

Abstract
α-Tocopherol (α-T) is the major form of vitamin E (VE) in animals and has the highest activity in carrying out the essential antioxidant functions of VE. Because of the involvement of oxidative stress in carcinogenesis, the cancer prevention activity of α-T has been studied extensively. Lower VE intake or nutritional status has been shown to be associated with increased cancer risk, and supplementation of α-T to populations with VE insufficiency has shown beneficial effects in lowering the cancer risk in some intervention studies. However, several large intervention studies with α-T conducted in North America have not demonstrated a cancer prevention effect. More recent studies have centered on the γ- and δ-forms of tocopherols and tocotrienols (T3). In comparison to α-T, these forms have much lower systemic bioavailability, but have shown stronger cancer preventive activities in many studies in animal models and cell lines. γ-T3 and δ-T3 generally have even higher activities than γ-T and δ-T. In this article, we review recent results from human and laboratory studies on the cancer preventive activities of different forms of tocopherols and tocotrienols, at nutritional and pharmacological levels. We aim to elucidate the possible mechanisms of the preventive actions and discuss the possible application of the available information for human cancer prevention by different VE forms.
Keywords: Vitamin E, tocopherols, tocotrienols, cancer prevention, humans
1. INTRODUCTION
Vitamin E (VE) activity is found in a group of eight fat-soluble compounds consisting of α, β, γ and δ-forms of tocopherols (T) and tocotrienols (T3). Among the VE forms, α-tocopherol (α-T) is most abundant in animal tissues, has the highest activity in the classical fertility restoration assay and is generally considered “the” VE in nutrition1. Because reactive oxygen species (ROS) are involved in many chronic diseases, such as cancer, cardiovascular diseases and neurodegenerative diseases, the possible preventive effects of the antioxidant nutrient VE against these diseases have been studied extensively. For example, lower VE intake or nutritional status was found to be associated with higher risk of different types of cancers in many studies2. However, several large-scale randomized trials with α-T carried out in North America during the past 20 years3–6, such as the Selenium and Vitamin E Cancer Prevention Trial (SELECT)5, failed to demonstrate a preventive effect against cancer. Because of these negative results, α-T is considered to have no cancer preventive activity in populations that are sufficient in VE nutrition. However, the non-α-T VE forms – the γ- and δ-forms of tocopherols and tocotrienols –have subsequently been shown to have cancer preventive activities in many laboratory studies. In 2010, we reviewed the cancer-preventive activities of tocopherols and tocotrienols2. Since then, this topic has been studied by a large number of investigators and reviewed by different authors7–12.
A literature search on PubMed in June 2019 yielded 1804 articles on “vitamin E and cancer”, 539 articles on “tocopherols and cancer”, and 156 articles on “tocotrienols and cancer” for the past 10 years. The present article is an updated review on the cancer preventive activities of different forms of VE in human and laboratory studies. In some instances, results from our own studies are discussed in more detail to illustrate the nature of the studies and challenges involved. In order to focus on prevention, studies on the possible therapeutic effects of VE and related synthetic derivatives are not covered in this article. We intend to elucidate the mechanisms of actions of different VE forms and to integrate laboratory and human studies in order to assess the possible use of γ- and δ-forms of tocopherols and tocotrienols for cancer prevention.
2. CHEMISTRY, ABSORPTION AND BIOTRANSFORMATION OF TOCOPHEROLS AND TOCOTRIENOLS
Tocopherols are widely occurring in dietary oils, such as corn, soybean, sesame and canola oils, as well as nuts. In these oils, γ-T is usually 3–5 times more abundant than α-T. δ-T is also abundant in some oils, while the contents of β-T in the dietary sources are very low. Tocotrienols are present in trace amounts in oils derived from rice bran, barley, wheat germ and rye and are not generally consumed in large quantities worldwide. Tocotrienols, however, are plentiful in palm oil (up to 800 mg/kg), mainly consisting of γ-T3 and α-T3, and are consumed mostly in Southeast Asia16.
All four tocopherols (α, β, γ and δ-T) consist of a chromanol ring and a 16-carbon phytyl side chain, but they differ in the number and position of the methyl group on the ring1 (Figure 1). α-T is tri-methylated at the 5-, 7- and 8-positions of the chromanol ring, whereas γ-T is dimethylated at the 7- and 8-positions, and δ-T is methylated at the 8-position. Tocotrienols (α, β, γ and δ-T3) have the same substitution pattern of methyl groups on the chromanol ring as tocopherols, but they have an unsaturated 16-carbon side chain with double bonds at the 3`-, 7`- and 11`- positions (Figure 1). The hydrocarbon tail and ring structure provide the lipophilicity for VE molecules to be incorporated into the lipid bilayers of biological membranes or in lipoproteins10. The phenolic group in the chromanol moiety effectively quenches reactive free radicals by one electron reduction and prevents the propagation of free radical reactions in lipid peroxidation. The resulting tocopherol phenoxy radical can be reduced by ascorbic acid or glutathione to regenerate the tocopherol molecule. This is a well-established physiological antioxidant mechanism for VE to protect the integrity of biological membranes. Since the antioxidant activities of different VE forms studied in vitro are dependent on the assay systems used, it is important to determine their antioxidant activities in vivo. F2-isoprostanes, isomers of prostaglandin F2, have been suggested as a reliable marker of in vivo free radical generation and oxidative lipid damage13.
Figure 1.
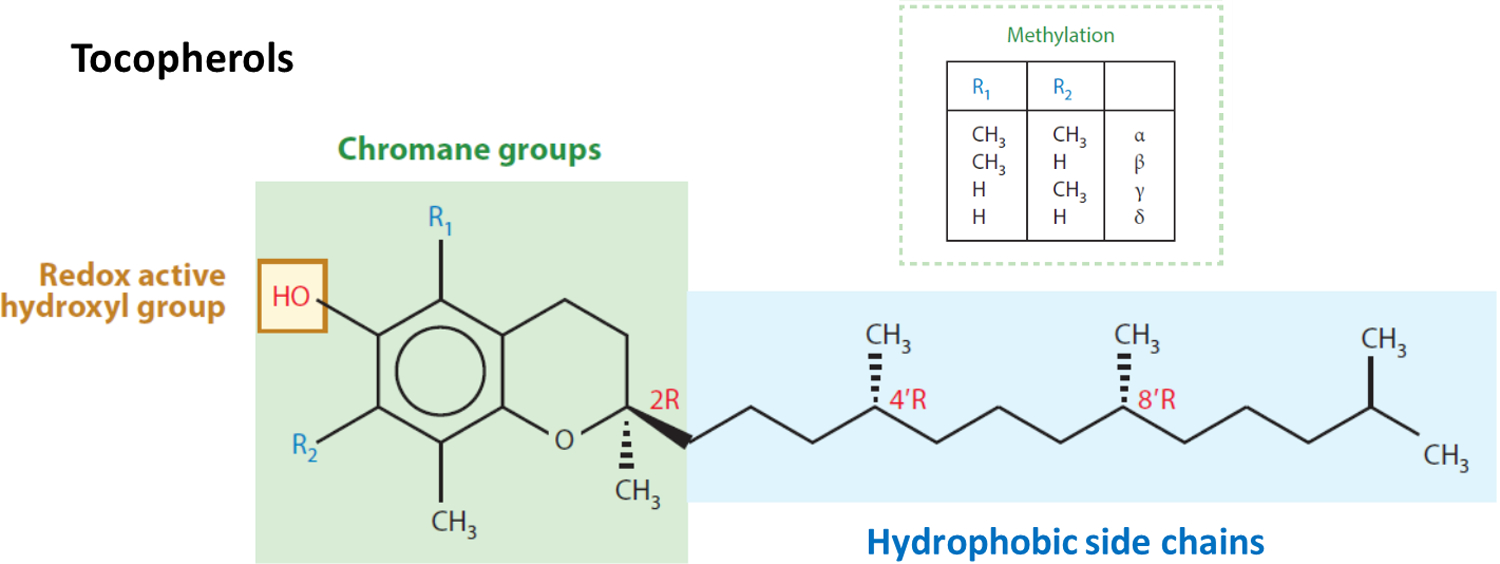
Structures of vitamin E analogues. The structures shown are the naturally occurring α, β, γ and δ-tocopherols (RRR) (modified from reference11). For the corresponding tocotrienols, each contains 3 double bonds on the side chain at 3`, 7` and 11` positions.
The unmethylated carbons at 5- and 7-positions are electrophilic centers that can effectively trap ROS and reactive nitrogen species (RNS). The formation of 5-nitro-γ-T, 5-nitro-δ-T, 7-nitro-δ-T, and 5,7-dinitro-δ-T has been reported1. 5-Nitro-γ-T formation was increased in the blood immediately after induction of acute inflammation in rats14. γ-T, but not α-T, reduces nitrogen dioxide to nitric oxide or traps it to form 5-nitro-γ-T. Nitrogen dioxide is a reactive free radical; if not reduced, it reacts with unsaturated fatty acid moieties to yield nitrite esters capable of nitrosating amines to form nitrosamines. Nitrogen dioxide can also induce single-strand DNA breaks in V79 cells, and the reaction is optimally inhibited by γ-T in comparison with other lipid soluble antioxidants15.
Dietary VE forms are absorbed from the intestinal mucosa in the free phenolic form, since esters are hydrolyzed by pancreatic esterases prior to absorption. VE forms are incorporated into the chylomicrons and transported to the liver via the lymphatic system. Dietary fat promotes VE absorption and their transfer into the lymphatic system. The uptake of different forms of VE into the liver is probably non-specific, but the transfer of these forms from the liver to very low-density lipoproteins is mediated by a specific α-T transfer protein (α-TTP)17,18. α-TTP in the liver preferentially transfers α-T to very low-density lipoproteins and therefore α-T is preferentially secreted into the circulation and transferred to non-hepatic tissues. Due to their low affinities for α-TTP, hepatic γ-T, δ-T and tocotrienols are less efficiently transferred to very low-density lipoproteins. Therefore, smaller portions of these VE forms are found in the blood and tissues, and most of these VE forms are degraded in the liver and other organs. The human serum plasma levels of α-T are generally 20–25 μM (8.6 – 10.8 mg/L), while those of γ-T are much lower at 1–3 μM8. However, upon supplementation with γ-T and δ-T3, their plasma concentration can be increased to higher than 20 μM and 5–10 μM, respectively, in healthy humans8.
The major route of VE metabolism is through side-chain degradation, initiated with hydroxylation of the ω-methyl group by cytochromes P450 (CYP) 4F or 3A and followed by five cycles of β-oxidation in the mitochondria, mainly in the liver, to cut off two-carbon units from the side-chain in each cycle19. Larger percentages of γ-T, δ-T and tocotrienols than α-T are degraded through this pathway20. This degradation pathway is illustrated in Figure 2 using γ-T as an example. The formation of long-chain metabolites, such as 13`-carboxychomanols (13`-COOHs) and 11`-COOHs, has been observed in different experimental systems8. Upon supplementation of γ-T (1 mg/kg diet), rather high levels of γ-T 13`-COOH, (~1 μmol/g of fecal samples) were observed8. Short side-chain metabolites, γ- and δ-carboxyethyl hydroxychroman (CEHC), as well as γ- and δ-carboxymethylbutyl hydroxychroman (CMBHC)21–23, are found at rather high concentrations in blood and tissues, and are excreted in the urine in conjugated forms as glucuronides and sulfates. Data from animal studies will be discussed in a later section. In our unpublished Phase 0 study, a commercial γ-T rich mixture (containing 248 mg α-T, 400 mg γ-T and 142 mg δ-T) was given to prostate cancer patients daily for 14 days before prostatectomy (CS Yang et al. unpublished results). Prostate tissue levels (μmol/kg) of α-T, γ-T and δ-T were 32.66, 4.77 and 0.60; the corresponding metabolites CEHCs were 0.22, 1.03 and 0.51; and the corresponding CMBHCs were 0.14, 0.22 and 0.11, respectively. These values were significantly higher than those in subjects taking placebo, except for the α-form of tocopherol and metabolites. The concentrations of urinary metabolites of the α-, γ- and δ-forms of CEHCs (μM) were much higher at 8.78, 128.82 and 94.38; and for the corresponding CMBHCs were 4.57, 52.23 and 43.69, respectively. These values were much higher than the pre-treatment values of the same subjects or the subjects taking placebo.
Figure 2.
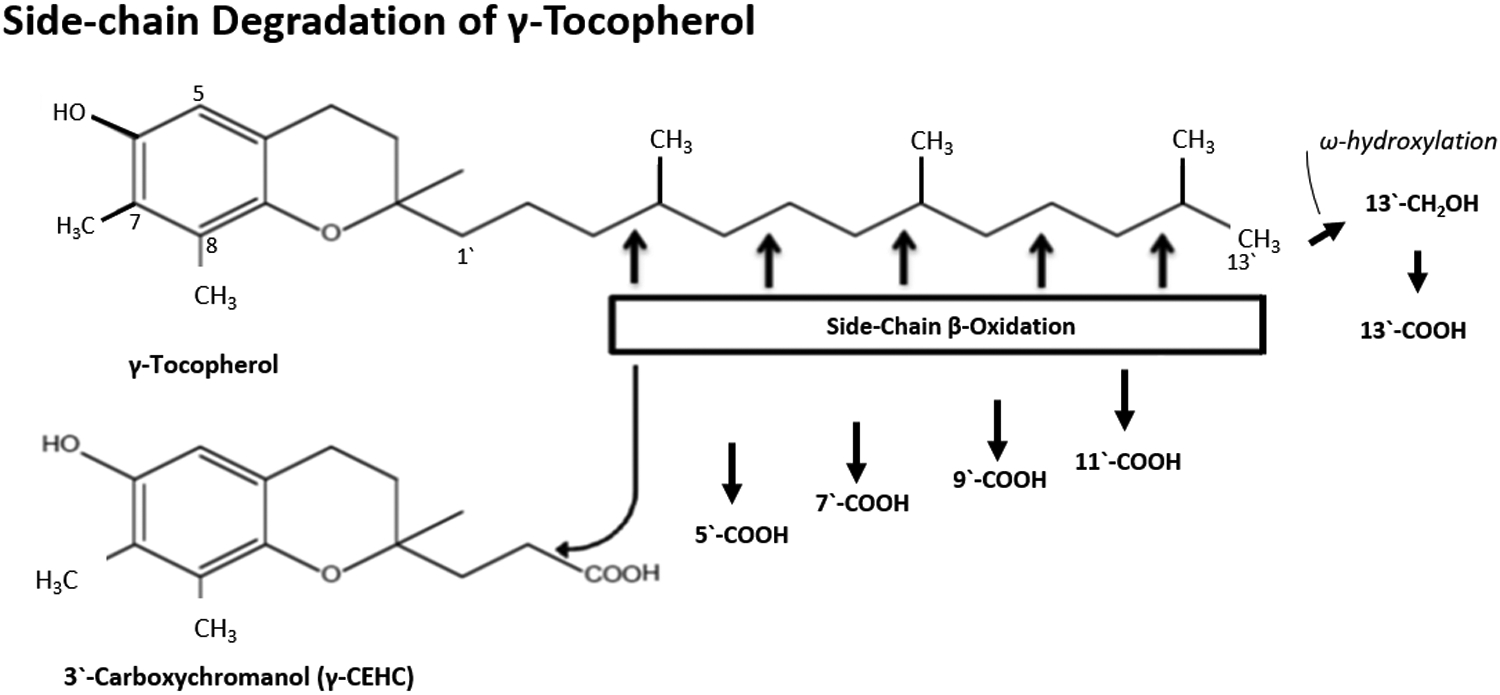
Side-chain degradation pathway of vitamin E using γ-tocopherol (γ-T) as an example for illustration (modified from reference24). The side-chain degradation is initiated by ω-oxidation and followed by five cycles of β-oxidation, each reducing the chain length by two carbons. The metabolites are named following reference8; for example, 13`-COOH is the metabolite of VE with carboxylic group at the 13’-position. The isomerization of double bond of tocotrienol side-chain; for example, in the conversion of carboxymethylhexenyl hydroxychroman to carboxymethylbutyl hydroxychroman (CMBHC or 5`-COOH), is catalyzed by the auxiliary enzymes 2,4-dienoyl-CoA reductase and 3,2-enoyl-CoA isomerase. The degradation of other forms of tocopherols and tocotrienols follows the same pathway.
In addition to degradation by mammalian enzymes, side-chain degradation of different forms of tocopherols and tocotrienols by intestinal microbes has been suggested by the following experiment24. In mice fed an AIN93 diet, the normal serum levels of α-T are usually 20–30 μM; those of other VE forms are very low and can be raised by dietary supplementation. When α-T, γ-T, δ-T, γ-T3 and δ-T3, each at a dose of 75 mg/kg body weight, were administered as a mixture i.g. to mice three hours before sacrifice, the serum level of α-T were not significantly changed, while those of γ-T, δ-T, γ-T3, and δ-T3 rose to approximately 2, 3, 6 and 5 μM, respectively24. The levels induced by the newly administered VE were increased more than 2-fold in animals that received antibiotics treatment, while that of α-T was increased to a much less extent24. The antibiotic treatment significantly decreased the urinary and fecal excretion of CEHCs and CMBHCs. The formation of side-chain degradation metabolites with different chain lengths was also decreased by antibiotics24. This result strongly suggests that intestinal microbes can degrade the VE side-chain; however, whether they can carry out the initial ω-hydroxylation reaction was not determined in this study. Considering the diverse activity of microbes in the oxygenation of different compounds, microbial ω-oxygenation (seen as ω-hydroxylation) of the VE side-chain is quite likely.
Some long-chain metabolites of VE, for example the 13`-COOH metabolites, have been shown to have interesting activities in inhibiting cyclooxygenase-2 (COX2) activity, inflammation and cell proliferation, as well as in inducing cell death8. Since larger proportions of γ- and δ-forms of VE are converted to these metabolites in comparison to α-T, these activities may contribute to the higher cancer preventive activities of γ- and δ-forms of VE forms. The short-chain VE metabolites, CEHCs and CMBHCs, existing at rather high concentrations in urine, may reflect dietary exposure of γ- and δ-forms of tocopherols and tocotrienols, as well as host and microbial degradation of these VE forms.
3. HUMAN STUDIES ON VE AND CANCER
The possible cancer prevention activities of VE have been studied extensively in humans. There are many studies demonstrating that lower VE intake or nutritional status is associated with increased risk of different types of cancer. However, there are also studies that did not show such an effect. In our previous review in 2010, the number of publications showing cancer risk reduction by VE in case-control, cohort and intervention studies was about the same as studies showing no association2. In this section, we will review some classical large-scale cancer prevention studies with VE to serve as a background. Then we will discuss new information gained from human studies conducted in the past decade.
3.1. Earlier large-scale intervention studies
3.1.1. The Linxian Nutrition Intervention Trial
In the 1970s, the association between diet, especially micronutrients, and human cancer attracted great research and public interest. Studies in animal models also showed that insufficiencies in certain vitamins or minerals could enhance carcinogenesis. The hypothesis that supplementation with micronutrients could reduce cancer risk in humans was tested in the 1980s in a large-scale intervention study on gastroesophageal cancer, the China-U.S. Cooperative Linxian Nutritional Intervention Trial (LNIT). The poor rural population in Linxian (now named Linzhou City), of Henan Province, China, sustained on a monotonous corn diet, had low intake of micronutrients; insufficiencies in some nutrients were indicated in blood nutrient analyses25. Because many micronutrients had been implicated in gastroesophageal cancer, nine nutrients were studied in LNIT. These nutrients were divided into 4 sets (Factors): (A) retinol, zinc; (B) riboflavin, niacin; (C) ascorbate, molybdenum; and (D) α-T, β-carotene, selenium (as selenium yeast), with each nutrient at 2–3 times the dose of the U.S. Recommended Daily Allowance. These sets of nutrients were combined in a factorial design in eight groups: placebo, AB, AC, AD, BC, BD, CD, and ABCD. The study involved 29,584 adults (aged 40 to 69) who were given daily supplementations as pills for 63 months (1985 to 1991). There were 2,127 deaths during the trial period; 32% were due to esophageal squamous cell carcinoma (ESCC) and gastric cancer (mainly gastric cardia cancer, which was commonly referred to as esophageal cancer in Linxian and accounted for 40% of the “esophageal cancer”). The result showed that daily supplementation with a combination of all-rac-α-tocopheryl acetate (50 mg), β-carotene (15 mg) and selenium (50 μg) (Factor D) to the general population (>40 years old) for 63 months decreased mortality due to gastric cancer (mainly gastric cardia cancer) by 20% and total cancer mortality by 13%26. Nested case-control studies showed that the baseline serum levels of α-T and selenium were each inversely associated with gastroesophageal cancer risk27,28. The mean baseline serum levels of α-T were approximately 8 mg/L, much lower than the median serum α-T levels (approximately 10 mg/L) in the Third National Health and Examination Survey in the United States. The result is consistent with the concept that the low antioxidant nutrition (VE and selenium) status makes the individuals susceptible to inflammation and carcinogenesis. Under these conditions, supplementation with VE and selenium attenuated carcinogenesis. Other nutrient combinations, however, did not show any significant effect on the endpoints measured26.
Furthermore, results from a 10-year follow-up showed that the preventive effect of the combination of α-T/β-carotene/selenium on gastric cardia cancer was sustained, and that this nutrient combination also protected against ESCC in younger subjects – when they were enrolled in the trial at ages 55 years old or younger (but not in those older than 55 years)29. It is possible that the intervention was ineffective in older subjects because they already had higher-grade precancerous lesions; however, it was effective in younger subjects because they had lower-grade or no precancerous lesions. This is consistent with the result of a parallel trial in Linxian on subjects with esophageal dysplasia, showing a lack of beneficial effect by supplementation with a combination of multiple micronutrients30. In the 25-year post-trial follow-up of the LNIT, the previously observed protective effects of α-T/selenium/β-carotene against total mortality was found lost 10 years post-intervention and the protective effects against gastric cancer was attenuated31.
The above cancer preventive effect of VE/selenium are supported by studies in a rat model demonstrating that a diet with insufficiencies in VE and selenium enhanced N-methylbenzylnitrosamine (MBNA)-induced esophageal carcinogenesis, and that supplementation of VE/selenium at the early stage of the experiment was more effective in preventing cancer formation than supplementation at a later stage32. These set of experiments demonstrate the cancer preventive activity of antioxidant nutrients (α-T and selenium) when supplemented to a human population or rats with low antioxidant nutritional status. Nevertheless, these two nutrients were not studied individually. The possible contribution of β-carotene (part of Factor D) in the preventive effect was not analyzed in the LNIT.
3.1.2. Large-scale intervention trials in North America
The results from several large-scale intervention studies with α-T conducted in North America, however, have been disappointing3–6. For example, in the Women’s Health Study with 39,876 healthy US women aged 45 years or older, the administration of 600 mg of α-T on alternate days did not significantly affect the incidence of colon, lung or total cancers3. In the Physicians’ Health Study II Randomized Control Trial, supplementation with VE (400 mg of α-T every other day) or vitamin C (500 mg synthetic ascorbic acid) to physicians for eight years did not reduce the risk of prostate cancer or all other cancers4.
The SELECT was launched based on the secondary endpoint analysis in the Alpha-Tocopherol Beta-Carotene (ATBC) Cancer Prevention Study on lung cancer in male smokers in Finland, showing that daily supplementation of 50 mg of α-T was significantly associated with lower incidence of prostate cancer and higher serum α-T was associated with a reduced risk of prostate cancer33,34. In the SELECT, 35,533 men (blacks > 50 years old; others > 55 years old) were randomized into four groups in a two-by-two design with 400 mg of all-rac-α-tocopheryl acetate and 200 μg selenium (from L-selenomethionine) daily, for an average of 5.5 years. Disappointingly, the results showed that the supplementations did not prevent prostate or other cancers5. It was noted that the α-T supplementation caused a 50% decrease in the median plasma γ-T levels5. A subsequent smaller randomized control trial (RCT) involving VE supplementation also did not find a significant protective effect35 (Table 1). No significant association between α-T supplementation and prostate cancer was found in the 18-year postintervention follow up of the ATBC36. The 7–12 year follow-up of the SELECT showed that subjects receiving the α-T supplementation had a hazard ratio of 1.17 for developing prostate cancer6. The conclusion “Dietary supplementation with vitamin E significantly increased the risk of prostate cancer among healthy men” could be misinterpreted by the public as “nutritional levels of VE increase prostate cancer risk”.
Table 1.
Human Studies on Vitamin E and Cancer
| Type of Study | Number of Subjects | Conclusion | Reference |
|---|---|---|---|
| RCT in Canada Prostate cancer |
156 participants receiving 400 IU VE supplement twice a day, 147 receiving a placebo | No significant effect association between VE supplementation and prostate cancer risk (HR: 1.03 CI: 0.67–1.60) | 35 (Fleshner et al., 2011) |
| Cohort, follow-up of ATBC Prostate cancer |
25,563 participants from original ATBC trial (18 years) | After 18 years of post-intervention follow-up, prostate cancer risk was not significantly affected by α-T supplementation (RR: 0.97 CI: 0.89–1.05) | 36 (Virtamo et al., 2014) |
| Case-Cohort, SELECT Prostate cancer |
1,746 prostate cancer cases, a subcohort of 3,211 controls (follow-up average of 5.5 years post-trial) | No significant association between serum levels of α-T (HR: 1.21 CI: 0.88–1.66) or γ-T (HR: 0.93 CI: 0.69–1.24) and the risk of prostate cancer | 40 (Albanes et al., 2014) |
| Observational study, Third NHANES Prostate cancer |
1,457 male participants | After variable adjustment, inverse association between serum α-T (highest vs. lowest quintile) and prostate cancer risk factors: testosterone, total estradiol and sex hormone binding globulin | 41 (Mondul et al., 2011) |
| Nested Case-control, CARET Prostate cancer |
684 cases of aggressive prostate cancer, 1441 controls | Among current smokers, inverse association between serum α-T (highest vs. lowest quartiles) with total prostate cancer risk (OR: 0.62 CI: 0.40–0.96); both α-T and γ-T inversely associated with aggressive prostate cancer risk (OR: 0.50 and 0.64, respectively) | 42 (Cheng et al., 2011) |
| Case-control, PLCO Prostate cancer |
680 cases of prostate cancer, 824 controls | Inverse relationship between serum α-T (highest vs. lowest quartiles) and prostate cancer (OR: 0.63 CI: 0.44–0.92), only found in current and former smokers. | 43 (Weinstein et al., 2012) |
| Observational, in the US Prostate cancer |
573 prostate cancer cases | Inverse association between serum γ-T (top vs. bottom quartiles) and high-grade prostate cancer (OR: 1.87 CI: 0.97–3.58, p = 0.02) | 44 (Bauer et al., 2013) |
| Cohort, PCaP Prostate cancer |
2,102 men with prostate cancer (1,023 African American and 1,079 European American) | Dietary intake of α-T (OR: 0.34 CI: 0.17–0.69) and δ-T (OR: 0.45 CI: 0.21–0.95) inversely associated with prostate cancer aggressiveness in European Americans, but not in African Americans | 45 (Antwi et al., 2015) |
| Nested Case-Control in the US Prostate cancer |
1025 participants (483 cases of prostate cancer, 542 controls) | SNP variations [BUD13, ZNF259, APOA5] which result in higher circulating α-T, correlated to lower risk of prostate cancer | 46 (Major et al., 2014) |
| Case-cohort from SELECT Prostate cancer |
1,424 men | SNPs (SEC14L2, SOD1, and TTPA) impacted on the association between VE and high-grade prostate cancer | 47 (Chan et al., 2016) |
| Cohort, SWHS Lung cancer |
72,829 female nonsmokers (1997–2010) | Lower lung cancer risk in those with adequate VE intake (14 mg/d or higher) than those with sub-adequate VE intake (HR: 0.78 CI: 0.60–0.99). VE supplementation positively associated with increased lung cancer risk (HR: 1.33 CI: 1.01–1.73) | 48 (Wu et al., 2015) |
| Prospective Cohort, ATBC Lung cancer |
22,781 male smokers from the original ATBC study (28 years follow-up) | Lower lung cancer risk in those with higher baseline α-T levels (5th quintile vs. 1st quintile HR: 0.76 CI: 0.66–0.87) | 49 (Huang et al., 2019) |
| Prospective Cohort, JPHC Lung cancer |
38,207 men and 41,498 women (from 1995–2013) | No relevant link between VE intake and risk of lung cancer (HR: 0.87 CI: 0.53–1.30 p = 0.115) | 50 (Narita et al., 2018) |
| Cohort, EPIC-Norfolk study Pancreatic cancer |
23,658 participants from the original EPIC-Norfolk study, 10 years follow-up | Inverse association between the combination of VE, vitamin C and selenium intake (lowest quartile vs. the sum of quartiles 2–4) with pancreatic cancer (HR: 0.33 CI: 0.13–0.84) | 53 (Banim et al., 2013) |
| Case-control in the US Pancreatic cancer |
384 pancreatic cancer patients, 983 controls | Higher VE intake (highest quintile vs. lowest quintile) (OR: 0.52 CI: 0.34–0.79) inversely associated to risk of pancreatic cancer | 54 (Jansen et al., 2013) |
| Case-Control in the US Pancreatic cancer |
811 patients with PDAC, 818 healthy controls | Risk of PDAC correlated with higher PhIP intake. Inverse correlation between total VE (OR: 0.60 CI: 0.43–0.83) and supplemental VE (OR: 0.59 CI: 0.43–0.81) with risk of PDAC | 55 (Li et al., 2019) |
| Secondary endpoint analysis, SELECT Bladder cancer |
34,887 participants from original trial, 7years after the original trial | No significant difference between control and intervention groups for cumulative bladder cancer risk (VE only- HR: 1.05 (0.64–1.73). VE + Selenium- 1.05 (0.63–1.70)) | 58 (Lotan et al., 2012) |
| Prospective Cohort, MCS Bladder cancer |
185,885 older adults (12.5 years after initial trial) | Highest quartile (≥5.9 mg/1000 kcal) of α-T intake in female patients had significantly lower risk of invasive bladder cancer (HR: 0.43 CI: 0.26–0.70). No correlation found for men | 59 (Park et al., 2013) |
| Case-Control, FCCS Colorectal cancer |
816 CRC patients, 815 controls | No significant association between VE intake and CRC after adjusting for calcium and polyunsaturated fatty acid intake | 64 (Wang et al., 2012) |
| Cohort, WHI Colorectal cancer |
5,477 women, 12 years follow-up | No significant association between baseline serum α-T or γ-T with risk of CRC or colon cancer | 65 (Kabat et al., 2012) |
| Secondary endpoint analysis, SELECT Colorectal cancer |
6,546 participants from original trial | VE supplementation did not affect colorectal adenoma risk (RR: 0.96 CI: 0.90–1.02) | 66 (Lance et al., 2017) |
| Case-control in China Colorectal cancer |
535 cases, 552 controls | Higher intake of VE (top vs bottom quartiles) associated with lower CRC risk (OR: 0.57 CI: 0.37–0.88). No statistically significant association between serum α-T and CRC risk | 67 (Luo et al., 2019) |
| Prospective Cohort in France Digestive cancer |
38,812 middle-aged subjects (7-year follow-up, 2009–2016) | Negative association between dietary VE or total VE (highest vs. lowest quartile) and risk of digestive cancer | 68 (Egnell et al., 2017) |
| Nested Case-Control, CSPPT Gastrointestinal (GI) cancer, Total cancers |
229 new cases, 229 controls | Higher serum α-T associated with decreased risk of GI cancer (OR: 0.83 CI: 0.73–0.95) and total cancer (OR: 0.91 CI: 0.84–0.99) for those with high baseline selenium levels. Low baseline selenium levels had opposite effect in increasing risk for total cancer by α-T (OR: 1.13 CI: 1.00–1.26) and non-GI cancers (OR: 1.25 CI: 1.03–1.50) | 69 (Wang et al., 2019) |
| Case-Control in China Cervical cancer |
458 cases of invasive cervical cancer, 742 controls | Serum VE inversely associated with risk of cervical cancer (highest vs. lowest quintile- OR: 0.53 CI: 0.37–0.74) | 72 (Guo et al., 2015) |
| SWH and SMH cohort studies Liver cancer |
132,837 subjects followed for an average of 10.9 years (women) and 5.5 years (men) | Dietary VE intake (including supplements) inversely associated with liver cancer risk | 77 (Zhang et al., 2012) |
| Cohort, ATBC Liver cancer |
29,046 men for baseline serum level, 22,805 for 3-year follow α-T serum level as subcohort | No significant association between serum levels of α-T and liver cancer risk (HR: 1.06 CI: 0.64–1.74) | 78 (Lai et al., 2014) |
| Prospective Cohort, VITAL Study Urothelial cancer |
77,050 participants | No significant association between serum α-T levels and risk of urothelial cell carcinoma | 79 (Hotaling et al., 2011) |
| Prospective Cohort, BNDNS Total cancer |
1054 participants (all elderly- 65 years old or higher over 13–14 years) | Dietary VE was a significant predictor of cancer mortality in males (4 models given- HR: 0.13 (0.04–0.42), HR: 0.23 (0.06–0.84), HR: 0.16 (0.04–0.65), HR: 0.19 (0.04–0.81)) | 81 (Bates et al., 2011) |
| JCCSECR Total cancer |
22,795 men and 35,539 women (19–21 years follow-up) | Multivariate analysis: inverse correlation between VE intake (highest vs. lowest quintile) and all-cause mortality (HR: 0.85 CI: 0.78–0.93) only in women | 82 (Ma et al., 2018) |
| Post-trial follow-up, PHS II Total cancer |
14,641 male physicians in the US (7,315 received 400 IU/day of α-T, 7,326 men received placebo) | VE supplementation had no long-term effect on cancer incidence HR: 0.99 (CI: 0.89–1.10) | 83 (Wang et al., 2014) |
Abbreviations: APOA5, Apolipoprotein A5; ATBC, Alpha-Tocopherol, Beta-Carotene Cancer Prevention (Trial); BNDNS, British National Diet and Nutrition Survey; BUD13, Budding-site selection protein 13; CARET, Carotene and Retinol Efficiency Trial; CI, 95% Confidence Interval; CRC, Colorectal Cancer; CSPPT, China Stroke Primary Prevention Trial; EPIC, European Prospective Investigation of Cancer; FCCS, Fukuoka Colorectal Cancer Study; GI, gastrointestinal; HR, hazard ratio; JCCSECR, Japan Collaborative Cohort Study for Evaluation of Cancer Risk; JPHC, Japan Public Health Center (study); LNIT, Linxian Nutrition Intervention Trial; MCS, Multiethnic Cohort Study; MOCDTBS, Malaysian Oral Cancer Database and Tissue Bank System; NHANES, National Health and Nutrition Examination Survey; PCaP, The North Carolina-Louisiana Prostate Cancer Project; PDAC, Pancreatic ductal adenocarcinoma; PhIP, 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline; PLCO, The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; PHS, Physicians Health Study; RCT, randomized controlled trial; RR, relative risk; SELECT, Selenium and Vitamin E Cancer Prevention Trial; SMHS, Shanghai Men’s Health Study; SNP, single nucleotide polymorphism; SWHS, Shanghai Women’s Health Study; TTCS, Tehran Thyroid Cancer Study; VITAL, Vitamins and Lifestyle; WHI, Women’s Health Initiative; ZNF259, Zinc finger protein 259
A possible interpretation for the lack of cancer prevention effect of α-T in the above studies is that the supplementation of a nutrient to a population that is already sufficient in this nutrient may not produce any beneficial effect. The mean baseline median plasma level of α-T in subjects of the SELECT was at an adequate level of 12.5 mg/L5. Since γ-T has been suggested to have strong anti-inflammatory and cancer preventive activities2,37–39, the decrease in blood and tissue levels of γ-T, caused by high doses of α-T, might have contributed to the increased prostate cancer risk in the SELECT. However, subsequent subcohort analysis of the SELECT showed no significant association between serum levels of α-T or γ-T and the risk of prostate cancer40 (Table 1). Another possibility is that some of these subjects already had preneoplastic lesions when entering the trial, and the supplementation with high doses of α-T promoted prostate cancer development. The exact reasons for these negative results from the SELECT and other trials are still not known. Nevertheless, the disappointing outcome of these large-scale trials reflects our insufficient understanding of the biological activities of VE and points to the need for systematic studies of the disease preventive activities of the different forms of VE.
3.2. Recent studies on VE and cancer in humans
Since the publication of the disappointing results of α-T from SELECT and other large-scale intervention trials, few intervention studies with α-T have been conducted. However, many case-control, nested case-control and prospective studies have been published between 2011–2019 (Table 1). Recent meta-analyses of VE and cancer are summarized in Table 2. The results from studies on different cancers are different, and some of them are discussed below.
Table 2.
Meta-analyses of Human Studies on Vitamin E and Cancer
| Studies included | Indicator: Relative Risk (95% CI) | Conclusion | Reference |
|---|---|---|---|
| 10 prospective, 1 case-control Lung cancer |
RR: 0.86 (0.74–0.99) p = 0.041 | Inverse association between VE intake and lung cancer risk | 51 (G. Chen et al., 2015) |
| 9 cohort studies Lung cancer |
VE intake (highest vs. lowest tertiles) RR: 0.84 (0.76–0.93). Every 2 mg/d increase in dietary VE associated with risk reduction by 5%, RR: 0.95 (0.91–0.99) | Higher levels of dietary VE associated with a lower lung cancer risk | 52 (Zhu et al., 2017) |
| 6 case-control, 4 cohort Pancreatic cancer |
Highest vs. lowest category RR: 0.79 (0.73–0.89) | Inverse association between VE intake and pancreatic cancer risk | 56 (Peng et al., 2015) |
| 4 case control, 7 cohort Pancreatic cancer |
OR: 0.70 (0.62–0.81) for overall OR: 0.63 (0.53–0.75) for case-control, OR: 0.85 (0.68–1.06) for cohort |
VE intake inversely associated with pancreatic cancer risk in case-control studies; no significant association in cohort studies | 57 (Chen et al., 2016) |
| 11 case-control, 9 cohort Bladder cancer |
RE: 0.82 (0.72–0.90) | Inverse association between VE intake and bladder cancer risk | 60 (Wang et al., 2014) |
| 3 RCT, 8 cohort Bladder cancer |
RR: 0.89 (0.78–1.00) | VE consumption inversely associated with risk of bladder cancer | 61 (Lin et al., 2019) |
| 8 case-control, 9 cohort Bladder cancer |
RR (unit increment): VE from diet, 0.83 (0.72–0.95); serum α-T, 0.84 (0.76–0.94) | Inverse association between VE intake or α-T serum level and bladder cancer risk | 62 (F. Chen et al., 2015) |
| 9 nested case-control Bladder cancer |
Highest vs. lowest categories RR: 0.79 (0.68–0.91) for serum α-T | Inverse association between α-T blood level and bladder cancer risk. | 63 (Cui et al., 2014) |
| 10 case-control Head and neck cancer |
OR with p-trend < 0.001: oral/ pharyngeal cancer, 0.59 (0.49–0.71); laryngeal cancer, 0.67 (0.54–0.83) | Inverse association between VE intake and head and neck cancer risk | 70 (Edefonti et al., 2015) |
| 1 cohort, 11 case-control Esophageal cancer |
Comparing highest to lowest category, OR: 0.47 (0.36–0.60) for esophageal cancer (ESCC or EAC) | Higher dietary VE is associated with lower risk of esophageal cancer | 71 (Cui et al., 2018) |
| 15 case-control Cervical cancer |
OR: 0.58 (0.47–0.72) for cervical neoplasia risk | VE intake and serum VE levels are inversely correlated with cervical cancer risk | 73 (Hu et al., 2017) |
| 9 nested case-control, 7 case-control, 2 cohort Breast cancer |
For median lowest level subgroup, pooled OR: 0.42 (0.25–0.72) | Inverse association between α-T serum level and breast cancer risk among the subgroup of median lowest serum level | 74 (Hu et al., 2015) |
| 7 case-control Renal cancer |
Highest vs. lowest category OR: 0.75 (0.59–0.91) | Inverse association between VE intake and renal cancer risk | 75 (Shang et al., 2015) |
| 7 case-control, 6 cohort Renal cancer |
Pooled highest vs. lowest categories RR: 0.81 (0.69–0.94) | Inverse association between VE intake and renal cancer risk | 76 (Shen et al., 2015) |
| 12 studies, 8 relevant to VE Non-Hodgkin’s Lymphoma |
RR: 0.98 (0.88–1.10) for Non-Hodgkin’s Lymphoma | No association between VE intake and risk for Non-Hodgkin’s Lymphoma | 80 (Psaltopoulou et al., 2018) |
| 69 prospective studies Total cancer |
RR: 0.97 (0.93–1.02) per 5 μg/d | No relevant association between dietary VE intake and total cancer or mortality, but an increasement of 500 μg α-T/dL in blood linked to 9% and 6% reduction in risk for total cancer, and mortality | 88 (Aune et al., 2018) |
Abbreviations: CHD, Coronary Heart Disease; CI, confidence interval; CVD, Cardiovascular Disease; EAC, esophageal adenocarcinoma; ESCC, esophageal squamous cell carcinoma; HR, hazard ratio; OR, odds ratio; RCT, randomized control trials; RE, risk estimates; RR, relative risk
3.2.1. Prostate cancer
The potential link between VE and prostate cancer has received great attention (Table 1). The third National Health and Nutrition Examination Survey found that serum α-T level was inversely associated with prostate cancer risk factors: testosterone, total estradiol and sex hormone binding globulin41. An inverse correlation between serum levels of α-T and prostate cancer risk was found among smokers in studies by Cheng et al.42 and Weinstein et al.43. Serum levels of γ-T (highest vs. lowest quartiles) were also shown to be inversely associated with the risk of aggressive or high-grade prostate cancer in studies by Cheng et al.42 and Bauer et al.44. In the North Carolina-Louisiana Prostate Cancer Project (PCaP), a study involving 1,023 African American and 1,079 European American participants, however, dietary intake of both α-T and δ-T were inversely correlated with prostate cancer aggressiveness, but only in European Americans45.
The importance of nutritional levels of VE in influencing prostate cancer is also supported by a nested case-control study in the US, showing that three single nucleotide polymorphism (SNP) variations (involving BUD13, ZNF259 and APOA5), which resulted in higher circulating α-T levels, were correlated with a lowered risk of prostate cancer46. More recently, three additional SNP variations (involving SEC14L2, we SOD1 and TTPA) were found to be associated with lower risk of prostate cancer in subjects receiving VE in the SELECT47. The SNP variation for both SEC14L2 and SOD1 were associated with significantly lowered prostate cancer risk. The SNP of TTPA, which results in lower levels of α-TTP and presumably lower α-T, was associated with increased risk of prostate cancer in the absence of VE supplementation, and VE supplementation seemed to offset this increased risk.
3.2.2. Lung cancer
In the Shanghai Women’s Health Study (SWHS) with 72,829 female non-smokers and a follow-up period of 12 years, lung cancer risk was found to be lower in those who met dietary guidelines for Adequate Intake (AI) of tocopherols (14 mg/day or more), compared to those with lower intake than AI48 (Table 1). Surprisingly, VE supplement usage, mostly in the form of α-T, was associated with increased lung cancer risk48. In the ATBC study with 28 years of follow-up, higher baseline α-T levels were correlated with lowered risk of lung cancer49. However, no relevant link between VE and lung cancer was observed in the Japanese Public Health Center (JPHC) Cohort study, which involved 38,207 men and 41,498 women with 18 years of follow-up50. In a meta-analysis, Chen et al.51 found an inverse correlation between VE intake and lung cancer risk (Table 2). Zhu et al.52 also found a link between a higher VE intake (highest vs. lowest tertile, quartile, or quintile) and a lower risk of lung cancer, and an increase of dietary VE of 2 mg/day was associated with a 5% decreased lung cancer risk (Table 2).
3.2.3. Pancreatic cancer and bladder cancer
Earlier cohort-study53 and a case-control study54 showed a significant inverse association between pancreatic cancer and nutrient intake, including VE, vitamin C and selenium. In a recent case-control study of pancreatic ductal carcinoma (PDAC) with 811 cases and 818 controls, cancer risk was associated with the consumption of 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine (PhIP) (derived from well-done, grilled or barbecued meat), and the cancer risk was also associated with lower levels of VE (including supplement) intake55. Intake of vitamin C and other nutrients were also inversely associated with PDAC55. Meta-analyses by Peng et al.56 and Chen et al.57 (Table 2) also showed an inverse association between VE intake and pancreatic cancer risk; this association was mainly dependent on case-control studies, while the results from cohort studies were not consistent.
In a secondary endpoint analysis of the SELECT, there was no significant difference in incidence of bladder cancer between control and intervention groups58. However, in a multiethnic cohort study in Hawaii, which involved 185,885 older adults, with 12.5 years follow-up, women within the highest quartile of α-T intake were found to have significantly lowered risk of bladder cancer, but no correlation was found for men59. Meta-analyses by Wang et al.60 and Lin et al.61 both found an inverse association between VE intake and bladder cancer risk. In another two meta-analysis, Chen et al.62 and Cui et al.63 both noted an inverse association between α-T and bladder cancer risk.
3.2.4. Gastrointestinal cancers
A case-control study from the Fukuoka Colorectal Cancer Study (FCCS) found no significant association between VE intake and colorectal cancer (CRC)64. The conclusion of now association was also found in a cohort study from the Women’s Health Initiative (WHI)65. A follow-up involving 6,546 participants from the original SELECT similarly showed that VE supplementation had no effect on the risk of CRC66. However, a recent case-control study in China found that higher intake of VE (highest vs. lowest quartiles) was associated with lowered CRC risk, nevertheless there was no association between serum α-T and CRC risk67.
A prospective cohort study conducted in France involving 38,812 participants over 7 years found an inverse association between VE intake and digestive cancer risk68. The China Stroke Primary Prevention Trial (CSPPT) found that in subjects with high baseline selenium levels, those with high serum α-T levels had a decreased risk of gastrointestinal cancer and total cancer; but intriguingly for those with low baseline levels of selenium, α-T had the opposite effect on total or non-GI cancers69. Meta-analysis by Edefonti et al.70 found an inverse relationship between VE intake and head and neck cancer incidence rates. A meta-analysis recently completed by Cui et al.71 showed that higher levels of dietary VE was associated with lowered risk of esophageal cancer.
3.2.5. Other cancers
Higher VE intake was found to be associated with lower risk of invasive cervical cancer in a case-control study in China72. This finding was corroborated in a meta-analysis which involved 15 case-control studies and found an inverse relationship between VE intake and cervical cancer73. Also, in meta-analyses, inverse associations were observed between serum levels of α-T and breast cancer risk by Hu et al.74 as well as renal cancer by Shang et al.75 and Shen et al.76. In the combined Shanghai Women’s and Men’s Health Studies, dietary VE intake (including supplements) was inversely associated with liver cancer risk77. However, no link between serum α-T levels and risk of liver cancer was found in a follow-up of the ATBC trial78. The prospective Vitamins and Lifestyle (VITAL) study found serum α-T levels had no connection with urothelial cancer rates79. A recent meta-analysis also found no relationship between non-Hodgkin’s Lymphoma and VE intake80.
In the British National Diet and Nutrition Survey, which involved 1,054 elderly men followed for 13–14 years, it was found that dietary VE had a significant inverse association with total cancer mortality in four different models81. In the Japan Collaborative Cohort Study for Evaluation of Cancer Risk, involving over 58,000 men and women over a period of 19–21 years, it was also shown that higher dietary intake of VE was inversely associated with all-cause mortality82. On the other hand, in the Physicians Health Study II, VE supplement had no long-term effect on cancer incidence83.
3.2.6. Studies with tocotrienols
There is a lack of epidemiological studies with tocotrienols because of its low consumptions in most areas, except in Southeast Asia where palm oil is consumed in large quantities. However, a few human trials have been reported. In a Phase I window-of-opportunity trial of δ-T3 in 25 patients with pancreatic neoplasia, δ-T3 was given orally twice a day for 13 days before surgery, in a dose-escalation scheme with total daily doses from 200 mg to 3200 mg84. These doses were well-tolerated and pharmacokinetic and pharmacodynamic parameters were measured. δ-T3 at dose of 600 mg induced significant apoptosis in the neoplastic cells of patients. The percentage of apoptosis (cleaved caspase-3 positive cells) increased in patients given doses of 200–600 mg, but doses of 800–3200 mg were less effective. The biological response rate also had a pattern starting with 50% for the dose of 200 mg, 100% for 400 mg and 600 mg, and then decreased to 66% for 800 mg and 1600 mg and to 33% for 3200 mg, showing a bell-shaped dose response84.
The same group of investigators also studied the pharmacokinetics and safety of δ-T3 in 36 healthy subjects85. The subjects received orally 100–1600 mg of δ-T3 as a single dose or twice daily for 14 consecutive days. No drug related adverse events were observed. After a single dose of δ-T3, maximum blood concentrations appeared at 4.0–9.3 h, with maximum concentration of 0.80–3.7 mg/L, and δ-T3 was eliminated with a half-life of 1.7–5.9 h85.
In Malaysia, a tocotrienol-rich fraction of palm oil was used as an adjuvant therapy for tamoxifen in women with early breast cancer86. During the five years of study, however, the adjuvant tocotrienol therapy did not affect breast cancer survival. On the other hand, in a recent phase II clinical trial in Denmark on 23 advanced-stage ovarian cancer patients, 300 mg of tocotrienols (90% δ-T3) was used 3 times daily in combination with the standard therapy of bevacizumab (10 mg/kg i.v.)87. The median progression-free survival was prolonged to 6.9 months from the common 2–4 months (bevacizumab alone) and the median overall survival was increased to 10.9 months from 5–7 months due to tocotrienols. This study shows the great potential of using tocotrienols as adjuvant therapy.
3.3. Assessment of human studies and mechanistic considerations
As discussed above, most studies found an inverse association between VE nutritional status and risk for cancer. In a meta-analysis of 69 prospective studies by Aune et al.88, even though there was no relevant link between dietary VE intake and cancer incidence or total mortality, an increase of 5 μg/mL in serum α-T level was linked to a 9% or 6% decrease in total cancer rate or total mortality rate, respectively. Meta-analysis that focused on serum VE levels were more likely to find an inverse association62,63,73,74. Genetic SNP’s that resulted in an increase of serum α-T levels were shown to be associated with a decrease in cancer risk46,47. All these results support the concept that insufficiency in VE nutrition increases cancer risk.
The dietary VE intake in the above studies was mainly obtained from food frequency questionnaires. The food items rich in VE also contain vitamin C, selenium and other constituents that may contribute to a reduction in cancer risk. Thus, the effects of “dietary VE” would also include the effects of other constituents from the food, and commercial VE supplements would not be as effective. Besides, VE supplements generally contains only synthetic α-T, whereas VE from the diet also contains γ-T and δ-T, in several fold more abundance. As will be discussed in the next section, in animal models, γ-T and δ-T are effective in inhibiting carcinogenesis, while α-T is not.
In contrast to the observational epidemiological studies, many intervention studies with high doses of α-T have not yielded preventive effects against cancer. A possible explanation is that these populations were already sufficient in VE nutrition, and supplementation with α-T would not have any beneficial effects. In addition, the use of α-T at high doses may decrease the blood and tissue levels of γ-T which are known to have cancer preventive activities. It remains to be determined whether γ-T, δ-T or tocotrienols would do better in intervention studies.
4. LABORATORY STUDIES ON VE AND CANCER PREVENTION
4.1. Studies with Tocopherols
In our previous review2, we summarized 18 studies that used α-T as a cancer preventive agent in animal models. Although some studies had shown prostate and mammary cancer preventive activities, 9 out of 10 studies on colon cancer prevention yielded negative results. These and the negative results from large intervention trials with α-T discouraged further studies with α-T. On the other hand, γ-T, δ-T and a γ-T-rich mixture of tocopherols (γ-TmT, a mixture of tocopherols with 59.3% γ-T, 25.4% δ-T, 13.5% α-T and 1.6% β-T) have been studied in a large number of animal models. Extensive studies in different cancer cell lines have also been conducted to elucidate the molecular mechanisms by which tocopherols induce cell apoptosis and inhibit cancer cell growth and invasion. Some of these studies are summarized in Table 3 and discussed below.
Table 3.
Laboratory Studies on Tocopherols and Cancer
| Experimental Model | Treatment | Results | Reference |
|---|---|---|---|
| Prostate Cancer | |||
| Transgenic adenocarcinoma of the mouse prostate (TRAMP) | γ-TmT at 1g/kg diet | γ-TmT reduced incidence of high-grade PIN from 60% (in control) to 30% and increased mice without PIN lesions from 0 to 20%. | 89 (Barve et al., 2009) |
| Transgenic rat for adenocarcinoma of prostate (TRAP) | α-T at 50 mg/kg diet, or γ-T at 50, 100 or 200 mg/kg diet | γ-T dose-dependently suppressed prostate tumor progression from PIN to adenocarcinoma & activation of caspases 3 & 7 in ventral lobe, α-T was not effective. | 90 (Takahashi et al., 2009) |
| Ventral prostate of rats treated with MNU | γ-T at 20 mg/kg in diet | γ-T significantly attenuated adverse effects of MNU in ventral prostate; decreased incidence of epithelial dysplasia, cell proliferation index, GST-pi & COX2 expression. | 91 (Sanches et al., 2013) |
| Wistar-Unilever rats injected with MNU, & stimulated by testosterone | α-T at 2 or 4 g/kg diet | α-T was not effective for prostate cancer chemoprevention. |
92 (McCormick et al., 2010) |
| PhIP-induced prostate cancer in hCYP1A mice |
γ-TmT, α-T, γ-T or δ-T at 2g/kg diet | γ-TmT, γ-T & δ-T, significantly reduced the number & severity of mPIN lesions. γ-TmT decreased the highly elevated 8-oxo-dG & nitrotyrosine levels and the expression of COX2, p-AKT & Ki-67, as well as prevented the loss of PTEN & Nrf2 expression in mPIN lesions. | 93 (Chen et al., 2016) |
| Human prostate cancer cells: LNCaP, VCaP, CWR22Rv1 | α-T & δ-T at 3g/kg diet. Cells: 20, 50, 100 μM α-T, γ-T, δ-T or γ-TmT | δ-T had strongest inhibitory effect on growth of LNCaP cells & xenograft tumors, and in inducing apoptosis. δ-T inhibited AR activation, and DHT-induced increase in PSA. | 94 (Huang et al., 2014) |
| DU145, PC-3, LNCaP, CWR22Rv-1 human prostate cancer cell lines | 10, 20, 30, 40, 50, 60 μM α-T, γ-T or δ-T | δ-T was most effective in inducing cell cycle arrest & apoptosis and in inhibiting RTK-mediated ATK T308 phosphorylation by P13K. | 95 (Wang et al., 2016) |
| Prostate specific PTEN−/− mice | α-T, γ-T or δ-T at 2g/kg diet | δ-T reduced prostate adenocarcinoma multiplicity at week 40 by 53.3% or 42.7%, with treatment starting at weeks 6 or 12, respectively. | 96 (Wang et al., 2018) |
| Mammary Cancer | |||
| Female MMTV/ErbB2/neu transgenic mice & Female Sprague-Dawley rats with NMU injection | 0.3% of tocopherol (α-, δ-, or γ-) or 0.3% of a γ-TmT | δ- & γ-tocopherols inhibited hormone-dependent mammary tumorigenesis in NMU-treated female Sprague-Dawley rats. δ-T & γ-T significantly decreased tumor multiplicity. | 97 (Smolarek et al., 2012) |
| Female ACI rats implanted with estrogen (E2) pellets | 0.3 or 0.5% γ-TmT (58% γ-tocopherol) diet | γ-TmT reduced serum E2 levels, suppressed serum levels of PGE2 & 8-isoprostane, decreased expression of ERα mRNA, whereas ERβ & PPARγ mRNA levels were increased. | 98 (Smolarek et al., 2013) |
| Female ACI rats in August-Copenhagen Irish rat model with E2 implants | 0, 0.05, 0.1, 0.3 & 0.5% γ-TmT | 0.3 & 0.5% γ-TmT decreased tumor volume (52% & 42%, respectively) & multiplicity (in part through decreasing E2 availability). γ-TmT induced expression of CYP1A1, increased PPARγ, PTEN & CDKN1B. | 99 (Das Gupta et al., 2015) |
| Female ACI rats implanted with E2 pellets (9mg) | 0.3% γ-TmT | γ-TmT reduced liver weight, 8-oxo-dG & 8-isoprostane levels, but had no effect on mammary hyperplasia. γ-TmT stimulated Nrf2-dependent antioxidant response in mammary glands. | 100 (Das Gupta et al., 2015) |
| Estrogen-stimulated MCF-7 cells in vitro and in vivo (Female nu/nu mice) | 0.2% α-, γ-, δ-tocopherol, or γ-TmT for 5 weeks |
γ-TmT reduced tumor volumes and weights. γ- and δ-T inhibited expression on cyclin D1 and c-Myc in vitro, attenuated estrogen-induced increase in 8-oxo-dG & nitrotyrosine levels. | 101 (Bak et al., 2017) |
| Female ACI rat model with E2 implants | 0.2% α-T, γ-T, δ-T, or γ-TmT for 30 weeks | δ-T, γ-T, & γ-TmT reduced mammary tumor volume by 51%, 60%, & 59%, respectively. γ-TmT supplementation increased the α, δ, & γ-CEHC concentrations by 3-, 61-, & 36-fold, respectively. | 102 (Das Gupta et al., 2017) |
| MCF-7 cell line | Estrogen or α-T, γ-T & δ-T | Treatment of estrogen-stimulated MCF-7 tumorspheres with α-T, γ-T or δ-T decreased the size of large tumorspheres (by 23–25%) mainly by affecting the stem cell-like population and expression of stem cell markers returned to near control levels. γ-T decreased the ALDH activity by 28.5%. | 103 (Bak et al., 2018) |
| Colon Cancer | |||
| Male CF-1 mice treated with AOM/DSS | Exp 1: γ-TmT at 3 g/kg diet Exp 2: γ-TmT at 1.7 or 0.3 g/kg diet |
γ-TmT significantly decreased the number of colon tumors and levels of PGE2, LTB4 & nitrotyrosine, while enhanced apoptosis in the colon; and decreased levels of PGE2, LTB4 & 8-isoprostane in plasma. | 104 (Ju et al., 2009) |
| Male Balb/c mice treated with AOM/DSS | γ-T or γ-TmT at 1 g/kg diet | γ-T decreased tumor multiplicity by 60% only with one-cycle DSS (at 1.5%), but not with three DSS cycles (1.5–2.5%). γ-T, but not γ-TmT, attenuated DSS-induced colon inflammation. | 105 (Jiang et al., 2013) |
| Male Wistar rats treated with DMH | VE: 75, 225 or 1500 IU | Supplementation with a low dose of α-T (225 IU, 3 times the RDI) had beneficial effects in reducing colon ACF formation and COX2 expression, while a high dose of (1500 IU) did not | 106 (Cohen et al., 2014) |
| Male F344 rats treated with AOM | δ-T, γ-T, α-T, or γ-TmT at 2 g/kg diet | δ-T, γ-T & γ-TmT lowered total numbers of ACF per rat, but α-T did not. δ-T showed strongest inhibitory effect on ACF (by 62.3%). δ-T, γ-T & γ-TmT significantly decreased levels of cyclin D1, 4-HNE & nitrotyrosine, preserved expression of PPARγ in the colon, and decreased serum levels of PGE2 & 8-isoprostane. | 107 (Guan et al., 2012) |
| hCYP1A mice treated with PhIP/DSS | δ-T, γ-T, or α-T at 2 g/kg diet | δ-T & γ-T significantly reduced colon tumor formation & suppressed 8-oxo-dG, nitrotyrosine, NF-κB, p65 & p-STAT3 in tumors & adjacent tissues. | 108 (Chen et al., 2017) |
| Human colon cancer cells HCT116 & HT29; INT 407 cells | δ-T, γ-T, γ-TmT, tocopherol phosphates (TP), tocopherol quinones (TQ) | δ-T significantly reduced the number of viable cells compared to control, while γ-T was not as effective. δ-T was more effective in inhibiting cancer cells than normal colon INT407 cells. γ-TQ had the most potent inhibitory activity of all the compounds examined. | 109 (Dolfi et al., 2013) |
| Lung & other cancers | |||
| Female A/J mice induced by NNK | γ-TmT at 3 g/kg diet | 3 g/kg γ-TmT significantly lowered tumor multiplicity, tumor volume & tumor burden (by 30%, 50% & 55%, respectively. | 110 (Lu et al., 2010) |
| A/J mice with CL13 murine lung cancer cells | γ-TmT at 1 & 3 g/kg diet | γ-TmT inhibited the growth of CL13 tumors by 53.9% & 80.5%, respectively. | 111 (Lambert et al., 2009) |
| Ferrets with cigarette smoke-induced lung squamous metaplasia | 27 male adult ferrets, α-T at 22 mg/kg body wt/day, | α-T (22 mg/kg BW/day orally) plus ascorbic acid (3 mg/kg/day orally) prevented squamous metaplasia and the overexpression of CCDN1 in the lung as well as ameliorated the lowering of α-T and vitamin A levels in the lung caused by smoke. | 112 (Kim et al., 2012) |
| Pancreatitis induced in male Sprague-Dawley rats by infusion of TNBS into pancreatic duct | α-T at 300, 600 or 900 mg/kg/day orally for 4 weeks | α-T dose-dependently decreased pancreatic pseudocyst formation and ameliorated pancreatic weight loss in chronic pancreatitis. | 113 (Li et al., 2011) |
| NMBA-induced esophageal cancer in Fischer 344 rats | α-T, δ-T or γ-TmT at 1.5 g/kg diet | α-T, δ-T & γ-TmT inhibited carcinogenesis by inhibiting NF-kB & CSCR3 signaling: suppressed the production of pro-inflammatory cytokines and the infiltration of CXCR3+ effector T cells (CD4+ Th1 & CD8+ CTLs). | 114 (Yang et al., 2018) |
Abbreviations: ACF, aberrant crypt foci; ACI, August-Copenhagen Irish; ALDH, aldehyde dehydrogenase; AOM/DSS, azoxymethane/dextran sulfate sodium; AR, androgen receptor; DHT, dihydrotesterone; E2, estrodiol; LTB4, leukotriene B4; MNU, methylnitrosurea; NMBA, N-nitrosomethylbenzylamine; 8-oxo-dG, 8-oxo-deoxyguanosine; PGE2, prostaglandin E2; PhIP, 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine; PIN, prostatic intraepithelial neoplasia; PSA, prostate specific antigen; RTK, receptor tyrosine kinase; TNBS, trinitrobenzene sulfonic acid
4.1.1. Prostate cancer
The prostate cancer preventive activities of tocopherols have been studied in transgenic mouse and rat models89,90. In the transgenic adenocarcinoma of the mouse prostate (TRAMP) model, supplementation of γ-TmT in the diet (1 g/kg) significantly decreased the numbers of prostate tumors and prostatic intraepithelial neoplasia (PIN)89. In the transgenic rat for adenocarcinoma of prostate (TRAP) model, γ-T at low concentrations of 50, 100 and 200 mg/kg diet dose-dependently reduced the number of adenocarcinomas in the ventral lobe, but α-T did not90. The inhibition was associated with enhanced apoptosis and the activation of caspases 3 and 7. Similarly, γ-T (20 mg/kg diet) significantly decreased methylnitrosourea (MNU)-induced ventral prostate epithelia dysplasia, cell proliferation, GST-pi and COX2 expression91. On the other hand, supplementation with α-T (2 or 4 g/kg diet) to MNU-treated rats did not prevent prostate formation92. The cancer preventive activity of γ-TmT was also demonstrated in a novel prostate carcinogenesis model in the CYP1A-humanized mice induced by a dietary carcinogen, PhIP93. Dietary administration of γ-TmT (2 g/kg of the AIN93 diet) effectively prevented the development of mouse PIN (mPIN). It also reduced PhIP-induced elevation of 8-oxo-deoxyguanosine (8-oxo-dG) and nitrotyrosine (caused by ROS and RNS) and the pro-inflammatory enzyme COX2, inhibited the pro-growth Ki-67 and p-AKT signaling, and prevented the loss of PTEN and Nrf2 expression. Further studies with purified δ-T, γ-T and α-T (2 g/kg diet) showed that δ-T was more effective than γ-T or α-T in preventing mPIN formation and p-AKT elevation93.
In human prostate cancer cell lines LNCaP, VCaP and CWR22Rv1, δ-T showed a much stronger activity than α-T in inhibiting cell growth and inducing apoptosis94. The inhibitory activity was associated with the suppression of androgen receptor activity and prostate specific antigen levels. δ-T was also more potent than α-T in inhibiting the growth of LNCaP cell xenograft tumors in the immunodeficient SCID mice, and the inhibition was associated with lowered cell proliferation and enhanced cell apoptosis in tumors94. In further studies with these and other prostate cancer cell lines, δ-T was also found to be more effective than γ-T and α-T in inhibiting prostate cancer cell growth by inducing cell cycle arrest and apoptosis95. By profiling the effects of δ-T on cell signaling using a phospho-kinase array, we found that the most prominently inhibited target was the phosphorylation of AKT on T308. Further studies revealed that δ-T attenuated the EGF/IGF-induced activation of AKT (via the phosphorylation of AKT on T308 induced by the activation of PIK3). Expression of dominant active PI3K and AKT in prostate cancer cell line DU145, in which PIK3, AKT, and PTEN are wild type, caused the cells to be refractory to the inhibition of δ-T, suggesting that δ-T inhibits the PIK3-mediated activation of AKT. Our data also suggest that δ-T interferes with the EGF-induced EGFR internalization, leading to the inhibition of the receptor tyrosine kinase-dependent activation of AKT.
The inhibition of AKT signaling and prostate carcinogenesis by δ-T was demonstrated in vivo in prostate-specific PTEN−/− (PTENp−/−) mice, in which the activation of AKT is the major driving force for tumorigenesis. By feeding PTENp−/− mice with an AIN93M diet supplemented with δ-T (2 g/kg diet) starting at the age of 6 or 12 weeks, the treatment reduced prostate adenocarcinoma multiplicity by 53.3% and 42.7%, respectively, at the age of 40 weeks. However, α-T (2 g/kg diet) was not effective96. Dietary δ-T also reduced the phosphorylation of AKT (T308), decreased proliferation and enhanced apoptosis in prostate lesions. In this model, the involvement of oxidative stress in carcinogenesis and the antioxidant activity of δ-T in cancer prevention was not apparent96. All these results indicated that γ-TmT, γ-T and δ-T prevented prostate carcinogenesis, and the activity of δ-T was higher than γ-T (at a dose of 2 g/kg diet). This effective inhibiting dose was much higher than the reported effective doses of γ-T at 20, 50 or 100 mg/kg diet90,91. In our laboratory, these low doses were not effective.
4.1.2. Mammary cancer
Our work also showed that δ-T, γ-T and γ-TmT inhibited tumorigenesis in estrogen receptor (ER) positive in vivo models of breast cancer by downregulation of ERα and Akt signaling, activation of PPARγ, upregulation of Nrf2-mediated antioxidant response, inhibition of oxidative stress and inflammatory markers, and modulation of CYP1A1-mediated estrogen metabolism97–100. Dietary administration of δ-T and γ-T, but not α-T, was shown to inhibit tumorigenesis in ER-positive breast cancer, while did not provide protective effects against ER-negative and HER-2 positive breast cancer, suggesting that the chemopreventive effects are due to estrogen-mediated and ER-dependent mechanisms97. δ-T and γ-T have been shown to inhibit estrogen-stimulated breast cancer tumor growth101. In a breast cancer model, mice with MCF-7 xenograft tumors implanted with estrogen, treatment with γ-T, δ-T or γ-TmT (2 g/kg diet) significantly reduced tumor volume and tumor weight, and inhibited cell proliferation-related genes such as cyclin D1 and c-Myc as well as estrogen-related genes TFF/pS2, cathepsin D, and progesterone receptor101. Further, decreased the levels of estrogen-induced oxidative stress and nitrosative stress markers, 8-oxo-dG and nitrotyrosine, as well as the DNA damage marker, phosphorylated histone 2A variant X (γ-H2AX), were evident101. The differences in chemopreventive efficacy of α-T, δ-T, γ-T and γ-TmT (2 g/kg diet) were further evaluated in female August-Copenhagen Irish (ACI) rats receiving estrogen implants in an experiment of 30 weeks. δ-T, γ-T and γ-TmT all significantly reduced mammary tumor volume102. The biological activities of individual forms of tocopherols at the whole transcriptome level were evaluated using RNA sequencing analysis, and the results showed that δ-T and γ-T had superior cancer preventive properties compared to α-T, based on their effects on transcriptome – in regulating the expression of genes involved in cell proliferation, metastasis, and tumor progression102. Estrogen plays an important role in breast cancer development potentially via cell proliferation and in increasing stem cell-like properties in the tumors. Treatment of MCF-7 tumorsphere with estrogen resulted in an increase in the CD44+/CD24- subpopulation and ALDH activity as well as the number and size of the tumorspheres, and treatment with γ-T or δ-T inhibited the estrogen-induced actions103. Further, overexpression of a key stem cell transcription factor OCT4 increased CD44 and SOX2 levels and expression of matrix metalloproteinases (MMPs), tissue inhibitors of MMPs (TIMPs) and urokinase plasminogen activator (uPA) and significantly increased cell invasion, suggesting an OCT4-mediated mechanism in estrogen-induced stemness and cell invasion in breast cancer and its inhibition by γ-T and δ-T103.
4.1.3. Colon cancer
In a colon cancer model using male CF-1 mice treated with azoxymethane (AOM) and promoted with DSS, γ-TmT (3 g/kg), lowered colon inflammation and reduced the number of colon adenomas104. The treatment also lowered the levels of prostaglandin E2 (PGE2), leukotriene B4 (LTB4) and 8-isoprostane in the plasma, as well as those of PGE2, LTB4 and nitrotyrosine in the colon, and increased the apoptotic index in adenomas. In a similar study in AOM-treated male BALB/c mice, however, γ-T, but not γ-TmT, (1 g/kg diet) suppressed moderate colitis-promoted colon tumorigenesis, but neither agent was effective in inhibiting severe colitis and related colon tumorigenesis105. Interestingly, in a 1,2-dimethylhydrazine (DMH)-induced colon cancer model in male Wistar rats, supplementation with a low dose of VE (α-T) (225 IU, 3 times the RDI) had beneficial effects in reducing colon ACF formation and COX2 expression, while a high dose of α-T (1500 IU) did not106. A comparative study of the inhibitory activity of α-T, γ-T and δ-T (at 2 g/kg diet) was conducted in AOM-treated Fischer 344 rats107. After treatment for 8 weeks, a large number of aberrant crypt foci (ACF) were developed. δ-T treatment showed the strongest inhibitory effect in decreasing the numbers of ACF by 62%, while γ-T and γ-TmT were less effective, but α-T was not effective. Immunohistochemical (IHC) analysis showed that δ-T or γ-T treatment reduced the levels of 4-hydroxynonenal, nitrotyrosine and cyclin D1 (CCND1), and preserved the expression of PPAR-γ in the colon. It also decreased serum levels of PGE2 and 8-isoprostane. Supplementation with δ-T or γ-T markedly increased the levels of δ-T or γ-T (from very low to 6.69 or 21.20 μmol/L, respectively), and their side-chain degradation metabolites (with δ- or γ-CEHCs at 23.32 or 6.07 μmol/L, respectively) in the serum. Rather high concentrations of δ-T and δ-CEHC or γ-T and γ-CEHC, were found in colon tissues (with concentrations of 16.91 and 7.60 or 21.49 and 6.91 μmol/kg, respectively), suggesting there could be local action of those compounds in the inhibition of colon carcinogenesis107.
In a mouse colon carcinogenesis model induced by PhIP and promoted by DSS in CYP1A-humanized mice, we demonstrated that δ-T and γ-T (2 g/kg diet) inhibit colon carcinogenesis mostly by protection against early cellular and DNA damage caused by ROS and RNS, during the DSS treatment period108. Treatment with δ-T after this period was much less effective in inhibiting tumorigenesis. During the DSS-treatment period, dietary δ-T decreased the levels of 8-oxo-dG and nitrotyrosine, as well as pro-inflammatory mediators (NF-κB p65 and p-STAT3), in tumors and adjacent tissues. In contrast, α-T did not significantly decrease tumorigenesis. Of note is that, in this PhIP/DSS-induced mouse model, significant inhibition of colon tumorigenesis by δ-T or γ-T (2 g/kg diet) was not observed in female mice, possibly due to the lower number of tumors formed, and the decrease in tumor number by δ-T did not reach statistical significance. The gender differences in colon cancer formation has been shown in both rodents and humans. Whether sex hormones play a role in influencing the actions of tocopherols in colon carcinogenesis remains to be studied.
These studies clearly demonstrate the higher activity of δ-T than γ-T in inhibiting colon tumorigenesis, while α-T was ineffective. This conclusion is consistent with our studies on colon cancer cell lines HCT116 and HT29109, showing this same ranking order in inhibiting cell growth, decreasing cancer cell colony formation and inducing apoptosis, while α-T was rather ineffective. This cell line study also showed that the rate of cellular uptake also followed the same ranking order, and γ-tocopherol quinone and γ-tocopherol phosphate had much higher inhibitory activities than γ-T, and even higher than the corresponding δ-forms of these derivatives109.
4.1.4. Lung cancer and other cancers
In lung cancer models, we demonstrated that dietary γ-TmT reduced tumor multiplicity, tumor volume or tumor burden in 4-(methylnitrosoamino)-1-(3-pyridyl)-1-butanone (NNK) or NNK plus benzo[a]pyrene (B[a]P)-induced lung cancer in A/J mice, as well as human lung cancer H1299 xenograft tumors in nu/nu mice110. In cell culture, the growth of H1299 cells was inhibited by tocopherols with their effectiveness following the order of δ-T > γ-TmT > γ-T, whereas α-T was not effective. γ-TmT inhibited the growth of CL13 murine lung cancer cells in vitro. Dietary administration of γ-TmT (1 or 3 g/kg diet) also significantly inhibited the growth of CL13 xenograft tumors in A/J mice111. In both the carcinogenesis and tumor growth models, the inhibitory action of γ-TmT was associated with lowered levels of 8-oxo-dG, γ-H2AX and nitrotyrosine, as well as enhanced apoptosis, in the tumors. In another model, ferrets were exposed to cigarette smoke to induce lung squamous metaplasia112. Treatment with α-T (22 mg/kg body weight/day orally) plus ascorbic acid (3 mg/kg/day orally) prevented squamous metaplasia and overexpression of cyclin D1 in the lung as well as ameliorated the smoke-induced lowering of α-T and vitamin A levels in the lung. In a rat model for pancreatitis induced by trinitrobenzene sulfonic acid (TNBS), α-T (300, 600 and 900 mg/kg/day, orally) dose-dependently decreased pancreatic pseudocyst formation and ameliorated pancreatic weight loss in chronic pancreatitis113.
In order to mimic the insufficiency of VE and selenium of the population with high risk of esophageal cancer in the LNIT, we used a VE/selenium insufficient diet in a rat model for esophageal cancer32. Supplementation of the diet with VE/selenium inhibited NMBA-induced esophageal carcinogenesis, and supplementation during the early stage of carcinogenesis was more effective than in the late stage in preventing tumor formation32. Further studies with rats fed a normal diet and supplemented with α-T, δ-T and γ-TmT (1.5 g/kg diet) demonstrated that all these supplements inhibited esophageal carcinogenesis114. Mechanistic studies suggest that these tocopherols inhibited esophageal carcinogenesis through attenuating NF-kB activation and CXCR3-mediated inflammation, with δ-T appeared to be more effective114. These results support the hypothesis that in humans, VE and selenium insufficiency makes the individual more susceptible to esophagitis, and supplementation with VE and selenium blocked the inflammatory mediators and inhibited carcinogenesis. The relative contribution of VE and selenium was not discerned from this set of animal studies.
4.2. Studies with Tocotrienols
Most of the studies on tocotrienols (mostly γ-T3 and δ-T3) have been carried out in cell lines, demonstrating the inhibition of cell growth and induction of apoptosis, and some of the studies have carried into xenograft tumor models. There are also a few recent studies in animal models. Some recent studies (published in 2011–2019) are summarized in Table 4. Earlier work has been covered by some excellent recent reviews7–9,12 and a book115. Some of the proposed molecular targets or pathways are highlighted in Figure 3.
Table 4.
Laboratory Studies on Tocotrienols and Cancer
| Experimental Model | Treatment | Conclusion | Reference |
|---|---|---|---|
| Human colon cancer cells SW620 | 4–20 δ-T3 | Downregulated WNT pathway (↓CTNNB1, ↓WNT1, ↓CCND1) | 116 (Zhang et al., 2011) |
| Human colon cancer cells HT-29 | 45 & 60 μM γ-T3 | Inhibited β-Catenin/Tcf signaling (↓CTNNB1, ↓BIRC5, ↓CCND1, ↓c-MYC↓) | 117 (Xu et al., 2012) |
| Human colon cancer cells SW620 & HCT-8 | 15–60 μM | Induced parapoptosis-like cell death by inhibiting Wnt signaling pathway (↓c-JUN, ↓CTNNB1, ↓CCND1) | 118 (Zhang et al., 2013) |
| Human breast cancer cells MDA-MB-231 & T-47D | 2–6 μM γ-T3 | Inhibits EMT and proliferation by ↓canonical Wnt/β-catenin signaling pathway | 119 (Ahmed et al., 2016) |
| Human colon cancer cells SW620 cells; xenograft tumors | 5, 10 & 20 mg/kg bw TRF | Inhibited tumor growth by ↑WNT pathway | 120 (Zhang et al., 2015) |
| Human oral cancer cells B88 | 50 μM γ-T3 | Improved chemosensitivity to docetaxel by ↓BIRC5, ↓c-IAP-1, ↓cIAP-2, ↓XIAP, ↓BCL2 and downregulating the expression of NF-kB-mediated anti-apoptotic gene products | 121 (Kani et al., 2013) |
| Murine RAW 264.7 macrophages; A20−/− & A20+/+ mouse embryonic fibroblasts | 10, 20 μM δ-T3 | Inhibited activation of NF-κB and TAK1, ↓IL-6, ↑A20, ↑CYLD, induced cellular stress, ↑intracellular dihydroceramides | 122 (Yang and Jiang 2019) |
| Human lung cancer cells A549 & H1299 | 10–40 μM δ-T3 | Inhibited proliferation, cell invasion, cell aggregation and adhesion, ↓MMP-9, ↑miR-451, inhibited uPa/NOTCH1 pathway proteins, inhibited NF-κB DNA-binding activity |
123 (Rajasinghe et al., 2018) |
| Human lung cancer cells A549 & H1650 | 15 μM δ-T3 | Augmented cisplatin-induced inhibition of cell invasion via the suppression of Notch-1 signaling pathway (↓NOTCH1, ↓HES1, ↓pro-CASP-3, ↓PARP, ↓VEGF, ↓MMP9) | 124 (Ji et al., 2012) |
| Human lung cancer cells A549 & H1299 | 20–30 μM δ-T3 | Arrested G0/G1 cell cycle and inhibited cell invasion and migration by ↑miR-34a; ↓NOTCH1, HES1, PARP, Bcl-xL, BIRC5, NF-κB, VEGF & MMP9 | 125 (Ji et al., 2012) |
| Human non-small cell lung cancer cell line A549 and H1299 | 10, 20 and 30 μM δ-T3 | Dose- and time-dependent inhibition of cell growth, cell migration, tumor cell invasiveness and induction of apoptosis associated with decreases in NOTCH1, HES1, survivin, MMP9, VEGF and VCL-XL expression | 126 (Ji et al., 2011) |
| Human lung cancer A549 & H1299 cells | 10, 20, 30 μM δ-T3 | Inhibited glutamine transporters and mTOR pathway | 127 (Rajasinghe et al., 2019) |
| HUVEC; human liver cancer cells HCCLM3; xenograft tumor | 50 μM γ-T3; 3.25 mg γ-T3 daily, 5 days a week | Inhibited VEGF-induced potential of migration and invasion of HUVEC by downregulating AKT/mTOR signaling pathway; inhibited tumor growth and VEGF-induced angiogenesis | 128 (Siveen et al., 2014) |
| Human bladder cancer cells T24, 5637, J82 & UMUC-3 | 50, 100, & 150 μM δ-T3 | Inhibited growth, induced G1 arrest and apoptosis by ↑CDKN1A, ↑CDKN1B, ↓pro-CASP-3, ↑c-CASP-3, ↑BAX, ↓BCL2 | 129 (Ye et al., 2015) |
| Human prostate cancer cells PC3 | 24 & 48 μM δ-T3 | Induced cytotoxicity by ↓SRC, ↓p-SRC, ↓STAT3, ↓p-STAT3 | 130 (Sugahara et al., 2015) |
| Human prostate cancer cells PC3; xenograft tumor | 12 μM γ-T3 | Polysaccharopeptides enhanced the effect of γ-T3 in inhibiting cell colony formation by ↑p-AMPK and inhibiting tumor growth | 131 (Liu et al., 2014) |
| Human breast cancer cells T47D | 5 μM γ-T3 | Inhibited exosomes-dependent (ED) cell growth; ↓ED HER3 and HER4 levels; ↓HER3/HER4 heterodimer downstream signaling; disrupted the integrity of lipid raft microdomain; ↓heregulin and mitogenic biopotency | 132 (Alawin et al., 2017) |
| Human breast cancer cells SKBR3, BT474 | 4 μM γ-T3 | The anticancer effect of γ-T3 is associated with its accumulation in the lipid raft microdomain due to ↓HER2 dimerization and ↓p-HER2 | 133 (Alawin et al., 2016) |
| Human pancreatic cells MiaPaCa-2 | 50 μM δ-T3 | Induced apoptosis via ↑EGR-1/BAX | 134 (Wang et al., 2015) |
| Human breast cancer cells MDA-MB-231 & MDA-MB-468 | 30–100 μM δ-T3 | Inhibited proliferation and induced apoptosis by ↑XIAP and expression of miR-429 | 135 (Wang et al., 2015) |
| Human pancreatic cells MIAPaCa-2, SW1990 & BxPC-3; xenograft tumor | 50 μM δ-T3; 100 mg δ-T3/kg bw, daily | Induced CDKN1B expression and suppressed RAS-MEK-ERK signaling; inhibited tumor growth | 136 (Hodul et al., 2013) |
| Human prostate cancer cells PC-3, LNCaP, CRL-1740, & CRL-1435; grade IV prostate adenocarcinoma | 3, 5, 20 & 40 μM γ-T3 | Growth inhibition by γ-T3 partially depended on PPAR-γ (↑PPAR-γ1, phosphorylation of ↑PPAR-γ2, ↑15-LOX-2, ↓TGFβ2↓, ↓NF-κB activation) | 137 (Campbell et al., 2011) |
| Human leukemia cells HL-60 & NB-4; human lymphoblast cells Raji; human bone marrow cells SY-5Y | 10–30 μM γ-T3 | Induced apoptosis (DNA ladder formation and nuclear fragmentation; cleavage of BID upregulating caspase cascade) | 138 (Inoue et al., 2011) |
| Human lung cancer cells A549; human glioblastoma U87MG | 2, 3 μM β-T3 | Induced mitochondria-regulated apoptosis by ↑CASP-8 activity, ↑BID activity, ↑CYCS activity | 139 (Lim et al., 2014) |
| Human lung cancer cells A549; human glioblastoma U87MG | 2–100 μM α-, δ- & γ-T3 | Interfered mitochondrial membrane permeability, promoted apoptosis by ↑CASP-8, ↑BID, ↑BAX, ↑CYCS | 140 (Lim et al., 2014) |
| Human colon cancer cells HCT-116; pancreatic cancer cells PANC-1; breast cancer cells MCF-7 | 20 μM γ-T3 | Altered sphingolipid metabolism by ↓DEGS | 141 (Jang et al., 2017) |
| Human colon cancer cells cancer HCT-116, HT-29 & Caco-2; Balb/c mice injected with AOM |
δ-T313′-carboxychromanol, 10 & 20 μM in vitro; 0.022% in diet to mice | Suppressed cell growth, induced cell apoptosis and autophagy by ↓COX-1/COX-2, ↓5-LOX, ↓DEGS1 activity; decrease mouse colon tumor multiplicity | 142 (Jang et al., 2016) |
| Human breast cancer cells MCF-7 & MDA-MB-231 | 20, 40 & 80 μM γ-T3 | Mediated canonical signal transduction or metabolic pathways (e.g., Nrf-2-modulated oxidative stress response, TGF-β signaling and endoplasmic reticulum stress response) by ↓CCND1, ↓CCND3, ↓CDK4, ↑c-PARP, ↑c-CASP-7, ↑ATF3, ↑HSPA5, ↑CHOP | 143 (Patacsil et al., 2012) |
| Mouse mammary gland cancer cells +SA; human breast cancer cells MCF-7 & MDA-MB-231 | 20 & 40 μM γ-T3 | Induced autophagy of the cancer cells by ↑LC3B-I, ↑LC3B-II, ↑BECN1, ↓BCL2, ↑c-PARP, ↑c-CASP-3, ↓PI3K, ↓p-AKT, p-MTOR↓ | 144 (Tiwari et al., 2014) |
| Human prostate cancer DU145 & PC3 cells | 12–50 μM δ-T3 | Reduced cell viability, triggered apoptosis, ER stress, autophagy, and paraptosis | 145 (Fontana et al., 2019) |
| Human breast cancer cells MCF-7 & MDA-MB-231 | 40 μM γ-T3 | Induced ER stress for cell death by ↑LC3B, ↑BECN1, ↑ATG5-ATG12, ↑p-JNK1/2, ↑p-p38, ↓BCL2, ↑BAX, ↑c-CASP-3, ↑HSPA5, IRE1α, ↑p-PERK, ↑ATF-4, ↑CHOP, ↑TRB3 | 146 (Tiwari et al., 2015) |
| Human melanoma cells A375; immunodeficient mice injected with A375 stem-like cells | 100 μM δ-T3 | Inhibited cell proliferation and formation/growth of melanopheres | 147 (Marzagalli et al., 2018) |
| Human pancreatic cancer cells L3.6pl, MiaPaCa-2 Xenograft tumors |
10 μM δ-T3; 200 mg δ-T3/kg bw, daily | δ-T inhibited tumor growth and liver and lung metastasis and reduced markers of proliferation, angiogenesis and stemness in vivo; reduced EMT markers in vitro | 148 (Husain et al., 2017) |
| Human breast cancer cells MCF-7/Adr & MCF-7/TamR |
10 μM γ-T3 | Eliminated enriched cancer stem cells and inhibited expression of STAT-3 signaling mediators by ↓CCND1, ↓BCL-XL, ↓BIRC5, ↓p-STAT-3 and activating de novo ceramide synthesis pathway | 149 (Gopalan et al., 2013) |
| Human gastric cells SNU-5, SNU-16, & MKN45; xenograft tumors | 10 & 50 μM γ-T3; 1 mg γ-T3/kg bw, 3 times/week | Potentiated the anticancer effect of capecitabine by ↓CCND1, ↓BCL2, ↓MMP9, ↓ICAM-1, ↓CXCR4↓ | 150 (Manu et al., 2012) |
| Mouse mammary gland cancer cells 4T1 xenograft tumor | 1 mg TRF/day | Inhibited tumor growth and metastasis | 151 (Hafid et al., 2013) |
| LSL-KrasG12D, LSL-Trp53R127H, Pdx-1-Cre mice | 200 mg δ-T3/kg bw, twice a day | Inhibited tumor growth, prolonged survival, and reversed EMT in tumors by ↑CDH1, ↓VIM, ↓VEGF, ↓c-PARP1, ↑BAX, ↑CDKN1B, ↑CDKN1A, ↓p-AKT, ↓ERK, and ↓p-ERK | 152 (Husain et al., 2013) |
| Mouse mammary gland cancer cells +SA; human breast cancer cells MDA-MB-231 | 2–6 μM γ-T3 | Lowered metastatic phenotypic expression by ↓Rac1/WAVE2 signaling pathway | 153 (Algayadh et al., 2016) |
| Human colorectal cancer cells HCT-116, SW-620 & HT-29; rats injected with AOM |
50 μM δ-T3 to cells 200 mg/kg bw twice a day to rats |
Inhibited malignant transformation, cell migration, and invasion; ↓E-cadherin, matrix metalloproteinase 9, ↓VEGF, ↓NF-κB, ↓CTNNB1, ↓vimentin. Inhibited formation of colorectal polyps and cancer in rats |
154 (Husain et., 2019) |
| Human colorectal cancer cells DLD-1 | 20 μM δ-T3 | Induced apoptosis and suppressed angiogenesis by ↑CASP-3, ↑CASP-9, ↑CDKN1A, ↑ CDKN1B, ↓p-AKT | 155 (Shibata et al., 2015) |
| Human gastric cancer cells SGC-7901; HUVECs; chick chorioallantoic membrane | 10–40 μM γ-T3 | Induced HUVECs apoptosis, migration and tube formation by downregulating Wnt signaling pathway (↓CTNNB1, ↓CD44, CCND1, ↓p-VEGFR-2, ↓MMP9); inhibited angiogenesis in vivo | 156 (Li et al., 2011) |
| Human prostate cancer PC3 & C42B cells | 25 μM γ-T3 | ↓ANGPT1; when combined with Tie-2 inhibitor, inhibited cell growth, activated AMPK, enhanced cytotoxic effect, inhibited autophagic flux | 157 (Tang et al., 2019) |
| Human breast cancer cells MCF-7 & MDA-MB-231; mouse mammary gland cancer cells +SA | 3 μM γ-T3 | Sesamin potentiated the inhibitory effect of γ-T3 in inhibiting proliferation by ↑CDKN1B, ↑CDKN2A and ↑E2F1 | 158 (Akl et al., 2013) |
| Human prostate cancer cells DU-145; human breast cancer cells MCF-7; human pancreatic cells PANC-1 | 12.5 μM δ-T3 | Ferulic acid promoted δ-T3 in inhibiting proliferation and inducing G1 arrest by ↑CDKN1A | 159 (Eitsuka et al., 2014) |
| Human colorectal cancer cells HT-29 & SW837 | 49–244 μM γ-T3 | γ-T3 and 6-gingerol synergistically inhibited cell growth and apoptosis | 160 (Yusof et al., 2015) |
| LNCaP cells | 5–10 μM γ-T and δ-T3 | Prostate cancer cells respond to δ-T3 more effectively than γ-T. Combined treatment of δ-T3 & γ-T was found to have greater significant effect by marked reduction of cancer cell proliferation. | 161 (Sato et al., 2017) |
Abbreviations: AOM, azoxymethane; DEGS, dihydroceramide desaturase; dhCer, dihydroceramide; dhSph, dihydrosphingosine; EMT, epithelial-to-mesenchymal transition; ER, endoplasmic reticulum; FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate; HUVECs, human umbilical vein endothelial cells; RAS-MEK-ERK, RAS-MAP kinase pathway; TRF, tocotrienol-rich fraction; Sph, sphingosine
Figure 3.
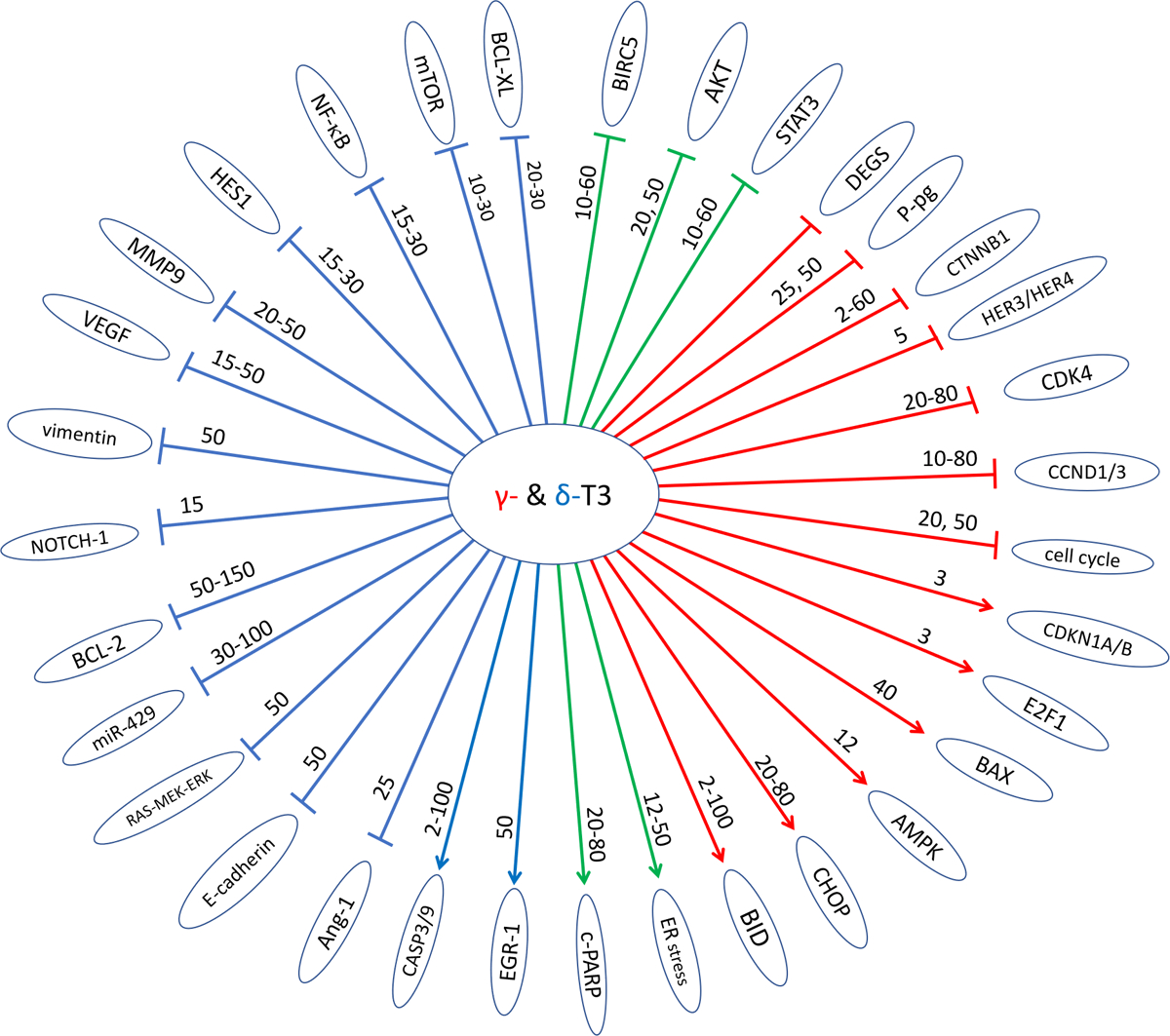
Proposed targets of for the cancer preventive activities of γ- and δ-T3 from studies in vitro. Red, blue, and green arrows indicate γ-T3, δ-T3, or both, respectively; → indicates activating/upregulating and ⊢ indicates inhibiting/downregulating a target (or pathway). The numbers on each arrow are the reported effective concentrations in μM. Abbreviations – AMPK, adenosine monophosphate-activated protein kinase; Ang-1, angiopoietin 1; AKT, protein kinase B; BAX, BCL2 associated X apoptosis regulator; BCL-2, B-cell lymphoma 2; BCL-XL, B-cell lymphoma-extra large; BID, BH2 interacting domain death agonist; BIRC5, baculoviral IAP repeat containing 5; CASP3/9, caspase-3, caspase-9; CCND1/3, cyclin D1/3; CDK4, cyclin-dependent kinase 4; CHOP, Ce/EBP homologous protein; c-PARP, cleaved poly (ADP-ribose) polymerase; DEGS, dihydroceramide desaturase; E2F1, E2F Transcription Factor 1; EGR-1, early growth response 1; ER stress, endoplasmic reticulum stress; HES1, hes family BHLH transcription factor 1; HER3/HER4, erb-b2 receptor tyrosine kinase 3/4; miR-429, microRNA-429; mTOR, mammalian target of rapamycin; MMP9, matrix metallopeptidase 9; NF-κB, nuclear factor of kappa light chain polypeptide gene enhancer in B cell 1; NOTCH-1, neurogenic locus notch homolog protein 1; P-pg, p-glycoprotein; RAS-MEK-ERK, RAS-MAP kinase pathway; STAT3, signal transducer and activator of transcription 3; VEGF, vascular endothelial growth factor.
4.2.1. Inhibition of cell proliferation and induction of apoptosis
δ-T3 and γ-T3 have been shown to inhibit growth and promote apoptosis in cell lines derived from gastric cancer, leukemia, breast cancer, oral cancer, lung cancer, glioblastoma, pancreatic cancer, prostate cancer, non-small cell lung cancer, bladder cancer and colorectal cancer (Table 4). Specific inhibitory patterns against cells from different cancer types are not observed, and the table is arranged based on the signaling pathways involved. These activities were observed commonly in the concentration range of 2–20 μM. The molecular mechanisms reported for the anti-proliferation, pro-apoptosis and other anti-cancer activities of δ-T3 and γ-T3, as depicted in Fig. 3, involve the inhibitory effects on WNT signaling116–120, 119NF-κB pathway121–123, NOTCH signaling123–126 and AKT/mTOR signaling127,128; and/or the activation of STAT3129,130, SRC kinase130, AMPK131 and receptor tyrosine kinases HER3/HER4132,133. The inhibition of proliferation and induction apoptosis were also reported to be mediated through tocotrienol-induced activation of EGR-1/BAX pathway134, upregulation of miR-34a125, miR-429135, p27/CDKN1B136 and PPAR-γ137, disruption of lipid raft132,133 and modification of mitochondria membrane and activation of pro-apoptosis genes138–140. Moreover, tocotrienols and their 13`-COOH metabolites modulate sphingolipid metabolism through inhibition of dihydroceramide desaturase (DEGS)122,141,142. Suppression of glutamine uptake127 were also suggested to be possible mechanisms. In addition, pathway analysis of the tocotrienol-induced gene expression profile changes suggests that γ-T3 and δ-T3 alter endoplasmic reticulum stress signaling and related autophagy and cell death143–146.
4.2.2. Suppression of stemness and inhibition of cell migration/invasion, tumor growth, metastasis and angiogenesis
The stemness of cancer cells has also been reported to be inhibited by tocotrienols. δ-T3 was shown to inhibit the formation of tumor spheres of human melanoma cells147. δ-T3 was also reported to inhibit the expression of stem cell transcription factors, including NANOG, OCT4 and SOX2, in pancreatic cancer cells and surface stemness marker CD44 in xenograft tumors of pancreatic cancer cells148. γ-T3 was reported to inhibit mevalonate pathway and activate de novo ceramide synthesis pathway, resulting in the reduced STAT-3 mediated signaling and leading to the inhibition on breast cancer stem-like cells149.
δ-T3 and γ-T3 have been demonstrated to inhibit tumor growth in xenograft models of pancreatic cancer cells136,148, gastric cancer cells150, mouse mammary cancer cells151, liver cancer cells128, and melanoma cells147, as well as the transgenic pancreatic cancer mouse models (LSL-KrasG12D;LSL-Trp53R127H;Pdx-1-Cre [KPC mice])152.
Other properties of cancer cells such as cell migration, cell invasion, and epithelial–mesenchymal transition have also been reported to be inhibited by tocotrienols in lung cancer cells, gastric cancer cells and breast cancer cells in vitro, in cell migration assay, wound healing assay, and cell invasive assay119,123,153. In γ-T3-treated breast cancer MDA-MB-231 cells, cell migration machinery regulators such as RAC1 and WAVE2 signaling were reduced153. In addition to the reduced cell migration, γ-T3-treated breast cancer MDA-MB-231 and T-47D cells showed epithelial–mesenchymal transition119. Using the same in vitro assays, δ-T3 was also found to suppress the migration and invasion of non-small lung cancer cells124 and colon cancer cells154. Studies in xenograft models showed that δ-T3 inhibited pancreatic cancer tumor growth and liver and lung metastasis148 and that tocotrienols-rich mixture inhibited lung metastasis of breast cancer cells151.
Tocotrienols were also found to display anti-angiogenesis activity in animal models. For examples, δ-T3 and γ-T3 suppressed the tumor angiogenesis of xenograft tumors of pancreatic cancer cells148, colorectal cells155 and liver cancer cells128 as well as pancreatic tumors developed in KPC transgenic mice152. The anti-angiogenesis effect of tocotrienols has been well demonstrated in typical angiogenesis tests such as tube formation assay of HUVEC128 and chick chorioallantoic membrane assays128,156. Reduced expression of angiogenesis factors such as VEGF and ANG1 was found in several studies and proposed to be the mechanism of anti-angiogenesis activities of tocotrienols128,150,152,157.
Many of the above activities were observed in a recent study (Husain, K. Accepted results in Scientific Reports). δ-T3 (10–100 μM) was found to inhibit colon cancer stem cell (CCSCs) survival and stemness (NANOG, OCT4 and SOX2) as well as markers for migration, invasion, inflammation (NF-kB), angiogenesis (VEGF) and metastasis (MMP9). In an orthotopic (cecum-injected CCSCs) xenograft model of metastasis, δ-T3 (200 mg/kg administered orally twice a day for 4 weeks) significantly retarded the CCSCs-derived tumor growth (Ki-67), inflammation (NF-kB), angiogenesis (VEGF and CD31) and β-catenin/CTNNB1, as well as induced apoptosis (cleaved-PARP) in tumor tissues. More importantly, it inhibited liver metastasis of CRC.
The inhibitory activities of tocotrienols against cancer have also been shown to be promoted by other natural products such as polysaccharopeptides140, sesamin158, ferulic acid159 and 6-gingerol160. The mechanisms of the synergistic effects remain to be further investigated. If these activities can be demonstrated in vivo, tocotrienols (or their combination with other agents) would have more potential for further exploration.
4.2.3. Studies in animal carcinogenesis models
The triple transgenic KPC mice, which mimic the genetic and histological changes observed in human pancreatic cancer, developed pancreatic cancer rapidly and the overall survival of the mice was 2.18 months. Treatment with δ-T3 (200 mg/kg pr os twice a day) significantly decreased tumor weight, cancer cell proliferation, CD31, VEGF and other growth-related signaling proteins152. It also increased the expression of E-cadherin and enhanced apoptosis (as reflected in CK18 and cleaved-CASP3). At week 16, δ-T3 treatment increased the survival to 70%, versus a survival of 10% in the vehicle control group. When δ-T3 treatment was combined with gemcitabine (100 mg/kg i.p.), the survival rate was 90%152. In a colorectal carcinogenesis model, rats were injected with AOM (15 mg/kg, sc) to induce cancer. δ-T3 treatment (200 mg/kg orally twice a day) for 20 weeks significantly inhibited colorectal polyps by 70% and CRC by 99% compared to the vehicle treatment group (P < 0.02, P < 0.001). The cancer inhibition effect was more potent than sulindac (50%)154.
An interesting study with a side-chain degradation product of δ-T3 was conducted in a mouse model for colon cancer induced by AOM/DSS. δ-T3–13`-COOH (0.22 g/kg diet) was found to significantly inhibit tumor formation, especially on tumors with size > 2mm2,142. This work demonstrates the cancer preventive activity of this δ-T3 metabolite, in agreement with its activities in inhibiting pro-inflammatory enzymes and DEGS to induce autophagy and apoptosis of human cancer cells142.
4.3. Assessment of laboratory studies and mechanistic considerations
The studies as reviewed above clearly demonstrated the cancer preventive activities of γ-T and δ-T in different animal models, while α-T was not effective. Cancer prevention activities of tocotrienols have also been demonstrated in a few studies in animals and many studies in cell lines. For mechanistic studies, treatment of cancer cell lines in culture or xenograft tumors with γ-T, δ-T, γ-T3 and δ-T3 compounds affects multiple cellular signaling molecules and pathways, leading to enhanced apoptosis and inhibition of cell proliferation, migration, invasion and metastasis and angiogenesis. It is unclear whether these signaling molecules are the direct targets of these VE forms – by direct physical binding, redox activities or affecting membrane structure – or consequences of the primary events.
Some of the VE form-induced signaling alterations were also observed in certain animal carcinogenesis models, which makes them relevant in vivo. Depending on the animal models used, different mechanisms have been proposed for the action. For example, quenching of ROS and RNS and inhibition of COX2 are commonly observed in many animal models, including PhIP-induced prostate carcinogenesis in CYP1A-humanized mice93, while in the PTENp−/− mouse model, inhibition of AKT phosphorylation appear to be the major mechanism and the antioxidant action of δ-T was not apparent in the inhibition of prostate carcinogenesis96. In the inhibition of mammary tumorigenesis, the anti-estrogenic activity of γ-T is likely to play a major role97–100. In these studies, γ-T was slightly more active than δ-T in inhibiting mammary tumorigenesis, while δ-T was more active than γ-T in the inhibition of prostate, colon and lung carcinogenesis.
With tocotrienols, there are many studies in cell lines, and some in animal models, demonstrating the potential activities in cancer prevention or therapy. In cell culture studies, γ-T3 and δ-T3 are generally more active than γ-T and δ-T8,161. The higher activities of tocotrienols may be due to 1) their higher fluidity of tocotrienols; and 2) their ability to also inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol biosynthesis162. In animal carcinogenesis models, direct comparison of tocopherols versus tocotrienols are lacking. Our unpublished result in the PhIP/DSS-induced colon tumorigenesis in CYP1A-humanized mouse model showed that δ-T3 (0.5 g/kg diet) produced 58% inhibition in tumor multiplicity (Yang et al., unpublished results), and this is comparable to an inhibition produced by δ-T at a dose of 2 g/kg diet108.
5. CONCLUDING REMARKS
As discussed in Section 3, the discrepancy between the cancer prevention results observed in observational epidemiological studies and the negative results from several large-scale human intervention trials with high doses of α-T raised the question “Does VE prevent cancer?”.
5.1. Does VE prevent cancer?
Our answer is “It depends on the VE nutritional status of the population and the form and dose of VE used”. From the results of human and animal studies reviewed above, the cancer preventive activity of VE can be analyzed at two levels:
At the nutritional level, insufficiency of VE could enhance carcinogenesis and increase cancer risk, and supplementation of VE to this population would likely reduce cancer risk. This concept is partially supported by the results from the LNIT25–27 and studies in animal models32. In this situation, α-T is more important than other forms of VE. For cancer prevention, consumption of diets rich in VE, mostly from plant-based food such as vegetables and whole grains, would be beneficial. Other constituents in these types of food could also contribute to the prevention of cancer and other diseases. Whereas in the large-scale intervention trials in North America3–5, the trial population mostly were VE sufficiency. Supplementation of a nutrient to a population that is already sufficient in this nutrient would not produce a beneficial effect.
Beyond the nutritional level, in individuals that are sufficient in VE nutrition, supplementation with γ-T, δ-T and tocotrienols, but not α-T, could reduce the cancer risk. There are sufficient data from animal studies for γ-T and δ-T; but only limited data for tocotrienols, even those tocotrienols have shown inhibitory activities in cancer cell lines in numerous studies. The question is whether these activities would manifest in humans, and what are the effective dose.
5.2. Molecular bases for the actions of different VE forms in cancer prevention
As discussed earlier, numerous signaling molecules and pathways have been shown to be up-regulated or down-regulated by different VE forms in a variety of cell lines. Some authors consider these molecules as “targets” of action of different VE forms. However, these changes could also be secondary events observed in the experimental system. The molecular basis of these alterations and their roles in cancer prevention remain unclear. Some of the molecular mechanisms of prevention by these compounds are discussed below.
5.2.1. Antioxidant activities
Since ROS and RNS are involved in inflammation and carcinogenesis, quenching of these free radicals would have anti-inflammatory and anti-carcinogenic effects. This action of all VE forms occurs at the nutritional level, and these forms without a methyl group on the 5-position of the chromanol ring (β, γ and δ forms) would have the advantage of being more active in trapping RNS. This action may contribute to the higher cancer preventive activity of these compounds over α-T.
5.2.2. Direct binding to target molecules
Although many molecules have been referred as targets of different VE forms, evidence for direct binding to these “targets” is lacking. In theory, specific targets via specific physical binding to VE forms could exist. An example is the specific binding of α-T to α-TTP. However, many attempts to identify direct targets for VE forms by molecular docking and physical binding studies have not yielded results that are convincingly relevant to cancer prevention.
5.2.3. Affecting membrane structure, lipid raft and fluidity
These actions have been suggested by several studies based on biophysical studies and investigations in cancer cells. In our studies, on the inhibition of prostate cancer cells, the possibility that δ-T binds to the membrane and influences tyrosine receptor kinase activity was proposed95. Membrane fluidity and lipid raft have been shown to be affected by VE132,133. The higher fluidity of tocotrienols163 may contribute to the higher rate of uptake and higher activity of tocotrienols over tocopherols in inhibiting cell growth. When applying the concept of influencing lipid raft to cancer prevention, however, a key issue to be resolved is the specificity of this action. That is, if certain VE forms affect lipid raft in pre-cancerous and cancer cells in early stages of carcinogenesis, then whether the same mechanism would affect the functions of normal cells is an important issue.
5.2.4. Action of VE degradation metabolites
γ- and δ-forms of VE have lower bioavailability with more side-chain degradation products formed, in comparison with α-T, but have higher anti-cancer activities. These results suggest that these metabolites may play an important role in cancer prevention, as discussed in previous sections. The side-chain degradation metabolites, 13`-COOHs, have shown cancer prevention activities8,142. We may speculate that even for the short chain metabolites, CEHCs and CHMBHCs, with the chromanol ring intake may still have antioxidant and other biological activities. With less lipophilicity, these metabolites could have accessibility different from the parent compounds. These properties may contribute to the cancer preventive activities of γ-T, δ-T, γ-T3 and δ-T3.
From the numerous “targets” or actions proposed by different authors, we may conclude that there is no single specific mechanism or target for the anti-cancer action. Depending on the experimental systems used, different mechanisms may play a more important role. This concept may also apply to humans exposed to different cancer etiological factors.
5.3. Issues on toxicity
One of the reported side effects of tocopherols is their interaction with vitamin K function in affecting blood clotting164. However, in our studies in animal models with tocopherols at 2 or 3 g/kg diet did not produce any observable toxic effect93,94,97–102,107,108,110,111. In our unpublished Phase 0 study, a commercial γ-T-rich mixture of tocopherols (790 mg) were given daily to prostate cancer patients two weeks before surgery (as described in Section 2), estimated blood loss was not significantly different from the placebo group and no adverse events were observed (Yang et al., unpublished results).
As discussed previously, in two human studies, one with pancreatic cancer patients84 and one with healthy volunteers85, δ-T3 at doses of 100 to 3200 mg per day for two weeks, did not show any side effects. In a recent study with tocotrienol supplements, 430 mg and 860 mg tocotrienol from DeltaGold 70 (70% tocotrienols of which 90% is δ-T3 and 10% is γ-T3) given to postmenopausal osteopenic women daily for 12 weeks, did not affect kidney and liver function parameters165. Supplementation for 6 weeks significantly raised serum level of δ-T3, and had no adverse effect on quality of life, body composition physical activity and nutrient intake.
5.4. Future studies
In order to provide a more clear understanding of VE and cancer prevention, the following future studies are suggested.
More mechanistic studies. Most of the mechanistic information on the action of VE forms, especially tocotrienols, are derived from studies in vitro, mostly with cancer cell lines. More innovative in vitro studies are needed to elucidate the basic molecular action of VE forms. It is unclear, however, whether the results from in vitro studies can be extrapolated to the situation in vivo. They may just be observations under those experimental conditions. Therefore, mechanistic studies in vivo are extremely important, especially if it can be pursued in humans. These studies would benefit from the information obtained from studies in vitro, as reviewed herein. In theory, if a VE form can inhibit certain key events that may occur in early carcinogenesis, then such activities could be translatable to cancer prevention. If the reported mechanisms are important in inhibiting pathways important in cancer growth and metastasis, then they may be useful for therapy to prevent metastasis.
Determine the dose-response of γ- and δ-forms of VE in cancer prevention studies in animal models. In our diet, α-T contributes to the majority of nutritional VE activity. Whether dietary levels of γ-T, δ-T and tocotrienols contribute to lowering cancer risk is unclear. Some results on γ-T dose-response studies in animal models were not consistent90,91,93,95. To provide more information on this issue, more dose-response studies on γ-T, δ-T and tocotrienols in animal models are needed. The information will help design human studies.
More human studies on the cancer preventive activities of γ-T, δ-T, γ-T3 and δ-T3. In epidemiological studies, a basic approach is to correlate the serum levels of these VE forms (and probably the urinary levels of corresponding CEHCs and CMBHCs) with cancer risk. Such studies on tocotrienols are important in populations consuming significant amounts of tocotrienols from palm oil or supplements. More human trials on the cancer preventive activity of these non-α-T VE forms, with relevant biomarker analyses, are even more important to provide more definitive answers on whether these VE forms can be used for cancer prevention, the mechanisms involved, and the doses required.
ACKNOWLEDGEMENTS
This work was supported by NIH Grants RO1 CA133021 and RO1 AT007036, as well as NCI Cancer Center support grant (P30 CA72720) and NIEHS Center grant (P30 ES005022). We thank Ms. Vi Dan for her important contribution in the preparation of this manuscript.
Abbreviations:
- α (β, γ or δ)-T
α (β, γ or δ)-tocopherol
- α (β, γ or δ)-T3
α (β, γ or δ)-tocotrienols
- α-TTP
α-T transfer protein
- ACF
aberrant crypt foci
- COX2
cyclooxygenase-2
- CRC
colorectal cancer
- CYP1A1
cytochrome P450 1A1
- DEGS
dihydroceramide desturase
- ER
estrogen receptor
- γ-H2AX
phosphorylated histone 2A variant X
- γ-TmT
γ-T rich mixture of tocopherols
- IHC
immunohistochemical
- KPC
triple transgenic LSL-KrasG12D
- LSL
Trp53R127H (mouse)
- Pdx-1-Cre
- LTB4
leukotriene B4
- MBNA
methylbenzylnitrosamine
- MMP
matrix metalloproteinases
- MNU
methylnitrosurea
- 8-oxo-dG
8-oxo-deoxyguanosine
- PDAC
pancreatic ductal carcinoma
- PGE2
prostaglandin E2
- PhIP
2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine
- PIN
prostatic intraepithelial neoplasia
- SNP
single nucleotide polymorphism
- VE
Vitamin E
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
References
- 1.Traber MG. Vitamin E regulatory mechanisms. Annu Rev Nutr. 2007;27:347–362. [DOI] [PubMed] [Google Scholar]
- 2.Ju J, Picinich SC, Yang Z, et al. Cancer-preventive activities of tocopherols and tocotrienols. Carcinogenesis. 2010;31(4):533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee IM, Cook NR, Gaziano JM, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. JAMA. 2005;294(1):56–65. [DOI] [PubMed] [Google Scholar]
- 4.Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2009;301(1):52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein EA, Thompson IM Jr., Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306(14):1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Constantinou C, Charalambous C, Kanakis D. Vitamin E and cancer: an update on the emerging role of gamma and delta tocotrienols. Eur J Nutr. 2019. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Q Natural forms of vitamin E and metabolites-regulation of cancer cell death and underlying mechanisms. IUBMB Life. 2019;71(4):495–506. [DOI] [PubMed] [Google Scholar]
- 9.Montagnani Marelli M, Marzagalli M, Fontana F, Raimondi M, Moretti RM, Limonta P. Anticancer properties of tocotrienols: A review of cellular mechanisms and molecular targets. J Cell Physiol. 2019;234(2):1147–1164. [DOI] [PubMed] [Google Scholar]
- 10.Zingg JM. Vitamin E: Regulatory Role on Signal Transduction. IUBMB Life. 2019;71(4):456–478. [DOI] [PubMed] [Google Scholar]
- 11.Zingg JM. Vitamin E: A Role in Signal Transduction. Annu Rev Nutr. 2015;35:135–173. [DOI] [PubMed] [Google Scholar]
- 12.Sailo BL, Banik K, Padmavathi G, Javadi M, Bordoloi D, Kunnumakkara AB. Tocotrienols: The promising analogues of vitamin E for cancer therapeutics. Pharmacol Res. 2018;130:259–272. [DOI] [PubMed] [Google Scholar]
- 13.Morrow JD, Awad JA, Kato T, et al. Formation of novel non-cyclooxygenase-derived prostanoids (F2-isoprostanes) in carbon tetrachloride hepatotoxicity. An animal model of lipid peroxidation. J Clin Invest. 1992;90(6):2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christen S, Jiang Q, Shigenaga MK, Ames BN. Analysis of plasma tocopherols alpha, gamma, and 5-nitro-gamma in rats with inflammation by HPLC coulometric detection. J Lipid Res. 2002;43(11):1978–1985. [DOI] [PubMed] [Google Scholar]
- 15.Bittrich H, Matzig AK, Kraker I, Appel KE. NO2-induced DNA single strand breaks are inhibited by antioxidative vitamins in V79 cells. Chem Biol Interact. 1993;86(3):199–211. [DOI] [PubMed] [Google Scholar]
- 16.Sen CK, Khanna S, Roy S. Tocotrienols in health and disease: the other half of the natural vitamin E family. Mol Aspects Med. 2007;28(5–6):692–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian J, Morley S, Wilson K, Nava P, Atkinson J, Manor D. Intracellular trafficking of vitamin E in hepatocytes: the role of tocopherol transfer protein. J Lipid Res. 2005;46(10):2072–2082. [DOI] [PubMed] [Google Scholar]
- 18.Morley S, Cecchini M, Zhang W, et al. Mechanisms of ligand transfer by the hepatic tocopherol transfer protein. J Biol Chem. 2008;283(26):17797–17804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sontag TJ, Parker RS. Cytochrome P450 omega-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J Biol Chem. 2002;277(28):25290–25296. [DOI] [PubMed] [Google Scholar]
- 20.Sontag TJ, Parker RS. Influence of major structural features of tocopherols and tocotrienols on their omega-oxidation by tocopherol-omega-hydroxylase. J Lipid Res. 2007;48(5):1090–1098. [DOI] [PubMed] [Google Scholar]
- 21.Swanson JE, Ben RN, Burton GW, Parker RS. Urinary excretion of 2,7, 8-trimethyl-2-(beta-carboxyethyl)-6-hydroxychroman is a major route of elimination of gamma-tocopherol in humans. J Lipid Res. 1999;40(4):665–671. [PubMed] [Google Scholar]
- 22.Chiku S, Hamamura K, Nakamura T. Novel urinary metabolite of d-delta-tocopherol in rats. J Lipid Res. 1984;25(1):40–48. [PubMed] [Google Scholar]
- 23.Parker RS, Swanson JE. A novel 5’-carboxychroman metabolite of gamma-tocopherol secreted by HepG2 cells and excreted in human urine. Biochem Biophys Res Commun. 2000;269(2):580–583. [DOI] [PubMed] [Google Scholar]
- 24.Ran L, Liu AB, Lee MJ, Xie P, Lin Y, Yang CS. Effects of antibiotics on degradation and bioavailability of different vitamin E forms in mice. Biofactors. 2019;45(3):450–462. [DOI] [PubMed] [Google Scholar]
- 25.Yang CS, Sun Y, Yang QU, et al. Vitamin A and other deficiencies in Linxian, a high esophageal cancer incidence area in northern China. J Natl Cancer Inst. 1984;73(6):1449–1453. [PubMed] [Google Scholar]
- 26.Blot WJ, Li JY, Taylor PR, et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst. 1993;85(18):1483–1492. [DOI] [PubMed] [Google Scholar]
- 27.Taylor PR, Qiao YL, Abnet CC, et al. Prospective study of serum vitamin E levels and esophageal and gastric cancers. J Natl Cancer Inst. 2003;95(18):1414–1416. [DOI] [PubMed] [Google Scholar]
- 28.Mark SD, Qiao YL, Dawsey SM, et al. Prospective study of serum selenium levels and incident esophageal and gastric cancers. J Natl Cancer Inst. 2000;92(21):1753–1763. [DOI] [PubMed] [Google Scholar]
- 29.Qiao YL, Dawsey SM, Kamangar F, et al. Total and cancer mortality after supplementation with vitamins and minerals: follow-up of the Linxian General Population Nutrition Intervention Trial. J Natl Cancer Inst. 2009;101(7):507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JY, Taylor PR, Li B, et al. Nutrition intervention trials in Linxian, China: multiple vitamin/mineral supplementation, cancer incidence, and disease-specific mortality among adults with esophageal dysplasia. J Natl Cancer Inst. 1993;85(18):1492–1498. [DOI] [PubMed] [Google Scholar]
- 31.Wang SM, Taylor PR, Fan JH, et al. Effects of Nutrition Intervention on Total and Cancer Mortality: 25-Year Post-trial Follow-up of the 5.25-Year Linxian Nutrition Intervention Trial. J Natl Cancer Inst. 2018;110(11):1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Fang J, Jia X, et al. Chemopreventive effects of early-stage and late-stage supplementation of vitamin E and selenium on esophageal carcinogenesis in rats maintained on a low vitamin E/selenium diet. Carcinogenesis. 2011;32(3):381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Virtamo J, Pietinen P, Huttunen JK, et al. Incidence of cancer and mortality following alpha-tocopherol and beta-carotene supplementation: a postintervention follow-up. JAMA. 2003;290(4):476–485. [DOI] [PubMed] [Google Scholar]
- 34.Weinstein SJ, Wright ME, Lawson KA, et al. Serum and dietary vitamin E in relation to prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1253–1259. [DOI] [PubMed] [Google Scholar]
- 35.Fleshner NE, Kapusta L, Donnelly B, et al. Progression from high-grade prostatic intraepithelial neoplasia to cancer: a randomized trial of combination vitamin-E, soy, and selenium. J Clin Oncol. 2011;29(17):2386–2390. [DOI] [PubMed] [Google Scholar]
- 36.Virtamo J, Taylor PR, Kontto J, et al. Effects of alpha-tocopherol and beta-carotene supplementation on cancer incidence and mortality: 18-year postintervention follow-up of the Alpha-tocopherol, Beta-carotene Cancer Prevention Study. Int J Cancer. 2014;135(1):178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Q, Christen S, Shigenaga MK, Ames BN. gamma-tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74(6):714–722. [DOI] [PubMed] [Google Scholar]
- 38.Campbell S, Stone W, Whaley S, Krishnan K. Development of gamma (gamma)-tocopherol as a colorectal cancer chemopreventive agent. Crit Rev Oncol Hematol. 2003;47(3):249–259. [DOI] [PubMed] [Google Scholar]
- 39.Hensley K, Benaksas EJ, Bolli R, et al. New perspectives on vitamin E: gamma-tocopherol and carboxyelthylhydroxychroman metabolites in biology and medicine. Free Radic Biol Med. 2004;36(1):1–15. [DOI] [PubMed] [Google Scholar]
- 40.Albanes D, Till C, Klein EA, et al. Plasma tocopherols and risk of prostate cancer in the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Cancer Prev Res (Phila). 2014;7(9):886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mondul AM, Rohrmann S, Menke A, et al. Association of serum alpha-tocopherol with sex steroid hormones and interactions with smoking: implications for prostate cancer risk. Cancer Causes Control. 2011;22(6):827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng TY, Barnett MJ, Kristal AR, et al. Genetic variation in myeloperoxidase modifies the association of serum alpha-tocopherol with aggressive prostate cancer among current smokers. J Nutr. 2011;141(9):1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinstein SJ, Peters U, Ahn J, et al. Serum alpha-tocopherol and gamma-tocopherol concentrations and prostate cancer risk in the PLCO Screening Trial: a nested case-control study. PLoS One. 2012;7(7):e40204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bauer SR, Richman EL, Sosa E, et al. Antioxidant and vitamin E transport genes and risk of high-grade prostate cancer and prostate cancer recurrence. Prostate. 2013;73(16):1786–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antwi SO, Steck SE, Su LJ, et al. Dietary, supplement, and adipose tissue tocopherol levels in relation to prostate cancer aggressiveness among African and European Americans: The North Carolina-Louisiana Prostate Cancer Project (PCaP). Prostate. 2015;75(13):1419–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Major JM, Yu K, Weinstein SJ, et al. Genetic variants reflecting higher vitamin e status in men are associated with reduced risk of prostate cancer. J Nutr. 2014;144(5):729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan JM, Darke AK, Penney KL, et al. Selenium- or Vitamin E-Related Gene Variants, Interaction with Supplementation, and Risk of High-Grade Prostate Cancer in SELECT. Cancer Epidemiol Biomarkers Prev. 2016;25(7):1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu QJ, Xiang YB, Yang G, et al. Vitamin E intake and the lung cancer risk among female nonsmokers: a report from the Shanghai Women’s Health Study. Int J Cancer. 2015;136(3):610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang J, Weinstein SJ, Yu K, Mannisto S, Albanes D. A Prospective Study of Serum Vitamin E and 28-Year Risk of Lung Cancer. J Natl Cancer Inst. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narita S, Saito E, Sawada N, et al. Dietary consumption of antioxidant vitamins and subsequent lung cancer risk: The Japan Public Health Center-based prospective study. Int J Cancer. 2018;142(12):2441–2460. [DOI] [PubMed] [Google Scholar]
- 51.Chen G, Wang J, Hong X, Chai Z, Li Q. Dietary vitamin E intake could reduce the risk of lung cancer: evidence from a meta-analysis. Int J Clin Exp Med. 2015;8(4):6631–6637. [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu YJ, Bo YC, Liu XX, Qiu CG. Association of dietary vitamin E intake with risk of lung cancer: a dose-response meta-analysis. Asia Pac J Clin Nutr. 2017;26(2):271–277. [DOI] [PubMed] [Google Scholar]
- 53.Banim PJ, Luben R, McTaggart A, et al. Dietary antioxidants and the aetiology of pancreatic cancer: a cohort study using data from food diaries and biomarkers. Gut. 2013;62(10):1489–1496. [DOI] [PubMed] [Google Scholar]
- 54.Jansen RJ, Robinson DP, Stolzenberg-Solomon RZ, et al. Nutrients from fruit and vegetable consumption reduce the risk of pancreatic cancer. J Gastrointest Cancer. 2013;44(2):152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li D, Tang H, Wei P, Zheng J, Daniel CR, Hassan MM. Vitamin C and Vitamin E Mitigate the Risk of Pancreatic Ductal Adenocarcinoma from Meat-Derived Mutagen Exposure in Adults in a Case-Control Study. J Nutr. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peng L, Liu X, Lu Q, Tang T, Yang Z. Vitamin E intake and pancreatic cancer risk: a meta-analysis of observational studies. Med Sci Monit. 2015;21:1249–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen JM, Jiang WX, Shao LM, Zhong DD, Wu YH, Cai JT. Association between intake of antioxidants and pancreatic cancer risk: a meta-analysis. Int J Food Sci Nutr. 2016;67(7):744–753. [DOI] [PubMed] [Google Scholar]
- 58.Lotan Y, Goodman PJ, Youssef RF, et al. Evaluation of vitamin E and selenium supplementation for the prevention of bladder cancer in SWOG coordinated SELECT. J Urol. 2012;187(6):2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park SY, Ollberding NJ, Woolcott CG, Wilkens LR, Henderson BE, Kolonel LN. Fruit and vegetable intakes are associated with lower risk of bladder cancer among women in the Multiethnic Cohort Study. J Nutr. 2013;143(8):1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang YY, Wang XL, Yu ZJ. Vitamin C and E intake and risk of bladder cancer: a meta-analysis of observational studies. Int J Clin Exp Med. 2014;7(11):4154–4164. [PMC free article] [PubMed] [Google Scholar]
- 61.Lin JH, Chen SJ, Liu H, Yan Y, Zheng JH. Vitamin E consumption and the risk of bladder cancer. Int J Vitam Nutr Res. 2019:1–8. [DOI] [PubMed] [Google Scholar]
- 62.Chen F, Li Q, Yu Y, Yang W, Shi F, Qu Y. Association of vitamin C, vitamin D, vitamin E and risk of bladder cancer: a dose-response meta-analysis. Sci Rep. 2015;5:9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cui R, Liu ZQ, Xu Q. Blood alpha-tocopherol, gamma-tocopherol levels and risk of prostate cancer: a meta-analysis of prospective studies. PLoS One. 2014;9(3):e93044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Z, Joshi AM, Ohnaka K, et al. Dietary intakes of retinol, carotenes, vitamin C, and vitamin E and colorectal cancer risk: the Fukuoka colorectal cancer study. Nutr Cancer. 2012;64(6):798–805. [DOI] [PubMed] [Google Scholar]
- 65.Kabat GC, Kim MY, Sarto GE, Shikany JM, Rohan TE. Repeated measurements of serum carotenoid, retinol and tocopherol levels in relation to colorectal cancer risk in the Women’s Health Initiative. Eur J Clin Nutr. 2012;66(5):549–554. [DOI] [PubMed] [Google Scholar]
- 66.Lance P, Alberts DS, Thompson PA, et al. Colorectal Adenomas in Participants of the SELECT Randomized Trial of Selenium and Vitamin E for Prostate Cancer Prevention. Cancer Prev Res (Phila). 2017;10(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo H, Fang YJ, Lu MS, et al. Dietary and serum vitamins A and E and colorectal cancer risk in Chinese population: a case-control study. Eur J Cancer Prev. 2019;28(4):268–277. [DOI] [PubMed] [Google Scholar]
- 68.Egnell M, Fassier P, Lecuyer L, et al. Antioxidant intake from diet and supplements and risk of digestive cancers in middle-aged adults: results from the prospective NutriNet-Sante cohort. Br J Nutr. 2017;118(7):541–549. [DOI] [PubMed] [Google Scholar]
- 69.Wang J, Guo H, Lin T, et al. A Nested Case-Control Study on Plasma Vitamin E and Risk of Cancer: Evidence of Effect Modification by Selenium. J Acad Nutr Diet. 2019;119(5):769–781. [DOI] [PubMed] [Google Scholar]
- 70.Edefonti V, Hashibe M, Parpinel M, et al. Vitamin E intake from natural sources and head and neck cancer risk: a pooled analysis in the International Head and Neck Cancer Epidemiology consortium. Br J Cancer. 2015;113(1):182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cui L, Li L, Tian Y, Xu F, Qiao T. Association between Dietary Vitamin E Intake and Esophageal Cancer Risk: An Updated Meta-Analysis. Nutrients. 2018;10(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo L, Zhu H, Lin C, et al. Associations between antioxidant vitamins and the risk of invasive cervical cancer in Chinese women: A case-control study. Sci Rep. 2015;5:13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu X, Li S, Zhou L, Zhao M, Zhu X. Effect of vitamin E supplementation on uterine cervical neoplasm: A meta-analysis of case-control studies. PLoS One. 2017;12(8):e0183395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu F, Wu Z, Li G, et al. The plasma level of retinol, vitamins A, C and alpha-tocopherol could reduce breast cancer risk? A meta-analysis and meta-regression. J Cancer Res Clin Oncol. 2015;141(4):601–614. [DOI] [PubMed] [Google Scholar]
- 75.Shang Y, Yi S, Cui D, Han G, Liu C. Vitamin E Intake and Risk of Renal Cell Carcinoma: A Meta-Analysis of 7 Case-Control Studies. J Ren Nutr. 2015;25(4):339–344. [DOI] [PubMed] [Google Scholar]
- 76.Shen C, Huang Y, Yi S, Fang Z, Li L. Association of Vitamin E Intake with Reduced Risk of Kidney Cancer: A Meta-Analysis of Observational Studies. Med Sci Monit. 2015;21:3420–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang W, Shu XO, Li H, et al. Vitamin intake and liver cancer risk: a report from two cohort studies in China. J Natl Cancer Inst. 2012;104(15):1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai GY, Weinstein SJ, Albanes D, et al. Association of serum alpha-tocopherol, beta-carotene, and retinol with liver cancer incidence and chronic liver disease mortality. Br J Cancer. 2014;111(11):2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hotaling JM, Wright JL, Pocobelli G, Bhatti P, Porter MP, White E. Long-term use of supplemental vitamins and minerals does not reduce the risk of urothelial cell carcinoma of the bladder in the VITamins And Lifestyle study. J Urol. 2011;185(4):1210–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Psaltopoulou T, Ntanasis-Stathopoulos I, Tsilimigras DI, Tzanninis IG, Gavriatopoulou M, Sergentanis TN. Micronutrient Intake and Risk of Hematological Malignancies in Adults: A Systematic Review and Meta-analysis of Cohort Studies . Nutr Cancer. 2018;70(6):821–839. [DOI] [PubMed] [Google Scholar]
- 81.Bates CJ, Hamer M, Mishra GD. Redox-modulatory vitamins and minerals that prospectively predict mortality in older British people: the National Diet and Nutrition Survey of people aged 65 years and over. Br J Nutr. 2011;105(1):123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma E, Iso H, Yamagishi K, Ando M, Wakai K, Tamakoshi A. Dietary Antioxidant Micronutrients and All-Cause Mortality: The Japan Collaborative Cohort Study for Evaluation of Cancer Risk . J Epidemiol. 2018;28(9):388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L, Sesso HD, Glynn RJ, et al. Vitamin E and C supplementation and risk of cancer in men: posttrial follow-up in the Physicians’ Health Study II randomized trial. Am J Clin Nutr. 2014;100(3):915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Springett GM, Husain K, Neuger A, et al. A Phase I Safety, Pharmacokinetic, and Pharmacodynamic Presurgical Trial of Vitamin E delta-tocotrienol in Patients with Pancreatic Ductal Neoplasia. EBioMedicine. 2015;2(12):1987–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mahipal A, Klapman J, Vignesh S, et al. Pharmacokinetics and safety of vitamin E delta-tocotrienol after single and multiple doses in healthy subjects with measurement of vitamin E metabolites. Cancer Chemother Pharmacol. 2016;78(1):157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nesaretnam K, Selvaduray KR, Abdul Razak G, Veerasenan SD, Gomez PA. Effectiveness of tocotrienol-rich fraction combined with tamoxifen in the management of women with early breast cancer: a pilot clinical trial. Breast Cancer Res. 2010;12(5):R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thomsen CB, Andersen RF, Steffensen KD, Adimi P, Jakobsen A. Delta tocotrienol in recurrent ovarian cancer. A phase II trial. Pharmacol Res. 2019;141:392–396. [DOI] [PubMed] [Google Scholar]
- 88.Aune D, Keum N, Giovannucci E, et al. Dietary intake and blood concentrations of antioxidants and the risk of cardiovascular disease, total cancer, and all-cause mortality: a systematic review and dose-response meta-analysis of prospective studies. Am J Clin Nutr. 2018;108(5):1069–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barve A, Khor TO, Nair S, et al. Gamma-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in TRAMP mice. Int J Cancer. 2009;124(7):1693–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takahashi S, Takeshita K, Seeni A, et al. Suppression of prostate cancer in a transgenic rat model via gamma-tocopherol activation of caspase signaling. Prostate. 2009;69(6):644–651. [DOI] [PubMed] [Google Scholar]
- 91.Sanches LD, Santos SA, Carvalho JR, et al. Protective effect of gamma-tocopherol-enriched diet on N-methyl-N-nitrosourea-induced epithelial dysplasia in rat ventral prostate. Int J Exp Pathol. 2013;94(6):362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McCormick DL, Rao KV, Johnson WD, Bosland MC, Lubet RA, Steele VE. Null activity of selenium and vitamin e as cancer chemopreventive agents in the rat prostate. Cancer Prev Res (Phila). 2010;3(3):381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen JX, Li G, Wang H, et al. Dietary tocopherols inhibit PhIP-induced prostate carcinogenesis in CYP1A-humanized mice. Cancer Lett. 2016;371(1):71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang H, He Y, Cui XX, et al. Potent inhibitory effect of delta-tocopherol on prostate cancer cells cultured in vitro and grown as xenograft tumors in vivo. J Agric Food Chem. 2014;62(44):10752–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang H, Hong J, Yang CS. delta-Tocopherol inhibits receptor tyrosine kinase-induced AKT activation in prostate cancer cells. Mol Carcinog. 2016;55(11):1728–1738. [DOI] [PubMed] [Google Scholar]
- 96.Wang H, Yang X, Liu A, Wang G, Bosland MC, Yang CS. delta-Tocopherol inhibits the development of prostate adenocarcinoma in prostate specific Pten−/− mice. Carcinogenesis. 2018;39(2):158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smolarek AK, So JY, Burgess B, et al. Dietary administration of delta- and gamma-tocopherol inhibits tumorigenesis in the animal model of estrogen receptor-positive, but not HER-2 breast cancer. Cancer Prev Res (Phila). 2012;5(11):1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smolarek AK, So JY, Thomas PE, et al. Dietary tocopherols inhibit cell proliferation, regulate expression of ERalpha, PPARgamma, and Nrf2, and decrease serum inflammatory markers during the development of mammary hyperplasia. Mol Carcinog. 2013;52(7):514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Das Gupta S, Sae-tan S, Wahler J, et al. Dietary gamma-Tocopherol-Rich Mixture Inhibits Estrogen-Induced Mammary Tumorigenesis by Modulating Estrogen Metabolism, Antioxidant Response, and PPARgamma. Cancer Prev Res (Phila). 2015;8(9):807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Das Gupta S, So JY, Wall B, et al. Tocopherols inhibit oxidative and nitrosative stress in estrogen-induced early mammary hyperplasia in ACI rats. Mol Carcinog. 2015;54(9):916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bak MJ, Das Gupta S, Wahler J, et al. Inhibitory Effects of gamma- and delta-Tocopherols on Estrogen-Stimulated Breast Cancer In Vitro and In Vivo. Cancer Prev Res (Phila). 2017;10(3):188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Das Gupta S, Patel M, Wahler J, et al. Differential Gene Regulation and Tumor-Inhibitory Activities of Alpha-, Delta-, and Gamma-Tocopherols in Estrogen-Mediated Mammary Carcinogenesis. Cancer Prev Res (Phila). 2017;10(12):694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bak MJ, Furmanski P, Shan NL, et al. Tocopherols inhibit estrogen-induced cancer stemness and OCT4 signaling in breast cancer. Carcinogenesis. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ju J, Hao X, Lee MJ, et al. A gamma-tocopherol-rich mixture of tocopherols inhibits colon inflammation and carcinogenesis in azoxymethane and dextran sulfate sodium-treated mice. Cancer Prev Res (Phila). 2009;2(2):143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiang Q, Jiang ZY, Jones-Hall Y, et al. Gamma-tocopherol attenuates moderate but not severe colitis and suppresses moderate colitis-promoted colon tumorigenesis in mice. Free Radical Bio Med. 2013;65:1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cohen C, Cardoso JF, Garcia SB, Vannucchi H. Vitamin E supplementation in chemical colorectal carcinogenesis: a two-edged knife. Nutrients. 2014;6(8):3214–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guan F, Li G, Liu AB, et al. delta- and gamma-tocopherols, but not alpha-tocopherol, inhibit colon carcinogenesis in azoxymethane-treated F344 rats. Cancer Prev Res (Phila). 2012;5(4):644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen JX, Liu A, Lee MJ, et al. delta- and gamma-tocopherols inhibit phIP/DSS-induced colon carcinogenesis by protection against early cellular and DNA damages. Mol Carcinog. 2017;56(1):172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dolfi SC, Yang Z, Lee MJ, Guan F, Hong J, Yang CS. Inhibitory effects of different forms of tocopherols, tocopherol phosphates, and tocopherol quinones on growth of colon cancer cells. J Agric Food Chem. 2013;61(36):8533–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu G, Xiao H, Li GX, et al. A gamma-tocopherol-rich mixture of tocopherols inhibits chemically induced lung tumorigenesis in A/J mice and xenograft tumor growth. Carcinogenesis. 2010;31(4):687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lambert JD, Lu G, Lee MJ, Hu J, Ju J, Yang CS. Inhibition of lung cancer growth in mice by dietary mixed tocopherols. Mol Nutr Food Res. 2009;53(8):1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim Y, Chongviriyaphan N, Liu C, Russell RM, Wang XD. Combined alpha-tocopherol and ascorbic acid protects against smoke-induced lung squamous metaplasia in ferrets. Lung Cancer. 2012;75(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li XC, Lu XL, Chen HH. alpha-Tocopherol treatment ameliorates chronic pancreatitis in an experimental rat model induced by trinitrobenzene sulfonic acid. Pancreatology. 2011;11(1):5–11. [DOI] [PubMed] [Google Scholar]
- 114.Yang H, Xu M, Lu F, et al. Tocopherols inhibit esophageal carcinogenesis through attenuating NF-kappaB activation and CXCR3-mediated inflammation. Oncogene. 2018;37(29):3909–3923. [DOI] [PubMed] [Google Scholar]
- 115.Tan B, Watson RR, Preedy VR. Tocotrienols : vitamin E beyond tocopherols. 2016. [Google Scholar]
- 116.Zhang JS, Li DM, He N, et al. A paraptosis-like cell death induced by delta-tocotrienol in human colon carcinoma SW620 cells is associated with the suppression of the Wnt signaling pathway. Toxicology. 2011;285(1–2):8–17. [DOI] [PubMed] [Google Scholar]
- 117.Xu W, Du M, Zhao Y, Wang Q, Sun W, Chen B. gamma-Tocotrienol inhibits cell viability through suppression of beta-catenin/Tcf signaling in human colon carcinoma HT-29 cells. J Nutr Biochem. 2012;23(7):800–807. [DOI] [PubMed] [Google Scholar]
- 118.Zhang JS, Li DM, Ma Y, et al. gamma-Tocotrienol induces paraptosis-like cell death in human colon carcinoma SW620 cells. PLoS One. 2013;8(2):e57779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ahmed RA, Alawin OA, Sylvester PW. gamma-Tocotrienol reversal of epithelial-to-mesenchymal transition in human breast cancer cells is associated with inhibition of canonical Wnt signalling. Cell Prolif. 2016;49(4):460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang JS, Zhang SJ, Li Q, et al. Tocotrienol-rich fraction (TRF) suppresses the growth of human colon cancer xenografts in Balb/C nude mice by the Wnt pathway. PLoS One. 2015;10(3):e0122175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kani K, Momota Y, Harada M, et al. γ-tocotrienol enhances the chemosensitivity of human oral cancer cells to docetaxel through the downregulation of the expression of NF-κB-regulated anti-apoptotic gene products. International journal of oncology. 2013;42(1):75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang C, Jiang Q. Vitamin E delta-tocotrienol inhibits TNF-alpha-stimulated NF-kappaB activation by up-regulation of anti-inflammatory A20 via modulation of sphingolipid including elevation of intracellular dihydroceramides. J Nutr Biochem. 2019;64:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rajasinghe LD, Pindiprolu RH, Gupta SV. Delta-tocotrienol inhibits non-small-cell lung cancer cell invasion via the inhibition of NF-kappaB, uPA activator, and MMP-9. OncoTargets and therapy. 2018;11:4301–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ji X, Wang Z, Sarkar FH, Gupta SV. Delta-tocotrienol augments cisplatin-induced suppression of non-small cell lung cancer cells via inhibition of the Notch-1 pathway. Anticancer Res. 2012;32(7):2647–2655. [PubMed] [Google Scholar]
- 125.Ji X, Wang Z, Geamanu A, Goja A, Sarkar FH, Gupta SV. Delta‐tocotrienol suppresses Notch‐1 pathway by upregulating miR‐34a in nonsmall cell lung cancer cells. International journal of cancer. 2012;131(11):2668–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ji X, Wang Z, Geamanu A, Sarkar FH, Gupta SV. Inhibition of cell growth and induction of apoptosis in non‐small cell lung cancer cells by delta‐tocotrienol is associated with notch‐1 down‐regulation. Journal of cellular biochemistry. 2011;112(10):2773–2783. [DOI] [PubMed] [Google Scholar]
- 127.Rajasinghe LD, Hutchings M, Gupta SV. Delta-Tocotrienol Modulates Glutamine Dependence by Inhibiting ASCT2 and LAT1 Transporters in Non-Small Cell Lung Cancer (NSCLC) Cells: A Metabolomic Approach. Metabolites. 2019;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Siveen KS, Ahn KS, Ong TH, et al. Y-tocotrienol inhibits angiogenesis-dependent growth of human hepatocellular carcinoma through abrogation of AKT/mTOR pathway in an orthotopic mouse model. Oncotarget. 2014;5(7):1897–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ye C, Zhao W, Li M, et al. Δ-Tocotrienol induces human bladder cancer cell growth arrest, apoptosis and chemosensitization through inhibition of stat3 pathway. PloS one. 2015;10(4):e0122712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sugahara R, UCHIDA A, SHIOZAWA S, VIRGONA N, Tomohiro Y. Annatto tocotrienol induces a cytotoxic effect on human prostate cancer PC3 cells via the simultaneous inhibition of Src and Stat3. J Nutr Sci Vitaminol. 2015;61(6):497–501. [DOI] [PubMed] [Google Scholar]
- 131.Liu J, Lau EY, Chen J, et al. Polysaccharopeptide enhanced the anti-cancer effect of gamma-tocotrienol through activation of AMPK. BMC Complement Altern Med. 2014;14:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Alawin OA, Ahmed RA, Dronamraju V, Briski KP, Sylvester PW. gamma-Tocotrienol-induced disruption of lipid rafts in human breast cancer cells is associated with a reduction in exosome heregulin content. J Nutr Biochem. 2017;48:83–93. [DOI] [PubMed] [Google Scholar]
- 133.Alawin OA, Ahmed RA, Ibrahim BA, Briski KP, Sylvester PW. Antiproliferative effects of γ-tocotrienol are associated with lipid raft disruption in HER2-positive human breast cancer cells. The Journal of nutritional biochemistry. 2016;27:266–277. [DOI] [PubMed] [Google Scholar]
- 134.Wang C, Husain K, Zhang A, et al. EGR-1/Bax pathway plays a role in vitamin E δ-tocotrienol-induced apoptosis in pancreatic cancer cells. The Journal of nutritional biochemistry. 2015;26(8):797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang C, Ju H, Shen C, Tong Z. miR-429 mediates delta-tocotrienol-induced apoptosis in triple-negative breast cancer cells by targeting XIAP. Int J Clin Exp Med. 2015;8(9):15648–15656. [PMC free article] [PubMed] [Google Scholar]
- 136.Hodul PJ, Dong Y, Husain K, et al. Vitamin E δ-tocotrienol induces p27 Kip1-dependent cell-cycle arrest in pancreatic cancer cells via an E2F-1-dependent mechanism. PloS one. 2013;8(2):e52526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Campbell SE, Rudder B, Phillips RB, et al. γ-Tocotrienol induces growth arrest through a novel pathway with TGFβ2 in prostate cancer. Free Radical Bio Med. 2011;50(10):1344–1354. [DOI] [PubMed] [Google Scholar]
- 138.Inoue A, Takitani K, Koh M, Kawakami C, Kuno T, Tamai H. Induction of apoptosis by gamma-tocotrienol in human cancer cell lines and leukemic blasts from patients: dependency on Bid, cytochrome c, and caspase pathway. Nutr Cancer. 2011;63(5):763–770. [DOI] [PubMed] [Google Scholar]
- 139.Lim SW, Loh HS, Ting KN, Bradshaw TD, Zeenathul NA. Antiproliferation and induction of caspase-8-dependent mitochondria-mediated apoptosis by beta-tocotrienol in human lung and brain cancer cell lines. Biomed Pharmacother. 2014;68(8):1105–1115. [DOI] [PubMed] [Google Scholar]
- 140.Lim SW, Loh HS, Ting KN, Bradshaw TD, Zeenathul NA. Cytotoxicity and apoptotic activities of alpha-, gamma- and delta-tocotrienol isomers on human cancer cells. BMC Complement Altern Med. 2014;14:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jang Y, Rao X, Jiang Q. Gamma-tocotrienol profoundly alters sphingolipids in cancer cells by inhibition of dihydroceramide desaturase and possibly activation of sphingolipid hydrolysis during prolonged treatment. J Nutr Biochem. 2017;46:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jang Y, Park NY, Rostgaard-Hansen AL, Huang J, Jiang Q. Vitamin E metabolite 13’-carboxychromanols inhibit pro-inflammatory enzymes, induce apoptosis and autophagy in human cancer cells by modulating sphingolipids and suppress colon tumor development in mice. Free Radic Biol Med. 2016;95:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Patacsil D, Tran AT, Cho YS, et al. Gamma-tocotrienol induced apoptosis is associated with unfolded protein response in human breast cancer cells. J Nutr Biochem. 2012;23(1):93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tiwari RV, Parajuli P, Sylvester PW. gamma-Tocotrienol-induced autophagy in malignant mammary cancer cells. Exp Biol Med (Maywood). 2014;239(1):33–44. [DOI] [PubMed] [Google Scholar]
- 145.Fontana F, Moretti RM, Raimondi M, et al. delta-Tocotrienol induces apoptosis, involving endoplasmic reticulum stress and autophagy, and paraptosis in prostate cancer cells. Cell Prolif. 2019;52(3):e12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tiwari RV, Parajuli P, Sylvester PW. gamma-Tocotrienol-induced endoplasmic reticulum stress and autophagy act concurrently to promote breast cancer cell death. Biochem Cell Biol. 2015;93(4):306–320. [DOI] [PubMed] [Google Scholar]
- 147.Marzagalli M, Moretti RM, Messi E, et al. Targeting melanoma stem cells with the Vitamin E derivative delta-tocotrienol. Sci Rep. 2018;8(1):587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Husain K, Centeno BA, Coppola D, Trevino J, Sebti SM, Malafa MP. δ-Tocotrienol, a natural form of vitamin E, inhibits pancreatic cancer stem-like cells and prevents pancreatic cancer metastasis. Oncotarget. 2017;8(19):31554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gopalan A, Yu W, Sanders BG, Kline K. Eliminating drug resistant breast cancer stem-like cells with combination of simvastatin and gamma-tocotrienol. Cancer letters. 2013;328(2):285–296. [DOI] [PubMed] [Google Scholar]
- 150.Manu KA, Shanmugam MK, Ramachandran L, et al. First evidence that gamma-tocotrienol inhibits the growth of human gastric cancer and chemosensitizes it to capecitabine in a xenograft mouse model through the modulation of NF-kappaB pathway. Clin Cancer Res. 2012;18(8):2220–2229. [DOI] [PubMed] [Google Scholar]
- 151.Hafid SRA, Chakravarthi S, Nesaretnam K, Radhakrishnan AK. Tocotrienol-adjuvanted dendritic cells inhibit tumor growth and metastasis: a murine model of breast cancer. PloS one. 2013;8(9):e74753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Husain K, Centeno BA, Chen D-T, Hingorani SR, Sebti SM, Malafa MP. Vitamin E δ-tocotrienol prolongs survival in the LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre (KPC) transgenic mouse model of pancreatic cancer. Cancer Prev Res. 2013;6(10):1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Algayadh IG, Dronamraju V, Sylvester PW. Role of Rac1/WAVE2 Signaling in Mediating the Inhibitory Effects of γ-Tocotrienol on Mammary Cancer Cell Migration and Invasion. Biological and Pharmaceutical Bulletin. 2016;39(12):1974–1982. [DOI] [PubMed] [Google Scholar]
- 154.Husain K, Zhang A, Shivers S, et al. Chemoprevention of Azoxymethane-induced Colon Carcinogenesis by Delta-Tocotrienol. Cancer Prev Res (Phila). 2019;12(6):357–366. [DOI] [PubMed] [Google Scholar]
- 155.Shibata A, Nakagawa K, Tsuduki T, Miyazawa T. delta-Tocotrienol treatment is more effective against hypoxic tumor cells than normoxic cells: potential implications for cancer therapy. J Nutr Biochem. 2015;26(8):832–840. [DOI] [PubMed] [Google Scholar]
- 156.Li Y, Sun W-G, Liu H-K, et al. γ-Tocotrienol inhibits angiogenesis of human umbilical vein endothelial cell induced by cancer cell. The Journal of nutritional biochemistry. 2011;22(12):1127–1136. [DOI] [PubMed] [Google Scholar]
- 157.Tang KD, Liu J, Russell PJ, Clements JA, Ling MT. Gamma-Tocotrienol Induces Apoptosis in Prostate Cancer Cells by Targeting the Ang-1/Tie-2 Signalling Pathway. Int J Mol Sci. 2019;20(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Akl MR, Ayoub NM, Abuasal BS, Kaddoumi A, Sylvester PW. Sesamin synergistically potentiates the anticancer effects of gamma-tocotrienol in mammary cancer cell lines. Fitoterapia. 2013;84:347–359. [DOI] [PubMed] [Google Scholar]
- 159.Eitsuka T, Tatewaki N, Nishida H, Kurata T, Nakagawa K, Miyazawa T. Synergistic inhibition of cancer cell proliferation with a combination of δ-tocotrienol and ferulic acid. Biochemical and biophysical research communications. 2014;453(3):606–611. [DOI] [PubMed] [Google Scholar]
- 160.Yusof KM, Makpol S, Jamal R, Harun R, Mokhtar N, Ngah WZ. gamma-Tocotrienol and 6-Gingerol in Combination Synergistically Induce Cytotoxicity and Apoptosis in HT-29 and SW837 Human Colorectal Cancer Cells. Molecules. 2015;20(6):10280–10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sato C, Kaneko S, Sato A, Virgona N, Namiki K, Yano T. Combination Effect of delta-Tocotrienol and gamma-Tocopherol on Prostate Cancer Cell Growth. J Nutr Sci Vitaminol (Tokyo). 2017;63(5):349–354. [DOI] [PubMed] [Google Scholar]
- 162.Song BL, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J Biol Chem. 2006;281(35):25054–25061. [DOI] [PubMed] [Google Scholar]
- 163.Serbinova E, Kagan V, Han D, Packer L. Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radic Biol Med. 1991;10(5):263–275. [DOI] [PubMed] [Google Scholar]
- 164.Takahashi O Haemorrhagic toxicity of a large dose of alpha-, beta-, gamma- and delta-tocopherols, ubiquinone, beta-carotene, retinol acetate and L-ascorbic acid in the rat. Food Chem Toxicol. 1995;33(2):121–128. [DOI] [PubMed] [Google Scholar]
- 165.Shen CL, Wang S, Yang S, et al. A 12-week evaluation of annatto tocotrienol supplementation for postmenopausal women: safety, quality of life, body composition, physical activity, and nutrient intake. BMC Complement Altern Med. 2018;18(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]