

Abstract
Insulin receptor (IR) and the epidermal growth factor receptor (EGFR) were the first receptor tyrosine kinases (RTKs) to be studied in detail. Both are important clinical targets—in diabetes and cancer, respectively. They have unique extracellular domain compositions among RTKs, but share a common module with two ligand‐binding leucine‐rich‐repeat (LRR)‐like domains connected by a flexible cysteine‐rich (CR) domain (L1‐CR‐L2 in IR/domain, I‐II‐III in EGFR). This module is linked to the transmembrane region by three fibronectin type III domains in IR, and by a second CR in EGFR. Despite sharing this conserved ligand‐binding module, IR and EGFR family members are considered mechanistically distinct—in part because IR is a disulfide‐linked (αβ)2 dimer regardless of ligand binding, whereas EGFR is a monomer that undergoes ligand‐induced dimerization. Recent cryo‐electron microscopy (cryo‐EM) structures suggest a way of unifying IR and EGFR activation mechanisms and origins of negative cooperativity. In EGFR, ligand engages both LRRs in the ligand‐binding module, “closing” this module to break intramolecular autoinhibitory interactions and expose new dimerization sites for receptor activation. How insulin binds the activated IR was less clear until now. Insulin was known to associate with one LRR (L1), but recent cryo‐EM structures suggest that it also engages the second LRR (albeit indirectly) to “close” the L1‐CR‐L2 module, paralleling EGFR. This transition simultaneously breaks autoinhibitory interactions and creates new receptor‐receptor contacts—remodeling the IR dimer (rather than inducing dimerization per se) to activate it. Here, we develop this view in detail, drawing mechanistic links between IR and EGFR.
Keywords: allostery, epidermal growth factor, insulin, negative cooperativity, receptor, receptor tyrosine kinase, structure
1. INTRODUCTION
Receptor tyrosine kinases (RTKs) are important regulators of key cellular processes, from metabolism to proliferation to survival and differentiation. 1 Accordingly, they are linked to numerous diseases—from diabetes to cancer—and are valuable therapeutic targets, with wide use of RTK agonists (such as insulin) and inhibitors (such as erlotinib) in the clinic, depending on the disease context. There are 58 human RTKs, which fall into 20 families based largely on domain composition of their ligand‐binding extracellular region (ECR). 1 In each case, a single transmembrane (TM) domain links the ECR to the tyrosine kinase domain‐containing intracellular region.
The mechanistic challenge in TM signaling by RTKs (and other single‐TM receptors) is that of transmitting extracellular signals through a single TM α‐helix. 2 Ligand‐induced receptor dimerization emerged early on as an elegant explanation for how single‐TM receptors signal, 3 and has now been visualized structurally in several cases. 1 The activating ligand is frequently bivalent (as a monomer or dimer), and binds to two receptors—thus effectively mediating receptor dimerization. In such cases, the receptor's ECR is often composed primarily of immunoglobulin‐like domains, and ligand‐induced dimerization appears to be ligand‐mediated and relatively straightforward. Examples of this are seen with stem cell factor/Kit, 4 nerve growth factor/TrkA, 5 platelet‐derived growth factor (PDGF)/PDGF receptor, 6 vascular endothelial growth factor (VEGF)/VEGF receptors, 7 and fibroblast growth factor (FGF)/FGF receptors. 8
The receptors for insulin and epidermal growth factor (EGF) are quite different. Insulin and EGF (Figure 1a–c) were among the first RTK ligands to be studied, 9 and the insulin receptor (IR) and EGF receptor (EGFR) families have since been subjects of a great deal of structural study. 10 IR and EGFR have a unique domain composition among RTKs 1 (Figure 1d). At their amino termini, both have two ligand‐binding leucine‐rich repeat (LRR)‐like “solenoid” or β‐helix domains (Figure 1d,e) that are separated by a cysteine‐rich domain (Figure 1d,f). Nomenclature differs between the two receptors. In IR, the LRR domains are called domains L1 and L2, whereas they are called domains I and III in EGFR. The intervening cysteine‐rich domain is called “CR” in IR and domain II in EGFR. 11 Following this shared L1‐CR‐L2 module (domain I/II/III in EGFR), the two receptor families diverge (Figure 1d). EGFR has a second CR domain (domain IV), whereas IR has three fibronectin type III (FNIII) domains (Figure 1d,g)—a domain type that is also seen in several other RTKs. 1 Perhaps most importantly, IR family members are disulfide‐linked dimers (Figure 1d). The IR is also processed by post‐translational cleavage within its second FNIII domain to yield an α‐chain and a β‐chain (Figure 1d), which are held together by a disulfide bond. In addition, disulfide bonds link the two α‐chains into a dimer—connecting both their first FNIII domains and the α‐chain C‐terminal region (αCT). IR is thus an (αβ)2 dimer regardless of ligand binding, arguing that simple ligand‐induced dimerization cannot explain its activation. Recent work has shown that the Caenorhabditis elegans EGFR is also dimeric in the absence of ligand 12 —although this is a noncovalent dimer. In the collective light of structural studies of RTKs, these observations argue that IR and EGFR are not prototypical RTKs as has been argued.13, 14 Indeed, IR family members are not known to change their oligomeric state upon ligand binding. Moreover, although human EGFR was among the first RTKs for which ligand‐induced dimerization was observed, 15 it does not share the ligand‐mediated dimerization mechanism 1 of Kit, PDGFR, TrkA, and others. Instead, ligand binding to EGFR induces conformational changes that promote “receptor‐mediated” dimerization. 16
FIGURE 1.
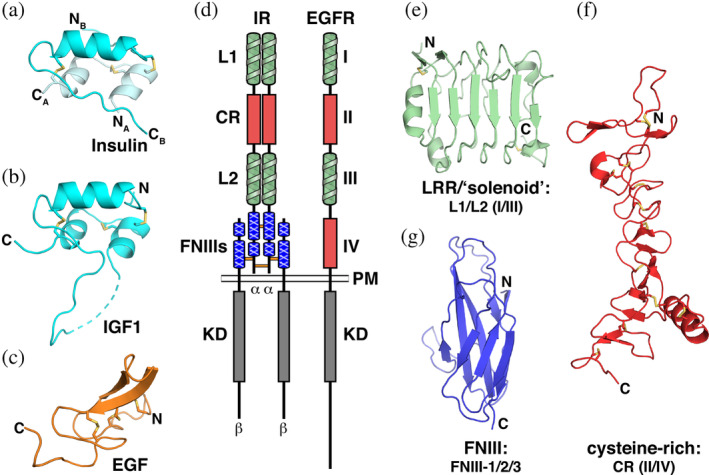
Structural components of the IR and EGFR families and their ligands. (a–c) Cartoon representations of the ligands. (a) Free insulin in the “T” state (pdb id 4ins), with the A and B chains colored pale cyan and cyan, respectively. (b) The single‐chain of IGF1 (pdb id 1imx). (c) The structurally distinct EGF molecule (pdb id 1jl9, chain B), colored orange. (d) Arrangement of domains in the IR and EGFR families. LRR‐like “solenoid” domains (L1/L2 and domains I/III for IR and EGFR, respectively) are colored green, cysteine‐rich domains (CR and domains II/IV) are red, the IR‐specific FNIII domains are blue, and the kinase domains (KD) are gray. The plasma membrane (PM) is shown, and locations of interchain disulfides in IR are marked with horizontal lines between chains. (e–g) Cartoon representations of structural domains found in IR and EGFR. (e) The IR L1 domain (pdb 2hr7, chain B, aa 4–158), an LRR‐like “solenoid” domain; (f) The IR CR domain (pdb 2hr7, chain A, aa 159–311), a cysteine‐rich (CR) domain; and (g) IGF1‐R FNIII‐1 (pdb id 5u8q, aa 460–578). Cartoons are colored the same as each domain in (d). Disulfides are shown as yellow sticks, and positions of N‐ and C‐termini are marked. The α and β chains of IR are also labeled. CR, cysteine‐rich; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FNIII, fibronectin type III; IR, insulin receptor; LRR, leucine‐rich‐repeat‐like
The complexity of the ECRs of IR family members has made them (even) more of a challenge for structural studies than EGFR family members. Exciting recent advances in cryo‐electron microscopy (cryo‐EM), however, have led to a series of new structures that begin to suggest a consensus for IR family signaling mechanisms.17, 18, 19, 20, 21 These structures also provide a valuable context for identifying similarities and differences between activation mechanisms for IR and EGFR family members. Structural descriptions of the two families to date have largely been presented separately—with a few exceptions.10, 22, 23 Our goal in this article is to illustrate intriguing similarities that have emerged, as we view recent developments in understanding IR family members from the perspective of the EGFR field.
2. THREE “STATES” OF IR AND EGFR FAMILY ECR STRUCTURES
A rich array of crystal and/or cryo‐EM structures now exists for ECRs of IR and EGFR family members. These structures have been determined either in the presence of cognate ligand or in its absence (frequently with antibodies bound). Across each family, the structures of most individual domains are not altered significantly upon ligand binding or dimerization (with the exception of the CR domain/domain II), but substantial domain/domain rearrangements occur in both families that appear to be crucial for receptor function.
Before discussing details of these domain rearrangements, it is helpful to ascribe the available structures to functional “states” if possible, something that we have previously been able to do for EGFR.14, 24, 25 By systematically overlaying currently available IR family ECR structures using the L2 domain as a reference, we find that the newly available cryo‐EM structures now allow three “states” to be defined for complete IR family ECR protomers. The first to be reported 26 was the “unliganded” state (Figure 2a), observed in crystal structures of (mostly) antibody‐stabilized IR family ECRs.26, 27, 28 The second is the new “ligand‐bound” state (Figure 2b), seen only recently—in cryo‐EM structures of IR and the insulin‐like growth factor 1 receptor (IGF1‐R).17, 18, 19, 20, 21 There are two main differences between the unliganded and liganded states of the protomer (discussed in detail later). The most obvious in comparing Figure 2a,b is a rotation about the axis marked with a blue circle in the linker between the L2 domain (used to overlay and orient the protomers) and FNIII‐1. This rotation substantially reorients the FNIII‐1/2/3 module, thought to be a key transition in receptor activation, 29 so that the angle between L2 and the FNIII‐1/2/3 module opens substantially upon ligand binding. The second notable change is a rotation of the L1/CR domain pair with respect to L2 about the axis marked by an orange circle in the CR/L2 linker in Figure 2a,b; this is induced directly by ligand binding, as discussed below. The unliganded state of IR shows significant heterogeneity between structures (Figure 2a) in L1 domain position—reflecting flexibility in the IR ECR that has been discussed before, 30 and which is checked by ligand binding. Indeed, recent efforts to obtain cryo‐EM structures of an unliganded insulin receptor ECR have failed, perhaps because of this heterogeneity.17, 21 The third state that we can define (Figure 2c) is also unliganded—but is found only in asymmetric dimers with a liganded protomer.18, 19 This state has only been seen in recent cryo‐EM structures, and under conditions where the long‐known negative cooperativity 31 of ligand binding to IR and IGF1‐R is preserved or restored.18, 19 We call this the “restrained unliganded” state. In this conformation, the L2 domain of the protomer retains the same relationship with the FNIII‐1 domain as in the unliganded state (Figure 2a), but L1 (which interacts with FNIII‐2′ of the opposing protomer) is brought closer to FNIII‐2 of its own chain, enabling direct intramolecular interactions that are absent in the unliganded state and apparently restrain the position of the L1 domain.
FIGURE 2.
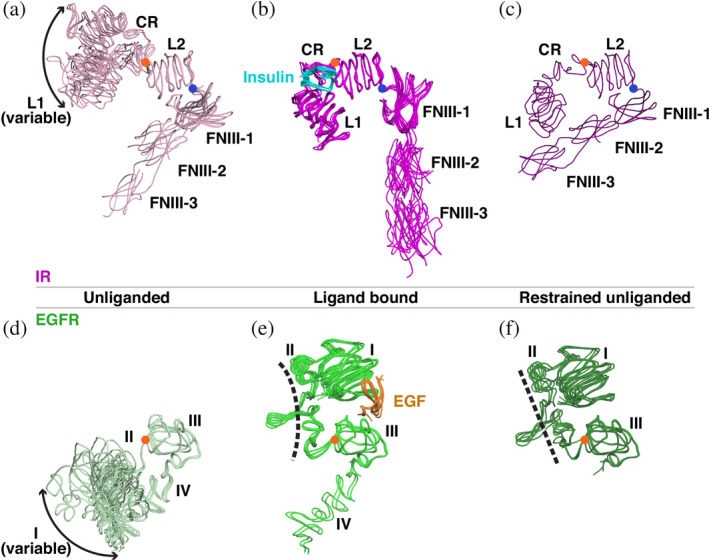
The three “states” of the IR and EGFR family ECR structures. (a–c) Structures of the IR ECR are grouped based on the orientations of the L1‐CR and FNIII‐1/2/3 modules relative to L2, which was used to align all structures. (a) The unliganded states (light pink) have an “open” L1‐CR‐L2 module and a “closed” L2‐FNIII‐1 relationship. The location of the L1‐CR module with respect to L2 is variable (denoted by the double‐headed arrow), reflecting flexibility in this region of the molecule. Orange circles mark the axes of rotation of L1‐CR with respect to L2, and blue circles mark the axis of rotation of L2 with respect to FNIII‐1—which define transitions between the different states. (b) When insulin (cyan) is bound, the L1‐CR‐L2 module is forced into a “closed” conformation, and the FNIII‐1/2/3 module becomes oriented away from L2 (magenta) in an “open” conformation. (c) The “restrained unliganded” state (purple), which is seen only in asymmetric dimers in which one protomer is liganded and the other not. The L1‐CR‐L2 module is “open” in this state, with L1 fixed by state‐specific interactions with FNIII‐2 in the same chain. This state is also seen in the asymmetric IGF1‐R structure. (d–f) The three states of EGFR ECR. (d) Unliganded structures (pale green) show “open” domain I–III conformations with a variable domain I location. (e) When EGF (orange) binds, domains I and III are drawn together into a “closed” conformation by interacting simultaneously with the same ligand. (f) The “restrained unliganded” conformation (dark green) seen in asymmetric dimers of EGFR. Note the straight domain II (dashed line) compared to the bent domain II in the ligand bound state shown in (e). CR, cysteine‐rich; ECR, extracellular region; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FNIII, fibronectin type III; IR, insulin receptor
Analysis of EGFR family member ECR structures similarly reveals three conformational states for the monomer—transitions between which satisfyingly explain ligand regulation of these receptors. 14 These states are shown in Figure 2d–f, again overlaid using the second LRR (domain III) as reference. In human EGFR, a single CR domain IV replaces the FNIII‐1/2/3 module seen in IR, and the spatial relationship between domain III (L2 equivalent) and domain IV is almost unchanged by ligand binding. As such, there is no equivalent of IR's L2/FNIII‐1 reorientation in going from the unliganded (Figure 2d) to the liganded (Figure 2e) protomer of EGFR family ECRs. On the other hand, the substantial reorientation of L1/CR with respect to L2 seen in IR (Figure 2a,b) is mirrored in EGFR (where these are domains I, II, and III). There is a substantial reorientation of the domain I/II pair about the axis marked with an orange circle between domains II and III in Figure 2d,e—analogous to the axis between CR and L2 in IR. As with L1 and L2 in IR, the two LRR domains of EGFR (I and III)—which both directly contact ligand (as discussed in more detail below)—are drawn together to “close” the domain I‐II‐III module. Also in common with the IR family, the first LRR domain of EGFR (domain I) is heterogeneous in its location in the unliganded monomer (suggesting substantial domain/domain flexibility), and this heterogeneity is lost upon ligand binding. Finally, a third “restrained unliganded” state can also be defined for EGFR (Figure 2f), again seen only in asymmetric dimers of the receptor's ECR in which the other protomer is liganded. Examples include a singly liganded asymmetric dimer of the Drosophila EGFR 32 and an asymmetric human EGFR dimer bound to a low affinity EGFR ligand. 33 Interestingly, the orphan ErbB2 receptor seems to represent a special case by forming this “restrained unliganded” state even as a monomer. 34
3. THE L1‐CR‐L2 MODULE
The conformational transitions seen in IR family ECR protomers (Figure 2a–c) clearly identify two modules—the L1‐CR‐L2 module (domain I–III in EGFR) and the FNIII‐1/2/3 module (which has no equivalent in EGFR). We begin with a discussion of the L1‐CR‐L2 (I‐II‐III) module, which seems to play a remarkably similar role in linking ligand binding to allosteric changes in both receptor families.
The L1‐CR‐L2 module of IGF1‐R was the first fragment of any of the receptors discussed here to be visualized structurally. 35 Despite little apparent contact between the three domains in this fragment, the spatial relationship between them is remarkably well conserved between ligand‐free IGF1‐R 35 and IR 36 and the unliganded ECRs from human EGFR, ErbB3, and ErbB4,37, 38, 39 even accounting for heterogeneity in L1/domain I position. Single examples are shown overlaid in Figure 3a to illustrate this similarity. Although there is flexibility in the L1/domain I position, the L1/CR (domain I/II) linkage is quite well conserved, and a loop connecting the end of CR (domain II) to L2 (domain III) seems to function as a flexible hinge.
FIGURE 3.
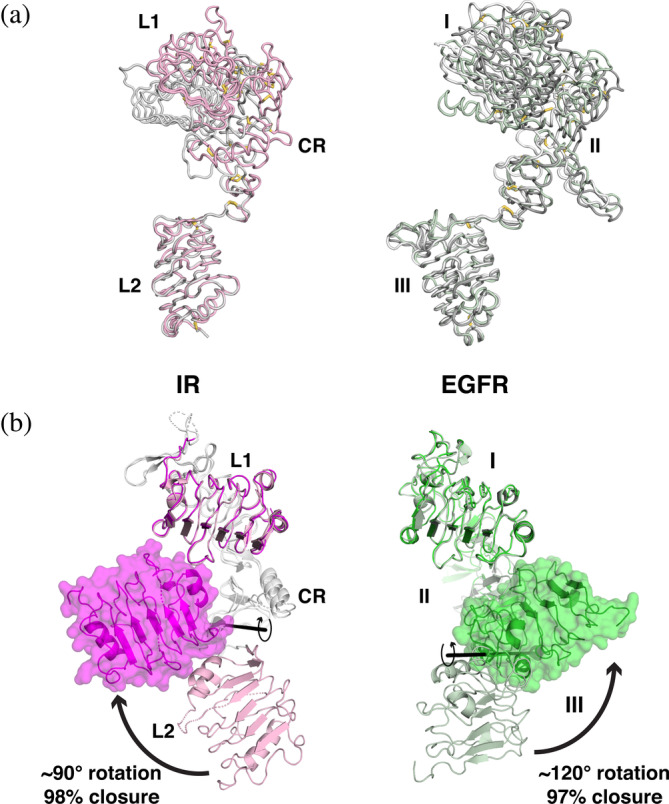
L1‐CR‐L2/I‐II‐III modules of IR and EGFR and their transitions. (a) Ribbon view depicting the similarity in L1‐CR‐L2/I‐II‐III module arrangements in crystal structures of unliganded IR and EGFR family members. Structures were aligned using the coordinates of L2 or domain III as reference (as in Figure 2a,d), and are depicted as backbone ribbons with disulfides in stick representation. The left‐hand panel shows representative examples of IR (light pink, pdb id 2hr7, chain A) and IGF1‐R (gray, pdb id 1igr). EGFR family members are shown on the right: EGFR in light green (pdb id 1yy9), ErbB3 in dark gray (pdb id 1m6b, chain A), and ErbB4 in gray (pdb id 5cus, chain B). (b) Changes in the relationship between L1 and L2 (domain I and domain III) upon ligand binding are shown. Left: A cartoon representation of the L1‐CR‐L2 module of IR—with the unliganded structure in light pink (pdb id 2hr7, chain B) and the ligand‐bound structure in magenta (pdb id 6pxw, chain A), with a transparent surface covering the L2 domain. Structures were aligned using the coordinates of the L1 domain. The CR domains are colored light gray. A clockwise ~90° rotation of the light pink L2 (unbound) about the axis indicated (black line, from DynDom3D webserver 40 ) brings L2 to the conformation seen in the ligand bound state (magenta), which has a “closed” L1‐CR‐L2 arrangement. Ligand (not shown for clarity) binds between L1 and L2—see Figure 2b—drawing them together. Right: A similar view of the domain I‐II‐III module of EGFR. The unliganded conformation is shown in light green (pdb id 1nql), and ligand bound conformation is darker green (pdb id 3njp, chain A). For EGFR, a counter‐clockwise rotation of ~120° about the indicated axis translates domain III from the unliganded (pale green) to the ligand bound (darker green) position to “close” the domain I‐II‐III module—driven by binding of EGF (not shown) to both domains I and III—see Figure 2e. CR, cysteine‐rich; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; IR, insulin receptor
3.1. Ligand binding induces “closure” of both I‐II‐III and L1‐CR‐L2 modules
Upon binding to EGFR family members, an activating ligand simultaneously engages both domains I and III in the I‐II‐III module (equivalent to IR's L1‐CR‐L2 module), and draws the two LRR domains close to one another by flexing the domain II/III hinge mentioned above (Figure 3b, right: note that ligand is not shown).11, 41, 42 This bivalent binding of a ligand to two sites within the same chain promotes a ~120° rotation of domain III about the hinge close to the end of domain II, and leads to a 97% “closure” of domains I and III as assessed by the DynDom program 40 (Figure 3b, right). Unexpectedly, the new cryo‐EM structures of ligand‐bound IR and IGF1‐R revealed an analogous change in the relationship between the L1 and L2 domains of these receptors upon ligand binding17, 18, 19, 20, 21 (Figure 3b, left), with a ~90° rotation of domain L2 about the same hinge at the end of CR. Although very similar in scale, leading to a 98% ligand‐induced closure of the IR L1‐CR‐L2 module as assessed by DynDom, this rotation occurs in the opposite direction in IR compared with EGFR (Figure 3b). Thus, the new IR/IGF1‐R structures reveal that ligand‐induced “closure” of the L1‐CR‐L2/I‐II‐III module—known to be key for regulation of EGFR by its ligands—is actually a feature common to both receptor families (although different in detail). Indeed, this step may be viewed as the most immediate consequence of ligand binding in both the EGFR and IR receptor families, relating them mechanistically in a clear manner for the first time. 17
3.2. Ligand binding to domain L1/domain I
Interestingly, the way in which the ligands dock onto the IR and EGFR LRRs are also very similar, with the differences in the closure direction shown in Figure 3b defined largely by the distinct locations of the two LRR‐binding sites on the bivalent insulin and EGF family ligands. Earlier structural studies of IR and IGF1‐R28, 43, 44, 45 firmly located the binding site for insulin and IGF1 on the L1 domain of their respective receptors. The mode of interaction here is unusual and interesting among RTKs (Figure 4a). Only the B chain of the two‐chain insulin molecule interacts directly with the L1 domain of the receptor, 43 and engages the central β‐sheet of the L1 domain. At the same time, insulin also interacts with a helix in the carboxy‐terminal segment of the α‐chain (αCT) of the adjacent protomer in the IR dimer (denoted αCT′ in Figure 4a because it comes from the other receptor protomer), and αCT′ mediates further interactions with the L1 domain (Figure 4a). The A chain of insulin (lighter cyan in Figure 4a) does not interact directly with the L1 domain—only contacting αCT′ and the insulin B chain. There is also a change in the insulin or IGF1 conformation upon receptor binding that has no equivalent in the EGFR family. In IR parlance, the L1 domain central β‐sheet and αCT′ together constitute “site 1,” or the primary binding site for insulin. The situation is very similar for IGF1 binding to the L1 domain of IGF1‐R. 28 One way of viewing the structural arrangement here is to consider that the bound ligand effectively comprises insulin (or IGF1) plus αCT′. EGFR and its relatives have no equivalent of αCT, and their ligands are structurally unrelated to insulin or IGF1 (Figure 1a–c). Like insulin + αCT, however, EGF docks onto the central β‐sheet of domain I (corresponding to L1)—binding to a location that closely matches the corresponding interface in IR or IGF1‐R (cf. Figure 4a,b). Just as structures of insulin or IGF‐1 bound only to L1 have been described,28, 43 so has EGF been observed bound only to domain I in an early crystal structure 38 determined at low pH. Moreover, the isolated domain I/II fragment of ErbB3 was reported to bind its ligand (neuregulin) with high affinity. 46
FIGURE 4.
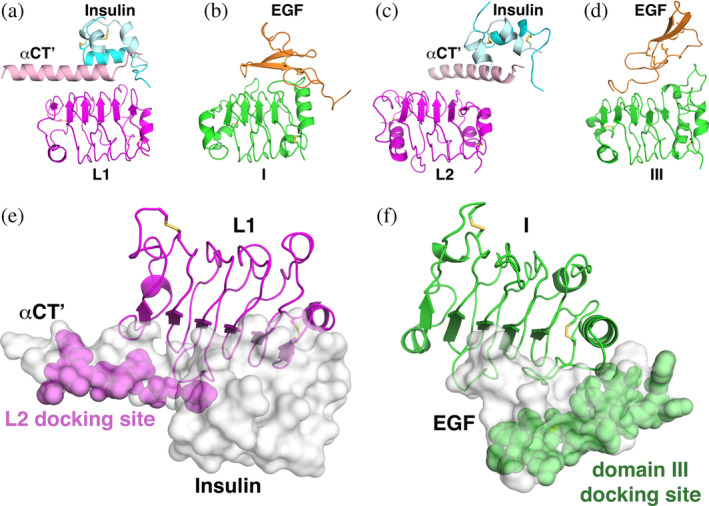
Similarities in the ligand docking sites across the IR and EGFR families. (a, b) The L1 domain of IR (a) and domain I of EGFR (b) were aligned and are shown in the same orientation (as magenta and green cartoons, respectively). Ligands bound to the main β‐sheet of the respective LRRs are shown, with insulin colored as in Figure 1a, the associated αCT′ colored light pink, and EGF colored orange. (c, d) Similar cartoons of the L2 domain of IR (c) and domain III of EGFR (d) are shown with bound ligands, with colors as in (a) and (b). Note that each ligand simultaneously engages two LRRs. (e) Surface representation of insulin plus αCT′ (gray) docked onto L1 (magenta cartoon). The location of the L2 docking site on the αCT′/insulin unit is highlighted with magenta spheres and labeled. The view is rotated approximately 90° about a horizontal axis relative to (a), and is the same orientation used in Figure 3b. (f) The domain III docking site on EGF (green spheres) is shown in a similar representation and orientation as in (e). Disulfides are shown in stick representation. IR structures are from pdb id 6pxw, and EGFR structures are from pdb id 3njp. EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; IR, insulin receptor; LRR, leucine‐rich‐repeat
3.3. Ligand binding to domain L2/domain III
One of the most intriguing findings from the recent cryo‐EM structures of insulin or IGF1 bound to their corresponding ECRs17, 18, 19, 20, 21 was the previously unappreciated involvement of the IR family L2 domain in ligand binding (Figure 4c). In EGFR family members, both LRR domains participate directly in ligand binding, and domain III seems to be the higher affinity ligand‐binding site.47, 48 Topologically equivalent regions of domains I and III from EGFR (Figure 4b,d) dock onto different parts of the same bivalent EGF‐family ligand molecule to drive the domain closure seen in Figure 3b for this receptor.11, 41, 42 Although neither insulin nor IGF1 contacts the L2 domain directly in the new ligand‐bound cryo‐EM structures, L2 is contacted directly by αCT′ (Figure 4c), which mediates significant interactions of the αCT′/ligand unit with the more N‐terminal part of the L2 domain's central β‐sheet (as well as the end of the domain). Thus, if one views αCT′ as a part of the ligand in the IR family as suggested above, it docks onto L2 in a manner that resembles EGF docking onto domain III. Alternatively, it could be argued that αCT′ extends the ligand‐binding surface of L2 in IR—onto which insulin docks. Either way, the argument can be made that the two LRR domains of IR are drawn together in Figures 2b and 3b by bivalent binding of ligand, in a similar manner as seen for EGFR, to “close” the L1‐CR‐L2 module. In both receptor families, ligand binding to the L1‐CR‐L2/domain I‐II‐III module substantially reorients the domains to “close” the module and to promote significant changes in receptor/receptor interactions that can drive signaling as outlined below.
3.4. Origin of the difference in L1‐CR‐L2 module domain closure direction
Considering the ligand/LRR interaction details outlined above, the origin of the difference in domain closure direction between IR and EGFR family members shown in Figure 3b can be explained. It simply reflects the large difference between the two small polypeptide ligands. Insulin and IGF1 are unrelated structurally to EGF (Figure 1a–c), and when docked onto the central β‐sheet of the L1 domain (with αCT′ in the insulin/IGF‐1 case), their respective L2/domain III binding sites face quite different directions (Figure 4e,f). The direction of closure of the two LRR domains is then defined by docking of the L2/domain III central β‐sheet (or adjacent surface) onto this second site—approaching from the left for IR in Figures 4e (magenta spheres) and Figure 3b, and from the right for EGFR in Figure 4f (green spheres) and Figure 3b.
4. L1‐CR‐L2 MODULE CLOSURE IS COUPLED TO ALTERED RECEPTOR DIMERIZATION IN BOTH EGFR AND IR FAMILY RECEPTORS
To drive signaling, the conformational transition in the L1‐CR‐L2 module upon ligand binding must trigger other changes in intra‐ and intermolecular domain/domain interactions in IR and EGFR—details of which differ substantially between the two families. The differences largely reflect the distinct complements of additional domains in the two receptor families. In the human EGFR family, the only additional domain is the second CR domain (domain IV), whereas in IR family receptors the L1‐CR‐L2 module is followed by three FNIII domains. To begin considering the respective activation mechanisms in EGFR and IR, it is instructive first to consider key intra‐ and intermolecular domain/domain interactions that occur in the unliganded and liganded receptors, and how these are affected by ligand binding.
4.1. EGFR family
An intramolecular domain II/IV interaction, considered autoinhibitory, is seen in the unliganded monomers of EGFR (Figure 5a, open orange circle), ErbB3, and ErbB4. This interaction is broken by the ligand‐induced closure of domains I and III described above. The domain I/II and domain III/IV relationships in the EGFR ECR remain relatively unchanged—so one half of the molecule can be said to rotate with respect to the other, effectively extending the ECR from the “tethered” state11, 37, 38 (Figures 2d and 5a) to an extended state (Figure 5b). Two dimerization interfaces—in domains II and IV—become exposed as a result of the domain I/III closure, and drive ligand‐induced self‐association of the receptor49, 50 as shown in Figure 5b. Importantly, the domain I/III closure also remodels the (intervening) domain II dimerization interface to promote intermolecular interactions mediated by this domain. 51 Thus, ligand binding both reveals and remodels dimerization interfaces that are buried in intramolecular interactions in the absence of ligand.
FIGURE 5.
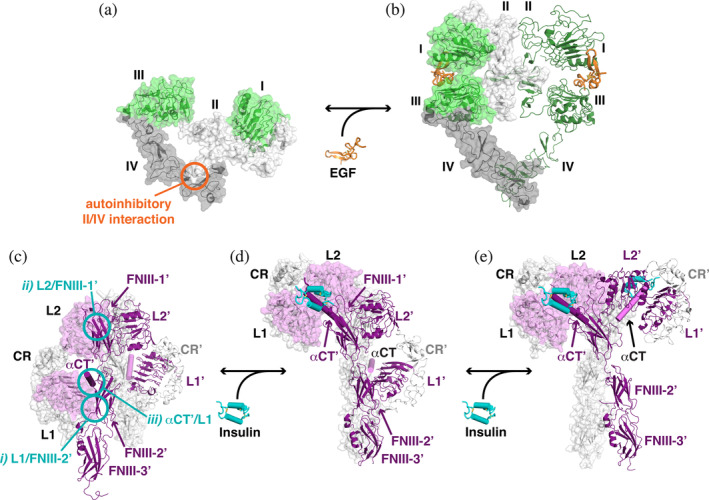
Coupling of L1‐CR‐L2 module closure to altered EGFR and IR dimerization. (a) An unliganded EGFR monomer (pdb id 1yy9) is shown as a cartoon with transparent surface. Domains I and III are green, domain II is light gray, and domain IV is darker gray. The autoinhibitory intramolecular interaction between domains II and IV is marked with an orange circle. (b) The EGF‐bound EGFR ECR dimer is shown with a transparent surface, adopting an extended conformation (pdb id 3njp). (c, d) To appreciate the changes that occur in IR upon ligand binding, structures of unliganded, singly liganded, and fully liganded dimers have been aligned using the coordinates of the FNIII‐1/FNIII‐1′ pair. (c) The unliganded symmetric IR dimer from X‐ray crystallographic studies (pdb id 4zxb) is shown. The left‐hand molecule has a transparent gray surface, with the exception of L1 and L2, for which the surface is colored light magenta. The αCT is also shown as a light magenta cylinder. The second molecule is shown with L1′, L2′, and the FNIII domains as darker purple cartoons, αCT′ as a dark purple cylinder, and CR′ as light gray cartoon. Intermolecular autoinhibitory interactions between (i) L1 and FNIII‐2′, (ii) L2 and FNIII‐1′, and (iii) L1 and αCT′ are highlighted with cyan circles (the symmetry equivalents are not shown). (d) The asymmetric singly liganded IR dimer (pdb id 6hn5 and 6hn4), with the same representation and coloring as in (c). The single bound insulin molecule is shown in cyan with cylindrical helices (and interacts with αCT′, shown in dark purple and labeled). The location of αCT (light magenta), which was not included in pdb id 6hn4 as it was not well ordered, is modeled based on its location relative to L1′ in the unliganded structure. (e) The symmetric IR dimer (pdb id 6pxv) is shown with two bound insulin molecules. For clarity, the additional two insulin molecules in this structure that bind near FNIII‐1 and FNIII‐1′ (see text) are not shown. CR, cysteine‐rich; ECR, extracellular region; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FNIII, fibronectin type III; IR, insulin receptor
4.2. IR family
The monomer/dimer situation is quite different in the IR family, because the receptors are disulfide‐linked (αβ)2 dimers regardless of whether ligand is bound. Moreover, in contrast to the dominant role of domain II as an “effector” of ligand‐induced conformational changes in EGFR, the corresponding CR domain in IR participates in no intra‐ or intermolecular interactions whether ligand is bound or not. Instead, the CR domain serves only as a flexible linker between the L1 and L2 domains in the IR family. Ligand‐induced changes in the conformation of IR and IGF1‐R can nonetheless be related conceptually to those in EGFR. They alter the nature of the dimer rather than the degree of dimerization per se. Indeed, this presumably must also be the case for EGFRs that dimerize constitutively (such as that from C. elegans 12 ) or under conditions in which EGFR forms much‐discussed “preformed” dimers. 52
As pointed out by Ward and Lawrence, 10 the unliganded IR dimer is “folded over” (Figure 5c), like the tethered ECR monomer of the EGF receptor. As with EGFR, interactions thought to be autoinhibitory hold IR in this folded‐over conformation. The autoinhibitory interactions in IR are quite different from those in EGFR, however. First, they are intermolecular, occurring between (rather than within) receptor protomers in IR. Second, IR autoinhibition involves domains that do not exist in EGFR family members. As marked in Figure 5c with open cyan circles, the autoinhibitory interactions in the unliganded IR dimer involve: (i) Association of the L1 domain (through its central β‐sheet) with FNIII‐2′ (of its neighboring protomer); (ii) Association of the L2 domain of each protomer with FNIII‐1′ (again of the neighboring protomer); and (iii) Docking of the αCT′ helix from the opposing protomer onto the L1 domain's central β‐sheet.
4.3. L1‐CR‐L2 transition in the IR dimer
Several key things are seen to happen upon binding of ligand to one of the protomers in the unliganded IR dimer17, 18 (Figure 5d)—noting that we do not intend to argue that they happen in this order temporally. First, by binding to the central β‐sheet of the L1 domain, insulin displaces L1 from its intermolecular interaction with the (relatively membrane proximal) FNIII‐2′ domain (interaction [i] in Figure 5c). L1 is drawn upward in the orientation shown in Figure 5c,d by 40–50 Å. Intriguingly, αCT′ remains (intermolecularly) associated with this L1 domain and is thus also drawn upward—although the L1/αCT′ interface is remodeled (as discussed in detail by Menting et al. 43 ). Bivalent binding of the insulin/αCT′ combination (considering this as a de facto ligand as discussed above) docks L1/insulin/αCT′ onto L2 of the same protomer—and promotes the domain “closure” depicted in Figure 3b. The L1/L2 domain “closure” is further augmented by direct interactions between L1 and L2 that are seen only in liganded IR/IGF1‐R (and have no equivalent in EGFR). The resulting remodeled, ligand‐bound, L1‐CR‐L2 module docks onto the top of FNIII‐1′ (from the opposing protomer in the dimer) as seen in Figure 5d. To achieve this, L2 has moved from the side of FNIII‐1′ (breaking autoinhibitory interaction [ii] in Figure 5c) to the top of that same domain. In addition, L1 has moved from the side of FNIII‐2′ (breaking autoinhibitory interaction [i]) to the top of FNIII‐1′. In effect, the L1‐CR‐L2 module appears to “present” insulin plus αCT′ to the top of the FNIII‐1′ domain in Figure 5d—with L2 having moved upward by about its own length and rotated ~90° about an axis perpendicular to the page in Figure 5.
The autoinhibitory intermolecular interactions within the IR dimer that are disrupted or remodeled upon ligand binding—(i) L1/FNIII‐2′, (ii) L2/FNIII‐1′, and (iii) αCT′/L1—are all mediated by the L1‐CR‐L2 module.26, 28, 29 Similarly, although intra‐ (rather than inter‐) molecular, the autoinhibitory interactions in EGFR that are disrupted upon ligand binding are also mediated by the corresponding domain I‐II‐III module. IR and EGFR use different parts of this module for autoinhibition, however—with IR using its LRRs, and EGFR its cysteine‐rich domain II. In both cases, though, the relevant parts of this module dock onto domains that are C‐terminal to L2/domain III (FNIII domains in IR; domain IV in EGFR). The new interactions acquired by the L1‐CR‐L2 module following ligand binding are intermolecular in both receptor families. In EGFR, they actually drive dimerization (Figure 5b). In IR (and IGF1‐R 19 ), they instead alter the arrangement of protomers within an already‐existing dimer. Again, different parts of the L1‐CR‐L2 module are used. EGFR uses its CR (domain II) to drive dimerization, whereas the opposite face of the L1‐CR‐L2 module mediates the new intermolecular interactions in ligand‐bound IR/IGF1‐R—and with a key contribution from the ligand itself (if one considers αCT′ as part of the ligand). This shift of intermolecular interactions within the IR dimer allows its rearrangement into a presumably active state (Figure 5d).
After the conformational changes described above, an asymmetric IR 18 or IGF1‐R 19 dimer results (Figure 5d), which may be related to the half‐of‐the‐sites negative cooperativity seen for these ligands as summarized below. Binding of a second ligand would promote essentially the same set of conformational changes in the unoccupied protomer (purple in Figure 5d) to yield a more symmetric dimer (Figure 5e), as was seen in the most recent cryo‐EM studies of IR.20, 21 In these structures, both of the L1‐CR‐L2 modules are occupied with insulin + αCT of the opposing protomer, and this module docks onto the top of the opposing FNIII‐1 domain to yield a “T”‐shape. 53 Remarkably, each of these new symmetric structures, which were determined in the presence of saturating insulin,20, 21 has two additional insulin molecules bound—for a total of four ligands per dimer. The additional insulins lie on the outer β‐sheet of each FNIII‐1 sandwich, and also contact a loop in L1′—so they bridge FNIII‐1 and L1′ as well as FNIII‐1′ and L1 (not shown). The precise role of these two additional insulins—observed in the same place in two independent studies20, 21—remains unclear, and possibilities are discussed at length in the original papers.
4.4. Influence of the L1‐CR‐L2 transition on the FNIII domain module
The L1‐CR‐L2 module is linked to the TM and kinase domains of the receptor by domain IV in EGFR family members, and by the FNIII‐1/2/3 module in IR. In EGFR, ligand binding induces dimerization of the ECR, bringing two copies of the membrane‐proximal domain IV—and thus the TM and kinase domains—together in an active dimer. 14 In IR, the two membrane proximal FNIII‐3 (and 3′) domains in a dimer similarly come close to one another upon ligand binding17, 18, 20, 21 (with direct contacts in some structures), likewise presumably allowing the TM and kinase domains to come together for receptor activation. 29 Rotation “inward” of the FNIII‐1′/2′/3′ module (purple in Figure 5c,d) is one key event in bringing these membrane proximal regions together. It appears to result from disengaging the pink L1 plus αCT′ module from the purple FNIII‐2′ domain in Figure 5c, to break autoinhibitory interaction (i). This also frees the FNIII‐1′/2′/3′ module for its upward rotation in going from Figure 5c to Figure 5d. For the close proximity between membrane‐proximal FNIII modules that is seen in the structures, the gray FNIII‐1/2/3 module must also be drawn inward (toward the viewer in Figure 5c). This might result in part from the ~90° reorientation of the ligand‐bound L2 domain (pink) mentioned above, and (indirectly) from αCT′ translocation (which alters the position of αCT/L1′ and thus of FNIII‐2). One result of all of these changes is that the conformation of the dark purple unliganded protomer in Figure 5d differs from that of the two unliganded protomers in Figure 5c. The dark purple protomer in Figure 5d adopts the “restrained unliganded” state shown in Figure 2c, in which the L1 domain and FNIII‐2 domain of the same (unliganded) protomer contact one another. 19
Although details are different between the two receptor families, ligand‐induced closure of the L1‐CR‐L2 module in IR or EGFR results in stabilization of a receptor dimer in which the membrane‐proximal domains are drawn close to one another. This theme seems to hold in nearly all RTKs, 1 and frequently involves membrane‐proximal FNIII domains.54, 55, 56
5. POSSIBLE COMMON ORIGINS OF NEGATIVE COOPERATIVITY
A final parallel that should be drawn between IR and EGFR relates to the potential origin of negative cooperativity. Negative cooperativity in binding of insulin to its receptor has long been known.23, 31 For EGFR, two classes of binding site (high‐ and low‐affinity) have also been the subject of much discussion,57, 58 and seem to be best explained by negative cooperativity.23, 59, 60 Structures of the Drosophila EGFR ECR (dEGFR)32, 61 showed how negative cooperativity could arise through half‐of‐the‐sites reactivity in an asymmetric dimer. The dEGFR ECR crystallizes as a dimer in the absence of ligand. 61 Binding of ligand between domains I and III of one receptor protomer in this dimer gives rise to an asymmetric dimer (Figure 6b) in which intimate interactions between domain II and domain II′ restrain domain II′ (in the right‐hand protomer) so that the movement of domains I′ and III' with respect to one another is restricted. The right‐hand unliganded protomer in Figure 6b corresponds to the “restrained unliganded” state in Figure 2f. Since ligand binding to the right‐hand protomer requires these domains to be pushed apart from one another, this restraint disfavors binding, leading to half‐of‐the‐sites negative cooperativity. 62 The conformation of the asymmetric dimer appears unchanged following (compromised) ligand binding to the right‐hand protomer in the case of the Drosophila EGFR. 32 Results indicating that a singly liganded EGFR dimer can signal 50 argue that similar negative cooperativity also operates in human EGFR. The dimer remains asymmetric when the second ligand to bind is the low‐affinity epiregulin ligand, 33 but becomes symmetric after binding of the second ligand with the higher affinity EGF and transforming growth factor‐α ligands (Figure 6c).41, 49
FIGURE 6.
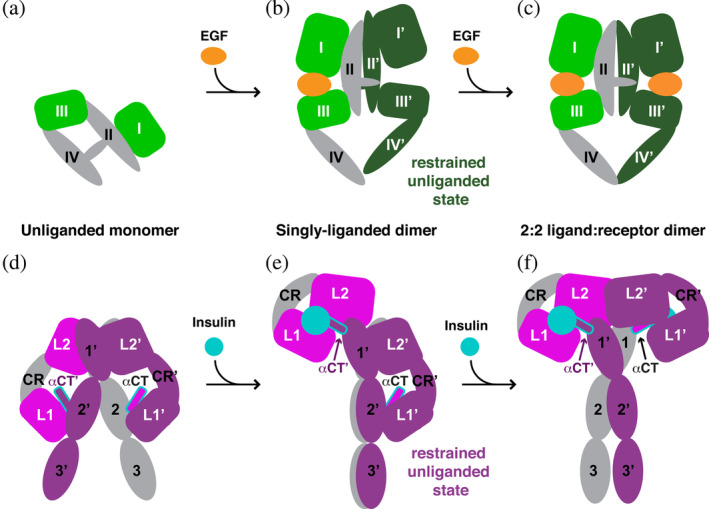
Models for the origin of negative cooperativity in the EGFR and IR families. (a–c) Model of negatively cooperative ligand binding to EGFR. (a) An unliganded EGFR monomer based on Figure 5a. (b) A singly liganded EGFR dimer based on the crystal structure of a 1:2 ligand: receptor complex seen for dEGFR (pdb id 3ltg). 32 The right hand protomer represents the restrained unliganded state shown in Figure 2f. (c) A symmetric 2:2 ligand: receptor dimer for EGFR, based on Figure 5b. The second ligand binds to the restrained dark green protomer in (b) with reduced affinity (as described in the text), and this is manifest as negative cooperativity. (d–f) Proposed model of negative cooperativity in the IR family. (d) An unliganded IR dimer is shown based on pdb id 4zxb. A similar dimer is seen for IGF1‐R in pdb id 5u8r. Colors are as in Figure 5c. (e) The singly liganded asymmetric dimer, based on recent cryo‐EM structures of IR and IGF1‐R (pdb ids 6hn4, 6hn5, and 6pyh).18, 19 L1 and L2 in the left‐hand molecule have been drawn together by ligand binding, and this module is “closed”. Adopting this structure imposes restraints on the right‐hand (purple) molecule—which represents the restrained unliganded state shown in Figure 2c. Binding of ligand between L1′ and L2′ of the right hand L1′‐CR′‐L2′ module requires that these restraints to be broken, which will cost energy—causing a second ligand binding event to have lower affinity. This could explain negative cooperativity in IR. (f) Symmetric 2:2 insulin:IR dimer based on pdb ids 6pxw, 6sof, and 6ceb,17, 20, 21 which would result after the second lower‐affinity binding event between L1′ and L2′. CR, cysteine‐rich; cryo‐EM, cryo‐electron microscopy; dEGFR, Drosophila epidermal growth factor receptor; EGFR, epidermal growth factor receptor; IR, insulin receptor
The mechanistic parallels between EGFR and IR described here suggest that negative cooperativity in the IR family may have an unexpectedly similar origin to that seen in EGFR. In the asymmetric IR dimer depicted in Figure 6e, ligand (insulin + αCT′) is bound to the L1‐CR‐L2 module of one protomer (which is “closed”). The L1′‐CR′‐L2′ module in the other protomer remains in an “open” arrangement (Figure 6e), overlaying well (but not perfectly) with the isolated unliganded module. 36 This unliganded L1′‐CR′‐L2′ module appears to be restrained, however, by intramolecular domain L1′/FNIII‐2′ interactions (seen only in the restrained unliganded state18, 19), and/or remodeling of the intermolecular L2′/FNIII‐1 interface. These restraints could impair rearrangement of the domains in the L1′‐CR′‐L2′ module upon ligand binding, so that the affinity of binding of the second ligand in going from the asymmetric dimer (Figure 6e) to the symmetric 2:2 (insulin:IR) dimer (Figure 6f) is reduced—which would be manifest as negative cooperativity in much the same way as described for EGFR. Li et al. 19 have also made a compelling argument that αCT is restrained in the asymmetric IGF1‐R dimer by its covalent linkage to αCT′ (which has been displaced upward by ~45 Å to the top of the dimer in Figure 6e). Since L1′ and αCT move together in progressing from the asymmetric dimer (Figure 6e) to the symmetric dimer (Figure 6f), this would certainly have the effect of restraining L1′‐CR′‐L2′ reorganization and contributing to negative cooperativity.
Thus, the origins of negative cooperativity in the IR and EGFR families may be unexpectedly similar. In both cases, the key appears to be restraint of the L1 (domain I) and/or L2 (domain III) positions. In IR, this appears to arise from their direct interactions with αCT and FNIII domains. In EGFR, they are instead restrained indirectly by restriction of the CR (domain II) conformation.
6. CONCLUSION
Although discussion of the activation mechanisms of IR and EGFR family members has typically been quite separate, new cryo‐EM views of ligand bound to presumed signaling‐competent forms of IR and IGF1‐R17, 18, 19, 20, 21 suggest ways of conceptually linking them. The “engine” of insulin or EGF‐induced change appears to be bivalent binding of ligand to the two LRRs (directly or indirectly) in both cases—resulting in a substantial reorientation of the two LRRs with respect to one another, and a “closure” of the L1‐CR‐L2 module. In EGFR, this transition breaks autoinhibitory interactions and exposes dimerization sites for new intermolecular interactions—resulting in ligand‐induced dimerization. Similarly, in IR, a ligand‐induced transition in the L1‐CR‐L2 module promotes new intermolecular interactions in the IR dimer that allow its remodeling into an active dimeric state ‐ again formed at the expense of autoinhibitory interactions seen only in the unliganded receptor. One key difference is that IR's autoinhibitory interactions are mostly intermolecular rather than intramolecular (as in EGFR)—because IR is always dimeric. In both IR and EGFR families, the L1‐CR‐L2 module dramatically changes its interaction properties upon ligand binding, and this is the “nucleus” of ligand‐induced change. A primary difference is that the “effector” of this change is the CR domain (domain II) in EGFR, whereas this function is shared by the L1 and L2 domains (plus bound ligand) in the IR family. This perspective on IR family regulation—informed by considerations that dominate the EGFR field—may help in unifying views of how these related receptor families function, and in understanding the origin of negative cooperativity that is such an important part of their biology.
7. METHODS
7.1. Structural alignments
All coordinates were obtained from the RCSB Protein Data Bank (PDB). Alignments were performed using Pymol (Schrödinger, LLC). All structures were aligned to 6pxv chain A, using the following domain boundaries: L1/domain I: IR, 1–155; IGF1‐R, 1–148; EGFR, 1–163; L2/domain III: IR, 309–468; IGF1‐R, 299–458; EGFR, 310–475; ErbB3, 309–474; ErbB4, 306–471; dEGFR, 301–467. FNIII‐1: IR, 471–591; IGF1‐R, 461–578.
7.2. Classification of the three “states” of IR and EGFR
Structures were superimposed using coordinates for L2/domain III of the structures of interest (amino acids listed above). Coordinates of IR and EGFR family members that contain the L1‐CR‐L2 module can be classified as listed below (chain ID is listed after the PDB ID where multiple chains are seen in the asymmetric unit). Only IR and human EGFR coordinates are shown in Figure 2, in italics below.
Unliganded: 2hr7A&B, 4zxb, 5kqvE&F, 6ce7B, 1igr, 5u8r, 5u8q, 1nql, 1yy9, 4krp, 3qwq, 4uv7, 1m6bA&B, 3p11, 4leo, 4p59, 2ahxA&B, 3u2p.
Ligand bound: 6pxvA&C, 6sofA&C, 6cebA&B, 6ce7A, 6hn5E, 6pyhD, 1moxA&B, 3njpA&B, 5wb7A&B, 3ltfC, 3ltgC, 3u7uA, 3u7uB.
Restrained unliganded: 6hn4F/6hn5F, 6pyhA, 5wb7C&D, 5wb8A&D, 3ltfA, 3ltgA. For 5wb7C&D, 5wb8A&D and 3tlfA a low affinity ligand is bound to the restrained unliganded state.
7.3. Analysis of the L1‐CR‐L2 conformational change
Domain motion analysis was performed using the DynDom3D webserver. 40 Coordinate files containing only the protein components for the L1‐CR‐L2 module were generated. Due to the variability in the relative orientation of the L1‐CR domains relative to L2 in the unliganded state, there is some variability in the angle of rotation for different unliganded and ligand bound pairs. In all cases, the axis is in a similar location near the CR/L2 (domain II/III) connection and the motion has over 90% closure character. The average rotation and closure for IR and EGFR are shown in Figure 3. For IR/IGF‐1R, most unliganded and ligand bound pairs can be related with rotation angles of between 75° and 99°, with the average around 90°. An outlier is with the unliganded 5kqv, where the rotation angle is 140°. For EGFR, the range is 100–140°.
AUTHOR CONTRIBUTIONS
Chun Hu: Conceptualization; writing‐review and editing. Mark Lemmon: Conceptualization; funding acquisition; supervision; visualization; writing‐original draft; writing‐review and editing. Kathryn Ferguson: Conceptualization; funding acquisition; supervision; visualization; writing‐original draft; writing‐review and editing.
ACKNOWLEDGEMENTS
We thank Irit Lax, Joseph Schlessinger, Daryl Klein, Abigail Lemmon, and members of the Ferguson and Lemmon labs for valuable discussions. This study was supported by National Cancer Institute grant R01‐CA198164 (to K. M. F. and M. A. L.).
Ferguson KM, Hu C, Lemmon MA. Insulin and epidermal growth factor receptor family members share parallel activation mechanisms. Protein Science. 2020;29:1331–1344. 10.1002/pro.3871
Funding information National Cancer Institute, Grant/Award Number: R01‐CA198164
Contributor Information
Kathryn M. Ferguson, Email: kathryn.ferguson@yale.edu.
Mark A. Lemmon, Email: mark.lemmon@yale.edu.
REFERENCES
- 1. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bormann BJ, Engelman DM. Intramembrane helix‐helix association in oligomerization and transmembrane signaling. Annu Rev Biophys Biomol Struct. 1992;21:223–242. [DOI] [PubMed] [Google Scholar]
- 3. Schlessinger J. Signal transduction by allosteric receptor oligomerization. Trends Biochem Sci. 1988;13:443–447. [DOI] [PubMed] [Google Scholar]
- 4. Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130:323–334. [DOI] [PubMed] [Google Scholar]
- 5. Wehrman T, He X, Raab B, Dukipatti A, Blau H, Garcia KC. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron. 2007;53:25–38. [DOI] [PubMed] [Google Scholar]
- 6. Shim AH, Liu H, Focia PJ, Chen X, Lin PC, He X. Structures of a platelet‐derived growth factor/propeptide complex and a platelet‐derived growth factor/receptor complex. Proc Natl Acad Sci U S A. 2010;107:11307–11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Markovic‐Mueller S, Stuttfeld E, Asthana M, et al. Structure of the full‐length VEGFR‐1 extracellular domain in complex with VEGF‐A. Structure. 2017;25:341–352. [DOI] [PubMed] [Google Scholar]
- 8. Schlessinger J, Plotnikov AN, Ibrahimi OA, et al. Crystal structure of a ternary FGF‐FGFR‐heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–750. [DOI] [PubMed] [Google Scholar]
- 9. Schlessinger J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb Perspect Biol. 2014;6:a008912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ward CW, Lawrence MC. Similar but different: Ligand‐induced activation of the insulin and epidermal growth factor receptor families. Curr Opin Struct Biol. 2012;22:360–366. [DOI] [PubMed] [Google Scholar]
- 11. Burgess AW, Cho HS, Eigenbrot C, et al. An open‐and‐shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;2:541–552. [DOI] [PubMed] [Google Scholar]
- 12. Freed DM, Alvarado D, Lemmon MA. Ligand regulation of a constitutively dimeric EGF receptor. Nat Commun. 2015;6:7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hubbard SR. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb Perspect Biol. 2013;5:a008946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2014;6:a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yarden Y, Schlessinger J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry. 1987;26:1443–1451. [DOI] [PubMed] [Google Scholar]
- 16. Lemmon MA, Bu Z, Ladbury JE, et al. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997;16:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scapin G, Dandey VP, Zhang Z, et al. Structure of the insulin receptor‐insulin complex by single‐particle cryo‐EM analysis. Nature. 2018;556:122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weis F, Menting JG, Margetts MB, et al. The signalling conformation of the insulin receptor ectodomain. Nat Commun. 2018;9:4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J, Choi E, Yu H, Bai XC. Structural basis of the activation of type 1 insulin‐like growth factor receptor. Nat Commun. 2019;10:4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uchikawa E, Choi E, Shang G, Yu H, Bai XC. Activation mechanism of the insulin receptor revealed by cryo‐EM structure of the fully liganded receptor‐ligand complex. Elife. 2019;8:e48630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gutmann T, Schäfer IB, Poojari C, et al. Cryo‐EM structure of the complete and ligand‐saturated insulin receptor ectodomain. J Cell Biol. 2020;219:e201907210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ward CW, Lawrence MC, Streltsov VA, Adams TE, McKern NM. The insulin and EGF receptor structures: New insights into ligand‐induced receptor activation. Trends Biochem Sci. 2007;32:129–137. [DOI] [PubMed] [Google Scholar]
- 23. De Meyts P. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem Sci. 2008;33:376–384. [DOI] [PubMed] [Google Scholar]
- 24. Dawson JP, Bu Z, Lemmon MA. Ligand‐induced structural transitions in ErbB receptor extracellular domains. Structure. 2007;15:942–954. [DOI] [PubMed] [Google Scholar]
- 25. Ferguson KM. Structure‐based view of epidermal growth factor receptor regulation. Annu Rev Biophys Biomol Struct. 2008;37:353–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKern NM, Lawrence MC, Streltsov VA, et al. Structure of the insulin receptor ectodomain reveals a folded‐over conformation. Nature. 2006;443:218–221. [DOI] [PubMed] [Google Scholar]
- 27. Croll TI, Smith BJ, Margetts MB, et al. Higher‐resolution structure of the human insulin receptor ectodomain: Multi‐modal inclusion of the insert domain. Structure. 2016;24:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu Y, Kong GK, Menting JG, et al. How ligand binds to the type 1 insulin‐like growth factor receptor. Nat Commun. 2018;9:821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kavran JM, McCabe JM, Byrne PO, et al. How IGF‐1 activates its receptor. Elife. 2014;3:e03772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ward CW, Menting JG, Lawrence MC. The insulin receptor changes conformation in unforeseen ways on ligand binding: Sharpening the picture of insulin receptor activation. Bioessays. 2013;35:945–954. [DOI] [PubMed] [Google Scholar]
- 31. De Meyts P. The structural basis of insulin and insulin‐like growth factor‐I receptor binding and negative co‐operativity, and its relevance to mitogenic versus metabolic signalling. Diabetologia. 1994;37(suppl 2):S135–S148. [DOI] [PubMed] [Google Scholar]
- 32. Alvarado D, Klein DE, Lemmon MA. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell. 2010;142:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Freed DM, Bessman NJ, Kiyatkin A, et al. EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell. 2017;171:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. [DOI] [PubMed] [Google Scholar]
- 35. Garrett TPJ, McKern NM, Lou M, et al. Crystal structure of the first three domains of the type‐1 insulin‐like growth factor receptor. Nature. 1998;394:395–399. [DOI] [PubMed] [Google Scholar]
- 36. Lou M, Garrett TPJ, McKern NM, et al. The first three domains of the insulin receptor differ structurally from the insulin‐like growth factor 1 receptor in the regions governing ligand specificity. Proc Natl Acad Sci U S A. 2006;103:12429–12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho HS, Leahy DJ. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002;297:1330–1333. [DOI] [PubMed] [Google Scholar]
- 38. Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–517. [DOI] [PubMed] [Google Scholar]
- 39. Liu P, Bouyain S, Eigenbrot C, Leahy DJ. The ErbB4 extracellular region retains a tethered‐like conformation in the absence of the tether. Protein Sci. 2012;21:152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poornam GP, Matsumoto A, Ishida H, Hayward S. A method for the analysis of domain movements in large biomolecular complexes. Proteins. 2009;76:210–212. [DOI] [PubMed] [Google Scholar]
- 41. Garrett TPJ, McKern NM, Lou M, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–773. [DOI] [PubMed] [Google Scholar]
- 42. Ogiso H, Ishitani R, Nureki O, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. [DOI] [PubMed] [Google Scholar]
- 43. Menting JG, Whittaker J, Margetts MB, et al. How insulin engages its primary binding site on the insulin receptor. Nature. 2013;493:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Menting JG, Yang Y, Chan SJ, et al. Protective hinge in insulin opens to enable its receptor engagement. Proc Natl Acad Sci U S A. 2014;111:E3395–E3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Menting JG, Lawrence CF, Kong GK, Margetts MB, Ward CW, Lawrence MC. Structural congruency of ligand binding to the insulin and insulin/type 1 insulin‐like growth factor hybrid receptors. Structure. 2015;23:1271–1282. [DOI] [PubMed] [Google Scholar]
- 46. Kani K, Park E, Landgraf R. The extracellular domains of ErbB3 retain high ligand binding affinity at endosome pH and in the locked conformation. Biochemistry. 2005;44:15842–15857. [DOI] [PubMed] [Google Scholar]
- 47. Lax I, Bellot F, Howk R, Ullrich A, Givol D, Schlessinger J. Functional analysis of the ligand binding site of EGF‐receptor utilizing chimeric chicken/human receptor molecules. EMBO J. 1989;8:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kohda D, Odaka M, Lax I, et al. A 40‐kDa epidermal growth factor/transforming growth factor alpha‐binding domain produced by limited proteolysis of the extracellular domain of the epidermal growth factor receptor. J Biol Chem. 1993;268:1976–1981. [PubMed] [Google Scholar]
- 49. Lu C, Mi LZ, Grey MJ, et al. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol Cell Biol. 2010;30:5432–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu P, Cleveland TE 4th, Bouyain S, Byrne PO, Longo PA, Leahy DJ. A single ligand is sufficient to activate EGFR dimers. Proc Natl Acad Sci U S A. 2012;109:10861–10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand‐induced conformational changes in the dimer interface. Mol Cell Biol. 2005;25:7734–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Purba ER, Saita EI, Maruyama IN. Activation of the EGF receptor by ligand binding and oncogenic mutations: The "Rotation Model". Cell. 2017;6:E13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gutmann T, Kim KH, Grzybek M, Walz T, Coskun Ü. Visualization of ligand‐induced transmembrane signaling in the full‐length human insulin receptor. J Cell Biol. 2018;217:1643–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph‐ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leppänen VM, Saharinen P, Alitalo K. Structural basis of Tie2 activation and Tie2/Tie1 heterodimerization. Proc Natl Acad Sci U S A. 2017;114:4376–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moore JO, Lemmon MA, Ferguson KM. Dimerization of Tie2 mediated by its membrane‐proximal FNIII domains. Proc Natl Acad Sci U S A. 2017;114:4382–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shoyab M, De Larco JE, Todaro GJ. Biologically active phorbol esters specifically alter affinity of epidermal growth factor membrane receptors. Nature. 1979;279:387–391. [DOI] [PubMed] [Google Scholar]
- 58. Magun BE, Matrisian LM, Bowden GT. Epidermal growth factor. Ability of tumor promoter to alter its degradation, receptor affinity and receptor number. J Biol Chem. 1980;255:6373–6381. [PubMed] [Google Scholar]
- 59. Wofsy C, Goldstein B, Lund K, Wiley HS. Implications of epidermal growth factor (EGF) induced EGF receptor aggregation. Biophys J. 1992;63:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Macdonald JL, Pike LJ. Heterogeneity in EGF‐binding affinities arises from negative cooperativity in an aggregating system. Proc Natl Acad Sci U S A. 2008;105:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alvarado D, Klein DE, Lemmon MA. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature. 2009;461:287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Koshland DE Jr. The structural basis of negative cooperativity: Receptors and enzymes. Curr Opin Struct Biol. 1996;6:757–761. [DOI] [PubMed] [Google Scholar]