

SUMMARY
All forms of diabetes mellitus (DM) are characterized by the loss of functional pancreatic β-cell mass, leading to insufficient insulin secretion. Thus, identification of novel approaches to protect and restore β-cells is essential for the development of DM therapies. Mesencephalic astrocyte-derived neurotrophic factor (MANF) is an endoplasmic reticulum (ER) stress inducible protein but its physiological role in mammals has remained obscure. We generated MANF-deficient mice that strikingly develop severe diabetes due to progressive postnatal reduction of β-cell mass, caused by decreased proliferation and increased apoptosis. Additionally, we show that lack of MANF in vivo in mouse leads to chronic unfolded protein response (UPR) activation in pancreatic islets. Importantly, MANF protein enhanced β-cell proliferation in vitro and overexpression of MANF in the pancreas of diabetic mice enhanced β-cell regeneration. We demonstrate that MANF specifically promotes β-cell proliferation and survival, thereby constituting a novel therapeutic candidate for β-cell protection and regeneration.
INTRODUCTION
Diabetes mellitus (DM) is a group of metabolic disorders characterized by the loss of functional pancreatic β-cell mass, leading to insufficient insulin secretion (Talchai et al. 2012; Weir and Bonner-Weir, 2013). Current diabetes therapies cannot prevent β-cell death or promote regeneration of remaining β-cells and rarely result in complete long-term metabolic normalization. Thus, one of the main strategies in improving current DM therapy is to define and validate novel approaches to protect and restore β-cell mass (Donath and Halban, 2004). In both rodents and humans, β-cells are formed by neogenesis from endocrine progenitor cells which proliferate extensively during the end of embryogenesis and early postnatal period to reach the proper adult β-cell mass (Dhawan et al., 2007; Meier et al., 2008).
A number of cellular insults can disrupt protein folding and cause accumulation of unfolded proteins triggering ER stress and if prolonged, lead to ER stress induced apoptosis (Szegezdi et al., 2006). Accumulation and aggregation of unfolded proteins results in dissociation of general ER stress chaperone GRP78/Bip from ER stress sensors PERK, ATF6 and IRE1, activation of downstream signaling UPR cascades, finally resulting in decreased protein synthesis, increased protein folding capacity and degradation of misfolded proteins (Szegezdi et al., 2006; Walter and Ron, 2011). Importantly, alterations in proteins involved in ER stress and UPR are linked to diabetes in humans and mice suggesting that unresolved ER stress is involved in the pathogenesis of β-cell loss in type 1 (T1D) and type 2 (T2D) diabetes (Delepine et al., 2000; Eizirik et al., 2008; Eizirik et al., 2013; Hetz, 2012).
MANF together with cerebral dopamine neurotrophic factor (CDNF) forms a new, highly evolutionarily conserved protein family, efficiently protecting and repairing midbrain dopaminergic neurons in animal models of Parkinso’s disease, protecting cardiac myocytes in myocardial infarction, and cortical neurons against ischemic stroke (Airavaara et al., 2009; Glembotski et al., 2012; Hellman et al., 2011; Lindholm et al., 2008; Lindholm and Saarma, 2010; Lindholm et al., 2007; Petrova et al., 2003; Voutilainen et al., 2009). However, the cytoprotective mechanisms of MANF are not known.
MANF mRNA and protein are widely expressed in most human and mouse organs with high levels in glandular cells of secretory tissues such as pancreas and salivary gland (Lindholm et al., 2008). Intracellularly MANF localizes to the luminal ER where it interacts with the chaperone GRP78 and is secreted in response to experimental ER stress in vitro (Apostolou et al., 2008; Glembotski et al., 2012; Lindholm and Saarma, 2010; Mizobuchi et al., 2007). Thus, recent studies suggest that MANF is an ER stress inducible protein for several cell populations.
To understand the physiological role of MANF in vivo, we generated MANF knockout mice (Manf−/−). Surprisingly, Manf−/− mice develop insulin deficient diabetes due to progressive postnatal reduction of β-cell mass caused by decreased β-cell proliferation and increased apoptosis. We also demonstrate that pancreatic islets of Manf−/− mice display activation of UPR genes and proteins, implicating unresolved ER stress as a primary cause of β-cell failure. Consistently, recombinant MANF protein enhanced β-cell proliferation in vitro. Furthermore, after streptozotocin-induced diabetes, we found enhanced β-cell proliferation and reduced β-cell death in the regions of pancreas that had been transduced to overexpress MANF. Our results demonstrate that MANF specifically promotes β-cell proliferation and survival thereby constituting a promising therapeutic agent for β-cell protection and regeneration.
RESULTS
Loss of MANF in mice results in growth retardation and diabetes
To understand the physiological role of MANF in vivo, we generated MANF knockout mice (Manf−/−) from an embryonic stem cell clone MANF_D06 (EPD0162_3_D06, C57Bl/6N-Manftm1a(KOMP)Wtsi), containing a β-galactosidase reporter cassette with a strong splice acceptor site inserted in the intron between exon 2 and exon 3 of the Manf gene, creating a constitutive null mutation through splicing of exon 2 to the reporter cassette (Figure 1A). We confirmed that MANF full-length mRNA and protein were not expressed in tissues of Manf−/− mice (Figure S1A–1B). We found that MANF-deficient mice display progressive gender independent growth retardation (Figure 1B). In addition, the body size and fat pad weights were significantly reduced in Manf−/− male mice compared to WT mice (Figure S1D–F). At 8–12 weeks of age, the health of the mutant mice quickly deteriorated and the animals were euthanized.
Figure 1. Retarded growth, hyperglycemia and hypoinsulinemia in Manf-deficient mice.
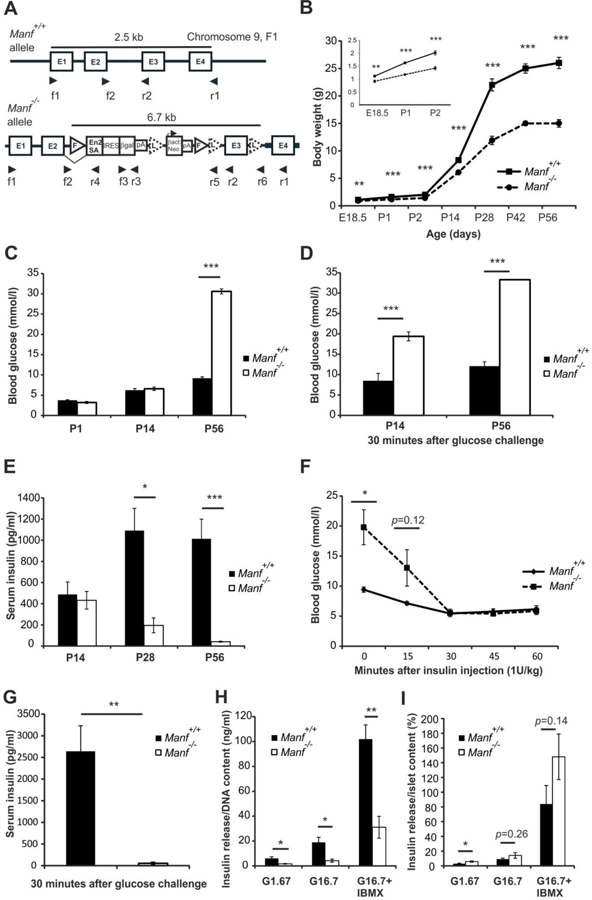
(A) Schematic illustration of the wild-type Manf+/+and mutant Manf−/− loci. A β-galactosidase reporter cassette (β-gal), strong splicing acceptor site (En2SA), exon (E), Frt (F)-site, LoxP (L)-site, bAct::Neo; human β-actin promoter driven neomycin resistance gene. Arrowheads indicate priming sites used in genotyping by PCR and in RT-PCR. (B) Growth curve of Manf+/+and Manf−/− littermates. Embryonic day (E) 18.5 to postnatal day (P) 2 (inset), n = 5–41, both sexes. P14-P56, n = 9–16, only males. (C) Ad libitum fed blood glucose levels, n = 16–34. (D) Blood glucose levels 30 minutes after glucose bolus injection, n = 4–12. (E) Serum insulin levels from ad libitum fed mice, n = 8–20. (F) Blood glucose levels measured after insulin injection, n = 5 per group. (G) Serum insulin levels in P56 mice measured 30 minutes after glucose bolus injection, n = 4. (H) In vitro insulin release from islets in response to low glucose (1.67 mmol/l; G1.67), high glucose 16.7 mmol/l; G16.7 and high glucose with IBMX 1 mmol/l, normalized to islet DNA content after 1 hour and (I) in vitro glucose stimulated insulin release compared to total islet insulin content, n = islets from 5–6 pancreases/group. Mean ± s.e.m., *p<0.05, **p<0.01, ***p<0.001 versus corresponding control. See also Figures S1 and S2.
The blood glucose levels of ad libitum fed animals were within normal range for Manf−/− mice compared to wild-type Manf+/+ measured at postnatal day 1 (P1) and at P14 (Figure 1C). However, at P56 the Manf−/− mice were severely hyperglycemic (Figure 1C) and they consumed significantly more water than Manf+/+ littermates (Figure S1G). Glucose tolerance test showed a severely compromised glucose clearance compared to Manf+/+ mice already at P14 (Figure 1D). In addition, significantly reduced levels of insulin were observed from sera of fed P28 Manf−/− mice compared to Manf+/+ littermates and at P56, insulin was barely detectable (Figure 1E). Insulin tolerance test suggested intact insulin sensitivity in the Manf−/− mice (Figure 1F). Circulating glucose-stimulated insulin levels were dramatically decreased in P56 Manf−/− mice (Figure 1G). Although remaining islets derived from diabetic P35 Manf−/− mice, secreted less insulin than WT islets after an overnight culture period (Figure 1H), their capacity to secrete insulin relative to cellular insulin content did not differ in response to glucose or glucose plus IBMX (Figure 1 I).
The diabetic phenotype was confirmed in independent conditional PGKCre/+:: Manfflox/flox(fl/fl) and pancreas specific Pdx1Cre/+:: Manfflox/flox(fl/fl) mouse lines (Supplementary Result Text and Figure S2A–1). Importantly, the diabetic phenotype was not observed when MANF was deleted mainly in central nervous system NestinCre/+:: Manf(fl/fl) mice, highlighting the indispensable role of pancreatic MANF in regulation of β-cell mass (Supplementary Result Text and Figure S2J–K).
Postnatal reduction in pancreatic β-cell mass in Manf−/− mice results from decreased β-cell proliferation and enhanced apoptosis
Immunohistological analysis revealed a progressive postnatal reduction in β-cell mass, loss of islet architecture and decreased intensity of insulin immunoreactivity in Manf−/− mice (Figure 2A–G). At P56 most of the islet insulin-positive β-cells were lost from Manf−/− pancreases (Figure 2E–F). Quantification of β-cell mass revealed no difference between genotypes at E18.5 whereas already by P1 the reduction in Manf−/− pancreases was 50% and by P56, 85% (Figure 2G). Proliferation is important for the β-cell expansion during late embryogenesis and in the neonatal period to reach the proper β-cell mass (Dhawan et al., 2007). There were no differences between genotypes in the number of proliferating (BrdU+ or Ki67+) insulin-positive β-cells quantified from pancreases at E18.5 (Figure 2H and Figure S3A–B), but a significant reduction in Ki67+ β-cells was observed in P1 and P14 Manf−/− mice (Figure 2H). Importantly, the number of proliferating exocrine acinar cells did not differ between genotypes (Figure 2I). Quantification of TUNEL+ β-cells revealed increased β-cell apoptosis in Manf−/− pancreas at P14 and P56 that coincided with the reduction of β-cell mass (Figure 2J and Figure S3C). Glucagon immunohistochemistry showed a normal alpha cell mass, indicating that the phenotype is β-cell specific (Figure S3D–J).
Figure 2. Decline in β-cell mass in Manf-deficient mice is caused by reduced β-cell proliferation and increased β-cell death.
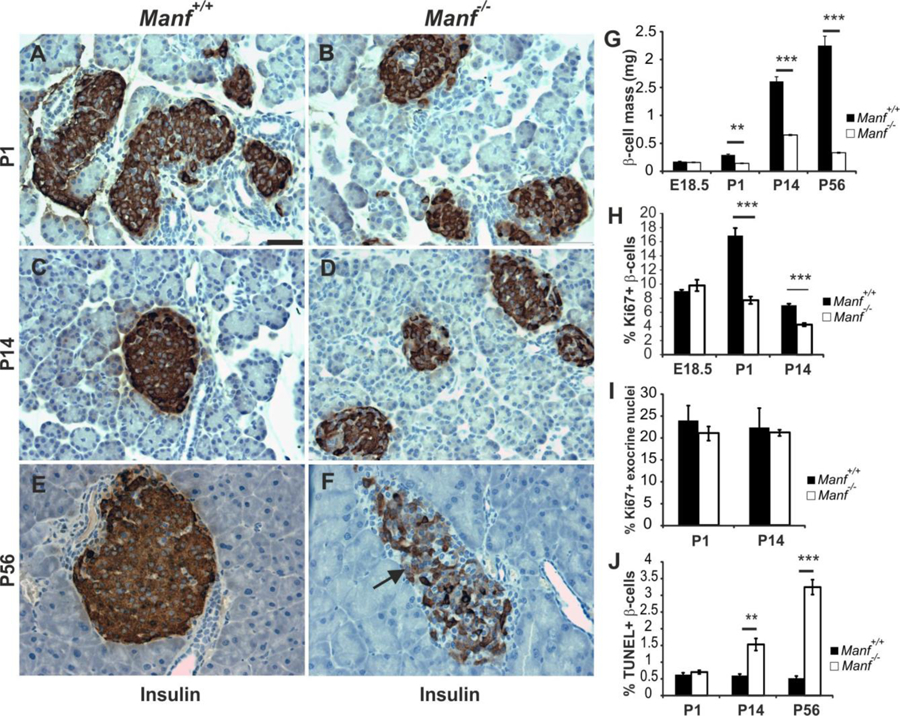
(A to F) Insulin immunohistochemistry on pancreas sections from Manf+/+ (A, C and E) and Manf−/− (B, D and F) animals at P1 (A and B), P14 (C and D) and P56 (E and F). Arrow in (F) points to β-cell with reduced insulin staining. Scale bar, 50 μm. (G) Progressive reduction in β-cell mass in Manf−/− mice from P1, n = 5 per group. (H) β-cell proliferation assessed by Ki67 and insulin staining, n = 3–5 per group. (I) Acinar cell proliferation assessed from Ki67-positive DAPI-stained nuclei from pancreatic exocrine tissue, n =5–6 per group. (J) Islet β-cell death assed by TUNEL and insulin double-staining, n = 5 per group. Mean ± s.e.m., **p<0.01, ***p<0.001 versus corresponding control. See also Figure S3.
Despite its universal expression pattern, MANF-deficiency affects β-cells
Expression of MANF has previously been detected in several tissues, including pancreatic acinar cells at E17 and in adult pancreatic islet cells (Lindholm et al., 2008; Mizobuchi et al., 2007). We studied in more detail the expression of MANF in mouse pancreas by double immunohistochemistry with insulin and MANF antibodies. High MANF expression was co-localized with insulin positive β-cells in mouse islets (Figure S4B, arrowhead). However, MANF-positive immunostaining was also detected in exocrine acinar cells (Figure S4B, arrow). Importantly, none to very weak background staining of MANF was detected in Manf−/− pancreas (Figure S4E). Similarly to expression in mouse, we detected MANF expression in the islets and exocrine tissue of human adult pancreas tissue (Figure S4G–I).
Consistent with progressively reduced β-cell mass from P1 Manf−/− pancreas, there was significantly reduced expression of Glucose transporter 2 (Glut2), insulin1/insulin2 (Ins1/2), and Pdx1, and a trend towards reduced Glucokinase (Gck) expression in islets of P1 Manf−/− mice (Figure 3B–D). Importantly, reduced expression of these β-cell markers was not observed in pancreases of E18.5 mice (Figure 3A). Along with other β-cell specific genes, also MafA mRNA expression was significantly reduced already in P1 Manf−/− islets (Figure 3B–D). GLUT2 immunohistochemistry showed clearly reduced membrane localization and expression of GLUT2 protein in P14 and P56 Manf−/− β-cells (Figure 3E–J). Taken together, our data indicates that the timing of the progressive loss of β-cell phenotype occurs postnatally in Manf−/− islets.
Figure 3. Expression of β-cell specific genes is reduced in Manf−/− islets.
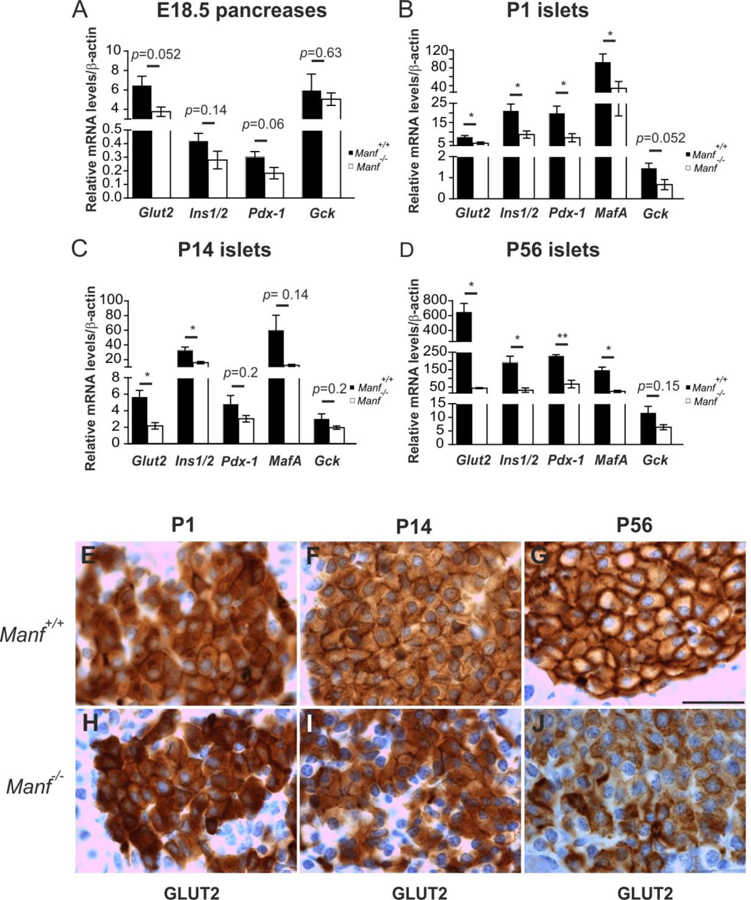
Quantitative RT-PCR for mRNA levels of β-cell specific genes Glut2, Ins1/2, Pdx-1, MafA and Gck in E18.5 pancreases (A) and islets from P1 (B), P14 (C) and P56 (D) pancreases, n = 4–10 per group. Mean ± s.e.m., *p<0.05, **p<0.01, versus the corresponding control. GLUT2 immunohistochemistry of pancreas sections from WT and Manf−/− mice at P1 (E, H), P14 (F, I) and P56 (G, J), scale bar, 100 μm. See also Figure S4.
Loss of MANF results in activated endoplasmic reticulum stress and unfolded protein response pathways
We next investigated ER stress and UPR pathways in Manf−/− E18.5 pancreases and isolated islets. Significantly higher levels of Chop and spliced Xbp1 but not Atf4, Grp78 or Atf6a were observed already in E18.5 Manf−/− pancreases compared to WT (Figure 4A). There was a trend towards higher levels of Atf4, Grp78, Chop, spliced Xbp1 and Atf6a mRNA expression in islets from P1 Manf−/− mice compared to WT (Figure 4B). A significant elevation of mRNA levels for Atf4, Grp78, Chop and Atf6a was found in the islets isolated from P14 Manf−/− pancreases compared to WT islets (Figure 4C). Taken together, our results demonstrate that loss of β-cell phenotype is preceded by upregulation of genes in IRE1 pathway starting at E18.5 followed by activation of PERK and ATF6 pathways (Figure 4F).
Figure 4. Unfolded protein response genes are upregulated and EIF2α protein is phosphorylated in islets from MANF-deficient mice.
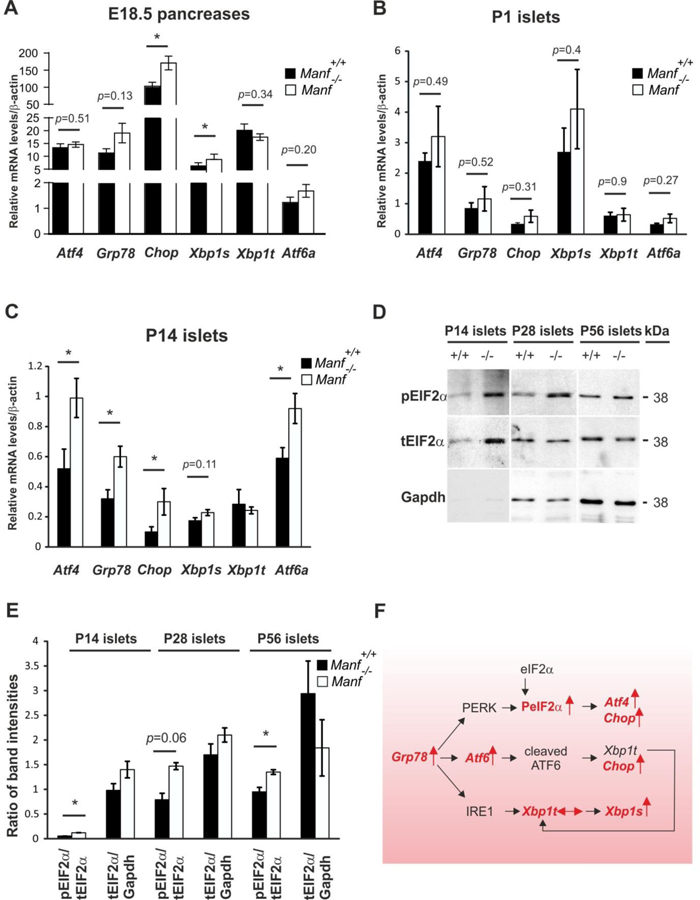
(A, B) Quantitative real-time PCR analysis of UPR genes Atf4, Grp78, Chop, Xbp1s, Xbpt and Atf6a in Manf+/+and Manf−/− E18.5 pancreases (A) and islets from P1 (B) and P14 (C) pancreases, n = 4–13 per genotype). (D) Western blotting with indicated antibodies on islet-lysates from P14, P28 and P56 mice. GAPDH (Glyceraldehyde 3-phosphate dehydrogenase). (E) Quantified intensities of Western blot bands of phosphorylated (p)EIF2α was compared to total amount of (t)EIF2α and tEIF2α to intensities of GAPDH, n = islets from 2–3 pancreases per genotype. Mean ± s.e.m., *p<0.05 versus corresponding Manf+/+control. (F) Analyzed ER stress pathways and UPR genes are indicated in red. Vertical arrow denotes increased, horizontal arrow unchanged expression in Manf−/− islets compared to WT animals. Upon accumulation and aggregation of unfolded proteins, GRP78 dissociates from ER stress receptors PERK, ATF6 and IRE1, activating downstream signaling UPR cascades. Phosphorylated PERK blocks global mRNA translation by phosphorylating eIF2α subunit. Transcription factor ATF4 escapes eIF2α translational control and in turn induces transcription of pro-apoptotic gene, Chop. Active ATF6 translocates to the nucleus where it induces chaperone genes such as Grp78 and Xbp1 and controls genes involved in the ER associated degradation (ERAD). Activated IRE1α removes by splicing an intron from Xbp1, generating spliced Xbp1 (Xbp1s) which translated to a transcription factor activates genes for ER-associated decay and chaperons for protein folding. Figure modified from (Szegezdi et al., 2006).
Furthermore, quantification of phosphorylated (p)eIF2α band intensities in relation to total levels of (t)eIF2α by Western Blot analysis revealed a higher level of phosphorylated eIF2α in P14, P28 and P56 Manf−/− islets (Figure 4D and E), demonstrating that the PERK pathway was constitutively activated in the postnatal Manf−/− islets (Figure 4F).
Recombinant MANF induces proliferation of pancreatic β-cells in vitro and in vivo
We assessed whether recombinant human MANF protein could directly affect mouse β-cell proliferation in vitro. Compared to islets cultured without added growth factors, MANF significantly increased β-cell proliferation (Figure 5A). Importantly, MANF together with placental lactogen (PL), a potent mitogen for β-cells, further increased the number of proliferating β-cells. Hence, extracellular MANF has a direct proliferative effect on mouse β-cells implying the presence of a yet unidentified receptor for MANF on the β-cells capable of mediating intracellular mitogenic signaling cascades.
Figure 5. MANF rescues islet size and selectively induces β-cell proliferation in vitro and in vivo.
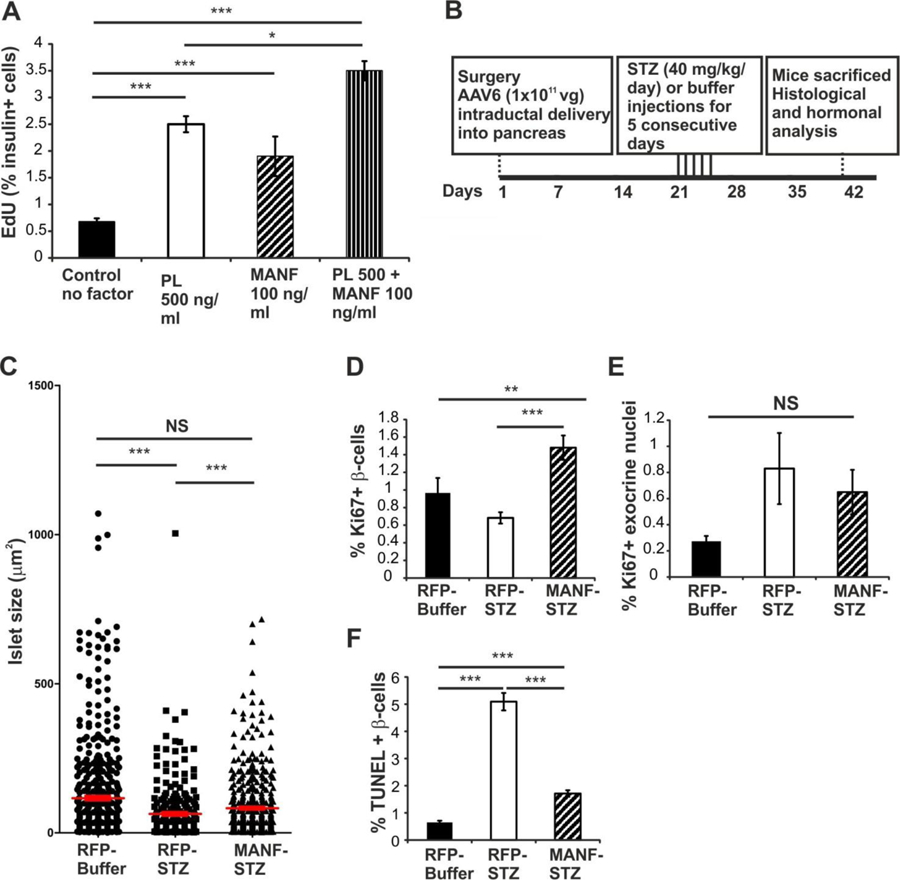
(A) MANF recombinant protein increases β-cell proliferation after five days in culture. Placental lactogen (PL), n = 5 wells per point. (B) Time course of the in vivo experiment. (C) Distribution of islet size in the AAV6-RFP-Buffer, AAV6-RFP-STZ and AAV6-MANF-STZ animals. Each symbol in the graph represents one islet and average islet size per group is shown by horizontal lines. n = 5–8 per group. (D) β-cell proliferation in AAV6-virus injected non-lesioned and STZ-treated mice assessed by Ki67 and insulin double–staining, n = 6 per group. (E) Acinar cell proliferation in AAV6-virus injected non-diabetic and STZ-treated mice, n = 6 per group. (F) Islet cell death assessed by TUNEL followed by insulin staining, n = 5–6 per group. Mean ± s.e.m., **p<0.01, ***p<0.001 versus corresponding control. See also Figure S5.
Our observed effects of MANF removal from β-cells in vivo and exogenous recombinant MANF protein on β-cells in vitro suggested that MANF might be therapeutic in a rodent model of diabetes. We therefore tested whether over-expression of MANF by intra-pancreatic delivery of a MANF-expressing adeno-associated virus serotype 6 (AAV6) in mice affects β-cell proliferation and survival after streptozotocin-induced β-cell depletion. Three weeks after AAV6-MANF or AAV6-RFP (a control red fluorescent protein) administration, animals were injected with low-dose streptozotocin (STZ) for 5 consecutive days to induce β-cell deficiency (Figure 5B). Robust, patchy expression of MANF, or RFP (Figure S5B, F and J) was observed in islet β-cells (transduction efficiency 4.3 ± 1.5%) and exocrine tissue (4.2 ± 1.5% cells transduced), confirming a successful delivery and over-expression of MANF and RFP. Consistent with elevated blood glucose levels and reduced serum insulin levels, STZ-induced β-cell loss was detected in the islets from mice treated with STZ (Figure S5H, L, M and N).
Insulin immunoreactive islets were larger in the STZ-injected, AAV6-MANF-treated pancreatic sections compared to the AAV6-RFP-treated mice (Figure 5C and Figure S5H and L). Although the blood glucose and insulin levels were comparable between AAV6-MANF and AAV6-RFP STZ-injected groups, we found a significantly higher β-cell proliferation rate in the STZ-injected, AAV6-MANF-treated groups compared to both the buffer injected (non-lesioned) AAV6-RFP and the STZ-injected, AAV6-RFP groups (Figure 5D and Figure S5M and N), demonstrating that over-expression of MANF could enhance β-cell proliferation and regeneration in vivo. Importantly, we found no significant difference in the number of proliferating exocrine acinar cells between treatment groups, suggesting that the proliferative effect of MANF was specific for endocrine pancreas (Figure 5E). Additionally, AAV6-MANF significantly protected against β-cell death compared to the AAV6-RFP STZ-injected group (Figure 5F). Taken together, our results demonstrate that gene therapy using MANF is able to induce β-cell proliferation and regeneration and protect β-cells from apoptosis in an experimental diabetes model.
DISCUSSION
In order to understand the physiological role of MANF in mammals, we generated MANF-deficient mice. Surprisingly, MANF-deficiency in mice leads to a progressive loss of β-cells resulting in diabetes mellitus due to reduced β-cell proliferation and enhanced β-cell death. The severe diabetic phenotype of global Manf−/− mice was unexpected since inactivation of MANF in fruitfly and knockdown of Manf mRNA expression in zebrafish causes a dopaminergic phenotype (Chen et al., 2012; Palgi et al., 2009). Conditional removal of MANF specifically from the pancreas in Pdx1Cre/+:: Manffl/fl mice lead to a diabetic phenotype similar to the global knockout mice. However, inactivation of MANF by Nestin-Cre expression did not result in a diabetic phenotype. In Nestin-Cre mice, Cre recombinase activity is detected in the central nervous system by E11, in mesenchymal and epithelial cells of the early pancreatic primordium, in scattered acinar cells of the exocrine pancreas in adults but not in islet endocrine cells (Delacour et al, 2004). Thus, our results obtained from conditional removal of MANF, strongly suggests that MANF produced locally in the islets is important for the β-cell proliferation and survival.
Previous studies have shown that MANF is upregulated in ER stress in vitro and can protect several cell populations from ER-stress induced cell death in vivo (Airavaara et al., 2009; Apostolou et al., 2008; Glembotski et al., 2012; Voutilainen et al., 2009). Interestingly, MANF protein level was also increased in the β-cells of diabetic Akita mice in which ER stress is caused by the accumulation of proinsulin in the ER (Mizobuchi et al., 2007). Furthermore, MANF total knockdown in fruitfly embryos has revealed increased expression of several genes involved in ER stress and increased eIF2α phosphorylation (Palgi et al., 2012).
Here we show that lack of MANF in vivo leads to ER stress and chronic UPR activation in pancreatic islets. The activation of UPR is clearly evident already at E18.5 by increased expression of spliced Xbp1 and Chop mRNA, followed by the upregulation of the general ER stress marker Grp78 and genes in the PERK and ATF6 pathways. Importantly, we found no difference in β-cell mass or in the number of proliferating β-cells quantified from WT and Manf−/− pancreases at E18.5. Similarly, β-cell specific genes Glut2, Ins1/2, Pdx1 and Gck were not significantly downregulated in Manf−/− pancreas at E18.5 indicating that ER stress precedes the impaired β-cell function.
As MANF is widely expressed in several tissues, the question arises why global MANF-knockdown results in such a robust β-cell phenotype? Increased phosphorylation of eIF2α which is known to lead to a global decrease in mRNA translation initiation is tolerated poorly by β-cells (Cnop et al., 2007). In addition, prolonged ATF6 activation down-regulates transcription factors PDX1 and MAFA, both critical for promoting expression of insulin and important for β-cell function (Artner et al., 2010; Seo et al., 2008). Thus, the current evidence suggests that UPR activation detected already at E18.5 in Manf−/− islets affects the expression of β-cell specific proteins leading to decreased insulin expression, reduced β-cell proliferation and increased β-cell death.
Several factors contributing to the regulation of β-cell mass, including insulin-like growth factors (IGF-I and IGF-II), glucagon-like peptide-1, prolactin, growth hormone and placental lactogens, are potential therapeutic targets for expansion of the human β-cell mass (Tarabra et al., 2012). However, therapeutic use of some of these hormones and growth factors has been limited by the lack of specificity, stimulation of uncontrolled β-cell growth and adverse effects on β-cell function. Furthermore, knockdown studies have revealed that many of these factors are not essential for physiological β-cell expansion and survival (Tarabra et al., 2012; Vasavada et al., 2006). Recently, a potential specific growth factor for β-cells, named betatrophin (alias ANGPTL8), was identified (Yi et al., 2013). However, the finding that deletion of ANGPTL8 did not affect β-cell mass or glucose metabolism (Wang et al., 2013), indicates that its physiological action on β-cells is very different from MANF.
Here we show that recombinant MANF is a potent stimulator of β-cell proliferation in vitro, constituting a novel protein with therapeutic potential for stimulating β-cell renewal. Additionally, our work provides novel tools for finding the mechanism of MANF action by identifying its receptor(s) and signaling pathways in β-cells.
Our in vivo gene therapy experiment using AAV-6 MANF showed that despite of low transduction efficiency (only ~4% of the β-cells) of pancreatic cells, MANF promotes normal islet morphology and specifically enhances β-cell proliferation and protects β-cells in mice in an experimental diabetes model. Future experiments using higher virus titers, longer MANF over-expression and different diabetic models are clearly needed to validate the therapeutic potential of MANF.
The severe postnatal decline in β-cell mass in the Manf−/− mice and our in vitro and in vivo data indicate that MANF may be one of the most potent secreted growth factors for regulating both β-cell proliferation and for maintaining β-cell mass in mice. Finally, high MANF expression in human pancreas suggests that MANF might have similar β-cell mitogenic effects also in humans. Future studies will evaluate the potential of MANF to protect and regenerate functional β-cells as a novel therapeutic agent for treating diabetes.
EXPERIMENTAL PROCEDURES
Manf-targeted ES clone
The targeted mouse embryonic stem (ES) cell clone MANF_D06 (EPD0162_3_D06, C57Bl/6N-Manftm1a(KOMP)Wtsi) was generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org). Genetically modified ES-cells were aggregated with morula-stage pre-implantation embryos (ICR strain) at the GM mouse unit of University of Helsinki.
Animals
All experimental procedures involving mice were approved by the Finnish Animal Ethics Committee of the State Provincial Office of Southern Finland. Mice were maintained in pathogen-free facility with a 12 h light/dark cycle and unlimited access to food (Harlan, Teklad Global, 16% protein rodent diet, 2916) and water. In all studies comparing Manf+/+ and Manf−/− mice, we used sex-matched siblings derived from crossings of Manf heterozygous (Manf+/−) animals in hybrid C57Bl6 x ICR mixed background. The day of vaginal plug was designated as E0.5. Age-matched NMRI male mice for the in vivo MLD-STZ experiment were obtained from Harlan Teklan, UK. Pancreatic islets were isolated from age-matched 8-week-old C57Bl/6JRccHsd (Harlan) female mice in β-cell proliferation assay.
Genomic DNA isolation and genotyping
DNA was isolated from ear-marks and genotyping was carried out by PCR using primers described in Supplemental Experimental Procedures.
Islet isolation and in vitro insulin release
Pancreases from mice were treated with collagenase P digestion (Collagenase P, Roche Diagnostics GmbH) followed by hand-picking of islets under a stereomicroscope (Miettinen et al., 2006). Human pancreatic tissue was obtained at autopsy at the Helsinki University Central Hospital and isolated human islets were received from Uppsala, Sweden, through the European Consortium for Islet Transplantation (ECIT). In vitro insulin release assay was performed as previously described (Miettinen et al., 2006).
RNA isolation, reverse transcription and quantitative PCR
RNA isolation, reverse transcription, quantitative PCR and primers are described in Supplemental Experimental Procedures.
Food and water intake and energy expenditure
Feeding and drinking, energy expenditure, (O2 consumption and CO2 production using indirect calorimetry), respiratory exchange ratio and locomotor activity was measured using CLAMS monitoring system (Columbus Instruments) in 6-week-old mice. For detailed description, see Supplemental Experimental Procedures.
Western analysis
Western blot were performed according to standard protocols and as described in the Supplemental Experimental Procedures.
Immunohistochemistry and quantification of β- and α-cell mass and islet size
Immunohistochemistry was performed according to standard procedures and beta- and alpha-cell mass analysis and islet size measurements were performed as described in the Supplemental Experimental Procedures.
Analysis of blood samples
Blood samples from mice were collected from the tail vein or terminal blood from heart and assayed for glucose (Accucheck Aviva Glucometer, Roche Diagnostics) and insulin (ultrasensitive mouse insulin ELISA, Crystal Chem.). For glucose challenge test P14 mice were fasted for 1 h and P56 mice 5–6 h before animals were injected i.p. with 2 g/kg body weight of glucose (in 0.9% NaCl). Insulin tolerance test was performed on 2 h fasted P42 male mice by injection of 1U/kg i.p. insulin (diluted in 0.9% saline, Humulin® Regular, Eli Lilly) and monitoring of blood glucose levels from tail vein every 15 minutes for 60 minutes.
AAV-vector construction and in vivo administration of AAV-vectors
The construction of the AAV packaging plasmids, generation of AAV vectors and retrograde pancreatic duct injections were carried out as described in Supplemental Experimental Procedures. Three weeks after AAV administration, AAV6-MANF and AAV6-RFP animals were injected on 5 consecutive days with a low-dose of streptozotocin (40 mg/kg/day, i.p., freshly dissolved in 0.1 M citrate buffer, pH 4.5).
In vitro β-cell proliferation assay
Islets from female, virgin 8 weeks old mice were isolated as described above. Equal numbers of islets per well were treated for 5 days with recombinant human placental lactogen (500 ng/ml, Affiland) or recombinant human MANF (100 ng/ml, Icosagen) or a mixture of both. The relative numbers of proliferating β-cells were quantified from wells of five repeats per treatment (details in Supplemental Experimental Procedures).
Statistical analysis
Unless otherwise stated significance of differences between groups was analyzed by Studentś unpaired two-tailed t test using Microsoft Excel software. Differences between more than two groups were calculated by one-way ANOVA followed by appropriate post-hoc test, using SPSS Inc. PASW Statistics 18 program. For statistical analysis of islet size distribution we used GraphPad Prism 5 and data were subjected to Kruskal-Wallis one-way analysis of variance test and differences were evaluated by Dun’s Multiple Comparison Test. Results are expressed as mean ± s.e.m. Results were considered significant at P< 0.05.
Supplementary Material
HIGHLIGHTS.
MANF-deficiency results in pancreatic β-cell depletion and diabetes in mice
MANF is highly expressed in mouse and human islet β-cells and exocrine acinar cells
MANF-deficiency leads to ER stress and chronic unfolded protein response in β-cells
MANF protein enhances proliferation of β-cells in vitro and in vivo in diabetic mice
ACKNOWLEDGEMENTS
S. Åkerberg, S.Wiss and D. Howard are thanked for excellent technical assistance. We thank Frank Grosvelt for CAG-Flp mice and Edgar Kramer for Nestin-Cre mice. We acknowledge NIH Knock-out Mouse Program (KOMP) for the MANF-targeted ES cell clone. M.L. was supported by Grant nr 117044 and J.R. by Grant nr 260030 from the Academy of Finland. This work was supported by grants from the Commission of the European Union, Collaborative project MOLPARK, nr 400752, The Lundbeck Foundation, personal price Mart Saarma, 2009, nr 7519003, Sigrid Jusélius Foundation nr 4701537 and the Juvenile Diabetes Research Foundation nr 17-2013-410.
University of Helsinki has filed a patent application based on these data, with M.L., T.D., E.P., P.L., J.R., and M.S. as inventors. University of Helsinki owns intellectual property rights.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five Supplemental Figures, one Supplemental Result Text, Supplemental Experimental Procedures and Supplemental References.
REFERENCES
- Airavaara M, Shen H, Kuo CC, Peranen J, Saarma M, Hoffer B, and Wang Y (2009). Mesencephalic astrocyte-derived neurotrophic factor reduces ischemic brain injury and promotes behavioral recovery in rats. J Comp Neurol 515, 116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou A, Shen Y, Liang Y, Luo J, and Fang S (2008). Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res 314, 2454–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, and Stein R (2010). MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 59, 2530–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Sundvik M, Rozov S, Priyadarshini M, and Panula P (2012). MANF regulates dopaminergic neuron development in larval zebrafish. Dev Biol 370, 237–249. [DOI] [PubMed] [Google Scholar]
- Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, Flamez D, Boyce M, Yuan J, and Eizirik DL (2007). Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem 282, 3989–3997. [DOI] [PubMed] [Google Scholar]
- Delacour A, Nepote V, Trumpp A and Herrera PL (2004). Nestin expression in pancreatic exocrine cell lineages. Mech Dev 121, 3–14. [DOI] [PubMed] [Google Scholar]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, and Julier C (2000). EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 25, 406–409. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Georgia S, and Bhushan A (2007). Formation and regeneration of the endocrine pancreas. Curr Opin Cell Biol 19, 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, and Halban PA (2004). Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 47, 581–589. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, and Cnop M (2008). The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29, 42–61. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Miani M, and Cardozo AK (2013). Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56, 234–241. [DOI] [PubMed] [Google Scholar]
- Glembotski CC, Thuerauf DJ, Huang C, Vekich JA, Gottlieb RA, and Doroudgar S (2012). Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem 287, 25893–25904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman M, Arumae U, Yu LY, Lindholm P, Peranen J, Saarma M, and Permi P (2011). Mesencephalic astrocyte-derived neurotrophic factor (MANF) has a unique mechanism to rescue apoptotic neurons. J Biol Chem 286, 2675–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13, 89–102. [DOI] [PubMed] [Google Scholar]
- Lindholm P, Peranen J, Andressoo JO, Kalkkinen N, Kokaia Z, Lindvall O, Timmusk T, and Saarma M (2008). MANF is widely expressed in mammalian tissues and differently regulated after ischemic and epileptic insults in rodent brain. Mol Cell Neurosci 39, 356–371. [DOI] [PubMed] [Google Scholar]
- Lindholm P, and Saarma M (2010). Novel CDNF/MANF family of neurotrophic factors. Dev Neurobiol 70, 360–371. [DOI] [PubMed] [Google Scholar]
- Lindholm P, Voutilainen MH, Lauren J, Peranen J, Leppanen VM, Andressoo JO, Lindahl M, Janhunen S, Kalkkinen N, Timmusk T, et al. (2007). Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448, 73–77. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, and Butler PC (2008). Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57, 1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Ustinov J, Ormio P, Gao R, Palgi J, Hakonen E, Juntti-Berggren L, Berggren PO, and Otonkoski T (2006). Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes 55, 3299–3308. [DOI] [PubMed] [Google Scholar]
- Mizobuchi N, Hoseki J, Kubota H, Toyokuni S, Nozaki J, Naitoh M, Koizumi A, and Nagata K (2007). ARMET is a soluble ER protein induced by the unfolded protein response via ERSE-II element. Cell Struct Funct 32, 41–50. [DOI] [PubMed] [Google Scholar]
- Palgi M, Greco D, Lindstrom R, Auvinen P, and Heino TI (2012). Gene expression analysis of Drosophilaa Manf mutants reveals perturbations in membrane traffic and major metabolic changes. BMC Genomics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palgi M, Lindstrom R, Peranen J, Piepponen TP, Saarma M, and Heino TI (2009). Evidence that DmMANF is an invertebrate neurotrophic factor supporting dopaminergic neurons. Proc Natl Acad Sci U S A 106, 2429–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova P, Raibekas A, Pevsner J, Vigo N, Anafi M, Moore MK, Peaire AE, Shridhar V, Smith DI, Kelly J, et al. (2003). MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J Mol Neurosci 20, 173–188. [DOI] [PubMed] [Google Scholar]
- Seo HY, Kim YD, Lee KM, Min AK, Kim MK, Kim HS, Won KC, Park JY, Lee KU, Choi HS, et al. (2008). Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology 149, 3832–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, and Samali A (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, and Accili D Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150, 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabra E, Pelengaris S, and Khan M (2012). A simple matter of life and death-the trials of postnatal Beta-cell mass regulation. Int J Endocrinol 2012, 516718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang Y, Quagliarini F, Gusarova V, Gromada J, Valenzuela DM, Cohen JC, and Hobbs HH (2013). Mice lacking ANGPTL8 (Betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc Natl Acad Sci U S A 110, 16109–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, and Garcia-Ocana A (2006). Growth factors and beta cell replication. Int J Biochem Cell Biol 38, 931–950. [DOI] [PubMed] [Google Scholar]
- Weir GC, and Bonner-Weir S (2013). Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci 1281, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutilainen MH, Back S, Porsti E, Toppinen L, Lindgren L, Lindholm P, Peranen J, Saarma M, and Tuominen RK (2009). Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson’s disease. J Neurosci 29, 9651–9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi P, Park JS, and Melton DA (2013). Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 153, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.