

Abstract
Regorafenib (Stivarga) is an oral small molecule kinase inhibitor used to treat metastatic colorectal cancer, hepatocellular carcinomas, and gastrointestinal stromal tumors. Diarrhea is one of the most frequently observed adverse reactions associated with regorafenib. This toxicity may arise from the reactivation of the inactive regorafenib-glucuronide to regorafenib by gut microbial β-glucuronidase (GUS) enzymes in the gastrointestinal tract. We sought to unravel the molecular basis of regorafenib-glucuronide processing by human intestinal GUS enzymes and to examine the potential inhibition of these enzymes. Using a panel of 31 unique gut microbial GUS enzymes derived from the 279 mapped from the human gut microbiome, we found that only four were capable of regorafenib-glucuronide processing. Using crystal structures as a guide, we pinpointed the molecular features unique to these enzymes that confer regorafenib-glucuronide processing activity. Furthermore, a pilot screen identified the FDA-approved raloxifene as an inhibitor of regorafenib reactivation by the GUS proteins discovered. Novel synthetic raloxifene analogs exhibited improved potency in both in vitro and ex vivo studies. Taken together, these data establish that regorafenib reactivation is exclusively catalyzed by gut microbial enzymes and that these enzymes are amenable to targeted inhibition. Our results unravel key molecular details of regorafenib reactivation in the GI tract and provide a potential pathway to improve clinical outcomes with regorafenib.
Graphical Abstract
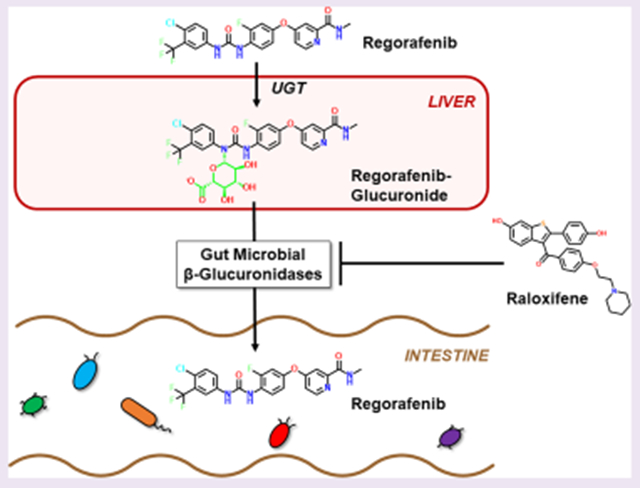
Regorafenib (Stivarga) is an oral small molecule kinase inhibitor whose antioncology effects are driven by inhibition of the tyrosine kinases VEGF and TIE2. It is indicated worldwide for patients with metastatic colorectal cancer (mCRC), hepatocellular carcinomas, and gastrointestinal (GI) stromal tumors. In the US and Europe, it is most often designated for patients with mCRC who have failed previous therapies or who are not candidates for other approaches. Although regorafenib is an effective antineoplastic compound, diarrhea is one of the most frequently observed adverse reactions.1–6 Regorafenib and its close structural homologue sorafenib, which is used for kidney, liver, and thyroid cancer (and also causes diarrhea), are known to be glucuronidated in mice, rats, and humans and to reach the GI tract as inactivated glucuronide metabolites.7–10 Thus, one mechanism by which regorafenib may cause GI toxicity is the reactivation of the drug within the GI lumen by gut microbial β-glucuronidase enzymes.11–14
Gut microbial β-glucuronidase (GUS) enzymes have been shown to reverse compound inactivation catalyzed by host phase II glucuronidation by removing the glucuronic acid added to a wide variety of drugs and endobiotics. Drug-glucuronide processing by GUS enzymes can lead to toxic levels of the reactivated drug in the GI tract. This process has been extensively studied for the colorectal and pancreas cancer drug irinotecan. Irinotecan’s active metabolite SN-38 is processed in the liver by phase II uridine diphosphate glucuronosyltransferase (UGT) enzymes to create the inactive metabolite SN-38-G, which is sent to the GI tract for excretion. In the gut, however, SN-38-G can serve as a substrate for microbial GUS enzymes that remove the glucuronide moiety to reactivate SN-38, which is toxic to intestinal epithelial cells and generates irinotecan’s dose-limiting diarrhea.15–18 A review of product package inserts has revealed that nearly 90% of oncology drugs known to be glucuronidated cause clinical gut toxicity. Nonsteroidal anti-inflammatory drugs (NSAIDs) are also metabolized by glucuronidation, reach the gut as inactive glucuronides, and are reactivated in the small intestine by gut microbial GUS enzymes to cause intestinal perforations and bleeding ulcers.19–24 Although this reactivation and subsequent toxicity has not been formally established for regorafenib, GUS enzymes may also play a role in regorafenib-induced toxicity.
GUS enzymes are capable of hydrolyzing a diverse array of glucuronides, but limited information is available on the specific types of gut microbial GUS enzymes that are most efficient at processing distinct drug glucuronides. To gain insight into the structural and functional diversity of GUS enzymes, we recently reported an atlas of 279 unique human gut microbial GUS enzymes identified from the stool sample catalogue in the Human Microbiome Project (HMP) and showed that these proteins clustered into six structural groups based on their active site loop features (e.g., Loop 1, Loop 2, No Loop, etc.).25 We have since created a representative panel of 31 of these enzymes for in vitro screening, and we have demonstrated that Loop 1 GUS enzymes are capable of processing the small standard glucuronide substrate p-nitrophenol-β-d-glucuronide (pNPG) and small molecule glucuronides including those of NSAIDs and SN-38 faster than non-Loop 1 GUS enzymes.15,25–27 However, this level of granularity has yet to be assigned to the other loop classes.
Here we investigate the role of a distinct subset of “No Loop” GUS enzymes in regorafenib-glucuronide reactivation. Regorafenib-glucuronide is unique among the drug-glucuronides examined to date because it is a “central glucuronide”; its glucuronic acid moiety is linked to the middle of the regorafenib molecule (Figure 1A). In contrast, SN-38-G and NSAID-glucuronides have their glucuronic acid sugars appended “terminally” to their chemical structure (Figure S1). Additionally, regorafenib-glucuronide is an N-linked glucuronide; all other substrates examined to date, including the standard GUS reporter substrates pNPG and 4-methylumbelliferyl glucuronide (4-MUG), have been O-linked. Thus, given these differences in chemical structure and glucuronide linkage, we hypothesized that a distinct set of gut microbial GUS enzymes would act on the central, N-linked regorafenib-glucuronide substrate compared to those that efficiently process compounds with terminal, O-linked glucuronides. To test this hypothesis, we screened our panel of 31 representative GUS enzymes using regorafenib-glucuronide and, validating our hypothesis, found that only four distinct enzymes are capable of reactivating regorafenib-glucuronide to regorafenib. Taken together, our data provide new information on drug-glucuronide processing by gut microbial GUS enzymes and suggest the need to discover new GUS inhibitors that have the potential to improve patient outcomes with regorafenib.
Figure 1.
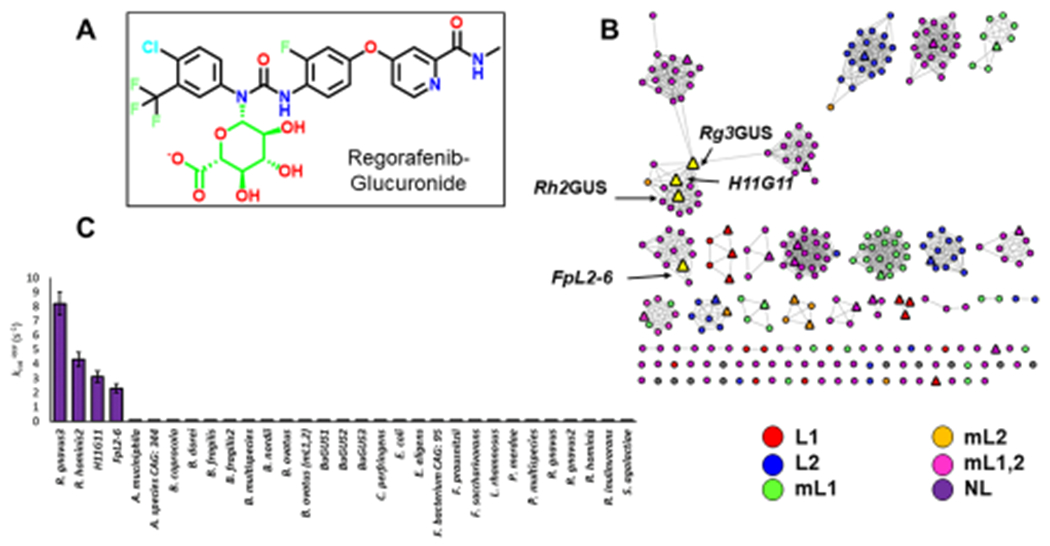
Identification of regorafenib-glucuronide processing GUS enzymes. (A) Structure of N-linked regorafenib-glucuronide with the glucuronide moiety highlighted in green. (B) Sequence similarity network (SSN) highlighting reactive GUS enzymes. SSN contains 279 GUS enzymes identified through the Human Microbiome Project (circles). Triangles are GUS enzymes tested. Yellow triangles are GUS enzymes that can reactivate regorafenib-glucuronide. SSN is color-coded by loop categorization defined previously. (C) rates of tested GUS enzymes.
RESULTS AND DISCUSSION
Identification of Regorafenib-glucuronide Processing Gut Microbial GUS Enzymes.
As members of the CAZy glycoside hydrolase 2 family, gut microbial GUS enzymes have been shown to share a common fold but exhibit unique active site structures and distinct substrate-processing functions.28,29 To gain greater insight into the specific sequence–structure–function relationships among GUS proteins, we generated a sequence similarity network (SSN) using sequences of β-glucuronidase enzymes found within the Human Microbiome Project. Using an alignment score of 10−220, the resultant SSN clusters the 279 unique protein sequences based on sequence identity and homology.30 We find that the human gut microbial GUS enzymes largely cluster based on the six previously defined active site loop architectures: Loop1 (L1), mini-Loop1 (mL1), Loop2 (L2), mini-Loop2 (mL2), mini-Loop1,2 (mL1,2), and No Loop (NL) (Figure 1B). Of the 279 unique GUS enzymes identified in the HMP, we have cloned, expressed, and purified 31 of these proteins for in vitro study. These enzymes were selected such that there is at least one representative enzyme from each major and minor clade in the SSN, as well as several singletons (Figure 1B). Enzymes were also chosen so that the prevalence of each loop category was comparable to what has been previously reported in the HMP.25 However, an exception to this is the Loop 1 enzymes, which are over-represented in our panel of 31 proteins as these have been previously shown to efficiently reactivate small molecule drug substrates, a key focus of our work.27 All 31 of these purified enzymes have been shown to be active with the small molecule GUS reporter substrate 4-MUG (Table S1). Of the 31 enzymes examined, crystal structures have been reported for 18 (Table S1), and these structural data correlate with the family groupings present in the SSN.
While most glucuronides tested to date are terminal, O-linked glucuronides regorafenib-glucuronide is unique in that it is a central N-linked glucuronide (Figure S1). For these reasons, we hypothesized that there would be a limited number of enzymes capable of processing this drug. To identify GUS enzymes capable of processing regorafenib-glucuronide, we incubated each of the 31 enzymes with 500 μM regorafenib-glucuronide to obtain a relative rate at one substrate concentration, a (s−1). For 27 of the 31 enzymes tested, no activity was observed (Figure 1C). However, four of the enzymes exhibited the ability to convert regorafenib-glucuronide to regorafenib (Figure 1C). These enzymes were Ruminococcus gnavus GUS3 (Rg3GUS [GenBank accession ID WP_118581144.1]), Roseburia hominis GUS2 (Rh2GUS [GenBank accession ID WP_118096903.1]), Faecalibacterium prausnitzii GUS L2-6 (FpL2-6 [GenBank accession ID CBK98066.1]), and H11G11 GUS (identified from an uncultured taxon [GenBank accession ID CBJ55484.1]). The four GUS enzymes that processed regorafenib-glucuronide are highlighted on the SSN (Figure 1B). Three of the four enzymes cluster in the same clade (H11G11, Rg3GUS, Rh2GUS) (Figure 1B), and the remaining enzyme (FpL2-6) is in an adjacent clade. These proteins are all NL enzymes and share between 42% and 69% sequence identity,31 and three have recently been shown to uniquely bind to the flavin cofactor FMN (FpL2-6, Rg3GUS, Rh2GUS).32 Thus, only an exclusive and small group of related GUS enzymes are capable of processing regorafenib-glucuronide.
We then determined catalytic efficiency (s−1 M−1) values with regorafenib-glucuronide for the four enzymes identified. These rates range from 1.21 × 102 to 9.94 × 103 s−1 M−1 (Table 1). In comparison, the catalytic efficiencies were also determined for the four enzymes with a small molecule reporter substrate, 4-methylumbelliferyl glucuronide (4-MUG). The rates for 4-MUG range between 8.92 × 104 and 5.40 × 105 s−1 M−1, 10–100 times faster than their rates with regorafenib-glucuronide (Table S2). Previous studies have examined the rate of Loop 1 GUS enzymes with the reporter substrate pNPG.27 These rates range between 2.0 × 103 and 9.2 × 105 s−1 M−1. Thus, the rates of regorafenib-glucuronide cleavage are significantly slower than what we have previously seen with other GUS enzymes and other GUS substrates. This is likely because the regorafenib-glucuronide substrate is significantly less accessible to the enzymes due to the steric hindrance a “central” glucuronide presents. Additionally, this is the first substrate examined that contains an N-linked glucuronide; the difference in electronic and structural characteristics of this linkage may contribute to the slower rates observed here. Despite their slower rates, though, we have identified the first microbiome-encoded enzymes from the GI tract capable of processing the inactive glucuronide conjugate of the anticancer drug regorafenib.
Table 1.
Rates of Regorafenib-Glucuronide Processing by FpL2-6, H11G11, Rg3, and Rh2 GUS Enzymes
| GUS | kcat/Km (s−1 M−1) |
|---|---|
| R. gnavus3 | 9900 ± 500 |
| R. hominis2 | 4870 ± 200 |
| H11G11 | 3540 ± 300 |
| FpL2-6 | 121 ± 10 |
Structural Interrogation of Regorafenib-glucuronide Processing.
The crystal structures of three of the four enzymes discovered to process regorafenib-glucuronide have been determined in their apo (unliganded) state; the exception to date is H11G11. Despite considerable effort, a regorafenib- or regorafenib-glucuronide-bound structure with the enzymes discovered has not been resolved. However, a model of regorafenib-glucuronide docked within the active site of Rg3GUS, the fastest enzyme with this substrate, was created based on extant structures and our knowledge of gut microbial GUS structure and function15,29,33 (Figure 2A). This model points toward key active site features that aid in the ability of these enzymes to process this substrate, including individual residues and the C-terminal domain (Figure 2A,B).
Figure 2.
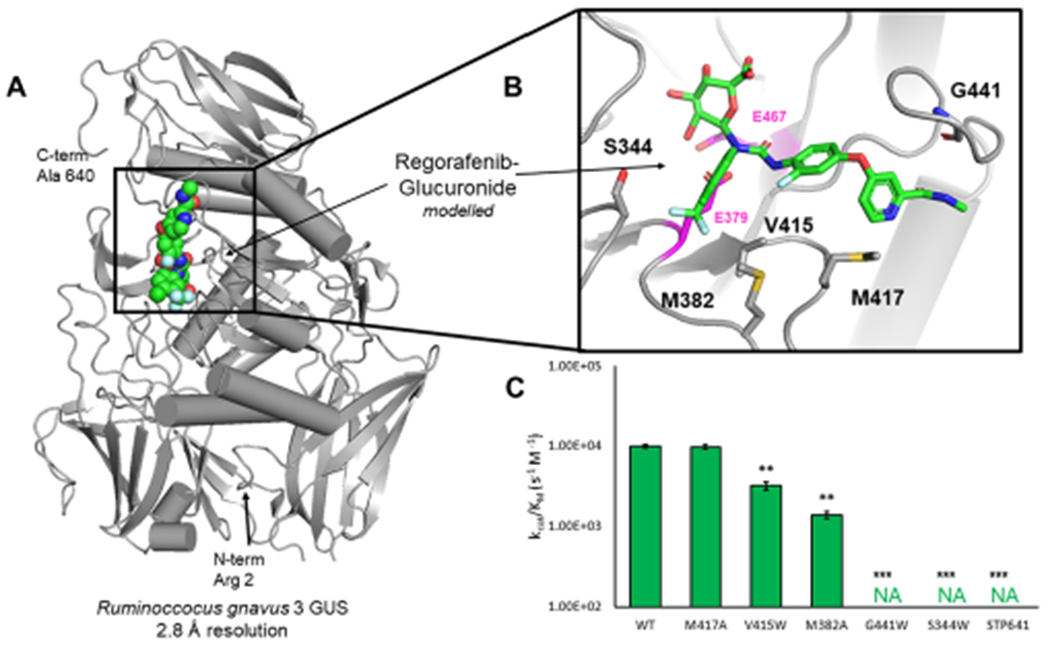
Structural basis for regorafenib-glucuronide processing. (A) Structure of Rg3GUS (PDB 6MVG) with regorafenib-glucuronide docked at active site (spheres). (B) Inset of Rg3GUS active site and modeled regorafenib-glucuronide. Residues of interest for mutagenesis are highlighted. Catalytic glutamates are highlighted in magenta. (C) Rates of Rg3GUS mutants in descending order from no effect to complete inactivation.
To test the validity of this model and to probe our understanding of the processing of regorafenib-glucuronide, mutations were introduced at key residues predicted to contact the regorafenib-glucuronide substrate at the active site. The catalytic glutamate residues are 2.3–6.7 Å from the residues examined by mutagenesis. Introduction of bulky tryptophan side chains at positions V415, G441, and S344 significantly reduced or eliminated regorafenib-glucuronide processing activity (Figure 2C). Furthermore, while mutating M417 to alanine had no effect on activity, mutating the adjacent M382 significantly reduced substrate-processing activity (Figure 2C). Importantly, most of the corresponding mutations had little effect on the processing of the smaller, standard GUS activity substrate 4-MUG (Figure S2). Specifically, V415W, M382A, and especially G441W, which significantly impacted regorafenib-glucuronide processing, had no effect on 4-MUG processing by Rg3GUS. Taken together, these data support the validity of the modeling of regorafenib-glucuronide into the active site of Rg3GUS and demonstrate that this unique central glucuronide substrate makes contacts to regions of the active site involved in 4-MUG processing.
The structures of Rg3GUS (PDB 6MVG), Rh2GUS (PBD 6MVH) and FpL2-6 (PDB 6MVF) have recently been reported by our group;32 these enzymes share high structural similarity, aligning with RMSD values of 1.6–1.8 Å over 624 equiv Cα positions. However, they all contain roughly 150 residues of missing density at their C-termini, potentially influencing the ability to visualize the complete active site architecture of these enzymes. We therefore modeled the C-terminal domain of Rg3GUS based on a previously resolved GUS structure with an intact C-terminal region present in its structure (5UJ6, a GUS from the human gut commensal microbe Bacteroides uniformis) (Figure 3A). We hypothesized that this region may be able to rotate into the active site and impact substrate turnover. To test this hypothesis, we created a form of Rg3GUS in which the C-terminal domain was eliminated by placing a stop codon following residue 641 (STP641). This mutant enzyme exhibits no activity with regorafenib-glucuronide and significantly reduced activity with 4-MUG, yet the structural integrity of this protein and all the mutant proteins examined here remains intact (Figure 2C, Figure 3B, Figure S3). These results indicate that the C-terminal domain of Rg3GUS is proximal to the active site and impacts substrate turnover. Although the exact role of this domain is unclear, recent work from our laboratory has demonstrated that similar C-terminal domains in other gut microbial GUS enzymes play roles in carbohydrate metabolism.27 Taken together, these data indicate that the C-terminal domains of the unique GUS enzymes identified here likely participate in the processing of both regorafenib-glucuronide and 4-MUG.
Figure 3.
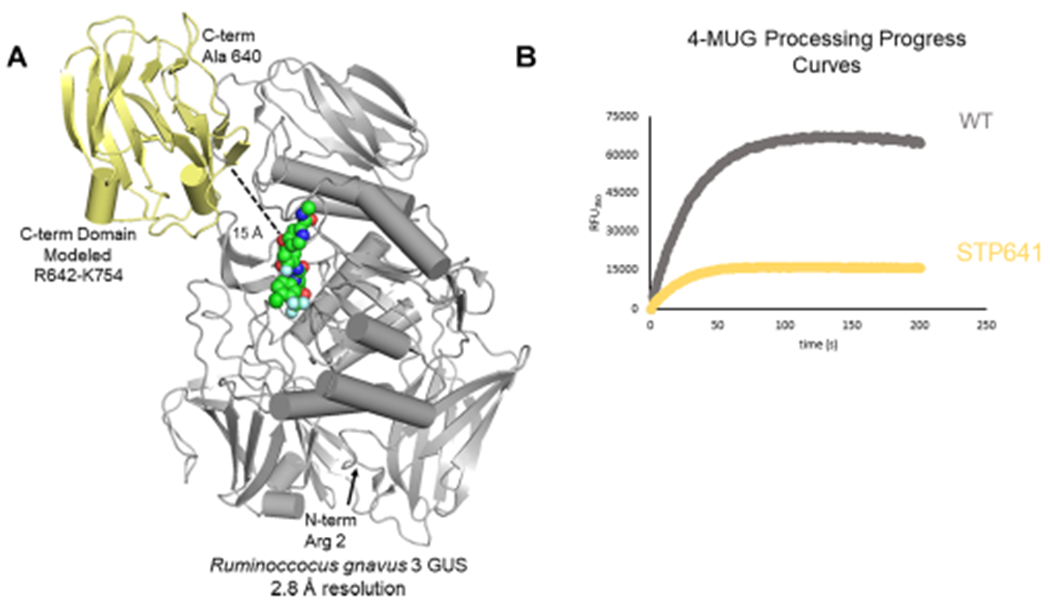
Previously unresolved domain of Rg3GUS. (A) Structure of Rg3GUS (PDB 6MVG) with regorafenib-glucuronide docked at active site (spheres in green). The yellow portion has not been resolved in the crystal structure and is modeled from extant GUS structure BuGUS2 (5UJ6). (B) Progress curves of WT and STP641 mutant with 4-MUG showing decreased activity of mutant.
Because Rg3GUS was the fastest of the four enzymes, we next sought to unravel differences between it and FpL2-6, the slowest enzyme. The two enzymes have an RMSD of 1.6 Å (Cα = 624) and thus have little difference in their overall secondary and tertiary structures. However, there is a unique and obvious difference in the individual residues at the active sites of these proteins: the key methionine in Rg3GUS (M382), Rh2GUS, and H11G11 is a glycine in FpL2-6 (G380) (Figure 4A). Note that the mutation of M382 to alanine significantly reduced the regorafenib-glucuronide processing activity of Rg3GUS. Thus, we hypothesized that G380 in FpL2-6 plays a role in the low regorafenib-glucuronide processing by this enzyme. Indeed, a G380M variant of FpL2-6 created to test this hypothesis exhibits enhanced activity from 1.21 × 102 to 4.56 × 103 s−1 M−1, a 40-fold increase in catalytic efficiency generated by a single point mutant (Figure 4B).
Figure 4.
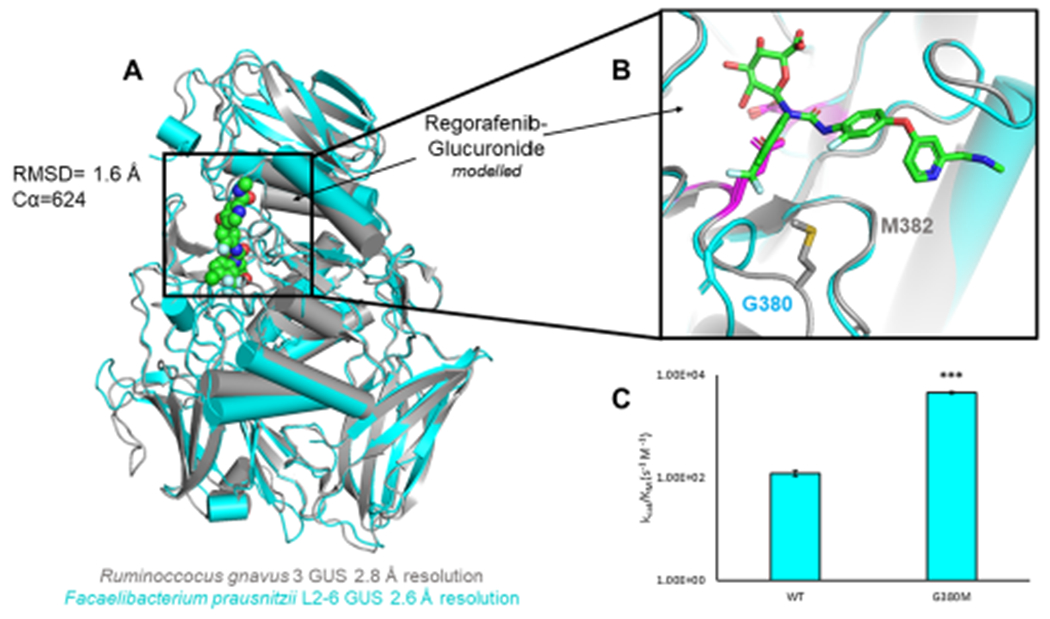
Significance of active site methionine. (A) Overlay of Rg3GUS (PDB 6MVG) and FpL2-6 (PDB 6MVF) the fastest and slowest GUS enzymes, respectively. (B) Inset of Rg3GUS and FpL2-6 GUS active sites and modeled regorafenib-glucuronide. Where Rg3GUS contains a methionine, FpL2-6 contains a glycine. Catalytic glutamates are highlighted in magenta. (C) Rates of FpL2-6 mutant G380M. Mutation to methionine increases the rate by 40-fold.
Finally, we examined the differences between the gut microbial enzymes capable of processing regorafenib-glucuronide and the many that were not able to catalyze the reactivation of this cancer drug. Using structural superpositions of the four enzymes discovered as active with regorafenib-glucuronide and the other orthologs of gut microbial GUS proteins (L1, mL1, L2, mL2, mL1,2, and other NL enzymes), we see that the active enzymes have considerably more open catalytic sites. Indeed, the inactive GUS enzymes form steric clashes with the regorafenib-glucuronide docked into the active site of Rg3GUS (Figure S4). These observations likely explain why 27 of the 31 enzymes examined had no activity with this substrate, as steric occlusion prevents productive substrate binding. In contrast, the four active enzymes uniquely have a more open platform at their active sites that allows this central glucuronide substrate to bind productively for catalysis. Taken together, these structural, modeling, and mutagenesis data begin to explain the molecular basis for the processing of regorafenib-glucuronide by specific gut microbial GUS enzymes.
Inhibition of Gut Microbial GUS Enzymes That Process Regorafenib-glucuronide.
We have previously developed selective, potent, and nonlethal inhibitors of gut microbial GUS enzymes that block the GI toxicity of irinotecan and poor outcomes with NSAIDs.15,16,18 However, to date, these inhibitors, which were discovered by high-throughput screening (HTS) using the Loop 1 GUS from E. coli, have been found to only be effective against Loop 1 gut microbial GUS enzymes.15,26 Thus, we sought to screen for inhibitors of the unique microbial GUS enzymes identified here that can process regorafenib-glucuronide. To accomplish this goal, we developed and validated a HTS-compatible assay using the Rg3GUS enzyme, the fastest with regorafenib-glucuronide. The assay contains three components: Rg3GUS, 4-MUG as the substrate, and compound (inhibitor) and exhibits a high-quality Z-prime (Z′) of >0.8. Using this assay, we conducted a preliminary screen with the Library of Pharmacologically Active Compounds (LOPAC1280; Sigma) to further validate the assay and identify potential inhibitor chemotypes of these NL GUS enzymes capable of reactivating regorafenib-glucuronide. Encouragingly, this initial proof-of-concept screen produced an excellent Z′ of 0.89 ± 0.02 and excellent correlation between duplicate runs (R2 = 0.98; Figure S5). Additionally, we obtained an initial hit, raloxifene, which inhibited Rg3GUS with an IC50 = 12.5 μM in follow-up dose–response studies (Table 2). This compound, which is a synthetic ligand for the estrogen receptor (ER) used in the treatment of osteoporosis, stood out to us because it is FDA approved, has been well-characterized, related compounds could be purchased commercially, and most side effects associated with administration of raloxifene HCl, including flu syndrome and hot flashes, are relatively minor.34–39 Additionally, by the calculations proposed in Maier et al.,40 the luminal concentration of raloxifene is roughly 40 μM in patients administered a low dose (60 mg/day) of the drug. This is above what is necessary to inhibit Rg3GUS in vitro. Furthermore, because the structure–activity relationship (SAR) around ER-binding has been well-characterized, it is expected that reducing off-target ER-binding would be highly feasible.
Table 2.
IC50 (μM) of Raloxifene and the Most Effective Analogsa
| FpL2-6 | H11G11 | Rg3GUS | Rh2GUS | |
|---|---|---|---|---|
| raloxifene | 78 ± 9 | 100 ± 5 | 13 ± 2 | 13 ± 3 |
| UNC7084 | 18 ± 3 | 16 ± 3 | 5 ± 1 | 7 ± 3 |
| UNC7087 | 15 ± 1 | 16 ± 1 | 4 ± 1 | 2 ± 1 |
| UNC7159 | 13 ± 1 | 14 ± 1 | 7 ± 2 | 3 ± 1 |
Data are presented in micromolar as the average of 3 biological replicates ± SEM.
Using raloxifene as a starting point, we next purchased analogs of raloxifene, as well as tamoxifen (the first-generation selective ER modulator, SERM), and other ER ligands (Figure S6) to perform an analog-by-catalog effort. In general, analogs of raloxifene were found to be more potent inhibitors of all four GUS enzymes in comparison to the other ER ligands tested (Table S3). Moreover, the commercial analog bazedoxifene showed minor potency improvements over raloxifene, suggesting that there is an opportunity to make even more potent compounds through rational medicinal chemistry. Thus, from the LOPAC screen and subsequent analyses, we identified raloxifene as an initial inhibitor compound capable of disrupting the catalytic activity of the gut microbial GUS enzymes that process regorafenib-glucuronide.
Synthesis of Novel GUS Inhibitor Analogs.
We next sought to create analogs of raloxifene that varied at four key positions to examine the SAR of this scaffold for inhibiting the four gut microbial GUS enzymes that process regorafenib-glucuronide (Figure 5A). Raloxifene was chosen due to its commercial availability and straightforward, modular synthesis. While we examined the two hydroxyl moieties and the ketone present in raloxifene, the piperidine group and its associated flexible linker were varied most extensively (Figure 5A). Given the low micromolar potency of bazedoxifene toward Rg3 and Rh2GUS, we were particularly interested in incorporating some of the structural features of bazedoxifene into the raloxifene scaffold (Figure S6). We synthesized a total of 20 raloxifene analogs that sample changes at each of these four positions (Figure S7, Figure S11–38), and we examined the ability of each of these analogs to inhibit the processing of regorafenib-glucuronide by the four gut microbial enzymes of interest, Rg3GUS, Rh2GUS, H11G11, and FpL2-6 (Table S4). Taken together, the data collected revealed that five novel analogs exhibited 2–6-fold improved potencies toward the four enzymes of interest compared to the starting molecule, raloxifene (Figure 5B, Table 2). First, we observed that reduction of the carbonyl group of raloxifene as in UNC7084, increased the overall flexibility of the molecule and led to a modest increase in potency for all four enzymes. Second, replacing the phenoxyethylpiperidine moiety with a phenyl-piperazine as in both UNC7087 and UNC7159 led to improvements in efficacy. Interestingly, UNC7268, which contains both of these modifications, demonstrated overall comparable potencies to both UNC7084 and UNC7159, and therefore an additive increase in potency was not observed. UNC7267, in which the piperidine is replaced with a larger azepane ring and the carbonyl group is reduced to more closely resemble bazedoxifene, is also more potent than raloxifene for the four enzymes tested but exhibits potencies comparable to the other improved analogs. Additionally, methylation of either a single or both hydroxyl groups did not result in consistent changes in potency across the four enzymes. The fact that the hydroxyl groups can be modified while still maintaining GUS inhibition is promising, as the 6′ hydroxyl group of raloxifene is necessary for ER binding.37 This suggests that methylation of this group can therefore decouple GUS inhibition from ER activity, if necessary.
Figure 5.
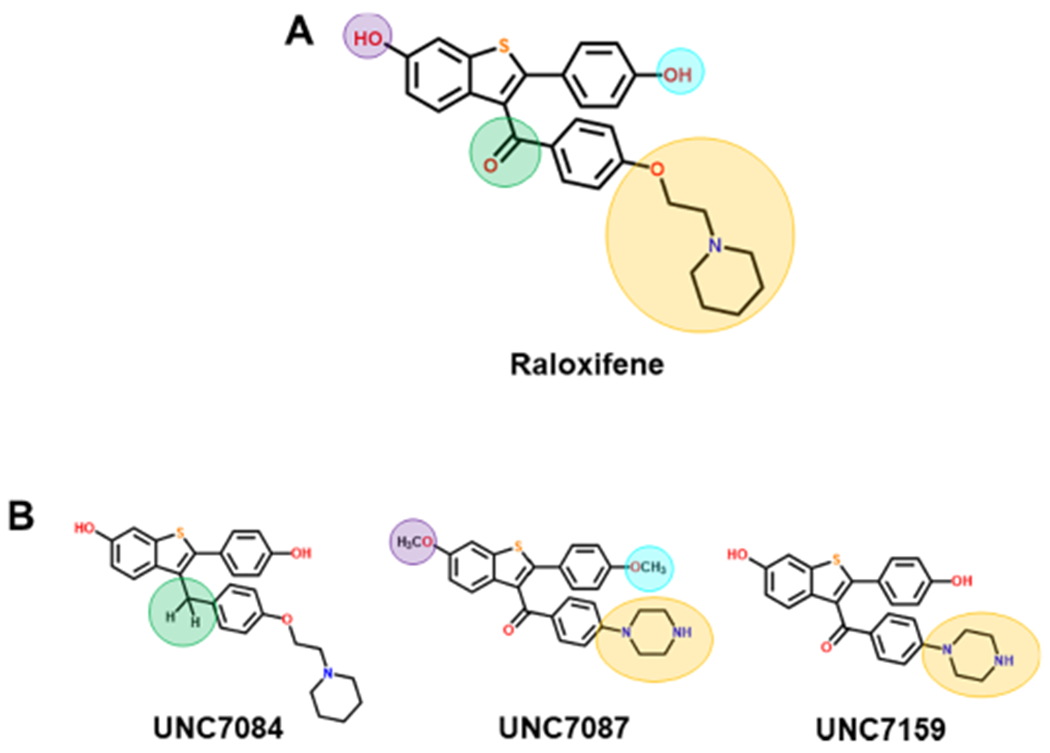
Inhibitor scaffolds. (A) Highlighted positions indicate modifications of raloxifene for SAR analysis. (B) Three analogs demonstrating improved potency over raloxifene.
We have previously shown for Loop 1 GUS enzymes that piperazine and piperidine rings are effective at potently inhibiting these particular gut microbial GUS enzymes;26 we were therefore intrigued to observe that a piperazine moiety enhanced inhibitor efficacy with the four enzymes screened. Thus, perhaps such ring structures with secondary amines may prove to be effective against a range of gut microbial GUS enzymes. Each chemotype appears specific, however, to a small clade of enzymes: UNC7084, UNC7087, and UNC7159, the three most potent analogs, show limited inhibition of the mammalian GUS enzyme, or other gut microbial GUS proteins (Figure S8), indicating that they are selective for the regorafenib-glucuronide processing proteins of interest in this study. Further, we show that they do not affect bacterial cell viability in either R. gnavus or E. coli (Figure S9). Taken together, these data reveal that small adjustments to the chemical structure of the screening hit raloxifene produced promising results with respect to targeted inhibition of gut microbial GUS enzymes capable of reactivating regorafenib from regorafenib-glucuronide.
Inhibition of Regorafenib-Glucuronide Reactivation in Mouse ex Vivo Intestinal Samples.
Finally, we sought to examine the reactivation of regorafenib-glucuronide by gut microbial GUS enzymes present in mammalian intestinal contents. We term these assays “in fimo” from the proper Latin word for excrement.41 We recently reported that the mouse GUSome contains 444 unique GUS enzymes, roughly 50% more than were found in the HMP.42 The mouse GUSome contains sequences with ≥80% identity to Rg3GUS, Rh2GUS, and FpL2-6, three of the four enzymes identified from the Human Microbiome Project capable of reactivating regorafenib-glucuronide in vitro. We therefore reasoned that mice would be an appropriate model of regorafenib-reactivation in fimo. Using material derived from the GI tract, we extracted the full set of active enzymes in each sample and examined them for substrate-processing activity. Typically, these assays take time points over the course of 1–3 h after addition of 1 mM substrate to monitor parent compound appearance or glucuronide disappearance. However, using both the fluorescence assay and HPLC methods we observed no conversion of regorafenib-glucuronide to regorafenib over 4 h in fecal samples. We tried 10 different mice that varied in strain (C57BL/6 and BALB/c), sex (M/F), and age (8–16 weeks). Similarly, these experiments were conducted with human feces from two donors, and again we see no regorafenib-glucuronide reactivation in feces alone.
Given the lack of activity observed in feces, we then dissected luminal samples from euthanized specific pathogen free (SPF) C57BL/6J mice into three portions: cecum, small intestine, and large intestine. We homogenized these intact samples to create lysates that would include microbial and host factors. We then repeated the assays using lysates from the homogenized samples, with time points up to 48 h. Although we see activity in every portion of the GI with these homogenates, we see relatively little conversion of regorafenib-glucuronide to regorafenib after 48 h in the small and large intestine (Figure 6A). This may be due to a combination of factors, including the abundance of total GUS proteins within the sample matrix and the fact that the quantity of these specific enzymes capable of hydrolyzing regorafenib-glucuronide is low. In contrast to the small and large intestinal samples, within the same 48 h time frame, almost all regorafenib-glucuronide is converted to regorafenib in the cecal extracts of the SPF mice tested (Figure 6A). These results are akin to those published previously with the N-glucuronide of the nearly isostructural cancer drug sorafenib, which was shown to be subject to reactivation by mouse cecal contents.14 We then replicated the regorafenib-glucuronide experiment using the ceca of germ-free (GF) mice to confirm that this reaction was not driven by host factors. We excised the cecum of 5 GF mice and dissected them into two parts: one contained the cecum and all of its contents (Figure 6B) and the second contained just the cecal contents (Figure 6C). After 48 h of incubation with regorafenib-glucuronide, we find that neither the cecum nor its contents were able to catalyze the reactivation of regorafenib from regorafenib-glucuronide. Thus, in the mouse cecum, the conversion of regorafenib to regorafenib-glucuronide is exclusively catalyzed by gut microbial enzymes.
Figure 6.
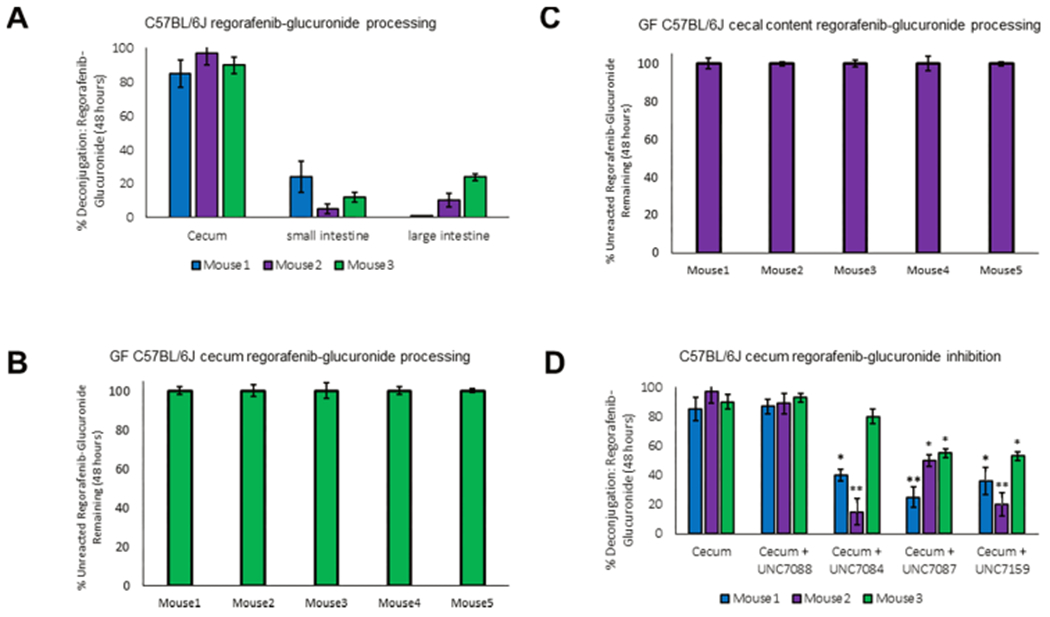
Cleavage and inhibition of regorafenib processing in mouse intestines. (A) Intestinal homogenate incubation with regorafenib-glucuronide in C57BL/6J mice after 48 h incubation at 37 °C. (B) In-tact cecum incubation with regorafenib-glucuronide in germ-free C57BL/6J mice shows no reactivation of regorafenib-glucuronide after 48 h incubation at 37 °C. X-axis delineates mouse (M) 1–5. (C) Cecal content incubation with regorafenib-glucuronide in germ-free C57BL/6J mice shows no reactivation of regorafenib-glucuronide after 48 h incubation at 37 °C. X-axis delineates mouse (M) 1–5. (D) Incubation (48 h) of cecal homogenates with addition of 20 μM inhibitor reduces regorafenib-glucuronide reactivation.
Focusing on the cecal homogenates from SPF mice that showed the largest percent conversion of regorafenib-glucuronide, we next sought to examine the ability of our novel analogs to inhibit this conversion. We tested three inhibitors, UNC7084, UNC7087, and UNC7159, and one negative control compound, UNC7088, that showed no inhibition in vitro. As expected, UNC7088 showed no inhibition in the cecal mixtures up to 100 μM (Figure 6D). However, at 20 μM inhibitor concentration, UNC7084, UNC7087, and UNC7159 significantly inhibited the conversion of regorafenib-glucuronide to regorafenib in two of the three samples tested (Figure 6D, Figure S10). UNC7084 failed to inhibit conversion by the cecal contents of one mouse, suggesting the possibility that additional regorafenib-glucuronide processing GUS enzymes exist that have yet to be discovered. In summary, however, we show that microbial GUS enzymes present in the GI tracts of mice, particularly in the cecum, are capable of converting regorafenib-glucuronide into regorafenib and are subject to inhibition by the small molecules presented here.
Taken together, we have identified a unique group of gut microbial GUS enzymes that process the unusual drug glucuronide of regorafenib, and have inhibited this reactivation both in vitro and ex vivo using novel analogs designed from a preliminary screening hit. However, there are several limitations to the current study. First, the connection between regorafenib-induced gut toxicity and the intestinal microbiota has not been established, and this will be a crucial focus of future work. If germ-free or antibiotic-treated mice still demonstrate regorafenib-induced gut damage, reversing such toxicity with inhibitors of gut microbial GUS enzymes would be unlikely to succeed. Second, in our experience, submicromolar inhibitors are necessary to observe protection against drug-induced gut toxicity in mice. Because we do not yet have this level of potency in the initial compounds presented here, we have not yet tested their efficacy against regorafenib-induced intestinal damage in vivo. Again, this will be performed as more potent compounds are developed. Third, the potential for raloxifene and the analogs described to alter the glucuronidation of regorafenib by mammalian UGT enzymes needs to be assessed. However, the data presented here demonstrate that unusual drug-glucuronide conjugates like regorafenib-glucuronide are processed by a unique small group of gut microbial GUS enzymes, which are in turn subject to targeted inhibition by compounds related to the SERM raloxifene. These results set the stage for the examination of the key questions outlined above toward the potential improvement of regorafenib and related kinase inhibitors like sorafenib in the clinical treatment of human malignancies.
Conclusion.
This study pinpointed a subset of distinct gut microbial GUS enzymes capable of reactivating regorafenib from regorafenib-glucuronide. We identified structural features of these enzymes that are essential to this reactivation, and we developed inhibitors that block this process both in vitro and in intestinal preparations from mice. Further, we show by comparing specific pathogen free and germ-free mice that reactivation is exclusively catalyzed by gut microbial enzymes. Thus, we have successfully initiated the development of the tools necessary to address regorafenib-induced gut toxicity. In addition, we provide a roadmap for the identification of gut microbial enzymes responsible for the toxic side effects of other tyrosine kinase inhibitors and the discovery of microbiome-targeted reagents to block these side effects.
METHODS
Full details for all materials and methods are provided in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Hardy and F. Potjewyd in the Center for Integrative Chemical Biology and Drug Discovery and the Chemistry Mass Spectrometry Core Laboratory at UNC Chapel Hill, especially D. Wallace, for help with identifying and validating compound hits. We thank A. Naziripour, D. Jenness, and L. Holt for technical support. And finally, we thank the National Institutes of Health (CA098468 and CA207416; M.R.R.) and the National Science Foundation GRFP (DGS-1650116; S.M.E.) for funding this research.
ABBREVIATIONS
- GUS
β-glucuronidase
- HMP
Human Microbiome Project
- FMN
flavin mononucleotide
- WT
wild-type
- 4-MUG
4-methylumbelliferyl glucuronide
- HTS
high-throughput screening
- SERM
selective estrogen receptor modulator
- ER
estrogen receptor
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- HSQC
heteronuclear single quantum correlation spectroscopy
- HMBC
heteronuclear multiple bond correlation spectroscopy
- LCMS
liquid chromatography–mass spectroscopy
- MeCN
acetonitrile
- TFA
2,2,2-trifluoroacetic acid
- THF
tetrahydrofuran
- GF
germ-free
- SPF
specific-pathogen-free
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publication Web site The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00663.
Materials and methods, activity of 31 GUS enzymes examined with 4-MUG, catalytic efficiency of 4-MUG by FpL2-6, H11G11, Rg3, and Rh2 GUS enzymes, IC50 (μM) for the 4 GUS enzymes involved in regorafenib-glucuronide processing of raloxifene related commercial compounds, SAR of raloxifene analogs, terminal and central glucuronides, structural basis for regorafenib-glucuronide processing, circular dichroism wavelength scan and melting temperature, steric occlusion by GUS orthologs, results of LOPAC HTS, structures of ER ligands, synthesized raloxifene analogs, targeted inhibition of regorafenib-glucuronide processing GUS enzymes, bacterial cell viability in the presence of synthesized compounds, HPLC results of mouse intestinal incubation, characterization (NMR and MS) of synthesized compounds (PDF)
The authors declare the following competing financial interest(s): Matthew Redinbo is a Founder of Symberix, Inc., which is developing microbiome-targeted therapeutics.
REFERENCES
- (1).Dhillon S (2018) Regorafenib: A Review in Metastatic Colorectal Cancer. Drugs 78, 1133–1144. [DOI] [PubMed] [Google Scholar]
- (2).Rey J-B, Launay-Vacher V, and Tournigand C (2015) Regorafenib as a single-agent in the treatment of patients with gastrointestinal tumors: an overview for pharmacists. Target. Oncol 10, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wilhelm S, Dumas J, Adnane L, Lynch M, Carter C, Schütz G, Zopf D, and Thierauch K-H (2011) Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 129, 245–255. [DOI] [PubMed] [Google Scholar]
- (4).Abou-Elkacem L, Arns S, Brix G, Gremse F, Zopf D, Kiessling F, and Lederle W (2013) Regorafenib Inhibits Growth, Angiogenesis, and Metastasis in a Highly Aggressive, Orthotopic Colon Cancer Model. Mol. Cancer Ther 12, 1322–1331. [DOI] [PubMed] [Google Scholar]
- (5).Bruix J, Tak W-Y, Gasbarrini A, Santoro A, Colombo M, Lim H-Y, Bolondi L, et al. (2013) Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur. J. Cancer 49, 3412–3419. [DOI] [PubMed] [Google Scholar]
- (6).Grothey A, Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Laurent D, et al. (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312. [DOI] [PubMed] [Google Scholar]
- (7).Stivarga: Highlights of prescribing information. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm (Accessed July, 15, 2019), Bayer Pharmaceuticals, 2012. [Google Scholar]
- (8).Tlemsani C, Huillard O, Arrondeau J, Boudou-Rouquette P, Cessot A, Blanchet B, Goldwasser F, et al. (2015) Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opin. Drug Metab. Toxicol 11, 785–794. [DOI] [PubMed] [Google Scholar]
- (9).Strumberg D, and Schultheis B (2012) Regorafenib for cancer. Expert Opin. Invest. Drugs 21, 879–889. [DOI] [PubMed] [Google Scholar]
- (10).Ettrich T, and Seufferlein T (2018) Regorafenib. Recent Results Cancer Res. 211, 45–56. [DOI] [PubMed] [Google Scholar]
- (11).Hotta K, Ueyama J, Tatsumi Y, Tsukiyama I, Sugiura Y, Saito H, and Hasegawa T (2015) Lack of Contribution of Multidrug Resistance-associated Protein and Organic Anion-transporting Polypeptide to Pharmacokinetics of Regorafenib, a Novel Multi-Kinase Inhibitor, in Rats. Anticancer Res. 35, 4681–4689. [PubMed] [Google Scholar]
- (12).Fu Q, Chen M, Anderson JT, Sun X, Hu S, Sparreboom A, and Baker SD (2019) Interaction Between Sex and Organic Anion-Transporting Polypeptide 1b2 on the Pharmacokinetics of Regorafenib and Its Metabolites Regorafenib-N-Oxide and Regorafenib-Glucuronide in Mice. Clin. Transl. Sci 12, 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Edginton AN, Zimmerman EI, Vasilyeva A, Baker SD, and Panetta JC (2016) Sorafenib metabolism, transport, and enterohepatic recycling: physiologically based modeling and simulation in mice. Cancer Chemother. Pharmacol 77, 1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vasilyeva A, Durmus S, Li L, Wagenaar E, Hu S, Gibson AA, Schinkel AH, et al. (2015) Hepatocellular Shuttling and Recirculation of Sorafenib-Glucuronide Is Dependent on Abcc2, Abcc3, and Oatp1a/1b. Cancer Res. 75, 2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wallace B, Wang H, Lane K, Scott J, Orans J, Koo J, Redinbo M, et al. (2010) Alleviating Cancer Drug Toxicity by Inhibiting a Bacterial Enzyme. Science 330, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wallace B, Roberts A, Pollet R, Ingle J, Biernat K, Pellock S, Redinbo M, et al. (2015) Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol 22, 1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Roberts A, Wallace B, Venkatesh K, Mani S, and Redinbo M (2013) Molecular Insights into Microbial β-Glucuronidase Inhibition to Abrogate CPT-11 Toxicity. Mol. Pharmacol 84, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Takasuna K, Hagiwara T, Hirohashi M, Kato M, Nomura M, Nagai E, and Kamataki T (1996) Involvement of beta-glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT-11) in rats. Cancer Res. 56, 3752–7. [PubMed] [Google Scholar]
- (19).Saitta K, Zhang C, Lee K, Fujimoto K, Redinbo M, and Boelsterli U (2014) Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 44, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li H, He J, and Jia W (2016) The influence of gut microbiota on drug metabolism and toxicity. Expert Opin. Drug Metab. Toxicol 12, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Boelsterli U, Redinbo M, and Saitta K (2013) Multiple NSAID-Induced Hits Injure the Small Intestine: Underlying Mechanisms and Novel Strategies. Toxicol. Sci 131, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Allison M, Howatson A, Torrance C, Lee F, and Russell R (1992) Gastrointestinal Damage Associated with the Use of Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med 327, 749–754. [DOI] [PubMed] [Google Scholar]
- (23).Duggan D, Hooke K, Noll R, and Chiu Kwan K (1975) Enterohepatic circulation of indomethacin and its role in intestinal irritation. Biochem. Pharmacol 24, 1749–1754. [DOI] [PubMed] [Google Scholar]
- (24).A. Boelsterli U, and Ramirez-Alcantara (2011) NSAID Acyl Glucuronides and Enteropathy. Curr. Drug Metab 12, 245–252. [DOI] [PubMed] [Google Scholar]
- (25).Pollet R, D’Agostino E, Walton W, Xu Y, Little M, Biernat K, Redinbo M, et al. (2017) An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 25, 967–977. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pellock S, Creekmore B, Walton W, Mehta N, Biernat K, Cesmat A, Redinbo M, et al. (2018) Gut Microbial β-Glucuronidase Inhibition via Catalytic Cycle Interception. ACS Cent. Sci 4, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Biernat K, Pellock S, Bhatt A, Bivins M, Walton W, Tran B, Redinbo M, et al. (2019) Structure, function, and inhibition of drug reactivating human gut microbial β-glucuronidases. Sci. Rep 9, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Little M, Pellock S, Walton W, Tripathy A, and Redinbo M (2018) Structural basis for the regulation of β-glucuronidase expression by human gut Enterobacteriaceae. Proc. Natl. Acad. Sci. U. S. A 115, E152–E161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pellock S, Walton W, Biernat K, Torres-Rivera D, Creekmore B, Xu Y, Redinbo M, et al. (2018) Three structurally and functionally distinct β-glucuronidases from the human gut microbe Bacteroides uniformis. J. Biol. Chem 293, 18559–18573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gerlt J, Bouvier J, Davidson D, Imker H, Sadkhin B, Slater D, and Whalen K (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta, Proteins Proteomics 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Altschul S, Gish W, Miller W, Myers E, and Lipman D (1990) Basic local alignment search tool. J. Mol. Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- (32).Pellock S, Walton W, Ervin S, Torres-Rivera D, Creekmore B, Bergan G, Redinbo M, et al. (2019) Discovery and Characterization of FMN-Binding β-Glucuronidases in the Human Gut Microbiome. J. Mol. Biol 431, 970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Combs SA, DeLuca SL, DeLuca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, and Meiler J (2013) Small-molecule ligand docking into comparative models with Rosetta. Nat. Protoc 8, 1277–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dadiboyena S (2012) Recent advances in the synthesis of raloxifene: A selective estrogen receptor modulator. Eur. J. Med. Chem 51, 17–34. [DOI] [PubMed] [Google Scholar]
- (35).Kalinin A, Reed M, Norman B, and Snieckus V (2003) Synthesis of Constrained Raloxifene Analogues by Complementary Use of Friedel–Crafts and Directed Remote Metalation Reactions. J. Org. Chem 68, 5992–5999. [DOI] [PubMed] [Google Scholar]
- (36).Jordan V (2003) Antiestrogens and Selective Estrogen Receptor Modulators as Multifunctional Medicines. 2. Clinical Considerations and New Agents. J. Med. Chem 46, 1081–1111. [DOI] [PubMed] [Google Scholar]
- (37).Martín-Santamaría S, Rodríguez J-J, de Pascual-Teresa S, Gordon S, Bengtsson M, Garrido-Laguna I, Ramos A, et al. (2008) New scaffolds for the design of selective estrogen receptor modulators. Org. Biomol. Chem 6, 3486–34956. [DOI] [PubMed] [Google Scholar]
- (38).Grese TA, Pennington LD, Sluka JP, Adrian MD, Cole HW, Fuson TR, Bryant H, et al. (1998) Synthesis and Pharmacology of Conformationally Restricted Raloxifene Analogues: Highly Potent Selective Estrogen Receptor Modulators. J. Med. Chem 41, 1272–1283. [DOI] [PubMed] [Google Scholar]
- (39).Evista: Highlights of prescribing information http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm (Accessed July 20, 2019), TEVA Pharmaceutical Industries, 1997. [Google Scholar]
- (40).Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson E, Typas A, et al. (2018) Extensive impact of nonantibiotic drugs on human gut bacteria. Nature 555, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bhatt A, Grillo L, and Redinbo M (2019) In Fimo: A Term Proposed for Excrement Examined Experimentally. Gastroenterology 156, 1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Creekmore BC, Gray JH, Walton WG, Biernat KA, Little MS, Xu Y, Redinbo MR, et al. (2019) Mouse Gut Microbiome-Encoded β-Glucuronidases Identified Using Metagenome Analysis Guided by Protein Structure. MSystems 4, e00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.