

Abstract
Alzheimer’s disease is typified by calcium dysfunction and neurofibrillary tangles of tau aggregates along with mitotic proteins. Using PC12 cells as a model system, we determined whether the Gαq/PLCβ/calcium signaling pathway impacts the manifestation of Alzheimer’s disease. Down-regulating PLCβ significantly increases tau protein expression and causes a large increase in tau aggregation. Stimulating Gαq to activate PLCβ results in a modest reduction in tau aggregation while inhibiting PLCβ activity results in a modest enhancement of tau aggregation. These results suggest that PLCβ may effect tau aggregation by an additional mechanism that is independent of its ability to transduce calcium signals. To this end, we found that a cytosolic population of PLCβ binds to a mitotic protein found in neurofibrillary tangles, CDK18, which promotes tau phosphorylation and aggregation. Taken together, our studies show that the loss of PLCβ1 can promote Alzheimer’s disease by a combination of its catalytic activity and its interaction with mitotic proteins thus offering an orthogonal method to control tau aggregation.
Keywords: phospholipase Cbeta, calcium signaling, tau aggregation, fluorescence imaging, mitotic hypothesis
Graphical abstract
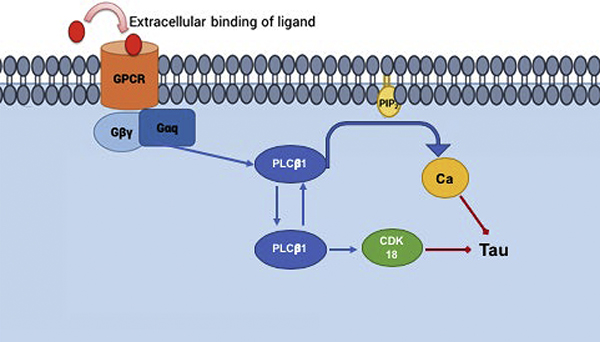
INTRODUCTION
Alzheimer’s Disease (AD) is characterized by a loss of neuronal tissue and the formation of neurofibrillary tangles that contribute to disruption of neuronal synapses, downstream atrophy of the cerebrum, and cognitive decline (see [1]). These neurofibrillary tangles are rich in hyperphosphorylated tau proteins. Normally, tau proteins assist in the organization of microtubule networks. Tau aggregation, initiated by phosphorylation, disrupts the cellular cytoskeleton ultimately leading to thinning of tissue in the cortex and cognitive decline [2, 3].
While tau aggregation is a hallmark of AD, the underlying causes of this disease are more complex. Recently, it was found that neurons in regions of the brain affected by AD have completed the S phase of the cell cycle, as evidenced by full or partial replication of DNA, but have not undergone mitosis (e.g [4]). This observation, along with the finding of mitotic proteins in neurofibrillary tangles, has led to the mitotic hypothesis. This theory posits that in AD, neurons move into the cell cycle but progression into mitosis is incomplete leading to apoptosis and subsequent neurodegeneration. Abnormalities in the cell cycle are a neuropathological quality conserved among early-onset familial AD and late-onset sporadic AD [5]. Other studies have shown that markers of cell cycle abnormalities may precede the accumulation of plaques and tangles and may account for biochemical, neuropathological, and cognitive changes seen in patients with AD.
Neurofibrillary tangles contain mitotic proteins that include cyclin-dependent kinase 18 (CDK18 also known as PCTAIRE3) [6]. CDK18 a member of the cdc2 protein kinase family and helps in gene stability during replication [7]. Although its precise cellular role in has not yet been determined, by analogy with its close family member, CDK16 (PCTAIRE-1) it is assumed to play a pre-mitotic as well as post-mitotic role. In disease patient samples, CDK18 is found to be concentrated within neurofibrillary tangles. CDK18 is found at elevated levels in the temporal cortex, the primary area of neurofibrillary tangles accumulation, of patients with AD as compared to healthy controls. Previously published data have suggested that overexpression of CDK18 may indirectly cause the T231 and S235 sites on tau to become phosphorylated, leading to accumulation of tau proteins into neurofibrillary tangles [6]. Overall, the presence of CDK18 in neurofibrillary tangles is in accord with re-entry of cells in the cell cycle and the mitotic hypothesis.
On the cellular level, Alzheimer’s disease is associated with calcium dysfunction in neurons (see [8]). Changes in cellular calcium initiated by hormones and neurotransmitters are mediated by the Gαq/ phospholipase Cβ signaling system. Upon binding of molecules such as acetylcholine, dopamine, serotonin, to their specific receptor, Gαq/ PLCβ becomes activated leading to a series of events that result in an increase in intracellular calcium and changes in the activity of a host of calcium-sensitive proteins [9, 10]. Of the four known members of the PLCβ family, PLCβ1 is most prominent in neuronal tissues and is the most sensitive to stimulation by Gαq. PLCβ1 deficiencies have been found in patients with neurological disorders such as epileptic encephalopathy [11], and knock-out mice confirm and recapitulate the findings [12, 13]. PLCβ1 is associated with the growth of neurites and synaptic plasticity in rat cortex [14, 15] and PLCβ1 mutations and deficiencies underlie a large number of neurological and psychiatric problems, including, memory loss, schizophrenia, impaired reality [16] and hot/cold sensitivity (see[17]). In knockout mice, PLCβ1 mutations impact spatial memory, social behavior, and the ability to filter redundant information from environmental stimuli [18]. Our own studies in cultured neuronal cells show that the loss of PLCβ1 promotes the aggregation of α-synuclein [19] whose intracellular inclusions fibrils give rise to Parkinson’s disease [20–29].
While the broad impact of PLCβ1 is thought to be traced to its role in mediating calcium signal from signals from Gαq activation, recent work suggests that the role of PLCβ1 is more complex. Besides its classic role in generating calcium signals through its localization on the plasma membrane, PLCβ1 also has an atypical cytosolic localization [30]. This cytosolic population interacts with unique partners that regulate RNA-induced silencing and the cell cycle [31–34]. PLCβ1 impacts the cell cycle of neuronal cells by binding to cyclin-dependent kinase 16 (CDK16 or PTCAIRE1) [32], a close relative of CDK18 [35]. In this study, we show that PLCβ1 has the ability to mediate tau aggregation through its plasma membrane population that transduces calcium signals from neurotransmitters, and also through its cytosolic population that binds CDK18. These two mechanisms work together to help reduce tau aggregation.
Materials and Methods
PC12 Cell Culture and Differentiation—
PC12 cells were purchased from American Tissue Cell Culture (ATCC) and cultured in DMEM (Gibco) with 10% horse serum from PAA (Ontario, Canada), 5% FBS (Atlanta Biological, Atlanta, GA), and 1% penicillin/streptomycin. Cells were incubated in 37 °C, 5% CO2, and 95% humidity. Differentiation was carried out a medium of 1% horse serum, 1% penicillin/streptomycin and initiated by the addition of nerve growth factor (NGF) from Novo protein. Medium was changed every 24 h.
Plasmids.
eGFP-tau-BiFC (pmGPF10C-Tau) was prepared by Henri Huttunen and purchased from Addgene, Cambridge, MA (plasmid #71433 and 71434; RRID:Addgene_71433 and 71434) as described [36]. siRNA(PLCβ1) was the pooled mixture of 4 siRNAs (GE Dharmacon, Lafayette, CO). The negative control siRNA, MISSION® siRNA Universal Negative Control, was from Sigma Aldrich cat# SIC001. CDK18 was prepared by William C Hahn MD, PhD from the Dana Farber Cancer Institute, and purchased from Addgene. The gene was cloned into pmCherry-C1 vector (Addgene) and designing new cloning sites at HINDIII and XbaI and inserting it into the vector using forward primer ttagtcGAATTCTatgatcatgaacaagatgaag and reverse primer agttgaTCTAGAtcagaagatgctctgccgcct.
Reagents—
All chemical reagents were diluted as per manufacturer’s specifications. In particular, carbamylcholine chloride (carbachol from Sigma Aldrich (St. Louis, MO)) was diluted using DI water for a final concentration of 5mM. Similarly, Calcium Orange (ThermoFisher Scientific, Waltham, MA) and [6-((17β−3-Methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione (U73122 from Sigma Aldrich) were diluted using DMSO for a final stock concentration of 2 mM and 1 mM respectively.
Transfection—
Plasmid transfections were accomplished using Lipofectamine 3000 (ThermoFisher Scientific, Waltham, MA) as recommended by the manufacturer. Transfection of siRNAs was also carried out using Lipofectamine 3000.
Western Blotting—
Samples were placed in 6-well plates and collected in lysis buffer that included Nonidet P-40 and protease inhibitors. After SDS-PAGE electrophoresis, protein bands were transfer to nitrocellulose or PVDF membranes (Hercules, CA). Primary antibodies to PLCβ1 (D-8), Tau (Tau 46) and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). After incubation with primary antibodies, the western blots were washed and incubated with anti-Mouse (Santa Cruz) or anti-Rabbit (Sigma) secondary antibody. The blots were then washed 3 times for 10 minutes and imaged using an Azure imager. Bands were measured at several sensitivities to ensure the intensities were in a linear range and avoid detector saturation. Data were analyzed using Image J in grayscale plot profile. Bands were normalized to loading control.
Confocal Imaging—
Cells were seeded in poly-D-lysine coated glass-covered dishes from MatTek (Ashland, MA). Images were acquired using Zeiss 510 meta confocal microscope. Data were analyzed using, LSM software and Image J software. Extended imaging was carried out using an environmental chamber set at 37°C and 5% CO2 where images were collected every 20 minutes. Carbamylcholine chloride (Carbachol) and [6-((17β−3-Methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl]-1H-pyrrole-2,5-dione (U73122) were obtained from Sigma Aldrich (St. Louis, MO).
Fluorescent plate reader assays.
Cell were seeded in 48 well tissue culture plates, well are separated in to 3 sections, and transfected with negative control siRNA, GFP-tau-BiFC and siRNA PLCβ1. After 48 hours of incubation, the fluorescence of the cells is quantified by a Perkin Elmer Victor3 Plate reader using an excitation of 488 nm at an emission of 525 nm.
Calcium imaging.
Cells were plated on glass bottom dishes (MatTek, Ashland, MA) and transfected with cells with siRNA(PLCβ1) or negative control siRNA. After 24 hours, cells were transfected with the two plasmids comprising eGFP-tau-BiFC. After 48 hours, cells were labeled with Calcium Orange (Thermo Fisher) at a final concentration of 5uM for 30 minutes at 37°C. Time series of cell images after stimulation with 5uM carbachol were carried out on a Zeiss LSM 510 Meta.
FRET/FLIM-Fluorescence imaging
FLIM measures Förster resonance energy transfer (FRET) by the decrease the fluorescence lifetime of a FRET donor molecule due to transfer of energy to an acceptor. Here, we excite the donor with intensity modulated light, and measure the shift in the phase of the emitted light, and the reduction in modulation for each individual pixel of the image and plot them on a phasor plot. These values change significantly in the presence of a FRET acceptor. The phasor plot is a combined graphical representation of all the raw FLIM data in a vector space. The phasor space is constructed by using two phasor vectors (G,S), where each component is represented as shown in Eq.1:
| (1) |
mx,y and φx,y are the modulation ratio and the phase delay measured for a particular modulation frequency (ω) at a pixel location (x,y). For a single lifetime population, the values from all of the pixels will fall on the phasor arc with longer lifetimes displayed to the left and shorter lifetimes, to the right. Since FRET shortens donor lifetimes, FRET will manifest itself by moving the (g(ω),s(ω)) data point to the right. For a mixed population of donor molecules, some of which undergoing FRET, the resulting phasor points localize inside the phasor arc [37]. Distributions inside the phasor arc therefore indicate FRET transfer and, hence, association of the two labeled proteins. The lifetime of an individual pixel can be calculated by:
| (2) |
For our experiments, we used green fluorescent donors (such as eGFP, enhanced Green Fluorescent Protein) or yellow fluorescent donor eYFP with red fluorescent proteins such as mCherry and dsRed acting as the FRET acceptors.
FLIM/FRET was performed by acquiring images of live cells plated on MatTek glass-bottom dishes using a 2-photon MaiTai laser (Spectra-Physics, Santa Clara, CA) (excitation 850 nm at 80 MHz) and a Nikon inverted confocal microscope in an ISS Alba System (Champaign, IL, USA).The Images were analyzed using ISS VistaVision and ImageJ software packages. Atto 425 fluorescent dye (t=3.61 ns) was used to calibrate the sample lifetimes.
Number and Brightness (N&B.
The Number and Brightness (N&B) analysis is a powerful tool that has been used previously to quantify graphically the aggregation state of diffusing proteins in living cells [38–41]. N&B analysis can determine of the number (N) of diff using particles within a given focal area and the intrinsic brightness (B) of each particle represented by pixels in an image and provides providing a map of brightness for every pixel using the following equation (3):
| (3) |
where I denotes the intensity of the signal, σ2 is the variance of the signal, σ02 is the readout noise variance of the detection electronics and offset is the detector offset. The analysis has been described in detail [41]. Higher variance in fluorescence is associated with higher-order oligomers. Therefore, the brightness vs intensity map can be used to determine the size of the aggregate at a given location as the brightness B can directly relate to the dimensions of the fluorescent molecules [38]. N&B studies were performed, by acquiring images of live cells plated on MatTek glass-bottom dishes using a 2-photon MaiTai laser (Spectra-Physics, Santa Clara, CA, USA), a Nikon inverted confocal microscope in an ISS Alba System (Champaign, IL,USA).The Images were analyzed using ISS VistaVision and SimFCS 4 (Irvine, CA, USA) software packages. The analysis has been described in more detail [39, 40]
Mass spectrometry –
Mass spectrometry measurements were carried out as previously described using the facility at the University of Massachusetts Medical School [42]. Briefly, cytosolic fractions were isolated from cells, and proteins bound to monoclonal anti-PLCβ1a (Santa Cruz, D-8) were separated by electrophoresis. Protein bands were isolated by cutting the gels, and proteins were extracted and typsinized, and loaded onto Magic C18AQ (Bruker-Michrom) and eluted. Ions were introduced by positive electrospray ionization via liquid junction into a Q Exactive hybrid mass spectrometer (Thermo Fisher). Mass spectra were acquired over m/z 300–1750 at 70,000 resolution (m/z 200) and data-dependent acquisition selected the top 10 most abundant precursor ions for tandem mass spectrometry by HCD fragmentation using an isolation width of 1.6 Da, collision energy of 27, and a resolution of 17,500.
Raw data files were peak processed with Proteome Discoverer (version 2.1, Thermo) prior to database searching with Mascot Server (version 2.5) against the Uniprot Rat database Search parameters included trypsin specificity with two missed cleavages or no enzymatic specificity. The variable modifications of oxidized methionine, pyroglutamic acid for N-terminal glutamine, phosphorylation of serine and threonine, N-terminal acetylation of the protein, and a fixed modification for carbamidomethyl cysteine were considered. The mass tolerances were 10 ppm for the precursor and 0.05Da for the fragments. Search results were then loaded into the Scaffold Viewer (Proteome Software, Inc.) for peptide/ protein validation and label free quantitation. These data were analyzed using percentage of total spectra in Scaffold4 software before plotting with Sigma Plot 14.
Statistical Analysis—
Data were analyzed using Microsoft excel and Sigma Plot 14 statistical packages that included Student’s t test and one way analysis of variance.
RESULTS
Loss of PLCβ promotes tau aggregation.
We first determined the effect of PLCβ1 down-regulation on the aggregation of endogenous tau in a rat neuronal-like cell line (PC12). We find that loss of PLCβ1 results in enhanced levels in the expression of at least two tau isoforms (Fig. 1A and Supplemental Fig. 1C). To support this observation, we followed the aggregation of tau tagged with bimolecular fluorescence complementation (BiFC) constructs in response to PLCβ1 knockdown (Supplemental Fig. 1A). In BiFC, one protein is tagged with a construct based on eGFP but missing four β strands and a second protein is tagged with the complementary strands. The two proteins are not fluorescent when separated but when they associate, the fluorophore is generated and the complex fluoresces.
Figure 1: Loss of PLCβ promotes tau expression:
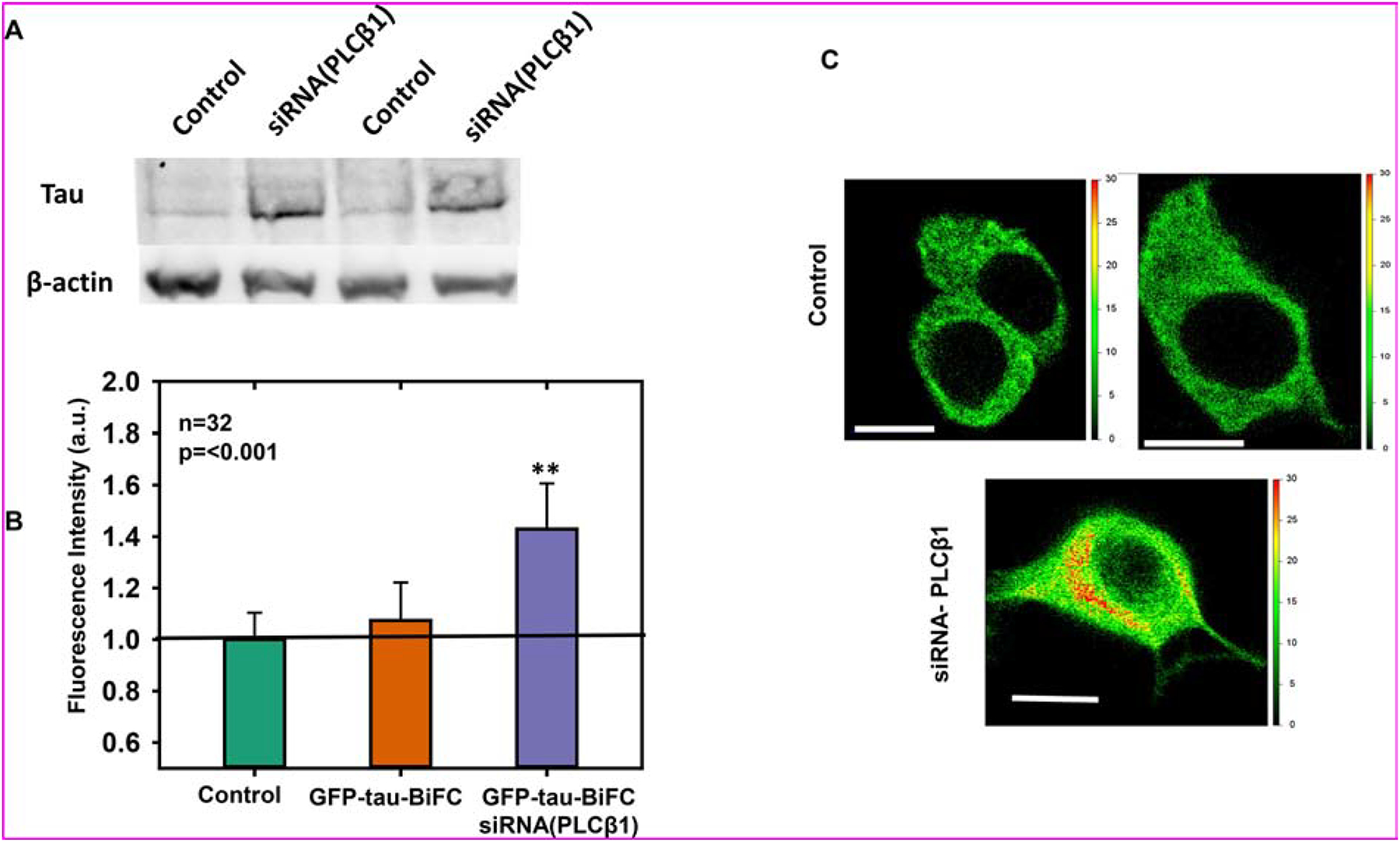
A) Western blot analysis of PC12 cells treated with negative control siRNA or siRNA(PLCβ1) and blotted with anti-tau and anti-actin antibodies. B) eGFP intensities in arbitrary units (a.u.) of PC12 cells expressing eGFP-Tau BiFC and treated with negative control siRNA or siRNA(PLCβ1) and quantified on a Perkins Elmer plate reader. The intensities are normalized to those obtained for PC12 cells overexpressing untagged eGFP (n=32, **= p=<0.001). C) Representative images of PC12 cells expressing eGFP-Tau-BiFC and treated with negative control siRNA or siRNA(PLCβ1) imaged on a Nikon Ti microscope equipped with a 2-photon Mai Tai laser. The scale bars to right of the images indicate a heat-map eGFP intensity of each individual pixel of the cell image. Scale bar= 10μm
We transfected the GFP-tau-BiFC proteins into undifferentiated PC12 cells and cells that have been treated with siRNA(PLCβ1), and monitored the amount of eGFP fluorescence on a microplate reader. Noting that GFP-tau-BiFC fluorescence only occurs with dimerization, these data (Fig 1B) show an increase in aggregation with loss of PLCβ1. Additionally, fluorescence microscopy images show that downregulating PLCβ1 results in increased BiFC fluorescence (Fig. 1C). Taken together, these studies show that loss of PLCβ1 increases in tau expression and results in enhanced tau aggregation.
To better define increased tau aggregation, we used number & brightness (N&B) analysis (see Materials and Methods). In this method, we measure both the intensity and brightness (B) values of eGFP-tau-BiFC molecules expressed in PC12 cells. Higher B values indicate the presence of oligomers whereas higher intensity values indicate molecules that fluorescence more efficiently, which also indicates increased formation of the eGFP-tau-BiFC complex. The results (Fig. 2 A–B) show that down-regulating PLCβ1 organizes tau molecules into both higher order oligomers and more strongly fluorescing species that are significantly compared to control cells (Fig. 2C–D).
Figure 2: Loss or inhibition of PLCβ1 promotes tau aggregation:
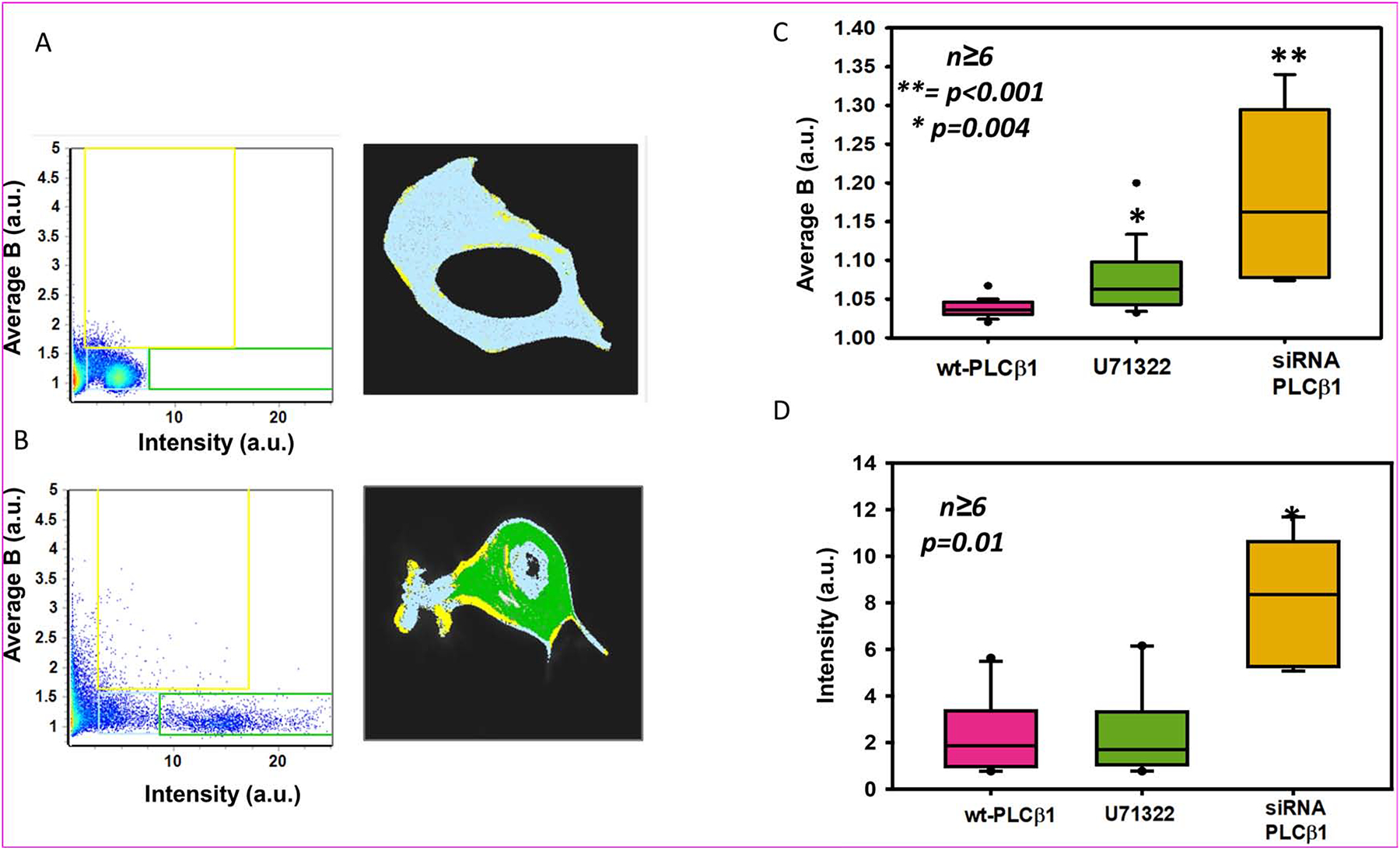
N&B results plotted as Brightness vs Intensity plot in arbitrary units (a.u.) of eGFP-tau-BiFC in PC12 cells treated with negative control siRNA (A) or siRNA(PLCβ1) (B) where individual pixels are highlighted either in a light blue (low intensity and brightness), green (high intensity) or a yellow box (high brightness) (left panels). The distribution of these highlighted pixels are overlaid on representative cells (right panels). C) The average brightness (B) or average intensity (D) of eGFP-tau-BiFC expressed in PC12 cells treated with negative control siRNA alone, with 1 uM PLC inhibitor U73122, or treated with siRNA(PLCβ1) (n≥6 across two individual experiments, **= p<0.001, * p=0.004 in (C) and p=0.01 in (D)).
Inhibition of PLCβ1 partially enhances tau aggregation.
Tau aggregation is initiated by phosphorylation at distinct sites and in a distinct sequence [7, 43]. The results of PLCβ1 knockdown indicate that tau phosphorylation and proteolysis may be modulated by a number of different calciumdependent enzymes that are either directly or indirectly linked to the Gαq/PLCβ1 signaling pathway, such as PKC, AMPK, calpain, etc. We tested whether the increase in tau aggregation with PLCβ1 knockdown is attributed to its role in mediating calcium signals using the PLC inhibitor U73122. Note this is chemical inhibits all cellular forms of PLC.
We first monitored changes in N&B of eGFP-BiFC-tau expressed in undifferentiated PC12 cells with U73122, and find that inhibition of PLCβ1 results in a significant increase in the average B values (Fig. 2B). This increase indicates a rise in the levels of tau oligomers. Although the trend is similar to the increase seen for PLCβ1 knockdown, it is not as large. Additionally, the average eGFP-tau-BiFC intensity with U73122 treatment did not differ from control (Fig. 2C). These studies suggest that PLCβ1 has additional mechanisms that affect tau aggregation.
Tau aggregation is diminished by Gαq activation / Ca2+ levels.
We carried out studies to determine whether tau aggregation is effected by neurotransmitter activation of Gαq. First, we measured changes in eGFP–tau-BiCF aggregation with the addition of carbachol, which is a stable form of acetylcholine. We find that carbachol diminishes the intensity of eGFP-tau-BiFC indicating a reduction in GFP-tau-BiFC dimers (Fig. 3A). Since this reduction may involve changes in calcium due to Gαq activation, we followed the correlation between eGFP-tau-BiFC intensity and intracellular calcium levels, as measured by a fluorescent calcium sensor, Calcium Orange, in real time. We find a clear inverse correlation between the two (Fig. 3B) suggesting that elevated calcium diminishes tau aggregation.
Figure 3: Tau aggregation is modulated in part by Gαq activity:
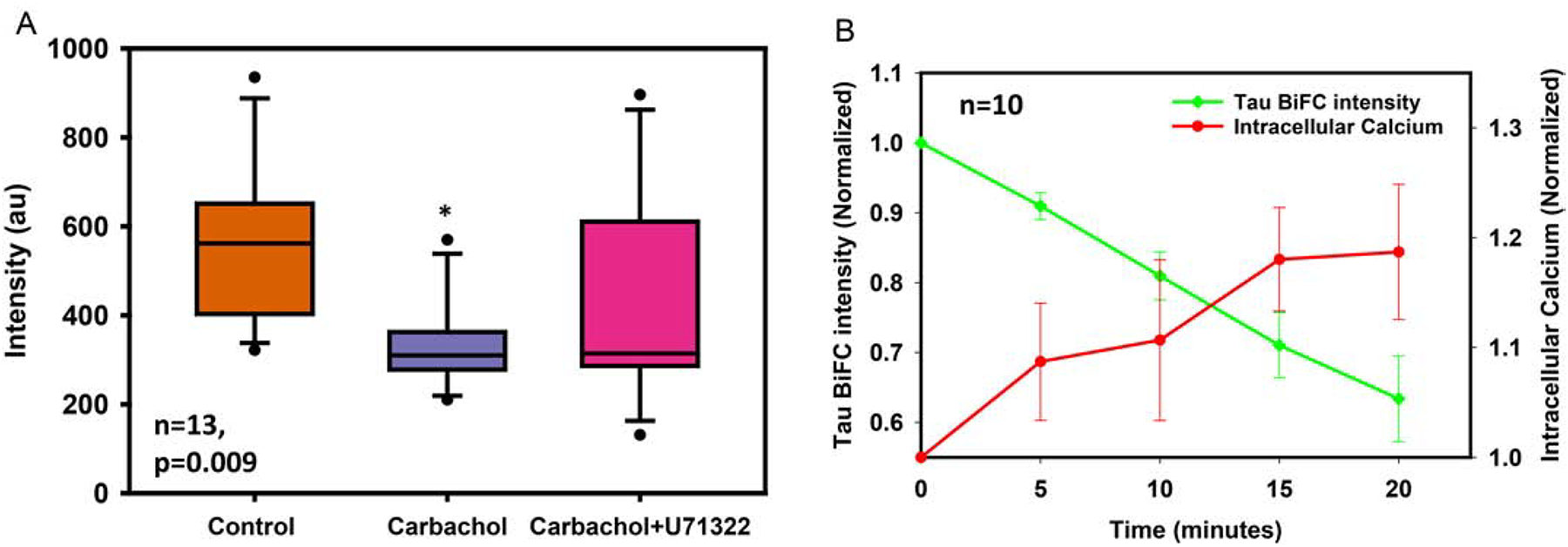
A) Intensities of eGFP-tau-BiFC expressed in PC12 cells under basal conditions, 10 minutes after treatment with 5 μM carbachol and 20 mins after a second treatment with 1 μM PLC inhibitor U73122 (n=13, p=0.009). B) eGFP-tau-BiFC intensity and Calcium Orange intensities in labeled PC12 cells after stimulation with 5 μM carbachol (n=10).
PLCβ1 interacts with tau during G2/M cell phase.
As noted in the Introduction, the neurofibrillary tangles of AD patients contain mitotic proteins. With this in mind, we tested whether the impact of PLCβ1 on tau aggregation differs in the G1 versus the G2/M phase. For these studies, we transfected cells with control siRNA or siRNA (PLCβ1) where the efficiency of down-regulation was slightly less than the studies above (compare Supplemental Fig. 1A versus 1B), which we believe is due to differences in lots of the reagents. After 48 hrs, we serum starved the cells to arrest them in the G1 phase, or treated them with the CDK1 inhibitor R0336 to arrest the cells in the G2/M phase. We find that down-regulating PLCβ1 increases tau aggregation in the G2/M phase but not in the G1 phase (Fig. 4A). This result is corroborated by a study where we isolated the cytosolic fraction of undifferentiated PC12 cells arrested in the G2/M phase and in the G1 phase, pulled-down PLCβ1 with a monoclonal antibody and identified the bound proteins by mass spectrometry. These results show that PLCβ1 pulls down tau in G2/M phase but not during G1 phase. (Fig 4B).
Figure 4: PLCβ1 interacts with tau during G2/M cell phase:
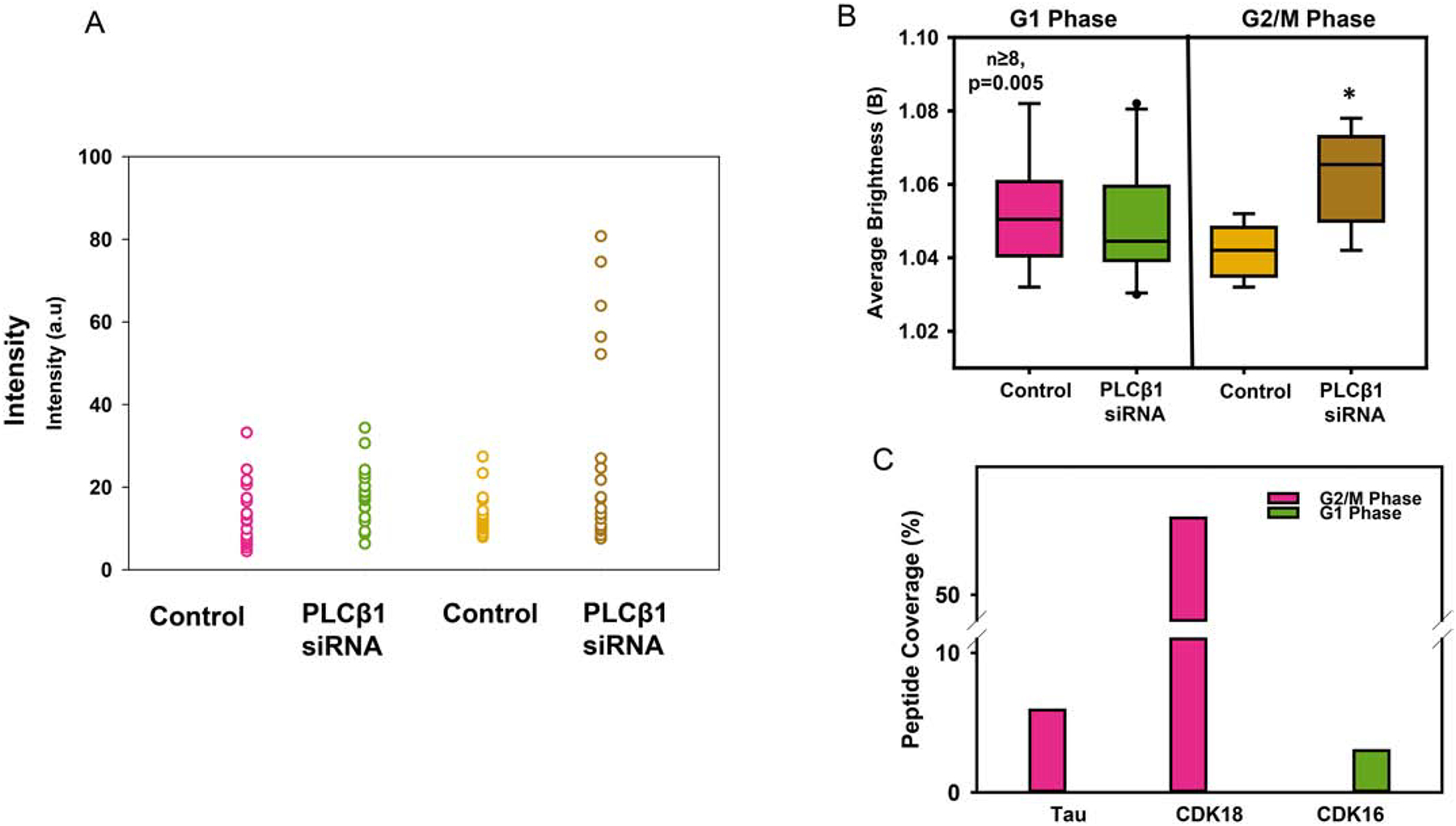
A) The average Brightness (B) values of PC12 cells expressing eGFP-tau-BiFC treated with either negative control siRNA or siRNA(PLCβ1) that were cell-cycle arrested in G1 or G2/M phases by serum-starvation or treatment with 10 μM CDK inhibitor R03306 (n≥8 across two individual experiments, * p=0.005). C) Result from a mass spectrometry analysis of tau and mitotic proteins complexed with PLCβ1 using an antibody pull-down. The graph shows the percent coverage of tau, CDK16 and CDK18, as calculated by dividing the number of amino acids in all found peptides by the total number of amino acids in the entire protein sequence, from PC12 cells arrested in G1 or in G2/M phases.
PLCβ1 binds to CDK18 during G2/M phase.
We screened the PLCb1 pull-down studies for mitotic proteins and found that in the G1 phase PLCβ1 pulls-down a kinase that plays a role in maintaining cell proliferation, CDK16 (see [32]), and in the G2/M phase PLCβ1 pulls down the mitotic protein CDK18 (Fig. 4B). CDK16 and CDK18 are highly homologous enzymes [35]. We have previously reported that PLCβ1 binds to CDK16 and inhibits kinase activity [32]. The high conservation of the kinase domains of CDK16 and CDK18 suggests that PLCβ1 will similarly inhibit CDK18. Although the exact function of CDK18 has yet to be described, a study reported that CDK18 indirectly promotes tau phosphorylation and that CDK18 is one of the mitotic proteins found in degenerative plaques in AD patients [6].
We verified that PLCβ1 and CDK18 associate in undifferentiated PC12 cells by Förster resonance energy transfer studies (FRET) as determined by fluorescence lifetime imaging (FLIM), see Materials and Methods. Here, we detect FRET by the reduction in the fluorescence lifetime of the donor (i.e. enhanced green fluorescent protein or eGFP) caused by the transfer of energy to FRET acceptor (i.e. mCherry). To this end, we transfected undifferentiated PC12 cells with eGFP-PLCβ1 and measured the reduction in lifetime in the presence of mCherry-CDK18 or free mCherry control. The lifetime measured in each pixel of the image can be plotted in polar coordinates to directly show reduced lifetimes that correspond to FRET by movement of the values inside the phasor arc (Fig.5A–C). The compiled lifetime data show that association between PLCβ1-CDK18 is very limited in unsynchronized cells that are predominantly in the G1 phase but becomes much higher when cells are arrested in the G2/M phase. This increased association is most likely due to the higher level of expression of both proteins in the M phase (Fig.5D, Supplemental Fig. 1D). These FRET results in conjunction with the previously reported FRET studies between PLCβ1 and CDK16 also corroborate the findings from mass spectrometry indicating that PLCβ1 binds to CDK16 during G1 phase and to CDK18 interactions occur primarily during G2/M phase (Fig.5E).
Figure 5: PLCβ1 binds to CDK18 during G2/M phase:
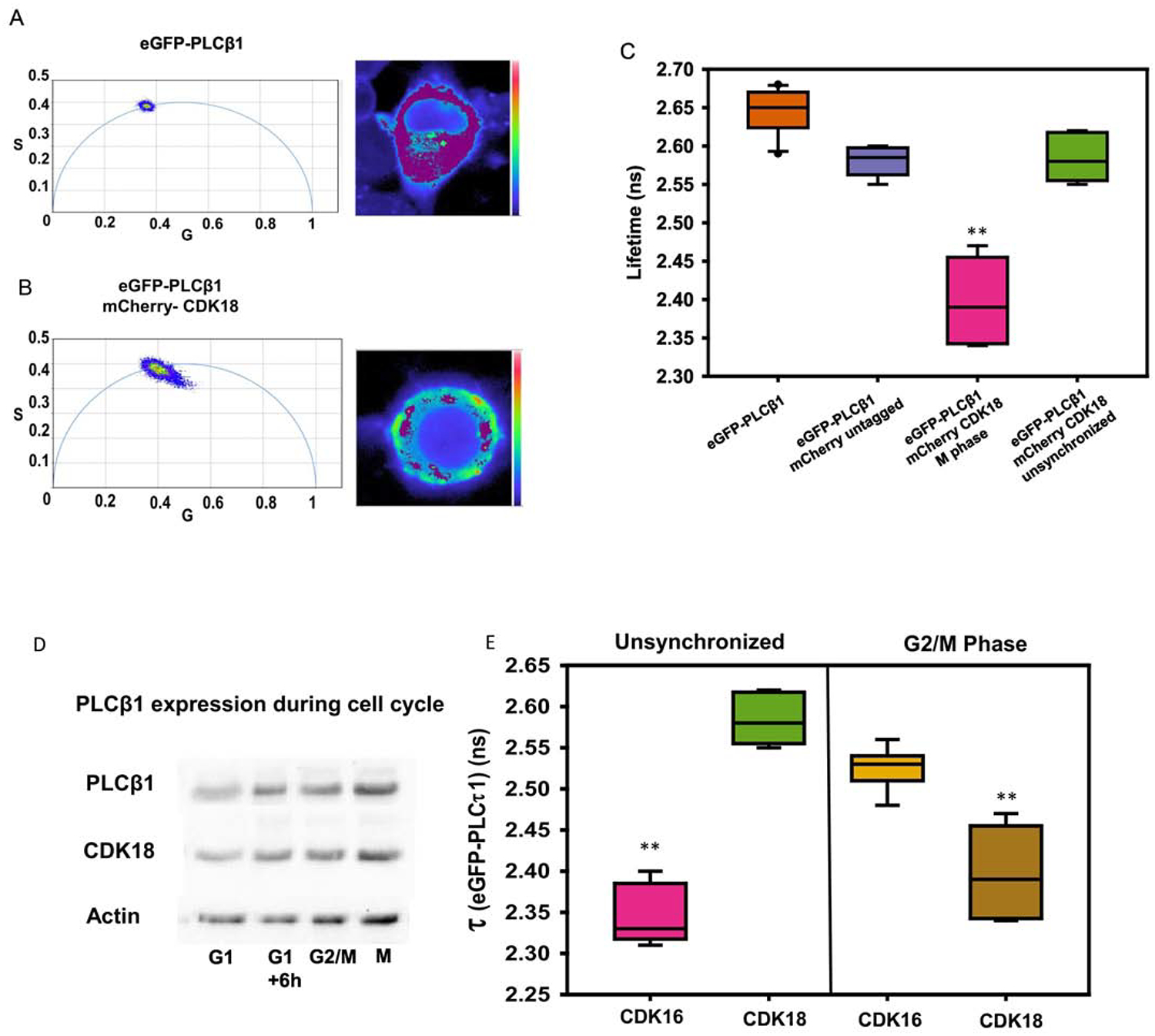
A) A phasor plot (left) and false color image of a representative PC12 cell (right) of eGFP-PLCβ1. The phasor plot shows that eGFP-PLCβ1 has a single fluorescent lifetime indicated by a homogenous population on the phasor arc. The pixels included in the magenta circle of the phasor plot (lifetime center= 2.56 ns) are false colored magenta and overlaid on an image of the cell (right), demonstrating a uniform lifetime throughout the cell. B) Similar study as in (A) except that in these cells, the FRET acceptor mCherry-CDK18 is co-expressed with eGFP-PLCβ1. The occurrence of FRET is seen by movement of the lifetimes of the pixel in the image towards the inside of the arc corresponding to reduced lifetime values due to FRET. The pixels included in the magenta circle of the phasor plot (lifetime center= 2.56 ns) are false colored magenta and overlaid on an image of the cell (right) showing the non-FRET populations. C) EGFP-PLCβ1 lifetimes of PC12 cells expressing eGFP-PLCβ1 alone, with untagged mCherry or with mCherry-CDK18 in unsynchronized cells and cells arrested in M phase where decreases in lifetimes are due to FRET (n=8, **= p<0.001). D) Western blot analysis of PC12 cells that are either unsynchronized or arrested at G1, G2/M, and M phases and blotted with anti-PLCβ1, anti-CDK18 and anti-actin antibodies. E) EGFP-PLCβ1 lifetimes of PC12 cells expressing both eGFP-PLCβ1 alone or with mCherry-CDK18, in either unsynchronized or arrested in G2/M phase. (**= p<0.001, n>=10).
DISCUSSION
In this study, we have shown that PLCβ1 impedes tau aggregation by two mechanisms: regulation of intracellular calcium and association with CDK18. Although not believed to be the determinant of Alzheimer’s disease, tau aggregation is main contributor. Tau has several truncated forms and its aggregation depends on the degree of phosphorylation and the pattern of the phosphorylation sequence [44]. Tau interacts with microtubules that stabilize the cell cytoskeleton including neuronal synapses [45]. Once phosphorylated, tau aggregates into intracellular particles that are thought to precede the formation of neurofibrillar tangles. Here, we monitored aggregation as an indirect effect of phosphorylation of a tagged tau construct.
Alzheimer’s disease involves complex and interconnected signaling pathways that involves multiple cell compartments including mitochondria [46]. Here, we focused on the Gαq/PLCβ pathway that is activated in response to neurotransmitters to increase intracellular calcium. Disruption of calcium homeostasis is associated with neuronal dysfunction. Our studies show a clear inverse correlation between cellular calcium levels and tau aggregation: stimulation of Gαq shifts tau to a less aggregated state, while inhibiting PLCβ1 shifts tau to a more aggregated state. Surprisingly, cells treated with siRNA(PLCβ1) showed a dramatic increase in eGFP-tau-BiFC aggregation that far exceeds the level seen by inhibiting PLCβ1 activity. This enhanced response led us to investigate other mechanisms through which PLCβ may contribute to tau aggregation.
Previous studies have shown that PLCβ1 has a cytosolic population that binds proteins involved in pathways that are distinct from Gαq. Our recent studies identified CDK16 as a binding partner of PLCβ1 in unsynchronized PC12 cells and found that PLCβ1 inhibits CDK16’s ability to phosphorylate and inactivate an inhibitor of cell cycle progression thus preventing progression through the cell cycle. Here, we show that when cells are arrested at the G2/M phase boundary, PLCβ1 no longer binds CDK16 but instead binds a close family member, CDK18. This transfer of PLCβ binding partners is thought to be due to changes in the local availability for PLCβ1 binding.
CDK18 is found in neurofibrillary tangles along with phosphorylated tau and has been implicated to indirectly promote tau phosphorylation [6]. While PLCβ1 is not expected to bind CDK18 in the G1 phase, if neurons reenter the cell cycle, then the activity of CDK18 could be inhibited by cytosolic PLCβ1. Down-regulating PLCβ1 would allow for CDK18 activity promoting tau aggregation, and reducing calcium levels may synergize these effects. Alternately, elevation of other PLCβ1 binding partners, such as Gαq may compete with CDK18 for PLCβ1 binding and enhance calcium signals. While mores studies are needed to understand the precise role of the plasma membrane and cytosolic populations of PLCβ1 on AD pathology, our studies show that approaches that increase PLCβ1 levels of may be beneficial in reducing the formation of neurofibrillary tangles.
Supplementary Material
HIGHLIGHTS.
Alzheimer’s disease is typified by calcium dysfunction and tau aggregates.
Gαq/PLCβ signaling pathway generate calcium signals.
Down-regulating PLCβ significantly increases tau protein expression and aggregation.
Inhibiting PLCβ activity results in a modest enhancement of tau aggregation.
PLCβ binds to CDK18 promoting tau phosphorylation and aggregation.
ACKNOWLEDGEMENTS.
The authors are grateful for the support of NIH GM116187.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Maccioni RB, Munoz JP, Barbeito L, The molecular bases of Alzheimer’s disease and other neurodegenerative disorders, Arch Med Res, 32 (2001) 367–381. [DOI] [PubMed] [Google Scholar]
- [2].Brion JP, Neurofibrillary Tangles and Alzheimer’s Disease, European Neurology, 40 (1998) 130–140. [DOI] [PubMed] [Google Scholar]
- [3].Mak E, Bethlehem RAI, Romero-Garcia R, Cervenka S, Rittman T, Gabel S, Surendranathan A, Bevan-Jones RW, Passamonti L, Vázquez Rodríguez P, Su L, Arnold R, Williams GB, Hong YT, Fryer TD, Aigbirhio FI, Rowe JB, O’Brien JT, In vivo coupling of tau pathology and cortical thinning in Alzheimer’s disease, Alzheimers Dement (Amst), 10 (2018) 678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Borysov SI, Granic A, Padmanabhan J, Walczak CE, Potter H, Alzheimer Aβ disrupts the mitotic spindle and directly inhibits mitotic microtubule motors, Cell Cycle, 10 (2011) 1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Atwood CS, Bowen RL, A Unified Hypothesis of Early- and Late-Onset Alzheimer’s Disease Pathogenesis, J Alzheimers Dis, 47 (2015) 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Herskovits AZ, Davies P, The regulation of tau phosphorylation by PCTAIRE 3: Implications for the pathogenesis of Alzheimer’s disease, Neurobiology of Disease, 23 (2006) 398–408. [DOI] [PubMed] [Google Scholar]
- [7].Barone G, Staples CJ, Ganesh A, Patterson KW, Bryne DP, Myers KN, Patil AA, Eyers CE, Maslen S, Skehel JM, Eyers PA, Collis SJ, Human CDK18 promotes replication stress signaling and genome stability, Nucleic Acids Research, 44 (2016) 8772–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Berridge MJ, Calcium hypothesis of Alzheimer’s disease, Pflugers Arch, 459 (2010) 441–449. [DOI] [PubMed] [Google Scholar]
- [9].Lyon AM, Tesmer JJG, Structural insights into phospholipase C-β function, Mol Pharmacol, 84 (2013) 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Suh P, Park J, Manzoli L, Cocco L, Peak J, Katan M, Fukami K, Kataoka T, Yun S, Ryu S, Multiple roles of phosphoinositide-specific phospholipase C isozymes, BMB reports, 41 (2008) 415–434. [DOI] [PubMed] [Google Scholar]
- [11].Schoonjans AS, Meuwissen M, Reyniers E, Kooy F, Ceulemans B, PLCB1 epileptic encephalopathies; Review and expansion of the phenotypic spectrum, Eur J Paediatr Neurol, 20 (2016) 474–479. [DOI] [PubMed] [Google Scholar]
- [12].Kim SW, Seo M, Kim DS, Kang M, Kim YS, Koh HY, Shin HS, Knockdown of phospholipase C-beta1 in the medial prefrontal cortex of male mice impairs working memory among multiple schizophrenia endophenotypes, J Psychiatry Neurosci, 40 (2015) 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Albert JL, Boyle JP, Roberts JA, Challiss RA, Gubby SE, Boarder MR, Regulation of brain capillary endothelial cells by P2Y receptors coupled to Ca2+, phospholipase C and mitogen-activated protein kinase, Br J Pharmacol, 122 (1997) 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Baxter RM, Cohen P, Obermeier A, Ullrich A, Downes CP, Doza YN, Phosphotyrosine residues in the nerve-growth-factor receptor (Trk-A). Their role in the activation of inositolphospholipid metabolism and protein kinase cascades in phaeochromocytoma (PC12) cells, European Journal of Biochemistry, 234 (1995) 84–91. [DOI] [PubMed] [Google Scholar]
- [15].Hannan AJ, Kind PC, Blakemore C, Phospholipase C-β1 expression correlates with neuronal differentiation and synaptic plasticity in rat somatosensory cortex, Neuropharmacology, 37 (1998) 593–605. [DOI] [PubMed] [Google Scholar]
- [16].Kim H.-j., Koh H-Y, Impaired Reality Testing in Mice Lacking Phospholipase Cβ1: Observed by Persistent Representation-Mediated Taste Aversion, PLoS One, 11 (2016) e0146376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].García del Caño G, Montaña M, Aretxabala X, González-Burguera I, López de Jesús M, Barrondo S, Sallés J, Nuclear phospholipase C-β1 and diacylglycerol LIPASE-α in brain cortical neurons, Advances in Biological Regulation, 54 (2014) 12–23. [DOI] [PubMed] [Google Scholar]
- [18].Udawela M, Scarr E, Boer S, Um JY, Hannan AJ, McOmish C, Felder CC, Thomas EA, Dean B, Isoform specific differences in phospholipase C beta 1 expression in the prefrontal cortex in schizophrenia and suicide, npj Schizophrenia, 3 (2017) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Narayanan V, Guo Y, Scarlata S, Fluorescence studies suggest a role for a-synuclein in the phosphatdiylinositol lipid signaling pathway, Biochem., 44 (2005) 462–470. [DOI] [PubMed] [Google Scholar]
- [20].Ahmad M, Attoub S, Singh MN, Martin FL, El-Agnaf OMA, γ-Synuclein and the progression of cancer, The FASEB Journal, 21 (2007) 3419–3430. [DOI] [PubMed] [Google Scholar]
- [21].Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM, Degradation of alpha-synuclein by proteasome, J Biol Chem, 274 (1999) 33855–33858. [DOI] [PubMed] [Google Scholar]
- [22].Duda JE, Lee VM, Trojanowski JQ, Neuropathology of synuclein aggregates, J Neurosci Res, 61 (2000) 121–127. [DOI] [PubMed] [Google Scholar]
- [23].El-Agnaf OM, Irvine GB, Review: formation and properties of amyloid-like fibrils derived from alpha-synuclein and related proteins, J Struct Biol, 130 (2000) 300–309. [DOI] [PubMed] [Google Scholar]
- [24].George JM, The synucleins, Genome Biol, 3 (2002) 3002.3001–3002.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hashimoto M, Masliah E, Alpha-synuclein in Lewy body disease and Alzheimer’s disease, Brain Pathol, 9 (1999) 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lundvig D, Lindersson E, Jensen PH, Pathogenic effects of α-synuclein aggregation, Molecular Brain Research, 134 (2005) 3–17. [DOI] [PubMed] [Google Scholar]
- [27].Recchia A, Debetto P, Negro A, Guidolin D, Skaper S, Giusti P, a-Synuclein and Parkinson’s disease, FASEB J, 18 (2004) 617–626. [DOI] [PubMed] [Google Scholar]
- [28].Scarlata S, Golebiewska U, Linking alpha-synuclein properties with oxidation: a hypothesis on a mechanism underling cellular aggregation, Journal of Bioenergetics and Biomembranes, 46 (2014) 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Guo Y, Rosati B, Scarlata S, α-synuclein increases the cellular level of phospholipase Cβ1, Cellular Signalling, 24 (2012) 1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dowal L, Provitera P, Scarlata S, Stable association between G alpha(q) and phospholipase C beta 1 in living cells, J Biol Chem, 281 (2006) 23999–24014. [DOI] [PubMed] [Google Scholar]
- [31].Garwain O, Scarlata S, Phospholipase Cβ-TRAX Association Is Required for PC12 Cell Differentiation, Journal of Biological Chemistry, 291 (2016) 22970–22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Garwain O, Valla K, Scarlata S, Phospholipase Cβ1 regulates proliferation of neuronal cells, The FASEB Journal, 32 (2018) 2891–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Philip F, Guo Y, Aisiku O, Scarlata S, Phospholipase Cβ1 is linked to RNA interference of specific genes through translin-associated factor X, The FASEB Journal, 26 (2012) 4903–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Philip F, Sahu S, Golebiewska U, Scarlata S, RNA-induced silencing attenuates G protein-mediated calcium signals, Faseb j, 30 (2016) 1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Okuda T, Cleveland JL, Downing JR, PCTAIRE-1 and PCTAIRE-3, two members of a novel cdc2/CDC28-related protein kinase gene family, Oncogene, 7 (1992) 2249–2258. [PubMed] [Google Scholar]
- [36].Brunello CA, Yan X, Huttunen HJ, Internalized Tau sensitizes cells to stress by promoting formation and stability of stress granules, Sci Rep, 6 (2016) 30498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Digman MA, Caiolfa VR, Zamai M, Gratton E, The phasor approach to fluorescence lifetime imaging analysis, Biophys J, 94 (2008) L14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Digman MA, Dalal R, Horwitz AF, Gratton E, Mapping the number of molecules and brightness in the laser scanning microscope, Biophys J, 94 (2008) 2320–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ossato G, Digman MA, Aiken C, Lukacsovich T, Marsh JL, Gratton E, A two-step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis, Biophys J, 98 (2010) 3078–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Plotegher N, Gratton E, Bubacco L, Number and Brightness analysis of alpha-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells, Biochim Biophys Acta, 1840 (2014) 2014–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Planes N, Digman MA, Vanderheyden P, Gratton E, Caballero-George C, Number and brightness analysis to study spatio-temporal distribution of the angiotensin II AT1 and the endothelin-1 ETA receptors: Influence of ligand binding, Biochim Biophys Acta Gen Subj, 1863 (2019) 917–924. [DOI] [PubMed] [Google Scholar]
- [42].Iwata S, Lee JW, Okada K, Lee JK Iwata M, Rasmussen B, Link TA, Ramaswamy S and Jap BK, Complete at of the 11 subunit bovine mitochondrial cytochrome bc1 complex., Science, 281 (1998) 64–71. [DOI] [PubMed] [Google Scholar]
- [43].Herskovits AZ, Davies P, The regulation of tau phosphorylation by PCTAIRE 3: implications for the pathogenesis of Alzheimer’s disease, Neurobiol Dis, 23 (2006) 398–408. [DOI] [PubMed] [Google Scholar]
- [44].Simic G, Babic Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milosevic N, Bazadona D, Buee L, de Silva R, Di Giovanni G, Wischik C, Hof PR, Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies, Biomolecules, 6 (2016) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, Wang Y-C, Swerts J, Beyens J, Miskiewicz K, Vilain S, Dewachter I, Moechars D, De Strooper B, Spires-Jones TL, De Wit J, Verstreken P, Tau association with synaptic vesicles causes presynaptic dysfunction, Nature Communications, 8 (2017) 15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shimohama S, Matsushima H, Signal transduction mechanisms in Alzheimer disease, Alzheimer Dis Assoc Disord, 9 Suppl 2 (1995) 15–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.