.; Dong M.; Glaeser R. M. Electron microscopy of biotinylated protein complexes bound to streptavidin monolayer crystals. J. Struct. Biol. 2012, 180, 249–253. 10.1016/j.jsb.2012.04.025. [DOI] [PubMed] [Google Scholar]
- Yu G.; Li K.; Huang P.; Jiang X.; Jiang W. Antibody-based affinity cryoelectron microscopy at 2.6-Å resolution. Structure 2016, 24, 1984–1990. 10.1016/j.str.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M.; Ara T.; Arifuzzaman M.; Ioka-Nakamichi T.; Inamoto E.; Toyonaga H.; Mori H. Complete set of ORF clones of Escherichia coli ASKA library (A Complete S et of E. coli K-12 ORF A rchive): Unique Resources for Biological Research. DNA Res. 2006, 12, 291–299. 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Schmidli C.; Albiez S.; Rima L.; Righetto R.; Mohammed I.; Oliva P.; Kovacik L.; Stahlberg H.; Braun T. Microfluidic protein isolation and sample preparation for high-resolution cryo-EM. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 15007–15012. 10.1073/pnas.1907214116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G. C.; Estrin E.; Matyskiela M. E.; Bashore C.; Nogales E.; Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ding Z.; Liu X.; Bao Y.; Huang M.; Wong C. C. L.; Hong X.; Cong Y. Architecture and subunit arrangement of the complete Saccharomyces cerevisiae COMPASS complex. Sci. Rep. 2018, 8, 17405. 10.1038/s41598-018-35609-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie G.; Chiu W. Comparison of Segger and other methods for segmentation and rigid-body docking of molecular components in Cryo-EM density maps. Biopolymers 2012, 97, 742–760. 10.1002/bip.22074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Goddard T. D.; Huang C. C.; Ferrin T. E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007, 157, 281–287. 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Trabuco L. G.; Villa E.; Mitra K.; Frank J.; Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 2008, 16, 673–683. 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs J. A.; Galkin V. E.; Wriggers W. Accurate flexible refinement of atomic models against medium-resolution cryo-EM maps using damped dynamics. BMC Struct. Biol. 2018, 18, 12. 10.1186/s12900-018-0089-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braitbard M.; Schneidman-Duhovny D.; Kalisman N. Integrative structure modeling: overview and assessment. Annu. Rev. Biochem. 2019, 88, 113–135. 10.1146/annurev-biochem-013118-111429. [DOI] [PubMed] [Google Scholar]
- Topf M.; Lasker K.; Webb B.; Wolfson H.; Chiu W.; Sali A. Protein structure fitting and refinement guided by cryo-EM density. Structure 2008, 16, 295–307. 10.1016/j.str.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel D.; Lasker K.; Webb B.; Velázquez-Muriel J.; Tjioe E.; Schneidman-Duhovny D.; Peterson B.; Sali A. Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012, 10, e1001244 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B.; Viswanath S.; Bonomi M.; Pellarin R.; Greenberg C. H.; Saltzberg D.; Sali A. Integrative structure modeling with the integrative modeling platform. Protein Sci. 2018, 27, 245–258. 10.1002/pro.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert G. C.; Melquiond A. S.; Bonvin A. M. Integrative modeling of biomolecular complexes: HADDOCKing with cryo-electron microscopy data. Structure 2015, 23, 949–960. 10.1016/j.str.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Fernandez-Martinez J.; Nudelman I.; Shi Y.; Zhang W.; Raveh B.; Herricks T.; Slaughter B. D.; Hogan J. A.; Upla P.; et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature 2018, 555, 475–482. 10.1038/nature26003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H.-T.; Apelt L.; Farrell D. P.; White S. R.; Woodsmith J.; Svetlov V.; Goldstein J. S.; Nager A. R.; Li Z.; Muller J.; et al. The molecular architecture of native BBSome obtained by an integrated structural approach. Structure 2019, 27, 1384–1394. 10.1016/j.str.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miled N.; Yan Y.; Hon W.-C.; Perisic O.; Zvelebil M.; Inbar Y.; Schneidman-Duhovny D.; Wolfson H. J.; Backer J. M.; Williams R. L. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007, 317, 239–242. 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- Schweppe D. K.; Chavez J. D.; Lee C. F.; Caudal A.; Kruse S. E.; Stuppard R.; Marcinek D. J.; Shadel G. S.; Tian R.; Bruce J. E. Mitochondrial protein interactome elucidated by chemical cross-linking mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 1732–1737. 10.1073/pnas.1617220114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K.; Elmlund H.; Kalisman N.; Bushnell D. A.; Adams C. M.; Azubel M.; Elmlund D.; Levi-Kalisman Y.; Liu X.; Gibbons B. J.; et al. Architecture of an RNA polymerase II transcription pre-initiation complex. Science 2013, 342, 1238724. 10.1126/science.1238724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A.; Joachimiak L. A.; Bracher A.; Mönkemeyer L.; Walzthoeni T.; Chen B.; Pechmann S.; Holmes S.; Cong Y.; Ma B.; et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 2012, 20, 814–825. 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Cimermancic P.; Viswanath S.; Ebmeier C. C.; Kim B.; Dehecq M.; Raman V.; Greenberg C. H.; Pellarin R.; Sali A.; et al. Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol. Cell 2015, 59, 794–806. 10.1016/j.molcel.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Fernandez-Martinez J.; Tjioe E.; Pellarin R.; Kim S. J.; Williams R.; Schneidman-Duhovny D.; Sali A.; Rout M. P.; Chait B. T. Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol. Cell. Proteomics 2014, 13, 2927–2943. 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S.; Alexander N.; Wötzel N.; Karakaş M.; Stewart P. L.; Meiler J. EM-fold: de novo atomic-detail protein structure determination from medium-resolution density maps. Structure 2012, 20, 464–478. 10.1016/j.str.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana P. C.; Hart G. T.; Nepusz T.; Yang H.; Turinsky A. L.; Li Z.; Wang P. I.; Boutz D. R.; Fong V.; Phanse S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk E.; Mih N.; Monk J.; Zhang Z.; O’Brien E. J.; Bliven S. E.; Chen K.; Chang R. L.; Bourne P. E.; Palsson B. O. Systems biology of the structural proteome. BMC Syst. Biol. 2016, 10, 26. 10.1186/s12918-016-0271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Sit A.; Christoffer C.; Chen S.; Kihara D. A global map of the protein shape universe. PLoS Comput. Biol. 2019, 15, e1006969 10.1371/journal.pcbi.1006969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickgauer J. P.; Grigorieff N.; Denk W. Single-protein detection in crowded molecular environments in cryo-EM images. eLife 2017, 6, e25648 10.7554/eLife.25648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R.; Bairoch A.; Wu C. H.; Barker W. C.; Boeckmann B.; Ferro S.; Gasteiger E.; Huang H.; Lopez R.; Magrane M.; et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S. D.; Das S.; Sillitoe I.; Orengo C. An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallographica Section D: Structural Biology 2017, 73, 628–640. 10.1107/S2059798317008920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks D. S.; Hopf T. A.; Sander C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012, 30, 1072. 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. T.; Bonneau R.; Ruczinski I.; Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins: Struct., Funct., Genet. 1999, 37, 171–176. . [DOI] [PubMed] [Google Scholar]
- Drew K.; Lee C.; Huizar R. L.; Tu F.; Borgeson B.; McWhite C. D.; Ma Y.; Wallingford J. B.; Marcotte E. M. Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 2017, 13, 932. 10.15252/msb.20167490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giurgiu M.; Reinhard J.; Brauner B.; Dunger-Kaltenbach I.; Fobo G.; Frishman G.; Montrone C.; Ruepp A. CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 2019, 47, D559–D563. 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández H.; Robinson C. V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2007, 2, 715. 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Skinner O. S.; Schachner L. F.; Kelleher N. L. The Search Engine for Multi-Proteoform Complexes: An Online Tool for the Identification and Stoichiometry Determination of Protein Complexes. Curr. Protoc. Bioinf. 2016, 56, 13.30.1–13.30.11. 10.1002/cpbi.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits A. H.; Vermeulen M. Characterizing protein-protein interactions using mass spectrometry: challenges and opportunities. Trends Biotechnol. 2016, 34, 825–834. 10.1016/j.tibtech.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Liu F.; Rijkers D. T.; Post H.; Heck A. J. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 2015, 12, 1179. 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. HADDOCK: a protein- protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Comeau S. R.; Gatchell D. W.; Vajda S.; Camacho C. J. ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. 10.1093/bioinformatics/btg371. [DOI] [PubMed] [Google Scholar]
- Gong X.; Wang P.; Yang F.; Chang S.; Liu B.; He H.; Cao L.; Xu X.; Li C.; Chen W.; Wang C. Protein–protein docking with binding site patch prediction and network-based terms enhanced combinatorial scoring. Proteins: Struct., Funct., Genet. 2010, 78, 3150–3155. 10.1002/prot.22831. [DOI] [PubMed] [Google Scholar]
- Demaine E. D.; Demaine M. L. Jigsaw puzzles, edge matching, and polyomino packing: Connections and complexity. Graphs Combin. 2007, 23, 195–208. 10.1007/s00373-007-0713-4. [DOI] [Google Scholar]
- McCafferty C. L.; Marcotte E. M.; Taylor D. W. Simplified geometric representations of protein structures identify complementary interaction interfaces. bioRxiv 2019, 880575. 10.1101/2019.12.18.880575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuth D. E.Dancing links. arXiv (Computer Science.Data Structures and Algorithms), November 15, 2000, cs/0011047, ver. 1. https://arxiv.org/abs/cs/0011047 (accessed 2019-12-17).
- Lodi A.; Martello S.; Vigo D. Heuristic algorithms for the three-dimensional bin packing problem. Eur. J. Oper. Res. 2002, 141, 410–420. 10.1016/S0377-2217(02)00134-0. [DOI] [Google Scholar]
- Khatib F.; Desfosses A.; Koepnick B.; Flatten J.; Popović Z.; Baker D.; Cooper S.; Gutsche I.; Horowitz S. Building de novo cryo-electron microscopy structures collaboratively with citizen scientists. PLoS Biol. 2019, 17, e3000472 10.1371/journal.pbio.3000472. [DOI] [PMC free article] [PubMed] [Google Scholar]