

Abstract
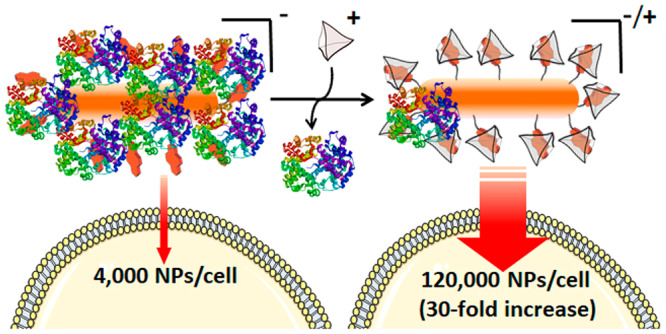
When nanoparticles (NPs) are exposed to biological media, proteins are adsorbed, forming a so-called protein corona (PC). This cloud of protein aggregates hampers the targeting and transport capabilities of the NPs, thereby compromising their biomedical applications. Therefore, there is a high interest in the development of technologies that allow control over PC formation, as this would provide a handle to manipulate NPs in biological fluids. We present a strategy that enables the reversible disruption of the PC using external stimuli, thereby allowing a precise regulation of NP cellular uptake. The approach, demonstrated for gold nanoparticles (AuNPs), is based on a biorthogonal, supramolecular host–guest interactions between an anionic dye bound to the AuNP surface and a positively charged macromolecular cage. This supramolecular complex effectively behaves as a zwitterionic NP ligand, which is able not only to prevent PC formation but also to disrupt a previously formed hard corona. With this supramolecular stimulus, the cellular internalization of AuNPs can be enhanced by up to 30-fold in some cases, and even NP cellular uptake in phagocytic cells can be regulated. Additionally, we demonstrate that the conditional cell uptake of purposely designed gold nanorods can be used to selectively enhance photothermal cell death.
Keywords: protein corona, gold nanoparticles, host−guest chemistry, cellular uptake, photothermal therapy
Nanoparticles (NPs) have gained great momentum in biomedicine, owing to their applications in diagnosis, drug delivery, and multimodal imaging, among other areas.1−4 Gold NPs have been of particular interest because of their low intrinsic cytotoxicity, easy customization of morphology and surface functionalization, and the possibility of using their plasmonic properties to unleash biological responses.5−7 In the area of cancer and related cellular diseases, a number of applications for detecting and/or killing cancer cells have been proposed.8,9 However, biomedical application of these NP-based strategies can be hampered by insufficient cellular uptake and reduced in vivo transport.10 Additional limitations are related to scavenging by the mononuclear phagocyte system, which results in NPs being trapped in the liver and spleen,11 as well as to the lack of spatial and temporal control over their biological activity.
Many such limitations are related to the well-established tendency of NPs to adsorb biomolecules, physiological proteins, in particular, forming the so-called “protein corona” (PC). When NPs are in a biological milieu such as the bloodstream, plasma proteins readily bind to their surfaces and form a PC that affects their physicochemical properties and compromises their transport, targeting, and cell uptake capabilities.12,13 Indeed, PC formation usually hampers direct contact of NPs with cell membranes, in turn inhibiting their internalization.14,15
Therefore, a major challenge in this field of biological chemistry involves the development of methods that provide control over PC formation on the NPs surface. In this context, research efforts have been mainly focused on two strategies to deal with the PC issue. The first one relies on the development of methods to control PC composition and improve NP biocompatibility,16 while the second strategy intends to prevent PC formation. Regarding the latter, the most common approach to reduce PC formation on NPs is based on precoating the NPs surface with hydrophilic polymers such as poly(ethylene glycol) (PEG).17,18 However, PEG coating can block protein adsorption only partially, and it has been reported that some plasma proteins (e.g., apolipoprotein E, IgG, etc.) can adsorb over PEG-coated surfaces.19 An alternative to PEG coating toward avoiding PC formation comprises the use of zwitterionic surface ligands. Strong electrostatic interactions between water molecules and zwitterionic ligands generate highly stable NPs, while minimizing nonspecific interactions with biomolecules.20 Zwitterionic ligands have been shown to inhibit PC formation completely in some specific cases,21 thereby leading to extended NP circulation lifetimes.22
Recently, an oligocationic covalent cage A, which enables the conversion of the negatively charged dye pyranine (pyr) into a positively charged host–guest complex, has been described (Figure 1A).23 This macromolecular cage has high selectivity and nanomolar affinity (Kd ≈ 1.2 nM) for pyr, and the resulting interaction has been used in biological applications.24,25 Additionally, its small size and straightforward functionalization render cage A a suitable candidate to be formulated into different dosage forms for use as a chemical stimulus.
Figure 1.

Supramolecular control over PC formation. (A) Chemical structures of the guest molecule pyr and the host cage A (the chemical structure of only one edge of the tetrahedral cage A is shown for clarity). (B) Schematic illustration of the reversible control over PC formation on AuNPs, using a supramolecular host–guest complex between negatively charged pyr and positively charged cage A.
We recently reported that small (2 nm) AuNPs carrying negatively charged pyranine molecules on their surface do not internalize into cells, whereas surface charge reversal by addition of cage A promotes their cellular uptake.26 This switch is effective with small NPs, which do not tend to form a PC, but a priori it might not work in the case of larger NPs. However, we envisioned that the host–guest interaction might be used as a handle to control the zwitterionic character of large NPs and, thereby, influence PC formation and cellular uptake. Herein we demonstrate that the PC formed on pyranine-functionalized 15 nm gold nanoparticles can be efficiently disrupted by judicious addition of the macromolecular cage A (Figure 1B). Importantly, the initial state (adsorbed proteins) can be recovered by adding a molecular guest that outcompetes for the host macromolecular cage, thereby providing reversible control over PC formation. This approach can be additionally used for the programmed cellular uptake of gold nanorods, which allows establishing a cell-selective photothermal therapy.
Results and Discussion
Citrate-stabilized Au nanospheres (AuNSs, 15 nm diameter) were initially coated with thiolated amino-polyethylene glycol (HS-PEG-NH2), resulting in AuNSs featuring exposed amino groups (NS1, Figure 2A). Subsequently, anionic pyranines bearing a carboxylic acid handle were covalently attached, to yield the expected pyr-derivatized nanoparticles NS2. Successful functionalization of NS1 with pyr was confirmed by zeta potential (Zp) measurements, since the Zp value shifted from +29 mV for NS1 to −37 mV for NS2 (Figure 2A). The colloidal stability of AuNPs during synthesis was verified by transmission electron microscopy (TEM) and UV–vis spectroscopy (Supporting Information Figures S1 and S2). Fluorescence spectroscopy after cyanide digestion of NS2 showed that the average number of pyr molecules per NP was ca. 400.
Figure 2.

NS2 synthesis and evaluation of its interaction with cage A. (A) Synthesis scheme and Zp characterization of NS1 and NS2. (B) Zp distributions for NS2 after addition of cage A. (black) 2 × 1011 particles/mL suspension of NS2 in Milli-Q water (−37 ± 2 mV). (red) After addition of 0.3 μM of A (+8 ± 2 mV). (green) After addition of 3 μM of A (+31 ± 2 mV). (purple) After addition of 5 μM of A (+32 ± 3 mV). (C) UV–Vis spectra of NS2 (1.5 × 1011 particles/mL) before and after addition of cage A. (black) NS2 in Milli-Q water. (red) After addition of 5 μM of A. (green) After addition of 10 μM of A. (D) Representative TEM corresponding to the sample with the red spectrum in (C).
Prior to studying PC formation, we evaluated the supramolecular interactions between NS2 and cage A using Zp measurements. We expected a shift of the initial negative Zp, arising from three negatively charged sulfonate groups in pyr, toward less negative or even positive values upon interaction with cage A. When an aqueous solution of cage A (0.3 μM, 2 equiv with respect to the concentration of pyranine groups) was added to an aqueous dispersion of NS2 (2 × 1011 particles/mL) Zp was found to shift from −37 ± 2 to +8 ± 2 mV, while further additions resulted in a further Zp shift up to +32 mV (Figure 2B). Importantly, absorbance spectra and TEM images confirmed that NS2 particles remained colloidally stable after completion of host–guest complexation (Figure 2C,D).
Two types of PC are commonly distinguished, which are known as “hard corona” and “soft corona”. On the one hand, the hard corona (HC) is formed by proteins with high affinity for the NP and is located next to the NP surface.27,28 On the other hand, the soft corona includes proteins with lower affinity, which get exchanged over time, and can be readily detached upon repeated washing. Importantly, because the HC remains adsorbed onto the NPs during biophysical events, it exerts a significant influence over the NP physiological properties.29 Therefore, we aimed at evaluating the effect of cage A on the formation and stability of a HC on NS2.
We prepared four dispersions of NS2 in 200 μL of diluted fetal bovine serum [FBS, 5% in phosphate-buffered saline (PBS)], containing different amounts of cage A (0, 5, 10, and 20 μM), which was added before FBS. Dynamic light scattering measurements confirmed the colloidal stability of NS2 in the presence of 5% FBS (Figure S3). After incubation for 30 min at room temperature, the nanoparticles were separated from nonadsorbed proteins by centrifugation and then extensively washed to remove all unbound proteins, until the supernatant was free of any detectable protein as indicated by the Bradford assay.30 Subsequently, the remaining adsorbed proteins forming the HC were detached from NS2 by a standard treatment with sodium dodecyl sulfate (SDS) (10%) and dithiothreitol (DTT; 0.5 mM) at 90 °C for 10 min.31 The protein content was analyzed by SDS polyacrylamide gel electrophoresis (PAGE), which showed that as little as 5 μM of cage is enough to reduce by more than half the amount of adsorbed HC proteins, whereas using 20 μM of A led to a fivefold reduction (Figure S4).
To confirm that the above effect is due to interactions between cage A and pyr on NS2, we performed control experiments using NPs lacking the pyranine moieties. We synthesized AuNPs coated with a thiolated poly(ethylene glycol) (NS-PEG) featuring a terminal methyl ether (HS-PEG5000-O–CH3, Figure S6). HC formation around these NPs was evaluated, both in the absence and in the presence of 10 μM A, using the above procedure. As expected, no significant differences in the amount of adsorbed protein were observed between both samples (Figure S4), thus excluding the possibility that a direct interaction between cage A and FBS proteins is the reason behind PC reduction. To rule out the possibility of a direct interaction between cage A and the NP surface, which might be hindered by the PEG polymer in the case of NS2, we studied another NP type, NS-Lac, with a short coating ligand bound to the NP surface. As expected, no difference was observed in the amount of PC detected upon addition of cage A or in its absence (Figure S7) To further confirm that the effect of cage A is mediated by interactions with pyranine, we used the intrinsic fluorescence of this chromophore, which is partially quenched upon formation of the host–guest complex.23 We observed that addition of increasing concentrations of cage A to a colloidal dispersion of NS2 in the presence of FBS (5% in PBS) does quench the fluorescence emission of pyranines, again evidencing host–guest complex formation, even in the presence of the proteins present in FBS (Figure S8).
While the above experiments demonstrate that the presence of cage A in the buffer affects the formation of a HC, the question arises whether addition of this macromolecule to NPs that are covered by a preformed HC can also disrupt the corona. We anticipated this goal to be challenging, since the HC is assumed to be tightly bound to the NPs surface. We additionally wondered whether the effect of cage A could be reversed by adding free pyr (8-hydroxypyrene-1,3,6-trisulfonic acid), which should compete for cage A and hence might restore the initial PC composition on the NPs.
Therefore, we incubated NS2 with diluted FBS (5% in PBS) for 30 min at 25 °C (total volume = 0.6 mL). We then separated a 0.4 mL aliquot of this dispersion and added cage A (20 μM), and the resulting mixture was kept at 25 °C for 10 min. Finally, to the 0.2 mL of the resulting dispersion, we added 50 μM pyranine, and the mixture was incubated at 25 °C for 10 min. After nonadsorbed proteins were removed, we applied the above-described protocol for HC release to the three resulting 0.2 mL solutions, followed by both SDS-PAGE analysis (Figure S5) and the bicinchoninic acid (BCA) assay (Figure 3A). The results revealed that addition of cage A (20 μM) to NS2 featuring a preformed PC resulted in removal of ca. 75%–79% of the HC after 10 min of incubation. This value is similar to that obtained in the experiment where the cage was added prior to FBS (82%). This result indicates that most of the proteins forming the HC are not irreversibly adsorbed to the surface of NS2, as they can be released under biocompatible conditions upon addition of the positively charged cage A. The BCA assay was also performed to quantify the protein recovered in the washing steps. Such experiments showed that, in the absence of cage A, only ca. 50% of the initial proteins used in the incubation were recovered in the washing steps, whereas in the presence of A the recovery increased up to 86% (Figure S9).
Figure 3.

Comparison of the amount and type of FBS proteins immobilized on NS2 particles upon addition of cage A and pyr. (A) (top) Schematic representation of the NP-protein system for each gel line. (bottom) NS2: BCA analysis for only NS2 (200 μL, 7.5 × 1011 particles/mL) diluted in FBS (5% in PBS). NS2 + A: same conditions as NS2 lane, followed by addition of 20 μM of cage A and 10 min of incubation. NS2 + A + pyr: same conditions as NS2 + A, followed by addition of 50 μM of free pyr and 10 min of incubation. (B) Cyclic addition to a colloidal dispersion of NS2 (1.5 mL, 7.5 × 1011 particles/mL) in diluted FBS (5% in PBS), with 10 μM of cage A (blue arrow) and 30 μM of free pyr (red arrow). After each addition, the dispersion was incubated for 10 min. Subsequently, the amount of adsorbed protein on 1.5 × 1011 particles was evaluated by stacking SDS-PAGE (bottom) and quantified using ImageJ (top). (C) List of proteins comprising the PC on NS2, for which the relative abundance increases or decreases more than threefold during the cage/pyranine cycle of the former SDS-PAGE assay, based on LC-MS/MS analysis. The ratio parameter is defined as the average relative abundance of the protein in steps 2, 4, and 6, (low protein immobilization) divided by the relative abundance in steps 1, 3, and 5 (high protein immobilization). Steps 1–6 are normalized to the abundance of the protein in each step.
The former assay also demonstrated that addition of pyr facilitates the recovery of the initial HC, likely because cage A is removed from the NP surface (Figure 3A). Moreover, reversible control of PC formation over NS2 could be repeatedly performed by successive additions of cage A and free pyr, thereby confirming the reversible character of this adsorption switching process (Figure 3B) and the possibility of regulating PC formation on AuNPs through external additives.
For completeness, NS2 was also incubated with 100% rat serum for 10 min. Subsequently, 100 μM of cage A was added, and the BCA assay was again performed to compare the amount of protein remaining on NS2, before and after cage addition. A decrease of 30% in protein adsorption was observed after cage addition, thereby proving that the supramolecular strategy can even be applied under in vivo-like, complex biological conditions (Figure S10).
As a control experiment to evaluate whether other positively charged polyamine molecules can be used instead of cage A, NS2 was incubated with 20 μM of spermine, in the presence of diluted FBS (5% in PBS). The BCA assay showed a reduction of 20% in the amount of protein adsorption, which is far less efficient than the 80% reduction observed for cage A under the same conditions (Figure S11). This result highlights the importance of a high affinity between both oppositely charged molecules, to obtain an efficient disruption of the PC.
Further insights regarding the effect of supramolecular interactions between A and NS2 on PC composition were obtained through a comprehensive comparison between electrospray liquid chromatography mass spectrometry (LC-MS/MS) profiles of protein coronas formed on NS2 dispersed in 5% FBS, before and after addition of 10 μM cage A. This assay showed significant differences in the relative abundance of several proteins after addition of cage A (see Table S1, steps 1 and 2), thereby proving that not all proteins are equally affected by host–guest complex formation. Furthermore, to evaluate whether the PC composition can also be reversibly controlled, five steps of cage A and pyr additions were performed with NS2, and an LC-MS/MS assay was performed for each step (Figure 3C and Tables S1 and S2). These results showed similar levels of relative abundance for each protein in the steps with high protein adsorption (steps 1, 3, and 5). Accordingly, the steps with low protein adsorption also gave similar values (steps 2, 4, and 6), thereby confirming the ability of our approach to reversibly regulate the composition and the amount of proteins of the HC on NPs.
A detailed analysis of PC composition in all steps allowed us to identify as many as 26 different proteins whose abundance after addition of cage A was reduced or increased by at least twofold (see Table S1) and 10 of them with a reduction by one-third (Figure 3C). The protein with the greatest reduction was β-enolase, which presents an overall reduction of 85% in the steps with low protein adsorption. The biological function of this protein is related to the development and regeneration of muscles. The only protein with more than twice increased presence after addition of cage A was the Tudor domain-containing protein 7, which is a component of specific cytoplasmic RNA granules involved in post-transcriptional regulation of several genes.
In an attempt to understand the variation in PC composition, we compared the isoelectric points, molecular weights, and aliphatic indexes of the different proteins. No correlation was observed between these parameters and variations in relative abundance. For instance, comparing the 10 proteins showing the most significant variations after cage A addition (Figure 3C), we found molecular weights between 50 and 500 kDa, isoelectric points between 5.3 and 8.7, and aliphatic indexes between 90 and 54. However, we also found that 8 of 26 proteins with the highest variation were involved in negative regulation of endopeptidase activity and 6 in blood coagulation. This unexpected specificity likely emerges from a structural similarity between local regions of proteins exhibiting similar biological roles32 and further proves the biological potential of our host–guest recognition strategy as a tool to control PC composition.
The above results confirmed that, via the use of host–guest interactions between cage A and exposed pyr, it is possible to disrupt protein adsorption on the NPs surface and that, by the addition of defined amounts of cage A and pyr, we can achieve efficient and reversible regulation on PC formation. While the precise molecular mechanism behind PC disruption is not entirely clear, it might be related to the presence of zwitterionic surface ligands, that is, ligands carrying both positive and negative charges, which favor the formation of a tight hydration layer around the NPs.33 Indeed, although the supramolecular nanocomposite formed by pyranine containing NS2 and cage A presents a similar Zp (+32 mV) as NS1 (+29 mV, one amino group per PEG ligand), 10 times more proteins were found to adsorb on NS1 than on the mixture of NS2 and 10 μM of A (Figures S12 and S4).
As mentioned in the introduction, PC formation hinders the contact between AuNPs and mammalian cell membranes, thereby preventing efficient internalization.34,35 We anticipated that our supramolecular approach might promote cellular uptake and eventually implement a stimuli-responsive control on the internalization. We performed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assays to assess the toxicity of NS2, both in the absence and in the presence of cage A, using human HeLa cells in Dulbecco’s Modified Eagle’s Medium (DMEM) medium containing 10% FBS. No cytotoxicity was observed for NS2 at concentrations up to 9 × 1010 particles/mL after 24 h of incubation (Figure S13). A slight decrease in cell viability (∼10%) was observed when incubation was performed in the presence of 5 μM cage A. This toxicity increase might be attributed to a superior NP uptake under these conditions (vide infra). Considering the low-to-absent levels of AuNP-induced cytotoxicity, we used the selected concentration (9 × 1010 particles/mL) for all subsequent experiments with HeLa cells.
Cell uptake of NS2 by HeLa cells, in the absence and presence of cage A, was studied using inductively coupled plasma-mass spectrometry (ICP-MS) to quantify the amount of internalized AuNPs. Incubation of HeLa cells with NS2 for 24 h, using DMEM with 10% FBS and appropriate washings, resulted in the internalization of 5800 ± 2000 particles/cell. When the same experiment was repeated in the presence of 5 μM cage A, cellular uptake increased by over an order of magnitude (74 000 ± 4000 particles/cell, Figure 4A). By using a larger amount of cage A (10 μM), we found a further increase in cellular uptake (up to 96 000 ± 2000 particles/cell, 17-fold with respect to the experiment without cage A). Additionally, in agreement with SDS-PAGE PC studies, when incubation was performed using 5 μM cage A and 10 μM free pyr, cellular uptake decreased by half (35 000 ± 10 000 particles/cell), as compared to the same experiment in the absence of pyr. Bio-TEM images of HeLa cells exposed to NS2 and cage A confirmed intracellular localization of AuNPs (Figure 4B), showing a large number of NPs clustered in cellular vesicles, as expected for an endosomal internalization pathway. In agreement with ICP-MS results, when the same incubation was performed in the absence of cage A, almost no NPs were found inside cells in TEM images (Figure S14).
Figure 4.

Uptake experiments in HeLa cells. (A) All NPs were incubated with cells using DMEM medium supplemented with 10% FBS, for 24 h, at 9 × 1010 NP/mL. For NS2 + cage A, 5 μM cage A was used. Zp values for each NP are indicated below the graph. The degree of internalization was quantified by ICP-MS. (B) TEM images of a HeLa cell incubated under the conditions described for NS2 + A.
One of the main limitations that hamper the application of NPs in medicine is the low delivery efficiency. For example, the delivery efficiency of purpose-designed NPs for solid tumors is usually less than 1% of the injected dose. The sequestration of NPs by macrophages of the mononuclear phagocytic system is one the main reasons behind this problem.10 To evaluate the generality of HC modulation as a means to regulate NP internalization, and the possibility of this strategy to avoid the mononuclear phagocytic system, cell uptake experiments were also performed using a phagocytic macrophage-like cell line (J774). It has been reported that the PC has a lower impact on the overall internalization efficiency for macrophage cell lines.36 Incubation of J774 cells with NS2 (3 × 1010 particles/mL), under the conditions described for HeLa cells, induced a relatively low internalization (4977 ± 1000 particles/cell). When the experiment was repeated in the presence of 5 μM cage A, NP uptake increased by 1 order of magnitude (50 000 ± 6000 particles/cell). These results confirm that the supramolecular strategy can be used to enhance cell internalization of relatively large NPs into various cell types, including those from the mononuclear phagocyte system, but they also indicate that the approach might eventually be used to hide NPs from the mononuclear phagocyte system.
In our previous work,26 we applied the cage/pyranine complex to trigger cellular internalization of 2 nm AuNPs, which have low nonspecific protein adsorption due to their small size,37 and concluded that the uptake switch of small NPs was related to the change in surface charge from negative to positive. We thus explored whether the switch in cell uptake for larger NS2 (15 nm) was associated with HC disruption, rather than to a mere inversion in surface charge. We therefore performed control uptake experiments with HeLa cells, using NS-PEG and amino-terminated NS1, in the presence of FBS. NS-PEG features neutral AuNP surfaces (Zp −3 mV), whereas NS1 particles are positively charged (Zp +29 mV), similar to NS2 in the presence of cage A (Zp +32 mV). Note that the size of the PC in these NPs was similar to that of NS2 in the absence of cage (Figures S4 and S12). Upon incubation, we determined internalization of 600 ± 400 particles/cell for NS-PEG and 2000 ± 1000 particles/cell for NS1, that is, a much lower uptake efficiency than for NS2 in the presence of cage A (74 000 ± 4000 particles/cell, Figure 4A). In agreement with the HC disruption mechanism, when the incubation of cells was performed in the absence of FBS, that is, without the PC effect, the uptake of NS2 in the presence of cage was only 3 times higher than that of NS1, 210 000 ± 40 000 and 74 000 ± 10 000 particles/cell, respectively. Taking into consideration these results and the poor cell internalization ability of cage A,25 we can safely conclude that the internalization of NS2 promoted by a supramolecular mechanism is related to the disruption of the HC. However, it has been shown that, in some cases, zwitterionic NPs display a lower cellular uptake than cationic NPs, even in the presence of FBS.38 Therefore, we propose that the dramatic increase in cellular uptake observed in our system stems from a synergistic effect between PC suppression and the charge switch from negative to positive.
Finally, considering that AuNPs induce local heating when illuminated by a resonant laser,39 we explored the application of the conditional cell uptake strategy to enhance photothermal therapy. Gold nanorods (AuNRs) are particularly interesting for hyperthermia applications, because their longitudinal localized surface plasmon resonance (LSPR) can be readily tuned within the near-infrared (NIR) region (700–850 nm), that is, within the first biological transparency window.40 Although AuNRs can efficiently absorb and scatter light, absorption largely dominates for small NRs,41 which is highly beneficial for efficient plasmonics-based photothermal therapy. In this context, we investigated the application of our conditional uptake strategy with small AuNRs (29 × 9 nm) featuring a longitudinal LSPR centered around 800 nm (Figure 5a). AuNRs (NR1) were synthesized by seeded growth,42 and their surface was subsequently decorated with pyranines, following the same protocol used for NS2, thus resulting in an equivalent NR3 (Figure S15).
Figure 5.
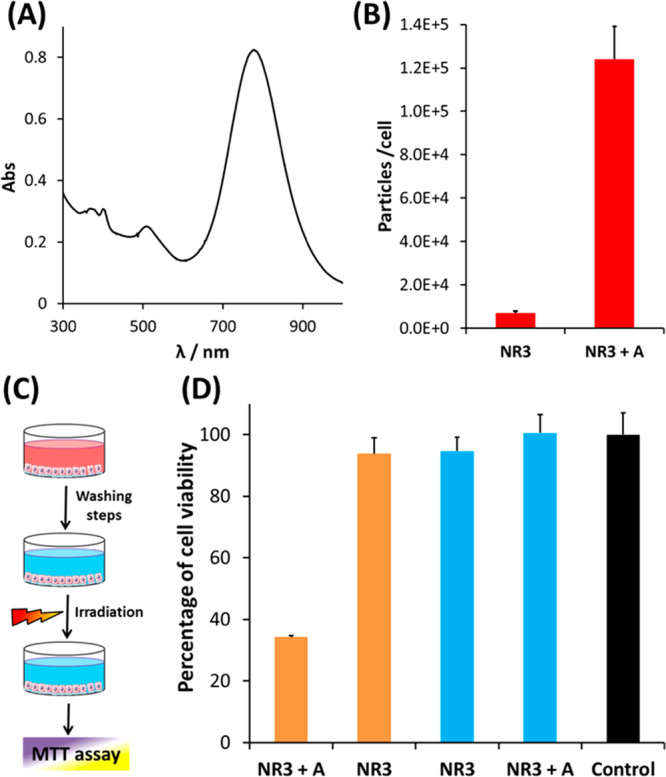
(A) UV–Vis–NIR spectrum of NR3. (B) Cell uptake of NR3 (9 × 1010 NR/mL) by HeLa cells, in the absence and in the presence of cage A (5 μM), as determined by ICP-MS. The incubation was performed in DMEM medium with 10% FBS for 24 h. (C) Schematic representation of the protocol followed in the NIR-laser hyperthermia experiments. HeLa cells were first incubated with NR3 (9 × 1010 NR/mL) for 24 h in DMEM medium with 10% FBS, with or without 5 μM cage A. Subsequently, noninternalized nanorods were removed by washing with PBS, and cells were irradiated with an 808 nm diode laser (Lumics, LU808T040) for 20 min at a power density of 3.2 W/cm2. Cell viability was calculated using the MTT assay (average of triplicate wells ± standard deviation). (D) Results of the irradiation experiment. The control bar (black) represents cell viability of the cells in the absence of cage A and NR3. Blue bars represent incubation without irradiation, proving that uptaken NR3 in the presence of cage A did not induce cytotoxicity.
As expected from our results with nanospheres, when cage A (5 μM) was mixed with NR3 in dilute FBS (5%), the amount of adsorbed proteins in the HC was reduced by more than half with respect to the system without cage (Figure S16). The cellular uptake behavior of NR3 was similar to that of NS2; that is, when a dispersion of 9 × 1010 NR/mL was added to HeLa cells and the mixture was incubated for 24 h using DMEM with 10% FBS, we determined a cellular uptake of only 4000 ± 1000 NR/cell, which increased to 126 000 ± 20 000 particles/cell (30-fold increase) in the presence of 5 μM cage A (Figure 5B).
We then explored whether the cage-responsive uptake of NR3 could be used for conditional control of cytotoxicity, in combination with NIR irradiation. HeLa cells were incubated with NR3 in DMEM with 10% FBS for 24 h, both in the absence and in the presence of cage A. Control experiments showed that NR3 is not cytotoxic at this concentration, regardless of the presence of cage A (Figure 5D, blue bars). After incubation, cells were washed twice with PBS to remove noninternalized AuNRs and then irradiated using a fiber-coupled 808 nm diode laser for 20 min. The viability of cells treated with NR3 only was roughly the same as that of non-irradiated cells. However, for those cells incubated with NR3 in the presence of cage A, the viability was reduced to 34% due to heat released from NR3 during laser irradiation (Figure 5D).
Conclusions
We have presented an efficient strategy for stimuli-responsive, reversible control of protein corona formation over gold nanoparticles. The approach is based on the supramolecular interaction between surface-exposed pyranines and positively charged supramolecular cages, resulting in the generation of PC-disrupting zwitterionic ligands. The strategy is fully reversible, so that the PC status can be fully controlled by varying amounts of cage A and externally added pyranines. Cellular uptake experiments with both cancer and phagocytic cells confirmed that this stimuli-responsive PC regulation provides an accurate handle over cell internalization of AuNPs, which is enhanced in the presence of the external cage. It was observed that PC composition after addition of the cage was preferentially impoverished in specific serum proteins with comparable functions. Therefore, this conditional release of specific proteins, which can be reversibly controlled by the addition of cage A and pyr, might find interesting biological applications. Finally, preliminary optical hyperthermia studies demonstrate the feasibility of regulating the laser-induced cytotoxicity of Au nanorods using external additives. This dual control on the toxicity using light and organic molecules is promising and deserves further studies, owing to its biomedical potential.
Methods
Synthesis of NS1
Citrate-stabilized Au nanospheres (Au NSs) of 14.8 nm of diameter were synthesized according to the standard Turkevich method.43 An aqueous suspension of as-synthesized citrate-stabilized Au NSs (0.4 mM in Au, 2 mL) was placed in a 2 mL centrifuge tube (1 mL solution in each tube). Then, a solution 10 mM of α-amino-ω-mercapto poly(ethylene glycol) hydrochloride (HS-PEG-NH2) (MW = 5000, 68 μL, calculated to provide 200 molecules per nm2) was added under stirring to each tube and incubated at room temperature for 1 h. Subsequently, AuNPs were centrifuged (18 500g, 30 min) and washed with water three times. Finally, the tubes were centrifuged again to increase the concentration, and the concentrated dispersions from both tubes were mixed.
Synthesis of NS2
Pyr-CO2H was covalently conjugated to NS1 with the aid of 1-ethyl-3-(3 dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS). Pyr-CO2H (7.0 mg, 0.012 mmol), EDC (3.4 mg, 0.018 mmol), and NHS (2.0 mg, 0.018 mmol) were dissolved in 0.6 mL of 2-ethanesulfonic acid (MES) buffer (0.1 M, pH = 6.4). The mixture was stirred for 15 min at room temperature to activate the dye. This solution was subsequently added to a 200 μL solution of NS1 ([Au] = 1.44 mM) and stirred at room temperature for 14 h. The AuNSs were then centrifuged (18 500g, 30 min) and washed several times with phosphate buffer (20 mM, pH 7.5) and Milli-Q water, until no Pyr-CO2H was detected in the washing phase. Finally, the nanoparticles were dispersed in 500 μL of Milli-Q water.
Synthesis of NR1
NR1 (longitudinal LSPR at 800 nm; length 29 nm; width 9 nm) was prepared using the seeded-growth method with some modifications, as previously described by Nikoobakht and El-Sayed.44 Seeds were prepared by the standard cetyl trimethylammonium bromide (CTAB)/NaBH4 procedure: 25 μL of a 0.05 M HAuCl4 solution was added to 4.7 mL of a 0.1 M CTAB solution, followed by injection of 300 μL of a freshly prepared 0.01 M NaBH4 solution, under vigorous stirring (1500 rpm). Excess borohydride was consumed by aging the seed solution for 30 min at room temperature prior to use. In a typical synthesis of a 50 mL Au NR solution, 500 μL of 0.01 M AgNO3, 1 mL of 1 M HCl, 500 μL of a 0.05 M HAuCl4, and 800 μL of 0.1 M ascorbic acid solution were added to 50 mL of 0.1 M CTAB. Then, 3 mL of the seed solution was added under stirring. The mixture was left undisturbed at 25 °C for at least 6 h. The resulting gold nanorods were centrifuged in 50 mL tubes (5000g, 60 min) to remove spherical impurities. The precipitate was discarded, and the supernatant was centrifuged at 18 500g for 45–60 min in 2 mL tubes. The precipitate was redispersed in 4 mL of a 2 mM CTAB solution. The concentrated Au NR colloid was centrifuged again at 18 500g for 45–60 min. This step was repeated twice.
Synthesis of NR2
An aqueous dispersion of NR1 (0.4 mM in Au, 1 mM of CTAB, 10 mL) was placed in 2 mL centrifuge tubes (1 mL solution in each tube). The dispersions were centrifuged (18 500g, 30 min) to reduce the volume to 100 μL. At this point, a 10 mM solution of HS-PEG-NH2 (MW = 5000, 71 μL, calculated to provide 200 molecules per nm2) was added under stirring to each tube. Subsequently, 800 μL of water was added, and the vials were stirred for 3 h. Finally, the AuNPs were centrifuged (18 500g, 30 min) and washed with water three times. Finally, the tubes were centrifuged again to increase the concentration, and the concentrated dispersions from both tubes were mixed.
Synthesis of NR3
Pyr-CO2H was covalently conjugated to NR2 with the aid of EDC and NHS. Pyr-CO2H (7.0 mg, 0.012 mmol), EDC (3.4 mg, 0.018 mmol), and NHS (2.0 mg, 0.018 mmol) were dissolved in 0.6 mL of MES buffer (0.1 M, pH = 6.4). The mixture was stirred for 15 min at room temperature to activate the dye. This solution was subsequently added to a 200 μL solution of NR2 ([Au] = 1.40 mM) and stirred at room temperature for 14 h. Finally, Au NRs were centrifuged (18 500g, 30 min) and washed several times with phosphate buffer (20 mM, pH 7.5) and Milli-Q water, until no Pyr-CO2H was detected in the washing phase. Finally, the NRs were dispersed in 500 μL of Milli-Q water.
Quantification of Pyranine in NS2
The amount of pyranine dye bound to NS2 was determined by ICP-MS, for the determination of the amount of gold. The average diameter was estimated from TEM micrographs of NS2 (14.8 ± 0.9 nm), and the number of gold atoms per NS was deduced according to the density of gold (1.932 × 10–20 g/nm3). The amount of pyr in solution was calculated using fluorescence spectroscopy after etching of NS2 with KCN (380 nm excitation wavelength). The fluorescence signal obtained at 430 nm after etching was used to calculate the amount of pyr in solution, using a calibration curve for Pyr-CO2H. The number of pyr molecules per NS was ca. 408.
Proteins Immobilized on NPs in the Presence and Absence of Cage A
To study the protein corona on gold nanoparticles, a dispersion of the corresponding NPs (7.5 × 1011 particle/mL) was prepared using diluted FBS [5% in 10 mM PBS] or 100% rat serum following the conditions described in the main text. Once the incubation was performed, AuNPs (1.5 × 1011) were separated from nonadsorbed proteins by careful centrifugation (10 °C, 19 000g, 30 min). In those cases in which the incubation was performed in the presence of cage A (with the exception of those incubations in which free pyr was subsequently added), the next washing step was performed using an aqueous solution of cage A (5 μM) to avoid the interaction between remaining FBS proteins and the NPs, after removal of A in the first washing step. Subsequently, AuNPs were extensively washed with PBS by centrifugation at 19 000g and 10 °C for 10 min, to remove all unbound proteins, until the supernatant was free of any detectable protein by Bradford assay. Absorbance values at 400 nm confirmed in all cases the quantitative recovery of the NPs during the washing steps. Subsequently, the remaining adsorbed proteins, forming the HC, were detached from AuNPs upon treatment with SDS (10%) and DTT (0.5 mM) at 90 °C for 10 min. The protein content was analyzed by SDS-PAGE or the BCA assay (see Supporting Information), and the chemical composition of HC was analyzed by LC-MS/MS.
SDS-PAGE Electrophoresis
Proteins immobilized on AuNPs (1.5 × 1011 particles) were mixed with 20 μL of Novex Tris-Glycine SDS buffer (1X Thermo Scientific), 5 μL of NuPAGE reducing buffer (10x, Thermo Scientific), and 10 μL of Milli-Q water. The mixture was boiled for 2 min at 95 °C. Treated samples were then loaded in Novex Tris-Glycine MiniGel of 10 wells 4–20% (Thermo Scientific), and the gels were run for 45 min at 120 mV in Novex Tris-Glycine SDS (1X) running buffer. Staining was performed with Coomassie Blue (Aldrich) for 2 h, followed by washing in Milli-Q water for 3–4 d. Proteins adsorbed onto AuNPs were quantified by using ImageJ.
Mass Spectrometry
Proteins immobilized onto AuNPs (1.5 × 1011 particles) were mixed with 20 μL of Novex Tris-Glycine SDS buffer (1X Thermo Scientific), 5 μL of NuPAGE reducing buffer (10X, Thermo Scientific), and 10 μL of Milli-Q water. The mixtures were boiled for 2 min at 95 °C. Treated samples were then loaded in Novex Tris-Glycine MiniGel of 10 wells 10% (Thermo Scientific), and the gels were run for 5 min at 120 mV in Novex Tris-Glycine SDS (1X) running buffer. Staining was performed with Coomassie Blue (Aldrich) for 2 h, followed by washing in Milli-Q water for 3–4 d. Proteins adsorbed onto AuNPs were quantified by using ImageJ. Bands of interest were washed in milli-Q water. Reduction and alkylation were performed using ditiothreitol (10 mM DTT in 50 mM ammonium bicarbonate) at 56 °C for 20 min, followed by iodoacetamide (50 mM iodoacetamide in 50 mM ammonium bicarbonate) for another 20 min in the dark. Gel pieces were dried and incubated with trypsin (12.5 μg/mL in 50 mM ammonium bicarbonate) for 20 min on ice. After rehydration, the trypsin supernatant was discarded; gel pieces were hydrated with 50 mM ammonium bicarbonate and incubated overnight at 37 °C. After digestion, acidic peptides were cleaned with trifluoroacetic acid (TFA) 0.1% and dried out in a RVC2 25 speedvac concentrator (Christ). Peptides were resuspended in 10 μL 0.1% formic acid (FA), sonicated for 5 min, and analyzed by LC-MS/MS. For a complete description on LC/MS/MS see Supporting Information.
Cell Cultures and Internalization Studies
HeLa or J774 cells were plated in 96-well microplates, 1.4 × 104 cells/well, and allowed to adhere overnight. The following day cell media was replaced with nanoparticle solution (final concentration 9 × 1010 particles/mL for HeLa cells and 3 × 1010 particles/mL for J774 cells) diluted in DMEM/FBS (two wells for each replicate). Cells were incubated with NPs for 24 h at 37 °C, followed by removal of the NPs and two washing steps with DMEM/FBS. Adherent cells were trypsinized, and samples were frozen overnight at −20 °C (at this point, the duplicate wells were combined). The resulting cell lysates were digested for 48 h using 500 μL of aqua regia. The sample solutions were then diluted to 3 mL with deionized water. The assays were performed in triplicate for each particle. The cells were counted prior to the addition of aqua regia to determine the number of cells.
The gold concentration in colloidal samples was determined by ICP-MS under the following operating conditions: Radio frequency power: 1550 W; plasma Ar flow rate: 14 L/min; nebulizer Ar flow rate: 1.12 L/min; auxiliary gas rate: 0.8 L/min; isotopes were monitored in kinetic energy determination (KED) mode: 197Au/193Ir (internal standard); dwell time: 10 ms; quartz cyclonic spray chamber.
Cellular Uptake Imaging by TEM
TEM was used to study the intracellular location of NP2, with and without cage A. HeLa cells were plated at 2 × 105 cells/well in a 12-well plate. Cells were allowed to adhere overnight, followed by addition of NP2 (9 × 1010 NP/mL), with and without cage A (5 μM). After 24 h of incubation, cells were washed and trypsinized. Cell pellets were washed with Sorensen’s buffer (0.1 M) and fixed with a 2% formaldehyde/2.5% glutaraldehyde solution in Sorensen’s buffer, initially for 10 min at room temperature, followed by replacement of the fixing solution and incubation at 4 °C for 4 h. Samples were washed with Sorensen’s buffer, stained with 1% OsO4, dehydrated, and embedded in Spurr’s resin. Once polymerized, 60 nm thick slices were cut using an ultramicrotome. Samples were imaged using TEM (JEOL JEM-1400PLUS, 40–120 kV).
NR3 Irradiation Assays
HeLa cells were plated in a 96-well plate at 1.4 × 104 cells/well and allowed to adhere overnight. The following day media was replaced with NR3, in the presence or absence of cage A. NRs were left overnight with cells (∼24 h) followed by laser irradiation after removing nonendocytosed NPs. Irradiation was performed with an 808 nm diode laser, illuminating the whole well (0.4 cm spot size) (t = 20 min, p = 3.2 W/cm2, VT = 200 μL/well). Cell viability was analyzed using the MTT assay, and absorbance was measured at 550 nm, showing both non-irradiated and irradiated wells, and in presence or absence of cage A.
Acknowledgments
Funding was received from MINECO (SAF2016-76689-R, MAT2017-86659-R), Xunta de Galicia (2015-CP082, ED431C 2017/19, Centro singular de investigación de Galicia accreditation 2019-2022, ED431G 2019/03), the European Union (European Regional Development Fund), and the European Research Council (ERC AdG No. 787510 to L.M.L.-M.; ERC AdG No. 340055 to J.L.M). J.M. and M.M.-C. thank MINECO for Juan de la Cierva fellowships (FJCI-2015-25080 and IJCI-2014-19326). The proteomic analysis was performed in the proteomics platform at CIC bioGUNE, which is supported by Basque Department of Industry, Tourism and Trade (Etortek and Elkartek programs), the Innovation Technology Department of the Bizkaia County; The ProteoRed-ISCIII (Grant No. PRB3 IPT17/0019); CIBERehd Network, and Severo Ochoa Grant (No. SEV-2016-0644). The authors thank G. González-Rubio for providing NR1. This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency, Grant No. MDM-2017-0720.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b08752.
General experimental procedures, nanoparticles characterization, protein corona studies, mass spectrometry analysis, cell viability assays (PDF)
Author Present Address
⊥ (J.M.) Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, The Netherlands.
The authors declare no competing financial interest.
Dedication
Dedication Dedicated to Professor E.W. “Bert” Meijer on the occasion of his 65th birthday.
Supplementary Material
References
- Mura S.; Nicolas J.; Couvreur P. Stimuli-Responsive Nanocarriers for Drug Delivery. Nat. Mater. 2013, 12, 991–1003. 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- Kim D.; Shin K.; Kwon S. G.; Hyeon T. Synthesis and Biomedical Applications of Multifunctional Nanoparticles. Adv. Mater. 2018, 30, 1802309. 10.1002/adma.201802309. [DOI] [PubMed] [Google Scholar]
- Lee D.-E.; Koo H.; Sun I.-C.; Ryu J. H.; Kim K.; Kwon I. C. Multifunctional Nanoparticles for Multimodal Imaging and Theragnosis. Chem. Soc. Rev. 2012, 41, 2656–2672. 10.1039/C2CS15261D. [DOI] [PubMed] [Google Scholar]
- Pelaz B.; Alexiou C.; Alvarez-Puebla R. A.; Alves F.; Andrews A. M.; Ashraf S.; Balogh L. P.; Ballerini L.; Bestetti A.; Brendel C.; Bosi S.; Carril M.; Chan W. C. W.; Chen C.; Chen X.; Chen X.; Cheng Z.; Cui D.; Du J.; Dullin C.; et al. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. 10.1021/acsnano.6b06040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; El-Sayed M. A. Gold Nanoparticles: Optical Properties and Implementations in Cancer Diagnosis and Photothermal Therapy. J. Adv. Res. 2010, 1, 13–28. 10.1016/j.jare.2010.02.002. [DOI] [Google Scholar]
- Giljohann D. A.; Seferos D. S.; Daniel W. L.; Massich M. D.; Patel P. C.; Mirkin C. A. Gold Nanoparticles for Biology and Medicine. Angew. Chem., Int. Ed. 2010, 49, 3280–3294. 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A.; Rivera Gil P.; Zhang F.; Zanella M.; Parak W. J. Biological Applications of Gold Nanoparticles. Chem. Soc. Rev. 2008, 37, 1896–1908. 10.1039/b712170a. [DOI] [PubMed] [Google Scholar]
- Ayala-Orozco C.; Urban C.; Knight M. W.; Urban A. S.; Neumann O.; Bishnoi S. W.; Mukherjee S.; Goodman A. M.; Charron H.; Mitchell T.; Shea M.; Roy R.; Nanda S.; Schiff R.; Halas N. J.; Joshi A. Au Nanomatryoshkas as Efficient Near-Infrared Photothermal Transducers for Cancer Treatment: Benchmarking against Nanoshells. ACS Nano 2014, 8, 6372–6381. 10.1021/nn501871d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovtzer R.; Agrawal A.; Kotov N. A.; Popovtzer A.; Balter J.; Carey T. E.; Kopelman R. Targeted Gold Nanoparticles Enable Molecular CT Imaging of Cancer. Nano Lett. 2008, 8, 4593–4596. 10.1021/nl8029114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S.; Tavares A. J.; Dai Q.; Ohta S.; Audet J.; Dvorak H. F.; Chan W. C. W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. 10.1038/natrevmats.2016.14. [DOI] [Google Scholar]
- Gustafson H. H.; Holt-Casper D.; Grainger D. W.; Ghandehari H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. 10.1016/j.nantod.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V. H.; Lee B.-J. Protein Corona: A New Approach for Nanomedicine Design. Int. J. Nanomed. 2017, 12, 3137–3151. 10.2147/IJN.S129300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvati A.; Pitek A. S.; Monopoli M. P.; Prapainop K.; Bombelli F. B.; Hristov D. R.; Kelly P. M.; Åberg C.; Mahon E.; Dawson K. A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137–143. 10.1038/nnano.2012.237. [DOI] [PubMed] [Google Scholar]
- Lesniak A.; Salvati A.; Santos-Martinez M. J.; Radomski M. W.; Dawson K. A.; Åberg C. Nanoparticle Adhesion to the Cell Membrane and Its Effect on Nanoparticle Uptake Efficiency. J. Am. Chem. Soc. 2013, 135, 1438–1444. 10.1021/ja309812z. [DOI] [PubMed] [Google Scholar]
- Mosquera J.; García I.; Liz-Marzán L. M. Cellular Uptake of Nanoparticles versus Small Molecules: A Matter of Size. Acc. Chem. Res. 2018, 51, 2305–2313. 10.1021/acs.accounts.8b00292. [DOI] [PubMed] [Google Scholar]
- Rana S.; Yeh Y.-C.; Rotello V. M. Engineering the Nanoparticle–Protein Interface: Applications and Possibilities. Curr. Opin. Chem. Biol. 2010, 14, 828–834. 10.1016/j.cbpa.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjidemetriou M.; McAdam S.; Garner G.; Thackeray C.; Knight D.; Smith D.; Al-Ahmady Z.; Mazza M.; Rogan J.; Clamp A.; Kostarelos K. The Human In Vivo Biomolecule Corona onto PEGylated Liposomes: A Proof-of-Concept Clinical Study. Adv. Mater. 2019, 31, 1803335. 10.1002/adma.201803335. [DOI] [PubMed] [Google Scholar]
- Mosquera J.; García I.; Henriksen-Lacey M.; González-Rubio G.; Liz-Marzán L. M. Reducing Protein Corona Formation and Enhancing Colloidal Stability of Gold Nanoparticles by Capping with Silica Monolayers. Chem. Mater. 2019, 31, 57–61. 10.1021/acs.chemmater.8b04647. [DOI] [Google Scholar]
- Aggarwal P.; Hall J. B.; McLeland C. B.; Dobrovolskaia M. A.; McNeil S. E. Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Adv. Drug Delivery Rev. 2009, 61, 428–437. 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro E.; Pons T.; Lequeux N.; Fragola A.; Sanson N.; Lenkei Z.; Dubertret B. Small and Stable Sulfobetaine Zwitterionic Quantum Dots for Functional Live-Cell Imaging. J. Am. Chem. Soc. 2010, 132, 4556–4557. 10.1021/ja1005493. [DOI] [PubMed] [Google Scholar]
- Moyano D. F.; Saha K.; Prakash G.; Yan B.; Kong H.; Yazdani M.; Rotello V. M. Fabrication of Corona-Free Nanoparticles with Tunable Hydrophobicity. ACS Nano 2014, 8, 6748–6755. 10.1021/nn5006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García K. P.; Zarschler K.; Barbaro L.; Barreto J. A.; O’Malley W.; Spiccia L.; Stephan H.; Graham B. Zwitterionic-Coated “Stealth” Nanoparticles for Biomedical Applications: Recent Advances in Countering Biomolecular Corona Formation and Uptake by the Mononuclear Phagocyte System. Small 2014, 10, 2516–2529. 10.1002/smll.201303540. [DOI] [PubMed] [Google Scholar]
- Mosquera J.; Zarra S.; Nitschke J. R. Aqueous Anion Receptors through Reduction of Subcomponent Self-Assembled Structures. Angew. Chem., Int. Ed. 2014, 53, 1556–1559. 10.1002/anie.201308117. [DOI] [PubMed] [Google Scholar]
- Mosquera J.; Zhao Y.; Jang H.-J.; Xie N.; Xu C.; Kotov N. A.; Liz-Marzán L. M. Plasmonic Nanoparticles with Supramolecular Recognition. Adv. Funct. Mater. 2020, 30, 1902082. 10.1002/adfm.201902082. [DOI] [Google Scholar]
- Rodríguez J.; Mosquera J.; Couceiro J. R.; Nitschke J. R.; Vázquez M. E.; Mascareñas J. L. Anion Recognition as a Supramolecular Switch of Cell Internalization. J. Am. Chem. Soc. 2017, 139, 55–58. 10.1021/jacs.6b11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosquera J.; Henriksen-Lacey M.; García I.; Martínez-Calvo M.; Rodríguez J.; Mascareñas J. L.; Liz-Marzán L. M. Cellular Uptake of Gold Nanoparticles Triggered by Host–Guest Interactions. J. Am. Chem. Soc. 2018, 140, 4469–4472. 10.1021/jacs.7b12505. [DOI] [PubMed] [Google Scholar]
- Monopoli M. P.; Åberg C.; Salvati A.; Dawson K. A. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol. 2012, 7, 779–786. 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- Hadjidemetriou M.; Kostarelos K. Nanomedicine: Evolution of the Nanoparticle Corona. Nat. Nanotechnol. 2017, 12, 288–290. 10.1038/nnano.2017.61. [DOI] [PubMed] [Google Scholar]
- Walkey C. D.; Chan W. C. W. Understanding and Controlling the Interaction of Nanomaterials with Proteins in a Physiological Environment. Chem. Soc. Rev. 2012, 41, 2780–2799. 10.1039/C1CS15233E. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A. Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- García I.; Sánchez-Iglesias A.; Henriksen-Lacey M.; Grzelczak M.; Penadés S.; Liz-Marzán L. M. Glycans as Biofunctional Ligands for Gold Nanorods: Stability and Targeting in Protein-Rich Media. J. Am. Chem. Soc. 2015, 137, 3686–3692. 10.1021/jacs.5b01001. [DOI] [PubMed] [Google Scholar]
- Petrey D.; Fischer M.; Honig B. Structural Relationships among Proteins with Different Global Topologies and Their Implications for Function Annotation Strategies. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 17377–17382. 10.1073/pnas.0907971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenoff J. B. Zwitteration: Coating Surfaces with Zwitterionic Functionality to Reduce Nonspecific Adsorption. Langmuir 2014, 30, 9625–9636. 10.1021/la500057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesniak A.; Fenaroli F.; Monopoli M. P.; Åberg C.; Dawson K. A.; Salvati A. Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells. ACS Nano 2012, 6, 5845–5857. 10.1021/nn300223w. [DOI] [PubMed] [Google Scholar]
- Hühn D.; Kantner K.; Geidel C.; Brandholt S.; De Cock I.; Soenen S. J. H.; Rivera Gil P.; Montenegro J. M.; Braeckmans K.; Müllen K.; Nienhaus G. U.; Klapper M.; Parak W. P. Polymer-Coated Nanoparticles Interacting with Proteins and Cells: Focusing on the Sign of the Net Charge. ACS Nano 2013, 7, 3253–3263. 10.1021/nn3059295. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Gause K. T.; Kamphuis M. M. J.; Ang C.-S.; O’Brien-Simpson N. M.; Lenzo J. C.; Reynolds E. C.; Nice E. C.; Caruso F. Differential Roles of the Protein Corona in the Cellular Uptake of Nanoporous Polymer Particles by Monocyte and Macrophage Cell Lines. ACS Nano 2013, 7, 10960–10970. 10.1021/nn404481f. [DOI] [PubMed] [Google Scholar]
- Piella J.; Bastús N. G.; Puntes V. Size-Dependent Protein–Nanoparticle Interactions in Citrate-Stabilized Gold Nanoparticles: The Emergence of the Protein Corona. Bioconjugate Chem. 2017, 28, 88–97. 10.1021/acs.bioconjchem.6b00575. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Huo S.; Mizuhara T.; Das R.; Lee Y.-W.; Hou S.; Moyano D. F.; Duncan B.; Liang X.-J.; Rotello V. M. The Interplay of Size and Surface Functionality on the Cellular Uptake of Sub-10 nm Gold Nanoparticles. ACS Nano 2015, 9, 9986–9993. 10.1021/acsnano.5b03521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Jain P. K.; El-Sayed I. H.; El-Sayed M. A. Plasmonic Photothermal Therapy (PPTT) Using Gold Nanoparticles. Lasers Med. Sci. 2008, 23, 217–228. 10.1007/s10103-007-0470-x. [DOI] [PubMed] [Google Scholar]
- Huff T. B.; Tong L.; Zhao Y.; Hansen M. N.; Cheng J.-X.; Wei A. Hyperthermic Effects of Gold Nanorods on Tumor Cells. Nanomedicine 2007, 2, 125–132. 10.2217/17435889.2.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.-H.; Murphy C. J. Mini Gold Nanorods with Tunable Plasmonic Peaks beyond 1000 Nm. Chem. Mater. 2018, 30, 1427–1435. 10.1021/acs.chemmater.7b05310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rubio G.; Kumar V.; Llombart P.; Díaz-Núñez P.; Bladt E.; Altantzis T.; Bals S.; Peña-Rodríguez O.; Noya E. G.; MacDowell L. G.; Guerrero-Martínez A.; Liz-Marzán L. M. Disconnecting Symmetry Breaking from Seeded Growth for the Reproducible Synthesis of High Quality Gold Nanorods. ACS Nano 2019, 13, 4424–4435. 10.1021/acsnano.8b09658. [DOI] [PubMed] [Google Scholar]
- Enustun B. V.; Turkevich J. Coagulation of Colloidal Gold. J. Am. Chem. Soc. 1963, 85, 3317–3328. 10.1021/ja00904a001. [DOI] [Google Scholar]
- Nikoobakht B.; El-Sayed M. A. Preparation and Growth Mechanism of Gold Nanorods (NRs) Using Seed-Mediated Growth Method. Chem. Mater. 2003, 15, 1957–1962. 10.1021/cm020732l. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.