

Abstract
Implantable and wearable biosensors that enable monitoring of biophysical and biochemical parameters over long durations are highly attractive for early and presymptomatic diagnosis of pathological conditions and timely clinical intervention. Poor stability of antibodies used as biorecognition elements and the lack of effective methods to refresh the biosensors upon demand without severely compromising the functionality of the biosensor remain significant challenges in realizing protein biosensors for long-term monitoring. Here, we introduce a novel method involving organosilica encapsulation of antibodies for preserving their biorecognition capability under harsh conditions, typically encountered during the sensor refreshing process, and elevated temperature. Specifically, a simple aqueous rinsing step using sodium dodecyl sulfate (SDS) solution refreshes the biosensor by dissociating the antibody–antigen interactions. Encapsulation of the antibodies with an organosilica layer is shown to preserve the biorecognition capability of otherwise unstable antibodies during the SDS treatment, thus ultimately facilitating the refreshability of the biosensor over multiple cycles. Harnessing this method, we demonstrate the refreshability of plasmonic biosensors for anti-IgG (model bioanalyte) and neutrophil gelatinase-associated lipocalin (NGAL) (a biomarker for acute and chronic kidney injury). The novel encapsulation approach demonstrated can be easily extended to other transduction platforms to realize refreshable biosensors for monitoring of protein biomarkers over long durations.
Keywords: refreshable biosensor, localized surface plasmon resonance, organosilica encapsulation, plasmonic biosensor, biosensor regeneration
Graphical Abstract

INTRODUCTION
Wearable and implantable biosensors have attracted extensive attention owing to their ability to provide continuous monitoring of biophysical and biochemical parameters in biofluids such as sweat, saliva, interstitial fluid, and tears.1–5 In the past few years, the frontiers and the possible applications of such devices are rapidly advancing from tracking physical activity and biophysical parameters to continuous monitoring of target molecular biomarkers at physiological and pathological concentrations. While there have been significant efforts in realizing wearable and implantable biosensors, there are still significant challenges that need to be overcome before such classes of biosensors can be widely used for making timely clinical interventions in pathological conditions that can rapidly manifest into life-threating events or chronic conditions. In fact, to date, only minimally invasive glucose monitoring devices have some commercial presence.6
Biosensors designed for continuous monitoring of biochemical analytes should be able to detect and quantify the target analyte (e.g., biomarker) for an extended duration. However, the number of analyte binding sites in most biosensors are limited, and once saturated with target analytes, it becomes insensitive to further changes in the concentration of the analyte as the analyte–recognition element interactions (especially antibody–antigen interactions) are virtually irreversible under normal conditions. Additionally, the long-term usage of the biosensor is limited by the poor stability of antibodies. A possible approach to overcome these problems is to design a strategy to preserve the biorecognition elements and refresh the sensor without compromising the sensitivity and specificity of the biorecognition elements. As a proof-of-concept, in this study, we employed plasmonic nanostructures as a transduction platform. Owing to their high refractive index sensitivity, plasmonic nanostructures are able to transduce biomolecular binding events (capture/release of the analyte) into measurable shift in the localized surface plasmon resonance (LSPR) wavelength.7–10 The refractive index sensitivity of plasmonic nanostructures has been harnessed to realize various chemical and biological sensors.11–15 Plasmonic biosensors relying on antibodies as recognition elements are highly promising as lab-on-chip devices for label-free protein detection in point-of-care and resource-limited settings.16–19
In most immunosensors, antigens are recognized and captured by antibodies via noncovalent interactions such as hydrogen bonds, van der Waals forces, electrostatic and hydrophobic interactions.20–23 These interactions can be disrupted by extremes of pH, high salt concentrations and surfactants.23 Under these harsh conditions, natural antibodies are unstable and are prone to lose their biorecognition capability owing to their poor chemical and environmental stability.24–27 We and others have reported various methods to preserve the biorecognition capability of antibodies under harsh conditions, which include encapsulation of immobilized antibodies with metallic–organic frameworks,16–18 silk,28 and sucrose,29 or addition of other preservatives.30,31 While these strategies successfully preserve the biorecognition capability of biodiagnostic reagents immobilized on plasmonic nanotransducers against harsh environmental conditions during storage and transportation, they do not protect the antibodies during sensor operation or during sensor refreshing.
In this report, we demonstrate the encapsulation of antibodies immobilized on plasmonic nanostructures with an organosilica layer, which renders refreshability to the biosensors by preserving the antibody biorecognition ability upon subjecting them to harsh chemical treatment for dissociating the antibody–antigen interactions. This novel method overcomes the challenges associated with poor stability of immobilized antibodies under harsh conditions and opens up opportunities for realizing wearable and implantable biosensors for continuous monitoring of protein biomarkers over long durations.
RESULTS AND DISCUSSION
AuNR–IgG-Based Biosensor.
We employed rabbit IgG and goat anti-rabbit IgG as a model of the antibody–antigen pair and gold nanorods (AuNRs) as plasmonic nanotransducers. We synthesized AuNRs using the seed-mediated method.32 Transmission electron micrograph (TEM) images revealed the length and diameter of the AuNRs as 57.5 ± 2.1 and 19.7 ± 2.6 nm, respectively (Figure 1A). The antibodies were conjugated to a bifunctional poly(ethylene glycol) (COOH-PEG-SH) chain to obtain IgG-PEG-SH.17,33,34 Subsequently, IgG-PEG-SH was anchored to the AuNR surface via a Au–S linkage. Poly(ethylene glycol) (PEG) chains offer two important advantages: (i) increased accessibility of IgG to target biomolecules by acting as a flexible linker34 and (ii) minimization of nonspecific binding owing to their high hydrophilicity.17 The immobilization of IgG-PEG-SH on AuNRs in solution resulted in an ~8 nm red shift in the longitudinal LSPR wavelength of AuNRs, corresponding to an increase in the refractive index of the medium surrounding AuNRs (Figure 1B). To realize plasmonic biosensors, the AuNR–IgG bioconjugates were uniformly adsorbed onto the 3-mercaptopropyl-trimethoxysilane (MPTMS)-functionalized glass substrates. Atomic force microscopy (AMF) images of the modified glass substrates revealed uniform distribution of AuNR–IgG bioconjugates with no signs of aggregation or patchiness (Figures 1C and S1). To minimize nonspecific binding, glass substrates with AuNR–IgG bioconjugates were exposed to thiol-terminated poly(ethylene glycol) (SH-PEG), which is expected to graft to the exposed regions of AuNRs and serve as a blocking layer. To investigate their biosensing performance, we exposed these plasmonic biochips to different concentrations of anti-IgG, which binds specifically to the IgG immobilized on the AuNRs. Consistent with our previous studies, the LSPR wavelength of AuNR exhibited a monotonic red shift with an increase in the concentration of anti-IgG (Figure S2).17,34 The limit of detection (LOD defined as: mean + 3 σ of the blank) of these biochips was found to be 240 pg/mL.
Figure 1.
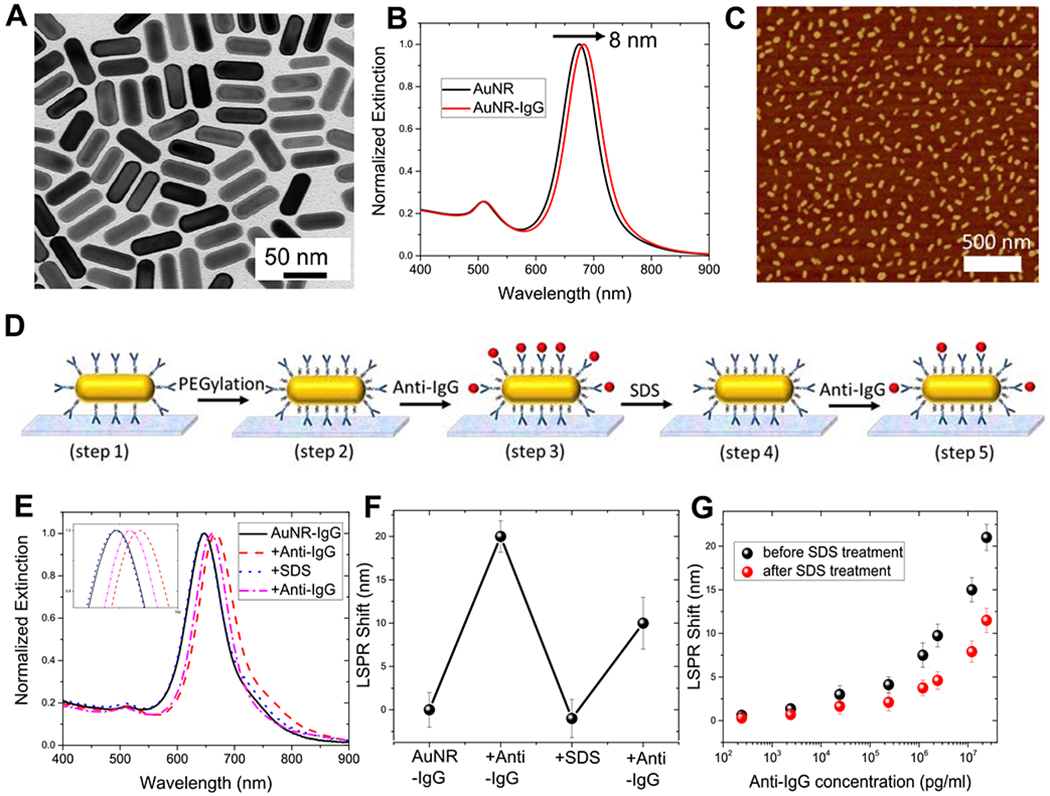
(A) Representative TEM image of AuNRs used as plasmonic nanotransducers. (B) Normalized vis-NIR extinction spectra of AuNR and AuNR conjugated with IgG, depicting an ~8 nm red shift in the LSPR wavelength. (C) Representative atomic force microscopy (AFM) image showing uniform distribution of AuNR–IgG bioconjugates on a glass substrate. (D) Schematic illustration of steps involved in the fabrication of the refreshable sensor. (E) Extinction spectra of AuNR–IgG bioconjugates obtained after each fabrication step shown in (D). The inset shows zoomed-in spectra highlighting the shifts in the LSPR wavelength. (F) LSPR shifts corresponding to biodetection and sensor refreshing. (G) LSPR shift upon exposure of plasmonic biochips to different concentrations of anti-IgG before (black) and after sodium dodecyl sulfate (SDS) (red) treatment. Error bars represent standard deviations from three different samples.
Refreshability of the AuNR–IgG Biosensor.
As mentioned above, antigens (anti-IgG in this case) are recognized and captured by antibodies (IgG conjugated to AuNRs in the present case) through noncovalent interactions, which can be disrupted by subjecting them to extreme pH or high concentrations of salt or surfactants.23,35 We used sodium dodecyl sulfate (SDS), an anionic surfactant frequently used in cleaning and hygiene products (e.g., toothpaste and mouthwash), as a chemical agent to disrupt the antigen–antibody interactions. SDS, known to disrupt protein conformation, binds relatively uniformly along the protein chain with its hydrophobic tail conferring net charge on proteins and thus exposing (unfolding) the otherwise buried regions of the protein.23,36–38 We posited that exposure of the bound antibody–antigen pair to SDS results in electrostatic repulsion between the antibody and antigen, thus overcoming the noncovalent interactions between them. Dissociation of the antibody–antigen pair essentially refreshes the binding sites and enables the reuse of the biosensor. We have harnessed the refractive index sensitivity of the AuNRs to monitor each step along this process (Figure 1D). Extinction spectra (Figure 1E) and the LSPR wavelength of AuNR (Figure 1F) were obtained following each step in the procedure: immobilization of AuNR-IgG bioconjugates on glass substrates (step 1); PEGylation of AuNR–IgG bioconjugates to minimize nonspecific binding (step 2); binding of anti-IgG to IgG (step 3); exposure to SDS solution (step 4); and rebinding of anti-IgG to IgG (step 5).
The LSPR wavelength of AuNR exhibited a ~20 nm red shift after exposure to 24 μg/mL anti-IgG (Figure 1E,F). After exposure to SDS, we observed a ~21 nm blue shift suggesting complete removal of anti-IgG and thus leaving behind AuNR–IgG bioconjugates on the plasmonic biochip, ready for another cycle of antigen detection (Figure 1E,F). However, when SDS-treated plasmonic biochips were exposed to the same concentration of anti-IgG, we observed that the biochip lost ~50% sensitivity as evident from only a ~10 nm red shift as opposed to ~20 nm observed in the pristine biochip (Figure 1E,F). The nearly 50% loss in biorecognition capability after SDS treatment was consistent over a broad range of anti-IgG concentration (Figure 1G). This is not surprising because in the process of dissociating anti-IgG:IgG complex, SDS partially denatures the immobilized IgG and thus results in the loss of its biorecognition capability. Consequently, with every cycle of SDS washing, we noted a progressive degradation in the biorecognition capability and by fourth cycle the plasmonic biochips exhibited only ~10% sensitivity compared to that in the pristine condition (Figure S3). Thus, this challenge of antibody denaturation and subsequent loss in biorecognition capability needs to be overcome to realize the refreshable sensor. Toward this goal, we explored antibody encapsulation as a strategy hypothesized to render protection against harsh and potentially denaturing conditions and ultimately leads to refreshable biosensors.
Polymer Encapsulation Strategy to Achieve Refreshability.
In a previous report, we have demonstrated an in situ polymerization technique for preserving the activity of enzyme, immobilized on plasmonic nanostructures, subjected to harsh conditions such as proteases and high temperature.39 Inspired by this biopreservation method, we posited that encapsulation of immobilized antibodies with an in situ formed polymer layer can preserve its biorecognition capability against SDS washing (Figure 2A). Following the immobilization of AuNR–IgG bioconjugates on glass substrates, a polymer encapsulation layer is formed through copolymerization of (3-aminopropyl) trimethoxysilane (APTMS) and trimethoxy(propyl)silane (TMPS) on AuNR and around immobilized IgG. The methoxy group of TMPS and APTMS undergoes rapid hydrolysis to form methanol and trisilanols.40‘41 Hydrolysis is followed by condensation of the silanols, which results in the formation of an amorphous aminopropyl functional polymer layer consisting of Si–O–Si bonds and functional end groups such as hydroxyl (−OH), amine (−NH3+), and methyl (−CH3).42,43 These end groups interact noncovalently via hydrogen bonding, hydrophobic, and electrostatic interactions with AuNR–IgG bioconjugates resulting in the formation of a stable organosilica layer around them. Next, bifunctional PEG (methoxy-PEG-silane) was covalently grafted on to the free regions of the organosilica layer. The methoxysilane group of PEG undergoes hydrolysis followed by condensation with the reactive silanol group present on the polymer surface, resulting in the formation of a stable covalent siloxane bond (Si–O–Si). PEG chains shield the functional groups present on the polymer layer, thus minimizing nonspecific binding.39,41,43 Biosensors with the organosilica protective layer were then exposed to a range of concentration of anti-IgG (240 pg to 24 μg) to allow antigen–antibody binding. The plasmonic biochips were subsequently exposed to an aqueous solution of SDS to overcome the noncovalent interactions, dissociate the antibody–antigen pair, and refresh the biochip. The restored biosensor was repeatedly exposed to anti-IgG to assess the refreshability of the biosensor.
Figure 2.
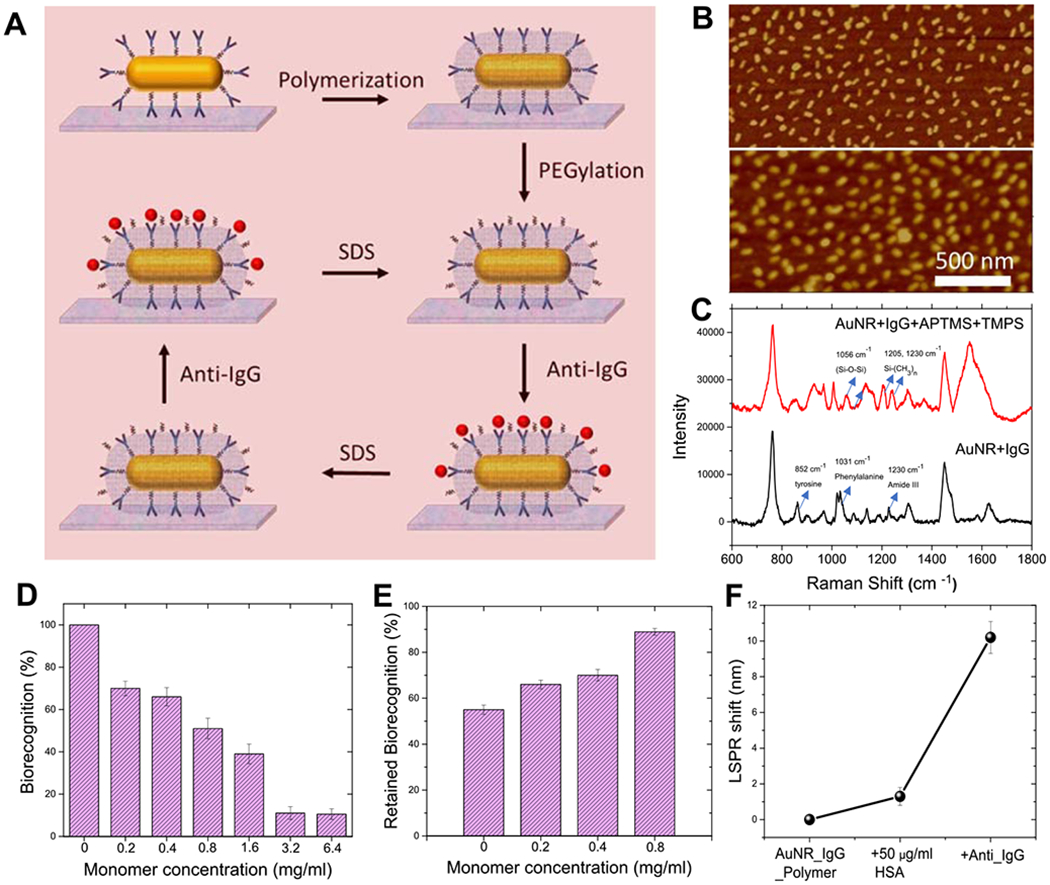
(A) Schematic illustration of the steps involved in the organosilica-based biopreservation of bioconjugates to realize refreshable biosensors. (B) Representative AFM image of AuNR–IgG bioconjugates on the glass substrate before (top) and after (bottom) polymer encapsulation with optimum monomer concentration (0.8 mg/mL). (C) Surface-enhanced Raman scattering spectra of AuNR–IgG bioconjugates before and after polymerization. (D) Biorecognition capability corresponding to different monomer concentrations, which determines the polymer thickness. (E) Retained biorecognition capability of AuNR–IgG bioconjugates corresponding to different monomer concentrations, after SDS treatment. (F) LSPR shift of the polymer-encapsulated biosensor after treatment with human serum albumin (HSA) and anti-IgG. Error bars represent standard deviations from three different samples.
Characterization and Optimization of the Organosilica Silica Layer Thickness.
Formation of the polymer encapsulation layer on AuNR–IgG bioconjugates was confirmed by redshift in the LSPR wavelength (Figure S4) and AFM imaging (Figure S6). The AFM image of the polymer-encapsulated bioconjugates revealed a change in the morphology corresponding to the formation of the organosilica polymer layer (Figures 2B, S1 and S6). The presence of an organosilica polymer layer on the AuNR–IgG bioconjugates was further confirmed by surface-enhanced Raman scattering (SERS) spectroscopy (Figure 2C). Pristine AuNR–IgG bioconjugates exhibited Raman bands at 852, 1031, 1230, and 1620–40 cm−1 corresponding to tyrosine, phenylalanine, amide III, and amide I of IgG.34 After the formation of an organosilica layer, we observed Raman bands at 1024, 1056, 1205, and 1230 cm−1 corresponding to Si–O–R stretching, Si–O–Si stretching, and −CH2 bending.39,43,44
It is important to note that if the entire antibody, including its antigen binding sites, is encapsulated within the polymer layer, it will severely compromise the biorecognition capability of IgG. On the other hand, if the encapsulation is insufficient, then the protection against SDS and long-term stability of the antibody will be limited. Therefore, the thickness of the encapsulating polymer layer is critical to provide both access for analyte binding and protection against harsh conditions. The thickness of the organosilica layer can be controlled either by varying the polymerization time or by changing the concentration of the APTMS and TMPS monomers. We varied the concentration of monomers while keeping the polymerization time constant (10 min), as it offers better control and repeatability over multiple batches. The red shift in the LSPR wavelength of the AuNR corresponding to the formation of the organosilica layer increased with an increase in the concentration of monomers, indicating a gradual increase in the thickness of the polymer layer (Figure S4). Pristine plasmonic biochips with no polymer encapsulation, which corresponds to the maximum availability of antibody binding sites, displayed a ~20 nm red shift (treated as 100% biorecognition capability). As the thickness of the polymer layer was gradually increased, the plasmonic biochips exhibited a progressive decrease in biorecognition capability (Figures 2D and S5). Here, biorecognition capability is defined as the percentage of the red shift upon specific binding of anti-IgG to IgG after encapsulation with a polymer layer compared with the red shift obtained from the same batch of biochips before encapsulation. An increase in the thickness of the polymer layer rendered biosensors increasingly stable against SDS washing, thus enabling their reusability. We used the percentage of retained biorecognition capability to quantitatively evaluate the preservation efficacy of the polymer encapsulation strategy. It was calculated as the percentage of the red shift upon specific binding of goat anti-rabbit IgG to the rabbit IgG on a restored biochip after one or more cycles of SDS treatment compared with the red shift obtained from the same batch of the biosensor before SDS treatment. For example, samples with no polymer encapsulation and before SDS treatment exhibited ~20 nm red shift, showing 100% biorecognition capability (Figure 2D), and after SDS treatment displayed ~10 nm red shift corresponding to only 50% retained biorecognition capability (Figure 2E). On the other hand, plasmonic biochips with polymer encapsulation corresponding to the monomer concentration of 0.8 mg/mL exhibited a red shift of 11.5 nm before SDS treatment and red shift of 10.5 nm following SDS treatment, corresponding to ~90% retained biorecognition capability (Figure 2E). This significant improvement in the biorecognition capability against SDS underscores the importance of polymer encapsulation of the antibody for the successful refreshability of the biosensors. By gradually changing the monomer concentration/thickness of the polymer layer, we found a balance between the loss of biorecognition capability and an increase in the preservation efficacy of polymer encapsulation of AuNR–IgG conjugates to achieve refreshability. Considering the retained biorecognition capability of ~90% for the monomer concentration of 0.8 mg/mL, we have employed this condition in subsequent experiments.
Specificity of the Polymer-Encapsulated AuNR–IgG Biosensor.
To determine the specificity of bioconjugates after polymer encapsulation, we measured the shifts in the LSPR wavelength of AuNR after the exposure of plasmonic biochips to high concentration (50 μg/mL) of interfering protein such as human serum albumin (HSA) (Figures 2F and S7). We found that the LSPR shift corresponding to the exposure of the polymerized biosensor to 50 μg/mL HSA was only ~1 nm, which is significantly lower than the ~10.5 nm red shift obtained upon exposure to 24 μg/mL anti-IgG. We attribute this low nonspecific binding to the covalently grafted PEG chains on the free surface of the organosilica polymer layer, which are known to resist nonspecific protein adsorption. Further, we probed the sensing capability of the polymer-encapsulated plasmonic biosensors by exposing them to different concentrations of anti-IgG and monitoring the LSPR shift of the AuNR. As expected, we observed a monotonic increase in the LSPR wavelength with an increase in the anti-IgG concentration (Figure S8). The limit of detection (defined as: mean + 3 σ of the blank) of these biochips was found to be 3.7 ng/mL.
Refreshability of the Polymer-Encapsulated AuNR–IgG Biosensor.
Next, we set out to investigate the refreshability of the polymer-encapsulated biosensors. Figure 3A shows the extinction spectra obtained after: immobilization of AuNR–IgG; formation of the organosilica layer; specific binding of anti-IgG (24 μg/mL) to IgG, which resulted in a ~10.5 nm red shift; refreshing the plasmonic biochip by SDS washing, which resulted in ~10.5 nm blue shift suggesting the dissociation of the anti-IgG:IgG pair; reuse of the refreshed biosensor by exposing it again to 24 μg/mL anti-IgG resulting in a ~10 nm red shift, thus depicting the refreshability of biosensors. All of the biosensors after polymerization and prior to exposure to the analyte were PEGylated. The PEGylation step after polymer encapsulation resulted in a small (~0.5 nm) red shift, which is not shown in Figure 3A. Figure 3B depicts sequential LSPR shifts obtained following each of the aforementioned steps suggesting that the polymer encapsulation strategy provides the ability to reuse the biosensors without significantly compromising the biorecognition capability. Similar results were observed for different concentrations of anti-IgG suggesting stability and refreshability of the biosensors over a large range of concentrations (Figure 3C).
Figure 3.
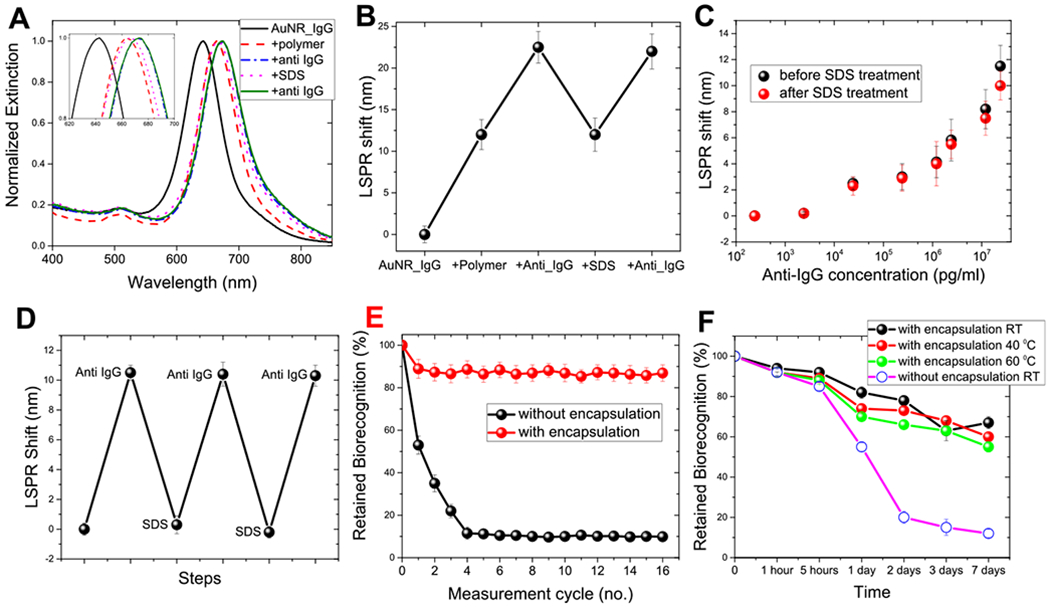
(A) AuNR extinction spectra corresponding to each step involved in the polymer encapsulation strategy. The inset shows zoomed-in spectra highlighting the shifts in the LSPR wavelength. (B) LSPR shift corresponding to each step involved in the polymer encapsulation strategy. (C) LSPR shift upon exposure of polymer-encapsulated biosensors to different concentrations of anti-IgG before (black) and after SDS (red) treatment. (D) LSPR wavelength shift after alternate exposure to anti-IgG and SDS. (E) Retained biorecognition capability of biosensors with and without polymer encapsulation over multiple capture/release cycles of the analyte. (F) Retained biorecognition capability of AuNR–IgG bioconjugates with and without polymer encapsulation stored at room temperature, 40 and 60 °C for different durations. Error bars represent standard deviations from three different samples.
We investigated the reusability of polymer-encapsulated plasmonic biochips over multiple cycles by subjecting the plasmonic biochips to repeated cycles of capture (exposure to anti-IgG) and release (exposure to SDS). Anti-IgG captured by the polymer-encapsulated AuNR–IgG conjugates was completely released with SDS treatment as confirmed by the LSPR blue shift, identical to the red shift observed during capture (Figure 3D). The refreshed biosensor was exposed to a fresh batch of anti-IgG (24 μg/mL) each time, resulting in a ~10 nm red shift suggesting the near-complete preservation of biorecognition capability of bioconjugates. The polymer-encapsulated AuNR–IgG preserved ~80% of biorecognition capability even after 16 cycles of SDS treatment (Figure 3E). On the other hand, biosensors without polymer encapsulation exhibited <40% of biorecognition capability after the second cycle and ~10% after the fourth cycle (Figure 3E). These results underscore the importance of a polymer encapsulation strategy to achieve refreshability without significantly compromising the biorecognition capability.
Long-Term Usability of the Polymer-Encapsulated AuNR–IgG Biosensor.
Another important aspect of deploying a refreshable biosensor over a long duration of time is the long-term stability of the biorecognition element under ambient and even harsh conditions. Therefore, we tested the efficacy of polymer encapsulation to preserve the biorecognition capability of AuNR–IgG bioconjugates against harsh conditions that, without polymer encapsulation, would lead to protein denaturation and consequent loss in biorecognition capability.24–27 The plasmonic biosensors with and without polymer encapsulation were stored at room temperature, 40 and 60 °C for different times (1 and 5 h and 1, 2, 3, and 7 days) to monitor the changes in the biorecognition capabilities of the antibodies (Figure 3F). After storage, plasmonic biochips were exposed to anti-IgG (24 μg/mL). Biochips with polymer encapsulation exhibited ~70% retention of biorecognition capability after storage at room temperature (25 °C) for 1 week compared to an almost complete loss in biorecognition capability for biochips without polymer encapsulation. Significantly, the biochips with polymer encapsulation retained ~60% of biorecognition capability even after storage at higher temperatures (40 and 60 °C) for a week. In contrast, pristine biochips lost more than 50% of biorecognition capability within 1 day and ~90% after 1 week. The remarkable stability of the polymer-encapsulated AuNR–IgG bioconjugates possibly stems from the restricted mobility of the biomolecules, and thus impeding protein denaturation even under extreme conditions.28,42 In other words, the noncovalent interactions between bioconjugates and organosilica layer impose steric hindrance on the antibodies, and thus restricting them from undergoing changes in secondary and tertiary structures (unfold).39
Biological Stability of the AuNR–IgG Biosensor.
In addition to the remarkable thermal stability, which allows long-term usability, biosensors are required to be stable against biological agents such as proteases in patient serum/urine samples, which can lead to proteolytic degradation of antibodies. Therefore, to probe the biological stability of polymer-encapsulated biosensors, we subjected AuNR–IgG-based pristine and organosilica-stabilized biosensors to different concentrations of protease dissolved in synthetic urine for different time periods at room temperature. We observed that the biorecognition capability AuNR–IgG bioconjugates decreased to ~8% for all conditions, while the polymer-encapsulated bioconjugates retained ~90, ~83, and ~70% of the biorecognition capability when subjected to 100 ng/mL, 1 μg/mL, and 10 μg/mL for 24 h, respectively (Figure 4A). These results suggest that the organosilica layer significantly lowers the accessibility of the immobilized antibody to the protease, rendering excellent biological stability against proteolytic digestion.
Figure 4.

(A) (%) Retained biorecognition capability of pristine and polymer-encapsulated AuNR–IgG-based biosensors after these had been subjected to different conditions of proteolytic degradation at room temperature. The LSPR wavelength shift after alternate exposure of polymer-encapsulated biosensors to anti-IgG and NaOH (B), PA (C), and glycine buffer (D). The error bar represents the standard deviation from three independent samples.
Compatibility of the Encapsulation Strategy with Other Chemical Regeneration Agents.
To further ascertain the universality of the polymer encapsulation strategy, we explored the compatibility of the polymer encapsulation with different regeneration techniques. We performed multiple capture/release cycles using the following chemical regeneration agents: (i) acid-mediated regeneration using 0.1 M phosphoric acid (PA) solution;45 (ii) base-mediated regeneration using 50 mM sodium hydroxide (NaOH);46 and (iii) 10 mM glycine/HCl buffer (pH 2.8).47 We investigated these chemical regeneration approaches by subjecting the polymer-encapsulated plasmonic biochips to repeated cycles of capture (exposure to the analyte) and release (10 min exposure to aforementioned regeneration agents). Figure 4B–D depicts the LSPR wavelength shift obtained after alternate exposures of the analyte and the regeneration agent. The polymer-encapsulated biosensors exhibited ~85% of biorecognition capability after treatment with different regeneration agents, including SDS, even after multiple wash cycles. We also exposed pristine plasmonic biosensors (i.e. unencapsulated) to these regeneration agents. As expected, these biosensors exhibited a significant loss in biorecognition capability after treatment with the aforementioned regeneration agents. These results underscore the universality of polymer encapsulation of the immobilized antibodies rendering stability against various regeneration agents for achieving refreshability.
Universality of the Polymer Encapsulation Strategy.
Finally, to verify the generality of the polymer encapsulation strategy for achieving the refreshable biosensor, we employed gold nanorattles (AuNRT) as plasmonic nanotransducers and neutrophil gelatinase-associated lipocalin (NGAL), a urinary biomarker for acute and chronic kidney injury, as target analytes.17 Figure 5A shows the TEM image of AuNRTs with an edge length of 34.2 ± 1.3 nm. Similar to IgG, anti-NGAL was conjugated to AuNRT and the conjugation was confirmed by a ~10 nm red shift in the LSPR wavelength of AuNRT (Figure S9). Subsequently, AuNRT–anti-NGAL bioconjugates were immobilized on MPTMS-functionalized glass substrates. After PEGylation, the plasmonic biochips were exposed to NGAL (5 μg/mL) resulting in a ~20 nm red shift in the LSPR wavelength of AuNRT (Figures 5B and S10). To determine the refreshability, the biochips were treated with SDS to release the captured NGAL from AuNRT–anti-NGAL bioconjugates. The complete removal of NGAL was evidenced by a ~20 nm blue shift in the LSPR wavelength. However, SDS treatment resulted in a ~56% loss in biorecognition capability. Following the SDS treatment, AuNRT exhibited the LSPR shift of only ~9.5 nm upon exposure to NGAL (5 μg/mL), as opposed to ~20 nm observed before the SDS treatment (Figure S11). This loss in the biorecognition capability was observed over a broad range of NGAL concentrations, which is consistent with that observed in the case of AuNR–IgG bioconjugates (Figure 5B). To overcome this loss in the biorecognition ability, we employed polymer encapsulation as a strategy to protect immobilized biorecognition elements against SDS treatment. The monomer concentration was optimized to attain a balance between biopreservation and antibody availability for the target antigen capture (Figure S12). The polymer-encapsulated AuNRT–NGAL antibody bioconjugates were exposed to different concentrations of NGAL. Although the LSPR shift exhibited by polymer-encapsulated biosensors was ~50% compared to those without polymer encapsulation, the polymer-encapsulated biosensors exhibited excellent preservation of biorecognition capability after SDS treatment (Figure 5C). For instance, polymer-encapsulated AuNRT–NGAL antibody bioconjugates exhibited nearly a similar LSPR red shift (~10 nm) upon exposure to NGAL (5 μg/mL), both before and after SDS treatment, suggesting excellent stability and refreshability. Similar results were observed for different concentrations of NGAL suggesting refreshability of the biosensors over a large range of concentrations (Figure 5D). The polymer-encapsulated bioconjugates retained nearly 80% of the biorecognition ability after 16 capture/release cycles, which is in stark contrast with less than 20% retained recognition capability of unencapsulated bioconjugates after just three capture/release cycles (Figure 5E). Finally, as for IgG, we probed the thermal stability of these biosensors by storing the biosensors at different temperatures (room temperature, 40 and 60 °C) for different durations (1, 2, 3, and 7 days). As expected, polymer-encapsulated biosensors retained >60% of biorecognition capability even after storage for 7 days at 60 °C, whereas pristine biochips lost more than 80% of the biorecognition capability within 1 day (Figure 5F). These results attest to the generality of the polymer encapsulation method in preserving and refreshing the biorecognition capabilities of immobilized antibodies.
Figure 5.

(A) Representative TEM image of Au nanorattles (AuNRT) used as nanotransducers. (B) LSPR shift upon exposure of AuNRT—NGAL antibody bioconjugates to different concentrations of NGAL before (black) and after SDS (red) treatment. (C) Extinction spectra corresponding to each step involved in the polymer encapsulation strategy of AuNRT—NGAL antibody bioconjugates. The inset shows zoomed-in spectra highlighting the shifts in the LSPR wavelength. (D) LSPR shift upon exposure of polymer-encapsulated AuNRT—NGAL antibody bioconjugates to different concentrations of NGAL before (black) and after SDS (red) treatment. (E) Retained biorecognition capability of biosensors with and without polymer encapsulation over multiple capture/release cycles of NGAL. (F) Retained biorecognition capability of AuNR–IgG bioconjugates with and without polymer encapsulation stored at room temperature, 40 and 60 °C for different durations. The error bar represents standard deviations from three different samples.
CONCLUSIONS AND OUTLOOK
In summary, we have introduced a facile and universal method based on in situ polymerization of an organosilica layer for preserving the biorecognition capabilities of immobilized antibodies. Polymer-encapsulated antibodies on plasmonic nanostructures exhibited remarkable stability over multiple capture/release cycles, and thus enabling refreshability of the biochips. The thickness of the polymer layer, controlled by the concentration of the monomers and the polymerization time, plays a critical role in determining the balance between the preservation of the recognition ability of the antibody and the availability of the antibody binding sites for antigen capture. Although in this study, we have employed a plasmonic biosensor as a transduction platform and SDS treatment as a method to overcome the antibody–antigen interaction, the encapsulation approach can be easily extended to other transduction platforms and other possible sensor refreshing methods. More specifically, although the SDS-based sensor refreshing strategy offers a rather narrow application window, primarily limited to implantables in the oral cavity, the polymer-based preservation method demonstrated here is universal. The encapsulation-based preservation method demonstrated overcomes a critical challenge in wearable and implantable biosensors and is expected to advance the design and implementation of wearable biosensors for long-term monitoring of protein biomarkers.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge support from the National Institutes of Health (R01-DE027098 and R01-CA141521). The authors thank the Nano Research Facility (NRF) and Institute of Materials Science and Engineering (IMSE) at Washington University for providing access to electron microscopy facilities.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.9b17506.
Experimental details, materials, and procedures; AFM images of bioconjugates and polymer-encapsulated bioconjugates immobilized on glass substrates; dose response of anti-IgG and the NGAL antibody-based plasmonic biosensor; control measurements (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.9b17506
The authors declare no competing financial interest.
Contributor Information
Rohit Gupta, Institute of Materials Science and Engineering and Department of Mechanical Engineering and Materials Science, Washington University in St. Louis, St. Louis, Missouri 63130, United States.
Jingyi Luan, Institute of Materials Science and Engineering and Department of Mechanical Engineering and Materials Science, Washington University in St. Louis, St. Louis, Missouri 63130, United States.
Shantanu Chakrabartty, Department of Electrical and Systems Engineering, Washington University in St. Louis, St. Louis, Missouri 63130, United States.
Erica L. Scheller, Department of Medicine, Division of Bone and Mineral Diseases, Washington University in St. Louis, St. Louis, Missouri 63110, United States
Jeremiah Morrissey, Department of Anesthesiology and Siteman Cancer Center, Washington University in St. Louis, St. Louis, Missouri 63110, United States.
Srikanth Singamaneni, Institute of Materials Science and Engineering and Department of Mechanical Engineering and Materials Science and Siteman Cancer Center, Washington University in St. Louis, St. Louis, Missouri 63130, United States.
REFERENCES
- (1).Gao W; Emaminejad S; Nyein HYY; Challa S; Chen K; Peck A; Fahad HM; Ota H; Shiraki H; Kiriya D; et al. Fully Integrated Wearable Sensor Arrays for Multiplexed In situ Perspiration Analysis. Nature 2016, 529, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bandodkar AJ; Jia W; Yardımcı C; Wang X; Ramirez J; Wang J Tattoo-based Noninvasive Glucose Monitoring: a proof-of-concept study. Anal. Chem 2014, 87, 394–398. [DOI] [PubMed] [Google Scholar]
- (3).Koh A; Kang D; Xue Y; Lee S; Pielak RM; Kim J; Hwang T; Min S; Banks A; Bastien P; et al. A soft, Wearable Microfluidic Device for the Capture, Storage, and Colorimetric Sensing of Sweat. Sci. Transl. Med 2016, 8, No. 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lee H; Choi TK; Lee YB; Cho HR; Ghaffari R; Wang L; Choi HJ; Chung TD; Lu N; Hyeon T; et al. A graphene-based electrochemical device with thermoresponsive microneedles for diabetes monitoring and therapy. Nat. Nanotechnol 2016, 11, 566–572. [DOI] [PubMed] [Google Scholar]
- (5).Mannoor MS; Tao H; Clayton JD; Sengupta A; Kaplan DL; Naik RR; Verma N; Omenetto FG; McAlpine MC Graphene-based Wireless Bacteria Detection on Tooth Enamel. Nat. Commun 2012, 3, No. 763. [DOI] [PubMed] [Google Scholar]
- (6).Kim J; Campbell AS; de Ávila BE-F; Wang J Wearable Biosensors for Healthcare Monitoring. Nat. Biotechnol 2019, 37, 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kelly KL; Coronado E; Zhao LL; Schatz GC The Optical Properties of Metal Nanoparticles: The Influence of Size, Shape, and Dielectric Environment. J. Phys. Chem. B 2003, 107, 668–677. [Google Scholar]
- (8).Stewart ME; Anderton CR; Thompson LB; Maria J; Gray SK; Rogers JA; Nuzzo RG Nanostructured Plasmonic Sensors. Chem. Rev 2008, 108, 494–521. [DOI] [PubMed] [Google Scholar]
- (9).Willets KA; Van Duyne RP Localized Surface Plasmon Resonance Spectroscopy and Sensing. Annu. Rev. Phys. Chem 2007, 58, 267–297. [DOI] [PubMed] [Google Scholar]
- (10).Mayer KM; Hao F; Lee S; Nordlander P; Hafner JH A Single Molecule Immunoassay by Localized Surface Plasmon Resonance. Nanotechnology 2010, 21 , No. 255503. [DOI] [PubMed] [Google Scholar]
- (11).Mayer KM; Lee S; Liao H; Rostro BC; Fuentes A; Scully PT; Nehl CL; Hafner JH A Label-free Immunoassay Based Upon Localized Surface Plasmon Resonance of Gold Nanorods. ACS Nano 2008, 2, 687–692. [DOI] [PubMed] [Google Scholar]
- (12).Olkhov RV; Fowke JD; Shaw AM Whole Serum BSA Antibody Screening Using a Label-free Biophotonic Nanoparticle Array. Anal. Biochem 2009, 385, 234–241. [DOI] [PubMed] [Google Scholar]
- (13).Mayer KM; Hafner JH Localized Surface Plasmon Resonance Sensors. Chem. Rev 2011, 111, 3828–3857. [DOI] [PubMed] [Google Scholar]
- (14).Kolluru C; Gupta R; Jiang Q; Williams M; Gholami Derami H; Cao S; Noel R; Singamaneni S; Prausnitz MR Plasmonic Paper Microneedle Patch for on-patch Detection of Molecules in Dermal Interstitial Fluid. ACS Sens. 2019, 4, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hu R; Gupta R; Wang Z; Wang C; Sun H; Singamaneni S; Kharasch ED; Morrissey JJ Bioplasmonic Paper–based Assay for Perilipin-2 Non-invasively Detects Renal Cancer. Kidney Int. 2019, 96, 1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang C; Sudlow G; Wang Z; Cao S; Jiang Q; Neiner A; Morrissey JJ; Kharasch ED; Achilefu S; Singamaneni S Metal-Organic Framework Encapsulation Preserves the Bioactivity of Protein Therapeutics. Adv. Healthcare Mater 2018, 7, No. 1800950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang C; Tadepalli S; Luan J; Liu KK; Morrissey JJ; Kharasch ED; Naik RR; Singamaneni S Metal-Organic Framework as a Protective Coating for Biodiagnostic Chips. Adv. Mater 2017, 29, No. 1604433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang C; Wang L; Tadepalli S; Morrissey JJ; Kharasch ED; Naik RR; Singamaneni S Ultrarobust Biochips with Metal–Organic Framework Coating for Point-of-Care Diagnosis. ACS Sens. 2018, 3, 342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Joshi GK; Blodgett KN; Muhoberac BB; Johnson MA; Smith KA; Sardar R Ultrasensitive Photoreversible Molecular Sensors of Azobenzene-Functionalized Plasmonic Nanoantennas. Nano Lett. 2014, 14, 532–540. [DOI] [PubMed] [Google Scholar]
- (20).Perelson AS; Oster GF Theoretical Studies of Clonal Selection: Minimal Antibody Repertoire Size and Reliability of Self-non-self Discrimination. J. Theor. Biol 1979, 81, 645–670. [DOI] [PubMed] [Google Scholar]
- (21).Frieden E Non-covalent Interactions: key to Biological Flexibility and Specificity. J. Chem. Educ 1975, 52, 754–762. [DOI] [PubMed] [Google Scholar]
- (22).Yao L; Xu S Force-induced Selective Dissociation of Noncovalent Antibody—Antigen Bonds. J. Phys. Chem. B 2012, 116, 9944–9948. [DOI] [PubMed] [Google Scholar]
- (23).Janeway CA Jr; Travers P; Walport M; Shlomchik MJ Immunological Memory In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science, 2001. [Google Scholar]
- (24).Brandts JF; Hunt L Thermodynamics of Protein Denaturation. III. Denaturation of Ribonuclease in Water and in Aqueous Urea and Aqueous Ethanol Mixtures. J. Am. Chem. Soc 1967, 89, 4826–4838. [DOI] [PubMed] [Google Scholar]
- (25).Turk BE; Huang LL; Piro ET; Cantley LC Determination of Protease Cleavage Site Motifs using Mixture-based Oriented Peptide Libraries. Nat. Biotechnol 2001, 19, 661–667. [DOI] [PubMed] [Google Scholar]
- (26).Nick Pace C; Trevino S; Prabhakaran E; Martin Scholtz J Protein structure, Stability and Solubility in Water and Other Solvents. Philos. Trans. R. Soc. Lond. B Biol. Sci 2004, 359, 1225–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Vermeer AW; Norde W The Thermal Stability of Immunoglobulin: Unfolding and Aggregation of a Multi-domain Protein. Biophys. J 2000, 78, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang C; Luan J; Tadepalli S; Liu K-K; Morrissey JJ; Kharasch ED; Naik RR; Singamaneni S Silk-Encapsulated Plasmonic Biochips with Enhanced Thermal Stability. ACS Appl. Mater. Interfaces 2016, 8, 26493–26500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).McKenzie KG; Lafleur LK; Lutz BR; Yager P Rapid Protein Depletion from Complex Samples using a Bead-based Microfluidic Device for the Point of Care. Lab Chip 2009, 9, 3543–3548. [DOI] [PubMed] [Google Scholar]
- (30).Crowe JH; Carpenter JF; Crowe LM The Role of Vitrification in Anhydrobiosis. Annu. Rev. Physiol 1998, 60, 73–103. [DOI] [PubMed] [Google Scholar]
- (31).Gubala V; Harris LF; Ricco AJ; Tan MX; Williams DE Point of Care Diagnostics: Status and Future. Anal. Chem 2012, 84, 487–515. [DOI] [PubMed] [Google Scholar]
- (32).Orendorff CJ; Gearheart L; Jana NR; Murphy CJ Aspect Ratio Dependence on Surface Enhanced Raman Scattering using Silver and Gold Nanorod Substrates. Phys. Chem. Chem. Phys 2006, 8, 165–170. [DOI] [PubMed] [Google Scholar]
- (33).Tadepalli S; Kuang Z; Jiang Q; Liu K-K; Fisher MA; Morrissey JJ; Kharasch ED; Slocik JM; Naik RR; Singamaneni S Peptide Functionalized Gold Nanorods for the Sensitive Detection of a Cardiac Biomarker using Plasmonic Paper Devices. Sci. Rep 2015, 5, No. 16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tian L; Morrissey JJ; Kattumenu R; Gandra N; Kharasch ED; Singamaneni S Bioplasmonic Paper as a Platform for Detection of Kidney Cancer Biomarkers. Anal. Chem 2012, 84, 9928–9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Goode JA; Rushworth JVH; Millner PA Biosensor Regeneration: A Review of Common Techniques and Outcomes. Langmuir 2015, 31, 6267–6276. [DOI] [PubMed] [Google Scholar]
- (36).Bhuyan AK On the Mechanism of SDS-induced Protein Denaturation. Biopolymers 2010, 93, 186–199. [DOI] [PubMed] [Google Scholar]
- (37).Michaux C; Pomroy NC; Prive GG Refolding SDS-denatured Proteins by the Addition of Amphipathic Cosolvents. J. Mol. Biol 2008, 375, 1477–1488. [DOI] [PubMed] [Google Scholar]
- (38).Xu Q; Keiderling TA Stop-flow Kinetics Studies of the Interaction of Surfactant, Sodium Dodecyl Sulfate, with Acid-denatured Cytochrome C. Proteins 2006, 63, 571–580. [DOI] [PubMed] [Google Scholar]
- (39).Tadepalli S; Yim J; Madireddi K; Luan J; Naik RR; Singamaneni S Gold Nanorod-Mediated Photothermal Enhancement of the Biocatalytic Activity of a Polymer-Encapsulated Enzyme. Chem. Mater 2017, 29, 6308–6314. [Google Scholar]
- (40).Abbas A; Tian L; Morrissey JJ; Kharasch ED; Singamaneni S Hot Spot-localized Artificial Antibodies for Label-free Plasmonic Biosensing. Adv. Funct. Mater 2013, 23, 1789–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Luan J; Xu T; Cashin J; Morrissey JJ; Kharasch ED; Singamaneni S Environmental Stability of Plasmonic Biosensors Based on Natural versus Artificial Antibody. Anal. Chem 2018, 90, 7880–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Correro MR; Moridi N; Schützinger H; Sykora S; Ammann EM; Peters EH; Dudal Y; Corvini PFX; Shahgaldian P Enzyme Shielding in an Enzyme-thin and Soft Organosilica Layer. Angew. Chem 2016, 128, 6393–6397. [DOI] [PubMed] [Google Scholar]
- (43).Luan J; Liu K-K; Tadepalli S; Jiang Q; Morrissey JJ; Kharasch ED; Singamaneni S PEGylated Artificial Antibodies: Plasmonic Biosensors with Improved Selectivity. ACS Appl Mater. Interfaces 2016, 8, 23509–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hu R; Luan J; Kharasch ED; Singamaneni S; Morrissey JJ Aromatic Functionality of Target Proteins Influences Monomer Selection for Creating Artificial Antibodies on Plasmonic Biosensors. ACS Appl. Mater. Interfaces 2017, 9, 145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Steegborn C; Skládal P Construction and Characterization of the Direct Piezoelectric Immunosensor for Atrazine Operating in Solution. Biosens. Bioelectron 1997, 12, 19–27. [DOI] [PubMed] [Google Scholar]
- (46).Bright FV; Betts TA; Litwiler KS Regenerable Fiber-optic-based Immunosensor. Anal. Chem 1990, 62, 1065–1069. [DOI] [PubMed] [Google Scholar]
- (47).Hong S-R; Choi S-J; Do Jeong H; Hong S Development of QCM Biosensor to Detect a Marine Derived Pathogenic Bacteria Edwardsiella Tarda using a Novel Immobilisation Method. Biosens. Bioelectron 2009, 24, 1635–1640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.