

Abstract
EGFR inhibition is efficacious in cancer therapy, but initially sensitive tumors often develop resistance. In this study, we investigated the potential to overcome acquired resistance to EGFR inhibitors with MEHD7945A, a monoclonal antibody that dually targets EGFR and HER3 (ErbB3). In cancer cells resistant to cetuximab and erlotinib, we found that MEHD7945A, but not single target EGFR inhibitors, could inhibit tumor growth and cell cycle progression in parallel with EGFR/HER3 signaling pathway modulation. MEHD7945A was more effective than a combination of cetuximab and anti-HER3 antibody at inhibiting both EGFR/HER3 signaling and tumor growth. In human tumor xenograft models, we confirmed the greater antitumor potency of MEHD7945A when compared to cetuximab or erlotinib. MEHD7945A retained potent activity in tumors refractory to EGFR inhibitor alone. Further, MEHD7945A also limited cross-resistance to radiation in EGFR inhibitor-resistant cells by modulating cell cycle progression and repair processes that control apoptotic cell death. Taken together, our findings confirm an important role of compensatory HER3 signaling in the development of acquired resistance to EGFR inhibitors and offer preclinical proof of concept that MEHD7945A can effectively overcome EGFR inhibitor resistance.
Keywords: MEHD7945A, HER3, Cetuximab, Radiation, Resistance
Introduction
Members of the ErbB/HER receptor family (EGFR, HER2, HER3, HER4) play an important role in tumorigenesis and have been studied intensively in cancer therapeutics. Blockade of the EGFR using either monoclonal antibody (mAb) or small molecule tyrosine kinase inhibitor (TKI) offers a promising approach that has been well validated over the last decade (1, 2). Unfortunately, many patients who initially respond to EGFR inhibitor treatments eventually manifest tumor progression (3–5). Hence, efforts to better understand underlying mechanisms of acquired resistance to EGFR inhibitors, and potential strategies to overcome resistance, are highly needed.
To understand underlying mechanisms of acquired resistance to EGFR inhibitors, we previously established a series of resistant clones to two different classes of EGFR inhibitors, cetuximab (mAb) and erlotinib (TKI), from sensitive tumor cell lines without EGFR and KRAS mutations following long-term EGFR inhibitor exposure (6, 7). Following systematic screening, we identified a significant increase of p-EGFR and p-HER3 in these resistant clones. Depletion of HER3 by siRNA restored sensitivity to the EGFR inhibitor cetuximab (8). Further analysis of these clones revealed an increase of EGFR-HER3 dimerization and subsequent EGFR-dependent activation of HER3. Consistent with this observation, several studies indicated that acquired resistance to EGFR inhibitors may derive in part from activation of HER3 to effectively bypass the effect of EGFR inhibition (9–11). Since HER3 is an obligate hetero-dimerization partner, these findings provide a rationale for the evaluation of combinatorial EGFR/HER3 targeting approaches in tumors manifesting acquired resistance to EGFR inhibitors.
As an inactive tyrosine kinase, HER3 is not amenable to inhibition with ATP analogs. MEHD7945A is a recently identified dual target antibody against EGFR and HER3 that exhibits dual action by inhibiting ligand dependent EGFR- and HER3-mediated downstream signaling (12). MEHD7945A shows profound antitumor activity in vitro and in vivo across a variety of tumor cell types when compared to the respective monospecific antibodies. In addition, MEHD7945A is effective in facilitating antibody-dependent cell-mediated cytotoxity, but appears to induce less skin toxicity in comparison to cetuximab in non-clinical studies. In the current study, we sought to investigate the capacity of MEHD7945A to overcome acquired resistance to EGFR inhibitors in our established cetuximab- and erlotinib-resistant tumor cells derived from lung and H&N cancers. In addition, since previous studies suggested cross-resistance to radiation in these resistant cells (7), we examined the effect of MEHD7945A in regulating radiation response in EGFR inhibitor-resistant cells.
Materials and Methods
Reagents and antibodies.
MEHD7945A and anti-HER3 (DL3.6b) were provided by Genentech, Inc (South San Francisco, CA). Cetuximab (Erbitux®) was provided by ImClone Systems Inc. (New York, NY) and erlotinib (Tarceva®) was provided by OSI Pharmaceuticals (Long Island, NY). Antibodies against EGFR, p-EGFR (Y1173), HER3 and Histone 3 were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA) and anti-p-DNAPK and Ku80 were obtained from Thermal Scientific Lab Vision (Kalamazoo, MI)-. Anti-α-tubulin was obtained from Calbiochem (San Diego, CA). All other antibodies were obtained from Cell Signaling Technology (Beverly, MA) and all other chemicals were purchased from Sigma (St. Louis, MO).
Primary and EGFR inhibitor-resistant tumor cells.
The primary human non-small cell lung carcinoma (NSCLC) H226 cells were provided by Drs John Minna and Adi Gazdar (University of Texas Southwestern Medical School, Dallas, TX) and were maintained in RPMI with 10% FBS. The human head and neck squamous cell carcinoma (HNSCC) SCC6 (UM-SCC6) cells were provided by Dr. Thomas E. Carey (University of Michigan, Ann Arbor, MI) and were cultured routinely in DMEM supplemented with 10% FBS and 1 μg/ml hydrocortisone. These cells were tested and authenticated by the provider. The acquired cetuximab- and erlotinib-resistant clones of H226, and SCC6 were developed following long-term exposure to cetuximab or erloinib as described previously (6, 7). All Cell culture media and supplements were obtained from Life Technologies, Inc. (Gaithersburg, MD).
Cell proliferation assay.
Viable growing cells was determined by crystal violet staining as described previously (7).
Cell cycle analysis.
Tumor cells were harvested by trypsin followed by ethanol fixation. After centrifugation, cells were incubated with phosphate-citric acid buffer (0.2 M Na2HPO4, pH 7.8, 4 mM citric acid) at room temperature for 45 min. Thereafter, cells were stained with a solution containing 33 μg/ml PI, 0.13 mg/ml RNase A, 10 mM EDTA and 0.5% Triton X-100 at 4 °C for 4 hrs. Stained nuclei were analyzed for DNA-PI fluorescence using a Becton Dickinson FACScan flow cytometer. Resulting DNA content was analyzed by Modfit (Verity Software House Inc., Topsham, ME) to determine the proportion of cells in subG0, Go/G1, S, and G2/M phases of the cell cycle.
EGFR inhibitor-resistant tumor xenografts.
Athymic nude mice (3-4-week-old males) were obtained from Harlan Bioproducts for Science (Indianapolis, IN) and maintained in a laminar air-flow cabinet under aseptic conditions. The care and treatment of experimental animals was in accordance with institutional guidelines. Cetuximab- or erlotinib-resistant tumor cells (~1 x 106) were injected subcutaneously into the dorsal flank area of the mice. Following the establishment of tumor, cetuximab or MEHD7945A was administered via i.p. injection twice per week, and erlotinib was given by oral gavage 5 days per week. Radiation treatment was delivered by a cabinet X-ray biological irradiator X-RAD 320 from Precision X-Ray, Inc. (North Branford, CT). Mouse was immobilized using custom-designed jigs that only exposed the dorsal flank with tumor xenograft to irradiation without exposing non-tumor bearing normal tissues. Tumor volume was determined by direct measurement with calipers and calculated by the formula; π/6 × (large diameter) × (small diameter)2.
Immunofluorescent staining of γH2AX Foci.
Cells were plated on chamber slides and exposed to 10 μg/ml of drugs for 1.5 hrs before irradiation. Twenty-four hrs following 3 Gy radiation, cells were fixed in 2% paraformaldehyde and permeabilized in 0.2% Triton X-100. The cells were then probed with anti-γH2AX antibody (Upstate, Billerica, MA) followed by Alexa Fluor 594-conjugated secondary antibody (Invitrogen, Carlsbad, CA). Fluorescent γH2AX foci were then captured using a Zeiss Axioplan fluorescent microscope. To quantitate γH2AX foci, visual scoring of foci in 200 randomly chosen intact nuclei from irradiated samples was determined after subtracting the background numbers of foci from un-irradiated samples.
Radiation survival.
Survival following radiation exposure was defined as the ability of the cells to maintain their clonogenic capacity and to form colonies. Briefly, after exposure to radiation, cells were trypsinized, counted, and seeded for colony formation in 35 mm dishes at 50-5000 cells/dish. Following 10-14 days, colonies were stained with crystal violet and manually counted. Colonies consisting of 50 cells or more were scored, and 4-10 replicate dishes containing 10-150 colonies/dish were counted for each treatment.
Cellular fractionation and immunoblotting analyses.
Cellular fractionation was performed as described previously (13). Detailed information is provided in the Supplementary Materials and Methods.
Apoptosis assessment.
Apoptosis was assessed by the loss of plasma membrane asymmetry as one of the earliest features of apoptosis using Annexin V/Propidium iodide (PI) kit from BD Biosciences Pharmingen (San Diego, CA). Detailed information is provided in the Supplementary Materials and Methods.
Statistical analysis.
Student t-test was used to evaluate the significance of differences between 2 samples, and ANOVA was used to evaluate differences among 3 or more groups in tumor xenograft studies. Differences between samples were considered statistically significant when p < 0.05. To assess additive or synergistic effects, we used the fractional product method as described previously (14). Briefly, the observed fractional tumor volume (FTV) is equal to the mean tumor volume of each treated group divided by the mean tumor volume of the control group at each time point. The synergy assessment was determined by calculating the ratio of FTV (drug) x FTV (radiation) / FTV (drug + radiation). A ratio greater than 1.0 suggests that the combined treatment effects are synergistic.
Results
MEHD7945A inhibits growth of cetuximab-resistant tumor cells.
We previously established acquired resistant clones to two distinct classes of EGFR inhibitor following long-term exposure to cetuximab or erlotinib in NSCLC and HNSCC tumor cells. We first compared the in vitro anti-proliferative effect of MEHD7945A with cetuximab in cetuximab-resistant clones from H226 and SCC6. As shown in the left panel of Fig 1A, both cetuximab and MEHD7945A exhibited similar capacity to inhibit the growth of parental H226 cells (small box figure). Notably, MEHD7945A significantly inhibited tumor growth of H226-CetR cells that remained refractory to cetuximab treatment. Similar results were observed in the cetuximab-resistant clone of SCC6 as shown in the right panel of Fig 1A. While parental SCC-6 cells responded well to both antibody treatments (small box), SCC6-CetR only responded to MEHD7945A. In analysis of cell cycle progression, we found that MEHD7945A induced a significant G0/G1 arrest accompanied by a reduction in the percentage of cells in S phase compared to control or cetuximab-treated cetuximab-resistant cells (Supplemental Fig 1A). These results corresponded well to the anti-proliferative effect of MEHD7945A shown in Fig 1A. Further immunobloting analysis revealed a significant inhibition of the MAPK and the PI3K-AKT signaling pathways by MEHD7945A, but not cetuximab (Supplemental Fig 1B). Maintenance of PI3K-AKT signaling was observed in our resistant cells and is known to be a critical factor for acquired resistance to EGFR inhibitors in several previous studies (15), this result suggests that MEHD7945A may overcome acquired resistance to cetuximab in part via inhibition of HER3-PI3K-AKT signaling.
Fig. 1. MEHD7945A inhibits growth of cetuximab-resistant cells.
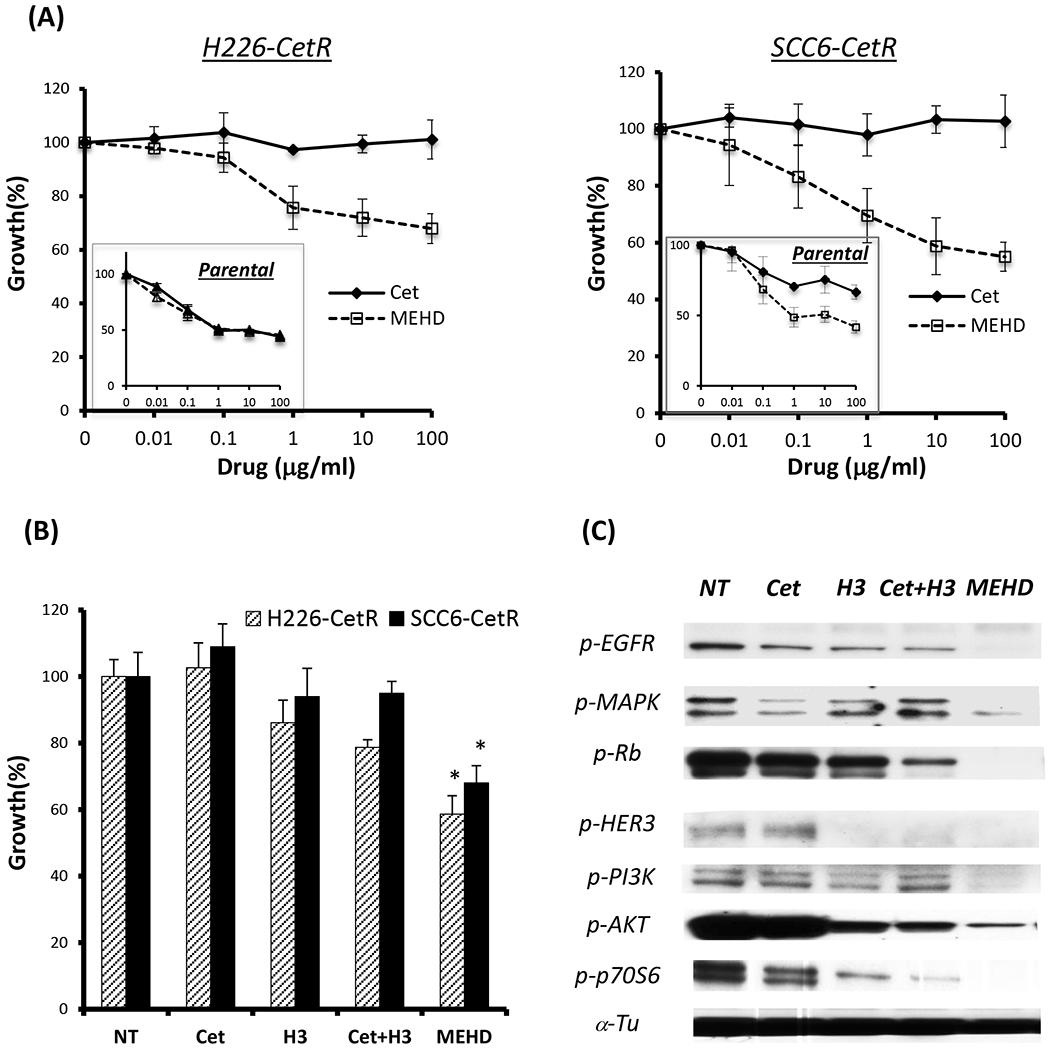
(A) cetuximab-resistant H226 (H226-CetR) or SCC6 (SCC6-CetR) cells were exposed to serial concentrations of cetuximab (Cet) or MEHD7945A (MEHD) for 72 hrs. Thereafter, growth of tumor cells was determined by cell proliferation analysis. Response of parental H226 to both drugs is shown in the box as a reference. (B) depicts a stronger anti-proliferative efficacy of MEHD7945A than the combination of cetuximab and anti-HER3 (H3) antibody. Results are expressed as percentage of cell growth relative to non-treated controls (NT) *p<0.05. (C) immunoblotting analysis showed that MEHD7945A is more efficient than the combination of cetuximab and anti-HER3 antibody to inhibit EGFR and HER3 signaling. The α-Tubulin (α-Tu) serves as a loading control.
We next compared the anti-proliferative effect of MEHD7945A against the combination of two monospecific antibodies, EGFR (cetuximab) and HER3 (DL3.6b) which is the corresponding HER3 antibody in MEHD7945A (12). Using the same concentration (10 μg/ml) of each antibody, we found MEHD7945A more potent to inhibit cellular proliferation than the combination of cetuximab and anti-HER3 antibody in both cetuximab-resistant clones (Fig 1B). Consistently, we found MEHD7945A more effective than the combination of cetuximab and HER3 antibody to inhibit EGFR-MAPK and HER3-PI3K-AKT signaling as shown in Fig 1C. These results suggest favorable clinical potential to investigate this dual target antibody approach rather than multidrug combination therapy with monospecific EGFR and HER3 antibodies.
To further explore if MEHD7945A could inhibit tumor cell growth in another EGFR inhibitor-resistant setting, we examined the effect of MEHD7945A on our erlotinib-resistant cells. As expected, erlotinib inhibited the growth of parental cells in a dose dependent manner, but had little effect on erlotinib-resistant H226 cells as shown in the left panel of Fig 2A. This confirmed the resistant phenotype of erlotinib-resistant cells. Interestingly, MEHD7945A inhibited the growth of both parental and erlotinib-resistant cells as shown in the right panel of Fig 2A. Similar results were observed in the parental and erlotinib-resistant SCC6 cells (Fig 2B). While both parental and erlotinib-resistant SCC6 cells responded differently to erlotinib, they both responded well to MEHD7945A with a similar pattern. As anticipated, parental cells responded better to MEHD7945A than erlotinib resistant cells likely reflecting the high expression of EGFR and HER3 in the resistant cells as shown in our previous study (8). Taken together, these results consistently demonstrate that MEHD7945A overcomes acquired resistant to both classes of EGFR inhibitors.
Fig. 2. MEHD7945A inhibits growth of erlotinib-resistant cells.
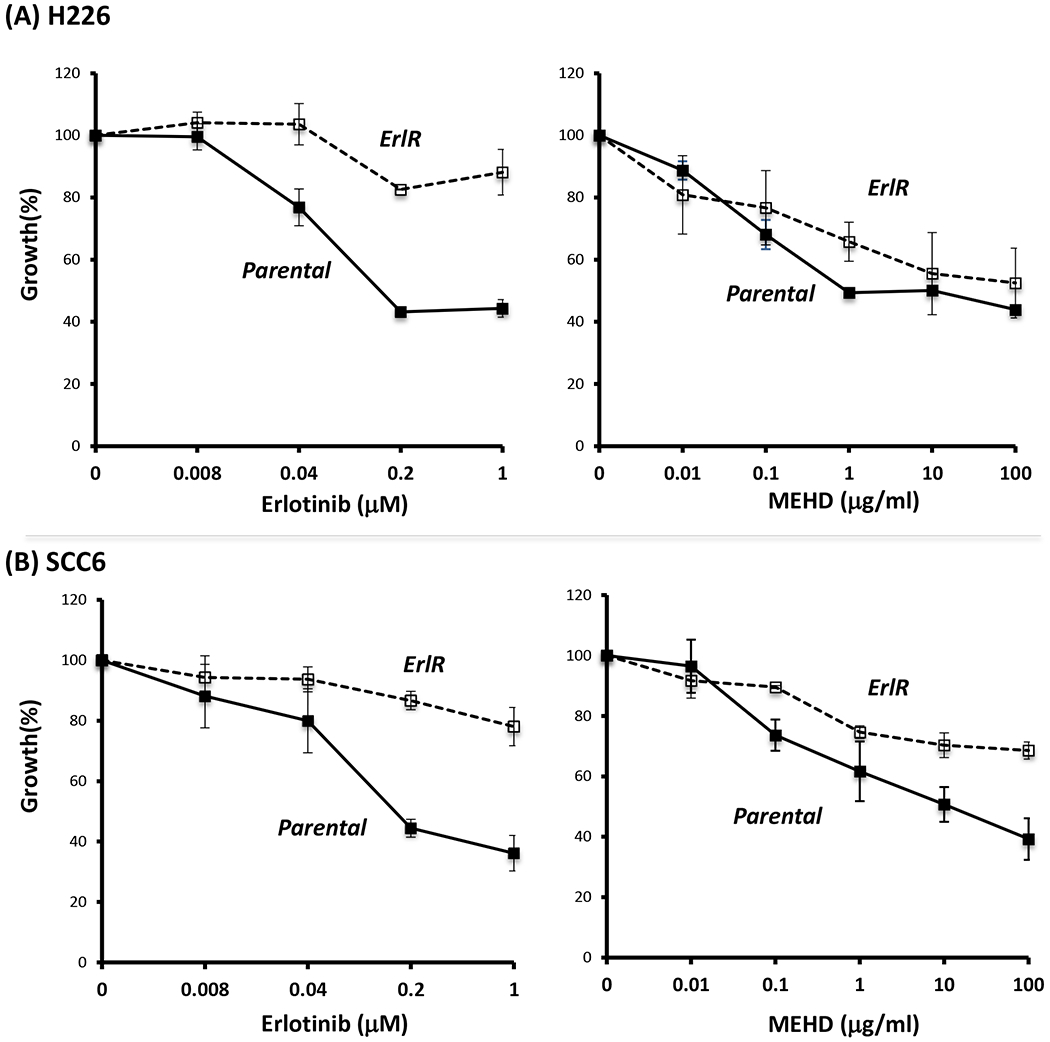
Parental or erlotinib-resistant H226 (A) or SCC6 (B) cells were exposed to serial concentrations of erlotinib (Erl) or MEHD7945A (MEHD) for 72 hrs. Thereafter, growth of tumor cells was determined by cell proliferation analysis.
MEHD7945A overcomes EGFR inhibitor resistance in human tumor xenografts.
To extend these in vitro findings, we inoculated cetuximab-resistant or erlotinib-resistant SCC6 tumor cells into athymic mice. Following establishment of tumors (150~200 mm3), mice were treated with the same dose of cetuximab or MEHD7945A. Treatment with MEHD7945A, but not cetuximab, induced significant growth delay of cetuximab-resistant tumors when compared to untreated controls as shown in the left panel of Fig 3. More importantly, MEHD7945A was found to induce regression of tumors that were highly refractory to cetuximab as shown at days 51~58. In contrast, cetuximab did not inhibit the growth of tumors that were previously treated with MEHD7945A. Similarly, erlotinib-resistant tumors responded with growth delay to initial challenge with MEHD7945A, but not erlotinib treatment. MEHD7945A also induce very brisk regression in those tumors that remained highly refractory to erlotinib treatment. These results confirm and extend the previous in vitro findings and indicate a profound capacity of MEHD7945A to overcome acquired resistance to both cetuximab and erlotinib in vivo.
Fig. 3. MEHD7945A overcomes resistance to cetuximab or erlotinib in human tumor xenografts.
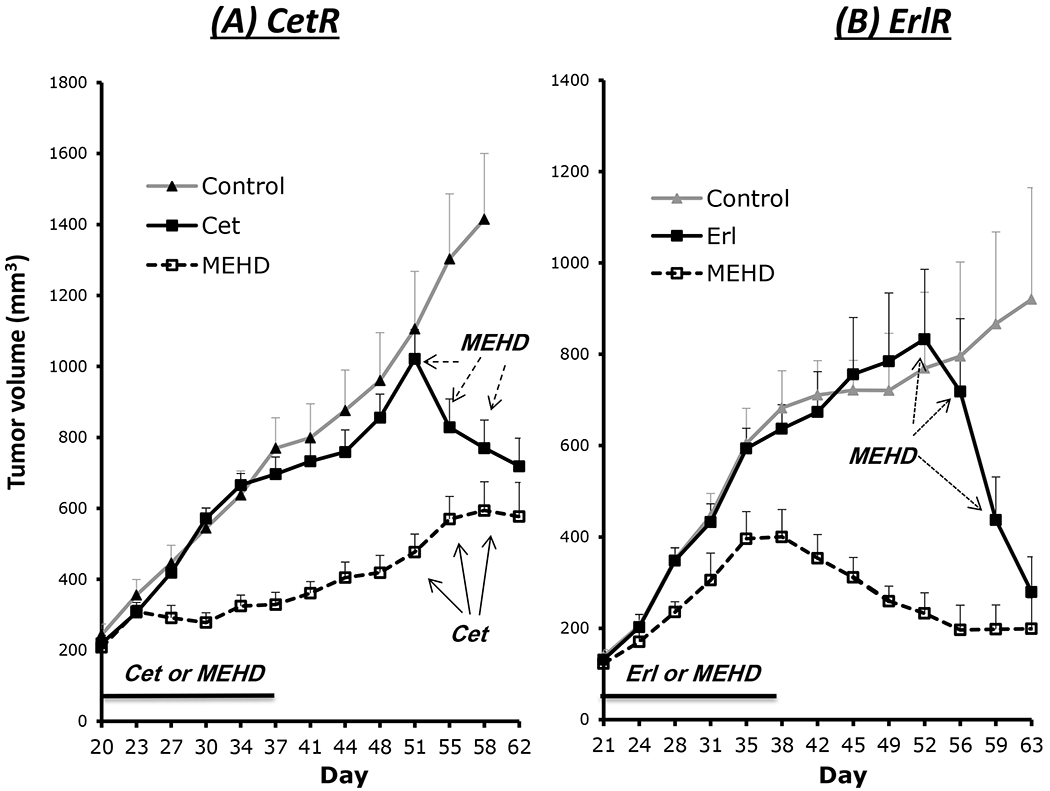
Cetuximab- or erlotinib-resistant SCC6 cells were inoculated into the dorsal frank of athymic mice. (A) Mice with CetR xenografts were initially treated with cetuximab or MEHD7945A at 2.2 mg/kg/dose twice weekly from day 20 - 37. Thereafter 4.4 mg/kg/dose of MEHD7945A was applied to mice that previously received cetuximab treatment, and cetuximab (4.4 mg/kg/dose) was applied to MEHD7945A-treated mice at day 51 for 3 consecutive doses. (B) Mice with ErlR xenografts were initially treated with 3 mg/kg/dose of MEHD7945A or 60 mg/kg/wk of erlotinib from day 21-38. At day 52, erlotinib-treated mice were challenged with 6 mg/kg/dose of MEHD7945A for 3 consecutive doses. Tumor volume was monitored and values represent mean tumor size (mm3) ± SEM (n=8 per group).
MEHD7945A overcomes cross-resistance to radiation
Our previous studies found a cross-resistance to radiation in EGFR inhibitor-resistant cells following long-term exposure to EGFR inhibitors (7). We therefore examined if MEHD7945A could overcome this cross-resistance to radiation in EGFR inhibitor-resistant cells. We compared the radiation response in cetuximab-resistant clones challenged with MEHD7945A or cetuximab. Using clonogenic survival analysis, we found that pretreatment of ceruximab did not change the response profile when compared to radiation alone control (NT). In contrast, treatment with MEHD7945A significantly reduced cell survival following radiation exposure in cetuximab-resistant H226 and SCC6 clones (Fig 4A). In addition, we characterized the DNA damage profile following 3 Gy radiation by examining the activation of the histone variant H2AX that becomes phosphorylated (γH2AX) following a reaction on radiation-induced DNA double-strand breaks (DSB). As shown in Fig 4B, a significant increase of γH2AX foci was observed in MEHD7945A-pretreated, but not cetuximab-pretreated cells by a factor of 1.2~2.2 when compared to untreated control. Interestingly, we found that MEHD7945A augmented radiation-induced DNA damage more significantly in SCC6-CetR than that of H226-CetR cells. This result is consistent with a more profound cell killing in MEHD7945A-treated SCC6-CetR cells when compared to H226-CetR cells determined by clonogenic survival analysis (Fig 4A). To further extend these in vitro findings, we compared the capacity of cetuximab and MEHD7945A to augment radiation response in our cetuximab-resistant tumor xenograft model system. As shown in Fig 4C, SCC6-CetR tumors respond modestly to treatment with cetuximab, MEHD7945A or radiation when compared to control tumors. As expected, the combination of cetuximab and radiation (XRT+C) did not produce a significant treatment benefit in the cetuximab-resistant tumors when compared to the corresponding single treatment with cetuximab (p>0.05) or radiation (p>0.05) up to day 66. However, tumor response to MEHD7945A was considerably stronger than that observed with cetuximab, and the combination of MEHD7945A and radiation (XRT+M) showed a significant tumor growth inhibition resulting in substantial growth delay when compared to the corresponding single modality treatment with radiation (p<0.01) or MEHD7945A (p<0.05) from day 44 or 51 respectively to the end of the experiment on day 66. Additional statistical analysis using the fractional product method (14) confirmed that the combined MEHD7945A and radiation treatment exhibited a strong synergy assessment ratio between 1.2 to 1.47 from day 37 to the end of the experiment on day 66.. Although a modest enhancement of radiosensitivity was observed in MEHD7945A-treated cells from the in vitro clonogenic survival analysis (Fig 4A), we observed a profound impact of MEHD7945A to augment radiation response in the cetuximab-resistant tumor xenografts. This result extends the in vitro findings and confirms that MEHD7945A can augment radiation response in cells that remain refractory to cetuximab.
Fig. 4. MEHD7945A is more potent than cetuximab to augment radiation response in EGFR inhibitor resistant cells.
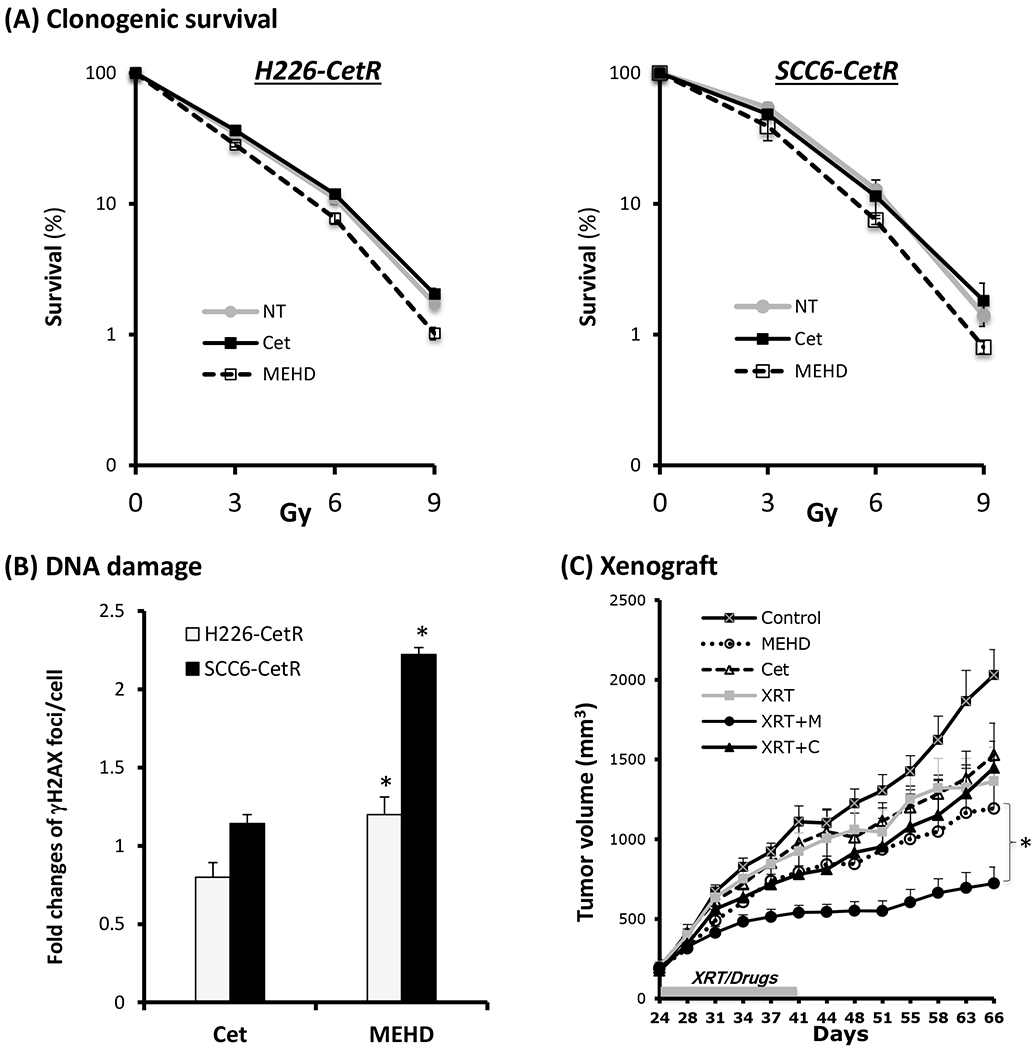
(A) Radiosensitivity of drug free control (NT), 10 μg/ml of cetuximab (Cet) or MEHD7945A (MEHD) pre-treated (72 hrs) CetR cells were examined by clonogenic survival analysis as described in “Materials and Methods”. Results were expressed as the percentage of colony formation relative to controls without radiation treatment. Data points are represented as mean ± SD (B) Radiation-induced DNA damage was determined by examining γH2AX foci in the nucleus by immunofluorescent staining as described in “Materials and Methods”. Results were expressed as the fold-change of foci relative to controls without drug treatment. Data points are represented as mean ± SD *p<0.05. (C) Radiation response was determined in a human xenograft model. Mice with SCC6-CetR tumor xenografts were treated with either single or combined drug and radiation (XRT) treatment starting at day 21. Cetuximab or MEHD7945A were delivered at a dose of 1 mg/kg and radiation was delivered at 2 Gy twice per week for 3 consecutive weeks. The grey box along the x-axis indicates the treatment interval from day 24 to 41. Tumor volume was monitored twice weekly and values represent mean tumor size (mm3) ± SEM (n=8 per group). *p<0.05 when compared with single modality treatment groups.
MEHD7945A inhibits radiation-induced survival and damage repair pathways
To further investigate underlying mechanisms for the effect of MEHD7945A on radiation response, immunoblotting analysis was conducted to examine the expression and activity of proteins involved in regulating survival and DNA damage repair. As shown in Fig 5A, we observed the activation of HER family members and their downstream MAPK and AKT signaling 24 hrs after exposure to 6 Gy radiation in cetuximab-resistant H226 cells. This radiation-induced survival signaling correlated well with an increase of p-Rb that serves as a key factor to stimulate G1-S phase transition. However, treatment with MEHD7945A was superior to cetuximab to inhibit radiation-induced survival signaling and the level of p-Rb. We also observed a significant increase of phosphorylated p53 that is critical in regulating cell cycle arrest and apoptosis in MEHD7945A-treated cells. Consistently, a significant increase of cleaved PARP and Caspase 3 that resulted from the activation of apoptosis was found in MEHD7945A-treated cells 48 hrs after radiation.
Fig. 5. MEHD7945A inhibits radiation-induced survival and damage repair signaling.
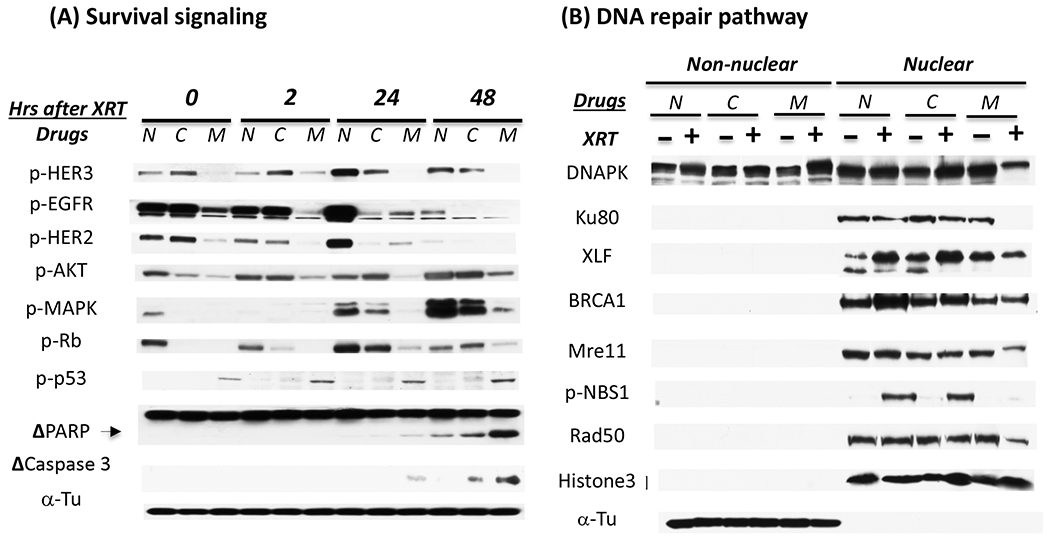
(A) H226-CetR cells were either non-treated (N) or pre-treated with 10 μg/ml of cetuximab (C) or MEHD7945A (M) for 24 hrs followed by 6 Gy radiation. Thereafter, cells were harvested at 0, 2, 24 or 48 hrs after radiation and lysed for western blot analysis. Δ represents cleaved fragment of PARP or Caspase 3. (B). depicts the effect of cetuximab and MEHD7945A on the expression of nuclear DNA damage proteins 48 hrs following radiation. Histone 3 and α-Tu serve as loading and purity controls of nuclear and non-nuclear fractions respectively. Figure is representative of 2~3 independent experiments with similar result.
Since DNA damage response is initiated with the recognition of damage and often results in cell cycle arrest for repair, we next examined if MEHD7945A could inhibit repair capacity by examining several key proteins involved in the repair of lethal DSB. DNAPK, Ku80 and XRCC-4 like factor (XLF) are involved in non-homologous end joining and BRCA1 is essential for initiating homologous recombination repair. Mre11, Rad50 and Nbs1 form a complex in DSB sites and act as crucial elements for DSB repair and cell cycle checkpoints (16). Inhibition of these molecules is known to sensitize tumor cells to radiation. Following nuclear fractionation, we found that the level of most repair proteins in the nucleus is increased following radiation exposure in the control non-treated cells (Fig 5B), such as XCF, BRCA1 and pNBS1. Treatment with MEHD7945A was more effective than cetuximab to reduce the level of all the tested repair proteins in cells exposed to radiation. Interestingly, we found that a significant decrease of nuclear DNAPK in the MEHD7945A-treated cells was accompanied by an increase of DNAPK in the non-nuclear fraction. This result is consistent with previous findings suggesting that EGFR agents inhibit repair capacity by disrupting nuclear import of functional DNAPK (17). The lack of nuclear DNAPK import inhibition in the cetuximab-treated cells is consistent with the cross-resistance to radiation observed in our cetuximab-resistant cells. Similar results were observed in cetuximab-resistant SCC-6 cells (data not shown). Taken together, these results suggest that MEHD7945A augments radiation response via the induction of cell cycle arrest followed by the induction of apoptosis and cell death likely reflecting inhibitory effects on DNA damage repair machinery.
MEHD7945A regulates cell cycle progression and apoptosis following radiation
To further investigate the effect of MEHD7945A on radiation-induced cell cycle progression, we examined the cell cycle phase distribution of cetuximab-resistant cells 1 or 2 days following exposure to 6 Gy radiation treatment. As shown in Fig 6A, the S phase cell cycle populations were enhanced 1 day after radiation in the untreated control of both EGFR inhibitor-resistant clones. This result correlates well with previous observations of an increase of proliferative and survival signaling 24 hrs after radiation (Fig 5A). However, treatment with MEHD7945A, but not cetuximab, induced a robust increase of cell arrest in G0/G1 phase. Interestingly, we found that MEHD7945A-treated cells resumed a similar cell cycle phase distribution to control cells 2 days after radiation. Further analysis of cells with sub-G0 DNA content comprising apoptotic cells and debris fractions, identifies a robust increase of cells in MEHD7945A-treated, but not cetuximab-treated cells 2 days after radiation (Supplemental Fig 2).
Fig. 6. MEHD7945A induces cells cycle arrest and apoptosis following radiation.
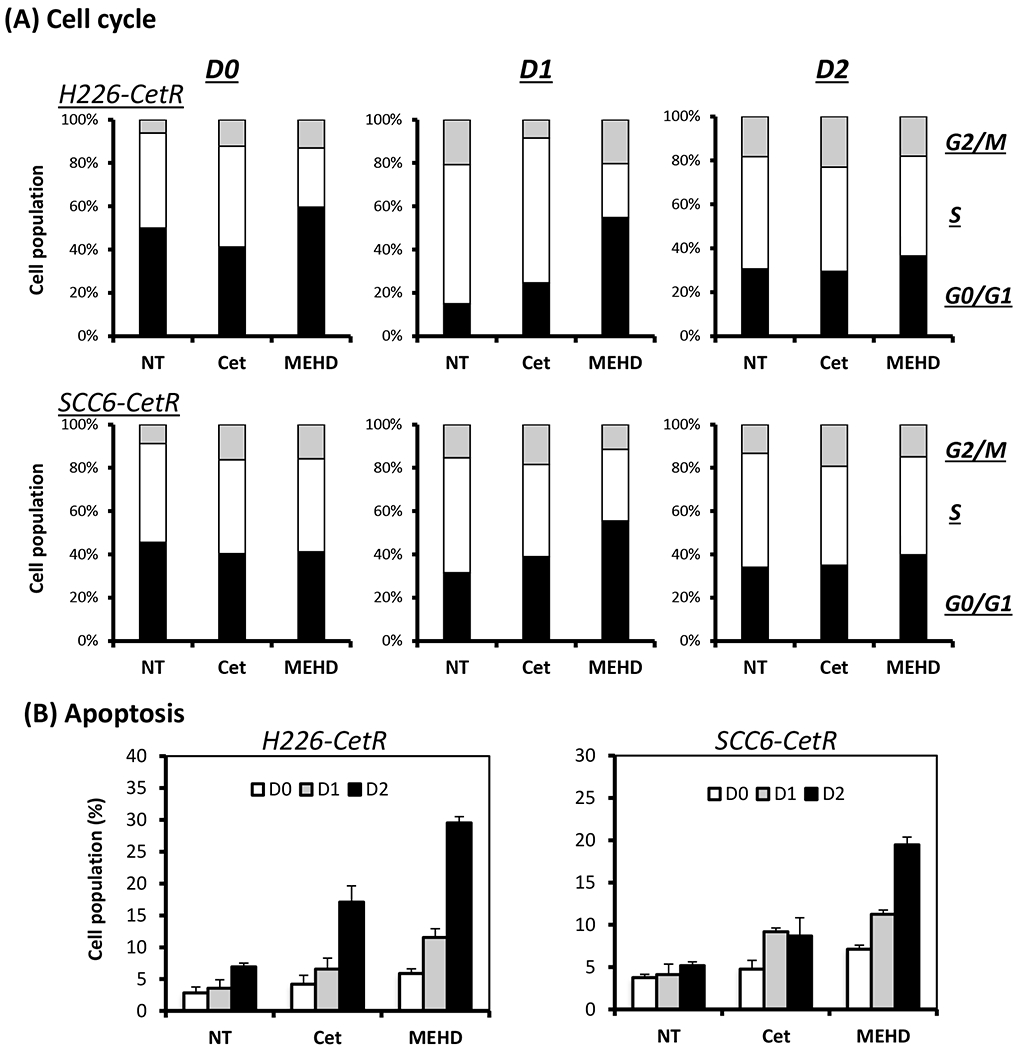
(A) CetR H226 or SCC6 cells were either non-treated (NT) or pre-treated with 10 μg/ml of cetuximab (Cet) or MEHD7945A (MEHD) for 24 hrs followed by 6 Gy radiation. Cells were then harvested at day 0 (D0), day 1 (D1) or day 2 (D2) following radiation and processed for cell cycle or Annexin V/PI apoptosis analysis by flow cytometry as described in “Materials and Methods”. (A) depicts cell populations in G0/G1, S and G2/M phase following radiation. (B) depicts the percentage of early apoptotic cells (Annexin V-positive; PI-negative). Columns, mean values of duplicate samples.
To further validate this observation of apoptosis, we applied another approach using Annexin V/PI flow cytometry analysis. Annexin V serves as a marker for the loss of plasma membrane asymmetry representing an early feature of apoptosis.As shown in Fig 6B, there is a significant increase of early apoptotic cells in the MEHD7945A-treated H226-CetR cells two days (D2) after radiation when compared to control and cetuximab-treated cells. There is only a modest increase of apoptotic cells in MEHD7945A-treated group one day (D1) after radiation. Similar results were observed in the cetuximab-resistant SCC-6 cells. These results again indicate that MEHD76945A is inducing cell cycle arrest early following exposure to radiation. With the capacity to inhibit DNA repair pathways, MEHD7945A appears to augment radiation effect via apoptosis induction in these unrepaired cells at a latter stage.
Discussion
In the current study, we provide evidence that MEHD7945A can overcome acquired resistance in two distinct EGFR inhibitor-resistant model systems. Using established cetuximab- or erlotinib-resistant cells from NSCLC and HNSCC, we find that MEHD7945A, but not EGFR inhibitors alone, effectively inhibits MAPK and PI3K-AKT survival pathways that play a key role in regulating acquired resistance to EGFR inhibitors (Fig 1). Further, MEHD7945A exhibits more potent antitumor capacity than cetuximab or erlotinib in human tumor xenograft systems, and effectively shrinks tumors that remain highly refractory to cetuximab or erlotinib (Fig 3). In addition, MEHD7945A overcomes cross-resistance to radiation in these EGFR inhibitor-resistant cells. This latter finding is noteworthy since most EGFR inhibitor combinations with cytotoxic chemotherapy have shown limited clinical benefit (5, 18). Although the combination of cetuximab and radiotherapy has shown improved 5-year survival in head and neck cancer patients, a substantial proportion of patients eventually manifest tumor recurrence (19). Since both EGFR and HER3 are activated after radiation (Fig 5A), MEHD7945A targeting of both receptors offers a promising approach to overcome acquired resistance in clinical therapy strategies that employ EGFR/radiation combinations.
Although specific mechanisms resulting in acquired clinical resistance to EGFR inhibitors and radiation are not fully understood, increasing evidence indicates that crosstalk among HER family members represents a major factor affecting clinical efficacy of HER-targeted therapy (20–22). Blockade of one HER receptor can be functionally compensated by another HER family member. Early studies in breast cancer cells demonstrated that trastuzumab inhibited signaling from HER2, but did not disrupt activation of dimerization between HER2 and other HER family members (11). Considerable evidence points to HER3 and/or EGFR as key contributors to acquired resistance against HER2 targeting agents (23). Similarly, we and other investigators have demonstrated that HER3 is involved in regulating acquired resistance to EGFR inhibitors (8, 9, 24). In addition, Engleman et al. identified that c-Met, another receptor tyrosine kinase, induced acquired resistance to gefitinib via coupling to HER3 and activation of HER3-PI3K-AKT signaling. (10). Interestingly, we found that the impact of MEHD7945A was more potent than dual agent blockade of EGFR by the combination of cetuximab and erlotinib (Supplemental Fig 3) which shuts down EGFR and has shown to be superior to either agent alone in our previous study (6). These findings reveal an important role of HER3 as a signaling hub for the HER family that results in compensatory pathways for EGFR inhibitors. These findings also highlight the potential value to inhibit functions of multiple HER family members to achieve the broadest clinical efficacy to overcome acquired resistance to EGFR inhibitor therapy. The capacity of MEHD7945A to overcome the primary (intrinsic) resistance to EGFR inhibitor therapy still needs to be examined since the role of HER3 in regulating intrinsic resistance to EGFR inhibitors is not yet well characterized. A separate study is near completion to investigate the effect of MEHD7945A on the growth and radiation response of primary lung and H&N tumor cell lines with variable sensitivities to cetuximab (25).
This two-in-one MEHD7945A targets both EGFR and HER3, thereby offering a combinatorial targeted therapy (12). Interestingly, we found that MEHD7945A was more effective than the combination of individual anti-EGFR and anti-HER3 antibody to inhibit tumor cell growth and MAPK and PI3K/AKT signaling pathways in cetuximab resistant cells (Fig 1B&C). Beyond a difference in binding affinity between MEHD7945A and the monospecific antibodies, the ability of MEHD7945A to simultaneously target EGFR/HER3 in close proximity or cluster could be another crucial factor to explain the superior effect of MEHD7945A over the combination of cetuximab and anti-HER3 antibody. Increasing evidence has shown the existence of EGFR dimers or clusters in cells even in the absence of ligand stimulation (26, 27). This alternate dimer is an important intermediate form in the transition of the inactive receptor to the active, untethered dimer (28). Since hetero-dimerization of EGFR was known to produce the most profound downstream signaling, MEHD7945A could then be more powerful than combination of individual targeting agents to shut down the proliferation signaling via its ability to bind to EGFR/HER3 clusters. Furthermore, it was recently established that combination of noncompetitive anti-EGFR Abs synergistically reduce surface receptor level and lead to enhanced tumor cell killing and prolonged survival in a variety of mouse models (29, 30). Friedman et al proposed that synergism results from the formation of large clusters of receptors on the cell surface following combination Ab treatment (29). With the potential of MEHD7945A to increase the formation of EGFR/HER3 clusters, it will be of interest to further investigate if MEHD7945A can enhance EGFR and HER3 internalization and degradation in tumor cells.
HER2 also appears to be involved in regulating acquired resistance to EGFR inhibitors. Following long-term exposure to cetuximab in vitro, Yonesaka found amplification of HER2 gene and/or increased neuregulin concentration in cetuximab-resistant clones of lung and colorectal cancers (31). Further analysis suggested that aberrant HER2 signaling, either through HER2 gene amplification or through autocrine neuregulin activation of HER3, led to persistent MAPK signaling and consequently to cetuximab resistance. Quesnelle also identified activated HER2, but not HER2 gene amplification as the underlying mechanism for acquired resistance to cetuximab in bladder tumor cells established from an in vivo model system (32). Interestingly, we found that MEHD7945A could also inhibit phosphorylation of HER2 in our resistant cells (Fig. 5A). It would be of interest to explore the capacity of MEHD7945A to overcome EGFR inhibitor resistance in tumors with activated HER2. In addition, it may also be valuable to explore if MEHD7945A is applicable to HER2-overexpressing breast cancers refractory to trastuzumab since EGFR and HER3 have been implicated as key factors to regulate trastuzumab resistance.
Acquired resistance presents a considerable challenge to the optimal clinical advancement of EGFR molecular targeting agents. In addition, resistance to EGFR agents may co-associate with resistance to other cancer drugs and radiation. Data from the current study suggests that MEHD7945A is able to overcome cross-resistance to radiation via inhibition of radiation-induced survival signaling and DNA damage repair that results in the induction of apoptosis (Fig 4~6). Treatment with MEHD7945A, but not cetuximab inhibited radiation-induced survival signaling and resulted in cell cycle arrest within 24 hrs after radiation. Following inhibition of DNA repair machinery following exposure to MEHD7945A, most unrepaired cells entered apoptosis by 48 hrs after radiation. Further, we identified a significant increase of p-p53 in cells treated with MEHD7945A (Fig 4B). These data are notable since recent work reveals p53 as a critical factor in regulating acquired resistance to EGFR inhibitors and radiation (15). Knocking down wild type p53 in sensitive H226 cells, we observe a reduction in the sensitivity to cetuximab and radiation. In contrast, with reconstitution of functional p53, cetuximab-resistant cells demonstrate sensitivity to both treatments (15). Hence, it is possible that MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation via p53-regulated pathways. Beyond radiotherapy, p53 and AKT pathways have been shown to regulate acquired resistance to several chemotherapeutic agents (33, 34). It will be of interest to examine the potential of MEHD7945A to overcome resistance to the combined administration of EGFR inhibitors and chemotherapy. Additional experiments are underway to explore this question.
In conclusion, MEHD7945A, a dual targeting antibody against both EGFR and HER3, demonstrates the capacity to overcome acquired resistance to EGFR inhibitors and radiation. Results from the current work suggest that MEHD7945A offers a promising therapeutic approach for combinatorial molecular target therapy. The single agent simplicity of MEHD7945A also provides an opportunity to combine with other agents that have been shown to regulate resistance to EGFR therapy. Our improved understanding of HER family signaling biology suggests that agents like MEHD7945A may prove highly valuable to advance the overall impact of EGFR therapy in cancer and help address the challenge of acquired resistance. Clinical trials are in development with MEHD7945A that will further investigate several of these important questions.
Supplementary Material
Acknowledgments
Special thanks to Gabriele Schaefer, Rob Akita and Andrea Pirzkall from Genentech for their critical review and suggestions for the manuscript. We also wish to thank ImClone, Genentech and OSI Pharmaceuticals for kindly providing cetuximab, MEHD7945A and erlotinib respectively for experimental studies.
Grant Support
This work was supported in part by NIH R01 CA 113448-01 (PMH) and a sponsored laboratory research agreement from Genentech (PMH).
Footnotes
Disclosure of Potential Conflicts of Interest: P.M. Harari holds a laboratory research agreement with Genentech. L.C. Amler and M.X. Sliwkowski are employed by Genentech, Inc, a member of the Roche Group, and are shareholders in Roche.
References
- 1.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol 2006; 33: 369–85. [DOI] [PubMed] [Google Scholar]
- 2.Harari PM, Allen GW, Bonner JA. Biology of interactions: Antiepidermal growth factor receptor agents. J Clin Oncol 2007; 25: 4057–65. [DOI] [PubMed] [Google Scholar]
- 3.Riely GJ, Kris MG, Zhao B, Akhurst T, Milton DT, Moore E, et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res 2007; 13: 5150–55. [DOI] [PubMed] [Google Scholar]
- 4.Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 2010; 28: 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with egfr-mutant lung cancer and acquired resistance to erlotinib or gefitinib: Implications for clinical trial design. Clin Cancer Res 2011; 17: 6298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (egfr): Combining anti-egfr antibody with tyrosine kinase inhibitor. Cancer Res 2004; 64: 5355–62. [DOI] [PubMed] [Google Scholar]
- 7.Benavente S, Huang S, Armstrong EA, Chi A, Hsu K-T, Wheeler DL, et al. Establishment and characterization of a model of acquired resistance to epidermal growth factor receptor targeting agents in human cancer cells. Clin Cancer Res 2009; 15: 1585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: Role of her (erbb) family members. Oncogene 2008; 27: 3944–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from her-family tyrosine kinase inhibitor therapy by the kinase-inactive her3. Nature 2007; 445: 437–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. Met amplification leads to gefitinib resistance in lung cancer by activating erbb3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 11.Jain A, Penuel E, Mink S, Schmidt J, Hodge A, Favero K, et al. Her kinase axis receptor dimer partner switching occurs in response to egfr tyrosine kinase inhibition despite failure to block cellular proliferation. Cancer Res 2010; 70: 1989–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, et al. A two-in-one antibody against her3 and egfr has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011; 20: 472–86. [DOI] [PubMed] [Google Scholar]
- 13.Hsu S-C, Hung M-C. Characterization of a novel tripartite nuclear localization sequence in the egfr family. J Biol Chem 2007; 282: 10432–40. [DOI] [PubMed] [Google Scholar]
- 14.Kimple RJ, Vaseva AV, Cox AD, Baerman KM, Calvo BF, Tepper JE, et al. Radiosensitization of epidermal growth factor receptor/her2‚äìpositive pancreatic cancer is mediated by inhibition of akt independent of ras mutational status. Clin Cancer Res 2010; 16: 912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S, Benavente S, Armstrong EA, Li C, Wheeler DL, Harari PM. P53 modulates acquired resistance to egfr inhibitors and radiation. Cancer Res 2011; 71: 7071–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolderson E, Richard DJ, Zhou B-BS, Khanna KK. Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin Cancer Res 2009; 15: 6314–20. [DOI] [PubMed] [Google Scholar]
- 17.Dittmann K, Mayer C, Rodemann H. Inhibition of radiation-induced egfr nuclear import by c225 (cetuximab) suppresses DNA-pk activity. Radiother Oncol 2005; 76: 157–61. [DOI] [PubMed] [Google Scholar]
- 18.Sequist LV, Lynch TJ. Egfr tyrosine kinase inhibitors in lung cancer: An evolving story. Annu Rev Med 2008; 59: 429–42. [DOI] [PubMed] [Google Scholar]
- 19.Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. The Lancet Oncol 2010; 11: 21–28. [DOI] [PubMed] [Google Scholar]
- 20.Shepard HM, Brdlik CM, Schreiber H. Signal integration: A framework for understanding the efficacy of therapeutics targeting the human egfr family. J Clin Invest 2008; 118: 3574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Z, Brdlik C, Jin P, Shepard HM. A pan-her approach for cancer therapy: Background, current status and future development. Expert Opin Biol Ther 2009; 9: 97–110. [DOI] [PubMed] [Google Scholar]
- 22.Vlacich G, Coffey RJ. Resistance to egfr-targeted therapy: A family affair. Cancer Cell 2011; 20: 423–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ. Trastuzumab-induced her reprogramming in “resistant” breast carcinoma cells. Cancer Res 2009; 69: 2191–94. [DOI] [PubMed] [Google Scholar]
- 24.Campbell MR, Amin D, Moasser MM. Her3 comes of age: New insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res 2010; 16: 1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Huang S, Armstrong EA, Sliwkowski MX, Amler LC, Harari P. Mehd7945a, dual specific antibody against egfr and her3, augments radiation response in head and neck squamous cell carcinoma. In: Proceedings of the 103rd Annual meeting of the American Association for Cancer Research: Abstract nr 2721 2012. [Google Scholar]
- 26.Saffarian S, Li Y, Elson EL, Pike LJ. Oligomerization of the egf receptor investigated by live cell fluorescence intensity distribution analysis. Biophysical J 2007; 93: 1021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I. Spatial control of egf receptor activation by reversible dimerization on living cells. Nature 2010; 464: 783–87. [DOI] [PubMed] [Google Scholar]
- 28.Gan HK, Walker F, Burgess AW, Rigopoulos A, Scott AM, Johns TG. The epidermal growth factor receptor (egfr) tyrosine kinase inhibitor ag1478 increases the formation of inactive untethered egfr dimers: Implication for combination therapy with monoclonal antibody 806. J Biol Chem 2007; 282: 2840–50. [DOI] [PubMed] [Google Scholar]
- 29.Friedman LM, Rinon A, Schechter B, Lyass L, Lavi S, Bacus SS, et al. Synergistic down-regulation of receptor tyrosine kinases by combinations of mabs: Implications for cancer immunotherapy. PNAS 2005; 102: 1915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, et al. Sym004: A novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res 2010; 70: 588–97. [DOI] [PubMed] [Google Scholar]
- 31.Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of erbb2 signaling causes resistance to the egfr-directed therapeutic antibody cetuximab. Sci Transl Med 2011; 3: 99ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quesnelle KM, Grandis JR. Dual kinase inhibition of egfr and her2 overcomes resistance to cetuximab in a novel in vivo model of acquired cetuximab resistance. Clin Cancer Res 2011; 17: 5935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: Drugging the p53 pathway. Nat Rev Cancer 2009; 9: 862–73. [DOI] [PubMed] [Google Scholar]
- 34.Dibble CC, Manning BD. A molecular link between akt regulation and chemotherapeutic response. Cancer Cell 2009; 16: 178–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.