

Abstract
Cyclooxygenase-2 [(COX-2) or prostaglandin endoperoxide H2 synthase-2 (PTGS-2)] induces the production of prostaglandins as part of the host-immune response to infections. Although a number of studies have demonstrated the effects of COX-2 promoter variants on autoimmune and inflammatory diseases, their role in malaria remains undefined. As such, we investigated the relationship between four COX-2 promoter variants (COX-2 −512 C > T, −608 T > C, −765 G > C, and −1195 A > G) and susceptibility to malaria and severe malarial anemia (SMA) upon enrollment and longitudinally over a 36-month follow-up period. All-cause mortality was also explored. The investigation was carried out in children (n = 1081, age; 2–70 months) residing in a holoendemic Plasmodium falciparum transmission region of western Kenya. At enrollment, genotypes/haplotypes (controlling for anemia-promoting covariates) did not reveal any strong effects on susceptibility to either malaria or SMA. Longitudinal analyses showed decreased malaria episodes in children who inherited the −608 CC mutant allele (RR = 0.746, P = 1.811 × 10−4) and −512C/−608T/−765G/−1195G (CTGG) haplotype (RR = 0.856, P = 0.011), and increased risk in TTCA haplotype carriers (RR = 1.115, P = 0.026). Over the follow-up period, inheritance of the rare TTCG haplotype was associated with enhanced susceptibility to both malaria (RR = 1.608, P = 0.016) and SMA (RR = 5.714, P = 0.004), while carriage of the rare TTGG haplotype increased the risk of malaria (RR = 1.755, P = 0.007), SMA (RR = 8.706, P = 3.97 × 10−4), and all-cause mortality (HR = 110.000, P = 0.001). Collectively, these results show that SNP variations in the COX-2 promoter, and their inherited combinations, are associated with the longitudinal risk of malaria, SMA, and all-cause mortality among children living in a high transmission area for P. falciparum.
Introduction
Malaria remains a significant global health burden, with an estimated 219 million (95% confidence interval [CI]: 203–262 million) cases reported worldwide in 2017 [1]. The African region accounted for 92% (200 million) of the cases, 99.7% of which were attributed to Plasmodium falciparum infections [1]. Consequently, an estimated 435,000 deaths were reported globally in 2017, with children under 5 years being the most vulnerable, accounting for 61% (n = 226,000) of the malaria mortalities worldwide, 93% of which occurred in the African region [1]. Malaria-related anemia was reported in 79% of children under 5 years, of which 21% were mild, 50% moderate and 8% severe [1]. In western Kenya, a holoendemic P. falciparum transmission region, falciparum malaria remains one of the leading causes of childhood morbidity and mortality [2]. The primary severe disease manifestation of P. falciparum infections in such areas is severe malarial anemia [SMA, hemoglobin (Hb) < 5.0 g/dL] [3], often with accompanying features of acidosis, respiratory distress, and acute renal failure, and in rare circumstances, hypoglycemia and cerebral malaria (CM) [4]. In addition, co-infections (e.g., bacteremia and HIV-1), nutritional deficiencies, and host genetic factors influence the severity of disease in malarial anemia [5–8].
The pathogenesis of SMA, at least in part, can be attributed to alterations in host-immune responses that suppress erythropoiesis and enhance hemolysis, resulting in profoundly low hemoglobin concentrations [9, 10]. Although the complex immunological cascades which promote the development of SMA are only partially defined, dysregulation in the cyclooxygenase-2 [(COX-2)]; prostaglandin endoperoxide H2 synthase-2 (PTGS-2)] pathway appears to be a central feature in the severity of P. falciparum infections [11, 12]. COX-2 (PTGS-2) is a rapidly responding gene that is induced by a wide variety of inflammatory and proliferative stimuli, with a fast mRNA turnover driven by a conserved element at the 3′-untranslated region [13]. The host immune response to microbial infections typically promotes increased expression of COX-2, which generates elevated levels of eicosanoid effector molecules, including prostaglandin E2 (PGE2) [14]. However, previous studies from our laboratory demonstrated that infection with P. falciparum suppressed COX-2 gene products and reduced PGE2 levels, a process which results in increased clinical severity in cerebral malaria, malaria in pregnancy, and malarial anemia [11, 15, 16].
The human COX-2 gene is ~8.3 kb in size and is located on chromosome 1 (q25.2-q25.3), containing 10 exons that synthesize a 4.6 kb mRNA product [17, 18]. Analysis of the promoter region reveals a number of regulatory elements, including a TATA box and transcription factor binding sites for, among others, stimulatory protein-1 (Sp1), nuclear factor-κB (NF-κB), nuclear factor for interleukin-6 (NF-IL6), glucocorticoid response element (GRE), activator protein 1 (AP-1), AP-2, CCAAT-enhancer binding protein (C/EBP), cyclic-AMP response element (CRE), cAMP response element binding protein (CREB), E2F transcription factor (E2F), MYB proto-oncogene (MYB) [17, 18].
There is vast literature on single nucleotide polymorphisms (SNPs) in the COX-2 promoter, which create inter-individual variability that alters susceptibility to a plethora of diseases. For example, COX-2 promoter SNPs and their haplotypes, can influence susceptibility to various cancers, cardiovascular disease, Parkinson’s disease, and viral hepatitis C infections [19–23]. Meta-analyses on some of the more commonly explored COX-2 promoter SNPs show that the COX-2 (−765 G > C) polymorphism is associated with the risk of prostate cancer, coronary artery disease, and chronic periodontitis [24–26], whereas variation at COX-2 (−1195 A > G) can influence susceptibility to hepatocellular carcinoma and other cancers [27, 28]. Meta-analyses of COX-2 haplotypes (−765 G > C and −1195 A > G) also reveal that these haplotypes alter susceptibility to gastric, esophageal, and breast cancers, and Alzheimer’s disease [29–32].
Variation at a number of polymorphic sites in the COX-2 promoter can alter transcriptional activity (reviewed in [33]), illustrating that altered susceptibility to various diseases is likely due to functional consequences on transcription regulation. For example, in both single constructs and haplotypic combinations, the C allele at COX-2 (−765) and the G allele at COX-2 (−1195) result in decreased luciferase activity through disruption of transcription factor binding sites [23, 34].
To our knowledge, the relationship between COX-2 promoter polymorphisms and malaria disease outcomes has not been reported. As such, we investigated the impact of four COX-2 promoter SNPs [−512 C > T (rs20420), −608 T > C (rs20419)], −765 G > C [rs20417], and −1195 A > G [rs689466]), and their haplotypes, on susceptibility to malaria and SMA during acute disease (cross-sectional) in Kenyan children (aged 2–70 months; n = 1081) residing in a holoendemic P. falciparum transmission region. In addition, the influence of the COX-2 promoter variants and haplotypes on susceptibility to malaria, SMA, and all-cause mortality were explored over a 36 months longitudinal follow-up period during the developmental phase of naturally acquired malarial immunity.
Methods
Study area
This study was conducted at Siaya County Referral Hospital (SCRH), a holoendemic P. falciparum transmission region in western Kenya. Details of the area have previously been published [35]. One of the primary causes of childhood mortality and morbidity in the Siaya community is falciparum malaria, which has remained stably transmitted [10]. The clinical manifestation of severe falciparum malaria in Siaya is SMA which can occur with other complications such as hyperparasitemia, respiratory distress, hypoglycemia, hyperlactatemia, renal dysfunction, electrolyte and fluid imbalances, and metabolic acidosis [36]. As with most holoendemic P. falciparum transmission regions, cerebral malaria as a manifestation of severe malaria is exceedingly rare in children who grow up in such regions [36]. Accordingly, SMA accounts for the highest percentage of hospital bed occupancy and results in significant in-hospital morbidity and mortality in the Siaya community [3]. Although hyperparasitemia can generate lysis and clearance of RBCs which can contribute to anemia in non-immune individuals [36], hyperparasitemia is a poor indicator of malaria disease severity given the complex and continuously evolving disease process [36]. Individuals inhabiting the study area are predominantly from the Luo ethnic group (>96%), a culturally homogeneous population [37] which provides an enhanced ability to investigate host-genetic factors that influence the pathogenesis of SMA.
Study design and participants
Since the overall goal of our longitudinal studies is to gain an improved understanding of how genes and gene pathways influence naturally-acquired malarial immunity, we identified children (primarily <12 months) who presented at the pediatric ward for their first ‘hospital contact’ (for any reason). After screening for malaria parasites, children with varying severities of malarial anemia were enrolled. Children were excluded from the study if they had previously been hospitalized and/or had reported use of antimalarials two weeks prior to presentation. Since CM is a rare manifestation of severe malaria in children who continuously reside within the region, the exclusion criteria also included a confirmed diagnosis of CM. However, none of the children presented with CM. Children presenting as outpatients for childhood vaccinations were also recruited. All study participants were scheduled for follow-up visits on day 14 after enrollment and quarterly for 36 months. Children who failed to return for scheduled visits were located using our GIS/GPS system. In addition, parents/legal guardians were asked to return with their child to the hospital during any acute illnesses between the scheduled visits (acute episodes). For all scheduled and acute episodes, patients were managed according to the Ministry of Health (MOH)-Kenya guidelines. Demographic and clinical laboratory measures for each participant were collected at each visit. Investigations presented here are for 1081 children followed for 36 months from 2002 to 2014. Details of the clinical laboratory measures for the study participants are presented below. Parents/legal guardians of the study participants provided informed written consent. The study was approved by the Ethics Committee of the Kenya Medical Research Institute, the University of New Mexico Institutional Review Board, the Los Alamos National Laboratory (LANL) Institutional Review Board and the Maseno University Ethics Review Committee. Informed written consents were obtained from the parents or legal guardians of all participating children.
Laboratory investigations
Heel or finger-prick blood samples (<100 μL) were obtained and used to determine key variables during recruitment and follow-up, namely parasite densities and Hb concentrations. To determine parasitemia, peripheral blood smears were prepared, stained with Giemsa reagent and examined under oil immersion. Asexual malaria parasites were counted against 300 leukocytes and densities estimated using total white blood cells counts. Upon recruitment, complete blood counts (CBC) were determined using a Beckman Coulter AcT diff2 (Beckman–Coulter Corporation, FL, USA). Based on Hb levels, children with any density parasitemia, were stratified into either non-SMA (Hb ≥ 5.0 g/dL; n = 670) or SMA (Hb < 5.0 g/dL; n = 150), according to the World Health Organization (WHO) definition of SMA [38]. The aparasitemic group (n = 261) comprised children with a P. falciparum negative blood smear who presented for childhood vaccinations, and those who presented at hospital for non-malarial diseases. Furthermore, to differentiate anemia caused by malaria from other causes, HIV-1, bacteremia, sickle-cell trait status, α3.7-thalassemia deletions, and Glucose-6-phosphate dehydrogenase (G6PD) deficiency were determined. HIV-1 status was determined as previously described [7]. Parents/legal guardians of the participating children received pre- and post-test HIV&AIDS counseling. At the time of enrollment, none of the children had been started on anti-retroviral therapy. Sickle-cell status was determined by alkaline cellulose acetate electrophoresis on Titan III plates (Helena BioSciences, Sunderland, UK) according to the manufacturer’s instructions. Bacteremia was determined by microbial cul-tivation according to our standard methods as described previously [8]. Alpha3.7-thalassemia variants were determined, as previously described [39]. Glucose-6-phosphate dehydrogenase (G6PD) deficiency was determined using TaqMan® polymerase chain reaction (PCR) assays, as previously published [40].
Selection of COX-2 SNPs
Selection of COX-2 promoter SNPs for genotyping was based on variants with minor allele frequencies (MAF) >8% from the International HapMap project or 1000 genomes project. Additional selection criteria included SNPs, which could potentially create or ablate transcription factor binding sites (TFBS) as determined using Alggen-Promo (v3.0.2) software. Analysis of linkage disequilibrium (LD) was also performed to ensure that the SNPs were not in strong LD using Multiallelic Interallelic Disequilibrium Analysis (MIDAS) software version 1.0 [41].
COX-2 genotyping
Genomic DNA was extracted from buccal swabs using the Buccal Amp™ DNA extraction kit (Epicentre Biotechnologies, Madison, USA) according to the manufacturer’s instructions and thereafter subjected to amplification using GenomiPhi® system (GE Healthcare, NJ, USA) to obtain sufficient quantities for genotyping. COX-2 −512 C > T (rs20420; [Assay ID: C_11997899_10]), −608 T > C (rs20419; [Assay ID: C_11997907_10]), −765 G > C (rs20417; [Assay ID: C_11997909_40]) and −1195 A > G (rs689466; [Assay ID: C_2517145_20]) promoter variants were individually genotyped using high through-put TaqMan® 5’ allelic discrimination Assay-By-Design method on a StepOnePlus Real Time system (Thermo Fisher Scientific, Carlsbad, CA, USA). PCR was performed in a total reaction volume of 10 μL with the following amplification cycles: initial denaturation (60 °C for 30 s and 95 °C for 10 min) followed by 40 cycles of (95 °C for 15 s and 60 °C for 1 min.) and a final extension (60 °C for 30 s). StepOne™ Software Version 2.3 was used for allelic discrimination. To validate results obtained with the Taqman® real-time genotyping assays, ~10% of the samples were randomly selected and genotyped using restriction fragment length polymorphism (RFLP) PCR. There was 100% concordance between the two methods for the randomly selected samples.
Haplotype combinations
Since haplotypes often reveal how combinations of different functional polymorphic alleles which interact to amplify, or moderate, their individual effects [36], haplotypes were constructed using the HPlus software program (Fred Hutchinson Cancer Research Center, WA, USA) with their distribution frequencies estimated based on a Bayesian algorithm. For this study, haplotypes were defined as a combination of four SNP alleles, where −512C/−608T/−765G/−1195A is equivalent to haplotype CTGA. As such, ‘carriage’ of haplotypes was defined as the co-inheritance of combinations of the four COX-2 SNP alleles (e.g., CTGA), while ‘non-carriers’ were defined as absence of the haplotype for a defined allele combination (e.g., non-CTGA haplotype).
Statistical analyses
Statistical software SPSS version 24.0 (IBM SPSS Inc., Chicago, IL, USA) was used for all cross-sectional (enrollment) data analyses. Demographic and clinical data between aparasitemic, SMA and non-SMA groups were compared using Pearson’s Chi square (χ2), and Kruskal Wallis tests. Differences in parasitological variables between SMA and non-SMA were computed using Mann–Whitney U test and Students t-test. Allele, genotype, and haplotype proportions of COX-2 promoter variants were compared across groups using Pearson’s Chi Square (χ2) and Fisher’s exact tests. Hardy–Weinberg Equilibrium (HWE) was computed using a χ2 goodness of fit test.
To investigate the relationship between genotypes or haplotypes and episodes of malaria or SMA at enrollment, binary logistic regression analysis was used to calculate the odds ratio (OR) and 95% confidence interval (CI). This model controlled for the potential confounding effects of age, sex, sickle-cell trait status, G6PD deficiency, α3.7-thalassemia deletions, bacterial infections, and HIV-1 positivity.
The association between COX-2 genotypes/haplotypes and longitudinal clinical outcomes were analyzed using R version 3.1.3 [42]. The impact of genotypes and haplotypes on longitudinal outcomes and number of malaria or SMA episodes were estimated using a bidirectional elimination stepwise Poisson regression model selected using the Akaike information criterion (AIC), accounting for covariates (age, sex, sickle-cell trait status, G6PD deficiency, α3.7-thalassemia deletions, bacterial infections, and HIV-1 positivity). For the loglinear models, we accounted for the varying length of observation, by treating the logarithm of the length of the observational window as an offset to the logarithm of the expected number of events. Further, survival analyses were conducted using Cox regression/survival analysis (R survival package [v. 2.38.2] coxph function) to model the risk of all-cause mortality associated with COX-2 genotypes/haplotypes over a 36-month follow-up period. Statistical significance was considered at P ≤ 0.050.
Results
Characteristics of the study participants
Children (n = 1081) were categorized into aparasitemic (n = 261), and parasitemic [non-SMA (Hb ≥ 5.0 g/dL, n = 670), and SMA (Hb < 5.0 g/dL, n = 150)] groups. The clinical, demographic, and laboratory characteristics of study participants are presented in Table 1. Although the sex distribution was comparable (P = 0.381), children with SMA were younger (P < 0.001), whereas glucose levels (P = 0.002) and admission temperature (P < 0.001) were lower in aparasitemic children.
Table 1.
Demographic, clinical, and laboratory characteristics of the study participants
| Characteristics | Aparasitemic (MPS Negative) | non-SMA (Hb ≥ 5.0 g/dL) | SMA (Hb < 5.0 g/dL) | P value |
|---|---|---|---|---|
| No. of participants (n = 1081) | 261 | 670 | 150 | |
| Sex, n (%) | ||||
| Male | 129 (49.4) | 345 (51.5) | 68 (45.3) | 0.381a |
| Female | 132 (50.6) | 325 (48.5) | 82 (54.7) | |
| Age (months) | 10.5 (12.7) | 13.8 (13.0) | 8.2 (9.2)*** | <0.001b |
| Glucose (mmol/L) | 5.0 (1.2) | 5.2 (1.6) | 5.2 (1.7) | 0.002b |
| Admission temperature (°C) | 37.0 (1.5) | 37.9 (1.9) | 37.5 (1.6) ** | <0.001b |
| Hematological parameters | ||||
| Hemoglobin (g/dL) | 10.3 (2.5) | 7.7 (2.9) | 4.4 (1.1)*** | <0.001b |
| Hematocrit (%) | 32.9 (6.4) | 25.0 (9.0) | 13.6 (3.5)*** | <0.001b |
| Red blood cells (×106/μL) | 4.7 (1.0) | 3.7 (1.4) | 1.9 (0.7)*** | <0.001b |
| Mean corpuscular volume (fL) | 69.7 (9.9) | 69.2 (11.8) | 72.8 (11.0)*** | <0.001b |
| Mean corpuscular hemoglobin (pg) | 22.3 (3.7) | 21.7 (3.8) | 22.7 (4.4)*** | <0.001b |
| Mean corpuscular hemoglobin concentration (g/L) | 31.8 (2.7) | 31.2 (2.6) | 31.6 (3.5) ** | 0.006b |
| Platelets (×103/μL) | 350.0 (220.0) | 153.0 (130.0) | 145.0 (101.0)*** | <0.001b |
| Mean platelet volume (fL) | 7.5 (1.5) | 8.1 (1.9) | 8.4 (1.5)* | <0.001b |
| WBCs (×103/μL) | 10.8 (7.5) | 11.2 (5.8) | 14.9 (9.7)*** | <0.001b |
| Lymphocytes (×103/μL) | 5.9 (4.2) | 5.0 (3.3) | 6.8 (5.2)* | <0.001b |
| Monocytes (×103/μL) | 0.8 (0.6) | 0.9 (0.8) | 1.6 (1.3)*** | <0.001b |
| Granulocytes (×103/μL) | 3.8 (3.1) | 5.0 (3.9) | 5.3 (4.9)** | <0.001b |
| Parasitological Indices | ||||
| Parasite density (MPS/μL) | 0.0 (0.0) | 25,891 (66,349) | 22,080 (55,516) | <0.001c |
| Geomean parasitemia (per μL) | 0 | 20,763 | 13,684* | <0.001d |
| HDP (≥10,000 parasites/μL), n (%) | 0 (0) | 460 (68.7) | 94 (62.7) | <0.001a |
| Genetic Variants | ||||
| Sickle cell trait, n (%) | 48 (18.8) | 114 (17.1) | 16 (10.7)** | 0.011a |
| G6PD deficiency, n (%) | 10 (4.4) | 32 (5.3) | 8 (5.5) | 0.235a |
| α+-Thalassemia deletion, n (%) | 0.462a | |||
| −α3.7/αα | 62 (27.4) | 169 (28.5) | 34 (24.6) | |
| −α3.7/−α3.7 | 60 (26.5) | 125 (21.1) | 32 (23.2) | |
| Endemic Co-infections | ||||
| HIV-1 exposure, n (%) | 38 (14.7) | 93 (13.9) | 28 (18.8)** | 0.011a |
| HIV-1 positive, n (%) | 12 (4.6) | 18 (2.7) | 12 (8.1)*** | 0.007a |
| Bacteremia positive, n (%) | 26 (10.0) | 48 (7.2) | 14 (9.3) | 0.318a |
Data are the median (interquartile range; IQR) unless otherwise noted. Children (n = 1081) were categorized into aparasitemic controls (n = 261; no parasitemia) and according to the WHO definition of SMA (63), into either non-SMA (n = 670; Hb ≥ 5.0 g/dL) or SMA (n = 150; Hb < 5.0 g/dL).
Statistical significance determined by the Chi-square analysis.
Statistical significance determined by Kruskal Wallis test, and where significant differences were observed, pairwise comparisons between the non-SMA and SMA groups were performed using Mann–Whitney U tests.
Statistical significance determined by Mann–Whitney U test.
Statistical significance determined by Students t-test. Bold indicates P ≤ 0.050.
Represents significant pairwise comparisons between the non-SMA and SMA groups at P ≤ 0.050,
represents P < 0.01, and
represents P < 0.001. MPS malaria parasites; HDP High density parasitemia (≥10,000 parasites/μL); G6PD Glucose 6 phosphate dehydrogenase deficiency
Based on a priori classification of the clinical groups, analyses of hematological indices demonstrated that children with SMA had significantly lower Hb levels (P < 0.001), relative to both non-SMA and aparasitemic groups. Consequently, relative to the non-SMA and aparasitemic groups, children with SMA group presented with decreased hematocrit (P < 0.001) and erythrocyte (P < 0.001) levels, and elevated mean corpuscular volume (MCV; P < 0.001) and mean corpuscular hemoglobin (MCH; P < 0.001). The mean cell hemoglobin concentration (MCHC; P = 0.006) was decreased in non-SMA group, and the platelet counts (P < 0.001) lower in the SMA group compared to the non-SMA and aparasitemic groups. Mean platelet volume (MPV) was elevated in the SMA group relative to non-SMA and aparasitemic children (P < 0.001). Similarly, children with SMA had elevated white blood cells (WBCs, P < 0.001), lymphocytes (P < 0.001), monocytes (P < 0.001), and granulocytes (P < 0.001) relative to the other two groups. Parasitological indices, including parasite densities (P < 0.001), geometric mean parasitemia (P < 0.001), and high-density parasitemia (HDP; parasites ≥ 10,000/μL; P < 0.001) were all lower among children with SMA compared to the non-SMA group.
Analyses of genetic variants that can influence anemia revealed more children with sickle-cell trait (HbAS) in the aparasitemic and non-SMA groups (P = 0.011), relative to the SMA group. However, glucose-6-phosphate (G6PD) deficiently (P = 0.235), as well as heterozygous (−α3.7/αα) and homozygous (−α3.7/−α3.7) α-thalassemia variants (P = 0.462) were comparable across the groups.
Since we have previously reported that HIV-1 and bacteremia influence the severity of malarial anemia, these two endemic co-infections were determined in the clinical groups [7, 8]. The SMA group had higher rates of HIV-1 exposure (P = 0.011) and HIV-1 (P = 0.007) than the non-SMA and aparasitemic groups. However, the number of children with blood-borne bacterial infections were comparable between the groups (P = 0.318).
COX-2 promoter SNP selection
The COX-2 gene and the position of the promoter SNPs (i.e., −512 C > T, −608 T > C −765 G > C, and −1195 A > G) selected for genotyping are detailed in Fig. 1a. Data from the International HapMap Project and 1000 Genomes Project were used to determine the allelic distributions of the four SNPs in different African populations [Luhya (LWK) and Maasai (MKK)], Yoruba (YRI; Nigeria), and African (AFR)] populations, and globally (Fig. 1b). Transcription factor binding site (TFBS) analyses revealed that all of the SNPs could potentially alter binding patterns for transcription factors (Fig. 1c). Additional analyses after genotyping the SNPs showed a modest amount of LD, based on the D′ score, between −512 C > T and −608 T > C (D′: −0.6646, r2: 0.00886) and −512 C > T and −765 G > C (D′: 0.6709, r2: 0.07556). However, the low r2 values indicate that these SNPs are not good proxies for one another [43]. Based on results from the different analyses, we included all four SNPs in the investigations presented here.
Fig. 1.
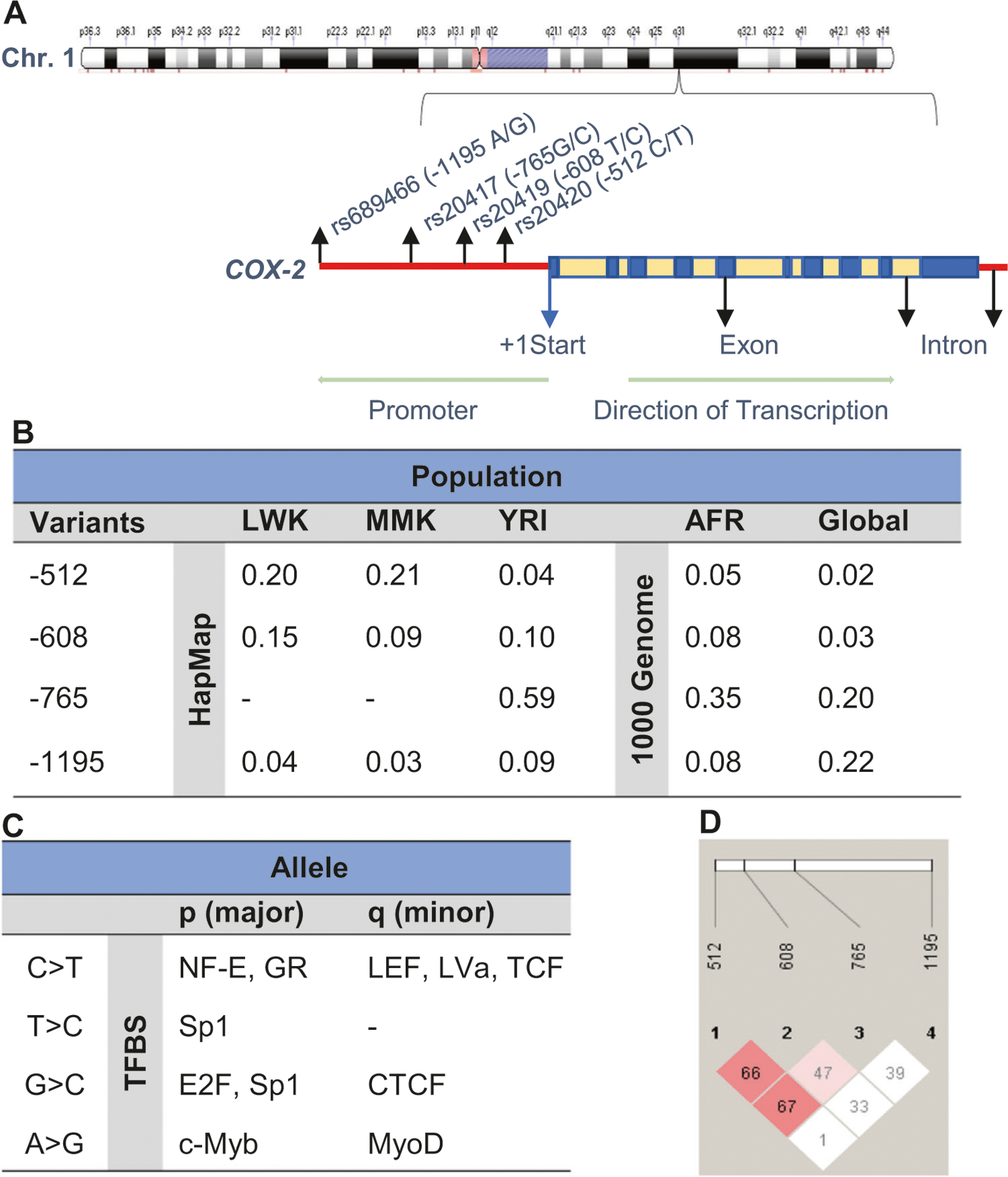
COX-2 chromosome location and linkage disequilibrium of SNPs. a Location of prostaglandin-endoperoxide synthase 2 (PTGS2; COX-2) on chromosome 1q31. COX-2 is 8,616 bp and is composed of 10 exons. Chromosome position (build GRCh38.p12) for the selected SNPs under investigation; rs20420 (−512 C > T) is located at 186680936, rs20419 (−608 T > C) located at 186681031, rs20417 (−765 G > C) located at 186681189 and rs689466 (−1195 A > G) located at 186681619. The alignment and gene structure of COX-2 are based the Golden Helix Genome Browse 1.0.1. b COX-2 genotypes minor allele frequencies (MAF) from the International HapMap project for the Kenyan [Luhya (LWK) and Maasai (MKK)] populations and Yoruba (YRI; Nigeria). MAFs from the 1000 Genome project shows African (AFR) ancestry and global allele frequencies. c Transcription factor binding analyses of the COX-2 variants. NF-E Eucaryotic nuclear factor, GR Glucocorticoid receptor, LEF Lymphocyte enhancing factor, LVa Lava lamp, TCF T-cell factor, SP1 stimulatory protein 1, E2F E2F transcription factor family, CTCF CCCTC binding factor, c-Myb MYB proto-oncogene, and MyoD myogenic differentiation. d Linkage disequilibrium between the selected COX-2 SNPs: −512 C > T and −608 T > C (D′: −0.6646, r2: 0.00886); −512 C > T and −765 G > C (D′: 0.6709, r2: 0.07556); −512 C > T and −1195 A > G (D′: 0.0148, r2: 0.00015); −608 T > C and −765 G > C (D′: −0.4787, r2: 0.02736); −608 T > C and −1195 A > G (D′: −0.3384, r2: 0.00154), and −765 G > C and −1195 A > G (D′: −0.3934, r2: 0.00517).
Distribution of COX-2 variants
The distribution of the COX-2 promoter SNPs [−512 C > T (rs20420), −608 T > C (rs20419), −765 G > C (rs20417) and −1195 A > G (rs689466)], allele frequencies, and Hardy–Weinberg Equilibrium (HWE) for each of the variants are shown in Table 2. There were no significant differences in the proportions of any of the four genotypic variants across the clinical groups. For each of the four SNPs, there was significant departure from HWE in the overall population, aparasitemic and non-SMA groups, but not in children with SMA (Table 2).
Table 2.
Distribution of COX-2 variants
| Variants | Total Population n (%) | Aparasitemic (MPS-) n (%) | Non-SMA (Hb ≥ 5.0 g/dL) n (%) | SMA (Hb < 5.0 g/dL) n (%) | P value | ||
|---|---|---|---|---|---|---|---|
| −512 | |||||||
| CC | 921 (85.2) | 217 (83.1) | 578 (86.3) | 126 (84.0) | 0.180a | ||
| CT | 139 (12.9) | 42 (16.1) | 76 (11.3) | 21 (14.0) | |||
| TT | 21 (1.9) | 2 (0.8) | 16 (2.4) | 3 (2.0) | |||
| MAF (p/q) | 0.916/0.084 | 0.912/0.088 | 0.919/0.081 | 0.911/0.089 | |||
| HWE (X2, P) | 28.327, <0.001 | 0.984, <0.001 | 36.877, <0.001 | 3.167, 0.075 | |||
| −608 | |||||||
| TT | 748 (69.2) | 186 (71.3) | 457 (68.2) | 105 (70.0) | 0.664a | ||
| TC | 277 (25.6) | 59 (22.6) | 179 (26.7) | 39 (26.0) | |||
| CC | 56 (5.2) | 16 (6.1) | 34 (5.1) | 6 (4.0) | |||
| MAF (p/q) | 0.820/0.180 | 0.826/0.174 | 0.816/0.184 | 0.830/0.170 | |||
| HWE (X2, P) | 18.746, 0.001 | 12.038, 0.001 | 8.335, 0.004 | 0.928, 0.335 | |||
| −765 | |||||||
| GG | 475 (43.9) | 113 (43.3) | 297 (44.3) | 65 (43.3) | 0.148a | ||
| GC | 450 (41.6) | 103 (39.5) | 275 (41.0) | 72 (48.0) | |||
| CC | 156 (14.4) | 45 (17.2) | 98 (14.6) | 13 (8.7) | |||
| MAF (p/q) | 0.648/0.352 | 0.630/0.370 | 0.649/0.351 | 0.673/0.327 | |||
| HWE (X2, P) | 8.375, 0.004 | 6.130, 0.013 | 6.657, 0.010 | 1.246, 0.264 | |||
| −1195 | |||||||
| AA | 973 (90.0) | 239 (91.6) | 595 (88.8) | 135 (92.7) | 0.271a | ||
| AG | 91 (8.4) | 17 (6.5) | 63 (9.4) | 11 (7.3) | |||
| GG | 17 (1.6) | 5 (1.9) | 12 (1.8) | 0 (0.0) | |||
| MAF (p/q) | 0.942/0.058 | 0.948/0.052 | 0.935/0.065 | 0.962/0.038 | |||
| HWE (X2, P) | 55.863, 0.001 | 29.471, 0.001 | 34.095, 0.001 | 0.224, 0.363 | |||
| Haplotypes | |||||||
| CTGA | 0 | 391 (36.2) | 100 (38.3) | 248 (37.0) | 43 (28.7) | 0.112a | |
| 1 | 690 (63.8) | 161 (61.7) | 422 (63.0) | 107 (71.3) | |||
| CCGA | 0 | 777 (71.9) | 195 (74.7) | 476 (71.0) | 106 (70.7) | 0.502a | |
| 1 | 304 (28.1) | 66 (25.3) | 194 (29.0) | 44 (29.3) | |||
| CTCA | 0 | 590 (54.6) | 137 (52.5) | 367 (54.8) | 86 (57.3) | 0.629a | |
| 1 | 491 (45.4) | 124 (47.5) | 303 (45.2) | 64 (42.7) | |||
| CTCG | 0 | 1072 (99.2) | 260 (99.6) | 662 (98.8) | 150 (100.0) | 0.228b | |
| 1 | 9 (0.8) | 1 (0.4) | 8 (1.2) | 0 (0.0) | |||
| TTCA | 0 | 952 (88.1) | 228 (87.4) | 597 (89.1) | 127 (84.7) | 0.292a | |
| 1 | 129 (11.9) | 33 (12.6) | 73 (10.9) | 23 (15.3) | |||
| TTGG | 0 | 1076 (99.5) | 260 (99.6) | 668 (99.7) | 148 (98.7) | 0.235b | |
| 1 | 5 (0.5) | 1 (0.4) | 2 (0.3) | 2 (1.3) | |||
| CCCA | 0 | 1,046 (96.8) | 250 (23.1) | 648 (96.7) | 148 (98.7) | 0.281a | |
| 1 | 35 (3.2) | 11 (4.2) | 22 (3.3) | 2 (1.3) | |||
| TTGA | 0 | 1059 (98.0) | 254 (97.3) | 657 (98.1) | 148 (98.7) | 0.622b | |
| 1 | 22 (2.0) | 7 (|2.7) | 13 (1.9) | 2 (1.3) | |||
| CTGG | 0 | 993 (91.9) | 242 (92.7) | 610 (91.0) | 141 (94.0) | 0.412a | |
| 1 | 88 (8.1) | 19 (7.3) | 60 (9.0) | 9 (6.0) | |||
| TCGA | 0 | 1077 (99.6) | 259 (99.2) | 668 (99.7) | 150 (100.0) | 0.414a | |
| 1 | 4 (0.4) | 2 (0.8) | 2 (0.3) | 0 (0.0) | |||
| CCGG | 0 | 1075 (99.4) | 260 (99.6) | 665 (99.3) | 150 (100.0) | 0.491b | |
| 1 | 6 (0.6) | 1 (0.4) | 5 (0.7) | 0 (0.0) | |||
| TTCG | 0 | 1075 (99.4) | 261 (100.0) | 664 (99.1) | 150 (100.0) | 0.157b | |
| 1 | 6 (0.6) | 0 (0.0) | 6 (0.9) | 0 (0.0) | |||
Data are presented as proportions [n (%)] of genetic variants within the study groups. Children (n = 1081) were categorized into aparasitemic controls (n = 261; no parasitemia) and based on WHO definition of SMA into either non-SMA (n = 670; Hb ≥ 5.0 g/dL) or SMA (n = 150; Hb < 5.0 g/dL). Minor Allele frequencies (MAF; p/q) were computed for variants within study groups. Hardy–Weinberg Equilibrium (HWE) was computed using a χ2 goodness of fit test. Haplotypes were constructed using the HPlus software program (Fred Hutchinson Cancer Research Center, WA, USA), and their distribution frequencies estimated using haploview software (version 4.2; Broad Institute, MA, USA). (0) = non-carriers of haplotypes and (1) = carriers of haplotypes.
Statistical significance determined by Pearson’s χ2 test.
Statistical significance determined by Fisher’s exact tests
Since haplotypic combinations offer important information about patterns of inheritance that influence susceptibility to SMA [36], their frequencies across groups were determined. The proportional distribution of non-carriers and carriers of the haplotypes in the overall population and clinical groups is shown in Table 2. Comparison of non-carriers versus carriers did not reveal any significant differences between any of the COX-2 haplotypes.
Relationship between COX-2 genetic variants, malaria, and SMA at enrollment
The cross-sectional influence of the COX-2 SNPs on susceptibility to malaria and SMA was determined using binary logistic regression analyses controlling for potential confounders (i.e., age, sex, sickle-cell trait status, G6PD deficiency, α3.7-thalassemia deletions, bacterial infections, and HIV-1 positivity) [5, 7, 8, 44] (Table 3). The wild-type variants (COX-2 −512 CC, −608 TT, −765 GG, and −1195 AA) were used as the reference groups in the analyses.
Table 3.
Impact of COX-2 variants on malaria and SMA at enrollment
| Variants | Malaria | SMA | ||||||
|---|---|---|---|---|---|---|---|---|
| Events | OR | 95% CI | P value | Events | OR | 95% CI | P value | |
| −512 | ||||||||
| CC | n = 704 | REF | – | – | n = 126 | REF | – | – |
| CT | n = 97 | 0.648 | 0.419–1.000 | 0.050 | n = 21 | 1.368 | 0.771–2.247 | 0.284 |
| TT | n = 19 | 2.427 | 0.497–11.859 | 0.273 | n = 3 | 1.106 | 0.287–4.265 | 0.883 |
| −608 | ||||||||
| TT | n = 562 | REF | – | – | n = 105 | REF | – | – |
| TC | n = 218 | 1.155 | 0.790–1.689 | 0.456 | n = 39 | 1.141 | 0.720–1.809 | 0.574 |
| CC | n = 40 | 0.644 | 0.326–1.275 | 0.207 | n = 6 | 0.904 | 0.324–2.522 | 0.848 |
| −765 | ||||||||
| GG | n = 362 | REF | – | – | n = 65 | REF | – | – |
| GC | n = 347 | 1.076 | 0.759–1.528 | 0.681 | n = 72 | 0.859 | 0.562–1.313 | 0.483 |
| CC | n = 111 | 0.966 | 0.595–1.569 | 0.890 | n = 13 | 1.805 | 0.902–3.610 | 0.095 |
| −1195 | ||||||||
| AA | n = 730 | REF | – | – | n = 135 | REF | – | – |
| AG | n = 74 | 0.753 | 0.230–2.463 | 0.638 | n = 11 | 0.851 | 0.422–1.716 | 0.652 |
| GG | n = 12 | 0.672 | 0.184–2.453 | 0.547 | n = 0 | – | – | |
| Haplotypes | ||||||||
| CTGA | n = 529 | 1.167 | 0.840–1.623 | 0.358 | n = 107 | 1.533 | 0.998–2.354 | 0.051 |
| CCGA | n = 238 | 1.022 | 0.716 – 1.458 | 0.906 | n = 44 | 1.021 | 0.655–1.590 | 0.927 |
| CTCA | n = 367 | 0.980 | 0.709 – 1.355 | 0.903 | n = 64 | 0.827 | 0.554–1.234 | 0.352 |
| CTCG | n = 8 | 2.474 | 0.307 –19.936 | 0.395 | n = 0 | – | – | – |
| TTCA | n = 96 | 1.254 | 0.796 –1.974 | 0.329 | n = 23 | 1.486 | 0.850–2.597 | 0.164 |
| TTGG | n = 4 | 1.611 | 0.127 –20.406 | 0.713 | n = 2 | 5.780 | 0.735–45.457 | 0.095 |
| CCCA | n = 24 | 0.882 | 0.342 –2.275 | 0.795 | n = 2 | 0.479 | 0.107–2.142 | 0.335 |
| TTGA | n = 15 | 0.796 | 0.252 –2.513 | 0.697 | n = 2 | 0.956 | 0.198–4.618 | 0.955 |
| CTGG | n = 69 | 0.949 | 0.548 –1.641 | 0.850 | n = 9 | 0.701 | 0.330–1.491 | 0.356 |
| TCGA | n = 2 | 0.146 | 0.013 –1.691 | 0.124 | n = 0 | – | – | – |
| CCGG | n = 5 | 1.348 | 0.154 –11.820 | 0.787 | n = 0 | – | – | – |
| TTCG | n = 5 | 1.611 | 0.127 –20.406 | 0.713 | n = 0 | – | – | – |
Data are presented as odds (OR) and 95% confidence intervals (CI) determined using binary logistic regression with the following covariates in the models: age at enrollment, sex, HIV-1 and bacteremia (presence/absence), and sickle cell trait, α+-thalassemia, and G6PD status. Children (n = 1081) were categorized into either malaria negative (n = 261; aparasitemic) or positive (n = 820; parasitemic) for determining susceptibility to malaria, and into either non-SMA (n = 670; Hb ≥ 5.0 g/dL) or SMA (n = 150; Hb < 5.0 g/dL) for determining susceptibility to SMA. Homozygous wild-type genotypes and non-carriers of haplotypes were used as references in the analyses. Bold indicates a P value ≤ 0.050
Inheritance of the heterozygous CT genotype at −512 decreased susceptibility to malaria by 35% [OR = 0.648 (95% CI = 0.419–1.000), P = 0.050, Table 3]. None of the other genotypes were significantly associated with susceptibility to malaria in the cross-sectional analyses (Table 3). Moreover, variation at the four SNPs did not significantly alter the cross-sectional risk of developing SMA (Table 3).
To determine the impact of the haplotypes on susceptibility to malaria and SMA (cross-sectionally), we performed binary logistic regression analyses (IBM SPSS Inc., Chicago, IL, USA), with non-carriage versus carriage as the predictor variables, controlling for identical covariates. None of the haplotypes revealed a significant risk profile for susceptibility to malaria (Table 3). However, inheritance of the CTGA haplotype increased the risk (53%) of developing SMA [OR = 1.533 (95% CI = 0.998–2.354), P = 0.051], while none of the other haplotypes significantly altered susceptibility to SMA (Table 3). Haplotypes with low frequencies and no SMA events (i.e., CTCG, TCGA, CCGG, and TTGG) were not run in the models.
Impact of COX-2 genotypes on the longitudinal risk of malaria and SMA
Children residing in holoendemic P. falciparum transmission regions typically experience repeated episodes of malaria prior to developing naturally-acquired immunity [10]. Prior to the development of malarial immunity, children can also experience one or more life-threatening episodes of SMA [10]. As such, we examined the impact of the COX-2 genotypes on susceptibility to malaria and SMA over 36 months of follow-up using Poisson regression modeling with age at enrollment, sex, sickle-cell trait status, G6PD deficiency, α3.7-thalassemia deletions, and bacteremia and HIV-1 positivity as covariates (Table 4). There were 5331 malaria episodes and 238 cases of SMA over the follow-up period. The number of malaria and SMA episodes for each of the genotypes is listed in Table 4.
Table 4.
Impact of COX-2 variants on malaria and SMA over 36 months
| Variants | Malaria | SMA | ||||||
|---|---|---|---|---|---|---|---|---|
| Events | RR | 95% CI | P value | Events | RR | 95% CI | P value | |
| −512 | ||||||||
| CC | n = 4591 | REF | – | – | n = 213 | REF | – | – |
| CT | n = 759 | 1.074 | 0.988–1.169 | 0.095 | n = 39 | 1.241 | 0.847–1.817 | 0.268 |
| TT | n = 109 | 1.072 | 0.866–1.329 | 0.522 | n = 6 | 1.088 | 0.449–2.637 | 0.852 |
| −608 | ||||||||
| TT | n = 3776 | REF | – | – | n = 184 | REF | – | – |
| TC | n = 1453 | 1.017 | 0.952–1.087 | 0.622 | n = 64 | 0.875 | 0.638–1.198 | 0.405 |
| CC | n = 230 | 0.746 | 0.640–0.869 | 1.811e−4 | n = 10 | 0.644 | 0.300–1.383 | 0.259 |
| −765 | ||||||||
| GG | n = 2290 | REF | – | – | n = 105 | REF | – | – |
| GC | n = 2322 | 0.980 | 0.919–1.045 | 0.537 | n = 1127 | 1.147 | 0.857–1.536 | 0.356 |
| CC | n = 847 | 1.035 | 0.943–1.136 | 0.466 | n = 26 | 0.654 | 0.397–1.076 | 0.095 |
| −1195 | ||||||||
| AA | n = 4963 | REF | – | – | n = 231 | REF | – | – |
| AG | n = 432 | 0.919 | 0.828–1.020 | 0.110 | n = 25 | 1.060 | 0.689–1.632 | 0.791 |
| GG | n = 64 | 0.798 | 0.615–1.035 | 0.089 | n = 2 | 0.303 | 0.042–2.168 | 0.235 |
| Haplotypes | ||||||||
| CTGA | n = 2390 | 1.077 | 0.987–1.174 | 0.096 | n = 123 | 1.464 | 0.971–2.207 | 0.069 |
| CCGA | n = 1411 | 1.017 | 0.939–1.101 | 0.682 | n = 64 | 1.085 | 0.743–1.584 | 0.674 |
| CTCA | n = 2140 | 1.026 | 0.952–1.105 | 0.508 | n = 99 | 1.049 | 0.746–1.477 | 0.782 |
| CTCG | n = 45 | 1.227 | 0.885–1.701 | 0.219 | n = 0 | – | – | – |
| TTCA | n = 676 | 1.115 | 1.013–1.227 | 0.026 | n = 38 | 1.320 | 0.842–2.068 | 0.226 |
| TTGG | n = 34 | 1.755 | 1.170–2.634 | 0.007 | n = 4 | 8.706 | 2.633–28.790 | 3.917e−4 |
| CCCA | n = 147 | 0.867 | 0.716–1.050 | 0.144 | n = 2 | 0.633 | 0.192–2.085 | 0.452 |
| TTGA | n = 113 | 1.032 | 0.842–1.265 | 0.763 | n = 3 | 0.830 | 0.259–2.664 | 0.754 |
| CTGG | n = 354 | 0.856 | 0.760–0.965 | 0.011 | n = 19 | 0.965 | 0.563–1.654 | 0.897 |
| TCGA | n = 16 | 0.670 | 0.401–1.119 | 0.126 | n = 0 | – | – | – |
| CCGG | n = 26 | 0.991 | 0.666–1.474 | 0.962 | n = 0 | – | – | – |
| TTCG | n = 31 | 1.608 | 1.091–2.371 | 0.016 | n = 4 | 5.714 | 1.725–18.930 | 0.004 |
Data are presented as relative risk (RR) and 95% confidence intervals (CI) determined using Poisson regression modeling performed on longitudinal data of >14,000 patient visits to assess the risk to malaria or SMA episodes. Children (n = 1081) were followed at day 14 after enrollment and 14 days after a malaria episode, quarterly, and at acute febrile visits for 36 months. There were 5,331 malaria episodes and 238 SMA (Hb < 5.0 g/dL) episodes. The log linear regression model controlled for age, sex, HIV-1 and bacteremia (presence/absence), and sickle cell trait, α+-thalassemia, and G6PD status. The homozygous wild-type genotypes and non-carriers of haplotypes were used as references in the analyses. Bold indicates a P value ≤ 0.050
Variation at −512 did not significantly alter the risk to either malaria or SMA. Although carriage of the TC allele at −608 did not impact on susceptibility to either malaria or SMA, inheritance of the CC allele decreased the longitudinal risk of malaria by 25% [RR = 0.746 (95% CI = 0.640–0.869, P = 1.811e−4], but had no influence on the development of SMA. None of the −765 genotypes significantly altered the risk profile for acquiring malaria or SMA. Although not significant, carriage of the CC allele at −795 decreased the risk of SMA by 35% [RR = 0.654 (95% CI = 0.397–1.076), P = 0.095]. Inheritance of the AG allele at −1195 did not affect the risk of either malaria or SMA. However, carriers of the GG allele at −1195 had a 20% decreased likelihood of developing repeated episodes of malaria during the follow-up [RR = 0.798 (95% CI = 0.615–1.035), P = 0.089], while the reduced risk of SMA in the small number of carriers was non-significant.
Secondary analyses were performed to determine if any of the SNPs showed an additive effect on susceptibility to malaria and SMA over the follow-up period. The additive models revealed a decreased risk of malaria for the −608 [RR = 0.945 (95% CI = 0.897–0.996), P = 0.034] and −1195 [RR = 0.911 (95% CI = 0.839–0.989), P = 0.026] genotypes, while the additive effect of the other SNPs did not alter the risk of developing either malaria or SMA.
Impact of COX-2 haplotypes on the longitudinal risk of clinical malaria and SMA
To further examine the relationship between COX-2 promoter variants and the risk of malaria and SMA, the impact of haplotypes on risk profiles was determined with Poisson regression analyses using identical covariates in which individuals without the haplotype were used as the reference group (Table 4). Stratification of malaria and SMA episodes for each of the haplotypes is listed in Table 4. Carriage of the CTGA haplotypes was associated with a marginal level of increased risk for both malaria [RR = 1.077 (95% CI = 0.987–1.174), P = 0.096] and SMA [RR = 1.464 (95% CI = 0.971–2.207), P = 0.069]. There was an enhanced risk of developing malaria in children who inherited the TTCA haplotype [RR = 1.115 (95% CI = 1.013–1.227), P = 0.026], but the enhanced risk of SMA (32%) was not significant [RR = 1.320 (95% CI = 0.842–2.068), P = 0.226]. Inheritance of the TTGG haplotype increased the risk of malaria by 75% [RR = 1.755 (95% CI = 1.170–2.634), P = 0.007], and the risk of developing SMA by 8.7-fold in the low number of carriers [RR = 8.706 (95% CI = 2.633–28.790), P = 3.97 × 10−4]. Similarly, carriage of the TTCG haplotype also enhanced susceptibility to malaria [RR = 1.608 (95% CI = 1.091–2.371), P = 0.016] and SMA [RR = 5.714 (95% CI = 1.725–18.930, P = 0.004).
The only haplotype in which inheritance significantly reduced the risk of malaria (14%) was observed for CTGG [RR = 0.856 (95% CI = 0.760–0.965), P = 0.011].However, carriage of this haplotype had no impact on the development of SMA [RR = 0.965 (95% CI = 0.563–1.654), P = 0.897]. Secondary analyses using an additive model of inheritance did not reveal any significant relationships between any of the haplotypes and either malaria or SMA.
Association between COX-2 promoter variants and mortality
Since SMA is a significant cause of childhood mortality in holoendemic transmission regions, we used Cox stepwise proportional hazards regression to investigate the relationship between COX-2 variants and survival over the follow-up period. For this analysis, we used identical covariates that were in the longitudinal malaria and SMA models. There were 68 deaths in the cohort of children. There were no associations between any of the COX-2 genotypes and mortality. However, carriage of the TTGG haplotype which increased susceptibility to SMA by 8.7-fold also substantially enhanced the risk of mortality [HR = 110.00 (95% CI = 6.420–1.88 × 103), P = 0.001]. None of the other haplotypes had a significant impact on the risk of mortality over the follow-up period.
Discussion
Historical presence of malaria in populations in endemic regions has exerted a substantial impact on the human genome, which includes the preservation of harmful variants, largely due to the advantageous protective effect against malaria fatalities in individuals with heterozygous alleles [44]. Although clinical immunity to malaria is largely mediated by the development of antibodies after repeated episodes of disease, immune-naïve children, who suffer the greatest burden of severe malaria, must rely on innate immune responses prior to the acquisition of naturally-acquired immunity [10, 36, 45]. As such, our efforts are focused on innate immune response genes and gene pathways that mediate the development of SMA in young children once an individual becomes infected [36]. To extend these findings, we investigated the impact of COX-2 promoter polymorphic alleles (−512 C > T, −608 T> C, −765 G > C, and −1195 A > G) on malaria and SMA cross-sectionally and longitudinally over a 36-month period during the development of malarial immunity. In addition, since SMA is a significant risk factor for childhood mortality in holoendemic P. falciparum transmission regions, we also determined the impact of inheritance of the COX-2 variants on all-cause mortality.
Prior to determining the relationship between genotypes/haplotypes and susceptibility to clinical outcomes of malaria, we compared the variant frequencies of the four loci independently. There were no significant variations in frequencies of the genotypes across the clinical groups (i.e., aparasitemic, non-SMA, and SMA). The allele frequencies in the Luo ethnic group (investigated) here most closely resemble those reported for the Yoruba population in the HapMap Project and the African population in the 1000 Genome Project. All four SNPs displayed significant departure from HWE in the overall study population. Although such departure could be attributed to genotyping error, this does not appear to explain the findings here since 10% of the samples from the overall population were randomly selected, and each polymorphic variant was genotyped using restriction fragment length polymorphism PCR, yielding identical results to the high-throughput assays. Since we largely sampled diseased individuals, the significant departure from HWE likely reflects an association of the variants with malaria [43, 46]. Given the genetic homogeneity of the population under study (>98% Luo), which resides in a holoendemic P. falciparum transmission region, it is feasible that deviation from HWE is due to the intense selection pressure from malaria. Collectively, these results suggest that the promoter variants investigated here have been largely maintained in African ethnic groups, potentially due to the selective pressure by life-threatening malaria.
To better understand the role of genetic variation in the pathogenesis of severe malaria, combinations of gene variants have been used to formulate multi-site haplotypes since they may reveal how combinations of different functional polymorphic alleles interact to amplify or moderate individual allelic effects. Since the combination of haplotypic constructs investigated here have not been previously reported, we performed LD analyses, which revealed low LD between the variants, particularly across the entire block. Comparison of genotype frequencies across groups revealed no variabilities in distribution. The genotypes displayed a significant departure from HWE in the aparasitemic and non-SMA groups, but not in the SMA group, suggesting possible selection for the mutations in the population. Further, comparison of haplotype frequencies revealed non-significant differences in proportions across groups.
Assessment of the relationship between individual genotypes and cross-sectional susceptibility to malaria and SMA, using a binary logistic regression model controlling for potential confounders, revealed that heterozygous carriage (CT) at the −512 locus decreased the risk of malaria, but not SMA. None of the other genotypes altered the risk to either malaria or SMA at enrollment. While none of the haplotypes significantly influenced the risk of malaria upon enrollment, inheritance of the CTGA haplotype moderately predisposed children to enhanced susceptibility to SMA. To our knowledge, there are no previous reports in human populations describing the relationship between the SNPs presented here and susceptibility to either malaria or SMA.
Children (<5 years) in high malaria transmission regions often experience multiple episodes of malaria before maturation of competent immunity [47]. Analysis of a single cross-sectional time point for genetic association studies investigating susceptibility to malaria and SMA may not suitably capture the changing dynamics of the disease burden and development of naturally-acquired immunity. To address this challenge, we performed longitudinal analysis to determine the influence of the COX-2 variants on malaria and SMA episodes across 36 months of follow-up, as well as all-cause mortality. Inheritance of the CC allele at the −608 loci decreased the longitudinal risk of malaria in the non-carriage vs carriage model, with the additive model at these loci also showing a protective effect. Variation at the −1195 loci was associated with protection against malaria only in the additive model. None of the COX-2 SNPs altered the risk to either SMA or all-cause mortality over the follow-up period.
Modeling of the longitudinal risk profiles associated with the haplotypes showed that inheritance of the TTCA haplotype significantly enhanced the risk of malaria, and marginally increased susceptibility to SMA. Carriage of the TTGG and TTCG haplotypes, however, significantly enhanced susceptibility to both malaria and SMA. The only haplotype associated with protective effects was CTGG, which decreased the risk of malaria, but not SMA. Collectively, these results show that haplotypic inheritance patterns of the four COX-2 promoter SNPs significantly influence the acquisition malaria across time, as well as the development of SMA once an individual becomes infected with falciparum. While the exact nature of the COX-2 inheritance patterns on the immune response to malaria remains to be determined, we postulate that the SNPs selected for investigation impart functional changes on COX-2 gene expression, and hence, the production of PGE2.
Our previous investigations showed that P. falciparum induces suppression of COX-2-derived PGE2, which in turn, enhances disease severity in malaria during pregnancy, and in children with either cerebral malaria or SMA [11, 15, 16]. As such, we speculate that the haplotypes, which enhanced the longitudinal risk of malaria and SMA, are associated with functionally less COX-2-derived PGE2 production. Unfortunately, there was not enough biological material available in the repository to test this hypothesis. Previous functional studies demonstrated that both single constructs and haplotypic combinations of the C allele at −765 and the G allele at −1195 have reduced COX-2 expression [23, 34]. As such, we selected these two variants, along with two others SNPs which have the potential to functionally alter COX-2 expression (see Fig. 1c). Although it is challenging to speculate about how combinatorial diverse of haplotypic constructs from the four SNPs functionally impact on COX-2 expression and PGE2 production without directly generating constructs, our hypothesis that enhanced risk to malaria and SMA in carriers of the rare TTGG and TTCG haplotypes is, at least in part, due to reduced COX-2-derived PGE2 production is consistent with previous functional promoter construct studies. Additional investigations aimed at defining the functional consequences of haplotypes derived from the four COX-2 SNPs are currently ongoing in our laboratory.
In western Kenya, SMA is among the leading causes of mortality of children below 5 years of age [2]. Our previous studies in this falciparum holoendemic region revealed that younger age, reticulocytosis, reduced erythropoiesis, elevated pigment-containing monocytes, and conjunctival and palmar pallor were significant predictors of SMA (reviewed in [36]). We and others have also shown that children with SMA have lower peripheral parasite densities than parasitemic children without anemia (Hb ≥11.0 g/dL) [35, 48], suggesting that acute hemolysis of RBCs is not likely the primary cause of low Hb levels in life-threatening SMA in this holoendemic transmission region [36]. Although CM is an important cause of mortality in children with malaria, such cases are rare is Siaya. This is largely because CM occurs in areas of low-to-moderate endemicity with seasonal variation, and primarily affects older children, adolescents, and adults with low levels of acquired immunity to malaria [49]. As such, CM cases are excluded from our studies since the presence of childhood CM in Siaya typically suggests recent migration into the study area. While CM is not a primary factor for childhood mortality in the region, our previous studies showed that HIV-1 and bacteremia substantially increase life-threatening anemia in Siaya [6–8]. Despite the burden of malaria and co-infections, the study cohort only had an all-cause mortality rate of 5.51%. Approximately 51% of the deaths occurred at the children’s homes and were recorded during the quarterly visits over the 36 months follow-up period. The mortality rates are much lower than those reported during a comparable time period for Nyanza Province, in which there were ~206 deaths per 1000 live births [50]. The lower number of deaths in our cohort can be attributed to: (1) enhanced and rapid laboratory diagnostics for common infectious causes of childhood diseases, including antimicrobial susceptibility testing, (2) improved clinical management with the availability of second-line antimicrobial agents (when required), and (3) a follow-up schedule which included the parent/guardian being asked to bring their child back to the hospital during acute febrile episodes for a complete clinical work-up and treatment. Cox stepwise proportional hazards regression modeling failed to find any associations between genotypes and all-cause mortality. However, analysis of the relationship between haplotypic carriage and all-cause mortality revealed that inheritance of the rare TTGG haplotype, which increased the longitudinal risk of malaria and SMA, also markedly enhanced the risk of mortality. The rarity of this haplotype may, therefore, be the result of selection pressure due to the substantial impact of TTGG inheritance on the development of severe malaria and all-cause mortality.
In conclusion, investigations reported here reveal novel relationships between COX-2 promoter variants and the longitudinal risk of malaria, SMA, and all-cause mortality. To our knowledge, this is the first study examining the impact of COX-2 genetic variation on malaria disease outcomes. It will be important for future studies to define the molecular consequences of the COX-2 variants presented here. Such information could be important for selective therapeutic interventions in individuals with particular COX-2 gene inheritance patterns, particularly since COX-2 altering pharmacological agents are widely used in clinical practice for other diseases.
Acknowledgements
We gratefully acknowledge the assistance of the University of New Mexico-Kenya team: Nicholas Otieno Ondiek, Vincent Odhiambo Otieno, Anne A Ong’ondo, Chrispine Wasonga Ochieng, Everlyne A Modi, Joan L A Ochieng, Jacob Oyoko Odeny, Joseph Oduor, Martin Ogalo, Moses Ebungure, Moses Lokorkeju, Rodney B Mongare, Stella Mariz Akinyi Oloo and Vincent Omanje. We are also grateful to all of the parents, guardians, and children who participated in the study. The content is solely the responsibility of the authors and does not represent the official views of the funding agencies. The content is solely the responsibility of the authors and the funders did not have any role in study design, data collection, data analysis, interpretation, or writing of the report. The work was supported by National Institutes of Health (NIH) Research Grants R01AI130473 and R01AI51305 (DJP), NIH Fogarty International Center Grants D43TW05884 (DJP, SBA) and D43 TW010543 (SBA, DJP), and LANL-LDRD 20150090DR (BHM, DJP).
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. World Malaria Report 2018 WHO Report. Geneva: World Health Organization; 2018. ISBN 978-92-4-156565-3 Contract No.: Licence: CC BY-NC-SA 3.0 IGO [Google Scholar]
- 2.Amek NO, Van Eijk A, Lindblade KA, Hamel M, Bayoh N, Gimnig J, et al. Infant and child mortality in relation to malaria transmission in KEMRI/CDC HDSS, Western Kenya: validation of verbal autopsy. Malar J. 2018;17:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Obonyo CO, Vulule J, Akhwale WS, Grobbee DE. In-hospital morbidity and mortality due to severe malarial anemia in western Kenya. Am J Trop Med Hyg. 2007;77(6 Suppl):23–8. [PubMed] [Google Scholar]
- 4.Breman JG, Egan A, Keusch GT. The intolerable burden of malaria: a new look at the numbers. Am J Trop Med Hyg. 2001;64 (1–2 Suppl):iv–vii. [DOI] [PubMed] [Google Scholar]
- 5.Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, et al. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet 2002;359:1311–2. [DOI] [PubMed] [Google Scholar]
- 6.Davenport GC, Ouma C, Hittner JB, Were T, Ouma Y, Ong’echa JM, et al. Hematological predictors of increased severe anemia in Kenyan children coinfected with Plasmodium falciparum and HIV-1. Am J Hematol. 2010;85:227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otieno RO, Ouma C, Ong’echa JM, Keller CC, Were T, Waindi EN, et al. Increased severe anemia in HIV-1-exposed and HIV-1-positive infants and children during acute malaria. AIDS. 2006; 20:275–80. [DOI] [PubMed] [Google Scholar]
- 8.Were T, Davenport GC, Hittner JB, Ouma C, Vulule JM, Ong’echa JM, et al. Bacteremia in Kenyan children presenting with malaria. J Clin Microbiol. 2011;49:671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fendel R, Brandts C, Rudat A, Kreidenweiss A, Steur C, Appelmann I, et al. Hemolysis is associated with low reticulocyte production index and predicts blood transfusion in severe malarial anemia. PLoS ONE. 2010;5:e10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perkins DJ, Were T, Davenport GC, Kempaiah P, Hittner JB, Ong’echa JM. Severe malarial anemia: innate immunity and pathogenesis. Int J Biol Sci. 2011;7:1427–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anyona SB, Kempaiah P, Raballah E, Davenport GC, Were T, Konah SN, et al. Reduced systemic bicyclo-prostaglandin-E2 and cyclooxygenase-2 gene expression are associated with inefficient erythropoiesis and enhanced uptake of monocytic hemozoin in children with severe malarial anemia. Am J Hematol. 2012;87:782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perkins DJ, Kremsner PG, Weinberg JB. Inverse relationship of plasma prostaglandin E2 and blood mononuclear cell cyclooxygenase-2 with disease severity in children with Plasmodium falciparum malaria. J Infect Dis. 2001;183:113–8. [DOI] [PubMed] [Google Scholar]
- 13.Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3’-untranslated region. J Biol Chem. 2000;275:11750–7. [DOI] [PubMed] [Google Scholar]
- 14.Agard M, Asakrah S, Morici LA. PGE(2) suppression of innate immunity during mucosal bacterial infection. Front Cell Infect Microbiol. 2013;3:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins DJ, Hittner JB, Mwaikambo ED, Granger DL, Weinberg JB, Anstey NM. Impaired systemic production of prostaglandin E2 in children with cerebral malaria. J Infect Dis. 2005;191:1548–57. [DOI] [PubMed] [Google Scholar]
- 16.Perkins DJ, Moore JM, Otieno J, Shi YP, Nahlen BL, Udhaya-kumar V, et al. In vivo acquisition of hemozoin by placental blood mononuclear cells suppresses PGE2, TNF-alpha, and IL-10. Biochem Biophys Res Commun. 2003;311:839–46. [DOI] [PubMed] [Google Scholar]
- 17.Kosaka T, Miyata A, Ihara H, Hara S, Sugimoto T, Takeda O, et al. Characterization of the human gene (PTGS2) encoding prostaglandin-endoperoxide synthase 2. Eur J Biochem. 1994; 221:889–97. [DOI] [PubMed] [Google Scholar]
- 18.Tazawa R, Xu XM, Wu KK, Wang LH. Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun. 1994;203:190–9. [DOI] [PubMed] [Google Scholar]
- 19.Cipollone F Cox-2 polymorphisms and cardiovascular disease: elucidating the hidden side of the disease. Atherosclerosis 2009;207:348–9. [DOI] [PubMed] [Google Scholar]
- 20.Hakansson A, Bergman O, Chrapkowska C, Westberg L, Belin AC, Sydow O, et al. Cyclooxygenase-2 polymorphisms in Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:367–9. [DOI] [PubMed] [Google Scholar]
- 21.Sakaki M, Makino R, Hiroishi K, Ueda K, Eguchi J, Hiraide A, et al. Cyclooxygenase-2 gene promoter polymorphisms affect susceptibility to hepatitis C virus infection and disease progression. Hepatol Res. 2010;40:1219–26. [DOI] [PubMed] [Google Scholar]
- 22.Wang W, Fan X, Zhang Y, Yang Y, Yang S, Li G. Association between COX-2 polymorphisms and lung cancer risk. Med Sci Monit. 2015;21:3740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Miao X, Tan W, Ning B, Liu Z, Hong Y, et al. Identification of functional genetic variants in cyclooxygenase-2 and their association with risk of esophageal cancer. Gastroenterology. 2005;129:565–76. [DOI] [PubMed] [Google Scholar]
- 24.Prakash G, Umar M, Ajay S, Bali D, Upadhyay R, Gupta KK, et al. COX-2 gene polymorphisms and risk of chronic period-ontitis: a case-control study and meta-analysis. Oral Dis. 2015; 21:38–45. [DOI] [PubMed] [Google Scholar]
- 25.Zhang L, Zhang Y, Zhang X, Hong B. Prostaglandinendoperoxide synthase 2 (PTGS2) rs20417 polymorphism and prostate cancer risk: a meta analysis. Int J Clin Exp Med. 2015; 8:20454–62. [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang MM, Xie X, Ma YT, Zheng YY, Yang YN, Li XM, et al. Association of COX-2 −765G>C genetic polymorphism with coronary artery disease: a meta-analysis. Int J Clin Exp Med. 2015;8:7412–8. [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Jiang H, Liu T, Tang W, Ma Z. Cyclooxygenase-2–1195G>A (rs689466) polymorphism and cancer susceptibility: an updated meta-analysis involving 50,672 subjects. Int J Clin Exp Med. 2015;8:12448–62. [PMC free article] [PubMed] [Google Scholar]
- 28.Xu W, Huang Y, Zhang T, Zhao L, Fan J, Li L. Cyclooxygenase-2 gene polymorphisms and susceptibility to hepatocellular carcinoma: A meta-analysis based on 10 case-control studies. J Cancer Res Ther. 2018;14(Supplement):S105–S13. [DOI] [PubMed] [Google Scholar]
- 29.Dai ZJ, Shao YP, Ma XB, Xu D, Tang W, Kang HF, et al. Association of the three common SNPs of cyclooxygenase-2 gene (rs20417, rs689466, and rs5275) with the susceptibility of breast cancer: an updated meta-analysis involving 34,590 subjects. Dis Markers 2014;2014:484729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang Y, Liu JL, Wu Y, Zhang ZY, Wu R. Cyclooxygenase-2 polymorphisms and susceptibility to esophageal cancer: a meta-analysis. Tohoku J Exp Med. 2011;223:137–44. [DOI] [PubMed] [Google Scholar]
- 31.Luo D, Long Y, Chen GJ. Cyclooxygenase-2 gene polymorphisms and risk of Alzheimer’s disease: a meta-analysis. J Neurol Sci. 2015;359:100–5. [DOI] [PubMed] [Google Scholar]
- 32.Luo MX, Long BB, Li F, Zhang C, Pan MT, Huang YQ, et al. Roles of Cyclooxygenase-2 gene −765G>C (rs20417) and −1195G>A (rs689466) polymorphisms in gastric cancer: a systematic review and meta-analysis. Gene 2019;685:125–35. [DOI] [PubMed] [Google Scholar]
- 33.Agundez JA, Blanca M, Cornejo-Garcia JA, Garcia-Martin E. Pharmacogenomics of cyclooxygenases. Pharmacogenomics 2015;16:501–22. [DOI] [PubMed] [Google Scholar]
- 34.Papafili A, Hill MR, Brull DJ, McAnulty RJ, Marshall RP, Humphries SE, et al. Common promoter variant in cyclooxygenase-2 represses gene expression: evidence of role in acute-phase inflammatory response. Arterioscler Thromb Vasc Biol. 2002;22:1631–6. [DOI] [PubMed] [Google Scholar]
- 35.Ong’echa JM, Keller CC, Were T, Ouma C, Otieno RO, Landis-Lewis Z, et al. Parasitemia, anemia, and malarial anemia in infants and young children in a rural holoendemic Plasmodium falciparum transmission area. Am J Trop Med Hyg. 2006;74:376–85. [PubMed] [Google Scholar]
- 36.Perkins DJ, Were T, Anyona S, Hittner JB, Kempaiah P, Davenport GC, et al. The Global Burden of Severe Falciparum Malaria: An Immunological and Genetic Perspective on Pathogenesis. Dynamic Models of Infectious Diseases: Springer; 2013. p. 231–83. [Google Scholar]
- 37.Bloland PB, Boriga DA, Ruebush TK, McCormick JB, Roberts JM, Oloo AJ, et al. Longitudinal cohort study of the epidemiology of malaria infections in an area of intense malaria transmission II. Descriptive epidemiology of malaria infection and disease among children. Am J Trop Med Hyg. 1999;60:641–8. [DOI] [PubMed] [Google Scholar]
- 38.WHO. Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Trans R Soc Trop Med Hyg. 2000;94(Suppl 1):S1–90. [PubMed] [Google Scholar]
- 39.Liu YT, Old JM, Miles K, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol. 2000;108:295–9. [DOI] [PubMed] [Google Scholar]
- 40.Hsu J, Fink D, Langer E, Carter ML, Bengo D, Ndidde S, et al. PCR-based allelic discrimination for glucose-6-phosphate dehydrogenase (G6PD) deficiency in Ugandan umbilical cord blood. Pediatr Hematol Oncol. 2014;31:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaunt TR, Rodriguez S, Zapata C, Day IN. MIDAS: software for analysis and visualisation of interallelic disequilibrium between multiallelic markers. BMC Bioinf. 2006;7:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing V, Austria. 2016 URL http://www.R-project.org 2018. [Google Scholar]
- 43.Hosking L, Lumsden S, Lewis K, Yeo A, McCarthy L, Bansal A, et al. Detection of genotyping errors by Hardy-Weinberg equilibrium testing. Eur J Hum Genet. 2004;12:395–9. [DOI] [PubMed] [Google Scholar]
- 44.Kwiatkowski DP, Luoni G. Host genetic factors in resistance and susceptibility to malaria. Parassitologia 2006;48:450–67. [PubMed] [Google Scholar]
- 45.Ryg-Cornejo V, Ly A, Hansen DS. Immunological processes underlying the slow acquisition of humoral immunity to malaria. Parasitology 2016;143:199–207. [DOI] [PubMed] [Google Scholar]
- 46.Eybpoosh S Hardy Weinberg equilibrium testing and interpretation: focus on infection. J Med Microbiol Infect Dis. 2018;6:35–6. [Google Scholar]
- 47.Griffin JT, Hollingsworth TD, Reyburn H, Drakeley CJ, Riley EM, Ghani AC. Gradual acquisition of immunity to severe malaria with increasing exposure. Proc Biol Sci. 2015;282:20142657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McElroy PD, ter Kuile FO, Hightower AW, Hawley WA, Phillips-Howard PA, Oloo AJ, et al. All-cause mortality among young children in western Kenya. VI: the Asembo Bay Cohort Project. Am J Trop Med Hyg. 2001;64(1–2 Suppl):18–27. [DOI] [PubMed] [Google Scholar]
- 49.Snow RW, Omumbo JA, Lowe B, Molyneux CS, Obiero JO, Palmer A, et al. Relation between severe malaria morbidity in children and level of Plasmodium falciparum transmission in Africa. Lancet 1997;349:1650–4. [DOI] [PubMed] [Google Scholar]
- 50.Otieno F, Omolo C Infant and Child Mortality Kenya Demographic and Health Survey 2003. Calverton, Maryland: 2004. [Google Scholar]