

Summary
Brain metastases are a common consequence of advanced lung cancer resulting in cranial neuropathies and increased mortality. Currently, there are no effective therapies to treat brain metastases due to a lack of actionable targets and failure of systemic therapies to penetrate the blood-brain barrier (BBB). Here we identify an autocrine signaling axis required for lung adenocarcinoma brain metastasis, whereby nuclear accumulation of the TAZ transcriptional co-activator drives expression of a panel of transcripts enriched in brain metastases, including ABL2 and AXL encoding for protein tyrosine kinases that engage in bidirectional signaling. Activation of ABL2 in turn promotes TAZ tyrosine phosphorylation and nuclear localization, establishing an autocrine AXL-ABL2-TAZ feed-forward signaling loop required for brain metastasis colonization. Notably, treatment with a BBB-penetrant ABL allosteric inhibitor or knockdown of ABL2, AXL or TAZ markedly decreases brain metastases. These findings suggest that ABL and AXL inhibitors might be effective against brain metastases.
Keywords: ABL2, TAZ, AXL, L1CAM, brain metastasis, lung adenocarcinoma, allosteric inhibitors
Graphical Abstract
eTOC Blurb
Brain metastases are a devastating complication of lung cancer with limited therapeutic options. Here Hoj et al. identify a TAZ-AXL-ABL2 autocrine signaling axis required for lung adenocarcinoma brain metastasis that is amenable to pharmacologic inhibition by BBB-penetrant ABL and AXL kinase inhibitors.
Introduction
Lung cancer is the leading cause of cancer mortality worldwide with lung cancer patients exhibiting the highest prevalence (~50%) of brain metastasis across all cancer types (Barnholtz-Sloan et al., 2004; Schouten et al., 2002). In contrast to other primary tumors, between 10-20% of lung cancer patients present with brain metastases at the time of diagnosis (Nayak et al., 2012; Schuette, 2004; Shin et al., 2014). Patients with lung adenocarcinoma, a subtype of non-small cell lung cancer (NSCLC), represent the highest group of patients with brain metastatic disease. Lung adenocarcinomas driven by mutations in the epidermal growth factor receptor (EGFR) are at particularly high risk for developing brain metastases (Shin et al., 2014). Despite early clinical successes with next-generation BBB-penetrant EGFR tyrosine kinase inhibitors (TKIs) such as osimertinib, relapses for lung adenocarcinoma patients with intracranial disease remain the rule rather than the exception (Kelly et al., 2018; Oxnard et al., 2018). Thus, the lack of durable treatment options for lung cancer patients suffering from brain metastases necessitates the study and discovery of novel therapeutic strategies.
Preclinical studies in mouse models have identified genes that mediate metastasis to the brain (Chen et al., 2016; Er et al., 2018; Sevenich et al., 2014; Valiente et al., 2014). Unfortunately, therapies targeting these and other metastasis regulators have not translated into effective therapies against brain metastasis. The ABL family of tyrosine kinases, ABL1 and ABL2, promote lung cancer metastasis to multiple organ sites in part through stabilization of the transcriptional co-activator TAZ (encoded by WWTR1) (Gu et al., 2016). Upon nuclear translocation, TAZ binds to the TEAD family of transcription factors to coordinate expression of target genes implicated in organ size (Yu et al., 2015), stemness (Kim et al., 2015), cell migration (Feng et al., 2016), and EMT (Moroishi et al., 2015). Here we report that expression of a constitutively-active, stable form of the TAZ in lung adenocarcinoma cells directs metastases predominantly to the brain following intracardiac injection. Further, we identify ABL2 as a previously unrecognized TAZ target gene and show that TAZ functions both downstream and upstream of ABL2 in metastatic lung cancer cells. Moreover, we find that the ABL2 non-receptor tyrosine kinase engages in bidirectional signaling with the AXL receptor tyrosine kinase (RTK), also a transcriptional target of TAZ. Activation of AXL can occur through both ligand-dependent and ligand-independent mechanisms that contribute to pro-invasive, metastatic, and therapy-resistance phenotypes across multiple tumor types (Goyette et al., 2018; Meyer et al., 2013; Rankin et al., 2014). Recently, expression of AXL and its ligand GAS6 were both shown to have correlative prognostic value for patients with lung adenocarcinoma brain metastases, however the molecular mechanisms by which ligand-activated AXL signaling contributes to the progression of this disease remain to be discovered (Wu et al., 2017). Our current findings reveal a feed-forward TAZ-AXL-ABL2 signaling axis that regulates expression of TAZ-dependent transcripts highly enriched in brain metastatic lung cancer cells. Importantly, we show that an allosteric inhibitor of the ABL kinases crosses the BBB and inhibits pathway signaling to impair brain metastasis outgrowth in mice. Our work thus uncovers actionable targets for the treatment of lung adenocarcinoma brain metastases.
Results
Active TAZ is necessary and sufficient for lung adenocarcinoma brain metastasis
We reported that activated ABL kinases are detected in some NSCLC tumor specimens and that ABL-regulated downstream targets are hyper-active in metastases isolated from lymph nodes compared to primary tumors (Gu et al., 2016). Unbiased transcriptome analysis of metastatic lung cancer cells harboring activated ABL kinases versus non-metastatic ABL knockdown cells revealed that among ABL-regulated pathways were those dependent on the transcriptional co-activator TAZ (Gu et al., 2016). Thus, we evaluated the metastatic phenotypes of lung cancer cells expressing an activated form of TAZ (TAZ4SA) resistant to ubiquitin-dependent degradation that translocates to the nucleus to drive transcription of target genes (Lei et al., 2008; Zhang et al., 2009). Unexpectedly we found that inducible expression of active TAZ4SA in EGFR mutant lung adenocarcinoma PC9 and HCC4006 cells predominantly promoted brain metastases following intracardiac injection into athymic nude mice relative to metastases at other organ sites (Figures 1A–E). Inducible expression of TAZ4SA in PC9 lung cancer cells revealed no measurable differences in cell viability compared to the non-induced PC9 cells (Figure S1A). Parental and TAZ4SA-expressing lung cancer cells labeled with a luciferase-TOMATO reporter were injected into athymic nude mice and monitored by bioluminescent imaging (BLI) (Figures 1A–D, S1B). Mice harboring TAZ4SA-expressing lung cancer cells exhibited markedly decreased brain metastasis-free survival (BMFS) compared to mice with control cells (Figures 1B and S1C). Quantification of a brain-metastatic index revealed a marked enrichment of brain metastases in mice harboring TAZ4SA-expressing PC9 and HCC4006 lung cancer cells compared to mice with control cells (Figures 1C and D). No significant difference was observed in overall whole-body metastatic burden between mice harboring parental and TAZ4SA cells (Figure S1D–E). These data suggest that active TAZ4SA promotes a brain-tropic phenotype without enhancing overall whole body metastasis. Notably, BLI analysis of the isolated brains of tumor-bearing mice (day 32 post-injection) revealed all mice injected with HCC4006-TAZ4SA cells exhibited brain metastasis, whereas mice injected with parental HCC4006 cells exhibited minimal disease burden (Figure 1E). Collectively, these findings show that stabilization and activation of TAZ in lung adenocarcinoma cells promotes brain metastases.
Figure 1. Activation of TAZ is necessary and sufficient to promote brain metastases of lung adenocarcinoma cells.
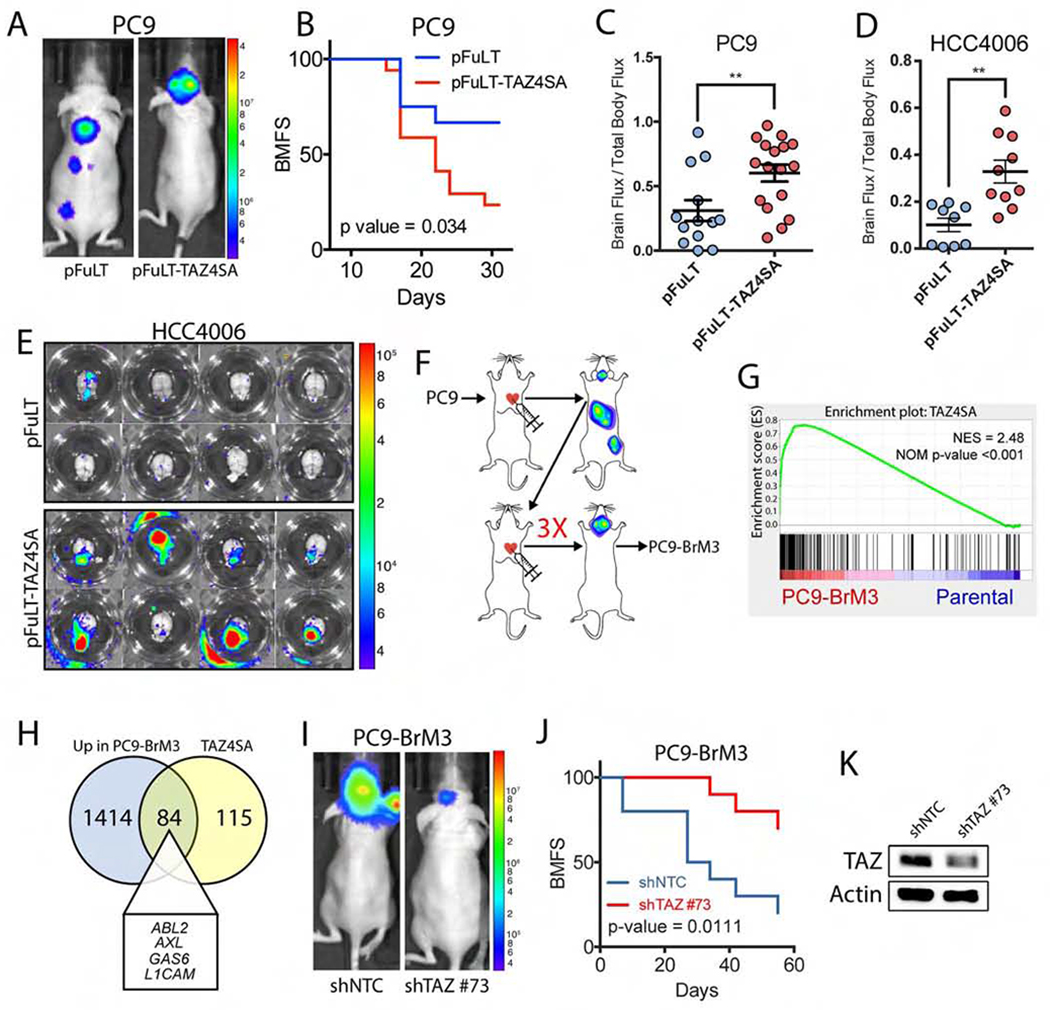
A) Representative images (day 30 post-injection) and B) analysis of brain metastasis-free survival (BMFS) in mice injected intracardially with PC9-pFuLT or pFuLT-Tet-TAZ4SA-expressing cells. Mice were given dox water for the duration of the study. Statistical analysis calculated by Log-rank (Mantel-Cox) test. Parental (n=13), TAZ4SA (n=17). C-D) Quantitative analysis (day 30 post-injection) of brain-metastatic index in mice injected intracardially with C) PC9-pFuLT (n=13) vs. PC9-pFuLT-Tet-TAZ4SA (n=17) or D) HCC4006-pFuLT (n=9) vs HCC4006-pFuLT-Tet-TAZ4SA (n=10) cells. Statistical analysis calculated by unpaired two-tailed t test. ** p-value < 0.01. E) Representative images of ex vivo mouse brains on day 32 post-injection with HCC4006-pFuLT or HCC4006-Tet-TAZ4SA cells. F) Diagram of iterative derivation of brain-metastatic cell lines. G) GSEA plot of TAZ4SA signature in PC9 parental vs PC9-BrM3 RNA-seq dataset. NES=normalized enrichment score. H) Venn diagram of overlapping transcripts upregulated in PC9-BrM3 and PC9-TAZ4SA cells. I) Representative images (day 42 post-injection) and J) BMFS in mice injected intracardially with PC9-BrM3 cells expressing non-target control (shNTC, n=10) or shTAZ (clone #73, n=10). K) Immunoblot to evaluate TAZ shRNA knockdown in PC9-BrM3 cells. Actin was used for protein loading control.
We next generated brain-metastatic PC9 cells (PC9-BrM3) by three rounds of serial intracardiac injection in athymic nude mice (Figure 1F). Approximately 80-90% of mice injected with PC9-BrM3 cells developed brain metastases. The PC9-BrM3 cells were then used to evaluate whether an activated TAZ transcriptional target gene signature might be enriched in brain metastatic cells relative to parental cells. To this end, we performed RNA sequencing (RNA-seq) and differential gene expression analysis comparing PC9-TAZ4SA versus parental PC9 cells to generate a TAZ4SA target gene signature. Genes from this list include previously described TAZ/TEAD target genes such as CYR61, THBS1 and CTGF in addition to previously uncharacterized TAZ gene targets (Table S1). Differential expression analysis was then performed on a publicly-available RNA-seq dataset comparing PC9-BrM3 versus parental PC9 cells (GEO: GSE83132) (Boire et al., 2017) followed by GSEA to identify pathway signatures enriched or depleted in PC9-BrM3 cells. We found that the TAZ4SA signature ranked among the top signatures enriched in PC9-BrM3 cells, which also included signatures for Epithelial-to-Mesenchymal Transition (EMT) and hypoxia, among others (Figures 1G and S1F). We identified 84 transcripts co-upregulated in both PC9-BrM3 cells and TAZ4SA-expressing cells (Figure 1H).
To investigate the mechanisms underlying activation of TAZ target genes in brain metastatic cells we evaluated TAZ expression. WWTR1 mRNA and TAZ protein levels were similarly expressed in parental and brain metastatic cells (Figures S1G and S1H). However, immunofluorescence staining revealed increased TAZ nuclear localization in PC9-BrM3 cells and PC9 cells expressing active TAZ4SA compared to parental cells (Figures S1I and S1J). These data suggest that enhanced nuclear TAZ accumulation, without increased TAZ expression, underlies the activation of TAZ transcriptional targets in brain metastatic cells.
We next evaluated whether TAZ is required for lung cancer brain metastasis by knockdown of TAZ in PC9-BrM3 cells. TAZ knockdown markedly impaired brain metastases and increased BMFS compared to control cells, (Figures 1I–K), revealing a requirement for TAZ in lung adenocarcinoma brain metastasis.
The receptor tyrosine kinase AXL and its ligand GAS6 are target genes of TAZ in lung adenocarcinoma
To identify actionable targets that could be exploited for the treatment of brain metastasis, we compared the top 1500 differentially-overexpressed genes from the PC9-BrM3 RNA-seq dataset to the known tyrosine kinome. We identified 23 tyrosine kinases whose corresponding transcripts were increased in brain metastatic PC9-BrM3 cells relative to parental cells and, among these, the most highly overexpressed tyrosine kinase was AXL (Figures 2A–C, 2E, S2A, and S2B). AXL transcript and protein levels were also markedly increased in brain-metastatic HCC4006-BrM and H1975-BrM3 lung adenocarcinoma cells compared to parental cells (Figures 2D, 2E, S2C, and S2D). AXL overexpression has been implicated as a mechanism of acquired therapy resistance to EGFR inhibitors in NSCLC, and correlates with adverse prognosis and decreased survival among NSCLC patients with brain metastases (Namba et al., 2019; Wu et al., 2017). RNA-seq analysis identified AXL among the most highly upregulated transcripts in both PC9-BrM3 and TAZ4SA-expressing PC9 cells (Figure 1H and Table S1), and RT-qPCR analysis of PC9, HCC4006 and HCC827 lung adenocarcinoma cell lines with inducible expression of TAZ4SA showed markedly increased AXL mRNA and protein (Figures 2F–H). Conversely, shRNA knockdown of TAZ (WWTR1) in PC9-BrM3 cells resulted in impaired expression of AXL mRNA (Figure 2I). Analysis of the AXL proximal promoter revealed TEAD DNA-binding consensus motifs (GGAATG) located 460, 559, 1259, and 1344 base pairs upstream of the AXL transcriptional start site (TSS) (Figure S2E).
Figure 2. The receptor tyrosine kinase AXL is overexpressed in brain-metastatic lung cancer cells and is a target of TAZ-dependent transcription.
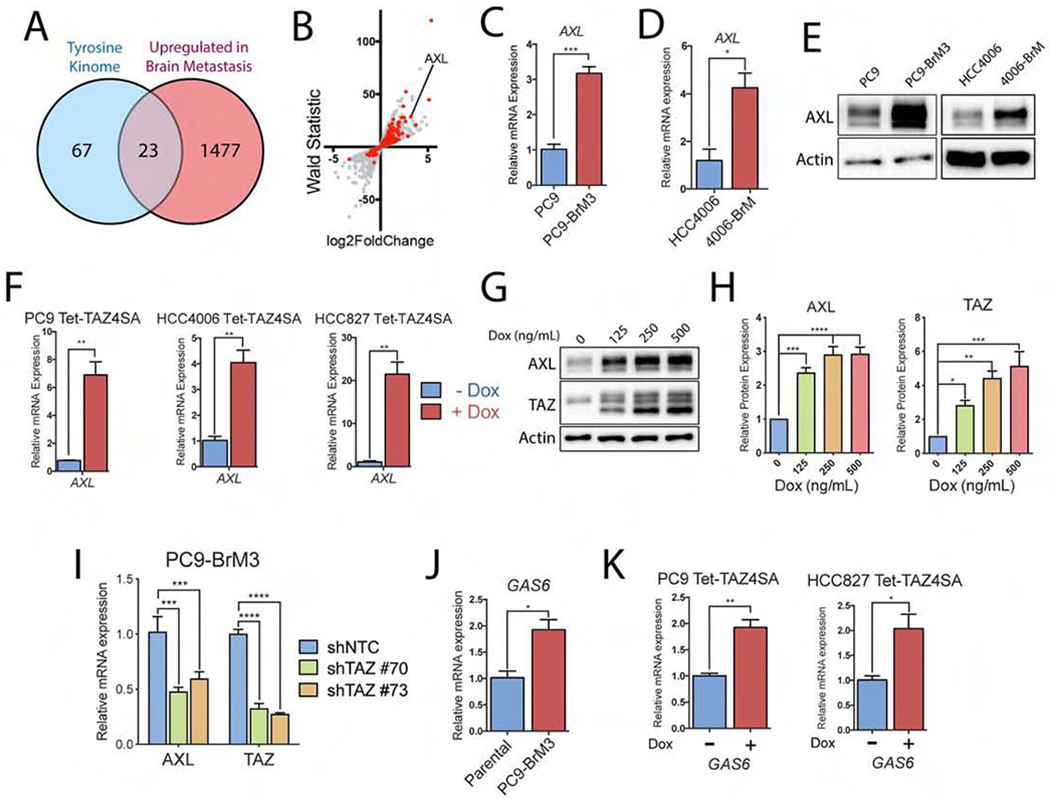
A) Venn diagram of the tyrosine kinome and top transcripts upregulated in PC9-BrM3 cells (RNA-seq). B) Scatter plot of RNA-seq dataset comparing PC9 parental vs BrM3 (GSE83132) mRNA expression. Red dots indicate transcripts in the TAZ4SA GSEA signature. C-D) RT-qPCR of AXL gene expression in C) PC9 parental vs BrM3 cells (n=3) and D) HCC4006 parental vs HCC4006-BrM cells (n=3). E) Immunoblot of AXL protein expression in PC9 parental vs PC9-BrM3 cells and HCC4006 parental vs HCC4006-BrM cells. F) RT-qPCR of AXL mRNA in three cell lines transduced with inducible TAZ4SA vector and treated with dox for 24 h (n=3). G) Immunoblots and H) quantification of PC9-Tet-TAZ4SA cells treated for 24 h with increasing concentrations of dox (n=3). I) RT-qPCR of PC9-BrM3 cells transduced with shRNAs against either shNTC or shTAZ (shTAZ #70 and shTAZ #73) (n=3). J) RT-qPCR of GAS6 mRNA in PC9 parental vs. BrM3 cells (n=3). K) RT-qPCR of GAS6 mRNA in PC9-Tet-TAZ4SA cells and HCC827-Tet-TAZ4SA cells treated with dox for 7 days prior to RNA harvest (n=3). Statistical analysis for pairwise comparisons performed by unpaired two-tailed t-test. Statistical analysis and multiple comparison testing performed by one-way ANOVA and Fisher post-hoc testing. * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001. For all graphs, error bars represent ± SEM.
Activation of the AXL RTK is regulated by both ligand-dependent and ligand-independent mechanisms (Graham et al., 2014). Heterodimerization of AXL with various RTKs has been shown to diversify downstream oncogenic signaling pathways in a ligand-independent fashion (Goyette et al., 2018). Receptor activation of AXL also occurs in a ligand-dependent manner mediated through binding of growth arrest-specific 6 (GAS6) to the AXL extracellular domain, which induces AXL receptor dimerization to promote autophosphorylation at tyrosine residues within its cytoplasmic domain (Meyer et al., 2013; Meyer et al., 2015; Varnum et al., 1995). We found that GAS6 mRNA expression was increased in PC9-BrM3 cells relative to parental cells (Figure 2J) and was also increased upon inducible expression of TAZ4SA (Figure 2K). Analysis of the GAS6 promoter revealed TEAD DNA-binding motifs located upstream and downstream of the GAS6 TSS (Figure S2F). These results show that TAZ mediates activation of AXL signaling in lung cancer cells through increased expression of transcripts for both the AXL receptor as well as its ligand GAS6 and suggest that activation of AXL-dependent signaling pathways might contribute to increased brain metastatic activity by lung cancer cells.
ABL2 is a transcriptional target of TAZ in lung adenocarcinoma
Unexpectedly, we found that ABL2 was one of 84 transcripts upregulated in both PC9-BrM3 cells and TAZ4SA-expressing cells (Figure 1H). Little is known regarding the transcriptional regulation of ABL2. RT-qPCR analysis of PC9 and HCC4006 cell lines transduced with a Tet-TAZ4SA construct showed that dox-inducible expression of TAZ4SA increased expression of ABL2 mRNA and protein, but not of the closely-related ABL1 kinase (Figures 3A–C). ABL2 was also identified among the 23 tyrosine kinases whose corresponding transcripts were increased in brain metastatic PC9-BrM3 cells versus parental cells (Figures 2A and S2A), which we confirmed by RT-qPCR analysis (Figure 3D). Promoter analysis of the ABL2 gene locus identified four putative TEAD DNA-binding consensus motifs within 2500 bp of the ABL2 TSS, including a proximally-located motif 49 base pairs upstream of the TSS (Figure 3E). We previously reported that inhibition of the ABL kinases with an ABL-specific allosteric inhibitor elicits TAZ protein degradation leading to impaired expression of TAZ target genes (Gu et al., 2016). To evaluate whether TAZ might bind to the ABL2 gene promoter and directly regulate its expression, PC9-BrM3 cells were treated with or without the ABL001 allosteric inhibitor (Asciminib) for 24 hours, followed by chromatin immunoprecipitation for TAZ and qPCR (ChIP-qPCR) of an amplicon encompassing both the 49 base pair upstream TEAD DNA binding motif and the ABL2 TSS. Treatment with ABL001 significantly reduced occupancy of TAZ at the ABL2 promoter (Figure 3F). Collectively these findings reveal a previously unappreciated role for active TAZ in the regulation of ABL2 transcription and show that ABL2 can function downstream of TAZ in addition to its role as an upstream regulator of TAZ protein stability.
Figure 3. ABL2 is a transcriptional target gene of TAZ in lung adenocarcinoma.

A-B) RT-qPCR of indicated mRNAs in A) PC9-Tet-TAZ4SA and B) HCC4006-Tet-TAZ4SA cells treated +/− dox for 7 days. Statistical analysis performed by one-way ANOVA and Fisher post-hoc testing (n=3). C) Immunoblots of PC9-Tet-TAZ4SA cells treated +/− dox for 24 h. D) RT-qPCR of ABL2 mRNA in PC9 parental vs. BrM3 cells (n=3). E) Schematic of ABL2 TSS and TEAD DNA binding motifs. F) ChIP-qPCR of PC9-BrM3 cells treated with DMSO or 10 uM ABL001 for 24 h (n=2). UTR: untranslated region control. Control IP: anti-rabbit IgG.
AXL engages in bidirectional signaling with ABL2
Tyrosine-phosphorylated AXL has previously been shown to recruit non-receptor tyrosine kinases, such as SRC and LCK, through interactions of their respective SH2 domains with tyrosine-phosphorylated Y821 on AXL (Braunger et al., 1997). We therefore evaluated whether ABL2 might also interact with activated AXL to regulate downstream signaling. To test this possibility, we performed co-immunoprecipitation of endogenous AXL and ABL2 proteins expressed in brain metastatic PC9-BrM3 cells in the absence and presence of the AXL ligand GAS6. We detected ligand-induced interaction of endogenous AXL with the ABL2 kinase by immunoprecipitation with an ABL2-specific antibody (Figure 4A). The finding that the interaction between AXL and ABL2 required ligand-induced AXL activation led us to evaluate whether these protein tyrosine kinases might engage in cross-activation. To inhibit the ABL kinases we employed the allosteric inhibitor GNF-5 which, like ABL001, binds with high affinity to the unique myristate-binding pocket of the ABL kinases (Deng et al., 2010; Zhang et al., 2010). AXL tyrosine phosphorylation, indicative of enhanced kinase activity, was inhibited in a dose-dependent manner upon inhibition of the ABL kinases (Figure 4B). Further, we found that knockdown of ABL2, but not ABL1, markedly decreased AXL tyrosine phosphorylation (Figure 4C). Knockdown of the ABL kinases also decreased phosphorylation of CRKL at Y207, a selective surrogate marker for ABL kinase activity (Figure 4C).
Figure 4. The AXL and ABL2 tyrosine kinases engage in bidirectional signaling in lung cancer cells.
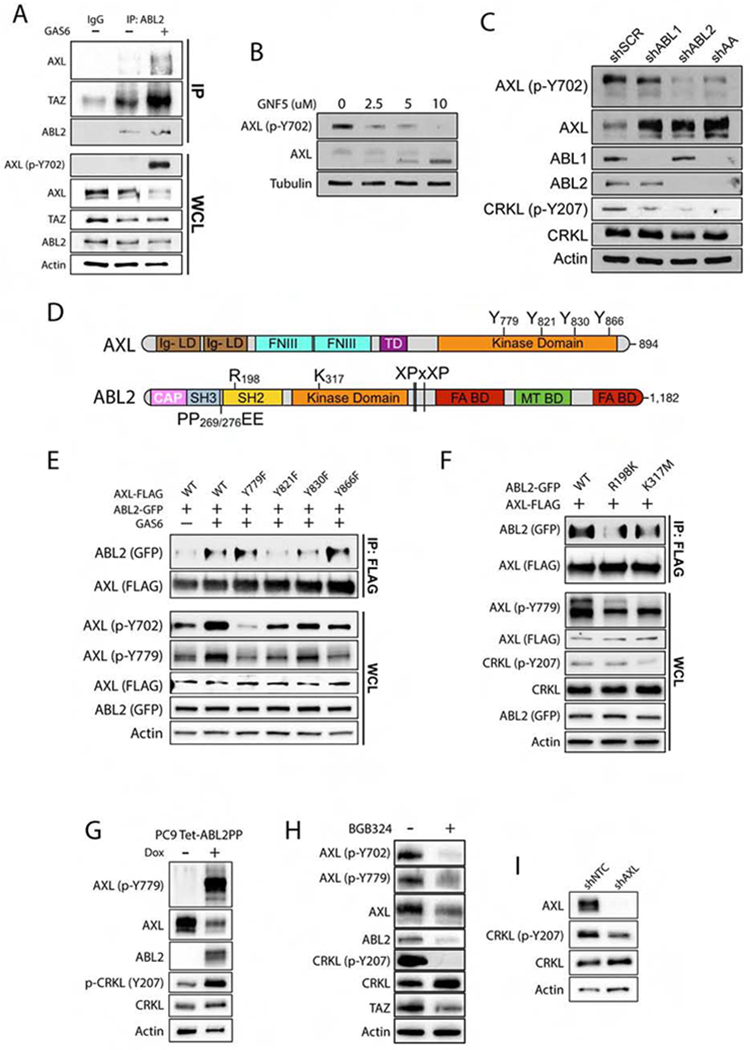
A) Immunoblots of PC9-BrM3 cells treated with 500 ng/mL recombinant human GAS6 for 1 h prior to immunoprecipitation (IP) with anti-ABL2 antibody (n=3). B) Immunoblots of AXL and p-AXL Y702 in PC9 cells treated with GNF-5 for 24 h. C) Immunoblots of PC9 cells transduced with shRNAs for scramble (SCR), ABL1, ABL2, and ABL1/ABL2 double knockdown (AA) (n=3). D) Schematic of AXL and ABL2 protein structural domains. E) Co-IP of indicated proteins in 293T cells co-transfected with FLAG-AXL WT or phospho-mutants and WT ABL2-GFP plasmids as indicated. Cells were serum-starved for 1 hr prior to addition of 500 ng/mL human GAS6 for 1 h. F) Immunoblots of 293T cells co-transfected with FLAG-AXL and ABL2-GFP wild-type (WT), R198K (SH2-dead) or K317M (kinase dead) mutants. Cells were serum-starved for 1 h prior to treatment with 500 ng/mL GAS6 for 1 h. G) Immunoblot of PC9 cells transduced with inducible active ABL2PP treated with dox for 24 h. H) Immunoblots of PC9-BrM3 cells treated ± 5 uM BGB324 for 24 h. I) Immunoblots of PC9-BrM3 cells transduced with shNTC or shAXL (n=3). WCL: whole cell lysate.
To further characterize the interaction between the AXL and ABL2 kinases, we performed site-directed mutagenesis of four tyrosine residues (Y779, Y821, Y830, Y866) within the intracellular domain of AXL to determine if the phospho-mutants impacted the receptor’s ability to engage with ABL2 (Figure 4D). Addition of GAS6 to serum-starved co-transfected cells resulted in a marked increase in the interaction between ABL2 and wild-type (WT) AXL, which was abolished with the AXL Y821F mutant and decreased with the AXL Y830F mutant, but was not affected by mutation of Y779 or Y866 on AXL (Figure 4E). To identify which domain of ABL2 was responsible for its interaction with AXL, we performed site-directed mutagenesis of the ABL2 SH2 (R198K) and SH1 kinase (K317M) domains (Figure 4D). We found that both the SH2 domain binding-deficient ABL2 R198K mutant and kinase-inactive ABL2 K317M mutant exhibited decreased interaction with the AXL receptor under ligand-activated conditions (Figure 4F). Conversely, inducible expression of an activated ABL2 protein (ABL2PP) in PC9 cells resulted in a profound increase in AXL tyrosine phosphorylation (Figure 4G). Moreover, pharmacologic inhibition of AXL with the small molecule inhibitor BGB324 (also known as R428) not only decreased phospho-AXL levels but was accompanied by reduced expression of ABL2, TAZ, and phospho-CRKL (Figures 4H). Knockdown of AXL in PC9-BrM3 cells with stably-transduced shRNA also decreased phospho-CRKL levels, indicative of reduced ABL kinase activity (Figure 4I). Together these data demonstrate that ABL2 promotes AXL tyrosine phosphorylation and, in turn, the activated AXL receptor interacts with ABL2 to elicit signaling through downstream effectors including TAZ.
Activation of ABL2 promotes TAZ nuclear localization
The pathways regulating activity of the TAZ and related YAP transcriptional co-activators are diverse (Moroishi et al., 2015; Varelas, 2014). Nucleocytoplasmic shuttling of TAZ/YAP is regulated downstream of Hippo pathway serine/threonine protein kinases, as well as by activation of protein tyrosine kinases and actomyosin contractility in response to extracellular ligands. Tyrosine phosphorylation of TAZ/YAP has been shown to regulate not only nucleocytoplasmic shuttling, but also protein stability and transcriptional activation (Ege et al., 2018; Feng et al., 2019; Kedan et al., 2018; Kofler et al., 2018; Rosenbluh et al., 2012; Shanzer et al., 2017). The Abl1 (c-Abl) kinase was reported to phosphorylate murine Taz on tyrosine (Y) 316 (corresponding to human TAZ Y321) in renal cells following hyperosmotic stress (Jang et al., 2012). We thus evaluated whether ABL2 might promote TAZ tyrosine phosphorylation in metastatic lung cancer cells. Dox-inducible expression of constitutively-active ABL2PP in PC9 cells resulted in increased TAZ tyrosine phosphorylation (Figure 5A). TAZ tyrosine Y321 is a predicted ABL SH2 binding site found within the TAZ transactivation domain, and is adjacent to a phosphodegron sequence that mediates recognition by the β-TrCP ubiquitin E3 ligase (Liu et al., 2010; Songyang et al., 1993). A recent report revealed that amino acids 290-345 of TAZ contain a nuclear localization signal import motif (NLS) (Kofler et al., 2018). The TAZ NLS contains acidic residues and is required not only for nuclear import but also for transcriptional activation of TAZ target genes. Tyrosine phosphorylated TAZ may regulate the interaction of TAZ with SH2-domain containing proteins including the ABL kinases. We found that endogenous ABL2 interacted with wild type (WT) TAZ (FLAG-tagged) in PC9-BrM3 cells, and the interaction of ABL2 and TAZ was markedly decreased by mutation of TAZ-Y321F, a TAZ mutant that exhibited decreased phospho-tyrosine levels compared to TAZ-WT (Figure 5B). Further, we showed that interaction of TAZ (FLAG-tagged) with ABL2 was markedly decreased by mutation of the ABL2 SH2 domain (R198K) (Figure 5C). Similarly, interaction of TAZ with kinase-inactive ABL2 (K317M) was decreased compared to ABL2 WT (Figure 5C), and expression of both ABL2 mutants resulted in decreased tyrosine phosphorylation of TAZ compared to that induced by co-expression of ABL2 WT (Figure 5C). These data show that active ABL2 promotes TAZ tyrosine phosphorylation, and that the ABL2 SH2 domain is required for interaction with phosphorylated Y321 on TAZ. Moreover, we observed increased nuclear localization of endogenous TAZ in PC9 lung cancer cells expressing active ABL2PP, which was maximal 1-2 hours after induction of ABL2PP protein expression (Figure 5D). To evaluate whether endogenous TAZ and ABL2 proteins interact, PC9-BrM3 cells were treated with the AXL ligand GAS6 to activate tyrosine phosphorylation signaling downstream of the AXL receptor. We detected basal interaction of TAZ and ABL2 in the absence of GAS6, but this interaction was increased after one hour of GAS6 stimulation of PC9-BrM3 cells (Figure 4A). Notably, we found that whereas endogenous ABL2 interacted with TAZ following co-immunoprecipitation, it did not interact with ABL1 (Figure 5E). Interestingly, neither ABL1 or ABL2 interacted with endogenous YAP in these cells (Figure 5E). Together, our data support the existence of a positive feedback loop where ligand-activated AXL promotes ABL2 activation and TAZ tyrosine phosphorylation leading to enhanced TAZ nuclear accumulation and transcription of TAZ target genes including AXL, ABL2 and GAS6 in brain-metastatic lung adenocarcinoma cells.
Figure 5. Activation of ABL2 signaling promotes tyrosine phosphorylation and nuclear localization of TAZ in brain-metastatic lung cancer cells.
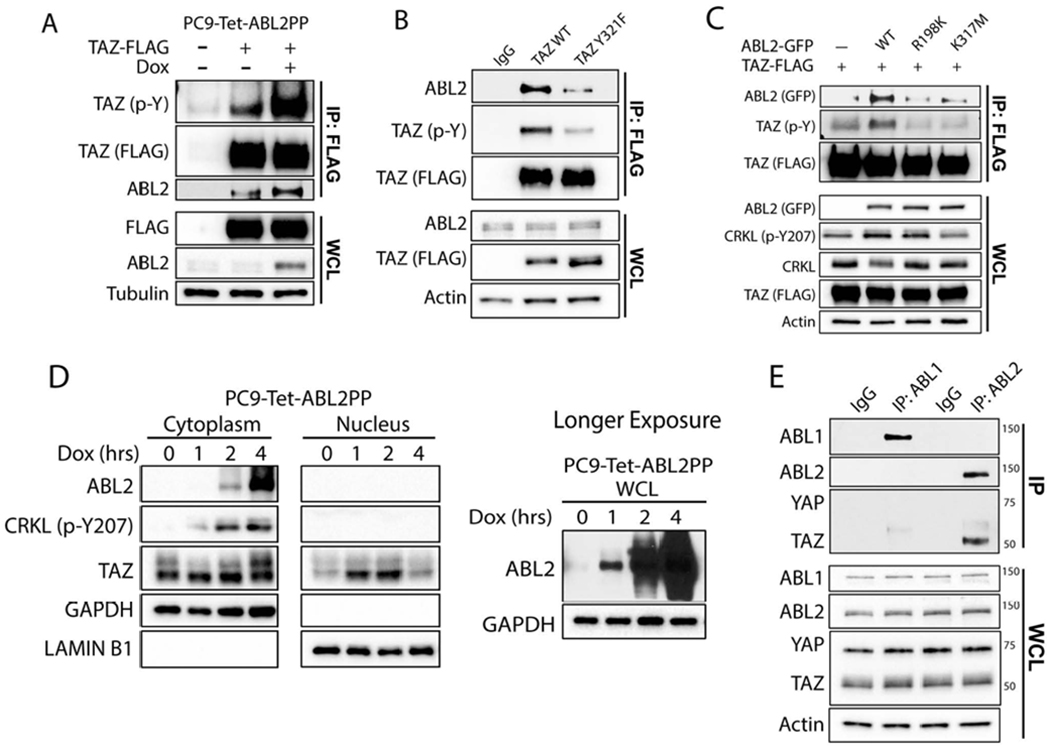
A) Co-IP of PC9-Tet-ABL2PP cells transfected with FLAG-TAZ wild-type (WT) and treated with dox for 1 hr prior to anti-FLAG pulldown. B) PC9-BrM3 cells were transfected with FLAG-TAZ-WT or FLAG-TAZ-Y321F, and following anti-FLAG pulldown, co-IP proteins were immunoblotted with indicated antibodies. C) Co-IP of indicated proteins in 293T cells co-transfected with TAZ-FLAG and ABL2-GFP WT, R198K or K317M mutants. D) Nuclear fractionation of PC9-Tet-ABL2PP cells treated with dox for indicated time points. GAPDH and Lamin B1 used for cytosolic and nuclear markers, respectively (n=3). E) Co-IP of PC9-BrM3 cells with antibodies against ABL1, ABL2, or IgG control. WCL: whole cell lysate.
AXL and ABL2 are necessary for lung adenocarcinoma metastasis to the brain
To assess whether interfering with ABL2 signaling might impair brain metastatic activity of lung adenocarcinoma cells, PC9-BrM3 cells expressing shRNAs against scramble control (shSCR) or ABL2 (shABL2) were injected intracardially into nude mice. We found that BMFS and overall survival (OS) were increased in mice injected with ABL2 knockdown PC9-BrM3 cells compared to control mice (Figures 6A and 6B). Quantification of brain flux at day 30 also revealed minimal disease burden in mice injected with ABL2 knockdown cells compared to those injected with control cells (Figures 6E and 6F). Similarly, stable depletion of AXL in PC9-BrM3 cells resulted in a marked increase in BMFS and OS (Figures 6C and 6D), and decreased brain-metastatic burden as quantified on day 30 post-intracardiac injection (Figures 6G and 6H). Together these data demonstrate that both AXL and ABL2 kinases are active signaling nodes required for lung cancer brain metastasis.
Figure 6. AXL and ABL2 tyrosine kinases are required for lung adenocarcinoma metastasis to the brain.
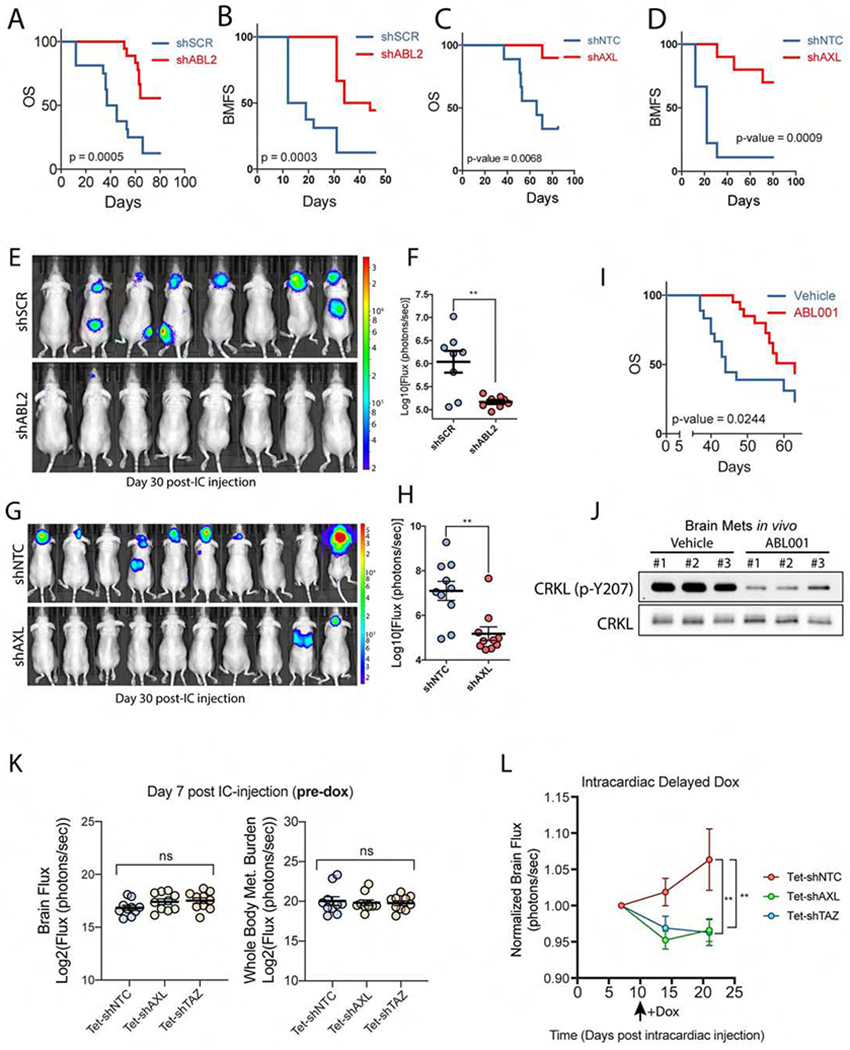
A) Overall survival (OS) and B) BMFS of mice injected intracardially with PC9-BrM3 cells expressing shSCR (n=16) or shABL2 (n=18). C) OS and D) BMFS of mice injected intracardially with PC9-BrM3 cells expressing shNTC (n=9) or shAXL (n=10). E) Representative images and F) quantification of brain met burden in mice on day 30 post-intracardiac injection of PC9-BrM3 cells expressing shSCR (n=8) or shABL2 (n=8). G) Representative images and H) quantification of brain met burden in mice on day 38 post-intracardiac injection of PC9-BrM3 cells with shNTC (n=10) or shAXL (n=10). I) OS of mice injected intracardially with PC9-BrM3 cells and treated with vehicle (n=18) or 100 mg/kg Q.D. ABL001 (n=20). Drug dosing began 24 h post-intracardiac injection after mice were separated into respective groups. J) Immunoblots of ABL kinase activity (p-CRKL-Y207) in established in vivo brain metastases from six mice injected with H1975 cells and treated with vehicle (n=3) or 100 mg/kg ABL001 (n=3). Mice were treated at 3, 12, and 24 h prior to harvesting brain metastases. K) Quantification of brain flux and whole-body flux in mice on day 7 post-intracardiac injection (pre-dox phase) with PC9-BrM3 cells transduced with inducible shNTC, shAXL or shTAZ. Statistical analysis performed by one-way ANOVA and Fisher post-hoc testing. L) Quantification of brain flux in the indicated mice before and after administering dox water starting on Day 10 (black arrow). Brain flux was normalized to the Day 7 baseline mean for each of the three groups with statistical analysis performed using a mixed-effects model. ** p-value < 0.01. For survival analysis, statistical testing performed by Log-rank (Mantel-Cox).
We next sought to determine if the brain-metastatic activity of lung cancer cells might be impaired by pharmacologic inhibition of this pathway using the ABL allosteric inhibitor ABL001 (Asciminib). ABL001 is currently in clinical trials for therapy-resistant patients with BCR-ABL+ chronic myeloid leukemia (Wylie et al., 2017). We selected the ABL allosteric inhibitors for the treatment of mice with brain metastases because they are ABL-specific and are predicted to cross the BBB (Figure S3). Mice were intracardially injected with PC9-BrM3 cells and imaged within 24 hr post-injection to assess successful injection of tumor cells prior to random assortment into vehicle or ABL001 (100 mg/kg Q.D.) groups. We observed a significant increase in OS in tumor-bearing mice treated with the ABL allosteric inhibitor (Figure 6I). Treatment with ABL001 increased median survival ~20 days over vehicle-treated mice (vehicle median survival = 44 days; ABL001 median survival = 63 days). Isolation of brain metastases from vehicle and ABL001-treated mice revealed effective inhibition of ABL kinases in brain metastases by ABL001 as shown by decreased phosphorylation of the ABL-specific site on CRKL (Figure 6J). These findings demonstrate that ABL001 traverses the BBB, effectively inhibits ABL kinases in brain metastases, and enhances survival of tumor-bearing mice.
It has been reported that following injection into the arterial circulation, brain metastatic lung cancer cells extravasate into the mouse brain within 7 days, followed by metastatic colonization of the brain parenchyma (Kienast et al., 2010; Valiente et al., 2014; Wingrove et al., 2019). To directly evaluate whether AXL-TAZ signaling is required for colonization of the brain by lung cancer cells, we employed dox-inducible shRNAs to knockdown AXL or TAZ in a temporally regulated manner (Figures S4A and S4B). Constitutive or inducible depletion of either AXL or TAZ did not inhibit PC9-BrM3 cell growth and viability and did not increase anoikis in vitro (Figures S4C–E). Stably transduced cells with dox-inducible shRNAs against either AXL or TAZ were intracardially injected into mice and, on day 7 post-intracardiac injection, BLI analysis was performed to confirm comparable levels of brain- and whole-body metastatic burden across treatment groups (Figure 6K). On day 10 post-intracardiac injection, mice were administered dox water ad libitum to induce shRNA expression. BLI analysis of brain metastatic burden across treatment groups revealed a significant decrease in metastatic growth in mice harboring AXL- and TAZ-depleted PC9-BrM3 cells relative to the control arm (Figure 6L). These data demonstrate that AXL and TAZ signaling is required for the colonization and early outgrowth of brain metastases.
Elevated expression of AXL, ABL2 and TAZ predicts poor survival in lung adenocarcinoma
Currently there is a lack of publicly available lung adenocarcinoma patient datasets comparing gene expression data from the primary lung tumor with that of corresponding brain metastases, in part due to the difficulty in obtaining operable or biopsy-accessible brain metastases. Enhanced AXL expression in lung adenocarcinoma brain metastases relative to primary tumors was reported to be prognostic of poor survival outcomes (Wu et al., 2017). Thus, we evaluated whether AXL, ABL2, and TAZ expression might have correlative prognostic value by analyzing overall patient survival (OS) and progression-free survival (PFS) using human lung adenocarcinoma gene expression datasets. Analysis of patients with lung adenocarcinoma correlating gene expression to patient survival revealed that high expression of AXL, ABL2 or WWTR1 (TAZ) individually predicted reduced OS and PFS (Figures S5A–F). Interestingly, these trends in survival were not observed for lung adenocarcinoma patients expressing high levels of ABL1 or YAP1, despite the functional overlap often ascribed with their respective paralogs ABL2 and WWTR1 (Figures S5I–L). Interestingly, mutual exclusivity analysis of lung adenocarcinoma gene expression datasets from the TCGA revealed that increased mRNA and protein expression of ABL2, AXL and WWTR1 frequently co-occurred in the same patients and was associated with decreased overall survival, a trend that did not hold true for ABL1 nor YAP1 (Figures S5G and S5H). These data suggest that enhanced expression of ABL2, AXL, and WWTR1 in lung adenocarcinoma patients might have prognostic value as an indicator of poor survival outcomes.
Activation of ABL2-TAZ-AXL signaling drives expression of L1CAM in brain metastatic lung cancer cells
We identified a TAZ-induced transcriptional program that promotes acquisition of brain metastatic phenotypes by lung adenocarcinoma cells. Further, active TAZ drives expression of target genes encoding for proteins implicated in neuronal differentiation and development (Figures S6A and S6C). Brain-metastatic cancer cells within the brain microenvironment have been shown to upregulate neuronal-related genes as an adaptation for efficient metastatic colonization of the brain (Neman et al., 2014; Zeng et al., 2019). Consistent with these reports, GSEA revealed significant enrichment of neuronal gene signatures in PC9-BrM3 cells compared to parental PC9 cells (Figures S6B and S6D). Among the neuronal transcripts shared between brain-metastatic and TAZ4SA-expressing cells were ABL2 and the neural cell adhesion molecule L1 (L1CAM), both of which have been implicated in axonogenesis and neuronal development among other processes (Lin et al., 2013; Schafer and Altevogt, 2010). Recent reports showed that L1CAM promotes breast and lung cancer brain metastasis by facilitating vascular co-option and colonization of tumor cells in the brain parenchyma (Er et al., 2018; Valiente et al., 2014). Despite the functional importance of this molecule in brain metastasis, the development of therapies targeting L1CAM has remained elusive.
As L1CAM mRNA was upregulated in both brain-metastatic and TAZ4SA-expressing lung cancer cells (Figure 1H), we evaluated whether manipulation of AXL-ABL2-TAZ signaling might regulate L1CAM expression in brain-metastatic tumor cells. L1CAM protein expression was markedly increased in PC9-BrM3 cells (Figure 7A), consistent with previous reports (Valiente et al., 2014). Inducible expression of TAZ4SA in PC9 and HCC4006 cells markedly upregulated expression of L1CAM protein (Figures 7B and S6E). Analysis of the L1CAM gene promoter revealed the presence of five TEAD DNA-binding motifs proximal to the L1CAM TSS (Figure S6F), and knockdown of TAZ markedly decreased expression of L1CAM mRNA (Figure 7C). These data show that L1CAM expression is regulated by TAZ-dependent transcription.
Figure 7. Activation of AXL-ABL2-TAZ signaling promotes expression of L1CAM in brain metastatic lung adenocarcinoma.
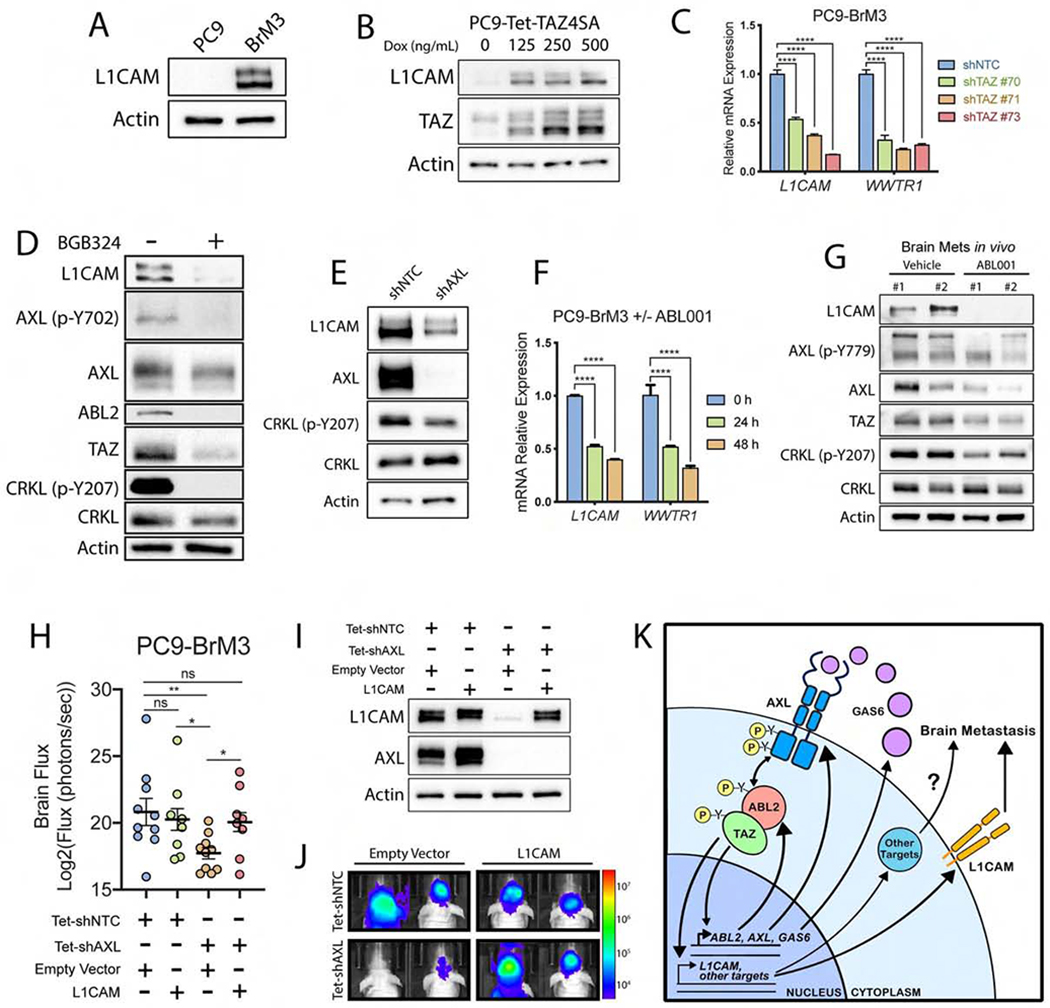
Immunoblot of L1CAM in A) PC9 parental vs BrM3 cells or B) PC9-Tet-TAZ4SA cells treated with increasing concentrations of dox for 24 h (n=3). C) RT-qPCR of L1CAM and WWTR1 mRNA in PC9-BrM3 cells transduced with shNTC control or shRNAs against WWTR1 (n=3). D) Immunoblots of PC9-BrM3 cells treated ± 5 uM BGB324 for 24 h. E) Immunoblots of PC9-BrM3 cells transduced with shNTC or shAXL. F) RT-qPCR of L1CAM and WWTR1 mRNA in PC9-BrM3 cells treated with 10 uM ABL001 for indicated time points (n=3). G) Immunoblots of indicated proteins in established in vivo brain metastases from mice injected with H1975 lung cancer cells and treated with vehicle (n=2) or 100 mg/kg ABL001 (n=2). Mice were treated at 3, 12, and 24 h prior to harvesting brain metastases. H) Quantification of brain met flux (day 34) in mice injected intracardially with PC9-BrM3 cells expressing Tet-shNTC with empty vector (EV) (n=10) or L1CAM (n=8), or Tet-shAXL with EV (n=10) or L1CAM (n=8). Mice were administered dox water starting 24 h prior to intracardiac injection. I) Immunoblots of PC9-BrM3 cells transduced in 7H. PC9-BrM3 cells were freshly harvested from mouse brains on Day 43 post-intracardiac injection. J) Representative images of mice from 7H. K) Proposed mechanism of autocrine AXL-ABL2-TAZ signaling axis in lung cancer brain metastasis.
We also sought to determine if L1CAM expression required AXL or ABL2 kinase activity in brain metastatic cells. Pharmacologic inhibition or knockdown of AXL elicited a profound decrease in L1CAM protein levels (Figures 7D and 7E). Treatment with ABL001 in vitro resulted in decreased expression of L1CAM and WWTR1 transcripts (Figure 7F). Notably, treatment of mice harboring established lung cancer brain metastases with ABL001 was highly effective at inhibiting expression of L1CAM, TAZ, AXL and p-AXL proteins in brain metastases in vivo (Figure 7G). Similar to decreased survival outcomes for patients with high-level expression of AXL, ABL2 and TAZ, elevated expression of L1CAM mRNA was predictive of reduced OS and PFS (Figures S6G and S6H).
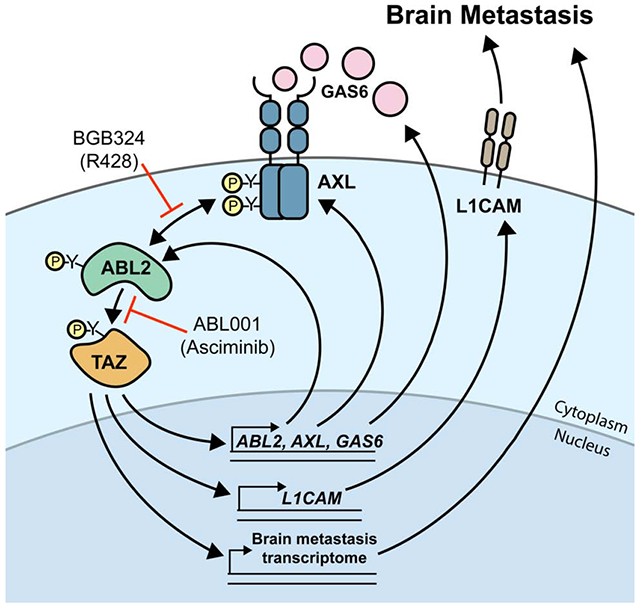
To assess whether L1CAM functions downstream of AXL signaling to promote brain metastases, L1CAM was ectopically expressed in PC9-BrM3 cells with dox-inducible knockdown of AXL, and these cells or corresponding vector control cells were intracardially injected in athymic nude mice (Figures 7H, I, J). Ectopic L1CAM rescued brain metastases in the AXL-deficient cells (Figures 7H and 7J). Both AXL and TAZ have been shown to promote tumor cell invasion (Graham et al., 2014; Zanconato et al., 2019). Expression of L1CAM rescued cell invasion in vitro in PC9-BrM3 cells following inducible knockdown of either AXL or TAZ (Figures S6I and S6J). Collectively, our data reveal that activation of a TAZ-AXL-ABL2 positive feedback loop drives expression of L1CAM to promote lung adenocarcinoma brain metastasis, and suggest that pharmacologic targeting of the AXL and ABL2 tyrosine protein kinases might therefore be employed to treat this deadly disease (Figure 7K).
Discussion
Current therapies to treat lung cancer brain metastases are ineffective due in part to the lack of mechanistic understanding of the molecular pathways that promote metastasis to the brain. Lung cancer cells that metastasize to the brain must acquire distinctive properties to penetrate the BBB and colonize the brain parenchyma, which is characterized by unique cellular and metabolic microenvironments distinct from those found at the site of the primary lung tumor. Experimental mouse models of brain metastasis have provided insights into the complex processes implicated in lung cancer brain metastases (Chen et al., 2016; Priego et al., 2018; Valiente et al., 2018). However, there is limited knowledge of the pathways critical for lung cancer brain metastases, and actionable targets are lacking among those pathways already implicated in this disease.
Here we report that expression of an activated form of the TAZ transcriptional co-activator (TAZ4SA) in lung adenocarcinoma cells promotes metastases predominantly to the brain relative to other organs. Unbiased RNA-seq analysis of parental and TAZ4SA-expressing cells found that a subset of the transcripts upregulated in these cells were also markedly elevated in brain metastatic lung adenocarcinoma. Among these transcripts were those encoding the ABL2 and AXL tyrosine kinases as well as the AXL ligand GAS6. Unexpectedly, we found inter-dependent regulation of the expression and activation of TAZ, ABL2, and AXL at the transcriptional and post-transcriptional levels. Inactivation of either AXL or ABL kinases decreased TAZ signaling, thus placing TAZ both upstream and downstream of these tyrosine kinases. Notably, inactivation of either ABL2 or AXL in lung adenocarcinoma cells markedly decreased brain metastases following intracardiac injection, and treatment of mice bearing lung cancer brain metastases in vivo with an ABL kinase allosteric inhibitor markedly decreased disease burden, inhibited ABL kinase substrates (p-CRKL), and decreased the levels of TAZ, AXL and phosphorylated AXL (p-AXL) proteins in the isolated metastases. Together our data show that the TAZ-regulated protein tyrosine kinases, ABL2 and AXL, can be targeted with available and BBB-permeable small molecule inhibitors for the treatment of lung adenocarcinoma brain metastases in mouse models.
We found that ABL2 protein but not ABL1, interacted with TAZ but not YAP in lung adenocarcinoma cells. While the mechanisms that regulate TAZ expression and activation downstream of the Hippo pathway LATS1/2 protein kinases have been well described, the regulation of TAZ downstream of protein tyrosine kinases remains poorly understood (Moroishi et al., 2015). Tyrosine phosphorylation of TAZ/YAP has been linked to the regulation of their protein stability, nucleocytoplasmic shuttling, and transcriptional activation (Ege et al., 2018; Feng et al., 2019; Kedan et al., 2018; Rosenbluh et al., 2012; Shanzer et al., 2017). We showed that activated ABL2 promoted TAZ nuclear accumulation, which may be mediated in part by disrupting the interaction of TAZ with the β-TrCP ubiquitin ligase (Gu et al., 2016). In this regard, TAZ tyrosine Y321 is found within the TAZ transactivation domain, adjacent to a phosphodegron sequence that mediates recognition by the β-TrCP ubiquitin E3 ligase. We showed that optimal interaction of ABL2 with TAZ required the presence of tyrosine Y321 on TAZ and the ABL2 SH2 and SH1 domains. Phosphorylation of TAZ Y321 by ABL2 may also regulate the acidic NLS found between amino acids 290-345 of TAZ (Kofler et al., 2018), which is required not only for nuclear import but also for transcriptional activation of TAZ target genes.
Acquisition of a neuronal phenotype by metastatic tumors is an adaptation that facilitates colonization and outgrowth of metastases in the brain (Fecci et al., 2019; Neman et al., 2014; Zeng et al., 2019). Our findings support this concept and reveal that activation of AXL-ABL2-TAZ signaling promotes expression of a subset of transcripts encoding neuronal-related proteins which are upregulated by brain-metastasizing lung adenocarcinoma cells. Among these transcripts we identified the L1CAM neuronal cell adhesion molecule as a target gene of TAZ-dependent transcription. L1CAM was shown to promote vascular co-option, migration and outgrowth of brain metastasizing tumor cells (Er et al., 2018; Valiente et al., 2014), however no studies thus far have uncovered effective therapies for the targeted inhibition of this molecule. We found that ectopic expression of L1CAM in AXL-deficient lung cancer cells rescued brain metastasis in mice. Thus, our discovery of a pharmacologically-amenable signaling pathway regulating L1CAM gene expression may have important clinical implications.
In summary, we have identified two actionable targets, AXL and ABL2, that function both downstream and upstream of TAZ in a previously uncharacterized autocrine feed-forward signaling axis required for lung adenocarcinoma brain metastasis. Activation of AXL has been implicated in acquired resistance to EGFR targeted therapies in lung cancer (Namba et al., 2019; Zhang et al., 2012). AXL inhibitors have been used in combination with targeted therapies to treat NSCLC patients with acquired resistance (NCT02424617), and in combination with docetaxel for the treatment of NSCLC (NCT02922777). We recently reported that ABL-specific allosteric inhibitors sensitize primary lung adenocarcinoma to chemotherapy by promoting tumor cell differentiation (Khatri et al., 2019). Because brain metastases often co-exist with systemic extra-cranial metastatic lung tumors, development of combination therapy regimens that include BBB-penetrant ABL2 or AXL kinase inhibitors may be effective for the treatment of both intracranial and extra-cranial lung tumors leading to increased survival and improved quality of life.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ann Marie Pendergast (ann.pendergast@duke.edu).
All unique and stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines.
Human NSCLC cell lines HCC4006, HCC827 and H1975 were purchased from ATCC. PC9 parental cells were a gift from Dr. Joan Massague (Memorial Sloan-Kettering Cancer Center, New York, NY, USA). PC9-BrM3, HCC4006-BrM, and H1975-BrM3 cell lines were derived in the Pendergast laboratory by serial intracardiac injection as described below. NSCLC cells were maintained in RPMI 1640 (Life Technologies) supplemented with 10% tetracycline-screened fetal bovine serum (FBS, Hyclone), 10 mM HEPES, 1 mM sodium pyruvate, and 0.2% glucose. H293T cells were used for transfection and virus production and were maintained in DMEM (Life Technologies) with 10% FBS (Corning). All cultures were maintained at 370 in humidified air containing 5% CO2. In studies with dox-inducible constructs, the dose of doxycycline (Millipore Sigma) administered was determined empirically depending on the vectors used. For experiments assessing effects of pharmacologic inhibitors in vitro (GNF-5, ABL001, BGB324), drugs were dissolved in DMSO and the final concentration of DMSO in culture media did not exceed 0.1% v/v. The ABL allosteric inhibitors GNF-5 and ABL001 (Asciminib) were synthesized by the Duke University Small Molecule Synthesis Facility and validated by LC-MS and 1H-NMR, as well as cell-based assays. The AXL kinase inhibitor BGB324 was obtained from MedKoo Biosciences and was validated with cell-based assays to evaluate cell signaling inhibitory activity.
Intracardiac injections.
All animal experiments were conducted in accordance with protocols approved by the Duke University Division of Laboratory Animal Resources Institutional Animal Care and Use Committee (IACUC). Cells were stably transduced with pFU-luciferase-Tomato (pFuLT) DNA prior to injection to allow for bioluminescent imaging (BLI) in vivo with or without shRNAs or expression constructs as described in figure legends. We used 8-12-week old age-matched female athymic nu/nu mice for all studies (Jackson Laboratory). Mice were anesthetized with 5% isoflurane prior to injections. For all studies, 4x105 lung cancer cells suspended in 100 uL PBS were injected into the left cardiac ventricle with a 30-gauge needle. Animals were monitored until full recovery from anesthesia and were subsequently imaged weekly to both confirm proper anatomical injection and to monitor for progression of disease burden using an IVIS XR bioluminescent imager. The ABL allosteric inhibitor ABL001 (Asciminib) was used for in vivo inhibition of the ABL kinases in tumor-bearing mice and was prepared as a suspension in sterile 0.5% methylcellulose/0.5% Tween-80 as described previously (Wylie et al., 2017). Mice were treated with either vehicle control or 100 mg/kg ABL001 via oral gavage once per day. The presence of brain metastases was confirmed either through ex vivo imaging of isolated mouse brains, or alternatively through in vivo BLI followed by isolation of brain metastases and cell culture expansion in vitro. Living Image software was used for analysis of BLI data. For intracardiac injections examining the effect of TAZ4SA induction in lung cancer cells, mice were administered 2 mg/mL doxycycline (Acros Organics) dissolved in 5% sucrose water (autoclave-sterilized) ad libitum starting one day prior to injections and continuing for the duration of the study. The brain-metastatic index was used to normalize the brain metastatic burden relative to overall metastasis between parental and TAZ4SA cohorts and was calculated by dividing the brain metastatic burden (flux, photons/sec) by the total whole-body metastatic burden (flux, photons/sec). For both of these studies, no significant difference was detected in overall whole-body metastatic burden between parental and TAZ4SA cohorts. For intracardiac injections of lung cancer cells transduced with dox-inducible shRNAs, mice were administered 3 mg/mL dox water ad libitum at the time points indicated in corresponding figure legends. For all studies, dox water was replenished every 48 h.
Derivation of brain-metastatic lung cancer cell lines.
Derivation of brain-metastatic cell lines was performed as described previously (Valiente et al., 2014). Human parental lung cancer cell lines PC9, HCC4006, and H1975 were injected intracardially (4 x 105 cells/injection) into 8 to 12-week old athymic nude mice and were monitored for progression of metastatic disease using an IVIS XR bioluminescent imager. When mice presented with advanced brain metastatic burden (~30 days post-intracardiac injection), animals were euthanized and whole brains were collected, minced, and digested in RPMI culture medium supplemented with 0.125% collagenase III and 0.1% hyaluronidase and placed on a rotator for 5 hours at room temperature. Following incubation, cells were centrifuged and resuspended in 0.25% trypsin for 10 minutes at 37C. Cells were then resuspended in culture media containing 1X Anti-Anti (Thermo Fisher). Cell lines were expanded to near-confluence in culture prior to sorting for Tomato-positive cells for future experimental use.
METHODS DETAILS
Analysis of ABL001 penetration across the BBB and inhibition of AXL-ABL2 pathway signaling in brain metastases.
To evaluate the pharmacologic effects of ABL001 on ABL kinase activity and downstream signaling in brain metastases in vivo, mice were injected with H1975 pFulT lung cancer cells and brain metastases were allowed to develop over the span of one month. After confirming the presence of established brain metastases in mice by BLI, mice were administered either vehicle or 100 mg/kg ABL001 via oral gavage at 3, 12, and 24 h prior to euthanasia. Immediately following euthanasia, mouse brains were dissected and tissue sections with visible brain metastases were excised and lysed in RIPA buffer containing protease-phosphatase inhibitor cocktail. Harvested samples were digested for 15 minutes on a rotator at 4°C, followed by centrifugation at 4°C for 15 minutes to clear the lysate of myelin and cell debris. Protein lysates were stored at −20°C prior to immunoblotting. To evaluate and confirm shRNA knockdown of AXL and ectopic expression of L1CAM protein from brain metastases directly isolated from mouse brains, PC9-BrM3 cells were isolated from freshly harvested mouse brains on Day 43 post-intracardiac injection and expanded in vitro in the presence of dox for three days prior to harvest for protein lysates.
RNA-sequencing analysis.
To generate the TAZ4SA gene expression signature we performed RNA-sequencing (RNA-seq) analysis of PC9 parental cells that were lentivirally-transduced with control pLVX-Tet-On-Advanced and pLVX-TP-3F-TAZ4SA plasmids. Expression of TAZ4SA was induced by treating subconfluent cell monolayers with 50 ng/mL dox for 24 h. Total RNA was collected using the RNeasy kit (Qiagen) and 1 μg total RNA input was used for each sample. Libraries were sequenced on an Illumina HiSeq 2000 sequencing system using 50-bp single-end reads by the Duke University Genome Sequencing facility. RNA-seq data was processed using the TrimGalore toolkit and reads that were 20nt or longer after trimming were used for further analysis. Reads were mapped to the GRCh37 version of the human genome and transcriptome using the STAR RNA-seq alignment tool (Dobin et al., 2013). Reads were used for subsequent analysis if they mapped to a single genomic location, and gene counts were compiled using the HTSeq tool (Anders et al., 2015). Only genes that had at least 10 reads in any given library were used in subsequent analysis. Normalization and differential expression were carried out using the DESeq2 Bioconductor package (Love et al., 2014) in the R statistical programming environment. The RNAseq accession number for this dataset deposited in GEO is GSE130425. For differential expression analysis of RNA-seq data comparing PC9 parental to brain metastatic PC9-BrM3 cells, raw paired-end sequencing files were obtained from a publicly-available dataset (GEO: GSE83132) (Boire et al., 2017). Reads were processed using TrimGalore followed by mapping to the GRCh37 version of the human genome and transcriptome obtained from Ensembl using the Hisat2 alignment tool (Kim et al., 2019). Read counts from aligned output files were determined with the Htseq-count tool using reference GTF files obtained from Ensembl. Normalization and differential expression of counts were then processed in R using the DESeq2 package.
Generation of TAZ4SA target gene expression signature.
Following analysis of TAZ4SA RNA-seq data, differential expression data were filtered and sorted to compile a ranked list of transcripts with enriched expression in cells expressing TAZ4SA. Transcripts with low or no expression counts in the TAZ4SA sample were filtered out of the gene list if counts fell below a preset threshold of <1.45 or if the log2 fold change of a gene fell below a preset threshold of +0.9 in the TAZ4SA group. The remaining genes were then sorted first by total transcript counts in the TAZ4SA sample (descending), then by log2FoldChange (descending). As a final filtering step, the top 200 enriched genes from this sorted list were included in the TAZ4SA signature and exported in gmx file format for inclusion in the GSEA software.
Gene set enrichment analysis.
Following analysis of RNA-seq differential expression data comparing PC9 parental to PC9-BrM3 cells, data were sorted by statistical rank and imported into the GSEA software tool from the Broad Institute (Subramanian et al., 2005). In addition to the TAZ4SA gene expression signature, the preranked gene list was processed under default settings and size filters for analysis across all signatures contained within the following mSigDB databases: Hallmark (v6.2, 50 gene sets), Positional (v6.2, 259 gene sets), Curated (v6.2, 3,648 gene sets), Motif (v6.2, 776 gene sets), Computational (v6.2, 782 gene sets), Gene Ontology (v6.2, 4,364 gene sets), Oncogenic Signatures (v6.2, 187 gene sets), and Immunologic Signatures (v6.2, 4,872 gene sets).
Real-time quantitative PCR.
RNA was isolated from subconfluent monolayers of cancer cells using the RNeasy RNA isolation kit (Qiagen), and cDNA synthesis was performed using oligo(dT) primers and M-MLV reverse transcriptase (Invitrogen). RT-qPCR was performed in triplicate wells using iTaq Universal SYBR Green Supermix (Bio-Rad). Primers used in this study were purchased from Sigma Aldrich and are listed in Supplemental Table S2. Analysis of real-time data was collected using a Bio-Rad CFX384 machine and CFX Maestro software. Expression levels of each gene were normalized to 18S or GAPDH control housekeeping genes using the ddCT algorithm.
Immunoblotting procedures.
Cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS) containing protease-phosphatase inhibitor cocktail (Cell Signaling). Cell suspensions were rotated at 4C for 15 minutes followed by microcentrifugation to remove cell debris, and protein concentration was quantified using the DC Protein Assay (Bio-Rad). Equal amounts of protein were separated by SDS/PAGE and transferred onto nitrocellulose membranes using the Transblot Turbo Transfer system (Bio-Rad). Membranes were incubated with primary antibody overnight at 4°C, followed by incubation with corresponding seco ndary antibody for 1 hr at room temperature. Blots were developed using SuperSignal West PLUS Chemiluminescent Substrate developing solution (Invitrogen) and imaged using either film or a ChemiDoc XRS+ imager (Bio-Rad). A list of all antibodies used is found in the Key Resources Table. The following antibodies used for immunoblot analysis were purchased from Cell Signaling: AXL (C89E7) (8661S), Phospho-AXL (Tyr702) (D12B2) (5724S), YAP/TAZ (D24E4) (8418S), TAZ (V386) (4883S), L1CAM (D5N9S) (89861S), beta-Actin (8H10D10) (3700S), Phospho-CrkL (Tyr207) (3181L), beta-Tubulin (D2N5G) (15115S), Lamin B1 (D9V6H) (13435S), Rabbit Anti-Mouse IgG (Light Chain Specific) (D3V2A) (58802S); R&D Systems: Phospho-AXL (Tyr779) (AF2228); Millipore Sigma: ABL1 (8E9) (MAB1130), FLAG (M2) (F1804), Phosphotyrosine (4G10) (05-321); Abnova: ABL2 (6D5) (H00000027-M03); Santa Cruz: CRKL (C-20) (sc-319), GAPDH (6C5) (sc-32233), Normal Mouse IgG (sc-2025); Roche Diagnostics: GFP (11814460001); Jackson Immunoresearch: Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) (115-035-003), Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) (115-035-144)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AXL (C89E7) | Cell Signaling | 8661S; RRID:AB_11217435 |
| Phospho-AXL (Tyr702) (D12B2) | Cell Signaling | 5724S; RRID:AB_10544794 |
| Phospho-AXL (Tyr779) | R&D Systems | AF2228; RRID:AB_2062560 |
| ABL1 (8E9) | Millipore Sigma | MAB1130; RRID:AB_2220996 |
| ABL2 (6D5) | Abnova | H00000027-M03; RRID:AB_828506 |
| YAP/TAZ (D24E4) | Cell Signaling | 8418S; RRID:AB_10950494 |
| TAZ (V386) (immunoblotting) | Cell Signaling | 4883S; RRID:AB_1904158 |
| L1CAM (D5N9S) | Cell Signaling | 89861S; RRID:AB_2800145 |
| beta-Actin (8H10D10) | Cell Signaling | 3700S; RRID:AB_2242334 |
| Phospho-CrkL (Tyr207) | Cell Signaling | 3181L; RRID:AB_331068 |
| CRKL (C-20) | Santa Cruz | sc-319; RRID:AB_631320 |
| beta-Tubulin (D2N5G) | Cell Signaling | 15115S; RRID:AB_2798712 |
| FLAG (M2) | Millipore Sigma | F1804; RRID:AB_262044 |
| GFP | Roche Diagnostics | 11814460001; RRID:AB_390913 |
| GAPDH (6C5) | Santa Cruz | sc-32233; RRID:AB_627679 |
| Lamin B1 (D9V6H) | Cell Signaling | 13435S; RRID:AB_2737428 |
| TAZ (M2-616) (immunofluorescence) | BD Biosciences | 560235; RRID:AB_1645338 |
| TAZ (ChIP-qPCR) | Millipore Sigma | HPA007415; RRID:AB_1080602 |
| Alexa Fluor 568 Phalloidin | Thermo Fisher | A12380 |
| Phosphotyrosine (clone 4G10) | Millipore Sigma | 05-321; RRID:AB_309678 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson Immunoresearch | 115-035-003; RRID:AB_10015289 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson Immunoresearch | 115-035-144 |
| Normal Mouse IgG | Santa Cruz | sc-2025; RRID:AB_737182 |
| Rabbit Anti-Mouse IgG (Light Chain Specific) (D3V2A) | Cell Signaling | 58802S; RRID:AB_2799549 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human GAS6 protein, CF | R&D Systems | 885-GSB-050 |
| GNF-5 (Abl kinase small molecule inhibitor) | Duke Small Molecule Synthesis Facility | N/A |
| ABL001 (Asciminib) | Duke Small Molecule Synthesis Facility | N/A |
| BGB324 (R428) | MedKoo Biosciences | 401810 |
| Doxycycline Hyclate ≥98% (for in vivo studies) | Acros Organics | 103516-794 |
| Doxycycline Hyclate (for in vitro studies) | Millipore Sigma | D9891 |
| Critical Commercial Assays | ||
| Cell Titer Glo | Promega | G7571 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S |
| Caspase-GLO 3/7 Assay | Promega | G8091 |
| Corning BioCoat Matrigel Invasion Chambers | Corning | 354480 |
| Deposited Data | ||
| PC9 Parental versus PC9-TAZ4SA RNA-seq | This paper | GEO: GSE130425 |
| PC9 Parental versus PC9-BrM3 RNA-seq | (Boire et al, 2017) | GEO: GSE83132 |
| Experimental Models: Cell Lines | ||
| PC9 | (Valiente et al, 2014) | N/A |
| PC9-BrM3 | This paper | N/A |
| HCC4006 | ATCC | CRL-2871; RRID:CVCL_1269 |
| HCC4006-BrM | This paper | N/A |
| NCI-H1975 | ATCC | CRL-5908; RRID:CVCL_1511 |
| H1975-BrM3 | This paper | N/A |
| HCC827 | ATCC | CRL-2868; RRID:CVC:_2063 |
| Experimental Models: Organisms/Strains | ||
| Mouse outbred athymic nu/nu | Jackson Laboratory | 007850; RRID:IMSR_JAX:007850 |
| Oligonucleotides | ||
| See Table S2 for a list of all primers and oligonucleotides. | ||
| Recombinant DNA & Plasmids | ||
| pLKO-puro Non-Target shRNA Control | Sigma Mission TRC1 | SHC016-1EA |
| pLKO-puro shAXL 1040 | Sigma Mission TRC1 | TRCN0000001040 |
| pLKO-puro shTAZ 70 | Sigma Mission TRC1 | TRCN0000019470 |
| pLKO-puro shTAZ 71 | Sigma Mission TRC1 | TRCN0000019471 |
| pLKO-puro shTAZ 73 | Sigma Mission TRC1 | TRCN0000019473 |
| TetO-FUW-pgk-puro | (Chowdhury et al., 2016) | Addgene #85747; RRID:Addgene_85747 |
| pLVX-Tight-Puro Tet-On Vector | Xaralabos Varelas, Boston University, Boston, MA, USA | N/A |
| pLVX-TP-3F-TAZ4SA | Xaralabos Varelas, Boston University, Boston, MA, USA | N/A |
| Scramble control shRNA | (Gu et al, 2016) | N/A |
| ABL1 shRNA | (Gu et al, 2016) | N/A |
| ABL2 shRNA | (Gu et al, 2016) | N/A |
| 3XFlag pCMV5-TOPO TAZ wild type | (Varelas et al., 2008) | Addgene #24809; RRID:Addgene_24809 |
| 3XFlag pCMV5-TOPO TAZ Y321F | This paper | N/A |
| Tet-pLKO-puro | (Wiederschain et al., 2009) | Addgene #21915; RRID:Addgene_21915 |
| Tet-pLKO-shNTC-puro | This paper | N/A |
| Tet-pLKO-shAXL-puro | This paper | N/A, shAXL clone TRCN0000001040 |
| Tet-pLKO-shTAZ-puro | This paper | N/A, shTAZ clone TRCN0000019470 |
| N174-MCS | http://n2t.net/addgene:81061 | Addgene #81061; RRID:Addgene_81061 |
| phL1A-pcDNA3 | (Hlavin and Lemmon, 1991) | Addgene #12307; RRID:Addgene_12307 |
| N174-L1CAM | This paper | N/A |
| pN1-eGFP | Anthony Koleske, Yale University, New Haven, CT, USA | N/A |
| pN1-ABL2-eGFP WT | Anthony Koleske, Yale University, New Haven, CT, USA | N/A |
| pN1-ABL2-eGFP R198K | This paper | N/A |
| pN1-ABL2-eGFP K317M | This paper | N/A |
| pWZL Neo Myr Flag AXL | (Boehm et al., 2007) | Addgene #20428; RRID:Addgene_20428 |
| pWZL Neo Myr Flag AXL Y779F | This paper | N/A |
| pWZL Neo Myr Flag AXL Y821F | This paper | N/A |
| pWZL Neo Myr Flag AXL Y830F | This paper | N/A |
| pWZL Neo Myr Flag AXL Y866F | This paper | N/A |
| Software and Algorithms | ||
| Prism 6 and 8 | Graphpad | http://graphpad.com/scientific-software/prism |
| ImageJ | (Schneider et al., 2012) | http://imagej.nih.gov |
| RStudio | R Foundation for Statistical Computing | http://rstudio.com |
| CBLigand Online BBB Predictor Tool v0.90 | (Liu et al., 2014) | http://cbligand.org |
| Living Image | Perkin Elmer | http://perkinelmer.com |
| GSEA 3.0 | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea |
| KM Plotter | (Gyorffy et al., 2013) | http://kmplot.com |
| CBioPortal v3.1.1 | (Cerami et al., 2012; Gao et al., 2013) | http://cbioportal.org |
| TrimGalore v0.4.4 | Babraham Bioinformatics | http://github.com/FelixKrueger/TrimGalore |
| STAR aligner | (Dobin et al., 2013) | http://github.com/alexdobin/STAR |
| HTSeq-count | (Anders et al., 2015) | http://htseq.readthedocs.io |
| DESeq2 v2.12 | (Love et al., 2014) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Hisat2 v2.1.0 | (Kim et al., 2019) | http://ccb.jhu.edu/software/hisat2/index.shtml |
Co-immunoprecipitation assays.
Cells were lysed with immunoprecipitation buffer (50 μM Tris-HCl pH 8.0, 150 mM NaCl, 1.0% NP-40, 1x protease inhibitor cocktail). Lysates were incubated for 30 min on ice and then cleared by spinning at 15,000 rpm for 10 min. Supernatants were incubated with antibodies overnight followed by incubation with protein A/G PLUS-agarose beads (Santa Cruz) for 3 h. Beads were washed three times with immunoprecipitation buffer afterwards. Bound proteins were eluted with 4x Laemmli Sample Buffer (BioRad) and visualized by SDS-PAGE on nitrocellulose membranes followed by immunoblot analysis. A portion of each protein lysate input was run concurrently to measure total expression for each protein. For experiments examining the effect of ligand-induced activation of the AXL receptor tyrosine kinase, recombinant human GAS6 (R&D systems) was added to serum-starved cells at 500 ng/mL for 1 h and as described in figure legends. Activation of the AXL receptor by GAS6 treatment was confirmed by immunoblot analysis using the Phospho-AXL Tyr702 antibody (Cell Signaling).
Nuclear/Cytosolic subcellular fractionation.
Fractionation of cells was performed following the REAP cell fractionation method as previously described (Suzuki et al., 2010). Briefly, cells plated in 10cm dishes were harvested in ice-cold PBS followed by a 10 second spin in a table-top micro-centrifuge. The supernatant was removed and cells were lysed and triturated in ice-cold 0.1% NP40 in PBS. A portion of the lysate was used for the whole cell lysate, and the remaining lysate was spun again for 10 seconds. A portion of the supernatant was then removed and designated as the cytosolic fraction. The remaining supernatant containing pelleted nuclei was washed and resuspended in NP40 lysis buffer followed by an additional 10 second-spin to re-pellet the nuclei. The supernatant was removed and nuclei were resuspended in 1X Laemmli sample buffer and designated as the nuclear fraction. Whole cell lysate and nuclear samples were sonicated twice for 5 seconds each on ice using a microprobe at level 2, followed by boiling in 1X Laemmli sample buffer for 1 min prior to immunoblot analysis. Antibodies against Lamin B1 (Cell Signaling) and GAPDH (Santa Cruz) were used for nuclear and cytoplasmic markers, respectively.
Kaplan-Meier Analysis and Mutual Exclusivity Analysis.
Lung adenocarcinoma patient microarray data were analyzed using the KMplot analysis tool (kmplot.com) (Gyorffy et al., 2013). Affymetrix identifiers for each gene were as follows: 202123_s_at (ABL1), 206411_s_at (ABL2), 202685_s_at (AXL), 204585_s_at (L1CAM), 202134_s_at (WWTR1), and 213342_at (YAP1). For all genes tested, patients were stratified into high (upper quartile) versus low (bottom three quartiles) gene expression. Mutual exclusivity analysis was performed with the CBioPortal online tool (Cerami et al., 2012; Gao et al., 2013) using the TCGA Provisional Lung Adenocarcinoma dataset. A user-defined list consisting of AXL, ABL1, ABL2, WWTR1 and YAP1 genes was filtered to include patients with corresponding mRNA expression (RNAseq, z-score threshold ± 2.0) and, if available, protein expression data (RPPA, z-score threshold ± 2.0) (n= 506). Mutation status and copy number alteration status were excluded from the analysis parameters. For survival analysis of the TCGA Provisional Lung Adenocarcinoma dataset, a user-defined gene list of AXL, ABL2, and WWTR1 was used under the same exclusion parameters as for the mutual exclusivity analysis.
Immunofluorescence and confocal microscopy.
To evaluate TAZ nuclear localization across cell lines by immunofluorescence (IF) staining and imaging, 3x104 lung cancer cells were plated on 18 mm glass coverslips in 12-well dishes for 72 h. For PC9-TAZ4SA conditions, 500 ng/mL dox was added to the culture media at the time of cell seeding. At endpoint, cells were fixed in 4% ice-cold PFA and blocked in 3% BSA in PBS (w/v) prior to staining with mouse anti-TAZ antibody (BD Biosciences). To visualize nuclei, fixed cells were stained with Hoechst 33342 diluted 1:10,000 in PBS. Outer cell boundaries were visualized by phalloidin staining using an Alexa Fluor 568 Phalloidin probe (Thermo Fisher). Cells were imaged using a Leica Inverted SP5 confocal microscope, followed by analysis in ImageJ software (Schneider et al., 2012). The nuclear/cytoplasmic ratio for each cell was calculated by quantifying the integrated density of stain overlapping with the nucleus followed by division of the integrated density of the stain corresponding to the cytoplasmic area of the cell. Six independent replicate wells were evaluated for each condition. A minimum of five cells were analyzed under blinded conditions from each of three non-overlapping images from each replicate sample (>90 cells analyzed per condition) which were then averaged to give a mean nuclear/cytoplasmic ratio per replicate (n=6).
Computational modeling.
For predictive modeling of blood-brain barrier (BBB) permeability, SDF files for each molecular compound were obtained from PubChem and loaded into the CBLigand Online BBB Predictor tool v0.90 (cbligand.org) (Liu et al., 2014). Each molecule was tested under both AdaBoost and SVM algorithms combined with either MACCS, Openbabel(FP2), Molprint 2D, or PubChem fingerprints individually.
ChIP-qPCR analysis.
PC9-BrM3 cells were grown in 10 cm dishes to 80% confluence, at which point cells were cross-linked with formaldehyde (1% final concentration) on ice for 5 min with gentle shaking. Formaldehyde was quenched by adding 2.5 M glycine in PBS (125 mM final concentration) for 5 min on ice with gentle shaking. Cells were then lysed in cell lysis buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS) containing protease and phosphatase inhibitors and transferred to a 15 mL conical tube for 15 min on ice. 1 mL of IP buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) was added, and lysates were then sonicated twice for 30 seconds each on ice using a microprobe on setting 5. Lysates were cleared by spinning down cellular debris at 13,000 RPM at 4°C. Samples containing antibody against TAZ (Millipore Sigma) or IgG control were rotated overnight at 4°C, after which Dynab eads Protein G were added to all samples and rotated at 4°C for 4 h. Beads were washed twice with IP buffer, twice with IP buffer + 0.5 M NaCl, and twice with TE Buffer (10 mM Tris, pH 7.5, 1 mM EDTA). Proteins were then eluted by adding TE buffer + 1% SDS and incubating at 65°C for 10 min, and beads were spun down at 14, 000 rpm for 10 min. Samples were then incubated overnight at 65°C to reverse cross-links, followed by treatment with proteinase K for 1.5 h at 37°C and purificatio n with the Qiagen PCR cleanup kit prior to analysis via qPCR. Primer sequences used for the ABL2 promoter region were: Forward 5’-GGCTGGGAGGGAGAGACC-3’, Reverse 5’-ATTGAAGCCGGTCTCTGTGG-3’. Negative control primer sequences corresponding to an untranslated region (UTR) downstream of the ABL2 coding sequence were: Forward 5’-GAGAAACCACAGAGCACCCA-3’, Reverse 5’-GGCGCCCAGGGTGATTTTTA-3’.
DNA plasmids.
The pLVX-Tet-On vector and pLVX-TP-3F-TAZ4SA (S66A, S89A, S117A and S311A) plasmids were provided by Dr. Xaralabos Varelas (Boston University, Boston, MA, USA). Sequences for shRNAs targeting the ABL kinases were as follows: scrambled shRNA (GGTGTATGGGCTACTATAGAA); ABL1 shRNA (GGTGTATGAGCTGCTAGAGAA); ABL2 shRNA (CCTTATCTCACCCACTCTGAA). Stable noninducible shRNAs against non-target control (NTC), TAZ and AXL in the pLKO.1 vector were from the Sigma Mission TRC1 Lentiviral shRNA library and were obtained through the Duke Functional Genomics Shared Resource Facility. Sequences and Sigma clone identifiers for each of these shRNAs were as follows: AXL shRNA (CGAAATCCTCTATGTCAACAT, cloneID TRCN0000001040); TAZ shRNA #70 (CCAGGAACAAACGTTGACTTA, cloneID TRCN0000019470); TAZ shRNA #71 (CAGCCAAATCTCGTGATGAAT, cloneID TRCN0000019471); TAZ shRNA #73 (CCTGCCGGAGTCTTTCTTTAA, cloneID TRCN0000019473). The constitutive active ABL2PP construct, described previously, was cloned into TetO-FUW-pgk-puro by restriction enzyme digest. The TetO-FUW-pgk-puro plasmid was a gift from Emily Dykhuizen (Addgene plasmid # 85747) (Chowdhury et al., 2016).
Cloning of shRNA or expression constructs into lentiviral vectors.
Dox-inducible shRNA sequences corresponding to AXL (TRCN0000001040) or TAZ (TRCN0000019470) were cloned into the all-in-one dox-inducible Tet-pLKO-puro vector kindly provided by Dmitri Wiederschain (Addgene plasmid #21915) (Wiederschain et al., 2009). Briefly, sense and anti-sense oligos for respective shRNA sequences flanked by 5’ Agel or 3’ EcoRI restriction site overhangs were mixed in 10X annealing buffer (1M NaCl, 100 mM Tris-HCl, pH=7.4) and annealed by placing in boiling water that was allowed to cool naturally to 30°0 on a lab b ench. Tet-pLKO-puro vector backbone was digested via restriction enzyme digest using Agel and EcoRl (NEB), followed by gel-purification using the QlAquick Gel Extraction Kit (Qiagen). Gel-purified vector and annealed oligos were then ligated using T4 DNA ligase (NEB) followed by transformation into One Shot Stbl3 chemically competent cells (ThermoFisher). For stable expression of L1CAM cDNA in L1CAM rescue experiments, L1CAM cDNA from the phL1A-pcDNA3 vector was cut by EcoRl restriction digest and cloned into the lentiviral N174-MCS vector. The phL1A-pcDNA3 plasmid was a gift from Vance Lemmon (Addgene plasmid #12307) (Hlavin and Lemmon, 1991), and the N174-MCS plasmid was a gift from Adam Karpf (Addgene plasmid #81061). For all cloning, plasmid DNA extracted from transformed colonies was validated by sequencing prior to transfection and lentiviral transduction into target cells.
Mutagenesis and transient transfection.
The 3XFlag pCMV5-TOPO-TAZ wild type (WT) plasmid was a gift from Jeff Wrana (Addgene plasmid # 24809) (Varelas et al., 2008). pWZL-Neo-Myr-Flag-AXL was a gift from William Hahn & Jean Zhao (Addgene plasmid # 20428) (Boehm et al., 2007). pN1-ABL2-eGFP (also known as pN1-ARG-eGFP) was a gift from Anthony Koleske (Yale University, New Haven, CT, USA). Mutants for 3XFlag pCMV5-TOPO-TAZ, pWZL-Neo-Myr-Flag-AXL, and pN1-ABL2-eGFP were generated by site directed mutagenesis using the Q5 Site-Directed Mutagenesis Kit (NEB) according to the manufacturer’s instructions. The primers used were as follows: TAZ Y321F FWD 5-TTGTGGGGACACTGAAGCACCCTAACCCC-3’, REV 5′-GGGGTTAGGTGCTTCAGTGTCCCCACAA-3’; AXL Y779F FWD 5’- ACATCAAGGCAAACAGTCCATCCAGACAGTCC-3′; REV 5’- GGACTGTCTGGATGGACTGTTTGCCTTGATGTC-3’, AXL Y821F FWD 5’- ATCCATGTTGACAAAGAGGATTTCGTCAGGCTCC-3’; REV5’ GGAGCCTGACGAAATCCTCTTTGTCAACATGGAT-3’, AXL Y830F FWD 5’- GGGGTTCAGGAAAACCTCCACCCTCATCC −3’; REV 5’- GGATGAGGGTGGAGGTTTTCCTGAACCCC -3’, AXL Y866F FWD 5’- CATCCTGCTGGACGCTTTGTCCTCTGCC-3′; REV 5’- GGCAGAGGACAAAGCGTCCAGCAGGATG -3′, ABL2 R198K 5’- GCTGCTCTCACTCTCTTTGACCAGGAAGCTGCCG-3′; REV 5′- GCGGCAGCTTCCTGGTCAAAGAGAGTGAGAGCAGC-3’, ABL2 K317M 5’-CTTCCTTCAGTGTCATCACAGCCACTGTAAGGCTGTACTT-3′; REV 5′- AAGTACAGCCTTACAGTGGCTGTGATGACACTGAAGGAAG-3’. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instruction. Cells were transfected with 1 μg of DNA.
Viral transduction procedures.
Lentiviral shRNAs against the ABL kinases (ABL1, ABL2 and AA double knockdown), N174-EV, N174-L1CAM, pLVX-Tet-On, and pLVX-TP-3F-TAZ4SA were packaged in 3rd generation lentiviral packaging vectors (pMDL, pCMV-VSV-G, pRSV-REV). For lentiviral expression of the TetO-FUW-ABL2PP plasmid and for shRNAs in the pLKO.1-puro backbone against non-target control (NTC), WWTR1 (TAZ) and AXL, 2nd generation lentiviral packaging vectors (pMD2.G and PSPAX2) were used. Briefly, 293T cells were transfected with packaging DNAs and corresponding DNAs using FuGENE6 reagent (Promega). Culture supernatants containing virus were harvested and filtered 24 hours and 48 hours after transfection and were added to NSCLC cell cultures in the presence of 8 μg/ml polybrene (Sigma-Aldrich) for a minimum of 6 h. Stably-transduced cells expressing N174-EV, N174-L1CAM, or pLVX-Tet-On vector were selected with G418 (100 ug/mL) for five days. Stably-transduced cells expressing TetO-FUW-ABL2PP, Tet-TAZ4SA or shRNAs cloned in the pLKO.1 backbone were selected with puromycin (1 μg/ml) for five days.
Cell viability assay.
Cells transduced with stable lentiviral shRNAs were seeded in 96-well plates in at least triplicate and measured each day using CellTiter-Glo reagent (Promega). Plates were read on a Tecan Infinite M1000 Microplate Reader and results were analyzed in GraphPad.
Caspase-GLO 3/7 assay.
PC9-BrM3 cells transduced with the indicated dox-inducible lentiviral shRNAs as indicated in figures and figures legends or PC9 parental cells lentivirally transduced with Tet-TAZ4SA expression vector were treated with 500 ng/mL dox for 0, 24, 48, 72 or 96 h prior to lysis in Caspase-GLO reagent according to manufacturer instructions (Promega). Each condition was run in at least duplicate wells each from two independent experiments, and dox and fresh media were replaced every 24 h prior to assay. PC9-BrM3 cells treated with 1 uM staurosporine for 24 h were used as a positive control. Plates were read on a Tecan Infinite M1000 Microplate Reader and results were analyzed in GraphPad.
Anoikis assay.
PC9-BrM3 cells transduced with the indicated dox-inducible lentiviral shRNAs as described in figures and figure legends or PC9 parental cells lentivirally transduced with Tet-TAZ4SA expression vector were seeded in ultra-low attachment 96-well plates (n=5 replicates per condition) and treated +/− 500 ng/mL dox for 72 h prior to cell lysis with CellTiter-GLO reagent. Plates were read on a Tecan Infinite M1000 Microplate Reader and results were analyzed in Graphpad.
3D Matrigel Invasion Assay.
PC9-BrM3 cells were transduced with dox-inducible shRNAs against either non-target control (Tet-shNTC), AXL (Tet-shAXL), or TAZ (Tet-shTAZ) as well as stable expression through lentiviral transduction of empty vector control or L1CAM cDNA (expressed in N174-MCS vector backbone). 72 h prior to seeding in Matrigel transwell invasion chambers (Corning), cells were treated with 500 ng/mL dox to induce shRNA expression. On the day of the assay, transwell filters were equilibrated for 2 h at 37°C with serum-free media prior to seeding 1x105 cells in 500 uL serum-free media containing 500 ng/mL dox (upper chamber). Complete culture media containing serum was added to the lower chamber to serve as a chemoattractant, and cells were allowed to invade for 24 h prior to fixation and staining with 0.5% crystal violet solution (0.5% w/v crystal violet, 25% v/v methanol, 75% v/v dH2O). The total number of invaded cells from eight non-overlapping brightfield images (10X) per condition from three independent experiments were counted and the means from each condition were analyzed in GraphPad (n=3).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 6 and GraphPad Prism 8 software. Mouse numbers per group were determined through statistical power calculations (α=0.05) where 10 mice per group allows for 90% power to detect inter-group differences of 50% and assuming intra-group variability of 25%. For Kaplan-Meier survival analysis, p values were calculated using log-rank (Mantel-Cox) testing. Statistical comparisons of 2 groups were conducted using Student’s t tests (unpaired, two-tailed). For comparisons involving more than two groups, data was evaluated by ANOVA followed by Fisher post-hoc testing or as described in figure legends. For comparisons between groups of unequal size, the mean value was used to allow for statistical analysis by ANOVA. For all tests, we considered a p value less than 0.05 as statistically significant. Data shown represent averages ± SEM unless otherwise indicated in figure legends.
DATA AND CODE AVAILABILITY
RNA sequencing raw data has been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE130425.
Supplementary Material
Highlights.
TAZ drives lung cancer brain metastases via transcription of ABL2, AXL and L1CAM
AXL, ABL2 and TAZ engage in an autocrine feed-forward loop during brain metastasis
Knockdown of AXL or ABL2 kinases decreases lung adenocarcinoma brain metastases
Allosteric ABL kinase inhibitor ABL001 crosses the BBB and impairs brain metastases
Significance.
Lung cancer is the leading cause of mortality among cancers worldwide with up to 50% of these patients progressing to brain metastasis. The majority of lung cancer patients with brain metastases exhibit cognitive impairment, seizures and neurological deficits. Currently, there are no effective therapies to treat brain metastases, in part due to the lack of actionable targets. Here we identify a targetable autocrine signaling axis required for lung adenocarcinoma brain metastasis driven by activation of the TAZ transcriptional co-activator which functions both downstream and upstream of the ABL2 and AXL protein tyrosine kinases. Depletion of ABL2 or AXL in lung cancer cells or treatment with specific kinase inhibitors markedly decreases brain metastasis and prolongs survival in mouse models. These findings suggest the use of available ABL and AXL kinase inhibitors for treating lung cancer brain metastases.
Acknowledgments
We thank Dr. Joan Massague (Memorial Sloan Kettering, New York, NY) for providing the PC9 parental cells, and Dr. Xaralabos Varelas (Boston University, Boston, MA) and Dr. Anthony Koleske (Yale University, New Haven, CT) for providing DNA plasmids. We thank the Duke University Genome Sequencing facility and the Duke Genomic Analysis and Bioinformatics core resource for providing assistance with RNA-sequencing data acquisition and analysis, the Duke Flow Cytometry Shared Resource for assistance with cell sorting, and the Duke Light Microscopy Core Facility. We thank Jill Hattaway for critical reading of the manuscript. This work was supported by National Institute of Health NCI grants R01CA195549 (A.M.P.), F31CA22496001 (J.P.H.), F99CA245732-01 (J.P.H.), 5T32GM007105-44 (J.P.H. and B.M.), the Lung Cancer Research Foundation-Free to Breathe Metastasis Research Grant (A.M.P.), the Emerson Collective, and the Duke SPORE in Brain Cancer grant (P50CA190991).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
A.M. Pendergast is a consultant/advisory board member for The Pew Charitable Trusts. A.M. Pendergast holds patent No.: US 9,931,342 B2 related to this work. The other authors declare no competing interests.
References
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, and Sawaya RE (2004). Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J Clin Oncol 22, 2865–2872. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL et al. (2007). Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129, 1065–1079. [DOI] [PubMed] [Google Scholar]
- Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, and Massague J (2017). Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 168, 1101–1113 e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, Bartram CR, and Janssen JW (1997). Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 14, 2619–2631. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson Ev et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu Kv et al. (2016). Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury B, Porter EG, Stewart JC, Ferreira CR, Schipma MJ, and Dykhuizen EC (2016). PBRM1 Regulates the Expression of Genes Involved in Metabolism and Cell Adhesion in Renal Clear Cell Carcinoma. PLoS One 11, e0153718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Okram B, Ding Q, Zhang J, Choi Y, Adrian FJ, Wojciechowski A, Zhang G, Che J, Bursulaya Bv et al. (2010). Expanding the diversity of allosteric bcr-abl inhibitors. J Med Chem 53, 6934–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, Foster C, Jenkins RP, and Sahai E (2018). Quantitative Analysis Reveals that Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst 6, 692–708 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, Griscom B, Rosenblum M, Boire A, Brogi E et al. (2018). Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol 20, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecci PE, Champion CD, Hoj J, McKernan CM, Goodwin CR, Kirkpatrick JP, Anders CK, Pendergast AM, and Sampson JH (2019). The Evolving Modern Management of Brain Metastasis. Clin Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, Lubrano S, Kishore A, Pachter JA, Konig GM et al. (2019). A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 35, 457–472 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Liu P, Zhou X, Li MT, Li FL, Wang Z, Meng Z, Sun YP, Yu Y, Xiong Yv et al. (2016). Thromboxane A2 Activates YAP/TAZ Protein to Induce Vascular Smooth Muscle Cell Proliferation and Migration. J Biol Chem 291, 18947–18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson Ev et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, Johnson RM, Carmeliet P, Basik M, Gaboury Lv et al. (2018). The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep 23, 1476–1490. [DOI] [PubMed] [Google Scholar]
- Graham DK, DeRyckere D, Davies KD, and Earp HS (2014). The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer 14, 769–785. [DOI] [PubMed] [Google Scholar]
- Gu JJ, Rouse C, Xu X, Wang J, Onaitis MW, and Pendergast AM (2016). Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI insight 1, e89647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, and Lanczky A (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavin ML, and Lemmon V (1991). Molecular structure and functional testing of human L1CAM: an interspecies comparison. Genomics 11, 416–423. [DOI] [PubMed] [Google Scholar]
- Jang EJ, Jeong H, Han KH, Kwon HM, Hong JH, and Hwang ES (2012). TAZ suppresses NFAT5 activity through tyrosine phosphorylation. Molecular and cellular biology 32, 4925–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedan A, Verma N, Saroha A, Shreberk-Shaked M, Muller AK, Nair NU, and Lev S (2018). PYK2 negatively regulates the Hippo pathway in TNBC by stabilizing TAZ protein. Cell Death Dis 9, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WJ, Shah NJ, and Subramaniam DS (2018). Management of Brain Metastases in Epidermal Growth Factor Receptor Mutant Non-Small-Cell Lung Cancer. Front Oncol 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri A, Gu JJ, McKernan CM, Xu X, and Pendergast AM (2019). ABL kinase inhibition sensitizes primary lung adenocarcinomas to chemotherapy by promoting tumor cell differentiation. Oncotarget 10, 1874–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, and Winkler F (2010). Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16, 116–122. [DOI] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Yang SJ, Hwang D, Song J, Kim M, Kyum Kim S, Kang K, Ahn J, Lee D, Kim M. Yv et al. (2015). A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat Commun 6, 10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler M, Speight P, Little D, Di Ciano-Oliveira C, Szaszi K, and Kapus A (2018). Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat Commun 9, 4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, and Guan KL (2008). TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Molecular and cellular biology 28, 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Yeckel MF, and Koleske AJ (2013). Abl2/Arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J Neurosci 33, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, Li T, Chan SW, Lim CJ, Hong Wv et al. (2010). The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem 285, 37159–37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang L, Lv M, Pei R, Li P, Pei Z, Wang Y, Su W, and Xie XQ (2014). AlzPlatform: an Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J Chem Inf Model 54, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AS, Miller MA, Gertler FB, and Lauffenburger DA (2013). The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Science signaling 6, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AS, Zweemer AJ, and Lauffenburger DA (2015). The AXL Receptor is a Sensor of Ligand Spatial Heterogeneity. Cell Syst 1, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, and Guan KL (2015). The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba K, Shien K, Takahashi Y, Torigoe H, Sato H, Yoshioka T, Takeda T, Kurihara E, Ogoshi Y, Yamamoto Hv et al. (2019). Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol Cancer Res 17, 499–507. [DOI] [PubMed] [Google Scholar]
- Nayak L, Lee EQ, and Wen PY (2012). Epidemiology of brain metastases. Curr Oncol Rep 14, 48–54. [DOI] [PubMed] [Google Scholar]
- Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, Li H, Hambrecht AC, Roberts E, and Jandial R (2014). Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A 111, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, et al. (2018). Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol 4, 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, Doglio L, Martinez L, Martinez-Saez E, Ramon YCS, et al. (2018). STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 24, 1024–1035. [DOI] [PubMed] [Google Scholar]
- Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A, Lindgren D et al. (2014). Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A 111, 13373–13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, et al. (2012). beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 151, 1457–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer MK, and Altevogt P (2010). L1CAM malfunction in the nervous system and human carcinomas. Cell Mol Life Sci 67, 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten LJ, Rutten J, Huveneers HA, and Twijnstra A (2002). Incidence of brain metastases in a cohort of patients with carcinoma of the breast, colon, kidney, and lung and melanoma. Cancer 94, 2698–2705. [DOI] [PubMed] [Google Scholar]
- Schuette W (2004). Treatment of brain metastases from lung cancer: chemotherapy. Lung Cancer 45 Suppl 2, S253–257. [DOI] [PubMed] [Google Scholar]
- Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, et al. (2014). Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 16, 876–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanzer M, Adler J, Ricardo-Lax I, Reuven N, and Shaul Y (2017). The nonreceptor tyrosine kinase c-Src attenuates SCF(beta-TrCP) E3-ligase activity abrogating Taz proteasomal degradation. Proc Natl Acad Sci U S A 114, 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DY, Na II, Kim CH, Park S, Baek H, and Yang SH (2014). EGFR mutation and brain metastasis in pulmonary adenocarcinomas. J Thorac Oncol 9, 195–199. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, and et al. (1993). SH2 domains recognize specific phosphopeptide sequences. Cell 72, 767–778. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, and Riabowol K (2010). REAP: A two minute cell fractionation method. BMC Res Notes 3, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Ahluwalia MS, Boire A, Brastianos PK, Goldberg SB, Lee EQ, Le Rhun E, Preusser M, Winkler F, and Soffietti R (2018). The Evolving Landscape of Brain Metastasis. Trends Cancer 4, 176–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, Chaft JE, Kris MG, Huse JT, Brogi E, and Massague J (2014). Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X (2014). The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141, 1614–1626. [DOI] [PubMed] [Google Scholar]
- Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW, and Wrana JL (2008). TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol 10, 837–848. [DOI] [PubMed] [Google Scholar]
- Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, Hunt RW, Trail G, Clogston C, Toso RJ, and et al. (1995). Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 373, 623–626. [DOI] [PubMed] [Google Scholar]
- Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer Cv et al. (2009). Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504. [DOI] [PubMed] [Google Scholar]
- Wingrove E, Liu ZZ, Patel KD, Arnal-Estape A, Cai WL, Melnick MA, Politi K, Monteiro C, Zhu L, Valiente M, et al. (2019). Transcriptomic Hallmarks of Tumor Plasticity and Stromal Interactions in Brain Metastasis. Cell Rep 27, 1277–1292 e1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Ma W, Zhou Q, Yan H, Lim ZF, Huang M, Deng C, Yu X, Su H, Komo S, et al. (2017). AXL-GAS6 expression can predict for adverse prognosis in non-small cell lung cancer with brain metastases. J Cancer Res Clin Oncol 143, 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu Wv et al. (2017). The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015). Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2019). YAP and TAZ: a signalling hub of the tumour microenvironment. Nat Rev Cancer 19, 454–464. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, McCabe BD, Galvan JA, Robinson HPC, Zlobec I, et al. (2019). Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, Xiong Y, Lei QY, and Guan KL (2009). TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem 284, 13355–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F et al. (2010). Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho J. Kv et al. (2012). Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 44, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing raw data has been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE130425.