

Abstract
The past decade has seen the emergence of immunotherapy as a prime approach to cancer treatment, revolutionizing the management of many types of cancer. Despite the promise of immunotherapy, most patients do not have a response or become resistant to treatment. Thus, identifying combinations that potentiate current immunotherapeutic approaches will be crucial. The combination of immune-checkpoint inhibition with epigenetic therapy is one such strategy that is being tested in clinical trials, encompassing a variety of cancer types. Studies have revealed key roles of epigenetic processes in regulating immune cell function and mediating antitumour immunity. These interactions make combined epigenetic therapy and immunotherapy an attractive approach to circumvent the limitations of immunotherapy alone. In this Review, we highlight the basic dynamic mechanisms underlying the synergy between immunotherapy and epigenetic therapies and detail current efforts to translate this knowledge into clinical benefit for patients.
Immune-checkpoint inhibition was introduced as a novel clinical paradigm of cancer therapy in March 2011 with the FDA approval of the anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) antibody ipilimumab for the treatment of advanced-stage melanoma. This treatment paradigm was further established upon the approval, in late 2014, of the anti-programmed cell death 1 (PD-1) antibodies nivolumab and pembrolizumab for the same indication. Since then, inhibitors targeting the CTLA-4 and PD-1 immune checkpoints have revolutionized the management not only of melanoma but also of non-small-cell lung carcinoma (NSCLC), renal cell carcinoma (RCC) and Hodgkin lymphoma, among other malignancies, as evidenced by improved survival outcomes in these patient populations1–15. Notably, the possibilities of immunotherapy as a cancer management strategy have long been recognized and pursued16. We are now in an age of renaissance of immunotherapy and immune-checkpoint inhibition is but one promising approach that has emerged; detailing aspects of the many other novel potentially efficacious immunotherapeutic strategies that are currently being explored is outside the scope of this Review, although examples include chimeric antigen receptor T cell therapy, vaccine-based approaches and natural killer (NK) cell-directed treatments17–24. Despite the early excitement regarding the promise of immune-checkpoint inhibitors (ICI), the majority of patients with cancer fail to derive clinical benefit from or ultimately develop resistance to such treatment25–28. Moreover, response rates vary between cancer types and are typically highest in patients with melanoma, urothelial cancer, NSCLC and colorectal cancers with microsatellite instability29; certain cancers, such as those of the pancreas, breast or ovaries, seem to be intrinsically resistant to ICI29–32, although patients with advanced-stage, programmed cell death 1 ligand 1 (PD-L1)-positive, triple-negative breast cancer have been shown to benefit from the addition of anti-PD-L1 antibodies to chemotherapy33. The variability in responsiveness to immune-checkpoint inhibition among cancer types has been attributed to several factors, including tumour mutational burden (TMB), immune phenotype of the tumour microenvironment (TME) and mechanisms of tumour immune evasion. Thus, the development of combinatorial strategies with ICI is needed to maximize clinical benefit, with several approaches being tested in clinical trials. These include dual ICI (for example, pairing anti-CTLA-4 and anti-PD-1 antibodies) as well as immunotherapy combined with chemotherapy, radiotherapy or epigenetic therapy. Indeed, dual immune-checkpoint inhibition with ipilimumab plus nivolumab is the most established combinatorial approach; this combination has been reported to improve progression-free survival (PFS) outcomes in patients with advanced-stage RCC and metastatic melanoma, compared with those associated with sunitinib and ipilimumab monotherapy, respectively, and is approved for the first-line treatment of these cancers34,35. The addition of pembrolizumab to chemotherapy has been shown to increase both PFS and overall survival (OS) in phase III trials involving patients with advanced-stage NSCLC, leading to FDA approval of this approach in the frontline setting36,37. The pairing of radiotherapy with ICI is currently being tested in a variety of settings across a range of solid tumour types (NCT02239900, NCT03700905, NCT03867175 and NCT03693014). Notably, patients with NSCLC receiving consolidation therapy with durvalumab (an anti-PD-L1 antibody) after chemoradiotherapy had a longer median PFS duration than those in a placebo group38. Beyond NSCLC, case reports describing the potential benefit of combined radiotherapy plus ICI have been published across a variety of solid cancers39–41.
In addition to the aforementioned combination regimens, the application of epigenetic therapy plus ICI is an emerging paradigm and an area of active clinical investigation (Supplementary Table S1). In this Review, we highlight the ‘roles’ of epigenetic regulation in both tumour and immune cell populations and the implications of epigenetic drugs in the perturbation of these compartments. We also summarize the current state of preclinical and clinical development of epigenetic-immunotherapy.
Overview of the ICI paradigm
Principles of ICI
The advent of ICI is the product of many years of basic science research seeking to discern why anticancer immunotherapy was not reaching the promise suggested for over a century since the original seminal insights provided by William B. Coley16. This breakthrough was made possible through an increased understanding of immune tolerance of cancer and centres upon targeting checkpoints in T cell priming and activation42–44, a concept that earned Allison and Honjo the 2018 Nobel Prize in Physiology or Medicine. Chronic interactions between tumour cells and subsets of immune cells induce this tolerance by rendering cytolytic CD8+ tumour-infiltrating lymphocytes ineffective in mounting antitumour responses45–49. The basic constituents of this mechanism, which is defined as immune checkpoint activation, are interactions between receptors on T cells, most notably PD-1 and CTLA-4, and their respective ligands, PD-L1 and CD80 or CD86, present on tumour cells42,50,51 and/or antigen-presenting cells52–54. Thus, the rationale for immune-checkpoint inhibition is the treatment with antibodies targeting PD-1, PD-L1 or CTLA-4 in order to reverse this inhibitory checkpoint action and facilitate antitumour effects1,51. In addition to the aforementioned checkpoints, a number of other immune checkpoint pathways have been identified and studies are ongoing to determine the feasibility of the component receptors and ligands as therapeutic targets. These targets include lymphocyte activation gene 3 protein (LAG3), T cell immunoglobulin mucin receptor 3 (TIM3; also known as HAVCR-2), B and T lymphocyte attenuator (BTLA), NK cell receptor 2B4, T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-type immunoglobulin domain-containing suppressor of T cell activation (VISTA), CD96 (also known as T cell surface protein tactile) and CD112 receptor (CD112R; also known as PVRIG), all of which are negative regulators of T cell activation55. In addition, targeting of co-stimulatory immune checkpoint proteins, such as the tumour necrosis factor receptor superfamily members 4–1BB (CD137), CD40, 0X40 and GITR, is another focus of immunotherapy drug development55.
Prerequisite for a response to ICI
A clinical response to CTLA-4 and/or PD-1 or PD-L1 ICI is dependent on the immune status of the tumour in the following ways. First, antigen-specific CD8+ lymphocytes must be present within the TME56–58. Second, the composition of resident immune cell populations must be polarized towards an immunopermissive state59–62. Third, tumours must be functionally competent for MHC class I-mediated antigen presentation to be receptive to immune attack and be dependent upon the PD-1-PD-L1 axis as the dominant mechanism of immune escape (reflected in the requirement for tumoural PD-L1 expression as a criteria for treatment with PD-1 or PD-L1 inhibitors, in some approved indications)63. A state of immune evasion arises if tumours lack these characteristics64–66, which enables a cancer to live under the ‘radar’ of immune detection. This evasive state characterizes what has been termed immune ‘cold’ tumours versus immune ‘hot’ tumours, which exhibit the defining characteristics detailed above58,67,68 (BOX 1; FIG. 1). Immune hot tumours often have higher mutational burdens than immune cold tumours and, relatedly, a greater number of neoantigens, which correlates with higher objective response rates to ICI across several common cancer types69. The association between TMB and responsiveness to ICI is well established, although the implications with regard to T cell behaviours, such as tumour infiltration, are nebulous and perhaps context dependent. Analyses of The Cancer Genome Atlas melanoma specimens revealed a lack of correlation between a T cell inflammation gene-expression signature and nonsynonymous somatic mutation burden70. By contrast, findings in RCC samples demonstrated a positive correlation between TMB and a T cell inflamed transcriptional signature71.
Box 1 |. Types of tumour immune microenvironments.
The past decade has seen the emergence of immunotherapy as one of the most promising treatment strategies for advanced-stage cancers. The ability of tumours to adapt in order to overcome innate and acquired immune mechanisms that would normally lead to recognition and killing of the tumour cells is a crucial aspect of cancer initiation and progression47,223. Four postulated states of immune landscapes have been observed in tumours and dictate the vulnerability of the tumours to different immunotherapeutic strategies224,225. This categorization is largely based on the expression of programmed cell death 1 ligand 1 (PD-L1) in the tumour microenvironment (TME) and the occurrence and distribution of CD8+ tumour-infiltrating lymphocytes (TILs) at the tumour site224.
Type I (adaptive immune resistance)
In the presence of abundant T cell infiltrates, tumours can develop adaptive immune-resistance mechanisms that often involve upregulation of PD-L1 (REFS44,226–229). Accordingly, these tumours are generally referred to as ‘hot tumours’ and are identified by the presence of CD8+ TILs along with expression of PD-L1 in the TME227. The expression of PD-L1 is a feedback response to IFNγ secreted by TILs that, via triggering of the T cell inhibitory receptor programmed cell death 1 (PD-1), diminishes the potential of those TILs to mount an antitumour response227. This TME is that most poised for clinical benefit from single-agent immune-checkpoint inhibition with anti-PD-1 or anti-PD-L1 antibodies, because this intervention can restore the cytolytic activity of CD8+ T cells230.
Type II (immunological ignorance)
Tumours with this immune microenvironment, generally referred to as ‘cold tumours’, are characterized by an absence of CD8+ TILs as well as a lack of expression of PD-L1 (REFS57,231). Patients with such tumours typically do not benefit from single-agent immune-checkpoint inhibition. Combinatorial therapeutic approaches using dual immune-checkpoint inhibition (typically with antibodies targeting PD-1 or PD-L1 and cytotoxic T lymphocyte antigen 4 (CTLA-4)), cancer vaccines, chimeric antigen receptor T cells or agents such as epigenetic drugs, which aid in recruiting key immune cells to the TME prior to the application of immune-checkpoint inhibition, are likely to be the most effective treatment strategies for these tumours11,35,144,178,232–234.
Type III (oncogenic pathway activation)
These tumours, in which expression of PD-L1 is often a result of constitutive oncogenic signalling, are termed innate immune-resistant tumours and include those that are PD-L1-positive in the absence of CD8+ T cells235–237. Such tumours underscore the importance of considering the presence of TILs in the TME in conjunction with PD-L1 status in order to predict the likelihood of a response to PD-1 or PD-L1 inhibition. Patients with tumours of this type will probably benefit from similar combinatorial therapeutic approaches to recruit TILs as those with type II tumours.
Type iV (immunological tolerance)
These tumours contain TILs that are rendered incapable of mounting antitumour responses despite a lack of PD-L1 expression in the TME. The development of immunological tolerance can result from immunoediting, which might involve suppression of the antigen processing and presentation machinery238, ineffective TIL activation owing to a lack of co-stimulatory signals or T cell exhaustion124. These tumours can also contain immunosuppressive cells such as regulatory T cells and myeloid-derived suppressor cells239. Therapeutic approaches for these tumours include targeting of immune checkpoint proteins other than PD-1 or PD-L1 or immunosuppressive pathways, such as immunometabolism (including the adenosine and indoleamine 2,3-dioxygenase pathways)240,241, adoptive transfer of immune effectors and cancer vaccine strategies242. Combination epigenetic therapy and immunotherapy approaches hold great promise in the treatment of these tumours, given the role of epigenetic events in regulating CD8+ T cell differentiation117,118,120,243 and the ability of epigenetic therapy to prevent CD8+ T cell exhaustion119and to shift CD8+ TILs to an effector and/or memory phenotype144.
Fig. 1 |. Effects of epigenetic therapy on the immune state of a tumour and rationale for the use of combination epigenetic and immunotherapy strategies in cancer.
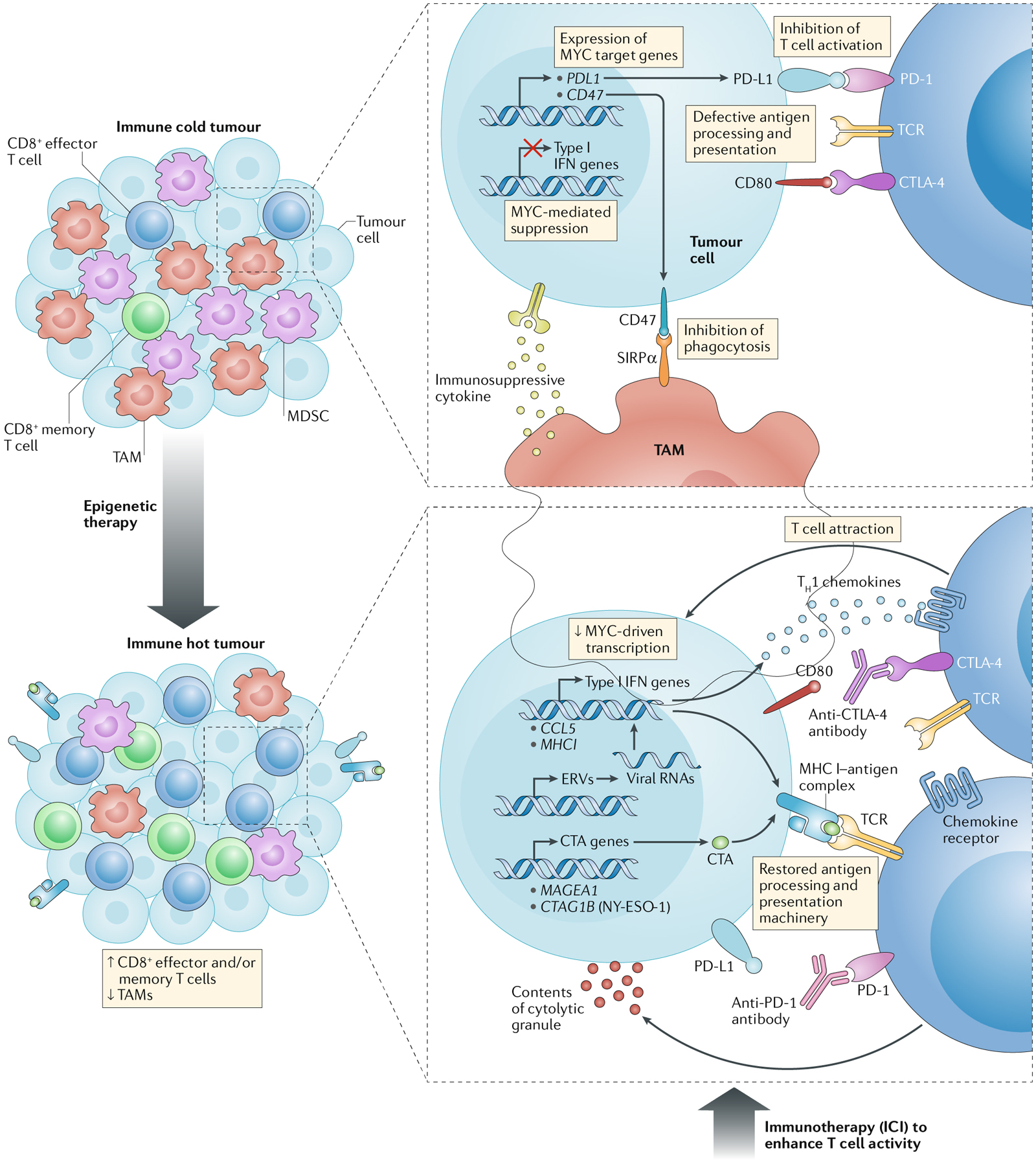
Epigenetic therapy has the potential to convert a tumour from an immune repressive (immune cold) to an immune permissive (immune hot) state through effects on several factors of the tumour microenvironment that normally impede the therapeutic activity of immune-checkpoint inhibition. Immune cold tumours are characterized by the absence of tumour-infiltrating lymphocytes, the presence of immunosuppressive cell populations, such as tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), and/or a lack of expression of programmed cell death 1 ligand 1 (PD-L1) by the tumour cells224,254. Epigenetic agents can modulate the immune composition of the tumour microenvironment by decreasing the abundance of TAMs and MDSCs and increasing the numbers of CD8+ effector T cells and memory T cells144,178. As well as having the potential to shift the differentiation of CD8+ tumour-infiltrating lymphocytes towards effector and/or memory phenotypes, epigenetic drugs can augment innate immune-related signalling and the expression of inflammatory proteins, such as chemokines142–144,146,147,164, which aid the recruitment of T cells to the tumour. In addition, epigenetic therapy can revert key aspects of cancer immunoediting via increased expression of tumour antigens, such as cancer/testis antigens (CTAs)148–151, and restoration of the MHC class I (MHC I) antigen processing and presentation machinery (which is often dysregulated in tumour cells)142–144,160,161, thus potentiating the immune recognition of tumours. Type I interferon (IFN) signalling is a major node of these immunological pathways and can be triggered in response to increased levels of cytoplasmic viral RNAs resulting from epigenetic de-repression of endogenous retroviruses (ERVs)146,147. Epigenetic therapy can also induce the repression of MYC and MYC-related signalling, thus counteracting the immunosuppressive functions of this oncogenic transcription factor, which include downregulation of type I IFN-mediated gene expression, for example, of the gene encoding the T cell-attracting chemokine CC-chemokine ligand 5 (CCL5)144; production of CCL9 that recruits immunosuppressive, PD-L1-positive macrophages to tumours and IL-23 that results in exclusion of T cells, natural killer cells and B cells (not shown); and upregulation of inhibitory immune-checkpoint proteins PD-L1 and CD47 in tumour cells, which suppress T cell activation and macrophage-mediated phagocytosis, respectively184,185. All of the above contribute to the activity of epigenetic agents in converting immune cold tumours into immune hot tumours224, such that the tumours become amenable to immunotherapeutic interventions. For example, the effectiveness of immune-checkpoint inhibitors (ICI) in unleashing an effective T cell-mediated immune response is likely to be enhanced in the context of re-establishment of effective antigen-presentation mechanisms, upregulation of PD-L1, a decreased abundance of TAMs, and increases in the numbers of effector and/or memory T cells within the tumour microenvironment224. CTLA-4, cytotoxic T lymphocyte antigen 4; PD-1, programmed cell death 1; SIRPα, signal-regulatory protein a; TCR, T cell receptor; TH1 cell, type 1 T helper cell.
Therapeutic strategies aimed at converting immune cold tumours into immune hot tumours are currently being intensely investigated. The implications of epigenetic mechanisms in the control of these states and how epigenetic therapy can be used to optimize this transition are discussed in detail in a later section of this manuscript.
Epigenetic mechanisms and therapeutics
Basic principles of epigenetics
As extensively outlined in multiple reviews72–77, epigenetics is the process by which changes mediating heritable patterns of gene expression are established without changing the sequence of DNA. Epigenetics can thus be viewed as a virtual ‘software package’ to control and utilize the information coded in the ‘hard drive’ of DNA. Thus, non-malignant cells and cancer cells have an epigenome’ constituted by regulation of the components of chromatin, which defines the interaction of DNA with proteins, principally histones. Nucleosomes, the 3D distribution of which throughout the genome essentially determines how DNA is packaged in a cell to regulate patterns of gene expression and chromosome structure, are the basic units of these interactions72,73,78–81. This packaging process is fine-tuned by interactions mediated by methylation of genomic DNA at CpG sites and by covalent marks, principally acetylation and methylation, of amino acids on histones in the context of the nucleosomes72,75,76,80–82.
Enzymes or ‘writers’ that establish DNA methylation (DNA methyltransferases (DNMTs)), histone acetylation (histone acetylases) and histone methylation (histone methyltransferases) control each of these processes. In turn, these epigenetic marks can be removed dynamically by enzymes referred to as ‘erasers’, which comprise the ten-eleven translocation enzymes that undo DNA methylation and histone deacetylases (HDACs) and histone demethylases that reverse histone acetylation and methylation, respectively83–85. Histone marks can be activating for RNA transcription, for example, lysine acetylation and some methylation modifications, whereas others, such as lysine deacetylation and certain methylation marks, mediate repressive states of gene expression. Finally, the DNA methylation and histone modifications are recognized by regulatory proteins, or ‘readers’, that enable these chromatin processes throughout the genome to modulate transcriptional profiles84–87.
Key specifics of the cancer epigenome
The cancer epigenome can be characterized by abnormalities in essentially every one of the epigenetic control features outlined in the preceding section73,75,84,85,88–90; the most investigated aspects to date are cancer-specific alterations in DNA methylation and histone acetylation, as has been extensively reviewed elsewhere73,76,79,85,91. The most common changes in DNA methylation found in cancer cells, as compared with their non-malignant counterparts, are global, genome-wide losses of methylation (hypomethylation) that could result in the upregulation of genes with pro-tumorigenic functions, accompanied by more focal, cancer-specific hypermethylation located at CpG rich sites or CpG islands in the promoter regions of hundreds of genes74,75,79,85. These hypermethylated promoters can be associated with repression of expression or prevention of inducibility of involved genes, providing an alternative suppressive mechanism to genetic aberrations for the loss of function of key tumour suppressor genes and is a central feature of carcinogenesis75,76,92–96. In addition, losses and gains of DNA methylation can involve other regulatory regions of the genome, such as gene enhancers, which often regulate networks of genes. The epigenetic alterations in these regulatory regions, which can be located distant from a given gene under their control, can influence cancer development97–101.
Abnormalities of histone acetylation, most commonly losses at gene promoter regions that result from increased activity of HDACs, can either accompany alterations of DNA methylation in mediating important effects on the cancer epigenome or constitute independent controlling effects85,102–104. Specifically, histone deacetylation, with or without coincident DNA methylation, can cause the repression of tumour suppressor genes105–108. Conversely, increased histone acetylase-mediated histone acetylation can constitute a cancer abnormality associated with abnormal upregulation of gene expression109. Thus, the targeting of DNA methylation and/or histone deacetylation (or acetylation), as extensively reviewed elsewhere104,110,111, is a major focus of epigenetic therapy and is central to its combination with ICI, as discussed later in this Review.
Roles in immune cell differentiation
In the past decade, a number of elegant studies in the field of immunology have led to a much more cohesive view of the epigenetic regulation of normal physiological subsets of key immune cell lineages. These studies provide insights into the functions and interactions of these cell populations within the TME. The epigenetic regulation of differentiation has been studied across several major immune cell populations, including myeloid cells, CD8+ T cells and CD4+ T cells. The epigenetics of CD4+ T cell differentiation have been reviewed extensively112,113, but remains an emergent paradigm; therefore, the following section is focused on the epigenetic regulation of CD8+ T cells and myeloid populations, the direct antitumour activities of which are potentially amenable to the actions of epigenetic drugs.
CD8+ T cell differentiation.
The activation and differentiation of CD8+ T cells are a result of stimulation following antigen presentation by professional antigen-presenting cells (Box 2). Epigenetic mechanisms have important roles in dictating the fate of T cells114 (FIG. 2). These mechanisms are essentially mediated by progressive, large-scale remodelling of chromatin, which modulates the accessibility of transcription factors to regulatory regions of target genes involved in T cell development, maturation and lineage commitment114–116. Transcription factor 7 (TCF7, also known as TCF1) has been identified as one of the key transcription factors involved in establishing the epigenetic identity of T cells during their differentiation and patterns the chromatin landscape, enabling T cell differentiation to evolve116. The pathways of naive CD8+ T cell differentiation to CD8+ effector T cells involve dynamic epigenetic changes in chromatin accessibility, with genome-wide gains and losses of DNA methylation and histone modifications observed during this process117–121 (FIG. 2). Similarly, epigenetic mechanisms also demonstrably regulate the dedifferentiation of CD8+ effector T cells to memory T cells118,122. This phenotypic switch is accompanied by a reversal of epigenetic repression of naive T cell-associated genes but with maintenance of demethylation of key genes expressed in CD8+ effector T cells118,119 (FIG. 2). Thus, CD8+ memory T cells have distinct patterns of chromatin accessibility associated with the capacity for rapid re-induction of effector functions123.
Box 2 |. Antigen-specific T cell activation.
The activation of naive T cells to form fully functional effector T cells is a highly regulated process that requires the simultaneous application of three distinct stimuli from professional antigen-presenting cells (APCs) such as dendritic cells or macrophages244. Signal 1 results from the interaction between the T cell receptor (TCR) and a peptide–MHC class II complex present on the APC or MHC class I in the setting of dendritic cell cross presentation245. This interaction triggers mitogen-activated protein kinase and phospholipase C signalling downstream of the TCR, inducing nuclear factor-κB and activator protein 1 activation, and culminating in transcription and expression of IL-2 (REF246). Although required for T cell activation, signal 1 alone is not sufficient to induce clonal expansion of these cells; TCR stimulation in the absence of a co-stimulatory signal can induce the formation of anergic T cells, leading to peripheral tolerance or defective effector T cell populations247. The co-stimulatory signal, or signal 2, can result from many potential receptor interactions, the most well-established of which is the CD28 receptor on T cells with B7 family ligands (CD80 or CD86) on APCs248,249. A crucial effect of signal 2 is increased transcription and stabilization of IL2 mRNA250. Signals 1 and 2 acting in concert drive the onset of T cell activation and proliferation, thus initiating an expansion phase. T cell responses must be fine-tuned to a particular function or immunological response and, therefore, a third signal provides the basis for cell polarization, optimal effector functionality and survival251. Signal 3 results from cytokines and/or chemokines present in the microenvironment, usually derived directly from APCs251. This signal has a potent effect on T cell differentiation, with factors such as IL-12 and IFNα, IFNβ and IFNγ skewing T cell fate towards cytotoxic T lymphocyte or type 1 T helper-type responses, whereas retinoic acid and transforming growth factor-β promotes the generation of regulatory T cells252,253. In the presence of sufficient antigen, proliferation of activated T cells will be initiated and sustained; upon clearance of antigen, T cell populations enter contraction, followed by a memory phase244. During this memory phase, stable numbers of long-lived, antigen-specific CD8+ T cells remain in the circulation; this cell population has a distinct phenotype that enables rapid reinduction of a robust cytotoxic activity upon antigen re-encounter118,123.
Fig. 2 |. Implications of DNA methylation-associated programmes on T cell differentiation.
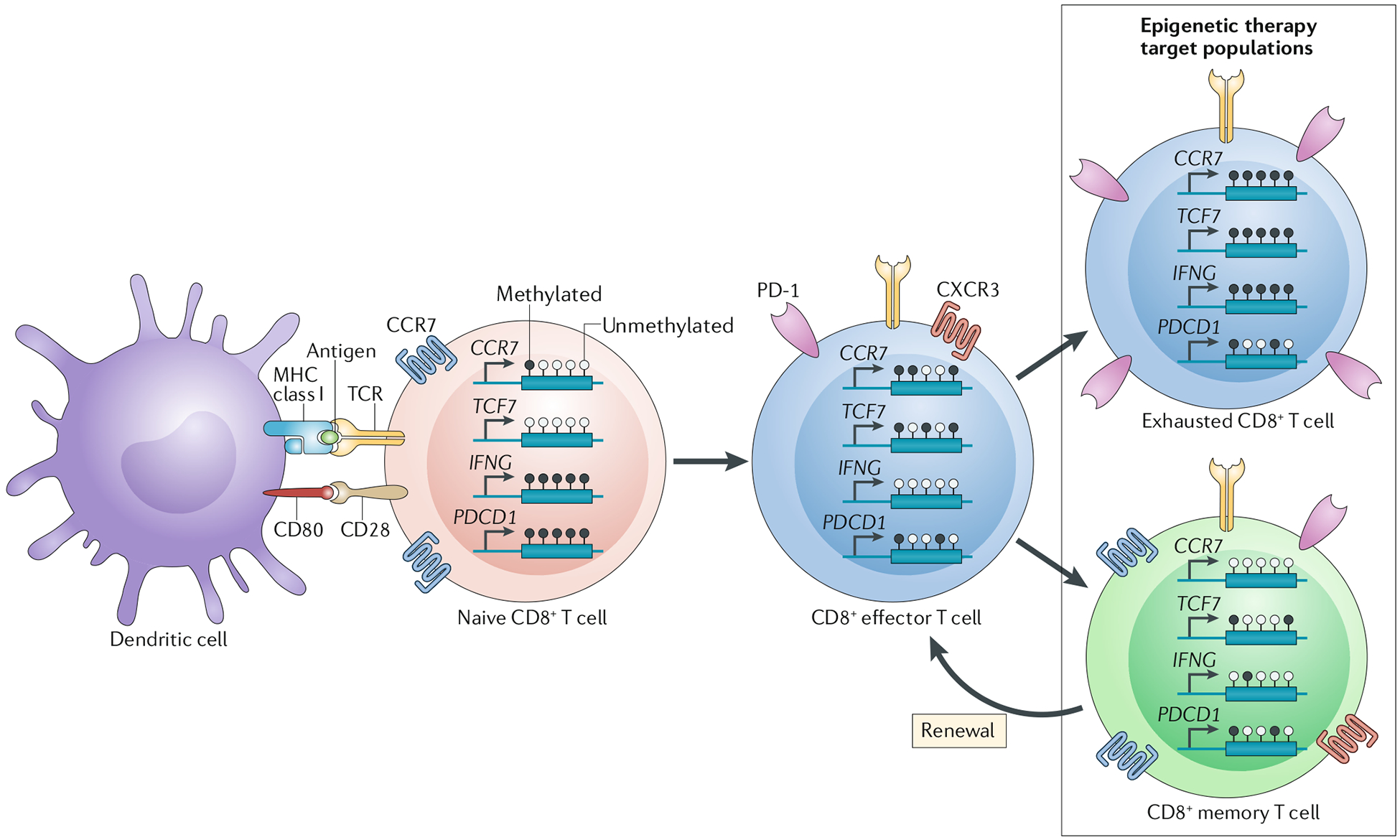
T cell activation from the naive to an effector state is induced by interaction between the T cell receptor (TCR) and corresponding MHC class II–peptide complex on professional antigen-presenting cells or MHC class I in the setting of dendritic cell cross presentation (the context shown in the figure) in concert with co-stimulatory molecule interactions and inflammatory stimuli255. Bone marrow-derived antigen-presenting cells — predominantly dendritic cells but also macrophages or B cells — are sufficient to induce CD8+ T cell priming, whereas CD4+T cells are unable to facilitate this process245. As elucidated in studies by Youngblood et al.118 and Ghoneim et al.119, among others, the methylation status of genes encoding several crucial mediators of T cell differentiation undergoes dynamic changes during the acquisition of major T cell phenotypes. For example, the transition from a naive to effector phenotype is characterized by the induction and repression of many distinguishing cell-surface markers, including the G protein-coupled chemokine receptors CXC-chemokine receptor 3 (CXCR3) and CC-chemokine receptor 7 (CCR7) and the inhibitory immune-checkpoint receptor programmed cell death 1 (PD-1). CXCR3 expression has been shown to be epigenetically regulated in antigen-specific CD4+ T cells, although it remains unclear whether the same is true in CD8+ T cells, and renders effector T cells responsive to interferon-inducible, type 1 T helper (TH1) cell-associated chemokines, such as CXC-chemokine ligand 9 (CXCL9), CXCL10 and CXCL11, which tend to emanate from sites of inflammation256. Augmentation of PD-1 expression through demethylation of the PDCD1 (PD-1) gene promoter and a regulatory region ~300 bp upstream of the transcription start site occurs rapidly following antigen stimulation of naive T cells as the direct result of activatory signalling from the TCR126,257. PD-1 signalling acts as a negative feedback regulator of the inflammatory activity of T cells by inhibiting TCR-mediated signalling258. Cell-surface expression of the homing receptor CCR7 is also dynamically regulated during the naive to effector phenotypic transition via DNA methylation and thus repression of the CCR7 gene118,119. CCR7 facilitates the recruitment of naive T cells from the bloodstream to lymphoid organs; therefore, downregulation of this receptor enables primed effector T cells to migrate from these organs to other tissues in surveillance of their cognate antigen259. In addition, effector T cells have an increase in methylation and thus repression of TCF7, which encodes transcription factor 7, as well as a loss of methylation and de-repression of IFNG, which encodes the inflammatory cytokine IFNγ119. The post-effector fate of CD8+T cell generally involves the acquisition of either of two major phenotypes, namely exhausted or memory260. The exhausted state is characterized by whole-genome gains in DNA methylation, including sites in TCF7, IFNG and CCR7 (REF119). These methylation gains result in reduced effector functionality in terms of both cytolytic activity and cell proliferation. In comparison with effector T cells, exhausted T cells have increased PD-1 expression and decreased CXCR3 expression, which act to sensitize T cells to inhibitory interactions with programmed cell death 1 ligand 1 and prevent chemotactic responses to (TH1) cell-associated chemokines, respectively. The acquisition of the memory phenotype in effector T cells is correlated with the demethylation and thus re-expression of CCR7 (REF.118), with retention of CXCR3 and PD-1 expression261–263. Memory T cells demonstrate increased IFNG methylation compared with that associated with the effector state, but do not demonstrate the high methylation levels of this gene found in naive or exhausted CD8+ T cells118,119. The T cell populations that seem to be most amenable to modulation with epigenetic therapies are those in the post-effector states of T cell differentiation (boxed area)119,144.
Another end point in the fate of effector T cells involves the acquisition of an ‘exhausted’ phenotype facilitated by chronic antigen stimulation. A hallmark of this cell state is the downregulation of effector functionality, as evidenced by a diminished capacity for induced production of tumour necrosis factor, IL-2 and IFNγ124. Exhausted T cells also have upregulation of inhibitory immune checkpoint molecules, such as PD-1, CTLA-4, LAG3 and TIM3, on the cell surface to levels exceeding those observed in effector T cells125. In the context of immunotherapy, PD-1 is an important target, the expression of which is regulated by both DNA methylation126 and alterations of chromatin accessibility127. Notably, exhausted CD8+ T cells regain minimal effector and/or memory functions following PD-1 inhibition in mouse models of chronic lymphocytic choriomeningitis virus infection119,128, thus suggesting the need to consider combination strategies to prevent the acquisition of or reverse the exhausted state. Crucially, when considering potential epigenetic-immunotherapy combinations, this finding was predominantly attributed to the epigenetic stability of these cells under PD-1 inhibition, in a state distinct from that of effector and memory T cells119,128; however, this epigenetic state could be counteracted, and T cells reinvigorated, through sequential inhibition of DNMTs followed by PD-L1, with similar findings observed in an immune-checkpoint inhibition-refractory mouse TRAMP-C2 model119. These data highlight the importance of considering epigenetic plasticity in dictating the effects of current immune-checkpoint inhibitors on CD8+ T cells.
Myeloid differentiation.
Epigenetic modifications also regulate the fate of cells of the myeloid lineage by orchestrating their differentiation and activation. These changes mainly involve various histone modifications that regulate the binding of lineage-specific transcription factors to their target genes, largely by modulating chromatin accessibility129,130. As discussed later in this manuscript, HDAC inhibitors, either alone or in combination with DNMT inhibitors, have proved to be effective in enhancing antitumour immunity across multiple preclinical models by depleting tumours of myeloid-derived suppressor cells (MDSCs) — a cell population known to induce peripheral T cell tolerance and to inhibit both T cell activation and proliferation131–133. Epigenetic mechanisms have also been implicated in the regulation of macrophage polarization134,135. Accordingly, in a mouse model of ovarian cancer, combination treatment with the DNMT inhibitor azacitidine and the ornithine decarboxylase inhibitor α-difluoromethylornithine results in depletion of pro-tumorigenic M2-like macrophages from the TME and enrichment with inflammatory, antitumour, M1-like macrophages136.
Targeting the cancer epigenome
In keeping with the crucial roles of epigenetic mechanisms in regulating the functions of non-malignant epithelial and immune cells as well as the epigenetic alterations associated with malignancy, strategies to target the cancer epigenome have proved effective in controlling tumour growth. The goal of such therapy is to reprogramme the epigenome of cancer cells in order to disrupt the self-renewal of stem-like cells, induce differentiation towards a non-malignant phenotype, block the invasive or metastatic behaviour of malignant cells, and/or sensitize tumours to other therapeutic interventions85,111,137–139. Excitement surrounding these concepts is increasing now that the pharmaceutical industry has developed different drugs with which to target virtually all of the writer, eraser and reader functions outlined above (TABLE 1). Many of these agents are undergoing testing in phase I and/or II clinical trials and the demonstration of acceptable toxicity profiles and promising efficacy is anticipated. These studies might therefore facilitate the future use of epigenetic agents as monotherapies or in combinatorial strategies. The only epigenetic drugs currently approved by the FDA for use in patients are DNMT inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukaemia (AML), in combination with the BCL-2 inhibitor venetoclax for the latter disease, and HDAC inhibitors for the treatment of cutaneous or peripheral T cell lymphoma and relapsed multiple myeloma (TABLE 1). DNMT inhibitors include azacitidine, which can result in the demethylation both DNA and RNA, as well as DNA-specific demethylating agents, such as decitabine and its derivative with a longer half-life, guadecitabine140. DNMT inhibitors and HDAC inhibitors are currently being studied alone and in combination with ICI across a variety of solid and haematological malignancies (TABLE 1). As the focus of this Review, we outline the promise of such combinations to enhance the efficacy of ICI and other immunotherapies in the following sections.
Table 1 |.
Summary of therapeutics targeting epigenetic modifiers
| Epigenetic agent | Stage of clinical development | Combination therapies |
|---|---|---|
| DNMT inhibitorsa | ||
| Azacitidine | FDA approved: MDS and AML | In combination with the BCL-2 antagonist venetoclax in AML |
| Phase I/II: various solid carcinomas, lymphomas and/or other haematological malignancies; Phase III: haematological malignancies | HDAC inhibitors, immune-checkpoint inhibitors and/or chemotherapeutic agents | |
| Decitabine or THU-DAC | FDA approved: MDS | NA |
| Phase I/II: various solid carcinomas, lymphomas and/or other haematological malignancies; Phase III: haematological malignancies and primary neoplasia of ovary | HDAC inhibitors, immunotherapies, PARP inhibitors and/or chemotherapeutic agents | |
| Guadecitabine | Phase I/II: various solid carcinomas and/or haematological malignancies; Phase III: haematological malignancies | HDAC inhibitors, immunotherapies, PARP inhibitors and/or chemotherapeutic agents |
| HDAC inhibitorsa | ||
| Entinostat | FDA breakthrough drug designation: advanced-stage breast cancer | Exemestane |
| Phase I/II: various solid carcinomas, lymphomas and/or other haematological malignancies; Phase III: hormone receptor-positive breast cancer | Immunotherapies | |
| Vorinostat | FDA approved: CTCL | NA |
| Phase I/II: various solid carcinomas, lymphomas and/or other haematological malignancies; Phase III: CTCL, multiple myeloma and ALL | Immunotherapies, PARP inhibitors or chemotherapeutic agents | |
| Romidepsin | FDA approved: CTCL | NA |
| Phase I/II: Hodgkin lymphoma, PTCL, multiple myeloma, NHL and/or solid carcinomas; Phase III: T cell lymphomas | Immunotherapies or chemotherapeutic agents | |
| Panobinostat | FDA approved: multiple myeloma | Bortezomib (proteasome inhibitor) plus dexamethasone |
| Phase I–III: various solid carcinomas, lymphomas and/or other haematological malignancies | Immunotherapies or chemotherapeutic agents | |
| Givinostat | Phase II: chronic myeloproliferative neoplasms | NA |
| Mocetinostat | Phase I/II: lymphoma, melanoma, NSCLC and/or other advanced-stage solid tumours | Guadecitabine and/or immune-checkpoint inhibitors, or brentuximab vedotin (anti-CD30 antibody–drug conjugate) |
| Valproic acid | Phase I–III: AML, MDS, various solid carcinomas and/or childhood ependymoma, virus-associated cancers | Azacitidine, chemotherapeutic agents or immune-checkpoint inhibitors |
| Belinostat | FDA approved: PTCL | NA |
| Phase I/II: T cell leukaemia or lymphoma, MDS or AML, glioblastoma or various other solid carcinomas and haematological malignancies | Zidovudine (±IFNα-2b), pevonedistat (NEDD8-activating enzyme inhibitor) or temozolomide (chemotherapy) plus radiotherapy | |
| HMT inhibitors | ||
| CPI-1205 (EZH2 inhibitor) |
|
|
| Tazemetostata (EZH2 inhibitor) |
|
|
| Pinometostat (DOT1L inhibitor) | Phase Ib/II: KMT2A-rearranged AML | Standard chemotherapies |
| LSD1 inhibitors | ||
| IMG-7289 | Phase I: myelofibrosis | NA |
| Seclidemstat (SP-2577) |
|
|
| INCB059872 |
|
|
| BET inhibitors | ||
| RO6870810 |
|
|
| INCB057643 |
|
|
| CPI-0610 |
|
|
| Molibresib (GSK525762) |
|
|
| ZEN003694 |
|
|
| BMS-986158 |
|
|
| MK-8628 | Phase I: AML (including de novo AML and AML secondary to MDS) or DLBCL | NA |
| AZD5153 |
|
|
ALL, acute lymphocytic leukemia; AML, acute myeloid leukaemia; ATRA, all-trans retinoic acid; BET, bromodomain and extra-terminal protein; BTK, Bruton tyrosine kinase; CTCL, cutaneous T cell lymphoma; CTLA-4, cytotoxic T lymphocyte antigen 4; DLBCL, diffuse large B cell lymphoma; DNMT, DNA methyltransferase; DOT1L, DOT1-like protein; EZH2, enhancer of zeste homologue 2; HDAC, histone deacetylase; HMT, histone methyltransferase; IDO1, indoleamine 2,3-dioxygenase 1; JAK, Janus kinase; LSD1, lysine-specific histone demethylase 1A; mCRPC, metastatic castration-resistant prostate cancer; MDS, myelodysplastic syndrome; NA, not applicable; NHL, non-Hodgkin lymphoma; NSCLC, non-small-cell lung cancer; PARP, poly(ADP-ribose) polymerase; PD-1, programmed cell death 1; PD-L1, programmed cell death 1 ligand 1; PTCL, peripheral T cell lymphoma; SCLC, small cell lung carcinoma; THU-DAC, tetrahydrouridine-decitabine.
Phase I-III studies of DNMT inhibitors and HDAC inhibitors, as well as phase II studies of tazemetostat monotherapy, are too numerous to list individually in separate bullet points and, for brevity, only broad descriptions of the disease setting and combination partners have been provided for these trials.
Rationale for epigenetic-immunotherapy
Epigenetic therapy modulates key regulatory features of both immune cells and tumour cells in ways that might overcome some of the current limitations of immunotherapy (FIGS 1; 2). For example, epigenetic drugs have the potential to reverse many processes that tumours engage to evade immune-mediated destruction (FIG. 1).
Epigenetic control of immune exhaustion
Much interest over the past several years has surrounded the state of immune cell ‘exhaustion’, which is a component of immune tolerance and evasion120,141. In the scenario of T cell exhaustion associated with cancer, tumour-targeting CD8+ T cells adopt a unique differentiation state, in which they are unable to mount effector functions and thus their cytolytic activity against tumour cells is impeded120. Importantly, as described above, this state is characterized by a complex programme of gene expression changes that are correlated with alterations in chromatin conformation and DNA methylation117,123. In mouse models, epigenetic therapy can reverse these changes in chromatin conformation and DNA methylation acquired during the transition to an exhausted T cell state, which is postulated to be induced by a DNMT3A-mediated de novo methylation programme119 (FIG. 2). Indeed, in these preclinical studies, prevention of the exhaustion state with DNMT inhibitors was associated with an increase in the efficacy of ICI with anti-PD-1 antibodies119.
Epigenetic reversion of immunoediting
DNMT inhibitors and HDAC inhibitors are known to promote innate immune-related signalling in cancer cells, which could potentially also enhance recognition of these cells by adaptive immune cell populations142–147 (FIG. 1). Early examples of this concept include the induction of tumour antigens, termed cancer/testis antigens (CTAs), consisting largely of proteins usually expressed exclusively in embryonic or germ cells during development148–156. The expression of CTA genes is controlled by transcriptional repression, including promoter DNA methylation and histone deacetylation in association with other histone modifications, thus making these genes likely targets of epigenetic therapy157–159. In addition to CTA upregulation, epigenetic therapy can potentiate tumour cell immune recognition through restoration of the MHC class I antigen processing and presentation machinery — deficiencies which can be selected for during cancer immunoediting and, indeed, are one of the defining characteristics of immune hot tumours142–144,160–163.
The list of actionable targets of epigenetic therapy has now expanded beyond CTAs, with a particular focus on DNMT inhibitor-mediated augmentation of signalling related to innate immunity and induction of inflammation-associated genes such as cytokines and chemokines142–144,146,147,164. Intriguingly, these effects are typically predicated on potentiation of type I and III interferon signalling invoked by increased levels of cytoplasmic viral RNAs, a phenomenon termed viral mimicry146,147. This response is largely centred on the transcriptional de-repression of endogenous retroviruses (ERVs)146,147. ERVs have been incorporated into the human genome over millennia, such that they now account for ~8% of the genome165, but are generally silenced in somatic cells through DNA methylation and repressive histone modifications166–169. Thus, DNMT inhibitors induce demethylation of ERV sequences, which enables ERVs to be transcribed into RNAs that fold into double stranded RNA structures. Subsequent interactions between these viral double stranded RNAs and cognate cytoplasmic sensors triggers a viral defence response, including induction of type I interferon signalling.
Taken together, these effects emphasize the demonstrable potential of epigenetic therapy to facilitate immune recognition of tumour cells, not least through augmentation of antigen expression, processing and presentation (FIG. 1). The following sections summarize the evidence accumulated to date that epigenetic therapy can overcome barriers to clinical responses to immunotherapy.
Combined epigenetic therapy and ICI
Preliminary clinical observations
Observations made in an early phase I/II clinical trial of combination epigenetic therapy with the DNMT inhibitor azacitidine and the HDAC inhibitor entinostat170 have helped to bring the concept of combined therapy with epigenetic drugs and ICI to the fore. These observations have guided the design of preclinical studies to explore the scientific underpinning for the promise of such approaches. Briefly, in this initial clinical trial involving 45 patients with advanced-stage refractory NSCLC170, two patients exhibited very durable, Response Evaluation Criteria in Solid Tumors (RECIST)-defined objective responses to epigenetic therapy and survived for 3–4 years after treatment. Additionally, five patients who had disease progression during the trial were subsequently enrolled in the first trials of anti-PD-1 antibodies; three of these patients achieved RECIST objective responses whereas the remaining two patients had stable disease for 24 weeks before progression142–170. Whilst these observations generated excitement, the underlying mechanism for the noted efficacy was not elucidated, thus spurring the initiation of multiple preclinical studies to evaluate the effects of epigenetic therapy on antitumour immune responses.
Promising preclinical data
DNMT inhibitor-based therapy.
Direct evidence of synergy between DNMT inhibition and immune-checkpoint inhibition has been established in the preclinical space across multiple model systems. In animal models of ovarian cancer or melanoma, the addition of a demethylating agent (decitabine and azacitidine, respectively) to anti-CTLA-4 antibody therapy increases the antitumour effect relative to that observed with immune-checkpoint inhibition alone, as evidenced by prolongation of survival146–171, with an enhancement of cytolytic CD8+ T cell accumulation within the tumours noted in the ovarian cancer model171. Other studies provide evidence that the effectiveness of anti-PD-1 antibodies can also be potentiated with the use of DNMT inhibitors. In the MMTV-Neu mouse breast cancer model, treatment with guadecitabine augments both MHC class I expression and T cell chemotaxis via the CXC-chemokine ligand 9 (CXCL9)/CXCL10/CXCL11-CXC-chemokine receptor 3 (CXCR3) axis, which has been correlated with enhanced tumour infiltration of CD8+ T cells and subsequent potentiation of responses to anti-PD-1 antibodies172. Additionally, Yu et al.173 delineated important decitabine-mediated immunological effects in a syngeneic mouse CT26 colon cancer model. These effects included mobilization of the antigen presentation machinery, intratumour accumulation of PD-1-positive CD8+ T cells and sensitization to anti-PD-1 antibody therapy. As mentioned above, in the mouse TRAMP-C2 model of immune-checkpoint inhibition-resistant prostate cancer, administration of decitabine was found to induce the sensitivity of CD8+ T cells to anti-PD-L1 antibodies through the prevention of the DNMT3A-mediated DNA methylation programme of exhausted T cells, thereby enhancing antitumour responses119. Together, these findings provide evidence supporting the effectiveness of combined DNMT inhibition and immune-checkpoint inhibition, which is mediated in part by epigenetic enhancements of the adaptive immune system.
HDAC inhibitor-based therapy.
The deployment of HDAC inhibitors in combination with immunotherapies has demonstrated efficacy across multiple animal models. The axes most amenable to perturbation by HDAC inhibitors are T cell chemoattraction gradients and myeloid cell populations, predominantly MDSCs. An early indication of the potential synergy between HDAC inhibition and immune-checkpoint inhibition was derived from a syngeneic mouse B16-F10 melanoma model174. In this study174, the concurrent application of the HDAC inhibitor panobinostat potentiated anti-PD-1 antibody therapy, thus resulting in slower tumour growth and longer survival than that observed with either treatment alone. The induction of PD-L1 after HDAC inhibition, mediated by a gain of PDL1 gene promoter acetylation, was one notable in vitro observation174. This induction of PD-L1 could constitute a possible resistance mechanism when HDAC inhibition is paired with ICI and should, therefore, be considered when evaluating PD-L1 tumour positivity in the context of epigenetic therapy. In a study using a subcutaneous mouse hepatocellular carcinoma model, administration of the HDAC inhibitor belinostat was demonstrated to enhance the efficacy of CTLA-4 inhibition, but not PD-1 inhibition, in association with increases in the abundance of M1-polarized tumour-associated macrophages and in IFNγ production by tumour-specific CD8+ T cells as well as decreased numbers of splenic regulatory T (Treg) cells175. Tumour antigen-presenting cells had upregulation of PD-L1 early after treatment with the HDAC and CTLA-4 inhibitors, with later upregulation of PD-1 on T cells also noted; simultaneous HDAC, PD-1 and CTLA-4 inhibition resulted in complete tumour rejection. Across multiple animal models of solid tumours, the HDAC inhibitor entinostat induced the depletion of MDSCs and enhanced the efficacy of anti-PD-1 therapy176,177. Specifically, Orillion et al. found that the application of entinostat reduced the levels of both MDSC-associated chemoattractants and MDSC suppressive activity177. Furthermore, evidence suggested that HDAC inhibition might promote the differentiation of this cell population177.
In addition to MDSC-directed immune effects, HDAC inhibitors have been found to have a variety of effects on T cell responses. For example, the HDAC inhibition using romidepsin increased levels of T cell chemoattractants and tumour infiltration in multiple lung adenocarcinoma models, with a correlated sensitization to anti-PD-1 therapy117. Building on these preclinical studies, HDAC inhibition in combination with immune-checkpoint inhibition is currently being explored in multiple clinical trials (TABLE 1; Supplementary Table S1) and might be most efficacious against cancers with a type IV TME, a hallmark of which is a demonstrably high level of MDSC infiltration (BOX 1).
DNMT inhibitor and HDAC inhibitor combinatorial paradigm.
The aforementioned concepts based on the combination of DNMT or HDAC inhibitors with ICI have subsequently been extended to combination paradigms founded on both DNMT and HDAC inhibition, with robust antitumour effects observed in multiple preclinical solid tumour models144,176,178. Importantly, in all cases, the efficacy of such combinations has been tightly tied to CD8+ T cell dependent mechanisms144 and/or associated with sensitization to immune-checkpoint inhibition176,178. In ovarian and lung cancer animal models, attraction of CD8+ T cells to the TME occurs in association with initiation of a type 1 T helper (TH1) cell chemokine axis involving CC-chemokine ligand 5 (CCL5) and CXCL10 (REFS194,197). These chemokines have the demonstrated ability to facilitate the attraction of CD8+ T cells through interaction with CC-chemokine receptor 5 (CCR5) and CXCR3, respectively, on these cells179–182. CCL5 seems of particular importance in patients with lung adenocarcinoma as this chemokine is an established hallmark of an active lymphocytic compartment in clinical samples and is associated with favourable survival outcomes183. Additionally, in animal models of NSCLC, the application of epigenetic therapy prevents the aforementioned exhausted phenotype in tumour-associated CD8+ T cells119, with acquisition of effector and/or memory phenotypes noted144.
In addition, preclinical studies in NSCLC models have identified MYC as a key target of combination DNMT plus HDAC inhibition (FIG. 1); suppression of MYC activity by such epigenetic therapy potentiates type I interferon signalling and the associated induction of CD8+ T cell-attracting chemokines, including CCL5 (REF144). A similar pattern of immune effects emerged from studies by another group after genetic manipulation of MYC expression in a mouse model of Kras-mutated lung adenocarcinoma184. In this study, MYC expression in tumour cells resulted in the production of CCL9 and IL-23; CCL9 was shown to mediate recruitment of PD-L1-positive macrophages and associated PD-L1-dependent expulsion of T and B cells, whereas IL-23 orchestrates exclusion of adaptive T cells and B cells and innate immune NK cells184. The implications of the potential epigenetic regulation of MYC on the tumour immune microenvironment are not limited to the setting of NSCLC. Casey et al.185 have established that MYC regulates the expression of both PD-L1 and CD47 in human T cell acute lymphoblastic leukaemia cells and mouse models of this disease. The regulation of these targets by MYC was found to be through direct binding and thus transcriptional induction, with MYC inactivation resulting in target downregulation and potentiation of antitumour responses noted in mouse models185. Notably, CD47, termed the ‘do not eat me’ antigen, facilitates antagonization of macrophage-dependent immune surveillance through interaction with signal-regulatory protein α (SIRPα) on macrophages. Targeting of the CD47–SIRPα axis is an emerging paradigm in immunotherapy186.
These preclinical studies establish, through a diverse set of mechanisms, the multifaceted potential utility of epigenetic therapy to enhance the efficacy of cancer immunotherapy (FIG. 1). These results have formed the basis of a growing number of clinical trials designed with the aim of translating the concept of epigenetic-immunotherapy into patient management.
Current clinical trials
The early clinical observations with combined DNMT and HDAC inhibition elucidated in the setting of advanced-stage, treatment-refractory NSCLC142,170, together with data obtained from the preclinical studies discussed above144, have prompted the initiation of two trials (NCT01928576 and NCT03220477; Supplementary Table S1). In these ongoing studies, DNMT plus HDAC inhibition is being combined with concurrent anti-PD-1 antibody therapy for patients with advanced-stage NSCLC. The protocols of these trials have been amended to focus on the potential of this combination in the treatment of both ICI-resistant and ICI-naive patient populations. Additionally, multiple clinical trials involving DNMT and/or HDAC inhibition plus immune-checkpoint inhibition are ongoing across a variety of solid tumours and myelodysplastic syndrome and/or AML (Supplementary Table S1). Notably, many of these trials include robust correlative studies, such as serial sampling of peripheral blood and tumour specimens for analyses of induced viral mimicry, interferon induction and T cell functional phenotypes (for example, NCT01928576, NCT03233724, NCT03220477, NCT03576963, NCT02901899 and NCT02397720). Although objective responses, disease stabilization and encouraging OS outcomes have been observed in a previous clinical trial of such a combination in patients with AML186,187, careful consideration of the optimal study populations and epigenetic agents are needed. This requirement is exemplified by results from a randomized, placebo controlled phase II trial of oral azacitidine (CC-486) added to pembrolizumab that failed to show a statistically significant difference in PFS (HR 1.374, 90% CI: 0.926–2.038; P = 0.179) or OS (HR 1.375, 90% CI: 0.830–2.276; P = 0.297) in patients with advanced-stage NSCLC188. Of note, the combination treatment group in this study received a median of two fewer cycles of therapy than the placebo group, in association with increases in the proportions of patients that had treatment-related adverse events, dose reductions and treatment interruptions188. This increased toxicity might be reflective, in particular, of the intestinal and haematological toxicities noted for the oral formulation of azacitidine, which have been associated with dose interruptions or reductions in 16% and 19% of patients (with myeloid neoplasms), respectively189. These findings highlight the need for careful selection of epigenetic modifying agents in order to maximize the potential synergy with specific ICI whilst limiting treatment-related toxicities.
Currently, it is too early to know whether clinically significant efficacy will emerge from the ongoing trials of combined epigenetic therapy and immune-checkpoint inhibition, thus warranting movement of these combinatorial approaches further towards formal clinical use. As the results of these trials are reported in the coming years, a focus on biomarkers will be essential to allocating these therapies to the patients who are likely to derive the greatest benefit.
Emerging epigenetic partners for ICI
The aforementioned preclinical studies demonstrating immunological effects of DNMT inhibitors and HDAC inhibitors have fostered a growing number of reports that epigenetic drugs with different targets can enhance the efficacy of ICI. Indeed, many of the alternative combinations are undergoing testing in early phase clinical trials. Emerging preclinical findings suggest that the effectiveness of ICI might be further enhanced by future strategies incorporating single or multiple epigenetic drugs with diverse targets.
EZH2 inhibitors
Therapeutics of this class inhibit the activity of the enzyme enhancer of zeste homologue 2 (EZH2), which is the histone-lysine N-methyltransferase subunit of the polycomb repressive complex 2 (PRC2). PRC2, via the activity of EZH2, is responsible for placing the transcriptionally repressive histone modifications H3K37me2 and H3K27me3 (REFS190–193). These forms of histone modifications are closely associated with genes vulnerable to cancer-specific DNA hypermethylation at gene promoter region CpG islands, which silences the expression or blocks the inducibility of the affected genes194–197. Multiple preclinical studies have demonstrated the potential of EZH2 inhibitors to augment the activity of immunotherapy or induce immunostimulatory effects. In mice harbouring tumours derived from patients with ovarian cancer together with adoptively transferred autologous CD8+ T cells derived from the same patients, the application of an EZH2 inhibitor in combination with a DNMT inhibitor led to the establishment a robust chemotactic gradient of the TH1-type chemokines CXCL9 and CXCL10, thus facilitating the attraction of CXC3R+ CD8+ effector T cells, with subsequent sensitization to ICI with anti-PD-L1 antibodies198. Moreover, Goswami et al.198,199 found that peripheral blood T cells from patients treated with ipilimumab had increased expression of EZH2; accordingly, they demonstrated that the use of an EZH2 inhibitor alone altered the phenotype and function of human Treg cells and enhanced the cytotoxic activity of human CD8+ effector T cells as well as sensitizing syngeneic mouse MB49 bladder cancer and B16-F10 melanoma to anti-CTLA-4 ICI198,199.
Additionally, EZH2 seems to have a specific role in mechanisms of adaptive resistance to immunotherapy (with anti-CTLA-4 antibodies or IL-2), whereby infiltrating tumour-reactive CD8+ T cells trigger induction of EZH2 in melanoma cells that leads to epigenetic silencing of antigen processing and presentation machinery as well as repression of TH1 cell-associated chemokines. EZH2 inhibition can reverse this adaptive resistance programme and enhances the efficacy of anti-CTLA-4 antibody therapy in mouse melanoma models200. Several EZH2 inhibitors have entered clinical testing (TABLE 1). Together, these studies provide initial indications of synergy between EZH2 inhibition and immune-checkpoint inhibtion and have provided the foundations for the initiation of phase I/II clinical trials of such combinations (Supplementary Table S1).
LSD1 inhibitors
Lysine-specific histone demethylase 1A (LSD1; also known as KDM1A) is the enzyme responsible for erasure of the key mono-methyl (me1) and di-methyl (me2) chromatin marks on histone H3, predominantly at lysines 4 and 9 (H3K4 and H3K9). This enzyme thereby functions as a transcriptional co-regulator in a context-dependent manner through demethylation of the repression-associated H3K9me1 and H3K9me2 marks or the activation-associated H3K4me1 and H3K4me2 marks201. LSD1 can also demethylate a number of nonhistone substrates, including DNMT1, and the loss of LSD1 expression is correlated with a decrease in DNMT 1 levels owing to increased methylation and destabilization of this protein202; therefore, LSD1 inhibition could potentially result in decreased global DNA methylation. Of note, LSD1 is also overexpressed in a number of malignancies and, thus, LSD1 inhibitors might be promising potential therapeutic options in a variety of cancers203–205. Moreover, The Cancer Genome Atlas data indicate that LSD1 expression is inversely correlated with CD8+ T cell infiltration into various cancers206,207. Accordingly, LSD1 inhibitors have been shown in multiple mouse cancer models to induce the viral mimicry-like response, with remarkable similarity to the effect observed with DNMT inhibitors206,207, which enhances the recruitment of T cells, increases antigen presentation and thereby sensitizes poorly immunogenic tumours, such as triple-negative breast cancers, to anti-PD-1 ICI206,207. Thus, LSD1 inhibitors now take a place among future, potential combinatorial epigenetic therapy strategies to enhance the efficacy of ICI. Pharmacological inhibitors are currently being tested in phase I/II clinical trials involving patients with various advanced-stage malignancies (TABLE 1), including in combination with ICI (Supplementary Table S1).
G9a inhibitors
The histone-lysine N-methyltransferase EHMT2 (also known as G9a) places the aforementioned repressive H3K9me2 mark in chromatin, including in the promoters of abnormally DNA hypermethylated genes (indeed, G9a and DNMT1 can function as part of a ternary complex)208. The gene encoding G9a can be overexpressed, with and without being amplified, in multiple tumour types, which has been associated with advanced-stage disease and an unfavourable prognosis209–211. Of note, knockout studies in mice have revealed a role of G9a in the maintenance of stem cell self-renewal212. Specific inhibitors of G9a are available for preclinical experiments, although a compound suitable for use in clinical trials has not yet been developed. However, an important role for G9a inhibition as a means of inducing viral mimicry has emerged — when used in combination with a DNMT inhibitor, a G9a inhibitor reduces H3K9me2 levels within the long terminal repeat regions of ERVs and thus augments ERV transcription in ovarian cancer cell lines213. Moreover, in cancer cells, repression of ERV sequences without DNA methylation is maintained, in part, by the presence of G9a and H3K9me2 at transcriptional start sites214. Thus, inhibition of G9a is an intriguing future candidate strategy for the enhancement of the therapeutic activity of ICI.
BET inhibitors
The bromodomain and extra-terminal (BET) family encompass a number of epigenetic readers, namely BRD2, BRD3, BRD4 and BRDT; these proteins generally recognize acetylated lysines in histones215, which accompany the open chromatin structures associated with active transcription, as described earlier. BRD4 is the most intensely investigated BET family member and inhibitors of this protein can suppress aberrantly active transcription in cancer216–218. Initially, this inhibition was thought to be focused specifically on targets of MYC oncogene activation, but other affected pathways have now also been identified219. A number of different BET inhibitors are being tested, including in combination with ICI in multiple phase I/II trials (TABLE 1; Supplementary Table S1), predominantly for the treatment of haematopoietic malignancies; a full assessment of efficacy of such agents is awaited.
Preclinical studies indicate that JQ1, one of the original bromodomain-targeted BET inhibitors, synergizes with anti-PD-1 antibodies in a mouse model of NSCLC with activating Kras mutation and Tp53 deletion220. As observed in studies of DNMT plus HDAC inhibition, JQ1 induced an increase in the abundance of activated, tumour-infiltrating T cells with a TH1-type cytokine profile as well as depletion of tumour-infiltrating Treg cells, and resulted in stronger, more-durable antitumour responses and improved survival compared with those observed with either agent alone220. Thus, use of BET inhibitors might provide yet another future combinatorial approach to enhancing the efficacy of ICI.
Conclusions
Epigenetic therapy has emerged as a promising combination partner for use with immunotherapy of advanced-stage malignancies. The potential of epigenetic therapy to enhance patient benefit when compared to immunotherapy alone is centred on its ability to overcome certain limitations of current immunotherapeutic strategies. The success of immunotherapy is dependent on the existence of a certain type of immune environment, principally the presence of tumour-infiltrating lymphocytes and PD-L1 expression in the TME. Epigenetic therapy has been shown to modulate various components of the TME, including augmentation of CTA expression and of antigen processing and presentation, increased attraction and infiltration of CD8+ T cells, and prevention or reversion of T cell exhaustion with a concurrent increase in the abundance of effector and/or memory T cells (FIG. 1). As focused upon in this Review, combining epigenetic therapy with ICI is, therefore, one of several possible combinatorial approaches to enhancing efficacy of the latter treatment strategy. In a growing number of clinical trials across multiple cancer types (Supplementary Table S1), emphasis has been placed upon testing the established epigenetic therapy agents, DNMT inhibitors and HDAC inhibitors (alone or in combination) together with ICI. These approaches are being investigated both in patients receiving their first line of immune-checkpoint inhibition and, more recently, in patients harbouring relapsed and/or refractory disease after prior immune-checkpoint inhibition with the aim of reversing resistance to the immunotherapy. The future adoption of these approaches as accepted cancer management strategies will be dependent on the observation of efficacy signals in these trials. The future will also see the development of novel approaches involving next-generation epigenetic drugs combined with emerging immunotherapy modalities, including vaccine-based and adoptive T cell therapies221,222.
Supplementary Material
Key points.
The past decade has witnessed the emergence of immune-checkpoint inhibition as the potential fourth pillar of anticancer therapy; however, combination therapeutic paradigms are needed to maximize benefits and overcome resistance to immune-checkpoint inhibition.
Epigenetic therapy has the ability to modulate the tumour microenvironment, for example, by inducing both the accumulation and infiltration of CD8+ lymphocytes through interferon-dependent, chemokine-mediated chemotaxis.
Epigenetic therapy can also prevent the emergence and/or acquisition of an epigenetic programme of T cell exhaustion and can facilitate the formation of CD8+ effector and/or memory T cells.
Histone deacetylase inhibitors can affect the tumour myeloid compartment by causing myeloid-derived suppressor cell depletion, differentiation and functional antagonism.
Epigenetic modulators can enhance tumour cell recognition and potentiate type I interferon responses through MYC and MYC-related target downregulation.
The combination of epigenetic drugs and immunotherapy is emerging as a crucial therapeutic paradigm across a variety of malignancies.
Acknowledgements
The work of the authors is supported by grants from The Dr Miriam and Sheldon G. Adelson Medical Research Foundation and the Defense Health Program through the Department of Defense Ovarian Cancer Research Program (Teal Innovator Award No. OC130454/W81XWH-14-1-0385). Opinions, interpretations, conclusions and recommendations presented in this manuscript are those of the author and are not necessarily endorsed by the Department of Defense. The authors also receive funding from The Hodson Trust (S.B.B.), the Commonwealth Foundation (S.B.B. and J.R.B.), the Emerson Cancer Research Award (S.B.B.), the Rising Tide Foundation for Clinical Research (S.B.B. and J.R.B.), the Stand Up To Cancer Jim Toth Sr Breakthrough Prize in Lung Cancer (S.B.B. and J.R.B.), the Van Andel Research Institute through the Van Andel Research Institute-Stand Up To Cancer Epigenetics Dream Team (to S.B.B.; Stand Up To Cancer is a program of the Entertainment Industry Foundation that is administered by AACR), and the NIH National Cancer Institute award number P30CA006973 (SKCCC Core Grant to S.B.B.). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests
S.B.B. is an inventor of the methylation-specific PCR platform, which is licensed to MDxHealth in agreement with Johns Hopkins University; S.B.B. and Johns Hopkins University are entitled to royalty sales shares. S.B.B. is on the Scientific Advisory Board for Mirati Therapeutics. J.R.B. is on advisory board/consultant for Amgen, BMS (uncompensated), Celgene, Genentech, Janssen Oncology, Lilly, Merck and Syndax. J.R.B. recieves grant research funding from AstraZeneca/MedImmune, BMS and Merck. K.A.M. is a consultant for AstraZeneca. All other authors declare no competing interests.
Peer review information
Nature Reviews Clinical Oncology thanks M. Maio and the other, anonymous, reviewers for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41571-019-0266-5.
References
- 1.Hodi FS et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med 364, 2517–2526 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Schadendorf D et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol 33, 1889–1894 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brahmer JR et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol 28, 3167–3175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamid O et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med 369, 134–144 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herbst RS et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Motzer RJ et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J. Clin. Oncol 33, 1430–1437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powles T et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515, 558–562 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Ansell SM et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med 372, 311–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolchok JD et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med 369, 122–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brahmer J et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med 373, 123–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borghaei H et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med 373, 1627–1639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forde PM, Chaft JE & Pardoll DM Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med 379, e14 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Hellmann MD et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decker WK et al. Cancer immunotherapy: historical perspective of a clinical revolution and emerging preclinical animal models. Front. Immunol 8, 829 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahmasebi S, Elahi R & Esmaeilzadeh A Solid tumors challenges and new insights of CAR T cell engineering. Stem Cell Rev. 15, 619–636 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Brown MP, Ebert LM & Gargett T Clinical chimeric antigen receptor-T cell therapy: a new and promising treatment modality for glioblastoma. Clin. Transl Immunology 8, e1050 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berzofsky JA et al. Cancer vaccine strategies: translation from mice to human clinical trials. Cancer Immunol. Immunother 67, 1863–1869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banday AH, Jeelani S & Hruby VJ Cancer vaccine adjuvants-recent clinical progress and future perspectives. Immunopharmacol. Immunotoxicol 37, 1–11 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Mougel A, Terme M & Tanchot C Therapeutic cancer vaccine and combinations with antiangiogenic therapies and immune checkpoint blockade. Front. Immunol 10, 467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang F, Xiao W & Tian Z NK cell-based immunotherapy for cancer. Semin. Immunol 31, 37–54 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Muntasell A et al. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol 45, 73–81 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Becker PS et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol. Immunother 65, 477–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heong V, Ngoi N & Tan DS Update on immune checkpoint inhibitors in gynecological cancers. J. Gynecol. Oncol 28, e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strasner A & Karin M Immune infiltration and prostate cancer. Front. Oncol 5, 128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auvray M et al. Second-1ine targeted therapies after nivolumab-ipilimumab failure in metastatic renal cell carcinoma. Eur. J. Cancer 108, 33–40 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Skoulidis F et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pham T et al. An update on immunotherapy for solid tumors: a review. Ann. Surg. Oncol 25, 3404–3412 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Torphy RJ, Zhu Y & Schulick RD Immunotherapy for pancreatic cancer: barriers and breakthroughs. Ann. Gastroenterol. Surg 2, 274–281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Royal RE et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother 33, 828–833 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brahmer JR et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmid P et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Motzer RJ et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med 378, 1277–1290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodi FS et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 19, 1480–1492 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Gandhi L et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med 378, 2078–2092 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Paz-Ares L et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med 379, 2040–2051 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Antonia SJ et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N. Engl. J. Med 377, 1919–1929 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Alomari AK et al. Possible interaction of anti-PD-1 therapy with the effects of radiosurgery on brain metastases. Cancer Immunol. Res 4, 481–487 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Haymaker CL et al. Metastatic melanoma patient had a complete response with clonal expansion after whole brain radiation and PD-1 blockade. Cancer Immunol. Res 5, 100–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagasaka M et al. PD1/PD-L1 inhibition as a potential radiosensitizer in head and neck squamous cell carcinoma: a case report. J. Immunother. Cancer 4, 83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leach DR, Krummel MF & Allison JP Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Okazaki T & Honjo T PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol 19, 813–824 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freeman GJ et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192, 1027–1034 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabinovich GA, Gabrilovich D & Sotomayor EM Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol 25, 267–296 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schreiber RD, Old LJ & Smyth MJ Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Marincola FM, Jaffee EM, Hicklin DJ & Ferrone S Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv. Immunol 74, 181–273 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Sakaguchi S, Yamaguchi T, Nomura T & Ono M Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Stamper CC et al. Crystal structure of the B7–1/CTLA-4 complex that inhibits human immune responses. Nature 410, 608–611 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Iwai Y et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamazaki T et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol 169, 5538–5545 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Kuang DM et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med 206, 1327–1337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pander J et al. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin. Cancer Res 17, 5668–5673 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Zahavi DJ & Weiner LM Targeting multiple receptors to increase checkpoint blockade efficacy. Int. J. Mol. Sci 20, E158 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pages F et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol 27, 5944–5951 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Galon J et al. Towards the introduction of the ‘immunoscore’ in the classification of malignant tumours. J. Pathol 232, 199–209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lanitis E, Dangaj D, Irving M & Coukos G Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol 28, xii18–xii32 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Peranzoni E et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl Acad. Sci. USA 115, E4041–E4050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shin JI & Ha SJ Regulatory T cells-an important target for cancer immunotherapy. Nat. Rev. Clin. Oncol 11, 307 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Woo EY et al. Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 61, 4766–4772 (2001). [PubMed] [Google Scholar]
- 62.Togashi Y, Shitara K & Nishikawa H Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol 16, 356–371 (2019). [DOI] [PubMed] [Google Scholar]
- 63.de Charette M, Marabelle A & Houot R Turning tumour cells into antigen presenting cells: the next step to improve cancer immunotherapy? Eur. J. Cancer 68, 134–147 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Beatty GL & Gladney WL Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res 21, 687–692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dustin ML The immunological synapse. Cancer Immunol. Res 2, 1023–1033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tamada K [Development of novel immunotherapy targeting cancer immune evasion]. Gan Kagaku Ryoho 41, 1062–1065 (2014). [PubMed] [Google Scholar]
- 67.Chen DS & Mellman I Elements of cancer immunity and the cancer-i mmune set point. Nature 541, 321–330 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Joyce JA & Fearon DT T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Yarchoan M, Hopkins A & Jaffee EM Tumor mutational burden and response rate to pd-1 inhibition. N. Engl. J. Med 377, 2500–2501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spranger S et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl Acad. Sci. USA 113, E7759–E7768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turajlic S et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 18, 1009–1021 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Jones PA & Baylin SB The epigenomics of cancer. Cell 128, 683–692 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baylin SB The cancer epigenome: its origins, contributions to tumorigenesis, and translational implications. Proc. Am. Thorac. Soc 9, 64–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baylin SB & Jones PA A decade of exploring the cancer epigenome — biological and translational implications. Nat. Rev. Cancer 11, 726–734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen H & Laird PW Interplay between the cancer genome and epigenome. Cell 153, 38–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baylin SB & Jones PA Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol 10.1101/cshperspect.a019505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Esteller M Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum. Mol. Genet 16, R50–R59 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Portela A & Esteller M Epigenetic modifications and human disease. Nat. Biotechnol 28, 1057–1068 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Esteller M Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet 8, 286–298 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Kouzarides T Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Struhl K & Segal E Determinants of nucleosome positioning. Nat. Struct. Mol. Biol 20, 267–273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allis CD, Jenuwein T, Reinberg D & Caparros M in Epigenetics 2nd edn (Cold Spring Harbor Laboratory Research Press, 2015). [Google Scholar]
- 83.Hyun K, Jeon J, Park K & Kim J Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49, e324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Allis CD & Jenuwein T The molecular hallmarks of epigenetic control. Nat. Rev. Genet 17, 487–500 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Dawson MA & Kouzarides T Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Zhou VW, Goren A & Bernstein BE Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet 12, 7–18 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Bonasio R, Tu S & Reinberg D Molecular signals of epigenetic states. Science 330, 612–616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pfister SX & Ashworth A Marked for death: targeting epigenetic changes in cancer. Nat. Rev. Drug Discov 16, 241–263 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Skulte KA, Phan L, Clark SJ & Taberlay PC Chromatin remodeler mutations in human cancers: epigenetic implications. Epigenomics 6, 397–414 (2014). [DOI] [PubMed] [Google Scholar]
- 90.Suva ML, Riggi N & Bernstein BE Epigenetic reprogramming in cancer. Science 339, 1567–1570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Easwaran H, Tsai HC & Baylin SB Cancer epigenetics: tumor heterogeneity, plasticity of stem-l ike states, and drug resistance. Mol. Cell 54, 716–727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kulis M & Esteller M DNA methylation and cancer. Adv. Genet 70, 27–56 (2010). [DOI] [PubMed] [Google Scholar]
- 93.Herman JG et al. Incidence and functional consequences of hMLH 1 promoter hypermethylation in colorectal carcinoma. Proc. Natl Acad. Sci. USA 95, 6870–6875 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong DJ, Barrett MT, Stoger R, Emond MJ & Reid BJ p16INK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res. 57, 2619–2622 (1997). [PubMed] [Google Scholar]
- 95.Klump B, Hsieh CJ, Holzmann K, Gregor M & Porschen R Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 115, 1381–1386 (1998). [DOI] [PubMed] [Google Scholar]
- 96.Jones PA & Laird PW Cancer epigenetics comes of age. Nat. Genet 21, 163–167 (1999). [DOI] [PubMed] [Google Scholar]
- 97.Kron KJ, Bailey SD & Lupien M Enhancer alterations in cancer: a source for a cell identity crisis. Genome Med. 6, 77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aran D & Hellman A DNA methylation of transcriptional enhancers and cancer predisposition. Cell 154, 11–13 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Hnisz D, Day DS & Young RA Insulated neighborhoods: structural and functional units of mammalian gene control. Cell 167, 1188–1200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aran D & Hellman A Unmasking risk loci: DNA methylation illuminates the biology of cancer predisposition: analyzing DNA methylation of transcriptional enhancers reveals missed regulatory links between cancer risk loci and genes. Bioessays 36, 184–190 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Loven J et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chi P, Allis CD & Wang GG Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 104.Cameron EE, Bachman KE, Myohanen S, Herman JG & Baylin SB Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet 21, 103–107 (1999). [DOI] [PubMed] [Google Scholar]
- 105.Verdone L, Caserta M & Di Mauro E Role of histone acetylation in the control of gene expression. Biochem. Cell Biol 83, 344–353 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Gallinari P, Di Marco S, Jones P, Pallaoro M & Steinkuhler C HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 17, 195–211 (2007). [DOI] [PubMed] [Google Scholar]
- 107.El-Osta A & Wolffe AP DNA methylation and histone deacetylation in the control of gene expression: basic biochemistry to human development and disease. Gene Expr. 9, 63–75 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Irvine RA, Lin IG & Hsieh CL DNA methylation has a local effect on transcription and histone acetylation. Mol. Cell Biol 22, 6689–6696 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Struhl K Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12, 599–606 (1998). [DOI] [PubMed] [Google Scholar]
- 110.Zahnow CA et al. Inhibitors of dna methylation, histone deacetylation, and histone demethylation: a perfect combination for cancer therapy. Adv. Cancer Res 130, 55–111 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Jones PA, Issa JP & Baylin S Targeting the cancer epigenome for therapy. Nat. Rev. Genet 17, 630–641 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Tripathi SK & Lahesmaa R Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol. Rev 261, 62–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shih HY et al. Transcriptional and epigenetic networks of helper T and innate lymphoid cells. Immunol. Rev 261, 23–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wilson CB, Makar KW & Perez-Melgosa M Epigenetic regulation of T cell fate and function. J. Infect. Dis 185, S37–S45 (2002). [DOI] [PubMed] [Google Scholar]
- 115.Hu G et al. Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity 48, 227–242.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Johnson JL et al. Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of t cells. Immunity 48, 243–257.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zheng H et al. HDAC inhibitors enhance T-cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin. Cancer Res 22, 4119–4132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Youngblood B et al. Effector CD8 T cells dedifferentiate into long-l ived memory cells. Nature 552, 404–409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ghoneim HE et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell 170, 142–157.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chang JT, Wherry EJ & Goldrath AW Molecular regulation of effector and memory T cell differentiation. Nat. Immunol 15, 1104–1115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carty SA et al. The loss of TET2 promotes CD8+ T cell memory differentiation. J. Immunol 200, 82–91 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scharer CD, Bally AP, Gandham B & Boss JM Cutting edge: chromatin accessibility programs CD8 T cell memory. J. Immunol 198, 2238–2243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wherry EJ T cell exhaustion. Nat. Immunol 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 125.Hashimoto M et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med 69, 301–318 (2018). [DOI] [PubMed] [Google Scholar]
- 126.Youngblood B et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity 35, 400–412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sen DR et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pauken KE et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alvarez-Errico D, Vento-Tormo R, Sieweke M & Ballestar E Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol 15, 7–17 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Ivashkiv LB & Park SH Epigenetic regulation of myeloid cells. Microbiol. Spectr 10.1128/microbiolspec.MCHD-0010-2015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Srivastava MK, Sinha P, Clements VK, Rodriguez P & Ostrand-Rosenberg S Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 70, 68–77 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang H et al. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol. Immunother 64, 1587–1599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nagaraj S, Schrum AG, Cho HI, Celis E & Gabrilovich DI Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol 184, 3106–3116 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rodriguez RM, Suarez-Alvarez B & Lopez-Larrea C Therapeutic epigenetic reprogramming of trained immunity in myeloid cells. Trends Immunol 40, 66–80 (2019). [DOI] [PubMed] [Google Scholar]
- 135.de Groot AE & Pienta KJ Epigenetic control of macrophage polarization: implications for targeting tumor-associated macrophages. Oncotarget 9, 20908–20927 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Travers M et al. DFMO and 5-azacytidine increase M1 macrophages in the tumor microenvironment of murine ovarian cancer. Cancer Res. 79, 3445–3454 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsai HC & Baylin SB Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 21, 502–517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tsai HC et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 21, 430–446 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Baylin SB & Ohm JE Epigenetic gene silencing in cancer — a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6, 107–116 (2006). [DOI] [PubMed] [Google Scholar]
- 140.Issa JJ et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 16, 1099–1110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wherry EJ et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 142.Wrangle J et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 4, 2067–2079 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li H et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 5, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Topper MJ et al. Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 171, 1284–1300.e21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Heninger E, Krueger TE & Lang JM Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol 6, 29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chiappinelli KB et al. Inhibiting DNA methylation causes an interferon response in cancer via dsrna including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roulois D et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Natsume A et al. The DNA demethylating agent 5-aza-2’-deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. Int. J. Cancer 122, 2542–2553 (2008). [DOI] [PubMed] [Google Scholar]
- 149.Moreno-Bost A et al. Epigenetic modulation of MAGE-A3 antigen expression in multiple myeloma following treatment with the demethylation agent 5-azacitidine and the histone deacetlyase inhibitor MGCD0103. Cytotherapy 13, 618–628 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Almstedt M et al. The DNA demethylating agent 5-aza-2’-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk. Res 34, 899–905 (2010). [DOI] [PubMed] [Google Scholar]
- 151.Goodyear O et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 116, 1908–1918 (2010). [DOI] [PubMed] [Google Scholar]
- 152.James SR, Link PA & Karpf AR Epigenetic regulation of X-l inked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene 25, 6975–6985 (2006). [DOI] [PubMed] [Google Scholar]
- 153.Weber J et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2’-deoxycytidine. Cancer Res. 54, 1766–1771 (1994). [PubMed] [Google Scholar]
- 154.De Smet C et al. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc. Natl Acad. Sci. USA 93, 7149–7153 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li J et al. Expression of BAGE, GAGE, and MAGE genes in human gastric carcinoma. Clin. Cancer Res 2, 1619–1625 (1996). [PubMed] [Google Scholar]
- 156.Sigalotti L et al. Promoter methylation controls the expression of MAGE2, 3 and 4 genes in human cutaneous melanoma. J. Immunother 25, 16–26 (2002). [DOI] [PubMed] [Google Scholar]
- 157.De Smet C, Lurquin C, Lethe B, Martelange V & Boon T DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol. Cell Biol 19, 7327–7335 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Maatouk DM et al. DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development 133, 3411–3418 (2006). [DOI] [PubMed] [Google Scholar]
- 159.Fratta E et al. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol. Oncol 5, 164–182 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ritter C et al. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep 7, 2290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ramsuran V et al. Epigenetic regulation of differential HLA-A allelic expression levels. Hum. Mol. Genet 24, 4268–4275 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Coral S et al. Prolonged upregulation of the expression of HLA class I antigens and costimulatory molecules on melanoma cells treated with 5-aza-2’-deoxycytidine (5-AZA-CdR). J. Immunother 22, 16–24 (1999). [DOI] [PubMed] [Google Scholar]
- 163.Nie Y et al. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis 22, 1615–1623 (2001). [DOI] [PubMed] [Google Scholar]
- 164.Karpf AR et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc. Natl Acad. Sci. USA 96, 14007–14012 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Griffiths DJ Endogenous retroviruses in the human genome sequence. Genome Biol. 2, REVIEWS1017 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lavie L, Kitova M, Maldener E, Meese E & Mayer J CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). J. Virol 79, 876–883 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Szpakowski S et al. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 448, 151–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brady T et al. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 23, 633–642 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jones PA, Ohtani H, Chakravarthy A & De Carvalho DD Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 19, 151–161 (2019). [DOI] [PubMed] [Google Scholar]
- 170.Juergens RA et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 1, 598–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang L et al. Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 blockade in a murine ovarian cancer model. Cancer Immunol. Res 3, 1030–1041 (2015). [DOI] [PubMed] [Google Scholar]
- 172.Luo N et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun 9, 248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yu G et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol. Immunol 16, 401–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Woods DM et al. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res 3, 1375–1385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Llopiz D et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother 68, 379–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kim K et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl Acad. Sci. USA 111, 11774–11779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Orillion A et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin. Cancer Res 23, 5187–5201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Stone ML et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl Acad. Sci. USA 114, E10981–E10990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kawai T et al. Selective diapedesis of Th1 cells induced by endothelial cell RANTES. J. Immunol 163, 3269–3278 (1999). [PubMed] [Google Scholar]
- 180.Schall TJ, Bacon K, Toy KJ & Goeddel DV Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 347, 669–671 (1990). [DOI] [PubMed] [Google Scholar]
- 181.Lederman MM, Penn-Nicholson A, Cho M & Mosier D Biology of CCR5 and its role in HIV infection and treatment. JAMA 296, 815–826 (2006). [DOI] [PubMed] [Google Scholar]
- 182.Stanford MM & Issekutz TB The relative activity of CXCR3 and CCR5 ligands in T lymphocyte migration: concordant and disparate activities in vitro and in vivo. J. Leukoc. Biol 74, 791–799 (2003). [DOI] [PubMed] [Google Scholar]
- 183.Moran CJ et al. RANTES expression is a predictor of survival in stage I lung adenocarcinoma. Clin. Cancer Res 8, 3803–3812 (2002). [PubMed] [Google Scholar]
- 184.Kortlever RM et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Casey SC et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Folkes AS et al. Targeting CD47 as a cancer therapeutic strategy: the cutaneous T-cell lymphoma experience. Curr. Opin. Oncol 30, 332–337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Daver N et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-iabel, phase II study. Cancer Discov 9, 370–383 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Levy BP et al. Randomised phase 2 study of pembrolizumab plus CC-486 versus pembrolizumab plus placebo in patients with previously treated advanced non-small cell lung cancer. Eur. J. Cancer 108, 120–128 (2019). [DOI] [PubMed] [Google Scholar]
- 189.Savona MR et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am. J. Hematol 93, 1199–1206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Genta S, Pirosa MC & Stathis A BET and EZH2 inhibitors: novel approaches for targeting cancer. Curr. Oncol. Rep 21, 13 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Kim KH & Roberts CW Targeting EZH2 in cancer. Nat. Med 22, 128–134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Margueron R & Reinberg D The polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yamagishi M & Uchimaru K Targeting EZH2 in cancer therapy. Curr. Opin. Oncol 29, 375–381 (2017). [DOI] [PubMed] [Google Scholar]
- 194.Schlesinger Y et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 39, 232–236 (2007). [DOI] [PubMed] [Google Scholar]
- 195.Widschwendter M et al. Epigenetic stem cell signature in cancer. Nat. Genet 39, 157–158 (2007). [DOI] [PubMed] [Google Scholar]
- 196.Easwaran H et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 22, 837–849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ohm JE & Baylin SB Stem cell chromatin patterns: an instructive mechanism for DNA hypermethylation? Cell Cycle 6, 1040–1043 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Goswami S et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Invest 128, 3813–3818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zingg D et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 20, 854–867 (2017). [DOI] [PubMed] [Google Scholar]
- 201.Hosseini A & Minucci S A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 9, 1123–1142 (2017). [DOI] [PubMed] [Google Scholar]
- 202.Wang J et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet 41, 125–129 (2009). [DOI] [PubMed] [Google Scholar]
- 203.Morera L, Lubbert M & Jung M Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 8, 57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Han H, Yang X, Pandiyan K & Liang G Synergistic re-activation of epigenetically silenced genes by combinatorial inhibition of DNMTs and LSD1 in cancer cells. PLOS ONE 8, e75136 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yang GJ, Lei PM, Wong SY, Ma DL & Leung CH Pharmacological inhibition of LSD1 for cancer treatment. Molecules 23, E3194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sheng W et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Qin Y et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 38, 390–405 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Esteve PO et al. Direct interaction between DNMT 1 and G9a coordinates DNA and histone methyl ation during replication. Genes Dev. 20, 3089–3103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Huang T et al. G9A promotes tumor cell growth and invasion by silencing CASP1 in non-small-cell lung cancer cells. Cell Death Dis. 8, e2726 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Casciello F, Windloch K, Gannon F & Lee JS Functional role of G9a histone methyltransferase in cancer. Front. Immunol 6, 487 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hu L et al. G9A promotes gastric cancer metastasis by upregulating ITGB3 in a SET domain-i ndependent manner. Cell Death Dis 9, 278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ma DK, Chiang CH, Ponnusamy K, Ming GL & Song H G9a and Jhdm2a regulate embryonic stem cell fusion-induced reprogramming of adult neural stem cells. Stem Cells 26, 2131–2141 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liu M et al. Dual inhibition of dna and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. 78, 5754–5766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Di Giacomo M, Comazzetto S, Sampath SC, Sampath SC & O’Carroll D G9a co-suppresses LINE1 elements in spermatogonia. Epigenetics Chromatin 7, 24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zeng L & Zhou MM Bromodomain: an acetyl-iysine binding domain. FEBS Lett. 513, 124–128 (2002). [DOI] [PubMed] [Google Scholar]
- 216.Filippakopoulos P et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Perez-Salvia M & Esteller M Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics 12, 323–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Xu Y & Vakoc CR Targeting cancer cells with bet bromodomain inhibitors. Cold Spring Harb. Perspect. Med 7, a026674 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tang Y et al. Epigenetic targeting of hedgehog pathway transcriptional output through bet bromodomain inhibition. Nat. Med 20, 732–740 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Adeegbe DO et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in KRAS-mutant non-small cell lung cancer. Cancer Immunol. Res 6, 1234–1245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lu D et al. Treatment with demethylating agent, 5-aza-2’-deoxycytidine enhances therapeutic HPV DNA vaccine potency. Vaccine 27, 4363–4369 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vo DD et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 69, 8693–8699 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gonzalez H, Hagerling C & Werb Z Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev 32, 1267–1284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Teng MW, Ngiow SF, Ribas A & Smyth MJ Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 75, 2139–2145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Galon J & Bruni D Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov 18, 197–218 (2019). [DOI] [PubMed] [Google Scholar]
- 226.Dong H et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med 8, 793–800 (2002). [DOI] [PubMed] [Google Scholar]
- 227.Taube JM et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med 4, 127ra37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ribas A Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov 5, 915–919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zou W & Chen L Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev Immunol 8, 467–477 (2008). [DOI] [PubMed] [Google Scholar]
- 230.Tumeh PC et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pages F et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139 (2018). [DOI] [PubMed] [Google Scholar]
- 232.Huang RR et al. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin. Cancer Res 17, 4101–4109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bald T et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 4, 674–687 (2014). [DOI] [PubMed] [Google Scholar]
- 234.Kershaw MH, Westwood JA & Darcy PK Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 13, 525–541 (2013). [DOI] [PubMed] [Google Scholar]
- 235.Parsa AT et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med 13, 84–88 (2007). [DOI] [PubMed] [Google Scholar]
- 236.Marzec M et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl Acad. Sci. USA 105, 20852–20857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Spranger S & Gajewski TF Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Schietinger A et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Spranger S Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 28, 383–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Leone RD & Emens LA Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 6, 57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yentz S & Smith D Indoleamine 2,3-dioxygenase (IDO) inhibition as a strategy to augment cancer immunotherapy. BioDrugs 32, 311–317 (2018). [DOI] [PubMed] [Google Scholar]
- 242.Makkouk A & Weiner GJ Cancer immunotherapy and breaking immune tolerance: new approaches to an old challenge. Cancer Res. 75, 5–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Scharer CD, Barwick BG, Youngblood BA, Ahmed R & Boss JM Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol 191, 3419–3429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Haring JS, Badovinac VP & Harty JT Inflaming the CD8+ T cell response. Immunity 25, 19–29 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Pozzi LA, Maciaszek JW & Rock KL Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J. Immunol 175, 2071–2081 (2005). [DOI] [PubMed] [Google Scholar]
- 246.Pennock ND et al. T cell responses: naive to memory and everything in between. Adv. Physiol. Educ 37, 273–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Appleman LJ & Boussiotis VA T cell anergy and costimulation. Immunol. Rev 192, 161–180 (2003). [DOI] [PubMed] [Google Scholar]
- 248.Allison JP CD28-B7 interactions in T-cell activation. Curr. Opin. Immunol 6, 414–419 (1994). [DOI] [PubMed] [Google Scholar]
- 249.Chen L & Flies DB Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev Immunol 13, 227–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Umlauf SW, Beverly B, Lantz O & Schwartz RH Regulation of interleukin 2 gene expression by CD28 costimulation in mouse T-cell clones: both nuclear and cytoplasmic RNAs are regulated with complex kinetics. Mol. Cell Biol 15, 3197–3205 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Curtsinger JM & Mescher MF Inflammatory cytokines as a third signal for T cell activation. Curr Opin. Immunol 22, 333–340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ramos HJ et al. Reciprocal responsiveness to interleukin-12 and interferon-a specifies human CD8+ effector versus central memory T-cell fates. Blood 113, 5516–5525 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Xiao S et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-p-driven Smad3 signaling and inhibiting IL-6 and Il-23 receptor expression. J. Immunol 181, 2277–2284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Binnewies M et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kaech SM & Wherry EJ Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27, 393–405 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hokeness KL et al. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J. Virol 81, 1241–1250 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ahn E et al. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl Acad. Sci. USA 115, 4749–4754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Okazaki T, Maeda A, Nishimura H, Kurosaki T & Honjo T PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl Acad. Sci. USA 98, 13866–13871 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Brinkman CC, Peske JD & Engelhard VH Peripheral tissue homing receptor control of naive, effector, and memory CD8 T cell localization in lymphoid and non-iymphoid tissues. Front. Immunol 4, 241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Angelosanto JM, Blackburn SD, Crawford A & Wherry EJ Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol 86, 8161–8170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Qin S et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Invest 101, 746–754 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ribas A et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol. Res 4, 194–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Gerlach C et al. The chemokine receptor cx3cr1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45, 1270–1284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.