

Abstract
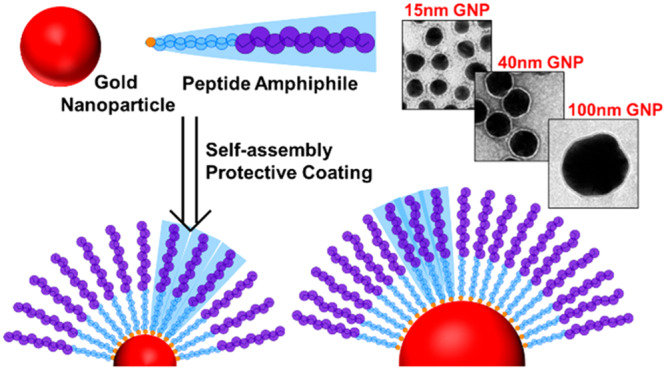
The functionalization of gold nanoparticles (GNPs) with peptidic moieties can prevent their aggregation and facilitate their use for applications both in vitro and in vivo. To date, no peptide-based coating has been shown to stabilize GNPs larger than 30 nm in diameter; such particles are of interest for applications including vaccine development, drug delivery, and sensing. Here, GNPs with diameters of 20, 40, and 100 nm are functionalized with peptide amphiphiles. Using a combination of transmission electron microscopy, UV–vis spectroscopy, and dynamic light scattering, we show that GNPs up to 100 nm in size can be stabilized by these molecules. Moreover, we demonstrate that these peptide amphiphiles form curvature-dependent, ordered structures on the surface of the GNPs and that the GNPs remain disperse at high-salt concentrations and in the presence of competing thiol-containing molecules. These results represent the development of a peptide amphiphile-based coating system for GNPs which has the potential to be beneficial for a wide range of biological applications, in addition to image enhancement and catalysis.
Keywords: gold nanoparticles, peptide amphiphiles, nanoparticle stability, β-sheet, self-assembled monolayer
Gold nanoparticles (GNPs) have been extensively studied in recent years and have found wide-ranging applications in areas including photothermal therapy,1,2 drug delivery,3,4 catalysis,5−7 biosensing,8,9 and vaccine development.10−12 GNPs are suited to such purposes due to their unique optical properties,13 ease of functionalization,14 facile synthesis, and programmable shape and size.15−17
One major disadvantage of GNPs is that they are toxic both in vitro and in vivo.18−20 GNPs also have a tendency to aggregate, resulting in limited mobility which restricts the possibility of targeted delivery and prevents a uniform size distribution of GNPs, properties which are crucial requirements for drug delivery and photothermal therapy for example.
To circumvent these issues, GNPs can be coated with a moiety that prevents aggregation. Examples of GNP coatings include silica,21 PEG,22,23 low molecular weight alkanethiols,24,25 and thiolated fatty acids.26,27 While these coatings reduce the toxicity of GNPs, they have inherent limitations, for example, silica coatings are porous and often heterogeneous,28,29 meaning they are reactive to components of cell culture media. Many polymers are large in size, therefore the diameters of polymer-coated GNPs are greatly increased, which limits cellular penetration. Additionally, polymers often exhibit packing defects that leave the gold surface exposed and cause aggregation.30 Alkanethiols and fatty acids are inherently hydrophobic, therefore they have poor solubility and can interact strongly with cellular membranes.31
A solution to many of these issues is to employ peptides as they are inherently biocompatible, can be programmed to possess well-defined folds, and can be facilely modified with targeting ligands or therapeutic molecules. Several peptide sequences have been developed, the most notable of which is CALNN, a five-amino acid peptide developed by the Levy group.32 When this peptide is attached to small (<30 nm) GNPs, the resulting conjugates have been shown to be stable at high-salt concentrations (>500 mM) and resistant to physical manipulations including size-exclusion chromatography (SEC), centrifugation, and lyophilization. Functional moieties have been appended to the peptide and have been demonstrated to remain active, for example, biotin-functionalized peptides bind streptavidin or avidin and act as a platform for biosensing;32,33 ligands such as abscisic acid are recognized by specific antibodies;34 and peptidic substrates are cleaved by their corresponding enzymes.35 There is no information available regarding the stability of larger GNPs (>30 nm) coated with CALNN.34,36 Similarly, other peptide sequences including CLPFFD37 and CGFAILSS38 have only been employed to stabilize small (i.e., < 30 nm) GNPs.
A second effective approach to GNP stabilization is to employ a short, thiolated, alkyl chain coupled to either a PEG molecule or a peptide.39−41 This method results in a thin GNP coating, and the packing of the alkyl chains is uniform. It is theorized that the success of this coating approach is due, in addition to the tight packing, to the hydrophobic nature of the alkyl chain, meaning water molecules are depleted at the GNP surface, which helps stabilize the particles. GNPs with such coatings have been equipped with targeting peptides and proven to fulfill the designed function.42 As with the peptide-coated GNPs, particles >35 nm in diameter were not stabilized effectively.39
Peptide amphiphiles are a class of molecules which possess both a hydrophilic peptidic portion and a hydrophobic alkyl tail.43,44 These molecules combine the favorable properties of alkyl chains and peptides, namely hydrophobicity, tight, ordered packing, and the formation of defined structures; therefore, we hypothesize that peptide amphiphiles could be used to effectively coat and stabilize GNPs.
We have developed a series of peptide amphiphiles using peptide sequences developed by the Stupp group.45 These have a sequence of (Val)v(Ala)a(Xxx)x where, usually, v = 3, a = 3, (Xxx)x = Glu3 or Lys3, although other combinations have been published.46 Alkyl chains of 11- or 16-carbons in length were attached to the peptide N-terminus. Such amphiphiles adopt a β-sheet conformation and form long cylindrical fibers in aqueous environments. Although peptide conformations have been shown to differ when in solution or when confined to a flat or curved surface,38,47 it has been hypothesized that the ability of peptides to self-assemble and adopt a defined conformation is important for GNP stabilization.38
To elucidate the impact of both domains, peptides were evaluated with and without alkyl chains attached, and the alkyl chain length was varied. In addition, our designed constructs were compared to state-of-the-art peptides. Initially, the structures and self-assembly properties of the peptides and amphiphiles were probed in solution. A coating protocol was subsequently devised, and the resulting GNP-peptide/amphiphile conjugates were assessed using UV–vis spectroscopy, dynamic light scattering (DLS), and transmission electron microscopy (TEM) to determine the aggregation degree, which provided insights as to how effectively the GNPs were stabilized. The secondary structure of the coatings on the surface of GNPs of different sizes was evaluated using Fourier transform-infrared (FT-IR) spectroscopy to determine if secondary structure affected stabilization. This technique also allowed us to probe whether confining the peptides and amphiphiles to surfaces of differing curvatures altered their secondary structure. In addition, the effects of high-salt concentrations and the presence of dithiothreitol (DTT), a competitive thiol-containing ligand, were also evaluated to determine the feasibility of employing such gold bioconjugates for applications in vitro and in vivo.
Results and Discussion
Peptide and Lipopeptide Design
Peptide amphiphiles comprising a thiolated alkyl tail of either 11 or 16 carbons in length and a peptide domain with the sequence V3A3E3 were designed and synthesized. A third construct with two C-11 alkyl chains was generated to investigate the effect of increased steric bulk within the hydrophobic domain and to probe the presence of a bidentate gold-binding motif. The sequences and structures of these three peptide amphiphiles, dubbed molecules 1–3 are shown in Scheme 1. For amphiphile 3, a fourth glutamic acid residue (Glu, E) was incorporated into the sequence to aid solubility.
Scheme 1. Sequences and Structures of Peptides and Amphiphiles Generated for This Study.

These designs are envisaged to stabilize GNPs for multiple reasons: Similar amphiphilic constructs adopt well-defined β-sheet structures,45 and the formation of an ordered structure is thought to be important for effective GNP coating.36 The strong negative charge of the amphiphiles aids GNP stability under physiological conditions, and there is evidence that negatively charged GNPs are less toxic than positively charged particles.20 The alkyl chain provides a dense, hydrophobic shell around the GNP, thereby aiding stability.30
In addition to molecules 1–3, a peptide without an alkyl chain but with a cysteine (Cys, C) residue was also designed (molecule 4, Scheme 1) to determine whether the peptide sequence alone was capable of stabilizing GNPs. State-of-the-art peptides CALNN, CVALNN, and CVVVT (5–7, Table S1) were synthesized and compared to molecules 1–4. Peptide 5 is the most commonly used sequence for the peptidic stabilization of GNPs, and its derivative, 6, was developed as there is evidence that additional steric bulk and hydrophobicity, provided by the valine (Val, V) residue, may provide additional stability.32 Peptide 7 has been demonstrated to be highly efficient at stabilizing small GNPs32 and was included to probe its stabilizing effects on larger GNPs.
All peptides were synthesized by solid-phase peptide synthesis (for full details see Materials and Methods). Briefly, after automated synthesis of the peptidic portion, both 1 and 2 were completed with an automated coupling of either 16-mercaptohexadecanoic acid (1) or 11-mercaptoundecanoic acid (2). For 3, Fmoc-Lys(Fmoc)-OH was coupled at the N-terminus of the V3A3E4 peptide, and upon Fmoc deprotection with 20% piperidine, 11-mercaptoundecanoic acid was coupled to both the N-terminus and the amine side-chain of the Lys moiety to yield a double-chain peptide amphiphile.
Assembly in Solution
The peptide amphiphiles developed by the Stupp group self-assembled into β-structured fibers in aqueous solution.45 To determine whether thiolation and alterations to the alkyl chain length affected the secondary structure of the peptide amphiphiles, their circular dichroism (CD) spectra were recorded (Figure 1) and compared to C16H31O-V3A3E3.45
Figure 1.
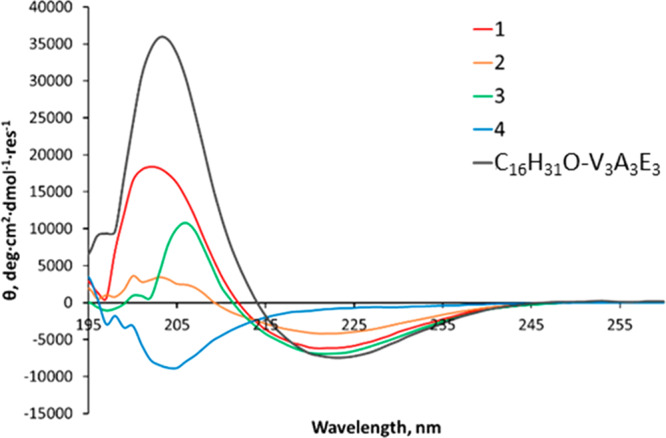
CD spectra of amphiphiles 1–3, the peptidic analogue 4, and C16H31O-V3A3E3, a peptide amphiphile developed by the Stupp group. All samples were prepared in PBS, pH 7.2, at a concentration of 100 μM for 1 and C16H31O-V3A3E3 and 250 μM for 2–4. The CD signal was converted to mean residue ellipticity which normalizes for peptide length and concentration (see Materials and Methods for details).
The spectra revealed that the introduction of a thiol, to generate amphiphile 1, did not significantly alter the secondary structure, as a spectrum corresponding to a β-structured species was observed. The magnitude of the CD signal was less intense than for C16H31O-V3A3E3, and we postulate that this could be due to intermolecular disulfide bond formation, or thiol oxidation, since both were observed by liquid chromatography-mass spectrometry (LC-MS), see Figure S1. Shortening the alkyl chain to produce 2 led to a reduction in the intensity of the CD signal, although a β-structure was still adopted. Interestingly, incorporation of two mercaptoundecanoyl chains, to form 3, produced a signal which was comparable in intensity to 1 and to C16H31O-V3A3E3. It should be noted that 1–3 exhibited a small shift in the position of the peak minimum when compared to C16H31O-V3A3E3. This could be due to a difference in the twist of the β-sheets,48 possibly as a result of disulfide bond formation. The peptide analogue 4 did not adopt an ordered structure, indicating the alkyl chain is important for self-assembly and the resultant formation of a defined secondary structure.
C16H31O-V3A3E3 forms β-sheets in aqueous media, and these β-sheets propagate into long, cylindrical fibers.45 The self-assembly properties of 1–4 were therefore investigated further with TEM (Figure 2). The TEM images show that 1–3 form fibers, although the fiber morphologies vary, which is likely to be due to the differences in the number or length of the alkyl chains. No defined assemblies were observed for 4, which is in agreement with the absence of a defined secondary structure, as determined by CD spectroscopy.
Figure 2.

Representative TEM images for (A) C16H31O-V3A3E3, (B) 1, (C) 2, (D) 3, and (E) 4. Samples were prepared in PBS, pH 7.2, and stained with 0.5% uranyl acetate. The scale bar is 200 nm for all images.
These studies confirm that peptide amphiphiles 1–3 self-assemble to form β-structured fibers in aqueous solution. While small differences in the secondary structures of the amphiphiles were observed, and the dimensions of the resulting fibers vary, it could be concluded that changing the alkyl chain length and adding a thiol to the N-terminus of these amphiphiles does not prevent their self-assembly.
Coating of GNPs
GNPs with diameters of 20, 40, or 100 nm were synthesized via sodium citrate reduction according to literature protocols15,49,50 (see Materials and Methods for further information). Twenty nm particles were evaluated as this size lies within the 10–30 nm range, which is commonly employed by other researchers when probing coating efficiency,26,29,30,32,37−39,47,51,40 and 100 nm GNPs were chosen as “large” GNPs (>35 nm) and have not previously been successfully coated with peptides or amphiphiles.
Ligand exchange is a technique commonly employed to generate covalently attached coatings on GNP surfaces via the displacement of citrate ions utilized in GNP synthesis.52 This strategy was employed to coat GNPs with our peptide amphiphiles. To circumvent undesired fiber formation during GNP coating, the thiolated peptide amphiphiles were dissolved in dimethyl sulfoxide (DMSO). This solution was mixed with an aqueous GNP suspension, yielding an initial GNP:peptide ratio of 1:100,000 and a final DMSO concentration of 25% (v/v). After 1 h of incubation, the samples were centrifuged to remove any unbound peptide before the pellet was resuspended in 5% DMSO (v/v). To eliminate any remaining unbound peptide or amphiphile and to exchange buffers, size exclusion chromatography (SEC) was performed using phosphate buffered saline (PBS) as the eluent.
The colloidal stability of the GNPs was initially studied using dynamic light scattering (DLS). Table 1 summarizes the average hydrodynamic diameters of the peptide- or peptide amphiphile-coated particles compared to the citrate-coated GNPs. 1 preserves the original size distribution for all three sizes of GNPs evaluated, and no aggregation was observed; although, the hydrodynamic diameter increased for all particle sizes due to the presence of the peptide amphiphile on the GNP surface (Table 1 and Figure S2). Amphiphile 2 successfully stabilized 20 and 40 nm GNPs, but 100 nm particles aggregated. Interestingly, 3, with two mercaptoundecanoyl alkyl chains, was capable of stabilizing all GNP sizes. To determine whether the enhanced stabilizing effect of 3, in comparison to 2, was due to the presence of two alkyl chains or to the additional Glu residue and the resulting increased negative charge, analogous amphiphiles with only one mercaptoundecanoyl chain were synthesized (for structures see Figure S3A). These peptide amphiphiles could not stabilize larger particles (Figure S3B), indicating that either the increased hydrophobicity or the bidentate nature of 3 was the reason for the improved GNP stability. Collectively, the behavior of 1–3 indicates that increased hydrophobicity of the amphiphile is critical for the stabilization of larger nanoparticles.
Table 1. Average Hydrodynamic Diameters of Citrate and Peptide/Amphiphile-Capped GNPsa.
| coating identity | hydrodynamic diameter, nm | ||
|---|---|---|---|
| citrate | 22.0 | 43.5 | 101 |
| 1 | 35.1 | 51.3 | 115 |
| 2 | 30.1 | 51.3 | 172 |
| 3 | 33.6 | 54.6 | 113 |
| 4 | 28.8 | 48.3 | 234 |
| 5 | 21.0 | 225 | x |
| 6 | 23.3 | 110 | x |
| 7 | 23.4 | 155 | x |
Values in red indicate aggregation was observed, and ‘x’ indicates that the coating experiments were not conducted as aggregation occurred for a smaller GNP size. Three independent measurements were recorded, and an average hydrodynamic diameter was calculated. For representative size distribution profiles, see Figure S2.
Molecule 4 was analyzed to investigate the stabilizing effect of the peptide domain alone. 4 could successfully stabilize 20 and 40 nm GNPs but not 100 nm particles (Table 1). The successful stabilization of the smaller particles is likely due to electrostatic repulsion induced by the highly negatively charged peptide sequence. Neither 5, 6, nor 7 were able to stabilize particles larger than 20 nm, which is in line with previous reports.34,36,38 It has been suggested that adding a bulky hydrophobic residue to 5, to generate 6, would enhance the stabilizing ability of the peptide. However, no evidence was found for this here, as 40 and 100 nm particles still aggregated. Equally, substituting Val for Ile, Leu, or Phe did not improve the stability of larger GNPs (Figure S3C). These results indicate that, to stabilize larger GNPs, either a significant increase in hydrophobicity or net charge of the stabilizing peptide is required or that the geometry of the peptide packing is not optimal for the stabilization of larger particles.
In addition to size distributions of the GNPs, their ζ potential was measured. Due to the peptide design, the surface charge of all samples was negative (Figure S4 and Table S2). The citrate-stabilized GNPs exhibited a tendency for the surface charge to increase with size: a ζ potential of −31 mV was measured for 20 nm GNPs, while values of −35 mV and −40 mV were recorded for 40 and 100 nm GNPs, respectively. Similar values have been reported by Piella et al.53 After coating, the ζ potential of the particles ranged between −15 and −25 mV for 20 nm GNPs. For all 40 nm GNPs, the ζ potential was approximately −25 mV, and for 100 nm GNPs, values between −34 and −40 mV were obtained. It is anticipated that this net negative charge will cause electrostatic repulsion between the particles, helping to prevent aggregation.
UV–vis spectroscopy and TEM were utilized to further probe the properties of the coated GNPs. Citrate-capped GNPs exhibit characteristic surface plasmon resonance (SPR) peaks: For our 20, 40, and 100 nm GNPs, peak maxima occur at 521, 530, and 577 nm, respectively (Figure 3). Coated GNPs that exhibited no aggregation by DLS showed no peak broadening or any significant red-shift (Figure 3); both features would be indicative of aggregation. Small red-shifts, between 1 and 7 nm, were observed and were attributed to an alteration in the dielectric constant at the GNP surface due to substitution of citrate by the peptides and amphiphiles. Some peak broadening was observed for 100 nm GNPs. This is likely a result of the natural sedimentation of these larger nanoparticles: The DLS data showed the samples were monodisperse, and this was confirmed by TEM analysis (vide infra), in which no GNP aggregates were observed. These UV–vis spectra confirm the results of the DLS experiments: Molecules 1–7 are capable of stabilizing 20 nm particles, 1–4 can stabilize 40 nm particles, but only 1 and 3 provide a stabilizing coating for 100 nm GNPs.
Figure 3.

Normalized UV–vis spectra of coated GNPs: (A) 20 nm GNPs coated with 1–7, (B) 40 nm GNPs with molecules 1–4, and (C) 100 nm GNPs coated with molecules 1 and 3. Conditions: PBS, pH 7.2.
All GNPs that had been demonstrated to have stable coatings were subsequently analyzed by TEM. The samples were negatively stained with uranyl acetate to determine whether the coating around the particles could be observed and also to ensure our coating method was effective at preventing fiber formation. Gratifyingly, a thin uniform shell around each particle was clearly visible, indicating the presence of an ordered coating.38 It should be noted that the observation of this surface layer around the 100 nm GNPs is in part hampered by the Fresnel fringes that arise from the underfocus imaging that is needed for their visualization in TEM.54
For all the samples analyzed, no GNP aggregation or fibers were observed (Figure 4 and Figures S5–S8), confirming the devised coating method was effective.
Figure 4.

TEM micrographs of peptide-capped GNPs. Top row: 20 nm GNPs coated with molecules 1–4; middle row: 40 nm GNPs coated with 1–4; and bottom row: 100 nm GNPs coated with molecules 1 and 3. Images for 20 nm GNPs coated with molecules 5–7 can be found in Figure S6. No TEM images were recorded for 100 nm GNPs coated with molecules 2 and 4, as DLS indicated these samples aggregated. Low-magnification images can be found in Figures S5–S8. Scale bars: 20 nm GNPs = 50 nm and 40 and 100 nm GNPs = 200 nm.
Coverage Density of GNP Coatings
As the TEM images appeared to indicate dense monolayers were formed on the surface of the GNPs, coverage densities were determined. High-coverage densities indicate a tightly packed ligand shell is formed around the GNP, leading to increased particle stability.36,38 Through incorporation of a tyrosine (Tyr, Y) residue, coverage densities were determined using UV–vis; a similar method was employed previously.6 Amphiphile 1 was therefore synthesized with a terminal Tyr residue (see Scheme S1 for the structure), and the GNPs were coated in the manner described in the previous section. The UV–vis absorbance of the supernatant, containing the unbound amphiphile, was measured after two cycles of centrifugation. As the initial concentration of peptide was known, it was possible to establish the amount of peptide attached to the GNPs. Combining this information with knowledge of the concentration and surface area of the GNPs allowed coverage densities to be calculated (for full details, see Materials and Methods). Using this method, the following coverage densities were determined: 3.72 peptides/nm2 for 20 nm GNPs, 2.47 peptides/nm2 for 40 nm GNPs, and 2.17 peptides/nm2 for 100 nm GNPs. These densities are high. Comparable densities determined by Shaw et al. for their 25 nm GNPs were 2.55 or 2.4 peptides/nm2 depending on the peptide sequence used,38 and a density of 0.72 peptides/nm2 for 5.4 nm GNPs was determined for samples prepared by Mikolajczak et al.6 It is likely that these high coverage densities are one of the reasons why the GNPs are so effectively stabilized.
Peptide and Amphiphile Assembly on GNP Surfaces
Several studies have highlighted that the surface curvature of nanoparticles can affect the secondary structure of covalently attached peptides.38,47,55 We have demonstrated that peptide amphiphiles 1–3 adopt defined β-sheet secondary structures in solution; therefore, their structures on the surface of GNPs were studied to determine whether they retain this structure when confined to a surface. Initially, the amphiphiles were probed using CD spectroscopy (Figure S9). A signal with a minimum at 215 nm, corresponding to the presence of β-structure, was detected on 20 nm GNPs coated with 1 even at GNP concentrations as low as 6 nM. Peptide amphiphile 2 behaved similarly but exhibited a minimum at 212 nm. The structure formed by 3 was more difficult to interpret, as the signal was very noisy due to both the low concentration of amphiphile and the scattering of the GNPs.
Analysis of secondary structure formation on larger GNPs using CD spectroscopy was problematic due to the strong scattering of the particles. Therefore, to probe the structures of 1–3 on the GNPs and to obtain quantitative data, Fourier-transform infrared (FTIR) spectra were obtained.
The data collected (Figure 5 and Figure S10) confirm that 1–3 do indeed form β-sheet structures, both in isolation and on GNP surfaces. The position of the peak maximum of the amide I region is suggestive of the predominant secondary structure type.6,47,56 Thus, an amide I peak located between 1629 and 1640 cm–1 is indicative of β-structure and 1649–1656 cm–1 is a typical position for α-structured peptides. Usually, the amide I peak is a superposition of different backbone conformations and can be deconvoluted to quantify the amount of different types of secondary structure present.
Figure 5.

Deconvoluted amide I peaks for 1–3 in powder form and on GNP surfaces. (A) 1, and as a coating on: (B) 20 nm, (C) 40 nm, and (D) 100 nm GNPs. (E) 3, and as a coating on: (F) 20 nm, (G) 40 nm, and (H) 100 nm GNPs. (I) 2, and as a coating on: (J) 20 nm and (K) 40 nm GNPs. Measurements were performed on lyophilized powders of lipopeptides and on hydrated films for GNPs conjugates.
Deconvolution and fitting of the amide I region showed that, in isolation, 1 and 3 almost exclusively adopted a β-sheet structure (Figure 5A,I and Table S3), while 2 exhibited 85% β-sheet content, (Figure 5E and Table S3). This high propensity to form β-structures in isolation was translated to the surface of 20 nm GNPs for all three molecules; they all self-assembled to form structures with >70% β-content (Figure 5B,F,J).
For 40 and 100 nm particles, the data were more challenging to interpret. The large sizes of the particles meant that surface selection rules, which state that vibrations whose dipole moment are parallel to the nanoparticle surface will be suppressed,57 strongly influencing the spectra obtained. The general trend, however, appeared to be that the amphiphiles displayed a lower amount of β-structure on these larger particles (Figure 5C–H and Table S3). This decrease in the amount of β-structure observed is in contrast to other studies. It should be noted that previous studies have examined particles between 5 and 25 nm in diameter,36,38 where this study looks at GNP sizes of ≥20 nm; therefore, to the best of our knowledge there is no precedent for structure formation on particles of this size. We hypothesize that, along with the detrimental effect of the surface selection rules, the decrease in propensity to form ordered structures observed may be related to several different factors. First, the coverage densities decrease with increasing particle size; therefore, the spacing between the amphiphiles is increased, making it more difficult for intermolecular hydrogen bonds, and therefore extended β-sheets, to form. Second, the amphiphiles are longer than the peptide sequences previously studied, meaning they have more conformational freedom which can affect self-assembly. This is supported by the fact that 2, which has a shorter alkyl chain than 1, is more ordered on both 20 and 40 nm GNPs. Finally, the low curvature of 100 nm GNPs means 1 is likely to be more tightly packed than 3, as it only has a single alkyl chain, meaning more amphiphiles can be attached to the surface; therefore, the level of self-assembly is increased. This is reflected in the relative percentages of β-structure observed.
While it can be concluded that the peptide amphiphiles do self-assemble to form defined β-structures on 20 nm particles and appear to do so on 40 and 100 nm GNPs, more detailed studies will be required to fully account for the effects of nanoparticle size and the surface selection rules.
In addition to analyzing the amide I band, analysis of the region between 2700 and 3500 cm–1 provides information about the packing of alkyl chains, (for spectra, see Figures S10–11). The band centered around 2850 cm–1 is attributed to the CH2 symmetric stretching mode and can be used to infer information regarding the alkyl chain packing.58,59 The well-defined and sharp bands observed for our molecules indicate they possess tightly packed alkyl chains when not conjugated to GNPs. Upon conjugation, a shift to higher wavenumbers and a broadening of the band is observed. This indicates the packing of the alkyl chains is less ordered, with the shift being more pronounced with larger GNP sizes. This is consistent with the observation that coverage density decreases with increasing particle size, meaning the molecules are further apart and therefore not optimally spaced to pack regularly.
Stability of Capped GNPs to Electrolytes
DLS, TEM, and UV–vis data have demonstrated that both 1 and 3 can stabilize a range of GNP sizes in PBS buffer. Particle stability at elevated salt concentrations provides information about the type of stabilizing interactions present. For example, if GNP stability is based solely upon repulsive electrostatic interactions, as with citrate-coated nanoparticles, then as the electrolyte concentration is increased, the magnitude of electrostatic repulsion is diminished as the surface charge of the GNPs is screened by the salt. As the stabilization of GNPs by both 1 and 3 is presumed to be largely due to hydrophobic effects, the GNPs should be resistant to high-salt concentrations.
Five different salt concentrations were tested: 0 mM, 150 mM, 500 mM, 1.5 M, and 3.0 M. The GNPs were incubated with these salt solutions, and their aggregation was monitored by UV–vis (Figure 6A–C and Figure S12). To compare aggregation tendencies, the optical density (O.D.) at each salt concentration was normalized to the intensity of the LSPR peak maximum at 0 mM NaCl (see Materials and Methods for full details).
Figure 6.

GNP stability to electrolytes and DTT. Aggregation tendencies in 0–3 M NaCl are shown for: (A) 20 nm, (B) 40 nm, and (C) 100 nm GNPs. The Y-axis represents O.D. at the LSPR peak maximum, the wavelength of which varies with GNP size and is as follows: (A) 525 nm, (B) 535 nm, and (C) 585 nm. The O.D. is normalized to the value for 0 mM NaCl in each case. A decrease in normalized O.D. indicates aggregation. Aggregation after exposure to 1 M DTT is expressed by the normalized aggregation factor (AF) for: (D) 20 nm, (E) 40 nm, and (F) 100 nm GNPs. Exposure to DTT was conducted in the presence of NaCl at the following concentrations: (D, E) 400 mM and (F) 150 mM.
While 1 was able to stabilize 20 nm GNPs at moderate salt concentrations, at concentrations >1.5 M, aggregation was evident. The same molecule could only stabilize 40 and 100 nm particles at low-salt concentrations. In contrast, 2 stabilized both 20 and 40 nm particles, even at high-salt concentrations (2 was not tested with 100 nm particles, as aggregation is observed in PBS). The final amphiphilic molecule, 3, proved highly effective at stabilizing the 20 nm particles, but some aggregation was present for 40 nm particles, and for 100 nm particles, severe aggregation occurred at salt concentrations >1.5 M. Our peptide molecule, 4, outperformed peptides 5–7 on 20 nm particles, and it was demonstrated to stabilize these particles even at high-salt concentrations (Figure 6A). However, 4 was not effective at stabilizing 40 nm particles.
What is particularly interesting is that 1 is less effective at high-salt concentrations in comparison to 2 and 3. It is assumed this is due to the longer alkyl chain length of 1, meaning the peptides have increased conformational freedom which may allow salt molecules to penetrate the coating, leading to destabilization.
Stability against Dithiothreitol-Induced Aggregation
GNP stability to dithiothreitol (DTT) was also probed, as DTT is a small molecule that is known to be capable of displacing thiol ligands from gold surfaces, leading to GNP aggregation.30,51 Resistance to thiol displacement indicates the gold surface is shielded from the surrounding solvent environment. Stability in the presence of DTT is important for GNPs with intended in vitro and in vivo functions, as intracellular thiol-containing molecules, such as glutathione, could displace the coating from the surface. In these experiments, DTT acts as a model for such molecules.
The experimental protocol was adapted from one devised by Mei et al.51 The GNP samples were incubated with 1 M DTT, and their UV–vis spectra were recorded over a period of 90 min (Figure S13). NaCl was added, as its presence leads to additional screening effects, speeding up the aggregation process. 400 mM NaCl was added to 20 and 40 nm GNP samples, but as this concentration, it led to aggregation of 100 nm GNPs, and a lower NaCl concentration of 150 mM was employed. To quantify the amount of aggregation as a result of ligand displacement, we used the normalized aggregation factor (AF) parameter.51 To calculate this parameter, two absorbance peaks were compared: the original plasmon band and an emerging peak found at higher wavelengths (615 nm for 20 and 40 nm GNPs; and 700 nm for 100 nm GNPs); the emergence of this second peak is indicative of aggregation. The latter was divided by the former to yield the AF, which in turn was normalized to the absorbance at 0 min (Figure 6D–F). If the normalized AF is higher than 1, it indicates aggregation is occurring.
The amphiphilic molecules 1–3 effectively insulated the surface of 20 nm GNPs from DTT, though particles coated with 2 showed a slight tendency to aggregate after 80 min (Figure 6D). This effect was more prominent when 40 nm GNPs were studied (Figure 6E). Aggregation of these particles with 2 was evident after 50 min, and aggregation was more pronounced after 90 min when compared to the 20 nm particles. 100 nm particles were coated with amphiphiles 1 and 3, and both appear to stabilize the particles over the time range tested. Although 3 does show a small increase in AF after 15 min, this does not increase significantly for the remainder of the experiment (Figure 6F).
When 4 was examined on 20 nm GNPs, aggregation in the presence of DTT was observed almost immediately, with severe aggregation being observed after 15 min (Figure 6D). In comparison, peptide 7, the best performing reference peptide from the electrolyte-induced aggregation experiments, showed significantly less aggregation than 4, although an AF of 1.5 was recorded after 90 min (Figure 6D).
The results of this DTT competition experiment highlight the importance of the alkyl chains and demonstrate that they can effectively shield the GNP surface from small molecules. This further reinforces the idea that these amphiphiles form dense, well-ordered monolayers on the GNP surface.
Conclusions
We have developed four molecules, three peptide amphiphiles and one peptide, and evaluated their ability to act as coatings for different sizes of GNPs. The DLS, UV–vis, and TEM data indicate that amphiphiles 1 and 3 are capable of stabilizing all three sizes of GNPs under biologically relevant conditions (Scheme 2), while 2 can stabilize 20 and 40 nm particles. FTIR analysis revealed that the amphiphiles retain their abilities to self-assemble on the surfaces of the GNPs, leading to the creation of dense, ordered monolayers.
Scheme 2. GNPs (to scale) and the Molecules Capable of Stabilizing Each Size.

Arrows represent the span of molecules that are capable of stabilizing each GNP size, that is, all molecules can act as coatings for 20 nm GNPs, 1–4 stabilize 40 nm GNPs, and 1 and 3 are effective coatings for the 100 nm GNPs.
The stabilizing effect of 1 is likely due to its long alkyl chain which provides increased hydrophobicity. For 3, stability is provided by a combination of hydrophobicity and the bidentate nature of the ligand, meaning that it has a stronger interaction with the GNPs and therefore an increased stabilizing effect.
Both 1 and 3 proved to be resistant to DTT over 90 min, and 3 outperformed 1 at high-salt concentrations. We hypothesize that this is to do with the packing of the peptides because the longer chain of 1 provides more conformational flexibility, meaning the peptides are not as ordered and the protective “shell” created by the amphiphile is more permeable. This is supported by the fact that 3 is more structured on 40 nm particles, suggesting it is better self-assembled than 1.
While 1 and 3 are universal sequences and stabilize all three GNP sizes tested, it should be noted that amphiphile 2 and our peptide, 4, could stabilize both 20 and 40 nm GNPs (Scheme 2). Therefore, in addition to creating two universal stabilizing sequences, we have added other designs to the “toolkit” of peptide-based stabilizing sequences for GNPs, and we have demonstrated that 4 outperforms the current “state-of-the-art” peptide sequences. Future work will focus on derivatizing these molecules with targeting and/or therapeutic moieties to probe their activities in vitro and in vivo.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich except where stated. TFA, piperidine, DMF, DCM, and acetonitrile were purchased from Biosolve. Oxyma pure was supplied by Carl Roth GmbH.
Gold Nanoparticle Synthesis
The 20 nm particles were prepared according to a combination of the Turkevich and Frens’ methods.49,50 In short, 25 mL of a 1 mM chloroauric acid (HAuCl4) solution was brought to a gentle boil while stirring. Five mL of a 1% trisodium citrate dehydrate solution was added to the boiling solution, and a condenser was attached to the flask. The mixture was boiled for 10 min before the sample was cooled to room temperature. The size of the particles was confirmed using DLS.
A seed-mediated growth was used to obtain particles of 40 and 100 nm diameter.15 Seeds were prepared by heating 15 mL of a 2.2 mM sodium citrate solution to boiling. 50 μL of 50 mM HAuCl4 was added, and the mixture boiled for 10 min, before the temperature of the solution was maintained at 90 °C. To perform controlled overgrowth of the seeds, the sequential addition of small amounts (1–3 mL) of 10 mM citrate solution was alternated with that of a 1 mM HAuCl4 solution (0.35–1 mL). It is critical to keep the molar ratio of citrate:HAuCl4 at 10:1. This manipulation was repeated as many times as required to obtain particles of the desired size, which was monitored by DLS. The sizes of the GNPs were confirmed by TEM and DLS (Figure S14).
Peptide and Amphiphile Synthesis
All peptide and lipopeptide sequences were synthesized by solid-phase peptide synthesis using standard Fmoc-chemistry protocols. 20% piperidine in DMF was used as the deprotection agent, and DIC/Oxyma were employed as activator/activator base. All sequences were synthesized on Wang resin preloaded with the corresponding C-terminal residue. All the molecules were cleaved from the resin using 1.5% deionized water, 2.5% triisopropylsilane (TIS), 2.5% phenol, and 2.5% 3,4-ethylenedioxythiophene (EDOT) in trifluoroacetic acid (TFA). The crude peptides and amphiphiles were precipitated into cold diethyl ether, pelleted by centrifugation, redissolved in water, and lyophilized prior to purification.
Peptide and Amphiphile Purification
HPLC purification was performed on a Shimadzu system equipped with two LC-20AR pumps, an SPD-20A UV–vis detector, and a Phenomenex Kinetex EVO C18 column. The mobile phases were water and acetonitrile, containing either 0.1% TFA, for peptides, or 0.1% NH3, for amphiphiles. The purity of the compounds was assessed using LC-MS (Figures S15–S25). All purified molecules were lyophilized and stored at −20 °C until required.
Preparation of Peptide-Capped GNPs
Coating of the GNPs was performed via a ligand exchange strategy. The desired coating molecule was dissolved in DMSO and added to a stirred GNP suspension. The volumes of coatings and GNPs were such that the final concentration of DMSO in the solution was 20–25% (v/v). After 1 h, the sample was centrifuged, (14,000 rpm, 45 min for 20 nm GNPs; 10 min for 40 nm GNPs; 5 min for 100 nm GNPs), and the supernatant removed and replaced with 5% DMSO. Size exclusion chromatography was performed to remove any remaining free ligand, and the elution buffer was phosphate buffered saline (PBS).
Dynamic Light Scattering Measurements
Size distributions of all particles in this study were obtained by dynamic light scattering (DLS) using a Zetasizer Nano-7 S (Malvern Instruments) with a 633 nm laser wavelength and a 173° fixed scattering angle. The temperature was maintained at 25 °C during the measurements. An aliquot of 500 μL for each sample was placed into a disposable plastic cuvette, and three separate measurements were recorded from which a mean hydrodynamic diameter was calculated.
Zeta Potential Measurements
All ζ potential measurements were performed on a Zetasizer Nano-7 S (Malvern Instruments) equipped with the same laser as described above. Samples in PBS were diluted 10 times with deionized water to reduce the salt concentration to <20 mM. Sample aliquots of 1 mL were placed in a universal dip cuvette, and the ζ potential was calculated from an average of three measurements.
UV–vis Spectroscopy
Spectra were recorded using a Cary 300 UV–vis spectrophotometer (Agilent). Samples were placed in 1 mm quartz cuvettes, and spectra were recorded between 900 and 350 nm unless otherwise stated. Samples were diluted with either MQ water or PBS as appropriate to provide an O.D. in the range of 0.6–1.0. All spectra were normalized to provide an O.D. = 1 to aid comparison, except for the spectra recorded for the electrolyte-induced aggregation experiments which were calculated as described below.
Attenuated Total Reflection-Infrared
Attenuated total reflection-infrared (ATR-IR) spectra were recorded on an Excalibur FTS 4000 setup equipped with a “golden gate”. For peptide-capped GNPs, the sample preparation was as follows: a 5 μL drop of a highly concentrated sample in D2O was placed on top of the crystal and blow-dried with air. After a uniform film was formed, a spectrum was recorded. For peptides, a lyophilized powder was used. Deconvolution and fitting of the Amide I peaks to the Lorenz function were performed using Origin Pro.
Transmission Electron Microscopy
For transmission electron microscopy (TEM) measurements, a 10 μL droplet of the sample of interest was placed on a continuous carbon grid on a copper support (200 mesh) and left for 20 min. The excess liquid was removed by manually blotting with filter paper. The grid was washed with deionized water once and blotted again. Uranyl acetate staining (0.5% for fiber samples and 1% for GNP samples) was applied, followed immediately by blotting. Images were collected on a JEM1400 Plus (JEOL) transmission electron microscope operated at 80 kV and equipped with a CCD camera or on the TU/e Sphera (TFS, www.cryotem.nl) operated at 200 kV, equipped with a 4096 × 4096 CDD camera.
Circular Dichroism Spectroscopy
Circular dichroism (CD) spectra of the amphiphiles and peptides were recorded using a JASCO J-815 spectropolarimeter, fitted with a Peltier temperature controller. All measurements were performed at 20 °C. Samples were measured in quartz cuvettes with a 2 mm path length, and spectra were recorded from 260 to 190 at 2 nm intervals with a 1 nm bandwidth. All spectra were converted to mean residue ellipticity (deg cm2 dmol·res–1) using eq 1:
| 1 |
where [θ]obs is the observed ellipticity in mdeg, c is the concentration of the sample in mM, n is the number of amino acids in the peptide or amphiphile, and l is the path length of the cuvette in cm.
Coverage Density Measurements
Coverage densities were determined for all three sizes of the GNPs used in this study. Lipopeptide 1 was modified with a Tyr residue and was used to prepare coated GNPs. The supernatant of the first centrifugation cycle was collected and combined with the supernatant of the following washes. As the concentration of Tyr-modified 1 added to the GNP solution was known, the concentration of peptide in the combined supernatants was determined using UV–vis. The average size and number concentration of the GNPs was also calculated,60 and coverage densities were determined from this information.
Stability to Electrolyte-Induced Aggregation
An aqueous GNP suspension was mixed with a 4.5 M NaCl solution to yield samples with the following final concentrations of salt: 150 mM, 500 mM, 1.5 M, and 3.0 M. The final volume of each sample and the GNP concentration remained constant for all samples tested. The optical densities of each sample at their maximum absorption wavelength were recorded using an Infinite M1000 plate-reader (Tecan). A spectrum from 650–400 nm was also recorded. This is because when GNPs aggregate, they exhibit a red-shift and often a broadening of the plasmon band. The O.D. values at the maximum absorption wavelength (O.D.LSPR) corresponded to the control sample (0 M NaCl), and this was taken as the value of a pristine, nonaggregated sample. The O.D. of the samples with increasing amounts of NaCl were compared to this sample, and a decrease in the value of the O.D. indicated aggregation. The LSPR peak maximums for GNPs of different sizes were as follows: 525 nm for 20 nm GNPs, 535 nm for 40 nm GNPs, and 585 nm for 100 nm GNPs.
Stability Study by Means of DTT-Induced Aggregation
The protocol adopted was adapted from that published by the Mattoussi group.51 4 M DTT, 2 M NaCl, deionized water, and coated GNPs were mixed to yield a solution with final concentrations of 1 M DTT and 400 mM NaCl, (150 mM for 100 nm GNPs). Addition of NaCl accelerates aggregation due to Debye screening effects. The O.D. of the final mixture was adjusted to a value of 0.35. Aggregation was monitored by UV–vis using a BioDrop Duo UV–vis spectrophotometer (Harvard Bioscience) for 90 min with a 5 min interval between the data points. The aggregation factor (AF) was expressed as a ratio between the O.D. at the peak maximum of the LSPR and the O.D. at 615 nm for 20 and 40 nm GNPs and the O.D. at 700 nm for 100 nm GNPs.
Acknowledgments
E.A. acknowledges the financial support of the Global Education Scholarship Program (Russia). A.K. acknowledges the support of an NWO (Netherlands Organization for Scientific Research) VICI grant (grant number: 724.014.001) which funded A.L.B. M.M.J.v.R. was funded by the NWO TOP-PUNT grant “Bi-Hy” (grant number: 718.016.003) which was awarded to N.S.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.0c01021.
Structures of reference peptides and additional amphiphiles; LCMS data; DLS size distributions, ζ potential values, low-magnification TEM images, additional CD spectra, complete FT-IR traces and spectra corresponding to the alkyl chains regions, complete UV–vis spectra for the electrolyte and DTT stability experiments; and characterization data for the citrate-coated GNPs (PDF)
Author Present Address
⊥ (Aimee L. Boyle) Department of Macromolecular Biochemistry, Leiden Institute of Chemistry, Leiden University, Leiden 2333 CC, The Netherlands
The authors declare no competing financial interest.
Supplementary Material
References
- Kennedy L. C.; Bickford L. R.; Lewinski N. A.; Coughlin A. J.; Hu Y.; Day E. S.; West J. L.; Drezek R. A. A New Era for Cancer Treatment: Gold-Nanoparticle-Mediated Thermal Therapies. Small 2011, 7, 169–183. 10.1002/smll.201000134. [DOI] [PubMed] [Google Scholar]
- Riley R. S.; Day E. S. Gold Nanoparticle-Mediated Photothermal Therapy: Applications and Opportunities for Multimodal Cancer Treatment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1449 10.1002/wnan.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreaden E. C.; Austin L. A.; Mackey M. A.; El-Sayed M. A. Size Matters: Gold Nanoparticles in Targeted Cancer Drug Delivery. Ther. Delivery 2012, 3, 457–478. 10.4155/tde.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G.; Ghosh P.; Rotello V. M. Functionalized Gold Nanoparticles for Drug Delivery. Nanomedicine 2007, 2, 113–123. 10.2217/17435889.2.1.113. [DOI] [PubMed] [Google Scholar]
- Corma A.; Garcia H. Supported Gold Nanoparticles as Catalysts for Organic Reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. 10.1039/b707314n. [DOI] [PubMed] [Google Scholar]
- Mikolajczak D. J.; Heier J. L.; Schade B.; Koksch B. Catalytic Activity of Peptide-Nanoparticle Conjugates Regulated by a Conformational Change. Biomacromolecules 2017, 18, 3557–3562. 10.1021/acs.biomac.7b00887. [DOI] [PubMed] [Google Scholar]
- Pengo P.; Baltzer L.; Pasquato L.; Scrimin P. Substrate Modulation of the Activity of an Artificial Nanoesterase Made of Peptide-Functionalized Gold Nanoparticles. Angew. Chem., Int. Ed. 2007, 46, 400–404. 10.1002/anie.200602581. [DOI] [PubMed] [Google Scholar]
- Gao J.; Liu D.; Wang Z. Microarray-Based Study of Carbohydrate-Protein Binding by Gold Nanoparticle Probes. Anal. Chem. 2008, 80, 8822–8827. 10.1021/ac8015328. [DOI] [PubMed] [Google Scholar]
- Jena B. K.; Raj C. R. Electrochemical Biosensor Based on Integrated Assembly of Dehydrogenase Enzymes and Gold Nanoparticles. Anal. Chem. 2006, 78, 6332–6339. 10.1021/ac052143f. [DOI] [PubMed] [Google Scholar]
- Almeida J. P. M.; Lin A. Y.; Figueroa E. R.; Foster A. E.; Drezek R. A. in vivo Gold Nanoparticle Delivery of Peptide Vaccine Induces Anti-Tumor Immune Response in Prophylactic and Therapeutic Tumor Models. Small 2015, 11, 1453–1459. 10.1002/smll.201402179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.; Ahn S.; Lee J.; Kim J. Y.; Choi M.; Gujrati V.; Kim H.; Kim J.; Shin E. C.; Jon S. Effects of Gold Nanoparticle-Based Vaccine Size on Lymph Node Delivery and Cytotoxic T-Lymphocyte Responses. J. Controlled Release 2017, 256, 56–67. 10.1016/j.jconrel.2017.04.024. [DOI] [PubMed] [Google Scholar]
- Niikura K.; Matsunaga T.; Suzuki T.; Kobayashi S.; Yamaguchi H.; Orba Y.; Kawaguchi A.; Hasegawa H.; Kajino K.; Ninomiya T.; Ijiro K.; Sawa H. Gold Nanoparticles as a Vaccine Platform: Influence of Size and Shape on Immunological Responses in vitro and in vivo. ACS Nano 2013, 7, 3926–3938. 10.1021/nn3057005. [DOI] [PubMed] [Google Scholar]
- Link S.; El-Sayed M. A. Size and Temperature Dependence of the Plasmon Absorption of Colloidal Gold Nanoparticles. J. Phys. Chem. B 1999, 103, 4212–4217. 10.1021/jp984796o. [DOI] [Google Scholar]
- Hakkinen H. The Gold-Sulfur Interface at the Nanoscale. Nat. Chem. 2012, 4, 443–455. 10.1038/nchem.1352. [DOI] [PubMed] [Google Scholar]
- Bastus N. G.; Comenge J.; Puntes V. Kinetically Controlled Seeded Growth Synthesis of Citrate-Stabilized Gold Nanoparticles of Up to 200 NM: Size Focusing versus Ostwald Ripening. Langmuir 2011, 27, 11098–11105. 10.1021/la201938u. [DOI] [PubMed] [Google Scholar]
- Grzelczak M.; Perez-Juste J.; Mulvaney P.; Liz-Marzan L. M. Shape Control in Gold Nanoparticle Synthesis. Chem. Soc. Rev. 2008, 37, 1783–1791. 10.1039/b711490g. [DOI] [PubMed] [Google Scholar]
- Piella J.; Bastus N. G.; Puntes V. Size-Controlled Synthesis of Sub-10-Nanometer Citrate-Stabilized Gold Nanoparticles and Related Optical Properties. Chem. Mater. 2016, 28, 1066–1075. 10.1021/acs.chemmater.5b04406. [DOI] [Google Scholar]
- Bhamidipati M.; Fabris L. Multiparametric Assessment of Gold Nanoparticle Cytotoxicity in Cancerous and Healthy Cells: The Role of Size, Shape, and Surface Chemistry. Bioconjugate Chem. 2017, 28, 449–460. 10.1021/acs.bioconjchem.6b00605. [DOI] [PubMed] [Google Scholar]
- Fraga S.; Brandao A.; Soares M. E.; Morais T.; Duarte J. A.; Pereira L.; Soares L.; Neves C.; Pereira E.; Bastos M. d. L.; Carmo H. Short- and Long-Term Distribution and Toxicity of Gold Nanoparticles in the Rat after a Single-Dose Intravenous Administration. Nanomedicine 2014, 10, 1757–1766. 10.1016/j.nano.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Goodman C. M.; McCusker C. D.; Yilmaz T.; Rotello V. M. Toxicity of Gold Nanoparticles Functionalized with Cationic and Anionic Side Chains. Bioconjugate Chem. 2004, 15, 897–900. 10.1021/bc049951i. [DOI] [PubMed] [Google Scholar]
- Liu S. H.; Han M. Y. Silica-Coated Metal Nanoparticles. Chem. - Asian J. 2009, 5, 36–45. 10.1002/asia.200900228. [DOI] [PubMed] [Google Scholar]
- Jokerst J. V.; Lobovkina T.; Zare R. N.; Gambhir S. S. Nanoparticle PEGylation for Imaging and Therapy. Nanomedicine 2011, 6, 715–728. 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuelfing W. P.; Gross S. M.; Miles D. T.; Murray R. W. Nanometer Gold Clusters Protected by Surface-Bound Monolayers of Thiolated Poly(ethylene Glycol) Polymer Electrolyte. J. Am. Chem. Soc. 1998, 120, 12696–12697. 10.1021/ja983183m. [DOI] [Google Scholar]
- Boal A. K.; Rotello V. M. Intra- and Intermonolayer Hydrogen Bonding in Amide-Functionalized Akanethiol Self-Assembled Monolayers on Gold Nanoparticles. Langmuir 2000, 16, 9527–9532. 10.1021/la0012384. [DOI] [Google Scholar]
- Templeton A. C.; Hostetler M. J.; Warmoth E. K.; Chen S. W.; Hartshorn C. M.; Krishnamurthy V. M.; Forbes M. D. E.; Murray R. W. Gateway Reactions to Diverse, Polyfunctional Monolayer-Protected Gold Clusters. J. Am. Chem. Soc. 1998, 120, 4845–4849. 10.1021/ja980177h. [DOI] [Google Scholar]
- Ansar S. M.; Chakraborty S.; Kitchens C. L. pH-Responsive Mercaptoundecanoic Acid Functionalized Gold Nanoparticles and Applications in Catalysis. Nanomaterials 2018, 8 (8), 339. 10.3390/nano8050339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laaksonen T.; Ahonen P.; Johans C.; Kontturi K. Stability and Electrostatics of Mercaptoundecanoic Acid-Capped Gold Nanoparticles with Varying Counterion Size. ChemPhysChem 2006, 7, 2143–2149. 10.1002/cphc.200600307. [DOI] [PubMed] [Google Scholar]
- Hanske C.; Sanz-Ortiz M. N.; Liz-Marzan L. M. Silica-Coated Plasmonic Metal Nanoparticles in Action. Adv. Mater. 2018, 30, 1707003. 10.1002/adma.201707003. [DOI] [PubMed] [Google Scholar]
- Knowles B. R.; Yang D.; Wagner P.; Maclaughlin S.; Higgins M. J.; Molino P. J. Zwitterion Functionalized Silica Nanoparticle Coatings: The Effect of Particle Size on Protein, Bacteria, and Fungal Spore Adhesion. Langmuir 2019, 35, 1335–1345. 10.1021/acs.langmuir.8b01550. [DOI] [PubMed] [Google Scholar]
- Schulz F.; Vossmeyer T.; Bastus N. G.; Weller H. Effect of the Spacer Structure on the Stability of Gold Nanoparticles Functionalized with Monodentate Thiolated Poly(ethylene Glycol) Ligands. Langmuir 2013, 29, 9897–9908. 10.1021/la401956c. [DOI] [PubMed] [Google Scholar]
- Atukorale P. U.; Guven Z. P.; Bekdemir A.; Carney R. P.; Van Lehn R. C.; Yun D. S.; Silva P. H. J.; Demurtas D.; Yang Y. S.; Alexander-Katz A.; Stellacci F.; Irvine D. J. Structure-Property Relationships of Amphiphilic Nanoparticles That Penetrate or Fuse Lipid Membranes. Bioconjugate Chem. 2018, 29, 1131–1140. 10.1021/acs.bioconjchem.7b00777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R.; Thanh N. T. K.; Doty R. C.; Hussain I.; Nichols R. J.; Schiffrin D. J.; Brust M.; Fernig D. G. Rational and Combinatorial Design of Peptide Capping Ligands for Gold Nanoparticles. J. Am. Chem. Soc. 2004, 126, 10076–10084. 10.1021/ja0487269. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Levy R.; Fernig D. G.; Brust M. The Peptide Route to Multifunctional Gold Nanoparticles. Bioconjugate Chem. 2005, 16, 497–500. 10.1021/bc050047f. [DOI] [PubMed] [Google Scholar]
- Zhou G. H.; Liu Y. Z.; Luo M.; Li X. H.; Xu Q. F.; Ji X. H.; He Z. K. Controlled Assembly of Gold Nanoparticles through Antibody Recognition: Study and Utilizing the Effect of Particle Size on Interparticle Distance. Langmuir 2013, 29, 4697–4702. 10.1021/la400404g. [DOI] [PubMed] [Google Scholar]
- Zhao C. L.; Qiu L. H.; Lv P.; Han A. L.; Fang G. Z.; Liu J. F.; Wang S. AuNP-Peptide Probe for Caspase-3 Detection in Living Cells by SERS. Analyst 2019, 144, 1275–1281. 10.1039/C8AN02145G. [DOI] [PubMed] [Google Scholar]
- Colangelo E.; Chen Q. B.; Davidson A. M.; Paramelle D.; Sullivan M. B.; Volk M.; Levy R. Computational and Experimental Investigation of the Structure of Peptide Monolayers on Gold Nanoparticles. Langmuir 2017, 33, 438–449. 10.1021/acs.langmuir.6b04383. [DOI] [PubMed] [Google Scholar]
- Olmedo I.; Araya E.; Sanz F.; Medina E.; Arbiol J.; Toledo P.; Alvarez-Lueje A.; Giralt E.; Kogan M. J. How Changes in the Sequence of the Peptide CLPFFD-NH2 Can Modify the Conjugation and Stability of Gold Nanoparticles and Their Affinity for Beta-Amyloid Fibrils. Bioconjugate Chem. 2008, 19, 1154–1163. 10.1021/bc800016y. [DOI] [PubMed] [Google Scholar]
- Shaw C. P.; Middleton D. A.; Volk M.; Levy R. Amyloid-Derived Peptide Forms Self-Assembled Mono layers on Gold Nanoparticle with a Curvature-Dependent beta-Sheet Structure. ACS Nano 2012, 6, 1416–1426. 10.1021/nn204214x. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Moyano D. F.; Parnsubsakul A.; Papadopoulos A.; Wang L. S.; Landis R. F.; Das R.; Rotello V. M. Ultrastable and Biofunctionalizable Gold Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 14096–14101. 10.1021/acsami.6b02548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson D.; Guida F.; Sologan M.; Boccardo S.; Pengo P.; Perissinotto F.; Iacuzzi V.; Pellizzoni E.; Polizzi S.; Casalis L.; Pasquato L.; Pacor S.; Tossi A.; Posocco P. Mixed Fluorinated/Hydrogenated Self-Assembled Monolayer-Protected Gold Nanoparticles: In Silico and in vitro Behavior. Small 2019, 15 (15), 1900323. 10.1002/smll.201900323. [DOI] [PubMed] [Google Scholar]
- Wilder L. M.; Fies W. A.; Rabin C.; Webb L. J.; Crooks R. M. Conjugation of an Alpha-Helical Peptide to the Surface of Gold Nanoparticles. Langmuir 2019, 35, 3363–3371. 10.1021/acs.langmuir.9b00075. [DOI] [PubMed] [Google Scholar]
- Jha S.; Ramadori F.; Quarta S.; Biasiolo A.; Fabris E.; Baldan P.; Guarino G.; Ruvoletto M.; Villano G.; Turato C.; Gatta A.; Mancin F.; Pontisso P.; Scrimin P. Binding and Uptake into Human Hepatocellular Carcinoma Cells of Peptide-Functionalized Gold Nanoparticles. Bioconjugate Chem. 2017, 28, 222–229. 10.1021/acs.bioconjchem.6b00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H. G.; Webber M. J.; Stupp S. I. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers 2010, 94, 1–18. 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamley I. W. Self-Assembly of Amphiphilic Peptides. Soft Matter 2011, 7, 4122–4138. 10.1039/c0sm01218a. [DOI] [Google Scholar]
- Pashuck E. T.; Cui H. G.; Stupp S. I. Tuning Supramolecular Rigidity of Peptide Fibers through Molecular Structure. J. Am. Chem. Soc. 2010, 132, 6041–6046. 10.1021/ja908560n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks M. P.; Sato K.; Palmer L. C.; Stupp S. I. Supramolecular Assembly of Peptide Amphiphiles. Acc. Chem. Res. 2017, 50, 2440–2448. 10.1021/acs.accounts.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal H. S.; Kraatz H. B. Effect of the Surface Curvature on the Secondary Structure of Peptides Adsorbed on Nanoparticles. J. Am. Chem. Soc. 2007, 129, 6356–6357. 10.1021/ja0703372. [DOI] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Kernya L.; Lee Y. H.; Goto Y.; Refregiers M.; Kardos J. Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3095–E3103. 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frens G. Controlled Nucleation for Regulation of Particle Size in Monodisperse Gold Suspensions. Nature, Phys. Sci. 1973, 241, 20–22. 10.1038/physci241020a0. [DOI] [Google Scholar]
- Turkevich J.; Stevenson P. C.; Hillier J. A Study of the Nucleation and Growth Processes in the Synthesis of Colloidal Gold. Discuss. Faraday Soc. 1951, 11, 55–75. 10.1039/df9511100055. [DOI] [Google Scholar]
- Mei B. C.; Oh E.; Susumu K.; Farrell D.; Mountziaris T. J.; Mattoussi H. Effects of Ligand Coordination Number and Surface Curvature on the Stability of Gold Nanoparticles in Aqueous Solutions. Langmuir 2009, 25, 10604–10611. 10.1021/la901423z. [DOI] [PubMed] [Google Scholar]
- Spicer C. D.; Jumeaux C.; Gupta B.; Stevens M. M. Peptide and Protein Nanoparticle Conjugates: Versatile Platforms for Biomedical Applications. Chem. Soc. Rev. 2018, 47, 3574–3620. 10.1039/C7CS00877E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piella J.; Bastus N. G.; Puntes V. Size-Dependent Protein-Nanoparticle Interactions in Citrate-Stabilized Gold Nanoparticles: The Emergence of the Protein Corona. Bioconjugate Chem. 2017, 28, 88–97. 10.1021/acs.bioconjchem.6b00575. [DOI] [PubMed] [Google Scholar]
- Friedrich H.; Frederik P. M.; de With G.; Sommerdijk N. Imaging of Self-Assembled Structures: Interpretation of TEM and Cryo-TEM Images. Angew. Chem., Int. Ed. 2010, 49, 7850–7858. 10.1002/anie.201001493. [DOI] [PubMed] [Google Scholar]
- White S. J.; Johnson S. D.; Sellick M. A.; Bronowska A.; Stockley P. G.; Walti C. The Influence of Two-Dimensional Organization on Peptide Conformation. Angew. Chem., Int. Ed. 2015, 54, 974–978. 10.1002/anie.201408971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goormaghtigh E.; Cabiaux V.; Ruysschaert J. M. Secondary Structure and Dosage of Soluble and Membrane Proteins by Attenuated Total Reflection Fourier-Transform Infrared Spectroscopy on Hydrated Films. Eur. J. Biochem. 1990, 193, 409–420. 10.1111/j.1432-1033.1990.tb19354.x. [DOI] [PubMed] [Google Scholar]
- Lee S. J.; Han S. W.; Kim K. Perfluorocarbon-Stabilized Silver Nanoparticles Manufactured from Layered Silver Carboxylates. Chem. Commun. 2002, 442–443. 10.1039/b111607j. [DOI] [PubMed] [Google Scholar]
- Beddoes C. M.; Gooris G. S.; Bouwstra J. A. Preferential Arrangement of Lipids in the Long-Periodicity Phase of a Stratum Corneum Matrix Model. J. Lipid Res. 2018, 59, 2329–2338. 10.1194/jlr.M087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R.; McElhaney R. N. Membrane Lipid Phase Transitions and Phase Organization Studied by Fourier Transform Infrared Spectroscopy. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 2347–2358. 10.1016/j.bbamem.2012.10.018. [DOI] [PubMed] [Google Scholar]
- Haiss W.; Thanh N. T. K.; Aveyard J.; Fernig D. G. Determination of Size and Concentration of Gold Nanoparticles from UV-Vis Spectra. Anal. Chem. 2007, 79, 4215–4221. 10.1021/ac0702084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.