

SUMMARY
Genes mutated in human neuronal migration disorders encode tubulin proteins and a variety of tubulin-binding and -regulating proteins have been identified, yet it is very poorly understood how these proteins function together to coordinate migration. Additionally, the way in which regional differences in neocortical migration is controlled is completely unknown. Here we describe a new syndrome with remarkably region-specific effects on neuronal migration in the posterior cortex, reflecting de novo variants in CEP85L. We show that CEP85L is required cell-autonomously in vivo and in vitro for migration, that it localizes to the maternal centriole, and that it forms a complex with many other proteins required for migration, including CDK5, LIS1, NDE1, KIF2A and DYNC1H1. Loss of CEP85L disrupts CDK5 localization and activation leading to centrosome disorganization and disrupted microtubule cytoskeletal organization. Together, our findings suggest that CEP85L highlights a complex that controls CDK5 activity to promote neuronal migration.
eTOC blurb:
Neuronal migration is essential for brain architecture during neurodevelopment. Kodani et al. demonstrate that the pachygyria gene, CEP85L is required to organize the centrosome and microtubule cytoskeleton to promote coordinated neuronal migration by activating the neuronal kinase, CDK5 at the centrosome.
INTRODUCTION
The orderly migration of neurons from the ventricular zone to the developing cerebral cortex is critical for the laminar organization of the cortex (Rakic, 1971), and disruptions to neuronal migration underlies the pathogenesis of lissencephaly (LIS), a disorder characterized by a reduction in cortical brain folds, with patients exhibiting a range of cognitive and motor defects (Di Donato et al., 2017). More than a dozen genes for neuronal migration disorders have been identified, with many of them encoding centrosomal proteins required for microtubule cytoskeletal organization (Di Donato et al., 2018), but many cases still remain unexplained. Moreover, how the LIS-associated proteins interact and organize at the centrosome is largely unknown. Here we describe a strikingly novel condition reflecting mutations in CEP85L, that causes posteriorly restricted pachygyria (reduced, coarse cerebral cortical folds) due to disrupted centrosome and microtubule cytoskeletal organization and we show that CEP85L represents a critical organizational link between many other centrosomal LIS-associated proteins.
RESULTS
De novo variants in CEP85L cause posterior specific pachygyria
Whole exome sequencing and targeted sequencing of a cohort of families with variable pachygyria identified seven individuals with variants in the CEP85L gene with a strikingly similar radiographic and clinical phenotype (Fig 1A and Table 1). The cortical malformation in all cases included bilateral posterior-predominant pachygyria consisting of a thin cortex, a thin cell-sparse zone underlying the cortex, and a thickened subcortical band, involving parietal, occipital and temporal lobes but completely sparing the cortex rostral to the central sulcus (Fig 1B and Video 1–2). All seven affected individuals had decreased white matter and a dysmorphic corpus callosum. Two of the individuals also exhibited Chiari I malformations. All exhibited developmental delay or intellectual disability but have learned to walk. Affected individuals suffered from seizures, either focal or epileptic spasms. Despite the dramatic posterior malformation, cortical visual impairment is not noted. Four individuals exhibited strabismus (three had esotropia, and one had exotropia) and one individual had convergence insufficiency. All individuals had head circumferences in the normal range and did not display consistent dysmorphic facial features.
Figure 1: Variants in CEP85L cause posterior-specific pachygyria.
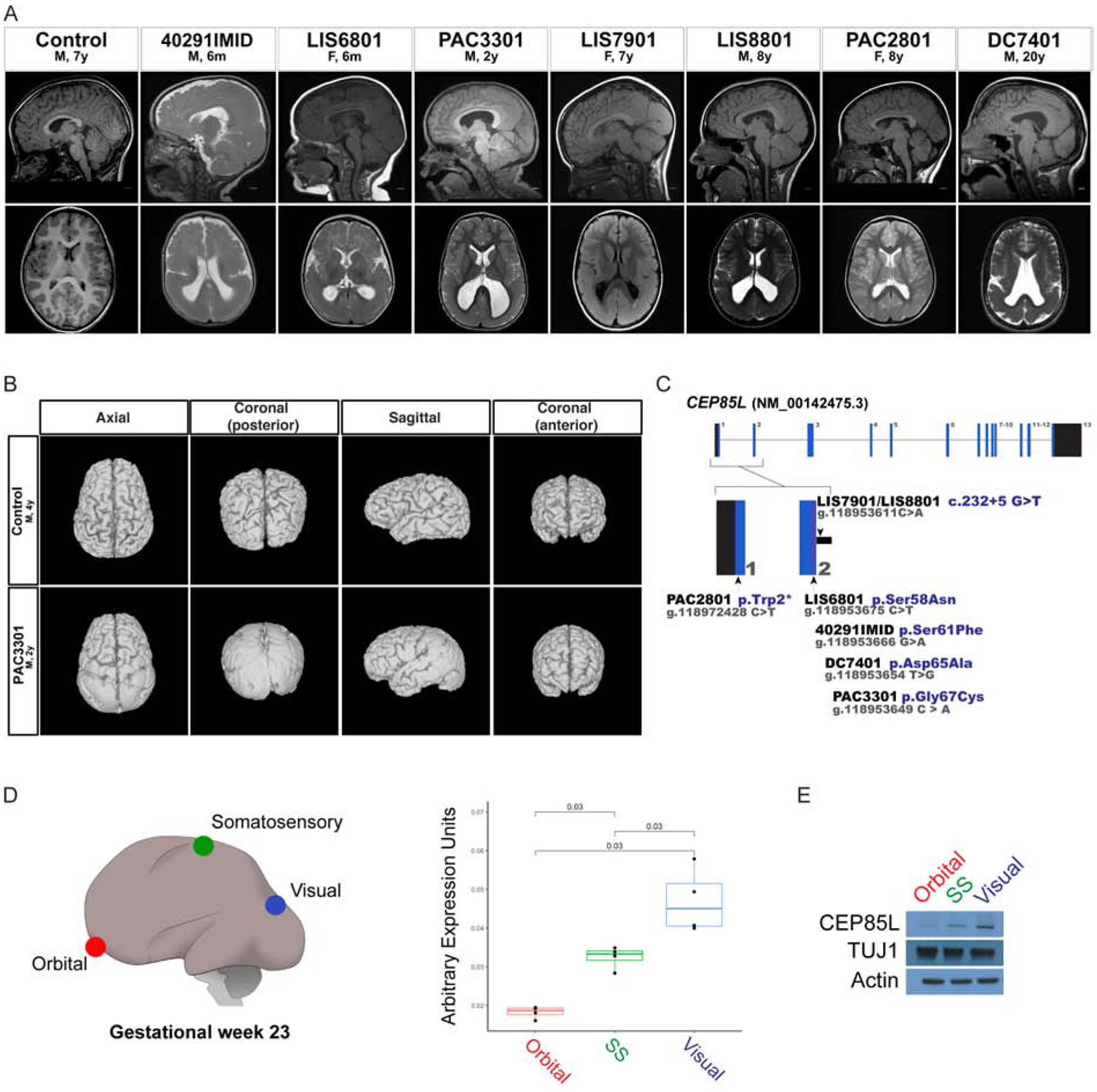
A. Sagittal and axial plane MRI images of a control and affected individuals with posterior reduced gyral folding. B. Three-dimensional MRI presentation of a control and PAC3301 patient with a de novo CEP85L variant. C. Schematic representation of exons of CEP85L shown as blue bars. The variants in CEP85L are found in exons 1 and 2. D. Brain region-specific qPCR of gestational week 23 cortex (GW), demonstrating the increasing rostral-to-caudal expression pattern of CEP85L normalized to β-actin. Orbital (red), somatosensory (green), and visual (blue) cortex. For quantifications, one brain region was analyzed in triplicate or quadruplicate (n=1). p < 0.03 (Student T-test). E. Whole cell lysate from the posterior frontal, parietal and occipital lobes of a GW 23 fetus blotted for CEP85L and the lissencephaly-associated protein, LIS1. Actin and TUJ1 served as a loading control and neuron-specific sampling control, respectively.
We initially identified three subjects (PAC2801, DC7401, LIS6801) from whole exome sequencing (WES) of 36 unrelated families with lissencephaly, pachygyria, or subcortical band heterotopia who lacked pathogenic variants in known lissencephaly genes (n=3/36). Two additional variants (LIS8801 and PAC3301) were identified by targeted sequencing of CEP85L in 11 individuals with posterior predominant lissencephaly. WES of LIS8801 and PAC3301 was performed to rule out other disease-causing variants. The two remaining individuals with CEP85L variants were identified using GeneMatcher and had been discovered by trio exome sequencing (40291IMID and LIS7901).
Four individuals carry missense variants in exon 2 of CEP85L, and three of these were confirmed to be de novo (c.182C>T, p.Ser61Phe; c.194A>C, p.Asp65Ala; c.199G>T, p.Gly67Cys). Parental samples were unavailable for LIS6801 (c.173G>A, p.Ser58Asn). PAC2801 has a de novo nonsense variant in exon 1 (c.5G>A, p.Trp2Ter) while two unrelated individuals LIS7901 and LIS8801 both share the same recurrent variant in the splice donor site of exon 2 (c.232+5 G>T) that is predicted to result in skipping of exon 2 (Fig 1C and Table 2). The variant in LIS7901 was confirmed de novo, but both parental samples were unavailable for LIS8801. All variants were verified by Sanger sequencing and were absent from normals in the 1000 Genomes and gnomAD databases. To test the enrichment of de novo CEP85L mutations in gyral disorders we compared the frequency of CEP85L mutations in our cohort of 36 exome sequenced lissencephaly cases (3/36) to 43,502 trios with various diagnoses sequenced at GeneDX, where 6 de novo CEP85L variants were found (6/43,502), demonstrating highly significant enrichment of de novo CEP85L mutations in patients with gyral abnormalities (p<6 × 10−8, Fisher’s exact test).
In the developing human and mouse, the NM_00142475.3 is the major isoform expressed in fetal brain (Johnson et al., 2015; Rie, 2017). Ensemble and Refseq denote a second isoform of CEP85L, NM_001178035, which differs from the NM_00142475.3 transcript in its alternative start codon and 5’UTR. The variants reported here all affect the NM_00142475.3 transcript. Although CEP85L is not severely constrained for missense or loss-of-function (LoF) variants, with many LoF changes in gnomAD, the two exons carrying the variants reported here show greater constraint than other exons (>95.9%) (Havrilla et al., 2019), and all disease-associated missense variants were clustered within 10 amino acids, suggesting that the first two exons are essential for CEP85L function.
The posterior-predominant malformation suggests that CEP85L expression is higher in the posterior cortex during development; this was confirmed by quantitative PCR (qPCR) of samples from the orbital frontal, somatosensory and visual cortex of human gestational week 23 (GW 23) brain (Fig 1D). CEP85L protein levels were also higher in the visual relative to the orbital cortex, whereas TUJ1, a marker shared by all neurons, was more uniform (Lee et al., 1990) (Fig 1E). The posterior-predominant malformation is similar to, but much sharper, than that seen with mutations in LIS1 (Guerrini et al., 2000) and DYNC1H1 (Jamuar et al., 2014). Although LIS1 levels were uniform across brain regions (Fig 1E), the similarity nonetheless suggested potentially close functional interactions of CEP85L with LIS1 and DYNC1H1.
CEP85L is required for neuronal migration.
A scratch wound healing assay suggested that CEP85L is required cell autonomously for migration. U2-OS cells transfected with siRNA directed against CEP85L or scrambled control (SC) demonstrated that (Fig 2A and Video 3–4) control cells filled the wounded area over 24 hrs, whereas CEP85L-depleted cells failed to migrate into the wound (Fig 2B). SC and CEP85L siRNA-transfected cells could properly orient their centrosomes toward the wound, which is the first step in the wound response, suggesting that the failure of migration is not due to defective cell polarization (Fig 2C–D).
Figure 2: CEP85L is required for neuronal migration.
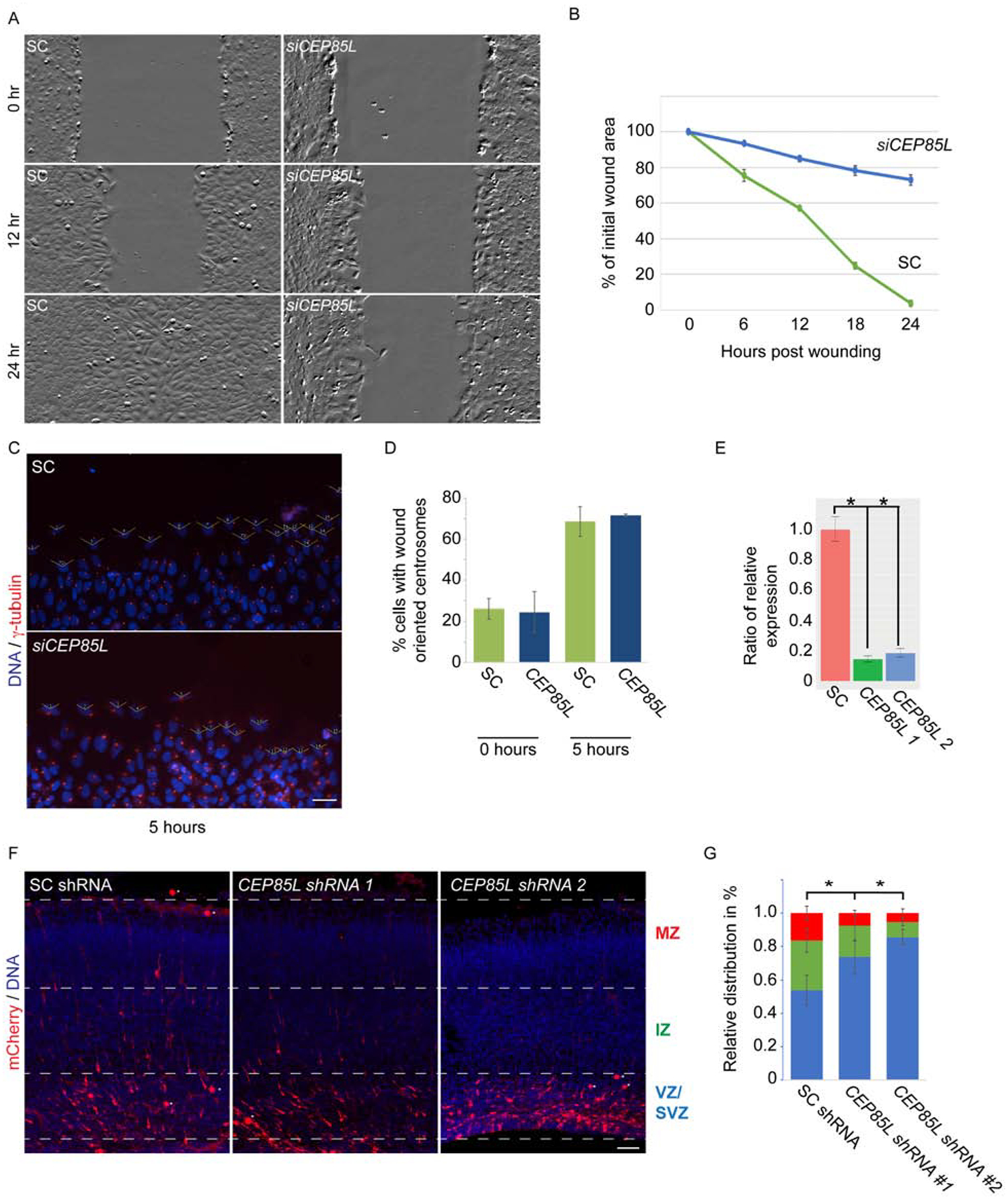
A. Time-lapse stills from scratch wound assays of scrambled control (SC) and CEP85L siRNA-transfected U2-OS cells. Confluent monolayers were wounded using a P200 tip and imaged over 24 hours using the Zeiss Celldiscoverer 7. Scale bar represents 200μm for all images. B. Quantifications of the areas of migration at the indicated time points of SC and CEP85L-depleted cells. For all quantifications, three distinct experiments were performed. C. Immunostaining of γ-tubulin (red) and DNA (blue) in SC and CEP85L siRNA transfected cells along the wound edge. Open-faced triangles are facing the wound. Scale bar represents 200mm for all images. D. Percentage of cells along the wound edge with centrosomes oriented toward the wound at 0 and 5 hrs. For all quantifications, 100 cells were analyzed per experiment (n=3). P<0.005 (Student’s t test). E. qRT-PCR of scrambled control (SC), CEP85L #1, CEP85L #2 shRNA transfected cells normalized to β-actin and represented as a ratio of the control. F. Embryonic day 14.5 mice were electroporated with mCherry and a SC, or with Cep85L #1 or #2 shRNAs and collected at E17.5. Scale bar 100mm. G. Percentage of electroporated SC or Cep85L shRNA transfected mCherry-positive cells in the ventricular and subventricular zone (VZ/SVZ), intermediate zone (IZ), or cortical plate (CP). At least three electroporated brains from each condition were quantified (n>3). *p < 0.005 (Student’s t-test).
Knockdown of Cep85L in mice using shRNA demonstrated a cell-autonomous requirement in migrating cortical neurons. We confirmed the efficiency of the Cep85L shRNA constructs by transfecting mouse Neuro-2a cells with the scrambled control (SC), or two nonoverlapping Cep85L shRNA constructs, observing that both targeting constructs suppressed mRNA levels >90% of controls (Fig 2E). We next examined whether CEP85L regulates cortical migration by electroporating an mCherry construct along with SC or Cep85L shRNA into embryonic day 14.5 wild-type mice. Electroporated brains analyzed 3 days post electroporation showed mCherry-positive SC-transfected cells in the ventricular, intermediate and marginal zones (Fig 2F), while Cep85L-depleted cells failed to migrate past the intermediate zone (Fig 2G), suggesting that Cep85L acts in migrating neurons.
CEP85L localizes to the maternal centriole to control microtubule organization
Immunohistochemistry showed that CEP85L localizes to one of the centrioles during the G1 phase of the cell cycle in U2-OS cells co-stained for Centrin, a centriolar protein (Fig 3A). We confirmed the specificity of the CEP85L antibody by immunofluorescence and western blotting using three non-overlapping siRNAs directed to CEP85L (Fig 3A–B). To confirm the presence of CEP85L at the centrosome, we isolated centrosomes from U2-OS cells and confirmed that CEP85L co-fractionated with the centrosomal component γ-tubulin (Fig 3C). Similar to cells in culture, CEP85L partially co-localized with the centrosomal protein, γ-tubulin, in GW23 fetal human brain tissue (Fig 3D). CEP85L protein levels are stable at the centrosome throughout the cell cycle (Fig S1A–B). Examination of endogenous CEP85L and GFP-tagged CEP85L relative to the proximal and subdistal appendage of the mother centriole showed partial co-localization with the proximal component CEP192 but not with the subdistal centriole component ODF2 (Fig 3E), demonstrating that CEP85L localizes to the proximal end of the mother centriole which is required for subdistal appendage organization (Mazo et al., 2016; Zhang et al., 2016).
Figure 3: CEP85L localizes to the mother centriole and regulates microtubule cytoskeletal organization.
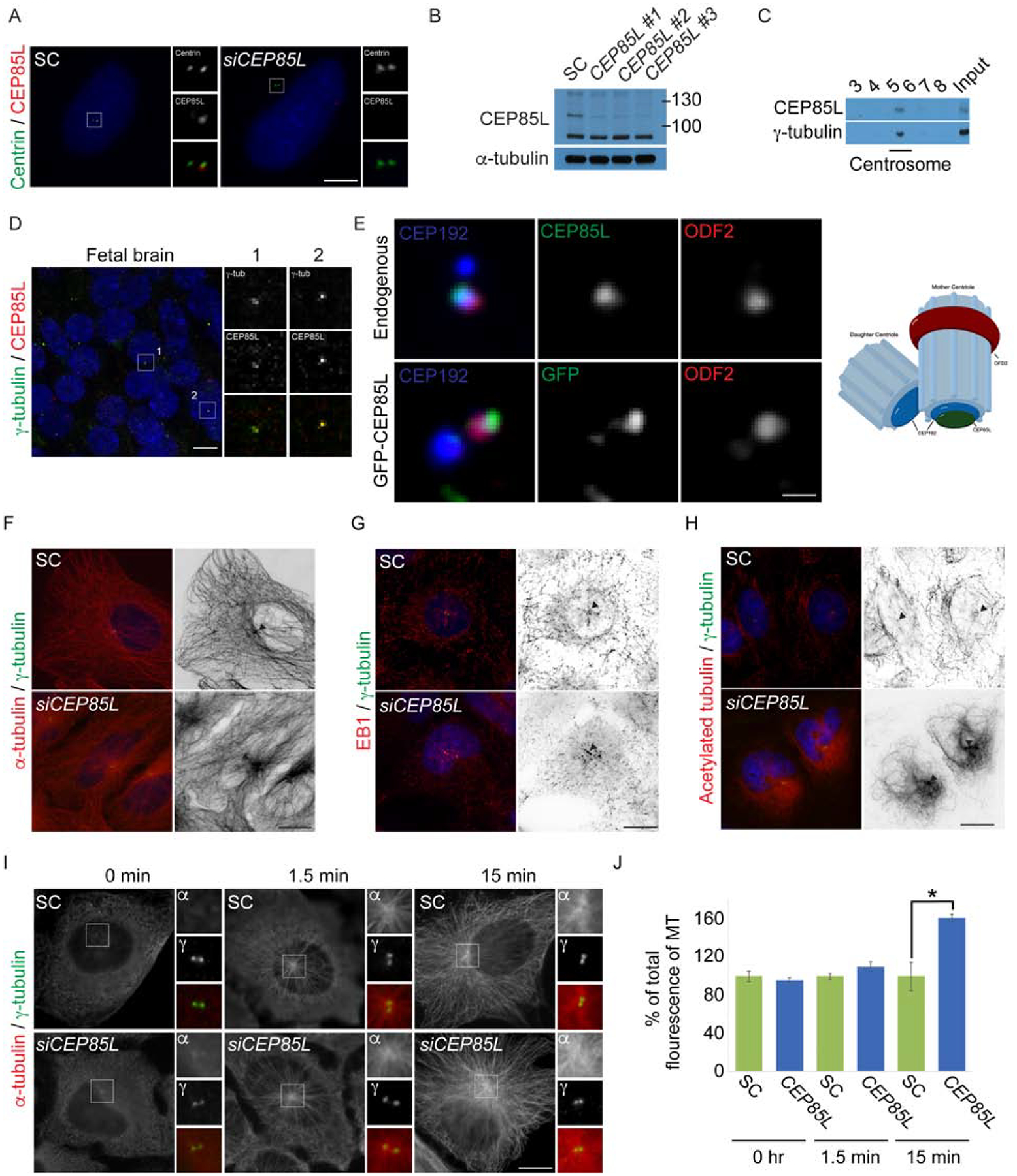
A. U2-OS cells treated with scrambled control (SC) or CEP85L siRNA co-stained with antibodies to Centrin (green) and CEP85L (red). Scale bar represents 5mm for all images. B. Whole-cell lysate from SC, CEP85L #1, #2 or #3 siRNA-treated U2-OS cells immunoblotted for CEP85L. Actin served as a loading control. C. Fractions from sucrose gradient-separated U2-OS cell lysates immunoblotted for CEP85L and γ-tubulin to identify the centrosomal fraction. D. Fresh frozen gestational week 23 fetal brains were co-stained for γ-tubulin (green) and CEP85L (red) Scale bar is 5mm for all images. E. Airyscan microscopy of U2-OS cells co-stained for CEP192 (blue) to mark the proximal centrioles, ODF2 (red) to mark subdistal appendages, and CEP85L (green) or GFP-CEP85L (green). Scale bar represents 1μm for Airyscan images. F-H. Immunofluorescence analysis of SC and CEP85L siRNA-treated U2-OS cells co-stained with γ-tubulin (green) and α-tubulin, EB1, or acetylated tubulin (red). Figures right of the merged image are inverted images of α-tubulin, EB1 or acetylated tubulin. Triangles denote the centrosome. Scale bars represent 10mm for all images. I. SC and CEP85L siRNA-treated U2-OS cells were subjected to a microtubule regrowth assay, fixed at the indicated time points and co-stained with α-tubulin (red) and γ-tubulin (green). Scale bar indicates 5mm for all images. J. Quantification of the mean fluorescence intensities ± s.d. of centrosomal α-tubulin in SC and CEP85L siRNA treated cells as expressed as the mean percentage ± s.d. of the fluorescence intensities of SC cells. For all quantifications, 10 cells were analyzed per experiment (n=3). * − p < 0.005 (Student’s t-test).
As subdistal appendages anchor microtubules to the mother centriole (Askham et al., 2002; Dammermann and Merdes, 2002; Delgehyr et al., 2005; Quintyne et al., 1999; Quintyne and Schroer, 2002) and the cytoskeleton is critical for neuronal migration (Lasser et al., 2018; Solecki et al., 2004), we examined whether CEP85L-depleted cells exhibited altered microtubule cytoskeletal organization. While SC-treated cells displayed a radial array of microtubules originating at the centrosome, CEP85L-depleted cells showed overly abundant centrosomally clustered microtubules (Fig 3F). In addition, we examined the plus-end-capping protein, EB1 that regulates the dynamic behavior of microtubules (Vitre et al., 2008) and found CEP85L-depleted cells had increased EB1 comets in the vicinity of the centrosome (Fig 3G and Fig S1C), indicating impaired microtubule dynamics. Cellular migration depends upon dynamic microtubules, therefore we examined whether the stabilized (Yan et al., 2018; Zuo et al., 2012), acetylated microtubule cytoskeleton was disrupted upon CEP85L-depletion. Relative to controls, CEP85L siRNA treated cells exhibited increased acetylated microtubules (Fig 3H) suggesting altered cytoskeletal dynamics may underlie the inability of CEP85L neurons to migrate in the developing cortex.
Given the increased centrosomal microtubules, we assessed the microtubule nucleating and anchoring ability of SC and CEP85L siRNA treated cells following microtubule depolymerization. After 15 minutes of regrowth, SC cells formed a normal radial microtubule array originating at the centrosome (Fig 3I). In contrast, CEP85L-depleted cells supported a dramatic increase in microtubules anchored at the centrosome (Fig 3J). These findings suggest that the defects in microtubule organization and dynamics may explain why cells depleted of CEP85L are incapable of migrating.
CEP85L localizes and is required to activate CDK5 at the mother centriole.
Immunoprecipitation of endogenous CEP85L from HeLa cells identified many co-precipitating proteins by LC-MS/MS (Table 3), including known centrosomal proteins (Jakobsen et al., 2011; Nogales-Cadenas et al., 2009) as well as products of genes essential for neuronal migration such as LIS1, NDE1, KIF2A, DYNC1H1 and CDK5 (Alkuraya et al., 2011; Bakircioglu et al., 2011; Lo Nigro et al., 1997; Magen et al., 2015; Poirier et al., 2013; Vissers et al., 2010) (Fig 4A). In addition, we identified CDK5RAP2, TUBGCP3, and NEDD1, proteins required for microtubule nucleation (Choi et al., 2010; Luders et al., 2006; Murphy et al., 1998; Tassin et al., 1998). CP110, a centriole protein (Spektor et al., 2007), served as a negative control. CEP85L interactors were sorted and prioritized based on centrosomal localization and associated mutations causing neuronal migration disorders. We confirmed specific interactions between CEP85L and LIS1, NDE1, KIF2A, DYNC1H1 by reciprocal co-immunoprecipitation (Fig 4B–C), suggesting potential links of CEP85L with other genes implicated in microtubule dynamics and neuronal migration.
Figure 4: CEP85L is required to localize and activate the lissencephaly protein, CDK5.
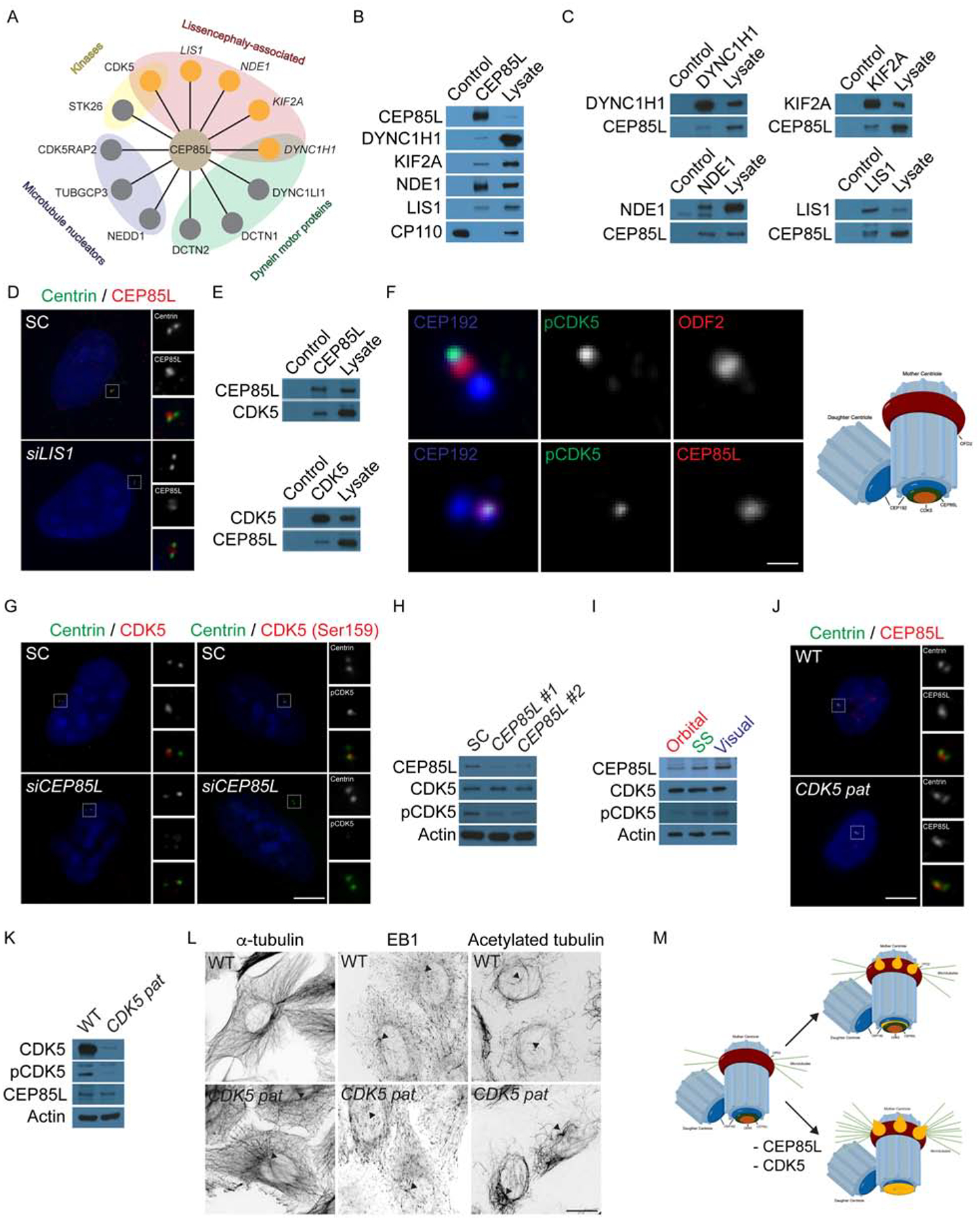
A. Schematic of centrosomal CEP85L interacting proteins identified by endogenous immunoprecipitation of CEP85L followed by LC-MS/MS analysis. Interactors were sorted and prioritized based on centrosomal localization and disease-association. B. Immunoprecipitated endogenous CEP85L and CP110 from HeLa cell lysates was immunoblotted for co-precipitating proteins for CEP85L, DYNC1H1, KIF2A, NDE1 and LIS1. CP110 served as a negative control throughout. Lysate represents 5% of the total cell lysate used in the immunoprecipitation assays. C. HeLa cell lysate was subjected to immunoprecipitation of DYNC1H1, KIF2A, NDE1 and LIS1. Precipitating proteins were immunoblotted for CEP85L, DYNC1H1, KIF2A, NDE1 and LIS1. D. U2-OS cells transfected with siRNA to SC or LIS1 co-stained with Centrin (green) and CEP85L (red). E. HeLa cell lysate was subjected to immunoprecipitation of CEP85L, CDK5, and CP110, which served as a negative control. Precipitating proteins were immunoblotted for CEP85L and CDK5. F. Airyscan maximum projections of U2-OS cells co-stained with antibodies to pCDK5 (green), ODF2 (red, to mark the subdistal appendages of mother centrioles), CEP85L (red), and CEP192 (blue) to mark the proximal domains of the centrioles. Scale bar represents 1μm for Airyscan images. G. Immunofluorescence of SC and CEP85L siRNA-transfected U2-OS cells co-stained for Centrin (green) and CDK5 (red) or pCDK5 (red). Scale bars represent 5mm for all images. H. Total cell lysates from U2-OS cells transfected with SC, CEP85L #1 or #2 probed with antibodies to CEP85L, CDK5, and pCDK5. Actin served as a loading control. I. Whole cell lysate from the posterior frontal, parietal and occipital lobes of a GW 23 human fetus blotted for CEP85L, CDK5 and pCDK5. Actin served as a loading control. J. WT and CDK5 patient fibroblasts (p.V162fsX19, CDK5 pat) co-stained with antibodies to Centrin (green) and pCDK5 (red). K. Whole cell lysate from WT or CDK5 patient fibroblasts probed with antibodies to CDK5, pCDK5, and CEP85L. Actin served as a loading control. L. Inverted images of WT and CDK5 patient cells stained with α-tubulin, EB1 or acetylated tubulin. Triangles denote the centrosome. Scale bars represent 10μm for all images. M. CEP85L (green) localizes CDK5 (beige) to the proximal end of mother centrioles (CEP192, blue) to be activated. At the centrosome CDK5 activity restricts the accumulation of LIS-associated proteins (orange) that localize to the proximal mother centriole and its subdistal appendages. Consequently, loss of CEP85L or CDK5 causes the excessive localization of LIS-proteins resulting in excessive anchoring of microtubules at the mother centriole leading to cells incapable of migrating.
CEP85L-depleted cells showed normal levels of LIS proteins although their pattern of centrosomal localization was disrupted (Fig S2A–C), suggesting that CEP85L functions to restrict the localization of these proteins at the centrosome. In contrast, depletion of LIS1 and NDE1 reduced the localization of CEP85L at the centrosome (Fig 4D and S2D–E) while loss of KIF2A or dynein inhibition with ciliobrevin did not disrupt CEP85L localization (Fig S2D–G), suggesting a potential model in which CEP85L plays roles downstream of the centrosomal proteins, LIS1 and NDE1 (which directly interact) (Derewenda et al., 2007), but upstream of the motor proteins, KIF2A and DYNC1H1. Depletion of LIS proteins or ciliobrevin treatment did not affect the stability of CEP85L (Fig S2H–I), suggesting that CEP85L and its interacting proteins are not interdependent for protein stability.
The relationship of the centrosomal LIS-associated proteins and Cyclin dependent kinase 5 (CDK5), which is also associated with cerebral cortical migration defects as well as cerebellar hypoplasia (Magen et al., 2015), has been unclear, but CEP85L may represent a key intermediary. We confirmed that CEP85L interacts with CDK5 using co-immunoprecipitation (Fig 4E) and used high resolution imaging to show that CDK5 and active CDK5 (pCDK5) (Sharma et al., 1999) both co-localize at the proximal end of mother centrioles with CEP85L (Fig 4F). We confirmed the specificity of the CDK5 and pCDK5 antibodies in cells transfected with SC or CDK5 siRNA (Fig S2J). As CEP85L disrupts the localization of LIS-associated proteins, we examined the localization of CDK5 and pCDK5 in CEP85L-depleted cells. Unlike the relationship between CEP85L and NDE1 and LIS1, CDK5 and pCDK5 were strikingly absent from the centrosome in CEP85L-depleted cells (Fig 4G), suggesting that CDK5’s localization requires CEP85L.
We next assessed the levels of CDK5 and pCDK5 following CEP85L depletion. Interestingly, CDK5 levels remained unchanged but pCDK5 was dramatically decreased upon CEP85L knockdown (Fig 4H). Remarkably, higher levels of pCDK5 in human visual, compared to frontal cortex, parallel the rostral to caudal increase in CEP85L expression (Fig 4I). Over-expression of GFP-CEP85L induces increased pCDK5 by western blot analysis and at the centrosome (Fig S2K–L), suggesting that CEP85L controls the localization and activation of CDK5 at the centrosome.
As CEP85L is required to localize CDK5 to the centrosome, we investigated whether disruption of CDK5 underlies the centrosomal and cytoskeletal changes in CEP85L-depleted cells. We confirmed that CDK5 was lost in patient fibroblasts with a homozygous splice site variant (p.V162fsX19) (Magen et al., 2015), but the localization of CEP85L was unaltered (CDK5 pat) (Fig 4J and S2M) suggesting that CEP85L is required to localize CDK5 but not conversely. As CDK5 interacts with LIS1, NDE1 and DYNC1H1 (Maskey et al., 2015; Pandey and Smith, 2011) we examined whether the loss of CDK5 could account for the over accumulation of LIS proteins at the centrosome in CEP85L-depleted cells. As in cells depleted of CEP85L, Cdk5−/− mouse embryonic fibroblasts (MEFs) displayed abnormal centrosomal accumulations of Dync1h1, Nde1, Kif2a and Lis1 (Fig S2N–O). Similar to depletion of CEP85L, protein levels of LIS-associated proteins were unchanged in Cdk5−/− cells (Fig S2P). The disorganization of the LIS proteins was also observed in CDK5 patient fibroblasts and CDK5-depleted U2OS cells (data not shown). These findings suggest a role for CDK5 in organizing LIS proteins at the centrosome downstream of CEP85L.
To confirm that cytoskeletal defects observed in CEP85L-depleted cells reflect disrupted CDK5, we examined microtubule organization in CDK5 patient and Cdk5−/− cells. Similar to CEP85L siRNA transfected cells, patient fibroblasts and Cdk5−/− MEFs exhibited increased centrosomal microtubules, EB1 and acetylated microtubules, strongly suggesting that the disruption to the cytoskeleton in CEP85L-depleted cells is due to disrupted CDK5 activity (Fig 4L–M and S2Q–R). To confirm that CDK5 activity is required to organize LIS proteins at the centrosome, we treated cells with the CDK5/1/2 inhibitor, Roscovitine at 20μM to selectively inhibit CDK5. Inhibition of CDK5 activity did not alter the localization of active CDK5 at the centrosome (Fig S2S and U). However, Roscovitine-treated cells exhibited increased centrosomal DYNC1H1, KIF2A, NDE1, and LIS1 similar to loss of CEP85L or CDK5 (Fig S2TU). Levels of the LIS proteins were unaltered due to the inhibition of CDK5 activity (Fig S2V). Taken together these findings suggest CEP85L localizes and activates CDK5 at the centrosome to control centrosome and cytoskeletal organization.
DISCUSSION
In summary, we present seven individuals from seven families with mutations in CEP85L with strikingly similar radiographical and clinical features. The missense mutations identified in CEP85L were constrained to a 10 amino acid stretch of a single constrained exon suggesting that this region is intolerant to alterations and may represent a highly critical domain for CEP85L function. As healthy individuals can tolerate loss-of-function and truncation mutations in other CEP85L exons, the missense mutations may affect a binding domain in CEP85L critical for function. Alternatively, this clustering of missense variants and the recurrent splicing variant suggest that some mutations could act by a dominant-negative mechanism. Additional studies are required to determine the pathogenic mechanism of CEP85L mutations.
CEP85L is an important component of the neuronal migration machinery, and disruption of CEP85L results in abnormal posterior cortical architecture, paralleling the higher levels of CEP85L expressed in the posterior cortex. We demonstrate that CEP85L associates with CDK5 at the centrosome to promote its activation and to organize centrosomal LIS-associated proteins (Fig 4N). Taken together, we demonstrate that CEP85L promotes CDK5 localization and activation at the centrosome to form a dynamic microtubule cytoskeleton required for neuronal migration in the developing cortex.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be addressed to the Lead Contact, Dr. Christopher A. Walsh (christopher.walsh@childrens.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
Peripheral blood samples from the affected individuals and parents were analyzed by whole-exome sequencing (WES). This study was approved by the institutional review boards of Boston Children’s Hospital and Beth Israel Deaconess Medical Center. Subjects were identified and evaluated in a clinical setting, and biological samples were collected for research purposes after obtaining written informed consent. 40291IMID was investigated via protocol approved by the institutional review boards for the protection of human subjects at the Institute of Mother and Child (Warsaw, Poland). The cases in this cohort were ascertained and processed using a variety of different methods.
Whole exome sequencing and data processing for PAC2801, LIS6801, DC7401, and PAC3301 was performed by the Genomics Platform at the Broad Institute of Harvard and MIT (Broad Institute, Cambridge, MA, USA). We performed whole exome sequencing on DNA samples (>250 ng of DNA, at >2 ng/ul) using Illumina exome capture (38 Mb target). Our exome-sequencing pipeline included sample plating, library preparation (2-plexing of samples per hybridization), hybrid capture, sequencing (150 bp paired reads), sample identification QC check, and data storage. Our hybrid selection libraries cover >90% of targets at 20x and a mean target coverage of ~100x. The exome sequencing data was de-multiplexed and each sample’s sequence data were aggregated into a single Picard BAM file. Exome sequencing data was processed through a pipeline based on Picard, using base quality score recalibration and local realignment at known indels. We used the BWA aligner for mapping reads to the human genome build 37 (hg19). Single Nucleotide Polymorphism (SNPs) and insertions/deletions (indels) were jointly called across all samples using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.4. Default filters were applied to SNP and indel calls using the GATK Variant Quality Score Recalibration (VQSR) approach. Lastly, the variants were annotated using Variant Effect Predictor (VEP). For additional information please refer to Supplementary Section 1 of the paper describing ExAC (Lek et al., 2016). The variant call set was uploaded on to Seqr and analysis was performed using the various inheritance patterns. A custom panel of genes known to be related to neuronal migration was generated and cases with variants in known genes were filtered out. Candidate variants were validated further by Sanger sequencing.
LIS7901 was enrolled through the Walsh laboratory, however sequencing was performed via GeneDx, Inc. GeneDx performed trio exome on this individual and a connection was made via Matchbox. Using genomic DNA from the proband and parents, the exonic regions and flanking splice junctions of the genome were captured using the IDT xGen Exome Research Panel v1.0. Massively parallel (NextGen) sequencing was done on an Illumina system with 100bp or greater paired-end reads. Reads were aligned to human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a custom-developed analysis tool. Additional sequencing technology and variant interpretation protocol has been previously described (Retterer et al., 2016). The general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/)
40291IMID’s DNA was isolated from clotted whole blood by using the Clotspin Baskets and the Gentra PureGene Blood kit (Qiagen) according to the manufacturer’s instructions. WES was performed at the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine through the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) initiative. Using 1 μg of DNA an Illumina paired-end pre-capture library was constructed according to the manufacturer’s protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC Illumina Barcoded Paired-End Capture Library Preparation protocol. Pre-capture libraries were pooled into 4-plex library pools and then hybridized in solution to the HGSC-designed Core capture reagent (52 Mb, NimbleGen) or 6-plex library pools used the custom VCRome 2.1 capture reagent (42 Mb, NimbleGen) according to the manufacturer’s protocol (NimbleGen SeqCap EZ Exome Library SR User’s Guide) with minor revisions. The sequencing run was performed in paired-end mode using the Illumina HiSeq 2000 platform, with sequencing-by-synthesis reactions extended for 101 cycles from each end and an additional 7 cycles for the index read. With a sequencing yield of 8.6 Gb, the sample achieved 94% of the targeted exome bases covered to a depth of 20× or greater. Illumina sequence analysis was performed using the HGSC Mercury analysis pipeline (https://www.hgsc.bcm.edu/software/mercury) which moves data through various analysis tools from the initial sequence generation on the instrument to annotated variant calls (SNPs and intra-read in/dels). The ACMG guidance for interpretation of sequence variants identified in known disease genes was applied (Table 1). Variants in candidate genes were considered pathogenic or potentially pathogenic based on: (i) variant frequency in the in-house and public mutation databases, (ii) bioinformatics analysis with application of predictive programs, (iii) genotype–phenotype correlation analysis, (iv) familial segregation studies, and (v) functional studies—if available. Identified variants were deposited into the ClinVar database (https://wwwncbi-nlm-nih-gov.ezp-prod1.hul.harvard.edu/clinvar/); consecutive accession numbers SCV000598581–SCV000598612.
Animal Use
Mouse experiments were carried out humanly and approved by Boston Children’s Hospital IACUC protocols. Mice were electroporated at embryonic day 14.5 and processed on day 17.5.
METHOD DETAILS
Quantitative PCR
RNA was isolated using the RNeasy kit (Qiagen) and reverse transcribed using SuperScript IV First-Strand Synthesis System (Life Technologies). Isolated cDNA was quantified using PowerUp SYBR Green Master Mix (Life Technologies) according to manufacturer’s instructions using a StepOnePlus Real-Time PCR System (Thermo Fisher). All primers (Thermo Fisher) for qPCR were generated using the Mass General Hospital/Harvard Medical School PrimerBank. All quantifications were normalized to β-actin.
Molecular biology
Human CEP85L cDNA (ENST00000368491) was PCR-amplified from HeLa cell cDNA and cloned into the eGFP-C1 plasmid (Clonetech). To generate the scrambled control and Cep85L shRNA constructs oligos were hybridized and closed into BLOCK-iT U6 RNAi Entry Vector Kit (Life Technologies). Constructs were subsequently cloned into pcDNA-DEST53 Vector Life Technologies). The mCherry-C1 construct was generated by PCR amplifying mCherry (gift of Dr. Roger Tsien) into the eGFP-C1 plasmid.
Cell culture
U2-OS and HeLa cells were maintained in Advanced DMEM (Life Technologies) supplemented with 3% fetal bovine serum (FBS, Life Technologies and Atlanta Biologics) and GlutaMax-I (Life Technologies). N2A cells were grown in DMEM supplemented with 10% FBS and GlutaMAX-I. Wild-type and Cdk5−/− mouse embryonic fibroblasts (gift from Drs. Douglas Lowy and Brajendra Tripathi, NIH) and wild-type and CDK5 patient fibroblasts (gift from Dr. Daniella Magen, Ruth Rappaport Children’s Hospital) were grown in AmnioMAX C-100 basal media supplemented with antibiotic-antimycotic (Life Technologies) and mycoplasma removal agent (Bio-Rad). Neuro-2a cells were maintained in EMEM (Life Technologies) supplemented with 10% FBS and GlutaMAX-I. U2-OS and HeLa cells were transfected using Lipofectamine RNAiMAX (Life Technologies) with 60pmol of STEALTH siRNA (Life Technologies) per six-well dish. Samples were analyzed 48 h post transfection. Plasmids were transfected using Lipofectamine3000 or Lipofectamine2000 (Life Technologies) according to manufacturer’s recommendations. In brief, 2.5μg of DNA and 5μl of P3000 and Lipofectamine3000 were used per six well transfection. Cells were analyzed at the described time points. Cells were synchronized using a double thymidine (Sigma) block and release to capture cells at various cell cycle stages. To inhibit Dynein, U2-OS cells were treated with DMSO or 50μM of Ciliobrevin in the dark for 1 hr at 37°C. CDK5 activity was inhibited using Roscovitine (Sigma) at a concentration of 20μM overnight.
Centrosome enrichment
Asynchronous U2-OS cells were treated with 2μM cytochalasin D and 1mg/ml of nocodazole for 1.5 hrs to depolymerize actin and microtubules, respectively. Centrosomes were enriched on a discontinuous sucrose gradient (70, 50 and 40% sucrose) and collected fractions were analyzed by western blotting.
Western blotting and immunoprecipitation
HeLa cells were incubated on ice with PBS (Life Technologies) for 5 min, harvested with a cell scraper (Corning) and lysed on ice in lysis buffer (1% IGEPAL630 (Sigma and Thermo Fisher), 50mM Tris pH7.4 (Life Technologies), 150mM NaCl (Ambion) in PBS) supplemented with protease and phosphatase inhibitor cocktail III (Sigma). For each immunoprecipitation 500μg of total lysate was incubated with 1–2μg of antibody for 2 h and then incubated with magnetic protein G-sepharose (GE Healthcare Life Sciences) for an additional 1.5 hours. Immunoprecipitating proteins were boiled in 2X Laemmli sample buffer with β-mercaptoethanol (Bio-Rad) or collected in pH2.0 Glycine (Life Technologies) and quenched in Tris pH9.0 (Ambion) for mass spectrometry analysis. Protein from flash frozen gestational week 23 brains were extracted using the NE-PER Kit (Thermo Fisher) followed by homogenization with a pellet pestle (Kimble). Reduced samples were separated on 4–15% TGX gels (Bio-Rad), transferred to supported BA85 Protran (GE Healthcare) and subjected to immunoblot analysis using ECL lightening Plus (Perkin-Elmer) or LiCOR Odyssey scanner for quantitative analysis.
Mass spectrometry analysis
Immunoprecipitations from HeLa cell lysates were analyzed as previously described (Kodani et al., 2015). Immunocomplexes were digested with trypsin (Promega) peptides were then analyzed using a LTQ Oribtrap Velos Pro ion-trap mass spectrometer (Thermo Fisher). Captured peptide identity was determined using Sequest software (Thermo Fisher) and filtered for peptide false discovery.
Immunostaining
Adherent cells were grown on sterilized cover glasses (Azer Scientific) and fixed with chilled methanol for 3 min to visualize centrosomal proteins and 2 min for microtubules. Fixed cells were blocked in blocking buffer (2.5% BSA (Sigma), 0.1% Triton X100 (Fisher) and 0.03% NaN3 in PBS (Life Technologies). Primary, secondary antibodies, and Hoechst33342 (Life Technologies) were diluted in blocking buffer and incubated with cells for at least 1 h at room temperature. To detect CEP85L, cells were blocked in 2.5% FBS instead of BSA. To immunolabel CEP85L and α-tubulin in fetal brain samples, sections were permeabilized using 0.3% Triton X100 in PBS, and incubated with antibody overnight in antibodies diluted in 300mM NaCl, 0.2% gelatin and 0.3% Triton X100 (Paridaen et al., 2013). Stained samples were mounted using Gelvatol and imaged on an inverted Zeiss Axio Observer Z1, LSM700 or LSM800 with Airyscan microscope. Flash frozen sections from a gestational week 23 brain were fixed in 4% PFA overnight, permeabilized using 0.3% Triton X100 in PBS and quenched in 0.1M glycine pH7.4. Sections were subsequently incubated with primary antibody in 0.3% Triton X100, 300mM NaCl and 0.2% gelatin. Subsequently, the samples were mounted using Flouromount-G (Southern Biotech).
Migration assay
siRNA transfected U2-OS cells were grown to confluency on uncoated plastic bottom 6 well dishes. Monolayers were scratched using a P200 Rainin pipette, rinsed and imaged continuously using a Zeiss Celldiscoverer 7 for 24 hrs. The Celldiscoverer chamber was set to 37°C with injected 5% CO2. Compiled images and videos were processed using the ZEISS ZEN software. Cell migration was calculated using the MRI Wound Healing Tool macro in FIJI.
Microtubule regrowth assay
U2-OS cells transfected with SC or CEP85L siRNA were treated with 200nM Nocodazole (Sigma) for 1.5 hours. Cells were washed with cold media and placed on ice for 30 min. Warm media was added to the cells and allowed to recover at 37°C for the indicated time periods prior to fixation in cold methanol.
In utero electroporation of mouse embryos
pCDNA DEST53 CMV-GFP-U6 scrambled control or Cep85l shRNA and mCherry were electroporated into the ventricles of embryonic day 12.5 and 14.5 mice as previously described (Saito, 2006; Yang et al., 2012). In brief, plasmids (1μg/ul) were injected into the telencephalic vesicle of embryos using a pulled micropipette. Five pulses of 30–50 V (950ms duration) were delivered across the embryo’s head using a BTX ECM830 pulse generator. Electroporated embryos were collected and analyzed by immunohistochemistry. mCherry positive cells in each cortical layer were quantified using FIJI and compared using a Chi-squared test.
QUANTIFICATION AND STATISTICAL ANALYSIS
To determine the statistical enrichment of mutations in CEP85L, we used the MedCalc’s a Fisher exact probability calculator to determine statistical significance.
For the migration assay, the wounded area over the time course was analyzed using the MRI Wound Healing Tool macro in FIJI.
For all immunofluorescence quantifications intensities were quantified using the ROI tool in the FIJI software. The fluorescence of the control was set as 100% and used to calculate the fluorescence of the treatment and represented as a percentage of the control.
DATA AND SOFTWARE AVAILABILITY
Data from the mass spectrometry of CP110 and CEP85L immunoprecipitations are available as Supplemental Table 3.
Supplementary Material
Supplementary videos 1 and 2, Related to Figure 1: A. 3D reconstruction of an age matched control and PAC3301 MRIs.
Supplementary videos 3 and 4, Related to Figure 2: A. Time lapses of scrambled control (SC) or CEP85L-depleted U2-OS cells imaged every five minutes for 24 hrs using a Celldiscoverer 7.
Table S3, Related to Figure 4: A. LC-MS/MS analysis of CP110 and CEP85L co-precipitating proteins.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CEP85L | Proteintech | Cat# 24588–1-AP |
| LIS1 | Sigma | Cat# SAB2500597; RRID: AB_10604255 |
| TUJ1 | Proteintech | Cat#66375–1-Ig |
| β-actin | Proteintech | Cat# 20536–1-AP; RRID: AB_10700003 |
| α-tubulin | Sigma | Cat# T6074; RRID: AB_261690 |
| Centrin | Sigma | Cat# 04–1624; RRID: AB_10563501 |
| ODF2 | Abnova | Cat# H00004957-M01; RRID: AB_1137338 |
| γ-tubulin | Sigma | Cat# T5192; RRID: AB_477582 |
| pHH3 | Cell Signal | Cat# 9701S; RRID: AB_331535 |
| CEP192 Alexa647 | Andrew Holland | PMID: 31115335 |
| Acetylated tubulin | Sigma | Cat# T6793; RRID:AB_477585 |
| EB1 | BD Biosciences | Cat# 610535; RRID: AB_397892 |
| DYNC1H1 | Proteintech | Cat# 12345–1-AP; RRID:AB_2261765 |
| DYNC1H1 | Bethyl Labs | Cat# A304–720A; RRID:AB_2620915 |
| KIF2A | Thermo Fisher | Cat# PA3–16833; RRID:AB_2131873 |
| CP110 | Proteintech | Cat# 12780–1-AP; RRID:AB_10638480 |
| NDE1 | Proteintech | Cat# 10233–1-AP; RRID:AB_2149877 |
| CDK5 | Cell Signal | Cat# 2506S; RRID:AB_2078855 |
| CDK5 | Santa Cruz Biotechnology | Cat# sc-6247; RRID:AB_627241 |
| pCDK5 (Ser159) | Santa Cruz Biotechnology | Cat# sc-377558 |
| pCDK5 (Ser159) | Thermo Fisher | Cat# PA5–64751; RRID:AB_2663116 |
| Centrin1 | Proteintech | Cat# 12794–1-AP; RRID:AB_2077371 |
| GFP-HRP | Cell Signal | Cat# 2037S; RRID:AB_1281301 |
| Native IgG HRP | Cell Signal | Cat# 5127S; RRID:AB_10892860 |
| Biological Samples | ||
| 23 week gestational fetal brain | Massachusetts General Hospital | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ciliobrevin D | Sigma | Cat# 250401 |
| Nocodazole | Sigma | Cat# M1404 |
| DMSO | Sigma | Cat# 472301 |
| Thymidine | Sigma | Cat# T1895 |
| Roscovitine | Sigma | Cat# R7772 |
| Mycoplasma Removal Agent | Bio-Rad | Cat# BUF035 |
| Lipofectamine3000 | Thermo Fisher | Cat# L30000 |
| Lipofectamine RNAiMAX | Thermo Fisher | Cat# 13778150 |
| Experimental Models: Cell Lines | ||
| U2-OS | ATCC | CVCL_0042 |
| HeLa | ATCC | CVCL_0030 |
| Neuro-2a | ATCC | CVCL_0470 |
| WT MEFs (mouse embryonic fibroblasts) | Dr. Douglas Lowy (NIH) | PMID: 25452387 |
| Cdk5−/− MEFs (mouse embryonic fibroblasts) | Dr. Douglas Lowy (NIH) | PMID: 25452387 |
| WT fibroblasts | Daniella Magen (Ruth Rappaport Children’s Hospital) | PMID: 25560765 |
| CDK5 patient fibroblasts | Daniella Magen (Ruth Rappaport Children’s Hospital) | PMID: 25560765 |
| Experimental Models: Organisms/Strains | ||
| Crl:CD-1 laboratory mouse | Charles River | Cat# 5652673; RRID: MGI:5652673 |
| Oligonucleotides | ||
| siRNA targeting sequence: CEP85L #1: GGCCACTTCGGAAATGGTCATCTTT | Thermo Fisher | Cat#: HSS139769 |
| siRNA targeting sequence: CEP85L #2: GGCCACTTCGGAAATGGTCATCTTT | Thermo Fisher | Cat#: HSS139770 |
| siRNA targeting sequence: CEP85L #3: GGCCACTTCGGAAATGGTCATCTTT | Thermo Fisher | Cat#: HSS180226 |
| siRNA targeting sequence: LIS1 #1: GGTACGTATGGTACGGCCAAATCAA | Thermo Fisher | Cat#: HSS107554 |
| siRNA targeting sequence: LIS1 #2: TGAAGCAACAGGATCTGAGACTAAA | Thermo Fisher | Cat#: HSS107555 |
| siRNA targeting sequence: LIS1 #3: CCAGAGACAACGAGATGAACTAAAT | Thermo Fisher | Cat#: HSS107556 |
| siRNA targeting sequence: NDE1 #1: GGAAACCATCAAGGAGAAGTTTGAA | Thermo Fisher | Cat#: HSS123304 |
| siRNA targeting sequence: NDE1 #2: GAGCAAGCAAATGACGACCTGGAAA | Thermo Fisher | Cat#: HSS123305 |
| siRNA targeting sequence: NDE1 #3: ACCGAGGACCCAGCTCAAGTTTAAA | Thermo Fisher | Cat#: HSS123306 |
| siRNA targeting sequence: KIF2A #1: CCCTGACCTTGTTCCTGATGAAGAA |
Thermo Fisher | Cat#: HSS105799 |
| siRNA targeting sequence: KIF2A #2: GAGACTTTAGAGGAAGTTTGGATTA | Thermo Fisher | Cat#: HSS105800 |
| siRNA targeting sequence: KIF2A #3: CCTAATGAAATGGTTTACAGGTTTA | Thermo Fisher | Cat#: HSS180178 |
| siRNA targeting sequence: CDK5 #1: GGTGACCTCGATCCTGAGATTGTAA | Thermo Fisher | Cat#: HSS101729 |
| siRNA targeting sequence: CDK5 #2: GGCAATGATGTCGATGACCAGTTGA | Thermo Fisher | Cat#: HSS101730 |
| siRNA targeting sequence: CDK5 #3: GATTCTGTCATAGCCGCAATGTGCT | Thermo Fisher | Cat#: HSS173470 |
| Scramble Control: AAACTAAACTGAGGCAATGCC | Thermo Fisher | N/A |
| CEP85L.EcoR1: GATTAGGAATTCGATGTGGGGGCGCTTCC | Thermo Fisher | N/A |
| CEP85L.BamH1: TCTTCTGGATCCTCACTGAGTAATGCAGTTGTCTCC | Thermo Fisher | N/A |
| shRNA.Scramble.F: CACCGAAACTAAACTGAGGCAATGCCCGAAGGCATTGCCTCAGTTTAG | Thermo Fisher | N/A |
| shRNA.Scramble.R: AAAACTAAACTGAGGCAATGCCTTCGGGCATTGCCTCAGTTTAGTTTC | Thermo Fisher | N/A |
| shRNA.Cep85l.1.F: CACCGCTTCCGTTTCCAAACATAGGCGAACCTATGTTTGGAAACGGAAGC | Thermo Fisher | N/A |
| shRNA.Cep85l.1.R: AAAAGCTTCCGTTTCCAAACATAGGTTCGCCTATGTTTGGAAACGGAAGC | Thermo Fisher | N/A |
| shRNA.Cep85l.2.F: CACCGCTGGGAATCCGATCAATGACGAATCATTGATCGGATTCCCAG | Thermo Fisher | N/A |
| shRNA.Cep85l.2.R: AAAACTGGGAATCCGATCAATGATTCGTCATTGATCGGATTCCCAGC | Thermo Fisher | N/A |
| qPCR.Cep85l.F: CAAGCCTAGTCGATCATTGGTC | Thermo Fisher | N/A |
| qPCR.Cep85l.R: AGATTCCCTATGTTTGGAAACGG | Thermo Fisher | N/A |
| qPCR.Actb: GGCTGTATTCCCCTCCATCG | Thermo Fisher | N/A |
| qPCR.Actb: CCAGTTGGTAACAATGCCATGT | Thermo Fisher | N/A |
| Recombinant DNA | ||
| pEGFP-C1 | Clontech | Cat# 6084–1 |
| pEGFP-CEP85L | Self | Self |
| mCherry-C1 | Self | Self |
| BLOCK-IT U6 | Thermo Fisher | Cat# K494500 |
| pcDNA-DEST53 | Thermo Fisher | Cat# 12288015 |
| Software and Algorithms | ||
| Adobe Illustrator 2019 | Adobe | RRID:SCR_010279 |
| Adobe Photoshop 2019 | Adobe | RRID:SCR_014199 |
| FIJI | FIJI | RRID:SCR_002285 |
Highlights.
Mutations in CEP85L cause posterior specific pachygyria
CEP85L is required for neuronal migration
Loss of CEP85L disrupts centrosome organization and function
CEP85L localizes and activates CDK5 at the centrosome
ACKNOWLEDGEMENTS
The authors thank the families for their invaluable participation in our study. We thank Drs. Douglas Lowy, Brajendra Tripathi, Daniella Magen and Andrew Holland for cell lines and antibodies. We also thank Drs. Meng-Fu Bryan Tsou and Laurence Pelletier for insightful discussions. CAW was supported by The Manton Center for Orphan Disease Research, R01NS035129 and R01NS032457 from the NINDS, and the Allen Discovery Center program through The Paul G. Allen Frontiers Group. CAW is an Investigator of the Howard Hughes Medical Institute. AK was supported by R21NS104633-01A1, the William Randolph Hearst Fund, and the Charles Hood Foundation. AO was supported by the K12 HD052896 Child Health Rearch Career Development Award Program. ES was supported by NIH T32GM007753 and the HCBI Simmons Award. Sequencing and analysis for PAC2801, LIS6801, DC7401, and PAC3301 were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to Daniel MacArthur and Heidi Rehm. AB was supported by GNT1113531 from the Australian Genomics Health Alliance and NHMRC, the Maurice de Rohan International Scholarship, and the Australian Government Research Training Program Scholarship. GeneDx Inc. performed a trio exome on LIS7901 and a connection was made via MatchMaker Exchange. The work done for 40291IMID was supported by the National Science Centre, Poland 2015/19/B/NZ2/01824 to WW, and exome sequencing was performed at the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine through the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) initiative. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
CAW. serves on Advisory Boards for the Allen Brain Institute, Third Rock Ventures, and Maze Therapeutics, and on Editorial Boards for the Annals of Neurology, Trends in Neurosciences, and neuroDEVELOPMENTS. MS. received research funding from Roche, Novartis, Pfizer, LAM Therapeutics, Quadrant Biosciences and has served on the Scientific Advisory Board of Sage Therapeutics, Roche, Takeda, Celgene and PTEN Research Foundation. In addition, he serves on the Board of the Tuberous Sclerosis Alliance. All of these activities are outside the submitted manuscript. JSC. is a consultant for Invitae. DV serves as a consultant to SK Life Science and Otsuka Pharmaceuticals, is on the speaker’s bureaus for UCB and Greenwich Pharmaceuticals, and conducts industry-supported clinical drug trials for SK Life Science, Biogen and UCB Pharmaceuticals. KM is an employee of GeneDX, Inc.
REFERENCES
- Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, et al. (2011). Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet 88, 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askham JM, Vaughan KT, Goodson HV, and Morrison EE (2002). Evidence that an interaction between EB1 and p150(Glued) is required for the formation and maintenance of a radial microtubule array anchored at the centrosome. Mol Biol Cell 13, 3627–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakircioglu M, Carvalho OP, Khurshid M, Cox JJ, Tuysuz B, Barak T, Yilmaz S, Caglayan O, Dincer A, Nicholas AK, et al. (2011). The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet 88, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Liu P, Sze SK, Dai C, and Qi RZ (2010). CDK5RAP2 stimulates microtubule nucleation by the gamma-tubulin ring complex. J Cell Biol 191, 1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammermann A, and Merdes A (2002). Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J Cell Biol 159, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgehyr N, Sillibourne J, and Bornens M (2005). Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci 118, 1565–1575. [DOI] [PubMed] [Google Scholar]
- Derewenda U, Tarricone C, Choi WC, Cooper DR, Lukasik S, Perrina F, Tripathy A, Kim MH, Cafiso DS, Musacchio A, et al. (2007). The structure of the coiled-coil domain of Ndel1 and the basis of its interaction with Lis1, the causal protein of Miller-Dieker lissencephaly. Structure 15, 1467–1481. [DOI] [PubMed] [Google Scholar]
- Di Donato N, Chiari S, Mirzaa GM, Aldinger K, Parrini E, Olds C, Barkovich AJ, Guerrini R, and Dobyns WB (2017). Lissencephaly: Expanded imaging and clinical classification. Am J Med Genet A 173, 1473–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donato N, Timms AE, Aldinger KA, Mirzaa GM, Bennett JT, Collins S, Olds C, Mei D, Chiari S, Carvill G, et al. (2018). Analysis of 17 genes detects mutations in 81% of 811 patients with lissencephaly. Genet Med 20, 1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini R, Barkovich AJ, Sztriha L, and Dobyns WB (2000). Bilateral frontal polymicrogyria: a newly recognized brain malformation syndrome. Neurology 54, 909–913. [DOI] [PubMed] [Google Scholar]
- Havrilla JM, Pedersen BS, Layer RM, and Quinlan AR (2019). A map of constrained coding regions in the human genome. Nat Genet 51, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg EA, et al. (2011). Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 30, 1520–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamuar SS, Lam AT, Kircher M, D’Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, et al. (2014). Somatic mutations in cerebral cortical malformations. N Engl J Med 371, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, and Walsh CA (2015). Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci 18, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani A, Yu TW, Johnson JR, Jayaraman D, Johnson TL, Al-Gazali L, Sztriha L, Partlow JN, Kim H, Krup AL, et al. (2015). Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasser M, Tiber J, and Lowery LA (2018). The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front Cell Neurosci 12, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, and Frankfurter A (1990). The expression and posttranslational modification of a neuron-specific beta-tubulin isotype during chick embryogenesis. Cell Motil Cytoskeleton 17, 118–132. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, and Ledbetter DH (1997). Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet 6, 157–164. [DOI] [PubMed] [Google Scholar]
- Luders J, Patel UK, and Stearns T (2006). GCP-WD is a gamma-tubulin targeting factor required for centrosomal and chromatin-mediated microtubule nucleation. Nat Cell Biol 8, 137–147. [DOI] [PubMed] [Google Scholar]
- Magen D, Ofir A, Berger L, Goldsher D, Eran A, Katib N, Nijem Y, Vlodavsky E, Tzur S, Behar DM, et al. (2015). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with a loss-of-function mutation in CDK5. Hum Genet 134, 305–314. [DOI] [PubMed] [Google Scholar]
- Maskey D, Marlin MC, Kim S, Kim S, Ong EC, Li G, and Tsiokas L (2015). Cell cycle-dependent ubiquitylation and destruction of NDE1 by CDK5-FBW7 regulates ciliary length. EMBO J 34, 2424–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazo G, Soplop N, Wang WJ, Uryu K, and Tsou MF (2016). Spatial Control of Primary Ciliogenesis by Subdistal Appendages Alters Sensation-Associated Properties of Cilia. Dev Cell 39, 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SM, Urbani L, and Stearns T (1998). The mammalian gamma-tubulin complex contains homologues of the yeast spindle pole body components spc97p and spc98p. J Cell Biol 141, 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales-Cadenas R, Abascal F, Diez-Perez J, Carazo JM, and Pascual-Montano A (2009). CentrosomeDB: a human centrosomal proteins database. Nucleic Acids Res 37, D175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey JP, and Smith DS (2011). A Cdk5-dependent switch regulates Lis1/Ndel1/dynein-driven organelle transport in adult axons. J Neurosci 31, 17207–17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paridaen JT, Wilsch-Brauninger M, and Huttner WB (2013). Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 155, 333–344. [DOI] [PubMed] [Google Scholar]
- Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 45, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintyne NJ, Gill SR, Eckley DM, Crego CL, Compton DA, and Schroer TA (1999). Dynactin is required for microtubule anchoring at centrosomes. J Cell Biol 147, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintyne NJ, and Schroer TA (2002). Distinct cell cycle-dependent roles for dynactin and dynein at centrosomes. J Cell Biol 159, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (1971). Neuron-glia relationship during granule cell migration in developing cerebellar cortex. A Golgi and electronmicroscopic study in Macacus Rhesus. J Comp Neurol 141, 283–312. [DOI] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet Med 18, 696–704. [DOI] [PubMed] [Google Scholar]
- Rie D.d. (2017). An integrated expression atlas of miRNAs and their promoters in human and mouse. [DOI] [PMC free article] [PubMed]
- Saito T (2006). In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc 1, 1552–1558. [DOI] [PubMed] [Google Scholar]
- Sharma P, Sharma M, Amin ND, Albers RW, and Pant HC (1999). Regulation of cyclin-dependent kinase 5 catalytic activity by phosphorylation. Proc Natl Acad Sci U S A 96, 11156–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki DJ, Model L, Gaetz J, Kapoor TM, and Hatten ME (2004). Par6alpha signaling controls glial-guided neuronal migration. Nat Neurosci 7, 1195–1203. [DOI] [PubMed] [Google Scholar]
- Spektor A, Tsang WY, Khoo D, and Dynlacht BD (2007). Cep97 and CP110 suppress a cilia assembly program. Cell 130, 678–690. [DOI] [PubMed] [Google Scholar]
- Tassin AM, Celati C, Moudjou M, and Bornens M (1998). Characterization of the human homologue of the yeast spc98p and its association with gamma-tubulin. J Cell Biol 141, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, et al. (2010). A de novo paradigm for mental retardation. Nat Genet 42, 1109–1112. [DOI] [PubMed] [Google Scholar]
- Vitre B, Coquelle FM, Heichette C, Garnier C, Chretien D, and Arnal I (2008). EB1 regulates microtubule dynamics and tubulin sheet closure in vitro. Nat Cell Biol 10, 415–421. [DOI] [PubMed] [Google Scholar]
- Yan B, Xie S, Liu Y, Liu W, Li D, Liu M, Luo HR, and Zhou J (2018). Histone deacetylase 6 modulates macrophage infiltration during inflammation. Theranostics 8, 2927–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, et al. (2012). Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151, 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, et al. (2016). Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166, 1147–1162 e1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Q, Wu W, Li X, Zhao L, and Chen W (2012). HDAC6 and SIRT2 promote bladder cancer cell migration and invasion by targeting cortactin. Oncol Rep 27, 819–824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary videos 1 and 2, Related to Figure 1: A. 3D reconstruction of an age matched control and PAC3301 MRIs.
Supplementary videos 3 and 4, Related to Figure 2: A. Time lapses of scrambled control (SC) or CEP85L-depleted U2-OS cells imaged every five minutes for 24 hrs using a Celldiscoverer 7.
Table S3, Related to Figure 4: A. LC-MS/MS analysis of CP110 and CEP85L co-precipitating proteins.
Data Availability Statement
Data from the mass spectrometry of CP110 and CEP85L immunoprecipitations are available as Supplemental Table 3.