

Abstract
Atg3‐catalyzed transferring of Atg8 to phosphatidylethanolamine (PE) in the phagophore membrane is essential for autophagy. Previous studies have demonstrated that this process requires Atg3 to interact with the phagophore membrane via its N‐terminal amphipathic helix. In this study, by using combined biochemical and biophysical approaches, our data showed that in addition to binding to the membranes, Atg3 attenuates lipid diffusion and enriches lipid molecules with smaller headgroup. Our data suggest that Atg3 promotes Atg8 lipidation via altering lipid diffusion and rearrangement.
Keywords: amphipathic helix, Atg3, Atg8, autophagy, lipid diffusion
Abbreviations
- ACF
autocorrelation function
- Atg3
autophagy‐related protein 3
- Atg5
autophagy‐related protein 5
- Atg7
autophagy‐related protein 7
- Atg8
autophagy‐related protein 8
- Atg12
autophagy‐related protein 12
- DPH
1,6‐diphenyl‐1,3,5‐hexatriene
- EGFP
enhanced green fluorescent protein
- FCS
fluorescence correlation spectroscopy
- GUV
giant unilamellar vesicle
- IM
isolation membrane
- LC3
microtubule‐associated proteins 1A/1B light chain 3
- LUV
large unilamellar vesicle.
- PE
phosphatidylethanolamine
- PI(3)P
phosphatidylinositol‐3‐phosphate
- PX
phox homology
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SNAP‐25
synaptosomal‐associated protein 25
- SNARE
soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor
- SUV
small unilamellar vesicle
- Vam7
vacuolar morphogenesis protein 7
1. INTRODUCTION
Autophagy, as an evolutionarily conserved pathway mediating bulk degradation of cytosolic proteins and organelles in eukaryotic cells, is vital for the maintenance of cell homeostasis.1, 2 Increasing evidence revealed that disorder of autophagy causes a variety of disease including neurodegeneration and cancer.2, 3, 4
Autophagy is featured by the formation of a double‐membrane structure called the autophagosome, which engulfs cytosolic cargoes and subsequently delivers the cargoes to the vacuole or lysosome for degradation and recycling.5, 6, 7 To date, over 40 autophagy‐related (Atg) proteins have been identified as crucial players for autophagy and autophagosome formation.8, 9 These proteins are capable of gathering together around the vacuole in yeast and the endoplasmic reticulum (ER) in mammals and act in cooperation with the membranes to generate a pre‐autophagosome structure (PAS), which develops into phagophore or isolation membrane (IM) with membrane elongation and eventually into autophagosome with membrane closure.5, 6, 10, 11
The conjugation of the ubiquitin‐like protein Atg8/LC3 to phosphatidylethanolamine (PE), an important phospholipid in the autophagosome membrane, is required for autophagosome formation. 12 For the conjugation reaction, the cysteine protease Atg4 first cleaves Atg8 at its C‐terminus to initiate the reaction.13, 14 Subsequently, the processed Atg8 is activated by the E1‐like enzyme Atg7 to form an Atg8–Atg7 thioester intermediate and is then transferred to the E2‐like enzyme Atg3 to form an Atg8–Atg3 thioester intermediate. Finally, Atg8 is conjugated to PE via an amide bond between its C‐terminal Gly and the amino group of PE. 15 In addition to these factors, a wealth of evidence has revealed that the E3‐like Atg5–Atg12–Atg16 complex promotes Atg8 conjugation to PE,16, 17, 18 via an Atg12–Atg3 interaction that leads to conformational changes of the Atg3 active center and/or more efficient recruitment of Atg3 to the autophagosome membrane. 19
Atg3 plays a central role in Atg8‐PE conjugation. 15 Atg3 contains an N‐terminal amphipathic helix (NH) responsible for membrane interaction and a core E2‐like enzyme domain that directly catalyzes Atg8 conjugation to PE (Figure 1a). The rate and efficiency of the conjugation reaction rely on the enzyme activity of Atg3 and the concentrations of the reaction components (i.e., the Atg3–Atg8 complex and PE). The Atg3 NH domain has been implicated in membrane binding and membrane curvature sensing, both essential for increasing local Atg3 concentration at specific sites on the autophagosome membrane.20, 21, 22, 23 In addition, in vitro studies showed that high PE concentration leads to efficient Atg8‐PE conjugation. However, the physiological concentration of PE on phagophore is much lower than the PE concentration used in in vitro lipidation assays.24, 25 Despite such findings, the mechanism by which Atg3 cooperates with the membrane components to promote Atg8‐PE conjugation has been unclear. In particular, whether Atg3 affects lipid fluidity and lipid arrangement on the membrane lipid matrix remains elusive.
FIGURE 1.
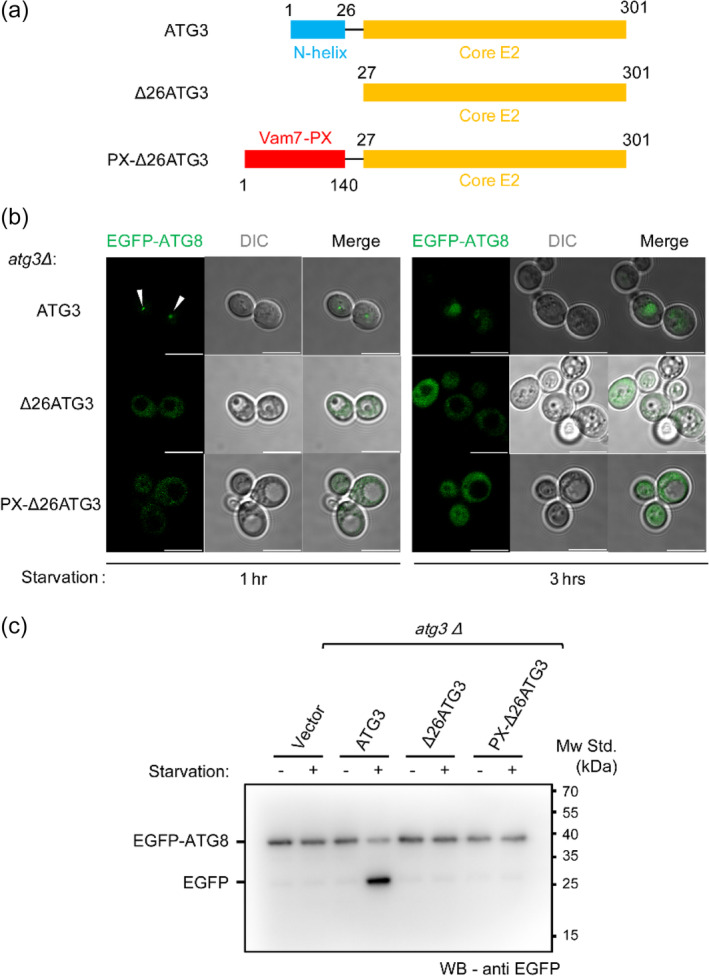
The N‐terminal amphipathic helix of Atg3 is required for autophagy initiation. (a) Illustration of the domain structures of Atg3 and variants used in this study. The N‐terminal helix and core E2 domain of Atg3 are colored with blue and orange, respectively. The genetically engineered PX domain of Vam7 (residues 1–140) is colored with red. (b) Representative laser scanning confocal microscopy (LSCM) graphs of yeast atg3Δ cells expressing EGFP‐Atg8, Atg3, and its variants upon 1‐ and 3‐hr starvation. The white arrows indicate EGFP‐Atg8 puncta during autophagy initiation. Scale bars, 10 μm. (c) EGFP‐Atg8 processing assay as shown by anti‐EGFP immunoblotting from the lysates of yeast atg3Δ cells expressing EGFP‐Atg8, Atg3, and its variants. The cells were grown to mid‐log phase and either harvested (−) or nitrogen starved for 3 hr (+). The degraded EGFP after starvation represents normal autophagy flux
In this study, we explored how Atg3 influences the property of the membrane lipid matrix under physiological PE concentration (20% molar ratio). Our results suggest that in addition to binding to the membranes, Atg3 can attenuate lipid diffusion and enrich small headgroup lipids (e.g., PE) in the membrane lipid matrix, which is important to promote Atg8 lipidation.
2. RESULTS
2.1. Chimeric PX‐Δ27Atg3 fails to restore autophagy flux in atg3Δ cells
Transfer of Atg8 to PE by Atg3 is the rate‐determining step in Atg8 lipidation.26, 27, 28, 29, 30 Binding of Atg3 to the phagophore membrane, which is dependent on its NH domain (Figure 1a), is required for Atg8 lipidation, but the underlying mechanism remains unclear. As the molecular beacon of the phagophore growing site, 31 phosphatidylinositol‐3‐phosphate [PI(3)P] plays an important role in the early PAS formation and in the subsequent phagophore expanding process via recruiting various Atg proteins that are responsible for Atg8 lipidation. PI(3)P on phagophore is produced by the autophagy‐specific PI3K complex upstream of Atg8 lipidation. 32 PI(3)P possesses abundant negative charges. Previous studies have shown that mutation of the positively charged residues on the Atg3‐NH domain severely blocks Atg8 lipidation both in vivo and in vitro. 20 It is conceivable that binding of Atg3‐NH to the PI(3)P‐containing membranes would recruit more Atg3 molecules to the membranes thus accelerating Atg8 lipidation. First, in our study, we investigated the importance of Atg3‐NH in Atg8 puncta formation and autophagosome‐vacuole fusion in yeast cells. Atg3 null cells display severe defects in autophagy. 33 Expression of Atg3 full‐length in atg3Δ yeast cells restored autophagy flux (Figure 1b), because we detected the formation of EGFP‐Atg8 puncta after 1‐hr nitrogen starvation and the EGFP fluorescent signal in the vacuoles in the following 3‐hr starvation that represents the fusion between the autophagosome and vacuole.
We next made a truncated Atg3 named Δ27Atg3 that lacks the NH domain, and a chimeric Atg3 construct named PX‐Δ27Atg3 that replaces the NH domain with the PX domain of Vam7 (a SNARE protein). As expected, Δ27Atg3 failed to restore autophagy flux in atg3Δ yeast cells (Figure 1b), which confirmed the important role of the NH domain. As a PI(3)P binding module, 34 the PX domain was expected to help recruit PX‐Δ27Atg3 to the phagophore membrane. However, similar to Δ27Atg3, chimeric PX‐Δ27Atg3 failed to rescue autophagy flux in atg3Δ yeast cells (Figure 1b).
To confirm the above observations obtained by fluorescent microscopy, we alternatively carried out a classical biochemical experiment, that is, the EGFP‐Atg8 processing assay, 35 to analyze autophagy flux. Since Atg8 can be distributed in both the inner and outer membrane of the autophagosome, the fusion of autophagosome with the vacuole would transfer the inner‐side Atg8 into the vacuole. If exogenously expressed Atg8 were tagged with EGFP, the vacuolar hydrolytic enzyme is expected to hydrolyze EGFP‐Atg8 and lead to the degradation of EGFP‐Atg8. Based on this assay, we found that Atg3 full‐length caused efficient EGFP‐Atg8 degradation, but Δ27Atg3 and chimeric PX‐Δ27Atg3 both failed to cause the degradation (Figure 1c), consistent with the imaging data shown above. These data confirmed that Atg3‐NH is required for maintaining autophagy and suggest that the NH domain functions beyond just serving as a membrane anchor, as replacement of NH with PX failed to support autophagy.
2.2. Membrane binding is not sufficient for Atg3 activity in Atg8 lipidation
To verify the membrane‐binding affinities of the Atg3 mutants we carried out liposome floatation experiments to analyze PI(3)P binding efficiency of Atg3 full‐length, Δ27Atg3 and PX‐Δ27Atg3. The liposomes contain basic membrane compositions with an additional 5% PI(3)P. The supernatants were collected and analyzed by SDS‐PAGE and immunoblotting. Under a short exposure time, only PX‐Δ27Atg3 was observed (Figure 2a). Atg3 full‐length was detected with prolonged exposure. In contrast, Δ27Atg3 only displayed background binding (Figure 2a). These results suggest that chimeric PX‐Δ27Atg3 indeed has a higher membrane binding affinity than Atg3 full‐length.
FIGURE 2.
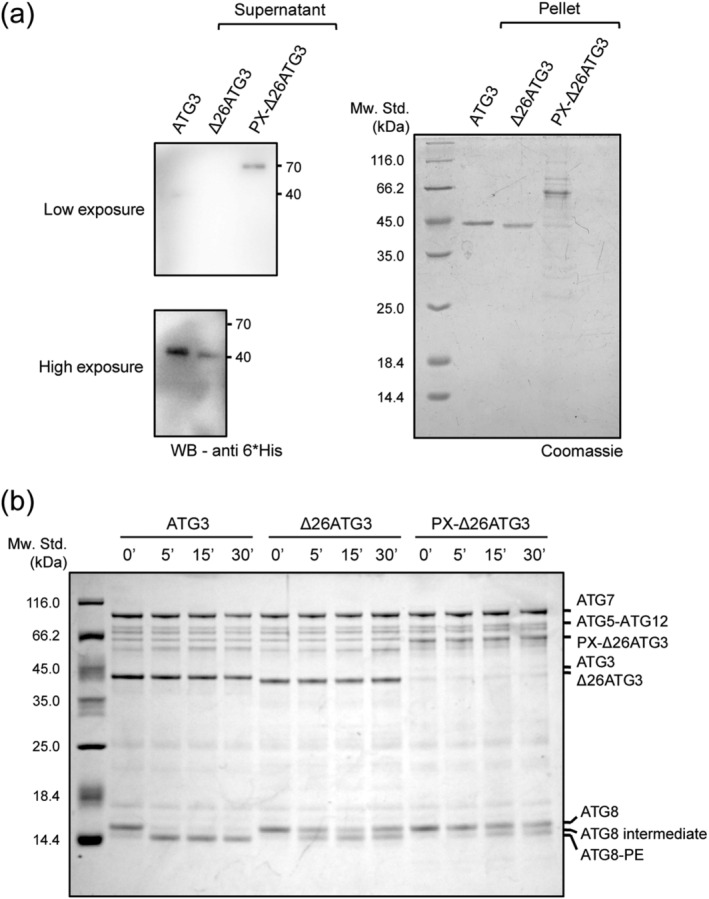
Binding of Atg3 to the membranes is not sufficient for Atg8 lipidation. (a) Binding of Atg3, Δ26Atg3, and PX‐Δ26Atg3 to phagophore‐mimic liposomes (POPC:POPE:DOPS:PI(3)P = 65:20:10:5) as measured by liposome floatation assay. The supernatants and pellets are shown by anti‐6*His‐tag immunoblotting and Coomassie brilliant blue (CBB) staining, respectively. At lower exposure, only PX‐Δ26Atg3 could be seen, which indicates the higher binding affinity. (b) in vitro Atg8 lipidation assay. Two micrometer GST‐Atg7, 2 μM Atg3, or its variants, 5 μM Atg8ΔR, 1 μM Atg5–Atg12 complex, 100 μM (total lipids) LUVs (POPC:POPE:DOPS:PI(3)P = 65:20:10:5), 1 mM ATP, 1 mM dithiothreitol (DTT), and 1 mM MgCl2 were mixed and incubated at 30°C for 0, 5, 15, and 30 min, respectively
Next, we monitored how the Atg3 mutants influence Atg8 lipidation in vitro using a previously established assay. 36 Compared to Atg3 full‐length, Δ27Atg3 and PX‐Δ27Atg3 both failed to catalyze Atg8 lipidation (Figure 2b), but displaying Atg8 intermediates as observed in the absence of liposome (Figure S1), which is consistent with previous studies. 23 Together, these data showed that replacement of the NH domain with a PI(3)P‐binding module supports Atg3 ability to bind the membrane but fails to support Atg3 activity in Atg8 lipidation, reinforcing the notion that the NH domain functions beyond serving as a membrane anchor in Atg8 lipidation.
2.3. Atg3 affects PE‐like lipid dynamics and rearrangement
It was unexpected that a stronger membrane‐binding PX domain could not support Atg3 function whereas the NH domain could. One possible explanation was that the NH domain, behaving as a typical amphipathic helix, alters lipid properties and dynamics via its membrane binding or insertion. It is well known that amphipathic helices generally participate in membrane shaping, membrane deformation, and lipid rearrangement.37, 38 We hence explored whether Atg3 has an ability to affect lipid dynamics and rearrangement on planner lipid bilayer by using z‐scan fluorescence correlation spectroscopy (FCS) (Figure 3a). Z‐scan FCS is an ideal tool to characterize lipid translational diffusion and lipid molecule dynamics. 39 To characterize lipid dynamics and rearrangements on the membranes, especially for PE, we utilized Alexa488 labeled hexa‐histidine‐tagged SNAP‐25‐derived peptide (SN2) as the fluorescent indicator and DGS‐NTA as the membrane anchor (Figure 3a). We assume that DGS‐NTA behaves like PE, as DGS‐NTA has a small effective headgroup like PE, followed by a NTA group floating in the solution (Figure 3a).
FIGURE 3.
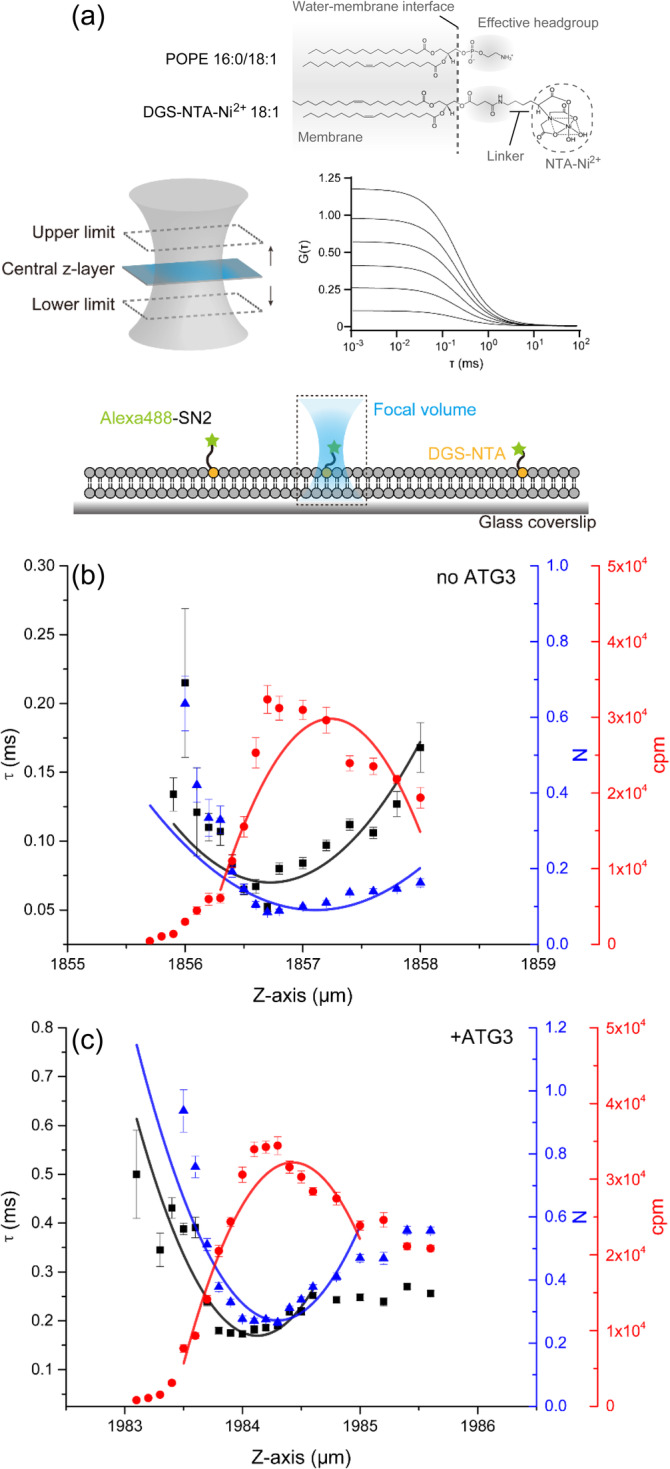
Atg3 attenuates lipid diffusion and enriches small headgroup lipids. (a) Illustration of bilayer z‐scan fluorescence correlation spectroscopy (FCS). Supported lipid bilayer (SLB) was made by LUV rupture on a glass coverslip. Hexa‐histidine tagged Alexa488‐labeled SN2 (green pentagrams) was immobilized on DGS‐NTA (orange circles). The enlarged focal volume and example autocorrelation curves are shown in the middle of the chart. The comparison of headgroups between POPE and DGS‐NTA2+ is shown on the top of the chart. Note that the scale of illustration is not the same as in reality. (b and c) Bilayer z‐scan FCS results of SLB‐bound Alexa488‐SN2 in the absence (b) and presence (c) of Atg3. SLB was prepared by LUV (POPC:POPE:DOPS:PI(3)P = 65:20:10:5) rupture. Curve fitting was achieved by using parabolic function. τ, the dwell time of fluorescent molecules in the confocal volume; N, number of fluorescent molecules in the confocal volume; cpm, counts per second per molecule. Individual plots are displayed as means ± SD
Each measurement was started from the bottom of the bilayer (i.e., the lower z‐limit) to the upper z‐limit (Figure 3a). Data acquired from each z‐coordinates were fitted to auto‐correlation function (ACF), which yields three parameters that include dwelling time of fluorescent molecule in the confocal volume (τ), number of fluorescent molecules in the confocal volume (N), and counts per second per molecule (cpm). Each parameter in different z‐coordinates was then plotted and fitted to parabolic function (Figure 3b,c). As our oil‐immersed 60× objective has a limited working distance and may cause reflection index mismatch, the cpm cannot drop to the background level at the upper z‐limit. When Atg3 was absent, the lateral diffusion of PE‐mimic DGS‐NTA‐Alexa488‐SN2 was measured with a dwell time of 0.068 ± 0.004 ms (Figure 3b, Table 1). However, the addition of Atg3 resulted in an increase of the dwell time to 0.197 ± 0.009 ms (Figure 3c, Table 1), which implies that Atg3 attenuates the lateral diffusion of PE‐like lipid (DGS‐NTA) on the lipid bilayer. Interestingly, the number of the fluorescent molecules in the confocal volume (N) was also increased (Figure 3c, Table 1), indicating that Atg3 causes a local enrichment of PE‐like lipid (DGS‐NTA) on the membranes. As a negative control, no significant binding between Atg3 and Alexa488‐SN2 was observed (Figure S2). Altogether, these data suggest that Atg3 exerts an effect on lipid dynamics and rearrangement, for example, attenuating lateral diffusion of PE and enriching local PE concentration on the membranes.
TABLE 1.
Z‐scan FCS results summary of the vertex value of parabolic fit
| 0% Chol | 0% Chol + Atg3 | 20% Chol | 40% Chol | |
|---|---|---|---|---|
| τ (ms) | 0.068 ± 0.004 | 0.197 ± 0.009 | 0.085 ± 0.002 | 0.107 ± 0.004 |
| N | 0.11 ± 0.02 | 0.26 ± 0.02 | 0.10 ± 0.03 | 0.11 ± 0.03 |
| cpm (×104) | 3.05 ± 0.03 | 3.32 ± 0.03 | 3.96 ± 0.03 | 4.11 ± 0.07 |
Note: Data are presented as means ± SD. *τ, the dwell time of fluorescent molecules in the confocal volume.
Abbreviations: cpm, counts per second per molecule; N, number of fluorescent molecules in the confocal volume.
2.4. Cholesterol attenuates lipid diffusion but exerts no influence on lipid enrichment
To verify the ability of Atg3 in attenuating lipid diffusion, we also performed DPH anisotropy assay because DPH can be embedded into the bilayer and it is a traditional membrane fluidity indicator. 40 Global lipid diffusion can be characterized by monitoring the anisotropy of DPH. When Atg3 was absent, the pure lipids displayed a much lower DPH anisotropy (Figure 4a). However, addition of Atg3 increased the DPH anisotropy and induced a lipid phase transition with elevated temperature (Tm = 15.50°C) (Figure 4a). These data are consistent with the z‐scan FCS results and suggest that Atg3 can attenuate lipid diffusion.
FIGURE 4.
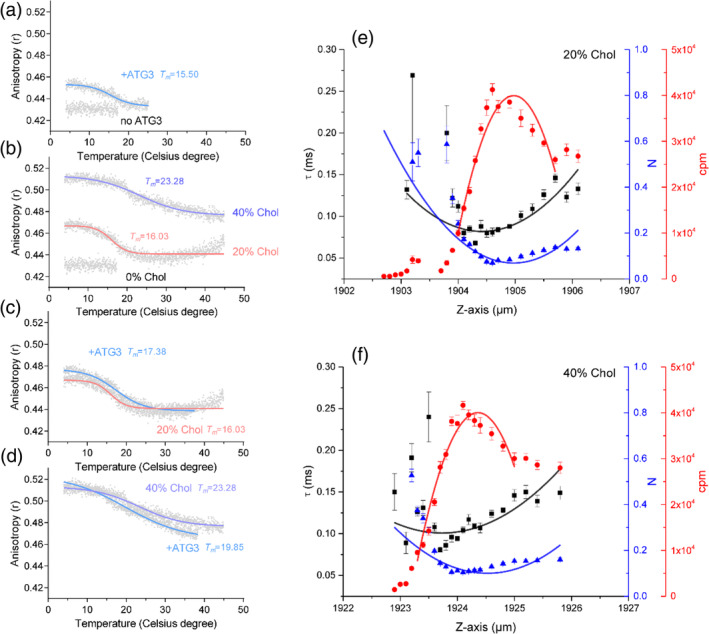
Cholesterol attenuates lipid diffusion but exerts no influence on lipid enrichment. (a) DPH anisotropy measurement of cholesterol‐free LUVs in the absence and presence of Atg3. Pure LUVs display no phase behavior. Addition of Atg3 induces a phase transition temperature of 15.50°C, which indicates attenuation of lipid translational diffusion. (b) Cholesterol induces phase behavior of the lipids as measured by DPH anisotropy. Incorporation of 20 and 40% cholesterol induce phase transition temperature of 16.03 and 23.28°C, respectively. Note that the absolute DPH anisotropy at high temperature in 40% cholesterol is larger than that in 0 and 20% cholesterol. (c and d) Atg3 has little effect on the phase behavior of LUVs contain 20% (c) and 40% (d) cholesterol. (e and f) Bilayer z‐scan FCS results of SLB‐bound Alexa488‐SN2 with additional 20% (e) and 40% (f) cholesterol. SLB was prepared by LUV (POPC:POPE:DOPS:PI(3)P = 65:20:10:5, additional cholesterols were supplied by replacing corresponding POPC ratio) rupture. Curve fitting was achieved by using parabolic function. τ, the dwell time of fluorescent molecules in the confocal volume; N, number of fluorescent molecules in the confocal volume; cpm, counts per second per molecule. Individual plots are displayed as means ± SD
Next, we explored whether the enrichment of small‐headgroup lipid molecules is driven by Atg3 or results from the altered lipid fluidity. To test this point, we applied cholesterol as a control in the experiments to investigate whether cholesterol likewise enriches small‐headgroup lipids, because cholesterol is able to decrease global lipid diffusion. As expected, 20 and 40% cholesterol indeed increased the DPH anisotropy and the temperature‐dependent lipid phase‐transition (16.03 and 23.28°C of Tm, respectively) (Figure 4b). In the presence of cholesterol, the addition of Atg3 had little influence on the DPH anisotropy and the temperature‐dependent lipid phase transition (Figure 4c,d). In addition, z‐scan FCS experiments showed that cholesterol indeed increased the dwell time of DGS‐NTA‐Alexa488‐SN2 in the lipid bilayer to 0.085 ± 0.002 ms (20% cholesterol) and to 0.107 ± 0.004 ms (40% cholesterol), respectively (Figure 4e,f, Table 1), which confirmed that cholesterol decreases global lipid diffusion. However, in contrast to Atg3, cholesterol displayed no significant effect on the number of fluorescent molecules in the focal volume (Figure 4e,f, Table 1). These results again suggest that Atg3 attenuates lipid diffusion and specifically enriches small‐headgroup lipid molecules.
Although Atg3 cannot increase the Tm of the lipid phase‐transition when the lipid bilayer already contains certain amounts of cholesterol, Atg3 still bound to cholesterol‐containing liposomes, shown by liposome floatation (Figure 5a). These data suggest that the binding of Atg3 to the membranes cannot further induce lipid rearrangement in the diffusion‐confined lipid bilayer. To verify this, we carried out in vitro Atg8 lipidation using giant unilamellar vesicles (GUVs) with different cholesterol molar ratios (Figure 5b). Intriguingly, a high ratio of cholesterol weakened Atg3 activity in Atg8 lipidation (Figure 5b). Hence, although cholesterol is able to decrease the global motilities of most lipid molecules, it seems unlikely to enrich certain lipid molecules (e.g., PE and PI(3)P). It is noteworthy that in most conditions Atg8 was selectively recruited to the small‐sized lipid structures (Figure 5b). We attributed this phenomenon to the curvature‐sensing ability of Atg3, as suggested by previous studies. 23 In all, these data suggest that in addition to the interaction with the membranes, Atg3 has an ability to attenuate lipid diffusing and enrich PE‐like lipid molecules in the membrane lipid matrix, which is important to promote Atg8 lipidation.
FIGURE 5.
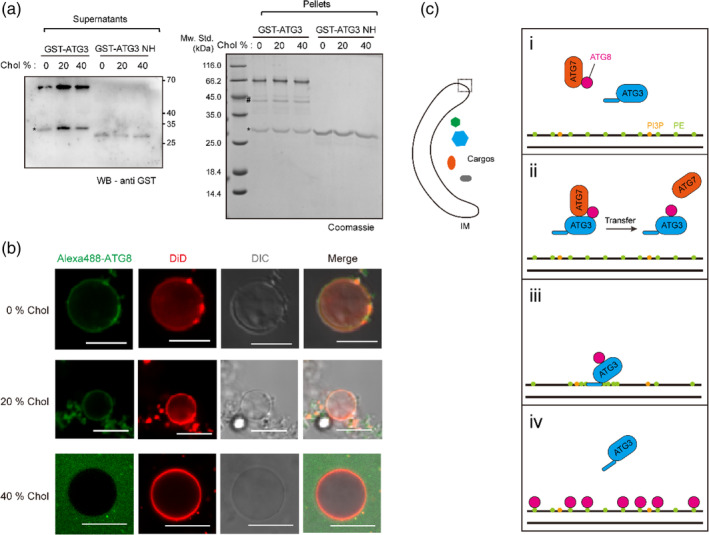
Atg3 mediates Atg8 lipidation by inducing potential lipid rearrangement. (a) Binding of GST‐Atg3 and GST‐Atg3‐NH to liposomes (POPC:POPE:DOPS:PI(3)P = 65:20:10:5, additional cholesterols were supplied by replacing corresponding POPC ratio) as measured by liposome floatation assay. The supernatants and pellets are shown by anti‐GST‐tag immunoblotting and Coomassie brilliant blue (CBB) staining, respectively. Asterisks indicate degraded GSTs. Hash indicates degraded Atg3 proteins. (b) in vitro Atg8 lipidation using giant unilamellar vesicles (GUVs). Two micrometer GST‐Atg7, 2 μM Atg3, 1.5 μM Alexa488‐labeled Atg8ΔR, 100 μM (total lipids) GUVs (POPC:POPE:DOPS:PI(3)P:DiD = 64:20:10:5:1, additional cholesterols were supplied by replacing corresponding POPC ratio), 1 mM ATP, 1 mM dithiothreitol (DTT), and 1 mM MgCl2 were mixed and incubated at 30°C for 1 hr. Scale bars, 10 μm. (c) Working model of Atg3‐mediated Atg8 lipidation. The overall view of the growing phagophore/isolation membrane (IM) is shown on the left. (i) Atg8 is first activated by E1‐like enzyme Atg7 (ATP‐dependent); (ii) The E1‐like complex (Atg7, Atg8) then transfer Atg8 to Atg3, thus forming the E2‐like complex (Atg3–Atg8); (iii) The E2‐like complex binds to phagophore membrane through N‐terminal amphipathic helix of Atg3. The N‐terminal amphipathic helix of Atg3 further induces local lipid rearrangement, where phosphatidylethanolamine (PE) and PI(3)P are enriched; (iv) Atg8s are conjugated to phosphatidylethanolamine (PE). Atg3 is recycled for another round of lipidation reaction
3. DISCUSSION
Atg8 lipidation is an essential step for autophagy initiation and phagophore growth. Previous studies have identified two ubiquitin‐like modification systems, Atg7–Atg3–Atg8 and Atg5–Atg12–Atg16, which synergistically conjugate Atg8 to PE. 15 It is also well known that Atg3, the E2‐like enzyme, transfers Atg8 to phagophore via binding of its NH domain to the highly curved membrane structures.21, 23 In this study, our results suggest that in addition to the membrane interaction, Atg3 attenuates lipid diffusion while simultaneously enriches local PE concentration to promote Atg8 conjugation to PE.
The most obvious effect of the peripheral membrane proteins is the obstruction of lipid diffusion. Indeed, the DPH anisotropy and z‐scan FCS results showed that both Atg3 and cholesterol attenuated lipid diffusion. However, unlike cholesterol, Atg3 specifically enriches small‐headgroup lipids (e.g., PE). Previous data have shown that increasing the molar ratio of PE on artificial liposome accelerates Atg8 lipidation in vitro,23, 24, 25, 41, 42 because the highly abundant amount of PE increases the probability of the contact between the E2‐complex (Atg3–Atg8 complex) and PE. Hence, the increased concentration of PE caused by Atg3 may account for the ability of Atg3 in promoting Atg8 lipidation. Although direct evidence for the interaction between Atg3 and PE is currently lacking, we assume that Atg3 functions via deforming the membranes,21, 43 which leads to lipid arrangement and PE enrichment.
Atg3 function is also dependent on PI(3)P. PI(3)P is functionally relevant to Atg8 lipidation via interacting with Atg3‐NH. 20 Compared to PE, PI(3)P has a quite larger headgroup. Considering that PI(3)P is the key lipid component of the autophagosome, we suppose that PI(3)P could also be enriched to the Atg3 binding site, which could compensate for the unfavorable free energy state caused by abundant PE (Figure 5c). Unfortunately, it is hard to estimate how much of the PI(3)P will be gathered at the ATG8 lipidation site. In addition, it was reported that Atg3 functions as a curvature sensor to mediate Atg8 lipidation. Consistent with this finding, our observation that Atg8 lipidation prefers to occur at small‐sized membrane structures, suggesting that the curvature sensing ability of Atg3 is necessary for Atg8 lipidation.
Our results that using DGS‐NTA to mimic PE is a compromising solution since the commercial fluorescent lipids usually contain a fluorophore‐conjugated directly to the headgroup of the lipids, which could inevitably enlarge the effective headgroup. Meanwhile, we could not find a proper fluorescent analogue for PI(3)P at present. Our future works will focus on molecular dynamic simulation of the PE and PI(3)P actions in the lipid bilayer and synthesizing fluorescent analogues that are more consistent with the physiological level. We believe that the subsequent efforts will provide more details for the ATG8 lipidation process.
4. MATERIALS AND METHODS
4.1. Cloning and mutagenesis
All the sequences used in this study are derived from Saccharomyces cerevisiae genome except for SN2 peptide, which is derived from human SNAP‐25 sequence (residues 140–206). For recombinant proteins expressed in Escherichia coli, Atg8ΔR (the last residues Arg116 was removed) and Atg7 were cloned into pGEX‐6p‐1 vector (GE Healthcare); SN2 peptide, Atg3, Δ26Atg3 (removal of first 26 residues), and PX‐Δ26Atg3 (PX domain was from Vam7, residues 1–125) were cloned into pGEX‐6p‐1 (GE Healthcare) and pET28a vector (Novagen), respectively. Atg7–Atg10 in pET11 vector and Atg12–Atg5 in pACYC184 vector were kindly gifts from Yoshinori Ohsumi. 17 For in vivo imaging and Atg8 processing assay, Atg8 was cloned into pRS415 II vector (from add gene) with its native promoter and terminator and an EGFP fluorescent protein at the N‐terminus. Atg3 and its mutants were cloned into pRS416 II vector (from addgene) with Atg3 native promoter and CYC1 (cytochrome c) terminator. Point mutations were achieved by QuickChange Site‐directed Mutagenesis Kit (Novagen).
4.2. Yeast strains
| Description | Genotype | Source |
|---|---|---|
| WT‐BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Thermo Fisher |
| atg3Δ | BY4741 atg3Δ::Kan | Thermo Fisher |
| atg3Δ EGFP‐ATG8 pRS416 II vector | BY4741 atg3Δ::Kan EGFP‐ATG8::LEU2 Ø::URA3 | This study |
| atg3Δ EGFP‐ATG8 ATG3 | BY4741 atg3Δ::Kan EGFP‐ATG8::LEU2 ATG3::URA3 | This study |
| atg3Δ EGFP‐ATG8 Δ26ATG3 | BY4741 atg3Δ::Kan EGFP‐ATG8::LEU2 Δ26ATG3::URA3 | This study |
| atg3Δ EGFP‐ATG8 PX‐Δ26ATG3 | BY4741 atg3Δ::Kan EGFP‐ATG8::LEU2 PX‐Δ26ATG3::URA3 | This study |
4.3. Recombinant protein purification
All of the recombinant proteins were expressed in E. coli BL21 DE3 cells. The cells were cultured to O.D.600 nm = 0.6 and induced with 0.2 mM isopropyl‐β‐d‐thiogalactoside (IPTG) at 20°C for 16 hr. Cells were collected by centrifugation at 4,200 rpm using a JM4.2 rotor (Beckman Coulter) at 4°C. Then the cells were resuspended by TBS300 (50 mM Tris‐Cl pH 8.0, 300 mM NaCl) supplied with 0.2% (vol/vol) Nonidet P‐40 (NP‐40), 1 mM phenylmethanesulfonyl fluoride (PMSF), and 0.5 mM tris(2‐carboxyethyl)phosphine (TCEP) and broken by AH‐1500 high‐pressure nano homogenizer (ATS) at 1,000 bar for 3 cycles. The broken cells were centrifuged at 16,000 rpm using a JA25.50 rotor (Beckman Coulter) at 4°C. Supernatants were then collected. For GST‐tagged proteins, the supernatants were mixed with glutathione Sepharose 4B resins (GE Healthcare, 1 ml bed volume used for 1 L cultures). The mixtures were gently rotated at 4°C for 2 hr followed by washing with TBS300 supplied with 0.2% (vol/vol) NP‐40 and 0.5 mM TCEP. The GST fusion tag was removed (if appropriate) by adding homemade recombinant HRV3C protease in HBS7.6 (25 mM HEPES‐K+ pH 7.6, 150 mM KCl) supplied with 0.2 mM TCEP. For hexa‐histidine tagged proteins, the supernatants were mixed with Ni‐NTA agarose resins (QIAGEN, 1 ml bed volume used for 1 L cultures). The mixtures were gently rotated at 4°C for 2 hr followed by washing with TBS300 supplied with 0.2% (vol/vol) NP‐40, 30 mM imidazole and 0.5 mM TCEP. The proteins were finally eluted by TBS150 (20 mM Tris‐Cl pH 8.0, 150 mM NaCl) supplied with 300 mM imidazole and 0.2 mM TCEP. After elution, additional 0.2 mM EDTA was added to prevent potential degradation. The affinity‐purified proteins were further purified by Superdex 200 pg 10/300 size‐exclusion chromatography (GE Healthcare). The concentrations of purified proteins were determined by the absorption at 280 nm and further analyzed by SDS‐PAGE.
4.4. Yeast cell culture
Saccharomyces cerevisiae strains stocked at −80°C in 30% (vol/vol) glycerol were first streaked on YPDA or SD agar plates. After culturing at 30°C for 36 hr, single colonies were picked and cultured in YPDA or SD medium at 30°C, 200 rpm to reach O.D.600 nm = 0.6. The cells were then washed by deionized water twice and transferred into SD‐N medium. Subsequent culturing was carried out at 30°C, 200 rpm for the desired time.
4.5. Imaging
Imaging was carried out with Nikon C2 laser scanning confocal microscopy (LSCM) equipped with a 20 mW Sapphire 488 nm solid‐state laser (Coherent) and a 40 mW Cubic 640 nm solid‐state laser (Coherent). The yeast cells were diluted with corresponding culture medium and dropped about 200 ul into Leb‐Tek 8‐well chambered coverglass (Thermo Scientific). For GUVs, Leb‐Tek 8‐well chambered coverglass was used as well. The observation was achieved using 60× (NA 1.40) or 100× (NA 1.49) oil‐immersed objective with a scanning rate of 1.9 μs per pixels. Generally, each figure was captured with a resolution of 1024*1024 pixels.
4.6. EGFP‐Atg8 processing assay
EGFP‐Atg8 processing assay was modified from a previous publication. 44 In brief, cultured yeast cells were collected by centrifuging at 1,000g at 4°C. The cell pellets were resuspended by TS buffer (100 mM Tris‐Cl pH 8.0, 200 mM sorbitol, 5 mM MgCl2, 1 mM CaCl2, and 2 mM DTT) supplied with 0.4% (vol/vol) Triton X‐100 and 10% (wt/vol) 0.5 mm‐diameter glass beads and broken by vortexing on ice. The mixtures were then centrifuged at 500g at 4°C to remove cell debris and glass beads. After centrifugation, 10% (wt/vol) trichloroacetic acid (TCA) was immediately added into the supernatants to extract total proteins. The samples were mixed gently and incubated on ice for 20 min followed by centrifuging at 20,000g at 4°C to collect the pellets. The pellets were washed by ice‐cold acetone twice and added 2× Laemmli buffer then boiling at 95°C for 5 min. Samples were injected into 12% (wt/vol) polyacrylamide SDS‐PAGE followed by immunoblotting using rabbit anti‐GFP polyclonal antibody (Proteintech).
4.7. Lipids
1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanolamine (POPE), 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (sodium salt) (DOPS), 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐(1′‐myo‐inositol‐3′‐phosphate) [PI(3)P], 1,2‐dioleoyl‐sn‐glycero‐3‐[(N‐(5‐amino‐1‐carboxypentyl)iminodiacetic acid) succinyl] (nickel salt) (DGS‐NTA) and cholesterol (ovine wool) were all purchased from Avanti Polar Lipids. 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindodicarbocyanine perchlorate (DiD) were obtained from molecular probes.
4.8. LUV preparation
Large unilamellar vesicles (LUVs) for in vitro Atg8 lipidation, liposome floatation assay, and DPH anisotropy measurement comprise POPC, POPE, DOPS, PI(3)P with the molar ratio of 65:20:10:5. LUVs for supported lipid bilayer formation comprise POPC, POPE, DOPS, PI(3)P, DGS‐NTA with molar ratio of 63:20:10:5:2. Cholesterol was supplied as indicated in the figure (replace corresponding POPC ratio). Lipids were mixed with the proper ratio in chloroform and dried under nitrogen flow followed by vacuuming for 2 hr. Lipid films were dissolved by freezing and thawing in HBS7.6 buffer with liquid nitrogen. Dissolved lipids were then extruded through a 200 nm polycarbonate film (Whatman) using a mini extruder (Avanti Polar Lipids). Prepared liposomes were stored at room temperature before using them.
4.9. GUV experiments
POPC, POPE, DOPS, PI(3)P, and DiD were mixed with a molar ratio of 64:20:10:5:1 in chloroform and spread onto 60 mm*24 mm indium tin oxide (ITO)‐coated glass slide. Excessive chloroform was removed by vacuuming for 2 hr. GUVs were prepared by electroformation with Vesicle Prep Pro (Nanion) at 1 V (sine wave) and 10 Hz in 300 mM sorbitol. For Atg8 lipidation, 2 μM Atg7, 2 μM Atg3, 5 μM Atg8ΔR, and 100 μM (total lipids) GUVs were mixed and incubated at 30°C.
4.10. In vitro Atg8 lipidation
Method for Atg8 in vitro lipidation was modified from previous publication. 45 The reaction system contains 2 μM GST‐Atg7, 2 μM Atg3 or its variants, 5 μM Atg8ΔR, 1 μM Atg5–Atg12 complex, 100 μM (total lipids) LUVs (POPC:POPE:DOPS:PI(3)P = 65:20:10:5), 1 mM ATP, 1 mM dithiothreitol (DTT), and 1 mM MgCl2. The mixtures were incubated at 30°C for 0, 5, 15, and 30 min. Reactions were stopped by adding 2× Laemmli buffer then boiling at 95°C for 5 min. Samples were injected into 12% (wt/vol) polyacrylamide urea SDS‐PAGE, run in Tris‐Tricine electrophoresis buffer followed by Coomassie brilliant blue (CBB) staining.
4.11. Liposome floatation
A total of 200 μM (lipids) LUVs were mixed with 5 μM purified Atg3 protein or its mutants and incubated for 1 hr at room temperature (25°C). The samples were then mixed with 80% (wt/vol) Histodenz (Sigma) dissolved in HBS7.6 to make 40% (wt/vol) Histodenz samples. Additional 30% (wt/vol) and 0% Histodenz were sequentially added to make discontinuous Histodenz density gradient. Samples were centrifuged at 200,000g for 1 hr using SW55Ti rotor (Beckman Coulter) at 20°C on L‐80XP ultracentrifuge (Beckman Coulter). After centrifugation, samples from the top 30 μL were taken and analyzed by SDS‐PAGE followed by immunoblotting using mouse anti‐6*His‐tag polyclonal antibody or rabbit anti‐GST polyclonal antibody (Proteintech).
4.12. Anisotropy measurements
1,6‐diphenyl‐1,3,5‐hexatriene (DPH, obtained from Sigma) dissolved in ethanol was first incubated with 1 mM (total lipids) LUVs at a final concentration of 1 μM at room temperature (25°C) overnight. Potential excess DPH molecules were removed by extensive dialysis. Fluorescence anisotropy measurements were carried out using PTI QM40 fluorescence spectroscopy equipped with motor‐driven polarizers and water circulator. Fluorescence anisotropy is given by:
where I is the fluorescent intensity, V and H are the polarizer angles of 90° (vertical) and 0° (horizontal), G is the grating factor:
The excitation and emission wavelengths of DPH were set to 360 and 450 nm, respectively.40, 46 The temperature‐anisotropy traces were collected from 4 to 45°C with the increasing step of 5°C per minute. Atg3 were added to 5 in 100 μM (total lipids) DPH‐LUVs.
For analyzing the binding between Alexa488‐labeled SN2 peptide and Atg3, the excitation and emission wavelengths were set to 485 and 520 nm, respectively.
4.13. Z‐scan FCS
Supported lipid bilayer (SLB) was prepared by LUV rupture as described previously. 47 Glass coverslips were treated by piranha solution (sulfuric acid: hydrogen peroxide = 3:1) and rinsed by deionized water extensively. Cleaned coverslips were dried under nitrogen flow and stored at room temperature (25°C). For SLB formation, 100 μM (total lipids) LUVs were added to coverslip supplied with an additional 2 and 1 mM CaCl2. After incubation at 37°C for 10 min, excess LUVs were washed out with HBS7.6 buffer supplied with 2 and 1 mM CaCl2. Hundred nanometer Alexa488‐labeled SN2 were then added to the SLB for 20 min' incubation at room temperature (25°C). Finally, 5 μM Atg3 were added to the SLB before FCS measurement.
Z‐scan FCS analysis was first described in previous publication. 39 Our setup contains Nikon Ti2 inverted microscopy equipped with 60× oil‐immerse objective (NA 1.40), LDH‐D‐C‐485485 nm picosecond laser diode (PicoQuant), PDL 800‐D pulsed diode laser driver (PicoQuant), hybrid photomultiplier (PMT) detector (PicoQuant), and a TimeHarp 260 TCSPC module (PicoQuant). The laser was set to 5 mW and 40 kHz. Raw data for each z‐layers were collected for 30 s. Raw data were fitted to autocorrelation function:
where T is the triplet fraction of molecules, τ trip is the lifetime of the triplet state, τ diff is the dwell time of diffusing molecules in the focal volume, κ is the shape factor of the focal volume. Number of molecules (N) in the focal volume is given by:
Autocorrelation function fitting was performed by using SymPhoTime (PicoQuant) and parabolic fitting of the z‐scan data were achieved by using OriginPro (version 2016 sr0, OriginLab).
4.14. Graphing and statistics
Prism 8.0.2 and OriginPro 2016 were used for graphing and statistical analysis. Methods used for hypothesis testing were indicated in the figure legends.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Shen Wang: Conceptualization; data curation; investigation; methodology; validation; visualization; writing‐original draft; writing‐review and editing. Yun Li: Conceptualization; data curation; investigation; methodology; visualization; writing‐original draft. Cong Ma: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; supervision; validation; visualization; writing‐review and editing.
Supporting information
Appendix S1. Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Yu Xue for sharing yeast strains and Dr. Yoshinori Ohsumi for providing constructs of pET11‐Atg7‐Atg10 and pACYC184‐Atg12‐Atg5. This work was supported by grants from the National Natural Science Foundation of China (31670846), the National Key Basic Research Program of China (2015CB910800), and Hubei Provincial Natural Science Foundation of China (2016CFA043).
Wang S, Li Y, Ma C. Atg3 promotes Atg8 lipidation via altering lipid diffusion and rearrangement. Protein Science. 2020;29:1511–1523. 10.1002/pro.3866
Funding information Hubei Provincial Natural Science Foundation of China, Grant/Award Number: 2016CFA043; National Key Basic Research Program of China, Grant/Award Number: 2015CB910800; National Natural Science Foundation of China, Grant/Award Number: 31670846
REFERENCES
- 1. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- 3. Kardideh B, Samimi Z, Norooznezhad F, Kiani S, Mansouri K. Autophagy, cancer and angiogenesis: Where is the link? Cell Biosci. 2019;9:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol. 2016;428:1681–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yin Z, Pascual C, Klionsky DJ. Autophagy: Machinery and regulation. Microbial Cell. 2016;3:588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Ann Rev Cell Dev Biol. 2011;27:107–132. [DOI] [PubMed] [Google Scholar]
- 9. Yao Z, Delorme‐Axford E, Backues SK, Klionsky DJ. Atg41/Icy2 regulates autophagosome formation. Autophagy. 2015;11:2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki K, Akioka M, Kondo‐Kakuta C, Yamamoto H, Ohsumi Y. Fine mapping of autophagy‐related proteins during autophagosome formation in Saccharomyces cerevisiae . J Cell Sci. 2013;126:2534–2544. [DOI] [PubMed] [Google Scholar]
- 11. Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre‐autophagosomal structure organization. Genes Cells. 2007;12:209–218. [DOI] [PubMed] [Google Scholar]
- 12. Nakatogawa H. Two ubiquitin‐like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013;55:39–50. [DOI] [PubMed] [Google Scholar]
- 13. Kim J, Huang WP, Klionsky DJ. Membrane recruitment of Aut7p in the autophagy and cytoplasm to vacuole targeting pathways requires Aut1p, Aut2p, and the autophagy conjugation complex. J Cell Biol. 2001;152:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kirisako T, Ichimura Y, Okada H, et al. The reversible modification regulates the membrane‐binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin‐like system mediates protein lipidation. Nature. 2000;408:488–492. [DOI] [PubMed] [Google Scholar]
- 16. Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin‐like conjugation systems in macroautophagy. 'Protein modifications: Beyond the usual suspects' review series. EMBO Rep. 2008;9:859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanada T, Noda NN, Satomi Y, et al. The Atg12‐Atg5 conjugate has a novel E3‐like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. [DOI] [PubMed] [Google Scholar]
- 18. Sakoh‐Nakatogawa M, Matoba K, Asai E, et al. Atg12‐Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol. 2013;20:433–439. [DOI] [PubMed] [Google Scholar]
- 19. Zheng Y, Qiu Y, Grace CRR, Liu X, Klionsky DJ, Schulman BA. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat Commun. 2019;10:3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanada T, Satomi Y, Takao T, Ohsumi Y. The amino‐terminal region of Atg3 is essential for association with phosphatidylethanolamine in Atg8 lipidation. FEBS Lett. 2009;583:1078–1083. [DOI] [PubMed] [Google Scholar]
- 21. Hervas JH, Landajuela A, Anton Z, Shnyrova AV, Goni FM, Alonso A. Human ATG3 binding to lipid bilayers: Role of lipid geometry, and electric charge. Sci Rep. 2017;7:15614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li YT, Yi C, Chen CC, et al. A semisynthetic Atg3 reveals that acetylation promotes Atg3 membrane binding and Atg8 lipidation. Nat Commun. 2017;8:14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nath S, Dancourt J, Shteyn V, et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane‐curvature‐sensing domain in Atg3. Nat Cell Biol. 2014;16:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nair U, Jotwani A, Geng J, et al. SNARE proteins are required for macroautophagy. Cell. 2011;146:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin‐like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. [DOI] [PubMed] [Google Scholar]
- 26. Hain AU, Weltzer RR, Hammond H, et al. Structural characterization and inhibition of the plasmodium Atg8‐Atg3 interaction. J Struct Biol. 2012;180:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Metlagel Z, Otomo C, Ohashi K, Takaesu G, Otomo T. Structural insights into E2‐E3 interaction for LC3 lipidation. Autophagy. 2014;10:522–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanida I, Sou YS, Minematsu‐Ikeguchi N, Ueno T, Kominami E. Atg8L/Apg8L is the fourth mammalian modifier of mammalian Atg8 conjugation mediated by human Atg4B, Atg7 and Atg3. FEBS J. 2006;273:2553–2562. [DOI] [PubMed] [Google Scholar]
- 29. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Intl J Biochem Cell Biol. 2004;36:2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamaguchi M, Noda NN, Nakatogawa H, Kumeta H, Ohsumi Y, Inagaki F. Autophagy‐related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3‐Atg8 interaction and is crucial for the cytoplasm‐to‐vacuole targeting pathway. J Biol Chem. 2010;285:29599–29607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noda T, Matsunaga K, Taguchi‐Atarashi N, Yoshimori T. Regulation of membrane biogenesis in autophagy via PI3P dynamics. Sem Cell Dev Biol. 2010;21:671–676. [DOI] [PubMed] [Google Scholar]
- 32. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. [DOI] [PubMed] [Google Scholar]
- 33. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy‐defective mutants of Saccharomyces cerevisiae . FEBS Lett. 1993;333:169–174. [DOI] [PubMed] [Google Scholar]
- 34. Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets the Vam7 t‐SNARE to vacuole membranes. Nat Cell Biol. 2001;3:613–618. [DOI] [PubMed] [Google Scholar]
- 35. Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279:29889–29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tanida I, Ueno T, Kominami E. In vitro assays of lipidation of mammalian Atg8 homologs. Curr Protoc Cell Biol. 2014;64(11.20):11–13. [DOI] [PubMed] [Google Scholar]
- 37. Drin G, Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010;584:1840–1847. [DOI] [PubMed] [Google Scholar]
- 38. Gimenez‐Andres M, Copic A, Antonny B. The many faces of amphipathic helices. Biomolecules. 2018;8:E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Humpolickova J, Gielen E, Benda A, et al. Probing diffusion laws within cellular membranes by Z‐scan fluorescence correlation spectroscopy. Biophys J. 2006;91:L23–L25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuhry JG, Duportail G, Bronner C, Laustriat G. Plasma membrane fluidity measurements on whole living cells by fluorescence anisotropy of trimethylammoniumdiphenylhexatriene. Biochim Biophys Acta. 1985;845:60–67. [DOI] [PubMed] [Google Scholar]
- 41. Ichimura Y, Imamura Y, Emoto K, Umeda M, Noda T, Ohsumi Y. In vivo and in vitro reconstitution of Atg8 conjugation essential for autophagy. J Biol Chem. 2004;279:40584–40592. [DOI] [PubMed] [Google Scholar]
- 42. Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE‐16. J Biol Chem. 2006;281:3017–3024. [DOI] [PubMed] [Google Scholar]
- 43. Knorr RL, Nakatogawa H, Ohsumi Y, Lipowsky R, Baumgart T, Dimova R. Membrane morphology is actively transformed by covalent binding of the protein Atg8 to PE‐lipids. PloS One. 2014;9:e115357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheong H, Klionsky DJ. Biochemical methods to monitor autophagy‐related processes in yeast. Methods Enzymol. 2008;451:1–26. [DOI] [PubMed] [Google Scholar]
- 45. Fracchiolla D, Zens B, Martens S. In vitro reconstitution of Atg8 conjugation and deconjugation. Methods Enzymol. 2017;587:377–390. [DOI] [PubMed] [Google Scholar]
- 46. van Blitterswijk WJ, van der Meer BW, Hilkmann H. Quantitative contributions of cholesterol and the individual classes of phospholipids and their degree of fatty acyl (un)saturation to membrane fluidity measured by fluorescence polarization. Biochemistry. 1987;26:1746–1756. [DOI] [PubMed] [Google Scholar]
- 47. Richter RP, Berat R, Brisson AR. Formation of solid‐supported lipid bilayers: An integrated view. Langmuir. 2006;22:3497–3505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary Material