

SUMMARY
Oxygen (O2) is both an indispensable metabolic substrate and a regulatory signal that controls the activity of Hypoxia-Inducible Factor 1α (Hif1a), a mediator of the cellular adaptation to low O2 tension (hypoxia). Hypoxic cells require Hif1a to survive. Additionally, Hif1a is an inhibitor of mitochondrial respiration. Hence, we hypothesized that enhancing mitochondrial respiration is detrimental to the survival of hypoxic cells in vivo. We tested this hypothesis in the fetal growth plate, which is hypoxic. Our findings show that mitochondrial respiration is dispensable for survival of growth plate chondrocytes. Furthermore, its impairment prevents the extreme hypoxia and the massive chondrocyte death observed in growth plates lacking Hif1a. Consequently, augmenting mitochondrial respiration affects the survival of hypoxic chondrocytes by, at least in part, increasing intracellular hypoxia. We thus propose that partial suppression of mitochondrial respiration is crucial during development to protect the tissues that are physiologically hypoxic from lethal intracellular anoxia.
Keywords: Hypoxia, HIF, Growth Plate, Chondrocyte, Fetal Development, Mitochondria, TFAM, Mitochondrial Respiration, Glycolysis, Cell Death
eTOC blurb:
The fetal growth plate is a hypoxic structure that gives rise to most of the skeleton. It is formed by cells known as chondrocytes. Yao et al. now show that impairment of mitochondrial respiration, and thus oxygen consumption, is crucial for the survival of hypoxic chondrocytes during fetal development.
Graphical Abstract
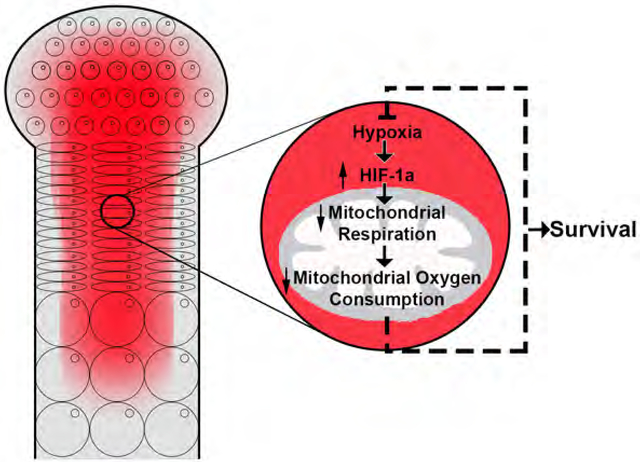
INTRODUCTION
A variety of cell types experience low oxygen (O2) tension or hypoxia during fetal development, in physiological settings of the adult organism, and in pathological conditions such as ischemia and cancer (Gandara and Wappner, 2018; Maes, et al., 2012a; Maes, et al., 2012b; Semenza, 2012; Dunwoodie, 2009). O2 controls the stability and activity of the transcription factor Hypoxia-Inducible Factor 1α (Hif1a) (Semenza, 2012). Hif1a is a mediator of the cellular adaptation to hypoxia and is required for survival of hypoxic cells (Maes, et al., 2012b; Semenza, 2012).
Mitochondrial respiration and non-oxidative glycolysis, namely the conversion of glucose to lactate, are the sources of intracellular ATP. Mitochondrial respiration is crucial for the development and homeostasis of tissues that are vascularized and well- oxygenated (Larsson and Rustin, 2001; Sorensen, et al., 2001; Wang, et al., 1999). Hif1a promotes non-oxidative glycolysis but inhibits mitochondrial respiration (LaGory, et al., 2015; Semenza, 2012; Kim, et al., 2006; Papandreou, et al., 2006). Since Hif1a is a survival factor, we hypothesized that, distinct from what has been reported in the context of well-oxygenated tissues (Larsson and Rustin, 2001; Sorensen, et al., 2001; Wang, et al., 1999), promoting mitochondrial respiration is detrimental to the survival of hypoxic cells because it increases O2 consumption and exacerbates their hypoxic status, thus leading to cell death.
For the testing of our hypothesis, we used the fetal growth plate as a model since this tissue, which is formed by cells known as chondrocytes, is avascular and hypoxic. The fetal growth plate recapitulates key events in cell biology with a very specific temporal and spatial pattern (Provot and Schipani, 2005). At first, chondrocytes, which originate from mesenchymal progenitors, proliferate at high rate and synthesize a characteristic extracellular matrix that is enriched in type II collagen and proteoglycans. Next, they exit the cell cycle and differentiate into hypertrophic cells that produce type X collagen and mineralize their surrounding matrix. Hypertrophic chondrocytes eventually die, and cartilage is invaded by blood vessels and replaced by bone with formation of a bone marrow cavity. Additionally, a few hypertrophic cells transdifferentiate into osteoblasts, which are the bone forming cells (Mizuhashi, et al., 2018; Yang, et al., 2014). The overall process is known as endochondral bone development and gives origin to most of the adult skeleton.
The fetal growth plate is a unique mesenchymal tissue since it is avascular for most of its length, although it requires an angiogenic switch for bone to be formed. Consistent with its avascularity, the fetal growth plate has an inner hypoxic region (Maes, et al., 2012b; Provot, et al., 2007; Schipani, et al., 2001), and Hif1a is an essential survival factor for fetal growth plate chondrocytes (Bentovim, et al., 2012; Maes, et al., 2012b; Schipani, et al., 2001).
In this study, we used mouse genetics to determine whether impairment of mitochondrial respiration is a requirement for the survival of hypoxic chondrocytes in vivo.
RESULTS
Mitochondrial respiration is not required for the survival of fetal growth plate chondrocytes
First, we established whether fetal growth plate chondrocytes require mitochondrial respiration for their survival. For this purpose, we conditionally inactivated Mitochondrial Transcription Factor A (Tfam) in the mesenchymal progenitors that give origin to chondrocytes by using a Prx1-Cre driver (Logan, et al., 2002), and a mouse homozygous for a floxed Tfam allele (TFAMfl/fl) (Ekstrand and Larsson, 2002; Larsson, et al., 1998). Tfam regulates expression of the mitochondrial genes encoding thirteen subunits of the electron transport chain (ETC) in the mitochondria and controls replication of mitochondrial DNA (Larsson, et al., 1998). All the other ETC subunits are products of the nuclear genomic DNA (Larsson, et al., 1998). The conditional deletion of Tfam is a powerful tool to efficiently inhibit mitochondrial respiration in vivo (Larsson, et al., 1998). Impairment of the ETC is the most consistent and best characterized biological consequence of loss of Tfam across a large variety of tissues and cell types (Vernochet, et al., 2012; Baris, et al., 2011; Sterky, et al., 2011; Weinberg, et al., 2010; Shi, et al., 2008; Ekstrand and Larsson, 2002; Larsson and Rustin, 2001; Sorensen, et al., 2001; Silva, et al., 2000; Wang, et al., 1999; Larsson, et al., 1998). Prx1-Cre;TFAMfl/fl mutant mice were born at the expected Mendelian frequency. Recombination of the floxed Tfam locus was virtually complete in Prx1-Cre;TFAMfl/fl mutant chondrocytes. As a result, Tfam mRNA, Tfam protein and mitochondrial DNA, used as a downstream target of Tfam activity, were all significantly decreased, or even undetectable, in mutant chondrocytes in vivo (Figure 1A–1D). Consistent with the loss of Tfam protein and activity and, consequently, with a severe impairment of the ETC, the mitochondrial membrane potential was also dramatically reduced in mutant chondrocytes cultured in vitro in either normoxic (20%O2) or hypoxic conditions (1%O2) (Figure 1E). However, as reported for other cell types (Wredenberg, et al., 2002), the mitochondrial mass was only modestly affected by the mutation (Figure S1E). Lastly, whereas levels of mitochondrial ROS as measured by MitoSOX Red were almost undetectable in mutant cells (Figure S1F), accumulation of total intracellular ROS was similar in control and mutant chondrocytes cultured in vitro in either normoxia or hypoxia (Figure S1G). This finding suggests that the virtual lack of signal observed in mutants upon use of MitoSOX Red was mainly secondary to an inability of the dye to cross the mitochondrial membrane as a consequence of a marked reduction of the mitochondrial membrane potential in mutant cells (Polster, et al., 2014).
Figure 1: Generation of mice lacking Tfam in the growth plate.

(A) 2-LoxP qPCR of genomic DNA extracted from newborn TFAMfl/fl (CTRL) or Prx1-Cre-TFAMfl/fl (TFAM) growth plates. Data were normalized to β2-microglobulin (n=3).
(B) Quantification of Tfam mRNA by qRT-PCR of total RNA extracted from newborn CTRL or TFAM growth plates. Data were normalized to TATA-box Binding Protein (TBP) (n=3).
(C) Quantification of Tfam protein by Western blot analysis of total protein lysate extracted from newborn CTRL or TFAM growth plates. A representative Western blot is shown on the left, and quantification of all biological replicates is provided on the right. Data were normalized to α-Tubulin (n=3).
(D) qPCR of mitochondrial DNA extracted from newborn CTRL or TFAM growth plates. Mitochondrial genes coding for Cytochrome b (mt-Cytb), 16S rRNA (mt-Rnr2) and Cytochrome c oxidase subunit 3 (mt-Co3) were analyzed. Data were normalized to β2- microglobulin (n=3).
(E) FACS analysis of mitochondrial membrane potential in chondrocytes isolated from CTRL or TFAM newborn growth plates and cultured in 20% or 1% oxygen (O2). A representative experiment is shown at the top; quantification of all biological replicates (n=4) is provided at the bottom. Results are expressed as percentage of CTRL 20%O2. See also Figure S1A–D.
(F) Whole mount Alcian Blue/Alizarin Red S staining of newborn forelimbs isolated from CTRL, Prx1-Cre-TFAM+/fl (Het-TFAM) and TFAM newborn mice. Alcian Blue detects sulfated glycosaminoglycans accumulation in the cartilaginous matrix; Alizarin Red S binds to calcium in the mineralized matrix (Mangiavini, et al., 2016). Representative specimens are shown on the left, and quantification of humerus length is provided on the right (n=7). Scale bars=2mm.
Data are presented as mean ± SEM; *p<0.05; ***p<0.01.
The Prx1-Cre driver mediates efficient Cre recombinase activity in the mesenchymal condensations that give origin to the forelimbs, hindimbs and sterna (Mangiavini, et al., 2015; Logan, et al., 2002). Moreover, in the Prx1-Cre transgenic mouse, Cre recombinase activity starts earlier in development and is higher in forelimbs than hindlimbs (Logan, et al., 2002). Therefore, throughout the study we systematically investigated the growth plates of forelimb stylopod (humerus), forelimb autopods and sterna.
Despite the loss of Tfam protein and activity, patterning of the limbs and development of the joints occurred normally in Prx1-Cre;TFAMfl/fl mutants (Figure 1F and Figure S2). Moreover, Prx1-Cre;TFAMfl/fl mutant limbs were similar in shape to Prx1-Cre;TFAMfl/+ and TFAMfl/fl control specimens, even if significantly shorter (Figure 1F). The overall structure of the developing growth plate was also not altered by the mutation, although hypertrophic differentiation was modestly delayed (Figure 2A and 2B), which explains, at least in part, the shortening of the limbs in the Prx1-Cre;TFAMfl/fl mutants. Chondrocyte proliferation rate was comparable in mutant and control samples (Figure 2C). More importantly, no dying cells could be identified in the proliferative layer of the mutant fetal growth plate (Figure 2D). This is a striking feature since the loss of Tfam has been associated with increased cell death in tissues other than cartilage (Wang, et al., 2001). In addition, a similar number of dying cells could be counted in the hypertrophic layer of both mutant and control growth plates (Figure 2D). Therefore, chondrocytes lacking Tfam do not die when they are proliferating but they retain a functional cell death machinery upon hypertrophic differentiation.
Figure 2: Loss of Tfam in the fetal growth plates.

(A) Safranin-O staining (representative brightfield images) and in situ hybridization for Sox9, type II collagen (Col2a1) and type X collagen (Col10a1) mRNAs (representative darkfield images) performed on histological sections of E13.5 TFAMfl/fl (CTRL) Prx1-Cre- TFAM+/fl (Het-TFAM) and Prx1-Cre-TFAMfl/fl (TFAM) humeri (n=3). Scale bars=200μm for full view panels and scale bars= 50μm for high magnification panels.
Safranin-O detects glycosaminoglycans accumulation in the cartilaginous matrix (Mangiavini, et al., 2016). Sox9 mRNA is exclusively expressed in the proliferative layer of normally developing growth plates (Provot and Schipani, 2005). Col2a1 mRNA is a marker of proliferative chondrocytes and early hypertrophic cells, whereas Col10a1 mRNA is a marker of hypertrophic chondrocytes (Provot and Schipani, 2005).
Loss of Tfam delayed chondrocyte hypertrophy as indicated by routine histology, by the uninterrupted expression of Sox9 mRNA, and by the smaller expression domain of Col10a1 mRNA.
(B) Safranin-O staining (representative brightfield images) and in situ hybridization analysis for Col2a1 and Col10a1 mRNAs (representative darkfield images) performed on histological sections of E15.5 CTRL, Het-TFAM and TFAM humeri (n=4). Scale bars=200μm.
CTRL and Het-TFAM were phenotypically identical. Conversely, the distance between the two Col10a1 mRNA domains and, thus, the extension of the bone marrow cavity, was modestly reduced in the E15.5 TFAM growth plate when compared to CTRL. This feature was most likely the consequence of the delayed appearance of hypertrophic chondrocytes and, thus, the delayed replacement of cartilage by bone in mutant specimens.
(C, D, E) Analysis of chondrocyte proliferation rate (n=3) by EdU assay (C), cell death (n=3) by TUNEL assay (D) and detection of hypoxia (n=5) by EF5 assay (E) in proximal growth plates of E15.5 CTRL and TFAM humeri. Representative images are shown at the top. EdU and TUNEL positive cells are shown in green, EF5 and DAPI positive cells in red and blue, respectively. Percentage of positive cells (C, D, E) and graphic representation of the EF5 signal (E) are provided at the bottom. In (E), the peripheral regions of the fetal growth plate and its inner core are indicated as “outer” and “inner”, respectively. Signal intensity is expressed in arbitrary unit (AU). Scale bars = 200μm. Data are presented as mean ± SEM; *p<0.05; ***p<0.01.
Lastly, no hypoxia was detected in the mutant fetal growth plate (Figure 2E), which indicates that mitochondrial respiration is a determinant of the inner hypoxic region present in the normal fetal growth plate. In agreement with these data, O2 consumption rate was significantly decreased in chondrocytes isolated from newborn TFAMfl/fl mice and transduced in vitro with Cre Adenovirus when compared to control cells (Figure S3A).
Taken together, our in vivo genetic experiment demonstrated that impairment of mitochondrial respiration is compatible with both differentiation of mesenchymal progenitor cells into chondrocytes and proliferation, survival, and physiological death of chondrocytes, albeit it delays their timely hypertrophy.
Impairment of mitochondrial respiration prevents the massive cell death of fetal growth plate chondrocytes lacking Hif1a
Next, we asked whether enhancing mitochondrial respiration leads to chondrocyte death and if this could be prevented by loss of Tfam. For this purpose, we took advantage of a loss-of-function mutation of Hif1a we had previously generated in mesenchymal progenitor cells of the limb bud (Prx1-Cre;HIF-1afl/fl mice) (Provot, et al., 2007).
Loss of Hif1a causes massive chondrocyte death in both the proliferative and hypertrophic layers of the fetal growth plate (Provot, et al., 2007; Schipani, et al., 2001). Expression of Hif1a in the developing growth plate ensures an adequate number of blood vessels in the perichondrium (Maes, et al., 2012a) and a proper accumulation of extracellular matrix in cartilage (Aro, et al., 2012; Bentovim, et al., 2012; Pfander, et al., 2003). However, these effects contribute only partially to the survival function of Hif1a in chondrocytes (Maes, et al., 2012a; Maes, et al., 2012b). More importantly, the viable chondrocytes present in Hif1a null growth plates are considerably more hypoxic than controls (Schipani, et al., 2001), and their extreme hypoxia cannot be corrected by augmenting delivery of O2 to the growth plate through an increase of the number of blood vessels in the soft tissue surrounding the growth plate (Maes, et al., 2012a). The degree of oxygenation of any cell is the net result of O2 availability and O2 consumption (Harvey, 1928). Since the severe hypoxia of Hif1a null chondrocytes could not be corrected by increasing O2 availability, it had to be the result of an increased O2 consumption. Consistent with this possibility, chondrocytes isolated from newborn HIF-1afl/fl mice and transduced in vitro with Cre Adenovirus displayed an increased O2 consumption rate when compared to control cells (Figure S3B).
We thus reasoned that the loss of Hif1a might cause the death of chondrocytes in vivo by augmenting mitochondrial respiration and, as a result, mitochondrial O2 consumption and intracellular hypoxia.
For the testing of our hypothesis, we generated mutant mice lacking both Tfam and Hif1a in mesenchymal progenitor cells of the limb bud. Of note, Tfam is not a direct downstream target of Hif1a (LaGory, et al., 2015). Prx1-Cre;TFAMfl/fl;HIF-1afl/fl double mutants and Prx1-Cre;TFAMfl/+;HIF-1afl/fl single mutants were born at the expected Mendelian frequency. Recombination of both the floxed Tfam locus and the floxed Hif1a locus was virtually complete in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl double mutant chondrocytes in vivo and Tfam and Hif1a activities were either undetectable or severely impaired in the same cells (Figure S4A–S4D). In addition, different from what we previously reported (Mangiavini, et al., 2014), in the current study no significant recombination of either the floxed Tfam locus or the floxed Hif1a locus could be detected in hepatocytes of Prx1-Cre;TFAMfl/fl;HIF-1afl/fl double mutants (Figure S4E), which excludes the presence of a significant and confounding liver phenotype in those mutant mice.
Like Prx1-Cre;TFAMfl/fl single mutant limbs, double mutant limbs lacking both Tfam and Hif1a were considerably shorter than controls but significantly longer than Prx1- Cre;TFAMfl/+;HIF-1afl/fl single mutants (Figure 3A). The shortening of the Prx1- Cre;TFAMfl/fl;HIF-1afl/fl double mutant limbs was most likely secondary to the delay of hypertrophic differentiation caused by loss of Tfam (Figure 3B, Figure 4, Figure S4F,G).
Figure 3: Loss of both Tfam and Hif1a in the growth plate.

(A) Whole mount Alcian Blue/Alizarin Red S staining of newborn forelimbs isolated from TFAMfl/fl;HIF-1afl/fl (CTRL), Prx1-Cre;TFAMfl/+;HIF-1afl/fl (HIF1) and Prx1- Cre;TFAMfl/fl;HIF-1afl/fl (TFAM/HIF1) mice. Representative specimens are shown on the left, and quantification of humerus length is provided on the right (n=4). Scale bars=2mm.
(B) In situ hybridization analysis for Col2a1 or Col10a1 mRNA on histological sections of E15.5 CTRL, HIF1 and TFAM/HIF1 humeri (n=3). Of note, some degree of hypocellularity is still present in the proximal growth plate of the TFAM/HIF1 humerus (arrow), which is indicative of persisting cell death. P=proximal; D=distal. Scale bars=200μm.
(C) H&E and Safranin-O staining performed on histological sections of distal growth plates of E15.5 CTRL, HIF1 and TFAM/HIF1 humeri (n=3). Scale bars=50μm.
(D) Quantification of cell death by TUNEL assay in CTRL, HIF1 and TFAM/HIF1 distal growth plates of E15.5 humeri. Representative images are shown on the left. Positive signal appears in green for TUNEL and blue for DAPI staining. Quantification of all biological replicates is provided on the right (n=3). Scale bars=100μm.
Data are presented as mean ± SEM; *p<0.05; ***p<0.01.
Figure 4: Loss of both Tfam and Hif1a in the developing autopod.

(A) Whole mount Alcian Blue/Alizarin Red S staining of autopods isolated from newborn TFAMfl/fl;HIF-1afl/fl (CTRL), Prx1-Cre;TFAMfl/+;HIF-1afl/fl (HIF1) and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl (TFAM/HIF1) mice (n=4). Scale bars=1mm. Of note, Alizarin Red S staining is decreased in the autopods of the TFAM/HIF1 mice compared to CTRL. This finding is consistent with the delay of hypertrophic differentiation caused by loss of Tfam (Figure 2).
(B) H&E staining (representative brightfield images) (low magnification at the top, high magnification at the bottom) and in situ hybridization analysis for type II collagen (Col2a1), type X collagen (Col10a1) and growth differentiation factor 5 (Gdf5) mRNAs (low magnification at the top, high magnification at the bottom) (representative darkfield images) performed on histological sections of autopods isolated from E15.5 CTRL, HIF1 and TFAM/HIF1 mice, respectively (n=3). Note the reduced intensity of Col10a1 mRNA signal in TFAM/HIF1 specimens, which is consistent with the delay of hypertrophic differentiation caused by loss of Tfam (Figure 2). Arrowheads point to the specified interzones in the developing joints. Scale bars=200μm for full view panels and scale bars= 50μm for high magnification panels.
The Prx1-Cre;TFAMfl/+;HIF-1afl/fl single mutant growth plate was wider, misshapen and severely hypocellular in its inner region as a consequence of a devastating cell death at this site when compared to controls (Figure 3A–D; Figure S4F,G). Of note, Prx1- Cre;TFAMfl/+;HIF-1afl/fl single mutant mice were phenotypically indistinguishable from the Prx1-Cre;HIF-1afl/fl mutants we had previously reported (Provot, et al., 2007) (Figure S5).
Loss of Tfam not only corrected the abnormal shape (Figure 3A) and the severely distorted architecture of the Prx1-Cre;TFAMfl/+;HIF-1afl/fl growth plate (Figure 3B), but also prevented the massive cell death observed in its inner core (Figure 3C–3D and Figure S4G). The “rescue” of the cell death was virtually complete in the growth plates of sternum and distal epiphysis of humerus, whereas modest signs of residual cell death continued to be present in the proximal epiphysis of humerus, which is thicker than the distal one (Figure 3B and Figure S4F,G). Similarly to Prx1-Cre;TFAMfl/fl single mutants, the percentage of proliferative chondrocytes in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl mutants (14% ± 2) and controls (12% ± 1) was not significantly different.
Taken together, our data indicate that in vivo loss of Tfam enabled to a large extent the survival of fetal growth plate chondrocytes lacking Hif1a.
Impairment of mitochondrial respiration prevents the delay of joint development observed in autopods lacking Hif1a
Like the developing growth plates, the mesenchymal condensations that give origin to those growth plates are also hypoxic (Amarilio, et al., 2007; Provot, et al., 2007). Loss of Hif1a in the mesenchymal condensation delays the differentiation of mesenchymal progenitor cells into chondrocytes in vivo (Amarilio, et al., 2007; Provot, et al., 2007). It also causes a joint segmentation defect that is secondary to a delay of joint specification (Amarilio, et al., 2007; Provot, et al., 2007). Different from the stylopod and zeugopod, in the autopod both events precede the massive chondrocyte death, which at that site occurs only late in development probably because of the unique geometry and reduced thickness of the skeletal segments forming the autopod (Provot, et al., 2007). Although the focus of our current study was not the role of mitochondrial respiration in mesenchymal cell differentiation and joint development, since the Prx1-Cre driver targets the whole limb (Logan, et al., 2002), to be thorough, we investigated the autopod of mutant mice lacking both Tfam and Hif1a. Indeed, concomitant loss of Tfam and Hif1a fully corrected the segmentation defect and the delayed joint specification observed in Prx1-Cre;TFAMfl/+;HIF-1afl/fl single mutant autopods, as indicated by whole mount Alcian Blue/Alizarin Red S staining, routine histology and in situ hybridization analysis for the detection of Growth Differentiation Factor-5 (GDF5) mRNA, which is an early marker of joint specification (Storm and Kingsley, 1999) (Figure 4A–4B). Moreover, the delayed differentiation of mesenchymal progenitor cells into chondrocytes secondary to the loss of Hif1a was also partially prevented in double mutant autopods lacking both Tfam and Hif1a, as shown by the increased expression of type II collagen mRNA in the Prx1-Cre;TFAMfl/fl;HIF-1afl/fl double mutant autopods when compared to Prx1-Cre;TFAMfl/+;HIF-1afl/fl single mutants (Figure 4B).
Taken together, our data indicate that in vivo loss of Tfam prevented the altered joint development observed in the mutant autopod lacking Hif1a and partially corrected the delayed differentiation of mesenchymal progenitor cells into chondrocytes at the same site.
Impairment of mitochondrial respiration leads to a compensatory increase of non- oxidative glycolysis in both control and mutant chondrocytes lacking Hif1a We then asked why, distinct from Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocytes, Prx1- Cre;TFAMfl/fl;HIF-1afl/fl cells do not die.
Hif2 is another member of the Hif family of transcription factors. Hif1 and Hif2 have been shown to have unique, overlapping and even opposing functions depending on the cellular context (Keith, et al., 2011). We determined that, distinct from Hif1a, Hif2 is dispensable for growth plate development (Araldi, et al., 2011). Moreover, conditional knockout of both Hif1 and Hif2 in mesenchymal progenitors generates growth plate abnormalities that are identical to the ones observed in specimens lacking only Hif1 (Merceron, et al., 2019). Nonetheless, to rule out the unlikely event that impairment of mitochondrial respiration could cause a compensatory increase of Hif2a activity in chondrocytes and this would be the reason why Prx1-Cre;TFAMfl/fl;HIF-1afl/fl cells do not die, we generated Prx1-Cre;TFAMfl/fl;HIF-1afl/fl;HIF-2afl/fl triple mutants (Gruber, et al., 2007). Efficient deletion of the Hif2a floxed locus was confirmed by qPCR, as previously reported (Merceron, et al., 2019). More importantly, as shown by histological analysis, Prx1-Cre;TFAMfl/fl;HIF-1afl/fl;HIF-2afl/fl triple mutant growth plates appeared to be indistinguishable from Prx1-Cre;TFAMfl/fl;HIF-1afl/fl double mutants (Figure S5C). This finding implies that the prevention of cell death in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl double mutant growth plates is not due to a compensatory increase of Hif2 activity. Next, we interrogated the metabolomic profile of mutant and control chondrocytes. Steady-state levels of intracellular ATP, as well as the ATP/ADP ratio, were similar in both Prx1-Cre;TFAMfl/fl;HIF-1afl/fl and Prx1-Cre;TFAMfl/+;HIF-1afl/fl mutant and TFAMfl/fl;HIF-1afl/fl control chondrocytes when cultured in vitro in either normoxia or hypoxia (Figure 5A). In agreement with these data, the pAMPK/AMPK ratio, which classically reflects the energy status of cells (Lin and Hardie, 2018), was comparable across the various experimental conditions (Figure 5B).
Figure 5: Energy levels in Prx1-Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF- 1afl/fl chondrocytes.

(A) Measurements of intracellular ATP, ADP, ATP/ADP ratio, intracellular lactate, intracellular glucose, lactate/glucose ratio, intracellular NADH, NAD and NADH/NAD ratio in chondrocytes isolated from TFAMfl/fl;HIF-1afl/fl (CTRL), Prx1-Cre;TFAMfl/+;HIF- 1afl/fl (HIF1) and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl (TFAM/HIF1) newborn growth plates and cultured in either 20% or 1%O2. Data were calculated as percentage of CTRL 20% O2 (n=3). Note that loss of Hif1a reduces intracellular lactate also in normoxia. However, this finding shows some degree of inter-assay variability (Figure S7B), most likely due to the very low levels of Hif1a protein in normoxia (Figure S6).
(B) Quantification of LDHA and pAMPK proteins by Western blot analysis of total protein lysate extracted from chondrocytes isolated from CTRL, HIF1 and TFAM/HIF1 newborn growth plates. Representative Western blots are shown on the left and quantification of all biological replicates is provided on the right. Data for LDHA and pAMPK were normalized to α-Tubulin and AMPK respectively, and calculated as percentage of the signal intensity of CTRL 20%O2 (n=3). Data are presented as mean ± SEM; *p<0.05; ***p<0.01. See also Figure S6A–B.
Consistent with the critical role for Hif1a in the regulation of non-oxidative glycolysis (Semenza, 2012), levels of intracellular lactate were significantly higher in control chondrocytes cultured at 1% O2 than in control cells at 20% O2. This increase did not occur upon loss of Hif1a in Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocytes but was restored by concomitant loss of Tfam in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl cells (Figure 5A). Of note, measurement of intracellular lactate has been shown to be a reliable indicator of lactate production in hypoxia (Lee, et al., 2015).
Intracellular glucose accumulation in both control and double mutant cells lacking Tfam and Hif1a was significantly lower than in Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocytes, whereas lactate over glucose ratio was significantly higher in control and Prx1- Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes than in Prx1-Cre;TFAMfl/+;HIF-1afl/fl cells but indistinguishable from controls (Figure 5A).
In agreement with the Hif1a-dependent impairment of the mitochondrial ETC, levels of intracellular NADH and the NADH/NAD ratio were higher in control chondrocytes cultured at 1% O2 than in control cells at 20% O2 (Figure 5A). This increase did not occur upon loss of Hif1a in Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocytes but was restored by loss of Tfam (Figure 5A). In addition, both parameters were significantly augmented in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes cultured in normoxia when compared to normoxic control and Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocytes (Figure 5A). NADH is an electron donor in both the mitochondrial ETC and the intracytoplasmic reaction that converts pyruvate to lactate.
Lastly, accumulation of lactic dehydrogenase-A (LDHA), which is the enzyme responsible for the conversion of pyruvate into lactate and is also a classical downstream target of Hif1a (Semenza, 2017), as expected, was increased in control cells cultured in hypoxia when compared to control cells in normoxia (Figure 5B). However, such an increase did not occur in either Prx1-Cre;TFAMfl/fl;HIF-1afl/fl or Prx1- Cre;TFAMfl/+;HIF-1afl/fl chondrocytes cultured in hypoxia.
Taken together, our findings demonstrate that loss of Hif1a did not lower levels of intracellular ATP, which suggests that growth plate chondrocytes have an excellent ability to generate ATP from either oxidative phosphorylation or non-oxidative glycolysis. Moreover, they show that loss of Tfam in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes led to a compensatory and Hif1a-independent increase of lactate accumulation. This increase was probably contributed by glucose and was not secondary to an up- regulation of LDHA expression but rather due to the high levels of intracellular NADH, which likely drove, at least in part, a compensatory augmentation of non-oxidative glycolysis.
Consistent with this interpretation, loss of Tfam per se led to a significant augmentation of lactate and NADH accumulation in both normoxia and hypoxia without affecting levels of intracellular ATP, LDHA and pAMPK/AMPK ratio when compared to control cells (Figure S7A).
Of note, accumulation of intracellular ATP and lactate were similar in Prx1- Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;HIF-1afl/fl cells (Figure 5 and Figure S7B), which implies that regarding those parameters, heterozygous loss of TFAM had not significantly modified the metabolic profile of chondrocytes lacking Hif1a. Loss of Tfam thus most likely maintained normal levels of intracellular ATP in chondrocytes lacking Hif1a by normalizing non-oxidative glycolysis and the accumulation of intracellular lactate at least in part through an increase of intracellular NADH. However, since Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocyte do not die because they are energy deficient, the compensatory increase of non-oxidative glycolysis was not the reason cell death was prevented in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl growth plates.
Impairment of mitochondrial respiration prevents the severe intracellular hypoxia of fetal growth plate chondrocytes lacking Hif1a
In agreement with the data obtained in chondrocytes exclusively lacking TFAM, a severe reduction of the mitochondrial membrane potential was detected in Prx1- Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes, whereas loss of Hif1a alone did not affect this parameter (Figure 6A). Mitochondrial mass was the same across the various genotypes and in both hypoxia and normoxia (Figure 6B). Likewise, levels of total ROS were not affected by either mutation or by cell exposure to hypoxia (Figure 6C). Consistent with the data obtained in chondrocytes exclusively lacking Tfam, mitoSOX Red signal was severely reduced in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes in both normoxia and hypoxia (Figure 6D). More importantly, however, Hif1a deficiency per se did not significantly affect the levels of mitochondrial ROS in either normoxia or hypoxia when compared to control cells in normoxia (Figure 6D). Lastly, exposure of control cells to hypoxia significantly decreased mitochondrial ROS in comparison to normoxic controls (Figure 6D). Of note, distinct from what occurs in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes, mitoSOX Red is a reliable indicator of mitochondrial ROS levels in control and Prx1-Cre;TFAMfl/+;HIF-1afl/fl mutant chondrocytes since mitochondrial membrane potential is normal in those cells (Figure 6A).
Figure 6: Mitochondrial parameters in chondrocytes isolated from Prx1-Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl chondrocytes.

(A, B, C, D) FACS analysis of mitochondrial membrane potential (A), mitochondrial mass (B), total ROS (C) and mitochondrial ROS (D) in chondrocytes isolated from TFAMfl/fl;HIF-1afl/fl (CTRL), Prx1-Cre;TFAMfl/+;HIF-1afl/fl (HIF1) and Prx1- Cre;TFAMfl/fl;HIF-1afl/fl (TFAM/HIF1) newborn growth plates and cultured in 20% or 1%O2. A representative experiment is shown at the top; quantification of all biological replicates is provided at the bottom (n=3). Signal intensity was calculated as percentage of CTRL 20%O2. In all the graphs, data are presented as mean ± SEM; *p<0.05; ***p<0.01. Of note, levels of mitochondrial ROS in hypoxic HIF1 chondrocytes were indistinguishable from normoxic controls, although significantly higher than controls at 1%O2.
Taken together, our findings show that, differently from the reports in other cell types (Semenza, 2012; Kim, et al., 2006), the chondrocyte death observed in the Prx1- Cre;TFAMfl/+;HIF-1afl/fl growth plate was not secondary to an augmentation of mitochondrial ROS accumulation.
Remarkably, the concomitant loss of Tfam and Hif1a prevented the dramatic increase of intracellular hypoxia observed in mutant growth plates lacking exclusively Hif1a (Figure 7A). In addition, levels of intracellular hypoxia were somewhat higher in those Prx1- Cre;TFAMfl/fl;HIF-1afl/fl growth plates such as the proximal epiphysis of humerus that still displayed histological evidence of ectopic cell death when compared to controls (Figure 7B). Accordingly, in Prx1-Cre;TFAMfl/fl;HIF-1afl/fl and Prx1-Cre;TFAMfl/+;HIF-1afl/fl humeri the percentage of chondrocytes positive for EF5 staining correlated with the percentage of positive cells for TUNEL with R2 values of 0.906 and 0.969 for proximal and distal growth plates, respectively.
Figure 7: Hypoxia in Prx1-Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl fetal growth plates.

(A, B) Detection of hypoxia on histological sections of TFAMfl/fl;HIF1fl/fl (CTRL), Prx1-Cre;TFAMfl/+;HIF-1afl/fl (HIF1) and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl (TFAM/HIF1) distal (A) and proximal (B) growth plates of E15.5 humeri using the marker of hypoxia EF5. Typical images are shown on the top and their graphic representation and quantification on the bottom (n=3). Signal intensity is expressed in arbitrary unit (AU). Scale bars=50μm. Consistent with the finding in the TFAM growth plate (Figure 2E), virtually no EF5 staining was detectable in the TFAM/HIF1 distal epiphysis of humerus.
(C) Measurements of O2 consumption rate (OCR) in chondrocytes isolated from HIF- 1afl/fl or TFAMfl/fl;HIF-1afl/fl mice and transduced in vitro with Cre Adenovirus (Ad-Cre). A representative experiment is shown at the top and quantification of all biological replicates at the bottom (n=6). FCCP = carbonyl cyanide-p- trifluoromethoxyphenylhydrazone. Data were calculated as fold over HIF-1afl/fl Ad-Cre cells at baseline. OCR was measured both at baseline and in “uncoupling” conditions. The “uncoupling” experiment was performed to maximize the O2 consumption by mitochondria (Brand and Nicholls, 2011). This is particularly important in primary chondrocytes as those cells have a low O2 consumption rate at baseline.
(D) Counting of dead cells by trypan blue staining in in vitro cultures of chondrocytes isolated from TFAMfl/fl;HIF-1afl/fl (CTRL), Prx1-Cre;TFAMfl/+;HIF-1afl/fl (HIF1) and Prx1- Cre;TFAMfl/fl;HIF-1afl/fl (TFAM/HIF1) newborn growth plates and maintained in 20% or 1%O2 for 4 days (n=4) (top panel). Western blot analysis of total protein lysate extracted from newborn CTRL or TFAM growth plates with an antibody specific for both full length (35kDa) and cleaved (17–19kDa) Caspase-3. A representative Western blot is shown (bottom panel). Data were normalized to α-Tubulin (n=3). Data are presented as mean ± SEM; *p<0.05; ***p<0.01
(E) Proposed model: Hif1a is essential for survival of hypoxic chondrocytes by negatively regulating mitochondrial respiration and mitochondrial O2 consumption and, thus, keeping hypoxia “in check”.
In agreement with those findings, loss of Tfam corrected the augmentation of the O2 consumption rate observed in chondrocytes lacking Hif1a (Figure 7C).
Lastly, cell death was dramatically increased in mutant chondrocytes lacking Hif1a and cultured in hypoxia when compared to both control cells either in normoxia or hypoxia and HIF1 mutants in normoxia (Figure 7D). More importantly, as it occurs in vivo, the death of HIF1 null chondrocytes was fully prevented by concomitant loss of Tfam (Figure 7D). The hypoxia-dependent cell death of Hif1a deficient chondrocytes occurred in a Caspase-3-independent manner (Figure 7D). Caspase-3 is the final effector of the apoptotic death machinery (Barman, et al., 2018).
Collectively, our data support the notion that a causative link exists between increased intracellular hypoxia and death in Prx1-Cre;TFAMfl/+;HIF-1afl/fl chondrocytes, In particular, they suggest that chondrocytes lacking Hif1a died as a result of their severe intracellular hypoxia; moreover, the cell death was prevented by concomitant loss of Tfam and was not mediated by Caspase-3.
DISCUSSION
In this study, we established that mitochondrial respiration is not required for survival of fetal growth plate chondrocytes. Moreover, we tightly associated the survival function of Hif1a to the inhibition of mitochondrial respiration in vivo during development.
We now propose the Hif1a-dependent suppression of mitochondrial O2 consumption during growth plate development is essential in order to prevent chondrocytes, which are physiologically hypoxic, from becoming virtually anoxic and dying (Figure 7E).
Hif1a is well known to reprogram metabolism by promoting non-oxidative glycolysis and inhibiting mitochondrial respiration with a variety of molecular mechanisms (LaGory, et al., 2015; Semenza, 2012; Kim, et al., 2006; Papandreou, et al., 2006). In particular, Hif1a increases expression of glucose transporters and numerous enzymes of the non- oxidative glycolytic pathway (Semenza, 2012). It also shunts pyruvate away from the mitochondria by activating the gene encoding pyruvate dehydrogenase (PDH) kinase 1 (PDK-1) (Kim, et al., 2006; Papandreou, et al., 2006). PDK-1 phosphorylates PDH, which is the enzyme that converts pyruvate to acetyl coenzyme A for entry into the TCA cycle. In addition, Hif1a decreases mitochondrial mass by promoting mitochondrial autophagy through the upregulation of Bcl2 Interacting Protein 3 (Bnip3) (Zhang, et al., 2008). Furthermore, it activates the transcription of the microRNA miR210, which has been shown to block expression of the iron-sulfur cluster assembly proteins that are required for the function of the TCA cycle and of the ETC (Chan, et al., 2009). Lastly, Hif1a impairs mitochondrial biogenesis by reducing levels of expression of proliferator- activated receptor-γ coactivator-1α(PGC-1α) (LaGory, et al., 2015).
In this study, consistent with the observation in every other cell type investigated thus far, we demonstrated that loss of Hif1a in growth plate chondrocytes augments mitochondrial O2 consumption with mechanisms that may involve, at least in part, regulation of mitochondrial autophagy. Previous papers reported a key role of Hif1a in regulating expression of PDK-1 in chondrocytes as well (Bentovim, et al., 2012). Moreover, we show that Hif1a in chondrocytes boosts non-oxidative glycolysis by, at least in part, up-regulating expression of LDHA.
It has been reported that loss of Hif1a induces cell death by promoting ROS accumulation upon stimulation of mitochondrial respiration (Kim, et al., 2006). We now propose that, distinct from what has been reported for other cell types, augmentation of intracellular hypoxia rather than intracellular ROS is the reason hypoxic chondrocytes lacking Hif1a die.
O2 is an indispensable metabolic substrate for numerous reactions that take place either in the cytosol or the nucleus; therefore, extreme hypoxia can affect cell viability by impinging on multiple aspects of cell biology and with modalities that are independent of either the generation of mitochondrial ATP or the regulation of Hif activity. In particular, among others, extreme hypoxia modulates, in presence of normal levels of ATP and in a Hif-independent manner, epigenetic modifications of histones, redox reactions in the endoplasmic reticulum, cytoplasmic translation of proteins, p53 activity and the DNA damage response (Salminen, et al., 2015; Olcina, et al., 2014; Fels and Koumenis, 2006; Liu and Simon, 2004; Graeber, et al., 1996). The identification of the cellular functions that are severely altered by extreme hypoxia and, as a result, cause chondrocyte death in the developing growth plate, warrants further investigations. Nonetheless, our genetic models prove that the use of non-oxidative glycolysis over mitochondrial respiration is crucial to protect embryos, which physiologically experience steeper gradients of oxygenation than adults (Gandara and Wappner, 2018), and, more generally, hypoxic tissues from lethal intracellular anoxia.
It is unlikely that a putative impairment of the mitochondrial apoptotic machinery contributes to preventing cell death in growth plates lacking both Tfam and Hif1a as Tfam deficiency per se has been associated with increased rather than reduced apoptosis (Wang, et al., 2001). Along those lines, in our study, percentage of dying hypertrophic chondrocytes, which are cells that normally undergo apoptosis during development (Provot and Schipani, 2005), was similar in control and mutant growth plates lacking Tfam. Lastly, the hypoxia-dependent cell death of Hif1a deficient chondrocytes in vitro was Caspase-3-independent.
In some developing growth plates such as the proximal epiphysis of the humerus both the cell death and the increased intracellular hypoxia secondary to loss of Hif1a were not fully corrected by the concomitant loss of Tfam. These data are in agreement with the additional roles played by other targets downstream of Hif1a as a survival factor including proteins that regulate angiogenesis or matrix accumulation (Aro, et al., 2012; Bentovim, et al., 2012; Maes, et al., 2012a).
Loss of Tfam also corrected the abnormal joint development observed in the mutant autopods lacking Hif1a and partially prevented the delayed differentiation of mesenchymal progenitor cells into chondrocytes at the same site. These findings show that mitochondrial respiration and thus mitochondrial O2 consumption are downstream of both the survival and differentiation functions of Hif1a in mesenchymal progenitor cells and chondrocytes during endochondral bone development. Our study provides an example of patterning and differentiation events in mammals that are affected by severe hypoxia, with yet unknown mechanisms.
Chondrocyte hypertrophy is a critical step of endochondral bone development and is a regulated by a complex interplay of numerous ligands, receptors and transcription factors (Liu, et al., 2017). Loss of Tfam delays terminal differentiation of growth plate chondrocytes. This finding is consistent with the notion that mitochondrial respiration is often a requirement for differentiated cells (Hamanaka, et al., 2013; Shyh-Chang, et al., 2013). Additional investigations are warranted to identify the molecular mechanisms mediating role of TFAM in chondrocyte hypertrophy.
Notably, loss of Hif1a did not decrease levels of intracellular ATP in growth plate chondrocytes, which is consistent with the notion that Hif1a deficiency impairs non-oxidative glycolysis and augments mitochondrial respiration. Even more remarkable, intracellular ATP was normal in chondrocytes lacking both Hif1a and TFAM, which was likely due to a compensatory increase of non-oxidative glycolysis as indicated by the measurements of lactate and glucose in those cells. These findings are consistent with previous reports showing that either loss of TFAM or mitochondrial DNA mutations could indeed be associated to compensatory increases of glycolysis (Wen, et al., 2019; Celotto, et al., 2011). However, reduced ATP consumption secondary to partial suppression of biosynthetic processes may also have contributed to keep the levels of intracellular ATP within the normal range in chondrocytes lacking both Hif1a and TFAM. Nonetheless, reprogramming of glycolytic metabolism also occurs in growth plate chondrocytes upon loss of Tfam and in a Hif1a-independent manner. This finding could pave the way to novel strategies to control metabolic pathways during skeletal development.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ernestina Schipani (eschipan@med.umich.edu).
Experimental model and subject details
Mouse strains
Generation and genotyping of TFAMfl/fl floxed (C57/B6), HIF-1afl/fl floxed (FVB/N), HIF- 2afl/fl floxed (C57/B6) and Prx1-Cre (FVB/N) transgenic mice have been previously described (Gruber, et al., 2007; Logan, et al., 2002; Schipani, et al., 2001; Larsson, et al., 1998). Sequences of primers used for genotyping are listed in the key resource table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti alpha-Tubulin | Cell Signaling Technology | Cat# 2125 |
| Rabbit polyclonal anti Tfam | Abcam | Cat# ab131607, RRID:AB_11154693 |
| Rabbit polyclonal anti HIF-1 alpha | Novus Biologicals | Novus Cat# NB100–479SS, RRID:AB_790147 |
| Rabbit polyclonal anti BNIP3 | Cell Signaling Technology | Cat# 3769, RRID:AB_2259284 |
| Rabbit polyclonal anti LDHA | Cell Signaling Technology | Cat# 2012, RRID:AB_2137173 |
| Rabbit polyclonal anti AMPK | Cell Signaling Technology | Cat# 2532, RRID:AB_330331 |
| Rabbit polyclonal anti pAMPK | Cell Signaling Technology | Cat# 2531, RRID:AB_330330 |
| Rabbit polyclonal anti Caspase-3 | Cell Signaling Technology | Cat# 9662, RRID: AB_331439 |
| Anti-EF5 antibody (ELK3–51) Cy3 | Hypoxia-Imaging.org | N/A |
| Bacterial and Virus Strains | ||
| Luciferase Adenovirus | Vector Biolabs | Cat# 1000 |
| Cre recombinase Adenovirus | Vector Biolabs | Cat# 1045 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hank’s Balanced Salt Solution | Thermo Fisher Scientific | Cat# 24020 |
| Dulbecco's Phosphate-Buffered Saline | Corning | Cat# 21–030-CV |
| DMEM, high glucose, GlutaMAX | Thermo Fisher Scientific | Cat# 10569 |
| HyClone™ Fetal Bovine Serum, Defined | Thermo Fisher Scientific | Cat# SH3007003HI |
| Trypsin-EDTA (0.25%) | Thermo Fisher Scientific | Cat# 25200056 |
| Trypsin-EDTA (0.05%) | Thermo Fisher Scientific | Cat# 25300 |
| Trypan Blue | Gibco | Cat# 15250–061 |
| Collagenase type 2 | Worthington Biochemical | Cat# 4176 |
| Phenol Chlorofom | Thermo Fisher Scientific | Cat# BP1750I |
| GoTaq Green Master Mix | Promega | Cat# M7122 |
| RNA-Bee (Tel Test Inc) |
Thermo Fisher Scientific | Cat# NC9850755 |
| Chloroform | Thermo Fisher Scientific | Cat# BP1145 |
| Isopropanol | Thermo Fisher Scientific | Cat# BP2618 |
| Ethanol | Thermo Fisher Scientific | Cat# BP2818 |
| DEPC water | Thermo Fisher Scientific | Cat# AM9906 |
| RNeasy Mini Kit | Qiagen | Cat # 74104 |
| RNase-Free DNase Set | Qiagen | Cat# 79254 |
| β-mercaptoethanol | Sigma-Aldrich | Cat# M3148 |
| Omniscript Reverse Transcription kit | Qiagen | Cat# 205111 |
| QuantiTect SYBR® Green PCR Kits | Qiagen | Cat# 204143 |
| NaCl 5M | Thermo Fisher Scientific | Cat# AM760G |
| Tris HCl pH8.0 | Thermo Fisher Scientific | Cat# BP1758 |
| Proteinase K | Roche | Cat# 03115828001 |
| Glycerol | Thermo Fisher Scientific | Cat# BP229 |
| Nonidet P-40 substitute | Sigma-Aldrich | Cat# 74385 |
| EDTA 0.5M | Thermo Fisher Scientific | Cat# BP2482 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Phosphatase Inhibitor Cocktail Tablets | Roche | Cat# 04 906 837 001 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| 10x Tris/Glycine/SDS | Bio-Rad | Cat# 1610732 |
| Tris | Thermo Fisher Scientific | Cat# BP152 |
| Glycine | Sigma-Aldrich | Cat# G8790 |
| Methanol | Sigma-Aldrich | Cat# 494437 |
| Laemmli sample buffer | Bio-Rad | Cat# 1610737 |
| Kaleidoscope protein marker | Bio-Rad | Cat# 1610324 |
| Tris Buffered Saline 10X | Thermo Fisher Scientific | Cat# BP 2471 |
| Tween-20 | Thermo Fisher Scientific | Cat# BP 337 |
| ECL™ Prime Western Blotting System | GE Healthcare | Cat# RPN2232 |
| Restore™ PLUS Western Blot Stripping Buffer | Thermo Fisher Scientific | Cat# 46430 |
| Acetone | Thermo Fisher Scientific | Cat# A18 |
| Alcian blue 8 GX | Sigma-Aldrich | Cat# A3157 |
| Alizarin Red S | Sigma-Aldrich | Cat# A5533 |
| Weigerťs iron hematoxylin solution PartA | Sigma-Aldrich | Cat# HT107 |
| Weigerťs iron hematoxylin solution Part B | Sigma-Aldrich | Cat# HT108 |
| Fast green FCF | JT Baker | Cat# M377–03 |
| Safranin O | Sigma-Aldrich | Cat# 58884 |
| Acetic Acid, Glacial | Thermo Fisher Scientific | Cat# A38 |
| Potassium hydroxide | Thermo Fisher Scientific | Cat# P250 |
| EF5 | Hypoxia-Imaging.org | N/A |
| Oligomycin | Sigma-Aldrich | Cat# 75351 |
| Carbonyl cyanide-trifluoromethoxyphenylhydrazone (FCCP) | Sigma-Aldrich | Cat# C2920 |
| Antimycin A | Sigma-Aldrich | Cat# A8674 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| Ammonium acetate | Sigma-Aldrich | Cat# A1542 |
| Cytoseal Xyl | Thermo Fisher Scientific | Cat # 8312 |
| DAPI, FluoroPure™ grade | Thermo Fisher Scientific | Cat # D21490 |
| Triton X-100 | Sigma-Aldrich | Cat # T8787 |
| Sodium citrate | Thermo Fisher Scientific | Cat # BP327 |
| Paraformaldehyde | Acros organics | Cat# 41678 |
| Ethylenediaminetetraacetic Acid, Disodium Na Salt | Thermo Fisher Scientific | Cat # BP120 |
| Critical Commercial Assays | ||
| In situ Cell Death Detection kit, fluorescein | Roche | Cat # 11684795910 |
| Click-iT EdU Alexa Fluor 488 Imaging Kit | Thermo Fisher Scientific | Cat # C10337 |
| MitoSOX™ Red Mitochondrial Superoxide Indicator, for live-cell imaging | Thermo Fisher Scientific | Cat # M36008 |
| MitoTracker™ Green FM | Thermo Fisher Scientific | Cat # M7514 |
| TMRE-Mitochondrial Membrane Potential Assay Kit | Abcam | Cat # ab113852 |
| ROS Detection Cell-Based Assay Kit | Cayman Chemical | Cat# 601290 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus: B6.Cg-Tg(Prrx1-cre)1Cjt/J | The Jackson Laboratory (Logan, et al., 2002) | Cat# JAX:005584, RRID:IMSR_JAX:005584 |
| Mus musculus: B6.Cg-Tfamtm1.1Ncdl/J | The Jackson Laboratory (Larsson, et al., 1998) | Cat# JAX:026123, RRID:IMSR_JAX:026123 |
| Mus musculus: B6.129-Hif1atm3Rsjo/J | The Jackson Laboratory (Schipani, et al., 2001) | Cat# JAX:005584, RRID:IMSR_JAX:005584 |
| Mus musculus: STOCK Epas1tm1Mcs/J | The Jackson Laboratory (Gruber, et al., 2007) | Cat# JAX:008407, RRID:IMSR_JAX:008407 |
| Mus musculus: Prx1-Cre;TFAM+/fl (mixed FVB/N-C57/B6) | This study | N/A |
| Mus musculus: Prx1-Cre;TFAMfl/fl (mixed FVB/N-C57/B6) | This study | N/A |
| Mus musculus: Prx1-Cre;HIF-1afl/fl (FVB/N) | This study | N/A |
| Mus musculus: Prx1-Cre;TFAMfl/+;HIF-1afl/fl (mixed FVB/N-C57/B6) | This study | N/A |
| Mus musculus: Prx1-Cre;TFAMfl/fl;HIF-1afl/fl (mixed FVB/N-C57/B6) | This study | N/A |
| Mus musculus: Prx1-Cre;HIF-1afl/fl (FVB/N) | This study | N/A |
| Mus musculus: Prx1-Cre; TFAMfl/fl;HIF-1afl/fl;HIF-2afl/fl (mixed FVB/N-C57/B6) | This study | N/A |
| Oligonucleotides | ||
| Prx1-Cre Forward: 5’-CAGGCGCCGCACAAAGGC-3’ Prx1-Cre Reverse: 5’-TGAACGAACCTGGTCGAAATC-3’ | (Logan, et al., 2002) | N/A |
| Tfam Floxed Forward (2-LoxP): 5’-CTGCCTTCCTCTAGCCCGGG-3’ Tfam Floxed Reverse (2-LoxP): 5’-GTAACAGCAGACAACTTGTG-3’ | (Larsson, et al., 1998) | N/A |
| Hif1a Floxed Forward (2-LoxP): 5’-TGATGTGGGTGCTGGTGTC Hif1a Floxed Forward (2-LoxP): TTGTGTTGGGGCAGTACTG-3’ | (Schipani, et al., 2001) | N/A |
| Hif2a Floxed Forward (2-LoxP): 5'-CAGGCAGTATGCCTGGCTAATTCCAGTT-3' Hif2a Floxed Forward (2-LoxP): 5'-CTTCTTCCATCATCTGGGATCTGGGACT-3'(Gruber, et al., 2007) | (Gruber et al PNAS 2007) | N/A |
| TBP Forward: 5’-AGAACAATCCAGACTAGCAGCA-3’ TBP Reverse: 5’-GGGAACTTCACATCACAGCTC-3’ | (Merceron, et al., 2019) | N/A |
| Tfam Forward 5’-CAAAGGATGATTCGGCTCAGG-3’ Tfam Reverse: 5’-TCGACGGATGAGATCACTTCG-3’ | This study | N/A |
| Hif-1a Forward: 5’-TCTCGGCGAAGCAAAGAGTC-3’ Hif-1a Reverse: 5’-AGCCATCTAGGGCTTTCAGATAA-3’ | (Merceron, et al., 2019) | N/A |
| Pgk1 Forward: 5’-ATGTCGCTTTCCAACAAGCTG-3’ Pgk1 Reverse: 5’-GCTCCATTGTCCAAGCAGAAT-3’ | This study | N/A |
| β2-Microglobulin Forward: 5’-TCATTAGGGAGGAGCCAATG-3’ β2-Microglobulin Reverse: 5’-ATCCCCTTTCGTTTTTGCTT-3’ | This study | N/A |
| mt-Cytb Forward: 5’-TGAGGGGGCTTCTCAGTAGA-3’ mt-Cytb Reverse: 5’-TAGGGCCGCGATAATAAATG-3’ | This study | N/A |
| mt-Rnr2 Forward: 5’-GGGATAACAGCGCAATCCTA-3’ mt-Rnr2 Reverse: 5’-ATTGGGATGTCCTGATCCAA-3’ | This study | N/A |
| mt-Co3 Forward: 5’-TAACCCTTGGCCTACTCACC-3’ mt-Co3 Reverse: 5’-ATAGGAGTGTGGTGGCCTTG-3’ | This study | N/A |
| Recombinant DNA | ||
| Sox9 ISH Probe | Mangiavini dev biol 2014 | N/A |
| Col2a1 ISH Probe | Merceron Plos One 2014 | N/A |
| Col10a1 ISH Probe | Merceron Plos One 2014 | N/A |
| Gdf5 ISH Probe | Provot J Cell Biol 2007 | N/A |
| Software and Algorithms | ||
| Bioquant (Bioquant Osteo 17.2.6) | Bioquant Image Analysis Corp. | RRID:SCR_016423 |
| Image J | Image J | RRID:SCR_003070 |
| FlowJo | FlowJo LLC | RRID:SCR_008520 |
| Photoshop CS6 | Adobe | RRID:SCR_014199 |
Colony maintenance
All animals were maintained in a vivarium monitored daily by the University of Michigan Unit for Laboratory Animal Medicine. The vivarium was kept on a 12:12 hour light and dark cycle at 22°C and a humidity level between 30–70%. Cages were kept on ventilated racks and provided with water and LabDiet 5LOD chow ad libitum.
Genotyping
Pups were genotyped from tail biopsies and identified by ear tags. Collected samples were incubated in lysis buffer (100mM Tris (pH8.0), 5mM EDTA (pH8.0), 0.2% SDS, 200mM NaCl) containing 150μg/mL proteinase K (Roche) overnight at 55°C. Genomi c DNA was precipitated with an excess of isopropanol and transferred into water before being used for genotyping. PCR reactions were performed using 1.5 μL of genomic DNA solution. The sets of primers used to for genotyping are listed the key resources table.
Ethical statement
All procedures involving mice were performed in accordance with the NIH guidelines for use and care of live animals and were approved by the University of Michigan Institutional Animal Care and Use Committee (IACUC) (Protocol number: PRO00007215).
METHOD DETAILS
Generation of Prx1-Cre;TFAMfl/fl, mutant mice.
TFAMfl/fl mice (Larsson, et al., 1998) were crossed with the Prx1-Cre transgenic line (Logan, et al., 2002) to generate Prx1-Cre;TFAMfl/+male mice, which in turn were bred with TFAMfl/fl females. TFAMfl/fl, Prx1-Cre;TFAMfl/+ and Prx1-Cre;TFAMfl/fl littermates were collected at E13.5, E15.5 or at birth for analysis. TFAMfl/fl and Prx1-Cre;TFAMfl/+ mice were phenotypically indistinguishable.
Generation of Prx1-Cre;HIF-1afl/fl mutant mice.
HIF1fl/fl mice (Schipani, et al., 2001) were crossed with the Prx1-Cre transgenic line (Logan, et al., 2002) to generate Prx1-Cre;HIF1fl/+male mice, which in turn were bred with HIF1fl/fl females. HIF1fl/fl and Prx1-Cre;HIF1fl/fl littermates were collected at birth for analysis.
Generation of Prx1-Cre;TFAMfl/fl;HIF-1afl/fl mutant mice.
TFAMfl/fl (Larsson, et al., 1998) and HIF-1afl/fl (Schipani, et al., 2001) mice were crossed to obtain TFAMfl/fl;HIF-1afl/fl mice. TFAMfl/fl;HIF-1afl/fl mice were bred with Prx1-Cre transgenic line (Logan, et al., 2002) to generate Prx1-Cre;TFAMfl/+;HIF-1afl/+ males, which in turn were bred with TFAMfl/fl;HIF-1afl/fl females. TFAMfl/fl;HIF-1afl/fl, Prx1- Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl littermates were collected at E15.5 or at birth for analysis.
Generation of Prx1-Cre;TFAMfl/fl;HIF1fl/fl;HIF2fl/fl
TFAMfl/fl mice (Larsson, et al., 1998), HIF-1afl/fl (Schipani, et al., 2001) and HIF-2afl/fl (Gruber, et al., 2007) mice were crossed to obtain TFAMfl/fl;HIF-1fl/fl;HIF-2afl/fl mice TFAMfl/fl; HIF-1fl/fl;HIF-2afl/fl mice were bred with Prx1-Cre;HIF-2afl/fl (Merceron, et al., 2019) mouse to generate Prx1-Cre;TFAMfl/+;HIF-1fl/+;HIF-2afl/fl males, which in turn were bred with TFAMfl/fl;HIF-1fl/fl;HIF-2afl/fl females. TFAMfl/fl;HIF-1fl/fl;HIF-2afl/fl, Prx1-Cre; TFAMfl/+;HIF-1fl/fl;HIF-2afl/fl and Prx1-Cre; TFAMfl/fl;HIF-1fl/fl;HIF-2afl/fl littermates were collected at birth for analysis.
At least three mutants and three controls were analyzed in each assay. For all experiments, both male and female mice were used.
Analysis of TFAM recombination in newborn liver
After sacrifice, livers of TFAMfl/fl;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl newborn pups were collected. Genomic DNA was extracted as described in the genotyping section. Recombination of the TFAM and HIF-1a floxed alleles was quantified by 2-LoxP qPCR of genomic DNA.
Isolation of growth plates from newborn control and mutant mice to document efficient deletion of Tfam and Hif1a activity in vivo.
Newborn growth plates of TFAMfl/fl, TFAMfl/fl;HIF-1afl/fl, Prx1-Cre;TFAMfl/fl, Prx1-Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF-1afl/fl forelimbs were dissected out and digested with collagenase type II as previously described (Mangiavini, et al., 2016). Next, genomic DNA (LaGory, et al., 2015), mitochondrial DNA (LaGory, et al., 2015), total RNA (Aro, et al., 2012) or total protein lysate (Aro, et al., 2012) were extracted from those digested growth plates as reported.
Efficient recombination of the Tfam or the Hif1a floxed allele was quantified by 2-LoxP qPCR of genomic DNA. Loss of Tfam or Hif1a mRNA was documented by qRT-PCR. Loss of Tfam protein was verified by Western blot analysis. Loss of Tfam activity was confirmed by qPCR of mitochondrial DNA. Loss of Hif1a activity was established by qRT-PCR for phosphoglycerate-kinase 1 (PGK1) mRNA, which is a downstream target of Hif1a (Aro, et al., 2012). Sequences of the primers used for qRT-PCR are listed in the key resource table.
Isolation and culture of growth plate chondrocytes from newborn mutant and control mice for the ex vivo analysis of mitochondrial and metabolomic parameters and cell death.
Primary chondrocytes were isolated from TFAMfl/fl, HIF1fl/fl, TFAMfl/fl;HIF-1afl/fl, Prx1-Cre;TFAMfl/fl, Prx1-Cre;HIF1fl/fl,Prx1-Cre;TFAMfl/+;HIF-1afl/fl and Prx1-Cre;TFAMfl/fl;HIF- 1afl/fl newborn growth plates of forelimbs and cultured for 4–7 days in 20%O2 or 1%O2 (H35 hypoxystation, Don Withley Scientific) as described (Mangiavini, et al., 2016). Upon in vitro culture, mitochondrial and/or metabolomic parameters and cell death were analyzed as described below.
Efficient recombination of the Tfam or the Hif1a floxed allele was quantified by 2-LoxP qPCR of genomic DNA as described above. Loss of Tfam or Hif1a protein was verified by Western blot analysis. Loss of Tfam activity was established by qPCR of mitochondrial DNA. Loss of Hif1a activity was confirmed by Western blot analysis for BCL2 Interacting Protein 3 (Bnip3), which is a downstream target of Hif1a (Zhang, et al., 2008). Sequences of the primers used for qRT-PCR are listed in the key resource table.
Isolation and culture of growth plate chondrocytes from newborn floxed mice for the in vitro analysis of O2 consumption rates.
Growth plate primary chondrocytes were isolated from newborn TFAMfl/fl, HIF-1afl/fl or TFAMfl/fl;HIF-1afl/fl floxed mice and transduced with Luciferase Adenovirus (Ad-Luc) or Cre recombinase Adenovirus (Ad-Cre) and cultured for 6–14 days prior to analysis. Recombination of the Tfam or the Hif1a floxed allele was quantified by 2-LoxP qPCR of genomic DNA. The efficiency of deletion ranged between 75% and 95%. Sequences of the primers used for qRT-PCR are listed in the key resource table.
Analysis of Mitochondrial DNA Content.
Mitochondrial DNA content was monitored using qRT-PCR. Mitochondrial DNA and genomic DNA were extracted in RIPA buffer from either newborn growth plates of control and mutant mice or ex-vivo cultured control or mutant chondrocytes. Mitochondrial specific primers (Cytochrome b, 16S rRNA and Cytochrome c oxidase subunit 3) were used to assess mitochondrial content and normalized to genomic DNA levels by amplification of the nuclear β2-microglobulin gene. Sequences of the primers used for qRT-PCR are listed in the key resource table.
Quantitative Real-Time PCR
Total RNA was isolated from newborn growth plates or cultured chondrocytes using RNA-Bee (Tel-Test Inc.) reagent according to manufacturer’s instructions. RNA quality control was performed using Agilent 2100 BioAnalyzer. Reverse transcription of mRNA was performed using Omniscript Reverse Transcriptase (Qiagen) as the manufacturer’s protocol recommended. Real-time PCR was performed as previously described (Aro, et al., 2012). The mRNA encoded by the TATA-box binding protein gene was amplified as an internal control. Relative mRNA levels were calculated using the 2−ΔΔCt methodology (Livak and Schmittgen, 2001). Sequences of the primers used for qRT-PCR are listed in the key resource table.
Analysis of mitochondrial membrane potential
To quantify mitochondrial membrane potential, the TMRE assay (Abcam) was used according to the manufacturer’s instructions. Briefly, primary chondrocytes isolated and cultured as above were incubated with 200nM TMRE probe for 25 minutes at 37°C, protected from light. After a brief trypsinization with Trypsin-EDTA (0.05%) at 37°C, cells were washed twice with FACS buffer (1x DPBS + 5% FBS), and flow cytometry was performed with a LSR Fortessa flow cytometer (Becton Dickinson). FlowJo Software was used for analysis.
Analysis of mitochondrial mass
To quantify mitochondrial mass, the Mitotracker assay (Invitrogen) was used according to the manufacturer’s instructions. Briefly, primary chondrocytes isolated and cultured as above were incubated with 100nM Mitotracker probe for 25 minutes at 37°C, protected from light. After a brief trypsinization with Trypsin-EDTA (0.05%) at 37°C, cells were washed twice with FACS buffer (1x DPBS + 5% FBS), and flow cytometry was performed with a LSR Fortessa flow cytometer (Becton Dickinson). FlowJo Software was used for analysis.
Analysis of total ROS
To quantify total ROS accumulation, the ROS detection cell-based assay kit (Cayman Chemicals) was used according to the manufacturer’s instructions. Briefly, primary chondrocytes isolated and cultured as above. After a brief trypsinization with Trypsin- EDTA (0.05%) at 37°C, cells were incubated with 5 μM dihydroethidium (DHE) probe for 30 minutes at 37°C, protected from light. Thereafte r, cells were washed twice with ROS staining buffer. Flow cytometry was performed with a LSR Fortessa flow cytometer (Becton Dickinson). FlowJo Software was used for analysis.
Analysis of mitochondrial ROS
To quantify mitochondrial ROS accumulation, the MitoSOX (Invitrogen) assay was used according to the manufacturer’s instructions. Briefly, primary chondrocytes isolated and cultured as above were incubated with 5 M MitoSOX probe for 20 minutes at 37°C, protected from light. After a brief trypsinization with Trypsin-EDTA (0.05%) at 37°C, cells were washed twice with FACS buffer (1x DPBS + 5% FBS), and next flow cytometry was performed with a LSR Fortessa flow cytometer (Becton Dickinson). FlowJo Software was used for analysis.
Analysis of O2 consumption rate
O2 consumption rate (OCR) was measured using an XF96 analyzer (Seahorse Biosciences) as described (LaGory, et al., 2015). Briefly, Ad-Luc or Ad-Cre transduced chondrocytes were plated at a density of 25,000 cells per well in a 96 multiwell plate on the day prior to analysis. Assays were performed in DMEM containing 25 mM glucose, 2 mM L-glutamine, and 1 mM pyruvate at 20%O2. At the indicated time points, oligomycin (Sigma) was added to a final concentration of 2.5 μg/mL, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; Sigma) to a final concentration of 5 μM, Antimycin A (Sigma) to a final concentration of 5 M and Rotenone (Sigma) to a final concentration of 1 μM. Measurement cycles consisted of a 3 minutes mixing step followed by a 5 minutes measurement step. The assays were performed at 20%O2. Raw OCR signals were normalized to cell number following completion of the experiment.
Measurements of intracellular ATP, ADP, Lactate, glucose, NADH, and NAD
Primary chondrocytes isolated from newborn control and mutant mice as described were cultured in vitro until they reached 90% confluency. Next, cells were washed with 1xPBS, followed by 150mM ammonium acetate for less than 30 seconds. Cellular activities were quenched by liquid nitrogen; upon evaporation of the liquid nitrogen, plates were wrapped in aluminum foil and stored at −80°C. Samples were analyzed by LC-MS as described (Lorenz, et al., 2011).
Counting of dead cells in vitro
Primary chondrocytes isolated from newborn control and mutant mice were cultured in vitro as described above. After a brief trypsinization with Trypsin-EDTA (0.05%) and staining with Trypan blue, the number of alive and dead cells was counted (Merceron, et al., 2012).
Western blotting
Proteins were extracted in RIPA buffer in the presence of protease and phosphatase inhibitors (Roche). Protein concentration was estimated by the BCA assay (Thermo Scientific). For each sample, 20 g of protein were resuspended in sample buffer and electrophoresed in a pre-cast 4–20% Tris gel (BioRad). After gel transfer to Polyvinylidene fluoride (PVDF) membranes using a BioRad Criterion system, blots were blocked in 5% non-fat milk/1x TBST for 1 hour at room temperature and incubated overnight at 4°C with the following primary antibod ies: Tfam (Abcam: ab131607) at 1:2000; Hif1a (Novus Biologicals: NB100479) at 1:1000, Bnip3 (Cell Signaling: CST3769) at 1:1000, LDHA (Cell Signaling: CST2012) at 1:1000, AMPK (Cell Signaling: CST2532) at 1:1000, pAMPK (Cell Signaling: CST2531) at 1:1000, caspase 3 (Cell Signaling: CST9662) at 1:1000, and α-Tubulin (Cell Signaling: #2125) at 1:10,000. The membranes were then incubated with a HRP-conjugated anti-Rabbit IgG (Santa Cruz: sc-2004) at 1:2000 for one hour at room temperature in 5% non-fat milk/1x TBST. Signal was detected by using enhanced chemiluminescence (ECL Prime Western Blotting System GE Healthcare). Protein molecular weight was determined using the Kaleidoscope protein marker. Western blot images were acquired and analyzed via the BioRad Image Lab system. Quantification was performed using ImageJ. The α-Tubulin signal was used to normalize for protein amount.
Routine histology, Alcian Blue and Alizarin Red whole-mount staining, H&E staining, Safranin-O staining and in situ hybridization analysis
For routine histology, H&E staining and Safranin-O staining, tissue was fixed in 4% PFA/PBS and processed as previously reported (Mangiavini, et al., 2016). The Alcian Blue and Alizarin Red S whole-mount staining were performed as previously described (Mangiavini, et al., 2016). In situ hybridization (ISH) analysis was performed using complementary 35S-labeled riboprobes as previously described (Mangiavini, et al., 2016). All images were acquired with an Eclipse E800 microscope (Nikon).
TUNEL, EdU and EF5 Assays
TUNEL assay was performed using an In situ Cell Death Detection kit (Roche) as previously reported (Mangiavini, et al., 2016). EdU proliferation assay was performed using Click-iT® EdU Alexa Fluor® 488 Imaging Kit (Invitrogen) according to the manufacturer’s instructions. Images for both TUNEL and EdU assays were captured using filters for FITC and DAPI. The EF5 assay was performed as previously described (Mangiavini, et al., 2016). Images for EF5 assay were captured using filters for Cy3 and DAPI. All images were acquired with Eclipse E800 microscope (Nikon).
Image processing and analysis
Signal distribution was analyzed using Image J. Positive signals for DAPI, EdU, TUNEL and EF5 assays were counted and quantified using BioQuant software (BIOQUANT OSTEO 17.2.6). Composite images presented in Figures 2 and 3 have been generated by merging individual images obtained from independent channels using Photoshop CS6 software.
Quantification and statistical analysis
Sample sizes were determined using power analyses with a minimal power value of 90%, an interval of confidence of 95% and assuming a normal distribution of the variables. P values are the result of ANOVA or Student’s t test provided by Microsoft Excel (*p<0.05; ***p<0.001). Significance of the data presented in Figures 1A–D, 2C–E and supplemental figures S1A–D, S4A–E, S6A (Tfam 2-LoxP) were calculated with Student’s t test. For all the other data, statistical analyses were performed using one- way ANOVA.
Supplementary Material
Highlights:
Mitochondrial respiration is dispensable for survival of fetal chondrocytes
Loss of Hif1a enhances mitochondrial respiration in fetal chondrocytes
Mitochondrial respiration augments intracellular hypoxia of fetal chondrocytes
Enhancing mitochondrial respiration is detrimental to survival of fetal chondrocytes
ACKNOWLEDGMENTS
This study was supported by NIH RO1AR065403 grant to ES. We thank Dr. Amy Chang at University of Michigan for helpful discussions, Dr. Yuji Mishina for critical review of the manuscript, and the Histology Core, the Flow Cytometry Core and the Metabolomics Core at University of Michigan-Medical School for providing us with excellent technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amarilio R, Viukov SV, Sharir A, Eshkar-Oren I, Johnson RS, and Zelzer E (2007). HIF1alpha regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development 134, 3917–28. [DOI] [PubMed] [Google Scholar]
- Araldi E, Khatri R, Giaccia AJ, Simon MC, and Schipani E (2011). Lack of HIF- 2alpha in limb bud mesenchyme causes a modest and transient delay of endochondral bone development. Nat Med 17, 25–6; author reply 27–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aro E, Khatri R, Gerard-O’Riley R, Mangiavini L, Myllyharju J, and Schipani E (2012). Hypoxia-inducible Factor-1 (HIF-1) but Not HIF-2 Is Essential for Hypoxic Induction of Collagen Prolyl 4-Hydroxylases in Primary Newborn Mouse Epiphyseal Growth Plate Chondrocytes. J Biol Chem 287, 37134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baris OR, Klose A, Kloepper JE, Weiland D, Neuhaus JF, Schauen M, Wille A, Muller A, Merkwirth C, Langer T, et al. (2011). The mitochondrial electron transport chain is dispensable for proliferation and differentiation of epidermal progenitor cells. Stem Cells 29, 1459–68. [DOI] [PubMed] [Google Scholar]
- Barman J, Kumar R, Saha G, Tiwari K, and Dubey VK (2018). Apoptosis: Mediator Molecules, Interplay with Other Cell Death Processes and Therapeutic Potentials. Curr Pharm Biotechnol 19, 644–663. [DOI] [PubMed] [Google Scholar]
- Bentovim L, Amarilio R, and Zelzer E (2012). HIF1alpha is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development 139, 4473–83. [DOI] [PubMed] [Google Scholar]
- Brand MD, and Nicholls DG (2011). Assessing mitochondrial dysfunction in cells. Biochem J 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celotto AM, Chiu WK, Van Voorhies W, and Palladino MJ (2011). Modes of metabolic compensation during mitochondrial disease using the Drosophila model of ATP6 dysfunction. PLoS One 6, e25823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, and Loscalzo J (2009). MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 10, 273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwoodie SL (2009). The role of hypoxia in development of the Mammalian embryo. Dev Cell 17, 755–73. [DOI] [PubMed] [Google Scholar]
- Ekstrand M, and Larsson NG (2002). Breeding and genotyping of Tfam conditional knockout mice. Methods Mol Biol 197, 391–400. [DOI] [PubMed] [Google Scholar]
- Fels DR, and Koumenis C (2006). The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 5, 723–8. [DOI] [PubMed] [Google Scholar]
- Gandara L, and Wappner P (2018). Metabo-Devo: A metabolic perspective of development. Mech Dev. [DOI] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, and Giaccia AJ (1996). Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 379, 88–91. [DOI] [PubMed] [Google Scholar]
- Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, and Simon MC (2007). Acute postnatal ablation of Hif-2alpha results in anemia. Proceedings of the National Academy of Sciences of the United States of America 104, 2301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Glasauer A, Hoover P, Yang S, Blatt H, Mullen AR, Getsios S, Gottardi CJ, DeBerardinis RJ, Lavker RM, et al. (2013). Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Science signaling 6, ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey E (1928). The oxygen consumption of luminous bacteria. Journal of General Physiology 11, 469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Johnson RS, and Simon MC (2011). HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, and Dang CV (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3, 177–85. [DOI] [PubMed] [Google Scholar]
- LaGory EL, Wu C, Taniguchi CM, Ding CK, Chi JT, von Eyben R, Scott DA, Richardson AD, and Giaccia AJ (2015). Suppression of PGC-1alpha Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell reports 12, 116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, and Rustin P (2001). Animal models for respiratory chain disease. Trends Mol Med 7, 578–81. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, and Clayton DA (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 18, 231–6. [DOI] [PubMed] [Google Scholar]
- Lee DC, Sohn HA, Park ZY, Oh S, Kang YK, Lee KM, Kang M, Jang YJ, Yang SJ, Hong YK, et al. (2015). A lactate-induced response to hypoxia. Cell 161, 595–609. [DOI] [PubMed] [Google Scholar]
- Lin SC, and Hardie DG (2018). AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab 27, 299–313. [DOI] [PubMed] [Google Scholar]
- Liu CF, Samsa WE, Zhou G, and Lefebvre V (2017). Transcriptional control of chondrocyte specification and differentiation. Semin Cell Dev Biol 62, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, and Simon MC (2004). Regulation of transcription and translation by hypoxia. Cancer Biol Ther 3, 492–7. [DOI] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–8. [DOI] [PubMed] [Google Scholar]
- Logan M, Martin J, Nagy A, Lobe C, Olsen E, and Tabin C (2002). Expression of Cre recombinase in the developing mouse limb bud driven by a Prx1 enhancer. Genesis 33, 77–80. [DOI] [PubMed] [Google Scholar]
- Lorenz MA, Burant CF, and Kennedy RT (2011). Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal Chem 83, 3406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Araldi E, Haigh K, Khatri R, Van Looveren R, Giaccia AJ, Haigh JJ, Carmeliet G, and Schipani E (2012a). VEGF-independent cell-autonomous functions of HIF-1alpha regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J Bone Miner Res 27, 596–609. [DOI] [PubMed] [Google Scholar]
- Maes C, Carmeliet G, and Schipani E (2012b). Hypoxia-driven pathways in bone development, regeneration and disease. Nat Rev Rheumatol 8, 358–66. [DOI] [PubMed] [Google Scholar]
- Mangiavini L, Merceron C, Araldi E, Khatri R, Gerard-O’Riley R, Wilson TL, Rankin EB, Giaccia AJ, and Schipani E (2014). Loss of VHL in mesenchymal progenitors of the limb bud alters multiple steps of endochondral bone development. Dev Biol 393, 124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiavini L, Merceron C, Araldi E, Khatri R, Gerard-O’Riley R, Wilson TL, Sandusky G, Abadie J, Lyons KM, Giaccia AJ, et al. (2015). Fibrosis and hypoxia- inducible factor-1alpha-dependent tumors of the soft tissue on loss of von Hippel-Lindau in mesenchymal progenitors. Am J Pathol 185, 3090–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiavini L, Merceron C, and Schipani E (2016). Analysis of Mouse Growth Plate Development. Curr Protoc Mouse Biol 6, 67–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merceron C, Portron S, Vignes-Colombeix C, Rederstorff E, Masson M, Lesoeur J, Sourice S, Sinquin C, Colliec-Jouault S, Weiss P, et al. (2012). Pharmacological modulation of human mesenchymal stem cell chondrogenesis by a chemically oversulfated polysaccharide of marine origin: potential application to cartilage regenerative medicine. Stem Cells 30, 471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merceron C, Ranganathan K, Wang E, Tata Z, Makkapati S, Khan MP, Mangiavini L, Yao AQ, Castellini L, Levi B, et al. (2019). Hypoxia-inducible factor 2α is a negative regulator of osteoblastogenesis and bone mass accrual. Bone Res. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, Nagasawa T, Kronenberg HM, and Ono N (2018). Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcina MM, Grand RJ, and Hammond EM (2014). ATM activation in hypoxia - causes and consequences. Mol Cell Oncol 1, e29903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, and Denko NC (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3, 187–97. [DOI] [PubMed] [Google Scholar]
- Pfander D, Cramer T, Schipani E, and Johnson R (2003). HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci 116, 1819–26. [DOI] [PubMed] [Google Scholar]
- Polster BM, Nicholls DG, Ge SX, and Roelofs BA (2014). Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species. Methods Enzymol 547, 225–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provot S, and Schipani E (2005). Molecular Mechanisms of endochondral bone development. Biochem Biophys Res Commun 328, 658–665. [DOI] [PubMed] [Google Scholar]
- Provot S, Zinyk D, Gunes Y, Kathri R, Le Q, Kronenberg HM, Johnson RS, Longaker MT, Giaccia AJ, and Schipani E (2007). Hif-1alpha regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol 177, 451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kauppinen A, and Kaarniranta K (2015). 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci 72, 3897–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, and Johnson RS (2001). Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev 15, 2865–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2017). Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J 36, 252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Burkart A, Nicoloro SM, Czech MP, Straubhaar J, and Corvera S (2008). Paradoxical effect of mitochondrial respiratory chain impairment on insulin signaling and glucose transport in adipose cells. J Biol Chem 283, 30658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ, and Cantley LC (2013). Stem cell metabolism in tissue development and aging. Development 140, 2535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, and Larsson NG (2000). Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 26, 336–40. [DOI] [PubMed] [Google Scholar]
- Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, and Larsson NG (2001). Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci 21, 8082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, and Larsson NG (2011). Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain- deficient dopamine neurons in vivo. Proceedings of the National Academy of Sciences of the United States of America 108, 12937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm EE, and Kingsley DM (1999). GDF5 coordinates bone and joint formation during digit development. Dev Biol 209, 11–27. [DOI] [PubMed] [Google Scholar]
- Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, An D, Lee KY, Ilkayeva OR, Zingaretti CM, et al. (2012). Adipose-Specific Deletion of TFAM Increases Mitochondrial Oxidation and Protects Mice against Obesity and Insulin Resistance. Cell Metab 16, 765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Silva JP, Gustafsson CM, Rustin P, and Larsson NG (2001). Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proceedings of the National Academy of Sciences of the United States of America 98, 4038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, et al. (1999). Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet 21, 133–7. [DOI] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, and Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America 107, 8788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YA, Xiong X, Scott T, Li AT, Wang C, Weiss HL, Tan L, Bradford E, Fan TWM, Chandel NS, et al. (2019). The mitochondrial retrograde signaling regulates Wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, and Larsson NG (2002). Increased mitochondrial mass in mitochondrial myopathy mice. Proceedings of the National Academy of Sciences of the United States of America 99, 15066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Tsang KY, Tang HC, Chan D, and Cheah KS (2014). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proceedings of the National Academy of Sciences of the United States of America 111, 12097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, and Semenza GL (2008). Mitochondrial autophagy is an HIF-1- dependent adaptive metabolic response to hypoxia. J Biol Chem 283, 10892–903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.