

Abstract
Genetic variation in the membrane trafficking adapter protein complex 4 (AP‐4) can result in pathogenic neurological phenotypes including microencephaly, spastic paraplegias, epilepsy, and other developmental defects. We lack molecular mechanisms responsible for impaired AP‐4 function arising from genetic variation, because AP‐4 remains poorly understood structurally. Here, we analyze patterns of AP‐4 genetic evolution and conservation to identify regions that are likely important for function and thus more susceptible to pathogenic variation. We map known variants onto an AP‐4 homology model and predict the likelihood of pathogenic variation at a given location on the structure of AP‐4. We find significant clustering of likely pathogenic variants located at the interface between the β4 and N‐μ4 subunits, as well as throughout the C‐μ4 subunit. Our work offers an integrated perspective on how genetic and evolutionary forces affect AP‐4 structure and function. As more individuals with uncharacterized AP‐4 variants are identified, our work provides a foundation upon which their functional effects and disease relevance can be interpreted.
1. INTRODUCTION
The transport of protein and lipid cargo between distinct cellular compartments is essential to life. This critical function is often carried out by large multisubunit coat protein complexes that facilitate formation of vesicles or tubules that contain specific cargoes. Vesicles bud from donor compartments and are then sorted to and fuse with acceptor compartments. 1 One such coat protein complex is adapter protein complex 4 (AP‐4). AP‐4 (ε/β4/μ4/σ4 subunits; Figure 1) is a member of the adapter protein (formerly Assembly Polypeptide) family, which comprises the closely related APs 1–5 and the COPI F‐subcomplex. Each member of the AP family is heterotetrameric in structure, and all play key roles in membrane trafficking pathways.2, 3 AP‐1 and AP‐2 are the most well characterized and closely associate with clathrin to form clathrin‐coated vesicles. 4 AP‐4 is recruited to the trans‐Golgi network (TGN) by the small GTPase, Arf1. 5 Unlike AP‐1 and AP‐2, AP‐4 does not associate with clathrin, suggesting AP‐4 must interact with additional proteins at the membrane to form a complete coat and initiate vesicle formation. AP‐4 was present in the last eukaryotic common ancestor (LECA), but it has been lost in multiple eukaryotic lineages. 6
FIGURE 1.
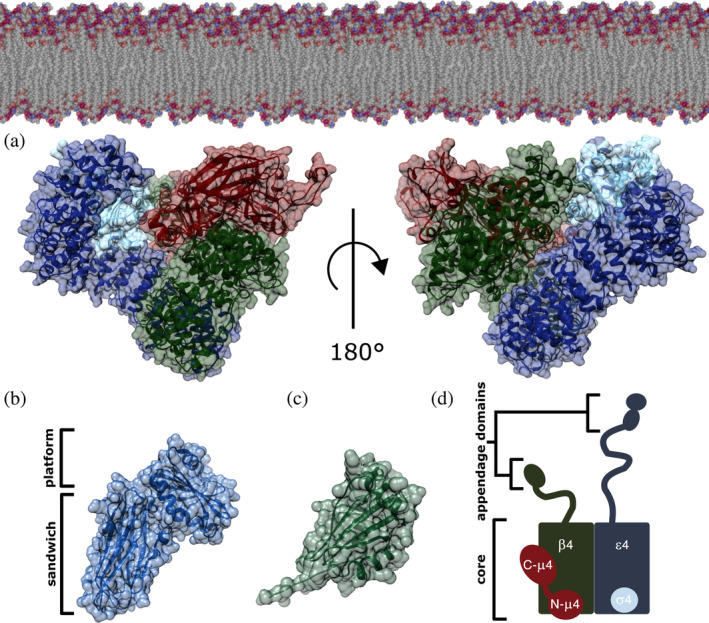
AP‐4 homology model. (a) AP‐4 core in its open conformation is depicted with ε, β4, μ4, and σ4 subunits shown in blue, green, red, and cyan, respectively. The complex is shown in the orientation thought to interact with the membrane, as based on related AP structures. The AP‐4 core homology model was generated using existing experimental structures of both AP‐2 (PDB: 2XA7) and AP‐4 (PDB: 3L81). (b) AP‐4 ε appendage domain homology model showing the platform and sandwich subdomains generated from AP‐2 α appendage (PDB: 1B9K). (c) β4 appendage domain (PDB ID: 2MJ7). (d) Schematic of the AP‐4 heterotetramer. The ε and β4 subunits contain C‐terminal appendage domains (20–30 kDa each) attached to the core (200 kDa) via long unstructured linkers. AP‐4, adapter protein complex 4
Recently, two groups showed AP‐4 colocalizes with accessory proteins tepsin, RUSC1, and RUSC2 at the TGN.7, 8 AP‐4 packages the three transmembrane proteins (ATG9A, SERINC1, and SERINC2) into vesicles transported to the cell periphery where ATG9A is believed to function in autophagosome formation.7, 8 The functions of the SERINC proteins remain poorly understood. AP‐4 is also implicated in neurological disease. The complex is expressed ubiquitously in cells, but AP‐4 seems especially important for neurological development.9, 10, 11, 12 Each of the four subunits can harbor mutations that cause hereditary spastic paraplegias (HSPs),13, 14, 15, 16 and current clinical research suggests patients have a specific subtype of HSP called AP‐4 deficiency syndrome. 17 Each AP‐4 gene (AP4E1/AP4B1/AP4M1/AP4S1) is also classified as a spastic paraplegia gene (SPG51/SPG47/SPG50/SPG52, respectively). Patients affected by AP‐4 deficiency syndrome typically present with early onset developmental delays, including delayed motor, muscle, and speech development. Other associated phenotypes include epilepsy, microencephaly, and brain malformations. Currently, there are about 150 known cases of AP‐4 deficiency syndrome. 17
Despite the importance of AP‐4, we still know relatively little about the precise pathway it mediates and why patients suffer severe neurological phenotypes. Current cell biology and clinical data suggests mistrafficking of ATG9A in the autophagy pathway inhibits the nucleation of autophagosomes in neurons, which prevents cargo degradation and recycling 18 in patients. In addition, we lack experimentally determined structural information about most of the complex. An NMR structure of the AP‐4 β4 appendage domain (PDB: 3L81, structure deposited, unpublished) (20 kDa; Figure 1) and X‐ray structures of the C‐terminal domain of the μ4 subunit 19 (30 kDa) have been experimentally determined, but the structure of most of AP‐4 (∼200 kDa) remains unknown. The lack of structural information makes it difficult to delineate and understand molecular mechanisms underlying known neurological and developmental deficiencies; it is also difficult to advise patients and families about potential clinical outcomes associated with specific mutations.
Advances in computational protein structure prediction and availability of sequencing data from diverse human populations and across species provide a variety of tools to investigate the evolutionary and functional constraints on AP‐4. We generated a structural model of AP‐4 by combining experimentally determined AP‐4 structures with homology models generated from AP‐2 crystal structures. AP‐2 was selected as the template structure because it was determined using experimental phasing methods, whereas the AP‐1 X‐ray structure was solved using molecular replacement from the AP‐2 structure. We therefore chose AP‐2 because it was the experimentally determined structure. We then used computational tools to analyze AP‐4 at both the gene and protein levels. We employed evolutionary biology techniques to analyze genetic variation, both common (defined as minor allele frequency, or MAF, >5%) and pathogenic. We show there are common genetic variants in AP‐4 that do not lead to disease. We further quantified evolutionary pressures on different regions of the protein complex, both within humans and across species, and correlated those regions with AP‐4 disease‐causing variants. Lastly, we integrated structural and genetic analyses to identify clusters of pathogenic variants in the AP‐4 complex and show putative “hot spots” that seem particularly susceptible to pathogenic mutations. Overall, our work provides a structural framework for interpreting the functional outcome of having a specific AP‐4 variant. It is imperative to provide both genetic and structural data to better understand how AP‐4 functions in the cell, as well as likely outcomes associated with newly identified AP‐4 variants in patients.
2. RESULTS
2.1. AP‐4 homology model
To analyze AP‐4 variants in a structural context, we generated homology models of the AP‐4 core in its open (Figure 1a) and closed (Figure S1A) conformations based on experimentally determined structures of AP‐2. We also generated a homology model of ε appendage (Figure 1b). The core includes the ε and β4 N‐termini, full‐length μ4, and full‐length σ4 (Figure 1a,d). The core and appendage domains of the open and closed conformations of AP‐2 were used as a template for one‐to‐one threading (Methods). Briefly, using the PHYRE2 server, we threaded the primary sequence of each AP‐4 subunit onto the analogous subunit of AP‐2. We specifically used crystal structures from the AP‐2 appendage domains (PDB IDs: 1B9K, 1E42) and both AP‐2 open and closed core structures (PDB IDs: 2XA7, 2VGL). 20 AP‐4 subunits ε, β4, μ4, and σ4 subunits demonstrate sequence similarities of 19, 29, 26, and 42% to respective AP‐2 counterparts. We selected the best predicted model (based on PHYRE2 confidence score) for each AP‐4 subunit and combined the models into a single structure by superimposing individual AP‐4 subunits onto the complete AP‐2 open core structure in Chimera (Figure 1). We combined these homology models with two experimentally determined AP‐4 structures, the β4 appendage domain (Figure 1b; PDB ID: 2MJ7) and C‐μ4 (PDB: 3L81), to generate a model of the entire AP‐4 complex. We used the AP‐4 open core homology model for subsequent structure‐based analyses, as the open core represents the active conformation that interacts with the membrane (Figure 1a). The cytosolic closed core AP‐4 homology model is shown for comparison in Figure S1. Given the relatively low sequence identity of the templates, our structural analyses focus on coarse‐grained spatial attributes. Electrostatics analysis in Chimera indicate AP‐4 is an acidic protein (Figure S1C,D).
2.2. Evolutionary conservation between species highlights functionally important AP‐4 regions
To identify regions of each AP‐4 subunit that are likely functionally important, we analyzed evolutionary conservation across vertebrates. We used ConSurf 21 to assign each residue a conservation score and mapped scores onto our AP‐4 model (Figures 2 and S2). ConSurf considers similarity across evolutionarily related proteins both within and between species, rather than explicitly testing for different modes of selection.
FIGURE 2.
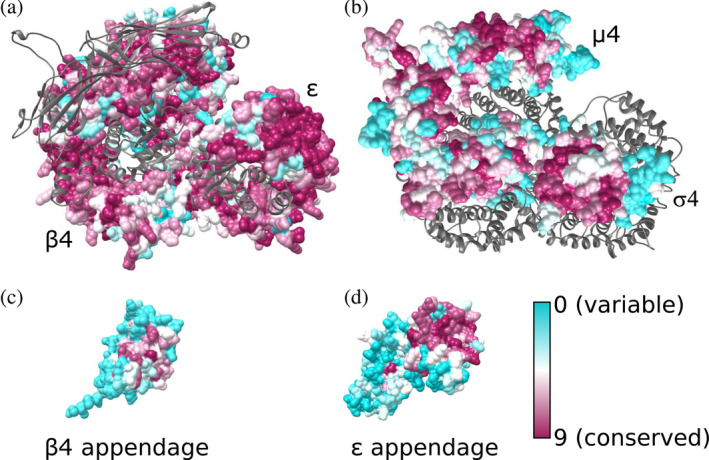
Evolutionary conservation of the AP‐4 core and appendage domains. Residues are colored from cyan to dark magenta based on conservation scores calculated using ConSurf. AP‐4 core with conservation scores mapped to residues are shown for (a) β4 and ε subunits with μ4 and σ4 shown as gray ribbons; (b) μ4 and σ4 subunits with β4 and ε subunits shown as gray ribbons; (c) the β4 appendage domain; and (d) the ε appendage domain. AP‐4 views in (a) and (b) are shown as top‐down views from the membrane. AP‐4, adapter protein complex 4
The two large subunits of AP‐4, β4, and ε, are the most conserved, with 51 and 52% of residues scoring an 8 or 9 on the conservation scale, respectively. This is likely as a result of constraint on the large surface interface between the two domains, which maintains overall secondary and tertiary structural integrity (Figure 2a,b). The appendage domains of β4 and ε have regions of conservation as well (Figure 2c,d). In other AP complexes, appendage domains are known to participate in protein–protein interactions at the membrane.22, 23, 24 In the β4 appendage, the most conserved region is the peptide binding groove identified by two groups as binding the LFxG[M/L]x[L/V] motif in the AP‐4 accessory protein, tepsin (Figure 2c).24, 25 The platform subdomain of ε appendage contains a larger patch of highly conserved residues (Figure 2d). The ε appendage binds the C‐terminus of tepsin 25 although the molecular mechanism has yet to be resolved structurally. ConSurf analysis suggests this highly conserved patch (F966, W970, I1009, W1039) is a strong candidate for binding the SAFAFLNA motif in tepsin. In contrast, the sandwich subdomain of the ε appendage is variable. In AP‐4 homologs, the equivalent appendage domain binds accessory proteins. For example, AP‐2 α appendage binds synaptojanin through its sandwich subdomain. 26 The lack of conservation in ε appendage may suggest this subdomain does not engage a protein partner, in contrast to AP‐2.
The C‐terminus of the μ4 subunit recognizes the Yxxφ and Yx[FYL][FL]E sorting motifs associated with lysosomal proteins and amyloid precursor protein (APP).19, 27 Both predicted binding pockets for these sorting motifs are highly conserved in our model, with 80% of residues scoring an 8 or higher in the canonical Yxxφ pocket and 56% scoring a 9 in the Yx[FYL][FL]E pocket. Additionally, the N‐terminus of the μ4 subunit and all of the σ4 subunit contain considerable regions of conservation. We attribute this conservation to overall folding requirements, as these domains stabilize the core by packing against and maintain key surface contacts with the ε and β4 core domains (Figure 2b). 28
Finally, AP‐4 σ4 contains a conserved hydrophobic patch (L65, V88, L98, and L103). This patch is analogous to the AP2 σ2 patch responsible for recognizing the dileucine cargo motif (D/E)XXXL(L/I), 29 and a binding pocket is present in our σ4 model. This suggests AP‐4 σ4 may also bind cargo. The SERINC proteins 7 have candidate dileucine motifs in their cytoplasmic regions, so σ4 may be a candidate for binding AP‐4 cargoes.
2.3. AP‐4 genes harbor both pathogenic variants and variants of uncertain significance
AP‐4 genes (AP4E1/AP4B1/AP4M1/AP4S1) harbor many genetic variants in human populations. In this section, we quantify the genetic and disease context of known AP‐4 variation to provide a reference frame for better interpreting the functional effects of newly identified variants. The ability to do this will be especially important for patients who require advice on potential clinical outcomes associated with genetic variation.
We report here for the first time common genetic variants found within each AP‐4 gene (Table 1). The detailed list of common variants including SNPs and genetic coordinates is shown in Table S1. We analyzed variants within the gene body of each AP‐4 gene from large databases of human genetic variation in over 140,000 individuals without severe genetic disease (1,000 Genomes and gnomADv2.1.1).30, 31 Hundreds of coding variants are present in each subunit colored bars, Figure 3a), but as expected, the majority of variants in all four AP‐4 genes occur within introns or untranslated regions (UTRs) (Figure 3a). Intronic variants are most common in the population (MAF > 5%), followed by UTR variants (Table 1). Coding variants in each of the four subunits have very low allele frequencies (MAF < 0.001%). Only four common coding variants were observed: one in AP4E1 and three in AP4B1.
TABLE 1.
Frequency of AP‐4 subunit human genetic variation from 1,000 Genomes and gnomAD in different genic contexts
| AP4E1 | AP4S1 | AP4B1 | AP4M1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Type | Allele frequency | Variant count | % within type | Variant count | % within type | Variant count | % within type | Variant count | % within type |
| UTR | Common | 6 | 0.8 | 5 | 2.7 | 3 | 3.5 | 4 | 0.7 |
| Rare | 6 | 0.8 | 12 | 6.4 | 3 | 3.5 | 8 | 1.3 | |
| Very rare | 703 | 98.3 | 170 | 90.9 | 79 | 92.9 | 589 | 98.0 | |
| Coding | Common | 1 | 0.5 | 0 | 0.0 | 3 | 0.5 | 0 | 0.0 |
| Rare | 1 | 0.5 | 0 | 0.0 | 1 | 0.2 | 0 | 0.0 | |
| Very rare | 214 | 99.1 | 127 | 100.0 | 568 | 99.3 | 124 | 100.0 | |
| Intron | Common | 237 | 8.3 | 184 | 9.4 | 7 | 1.8 | 11 | 2.1 |
| Rare | 138 | 4.8 | 131 | 6.7 | 8 | 2.1 | 9 | 1.7 | |
| Very rare | 2,485 | 86.9 | 1,650 | 84.0 | 368 | 96.1 | 509 | 96.2 | |
Note: Common variants have allele frequency (AF) > 5%; rare variants have AF <5% and > 1%; and very rare have AF <1%. Variant consequences are defined with respect to transcript annotations from Ensembl.
FIGURE 3.
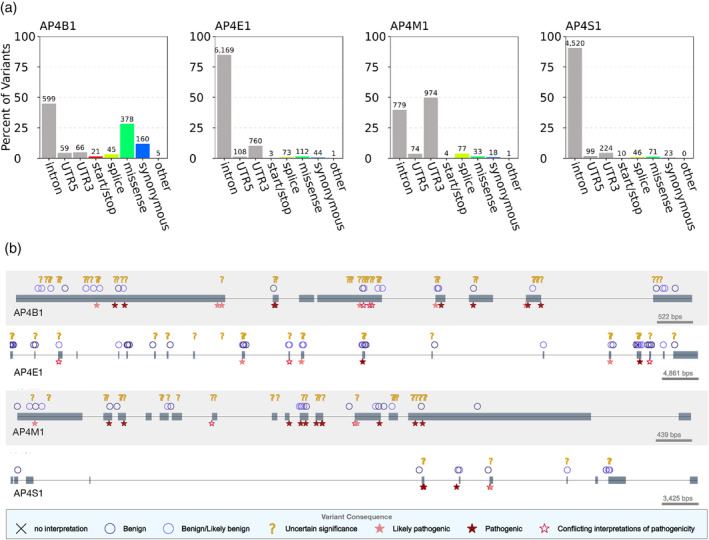
AP‐4 subunits harbor benign, disease‐associated, and uncharacterized human genetic variation. (a) Distribution of genic contexts of AP‐4 subunit genetic variants. We queried all variants occurring within the four AP‐4 genes in databases of human genetic variation across more than 140,000 individuals (1,000 Genomes and gnomAD). Variants were annotated based on their genic context (intron, UTR, coding) and functional impact on protein product (start/stop: gain or loss of transcription start or stop; splice: variant influencing splicing; missense; synonymous; or other: coding variants with missing label). The majority of variants are noncoding but many variants that influence coding sequence (missense, synonymous, splice) are present in each subunit. (b) Clinical consequences of AP‐4 variants. We plotted variants extracted from ClinVar along the gene body of each AP‐4 subunit (one row per AP‐4 subunit). Gray boxes represent exons occurring in known transcripts. Variants are displayed based on their predicted clinical consequence as reported in ClinVar. While several variants are known to be benign or pathogenic, more than 100 remain of uncertain significance. AP‐4, adapter protein complex 4
Next, we explored the clinical consequences of variants occurring within the gene body of all four AP‐4 genes using the ClinVar database (Figure 3b). 32 Briefly, ClinVar is a centralized database containing over 430,000 variants and their associated pathological interpretations submitted by clinical testing laboratories. 33 Many AP‐4 variants were classified as benign (5–27 variants per subunit), but hundreds of variants were classified as “pathogenic” or “likely pathogenic” (52 in AP4S1; 130 in AP4E1; Table 2). However, 160 variants across AP‐4 genes remain annotated as “variants of uncertain significance” (VUS; Table 2, Figure 3b) and hundreds more have no annotation. Interestingly, all four reported pathogenic missense mutations in patients (see next section) are annotated as VUS in ClinVar.
TABLE 2.
Clinical consequences for variants in the gene body of AP‐4 subunits as annotated in ClinVar
| AP4B1 | AP4E1 | AP4M1 | AP4S1 | |
|---|---|---|---|---|
| Benign | 8 | 27 | 11 | 5 |
| Benign/likely benign | 4 | 3 | 1 | 1 |
| Likely benign | 14 | 21 | 10 | 5 |
| Uncertain significance | 56 | 53 | 39 | 12 |
| Conflicting interpretations of pathogenicity | 3 | 3 | 2 | 1 |
| Likely pathogenic | 6 | 3 | 2 | 4 |
| Pathogenic | 19 | 20 | 29 | 25 |
| Total | 110 | 130 | 94 | 52 |
2.4. Pathogenic AP‐4 variants cluster in three‐dimensional space
Our previous work34, 35 has shown that evaluating the Euclidean distance in 3D space of uncharacterized variants relative to pathogenic and benign variants can aid in variant prioritization. Sivley et al.34, 35 developed a program called PathProx that evaluates the relative 3D proximity of a variant to known pathogenic and benign variants. To explore the spatial distribution of variants in AP‐4, we mapped onto the AP‐4 homology model four reported pathogenic variants (curated from the literature)13, 14, 15, 16, 36 and six variants annotated as likely pathogenic in ClinVar (Table 3). 32 We also identified 699 likely benign variants in AP‐4 from gnomAD database (Figure 4). 31 This metric predicted pathogenicity. Pathogenic variants were significantly more clustered in three‐dimensional space than expected by chance (Figure 4; p < .0001, Ripley's K at a range of distance thresholds from 15–45 Å). The regions most associated with pathogenic variation cluster in the α8, α9, and α10 helices in the core of the β4 subunit and in the C‐terminus of μ4 (Figure 4). The core of the β4 subunit also contains residues that exhibit the strongest association with neutral variation; all of these variants cluster in helices α14, α15, and α16.
TABLE 3.
List of likely and reported pathogenic AP‐4 variants used for PathProx analysis
FIGURE 4.
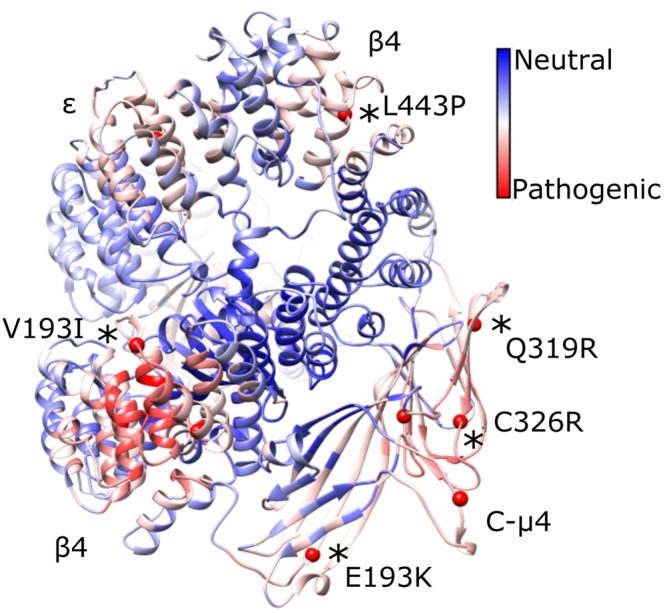
Pathogenic variants cluster in 3D space on the AP‐4 core. We mapped known pathogenic AP‐4 variants (red spheres; cf. Table 3) with 699 likely benign variants (gnomAD) onto the AP‐4 core model. The linkers and appendages do not contain known pathogenic variants, so we only show the core domain. Variants confirmed in patients are depicted by black asterisks and labeled. PathProx reveals pathogenic variants are significantly more clustered than expected if variants were randomly distributed across the protein (p < .0001, Ripley's K). We computed the difference in average proximity for each position to pathogenic and benign variants. Regions of the protein closer (on average) to pathogenic variation are shown in red, and regions more proximal to benign variants are shown in blue. These data suggest the β4/N‐μ4 interface and C‐μ4 are especially susceptible to pathogenic variation. AP‐4, adapter protein complex 4
We considered four pathogenic variants in AP‐4 β4. Three amino acid mutations (R107Q and R206Q) are positioned at the interface between the core domain of β4 and the N‐terminal domain of the μ4 subunit. Nonconservative mutations at this interface are likely to destabilize key interactions between the two subunits and impair complex assembly. A third mutation, V193I, is positioned in the same region of β4 as the previous mutations but is located in the linker between helix α10 and α11. A Val to Ile mutation at this position could possibly disrupt packing in this linker of β4. The fourth mutation in β4, L443P, is located on helix α25. The presence of a proline residue likely prohibits folding of the α helix and therefore leads to potential unfolding of the protein. AP‐4 ε has a single pathogenic variant in our model: V442I located on helix α26. The incorporation of a larger residue likely affects stability of the protein or drives unfolding.
AP‐4 μ4 contains five pathogenic variants in our model; these are dispersed throughout the C‐terminal subdomain. Three mutations (F416L, Q319R, and C326R) are located in the core of the μ4 domain and are expected to cause protein misfolding by disrupting β‐sheet formation. Mutation G250S is located on a loop region near helix α7. The substitution of a a larger polar Ser residue for a small, nonpolar Gly has a high probability of causing instability, especially because Gly residues can adopt conformations prohibited for other amino acids. Finally, mutation E193K is located near the Yxxφ cargo binding site. This charge reversal mutation may possibly affect the ability of the protein to form intra‐ and intermolecular interactions, including potential interactions with the membrane.
One potential challenge with our analysis is the relatively small number of reported pathogenic missense variants. To evaluate the utility of PathProx in evaluating VUS, we performed leave‐one‐out cross‐validation to quantify the predictive accuracy of the PathProx metric at distinguishing likely benign and pathogenic variants. PathProx identified pathogenic variants only slightly better than chance (Figure S3; ROC AUC of 0.57, PR AUC of 0.04). However, a simpler metric based on the distance to the nearest pathogenic variant performed significantly better (Figure S3; ROC AUC of 0.64, PR AUC of 0.18). This suggests that considering the spatial orientation of newly identified AP‐4 variants with respect to known variants may help clinicians interpret functional effects, but it will be very important to consider additional mutations as they are confirmed to improve our PathProx‐based model.
2.5. Structural insights from AP‐4 homology model
C‐μ4 cargo binding pockets. The C‐μ4 structure has been experimentally determined by X‐ray crystallography (PDB IDs: 3L81, 4MDR).19, 27 We compared both predicted cargo binding sites on C‐μ4. First, our analysis reveals the deep and hydrophobic Tyr binding pocket identified in all other μ subunits is absent. C‐μ4 cannot accommodate the Tyr residue found in a canonical Yxxφ motifs (Figure 5a), because residue Arg441 is positioned so that it occludes the space the Tyr residue would occupy. The binding pocket in C‐μ2 is shown for comparison (Figure 5b). However, conservation data reveal the Yxxφ binding pocket is more conserved than the YKFFE pocket, and PathProx predicts the Yxxφ pocket is more prone to pathogenic variation than is the YKFFE pocket (Figure 5c). Finally, the position of the YKFFE pocket is inconsistent with cargo binding in the open conformation (discussed further below).
FIGURE 5.
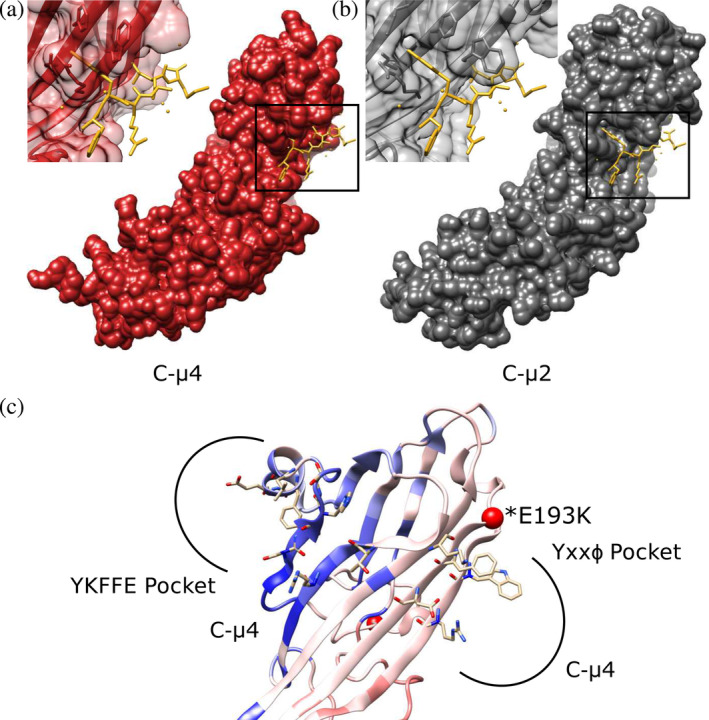
C‐μ4 cargo binding pockets. (a) The predicted Yxxφ binding pocket in C‐μ4 (PDB: 3L81) is shown with Yxxφ peptide from C‐μ2 superimposed (PDB: 1BW8). (b) The C‐μ2 Yxxφ cargo binding pocket with Yxxφ superimposed (PDB: 1BW8). C‐μ4 lacks the deep hydrophobic pocket known to accommodate the Tyr residue in canonical endocytic Yxxφ motifs. (c) Both YKFFE and Yxxφ binding pockets are shown mapped onto the PathProx C‐μ4 model. Residues important for motif binding in both pockets are shown as sticks. PathProx analysis indicates regions proximal to known pathogenic variation (mapped in red) and regions closer to benign variants (mapped in blue). Confirmed pathogenic mutation E193K shown as a red sphere. Current data suggest the Yxxφ binding pocket is more susceptible to pathogenicity, although the YKFFE site is believed to be important for sorting AP‐4 cargoes like ATG9. AP‐4, adapter protein complex 4
2.6. AP‐4 genes have experienced long‐term negative selection and recent positive selection
By analyzing patterns of missense genetic variation within and between species, we can identify regions evolving under different evolutionary constraints. Genetic variation in coding regions is often deleterious and negatively affects fitness. 37 To quantify long‐term evolutionary pressures on AP‐4 genes, we computed rates of nonsynonymous (dN) and synonymous (dS) substitutions for each amino acid site across vertebrates (Methods). As expected, across all four genes, the majority of sites have experienced higher rates of synonymous substitution (Figure S4), suggesting long‐term negative selection on many sites. Overall, 48.5% of AP4S1 sites (82/169) had significant evidence of negative selection (dN/dS < 1, p < .05, binomial test with Bonferroni correction), AP4E1 had the fewest sites (13/1,181, 1.1%), while AP4M1 and AP4B1 were intermediate, with 32.3% (240/743) and 29.6% (415/1,403), respectively. Only two sites across all genes (ε subunit residues Arg204 and Leu207, both located in the protein core) demonstrated evidence of positive selection (dN/dS > 1) at a nominal significance threshold (p < .05).
To provide recent evolutionary context for genetic variants observed in human AP‐4 genes, we evaluated coding variants for signatures of differentiation in frequency (FST) and recent positive selection (XP‐EHH) between European, Asian, and African ancestry superpopulations (Figure 6) from 1,000 Genomes (Methods). FST detects differences in the frequency of variants between populations, and the extended haplotype homozygosity test (XP‐EHH) detects population‐specific positive selection by considering the length and homozygosity of haplotypes. We evaluated the significance of the observed values by generating empirical null distributions to account for the allele frequency and linkage disequilibrium of tested variants (Methods).
FIGURE 6.
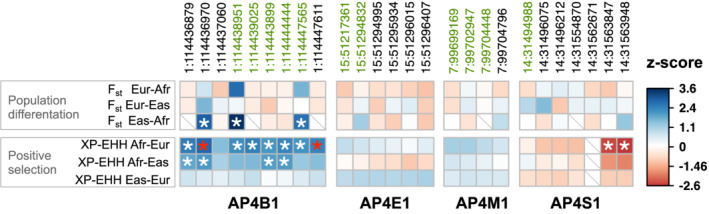
Exonic variants in AP‐4 genes exhibit evidence for recent population differentiation and positive selection. We tested variants in exons of any transcript of AP‐4 genes for evidence of evolutionary signatures of population differentiation (FST) and recent positive selection (XP‐EHH) within the 1,000 Genomes superpopulations (Methods). For a given evolutionary measure (rows) and exonic variant (columns, chromosome:position in hg19/GRCh37), the observed value was compared to a matched background distribution to derive a z‐score (indicated by the color of square per cell) and calculate an empirical p‐value. A positive (blue) or negative (red) z‐score indicates an increased or decreased value, respectively, compared to median value derived from matched background regions. Nominally significant (p < .05) and Bonferroni‐corrected statistically significant variant‐evolutionary measure pairs are denoted by white and red asterisks, respectively. Cells with missing data are indicated by a gray diagonal slash. Variants colored in green are classified “benign” by ClinVar. All exonic variants present in the SNPSNAP database and for which we could calculate a value for FST and XP‐EHH (see Methods) are shown. Overall, these data indicate AP4B1 exhibits differences among human populations and has undergone positive selection in African populations. AP‐4, adapter protein complex 4
Several AP4B1 variants had nominally significant evidence (p < .05) of recent positive selection (XP‐EHH ≥1.96) in African populations compared to non‐African populations (Figure 6). Three identified variants also had nominally significant differences in frequency between Asian and African populations (p < .05, FST ≥ 0.46). Two variants in AP4B1 (1:114436970 and 1:1144447611) passed Bonferroni correction for statistically significant evidence for positive selection in Africans compared with Europeans (XP‐EHH Afr‐Eur). Two variants in AP4S1 had nominally significantly negative XP‐EHH between Africans and Europeans populations, indicating positive selection in Africans. No variants in AP4E1 and AP4S1 had statistical evidence of recent evolutionary shifts between human populations. While several significant variants have been classified as benign by ClinVar (Figure 6, green), five are of unknown significance. These VUS primarily occur in exonic regions of AP‐4 genes.
3. DISCUSSION
AP‐4 remains one of the least understood members of the adaptor protein complex family. This is partially because AP‐4 is endogenously expressed at much lower levels than other adaptor proteins, and many model organisms (yeast, flies, worms) lack AP‐4. 2 Genetic variation in AP‐4 has been associated with rare spastic paraplegias and other neurological disorders, 36 but the association of both rare and common genetic variation in AP‐4 with disease has not been comprehensively investigated. To address the need for approaches to interpret new AP‐4 variants, we constructed a homology‐based structural model of the AP‐4 complex; mapped conservation and key AP‐4 variants into this model; and quantified the evolutionary history and structural location of known and predicted variants.
3.1. Insights into AP‐4 conservation and function from structural modeling
The AP‐4 core. The evolutionary conservation analysis indicated much of AP‐4 core is well conserved (Figures 2a and S2), especially the internal surfaces of subunits known to interact with each other to assemble the heterotetramer. Proteins in the AP family share similar domain architectures and thus conservation reflects the requirement to maintain the structure of the interactions between subunits within the core.2, 38, 39 The σ4 and N‐μ4 subunits contain longin domain protein folds. These domains play a structural role in stabilizing the conformations of AP complexes. Longin domains in multiple trafficking proteins are known to interact with protein binding partners, 40 including cargoes 29 and consequently this may contribute to high conservation in AP‐4. 28 We also note many surface‐exposed residues in the AP‐4 core homology model are well conserved (Figure S2). There are large patches of conservation on both large subunits, which may suggest these surfaces are functionally important and could be candidates for binding other protein partners.
Appendage domains. Appendage domains in AP complexes participate in protein–protein interactions. Conserved residues in AP‐4 appendage domains are also likely to mediate protein–protein interactions; we and others have shown the conserved patch on β4 appendage binds a LFxG[M/L]x[L/V] motif in tepsin.24, 25 Based on conservation scores, we predict the ε appendage platform subdomain likely binds the SAFAFLNA motif at the tepsin C‐terminus. 25 We attempted to test our idea experimentally by mutating conserved hydrophobic residues in the hypothesized tepsin binding pocket. We designed mutations based on AP‐2 α appendage, 26 and we attempted three separate mutations individually (F966A, W970A, and H1028A). Unfortunately, each mutation destabilized protein fold, and we were unable to obtain soluble protein. This suggests the hydrophobic pocket formed by these residues is critical to the stability of the appendage domain. The ε appendage sandwich subdomain exhibits less overall conservation than the platform subdomain, and this lack of conservation may suggest the ε sandwich subdomain is not critical for recruiting a binding partner.
Cargo binding. A major unresolved question in the field is how AP‐4 recognizes cargo at the molecular level. AP‐2 recognizes Yxxφ and acidic dileucine motifs in transmembrane proteins through its C‐μ2 and σ2 subunits, respectively.29, 41 C‐μ4 has been predicted to bind canonical Yxxφ‐based motifs 42 but reported binding is much weaker (Kd ∼ 100 μM) than is Yxxφ binding to C‐μ2 (Kd ∼ 1–10 μM). 29 Despite high conservation at the sequence level, experimentally determined X‐ray structures of C‐μ2 and C‐μ4 reveal these two domains have structurally diverged such that C‐μ4 is unlikely to accommodate Yxxφ motifs. One explanation for weak observed binding may be that C‐μ4 weakly binds canonical Yxxφ motifs at the YKKFE binding site (Figure 5c), located on the opposite face of the subdomain.27, 42
We would predict the Yxxφ pocket should be important for AP‐4 function: it is more conserved than the YKFFE pocket in vertebrates, and it may be more prone to pathogenic variation (cf. Figure 5c). The Yxxφ pocket is also positioned apposed to the membrane in an open AP‐4 conformation, where it would be accessible to transmembrane proteins. However, X‐ray structures clearly show the Tyr binding pocket is blocked. Instead, structural and biochemical data indicate C‐μ4 binds the YKFFE motif found in APP. 19 The Robinson and Bonifacino groups have now shown ATG9A is an important AP‐4 cargo.7, 8 ATG9A harbors a YKFFE‐like motif (YQRLE) in its N‐terminus, which is predicted to be cytosolic. Yeast two‐hybrid and immunoprecipitation data suggest this motif could interact with AP‐4, but the effect of mutating or removing the motif on ATG9A trafficking remains unknown. These data from cultured cells and cells derived from human patients thus suggest the YKFFE binding site may be critical for C‐μ4 function in cells. However, this binding pocket does not seem accessible in an AP‐4 open conformation (Figure S5). The YKFFE binding site is located between the C‐μ4 and β4 subunits, which are predicted to have extensive surface contacts in the open conformation. 41 Residues from β4 would likely occlude the YKFFE binding site if AP‐4 adopts the same open conformation that has been observed in multiple AP complexes.41, 43, 44 Since we anticipate AP‐4 will exist in a similar conformation, it remains very important to determine an experimental structure of AP‐4 in its membrane‐bound form. There are many important questions about the molecular mechanism by which AP‐4 recognizes cargo and engages the membrane.
3.2. Pathogenic proximity
Spatial analysis of known AP‐4 variants identified potential “hot spots” on the protein where pathogenic variants may cluster. Our analysis indicated specific locations on β4 and μ4 subunits may be particularly prone to pathogenicity. We compared the average pathogenic proximity of sites with their evolutionary conservation; this analysis revealed the most pathogenic regions generally overlap with the most conserved regions. We quantified this observation by plotting the conservation score versus the PathProx score of each residue for all four AP‐4 subunits (Figure 7). As expected, residues most susceptible to pathogenic variation tend to be more conserved. The regions highlighted by both conservation and PathProx analyses underscore the importance of stabilizing the AP‐4 core structurally. Variation in these important regions can drive pathogenicity. Of confirmed pathogenic variants, all but two on the μ4 subunit (Q319R and C326R) have conservation scores of 8 or 9. The benign missense variants identified by ClinVar have conservation scores of 5 or less, further highlighting the utility of combining conservation and structural analysis for variant interpretation. We require additional confirmed mutations to improve the PathProx model. However, as new variants are identified, we may be able to combine PathProx modeling with conservation to help clinicians and families analyze and understand specific variants identified in human patients.
FIGURE 7.
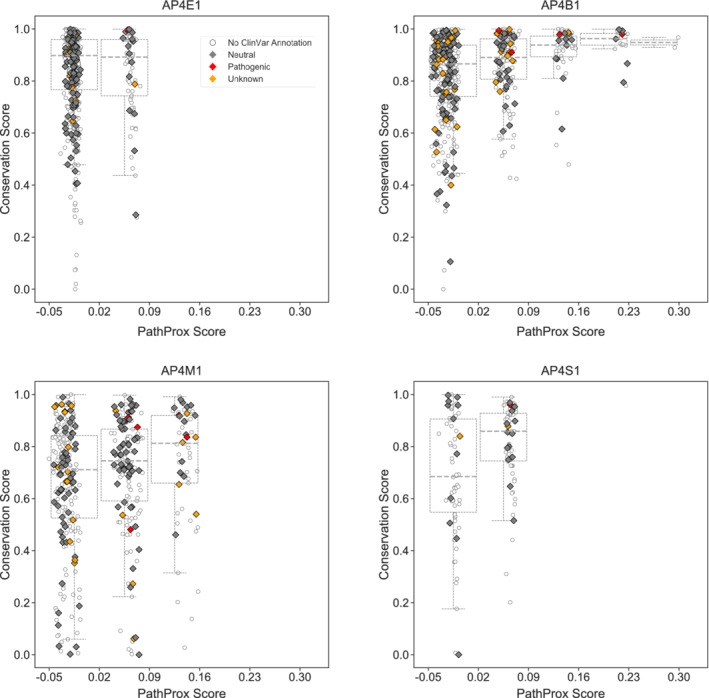
Conserved AP‐4 residues tend to exhibit higher PathProx scores. The PathProx score and a normalized conservation score derived from ConSurf for every residue in each AP‐4 subunit are plotted. Pathogenic variants are annotated in red; neutral/benign variants are shown in gray; and variants of unknown significance (VUS) are shown as empty circles. Boxplots are drawn after binning PathProx scores into equal size units of 0.07 to illustrate how conservation scores increase with increasing PathProx scores. Neutral/benign variants and VUS were compiled from ClinVar annotations. Conservation scores of 1 indicate the highest conservation while lower scores indicate highly variable positions across species. Higher PathProx scores indicate a greater average proximity to pathogenic variation compared with benign variation. AP‐4, adapter protein complex 4
3.3. Genetic variation and recent human evolution of AP‐4
Our analyses suggest there may be functionally relevant genetic differences in AP4B1 between different human populations. This is particularly notable since most identified AP‐4 patients have mutations in AP4B1 (the β4 subunit). In contrast, we do not find differences in the other AP‐4 genes. Specifically, there are differences between East Asian and African populations in exonic regions of β4. Furthermore, AP4B1 appears to have undergone positive selection in Africans. Overall, these analyses suggest AP‐4 genes experienced different evolutionary pressures during recent human evolution, and that some variation in these genes is likely to be tolerated and potentially even beneficial. Similar functional impacts based upon evolutionary pressure is present in other trafficking proteins, such as clathrin. 45 It will be interesting to determine whether differences in the β4 subunit contribute to different functional outcomes.
4. MATERIALS AND METHODS
4.1. Characterizing AP‐4 subunits variants by allele frequency and functional annotation
Variants from 1,000 Genomes and gnomAD (https://gnomad.broadinstitute.org/ ) in coding and regulatory regions of AP‐4 genes were downloaded from Ensembl GRC37 (Release 75) using BioMart.30, 31 We report the global minor allele frequency from either 1,000 Genomes, gnomAD or the mean across the two databases if the variant is common to both databases. We define variants with frequency >5% as common, frequency ≤5% and >1% as rare, and frequency ≤1% as very rare. To characterize the functional consequence of variants in the gene body of AP‐4 subunits, we mapped Ensembl functional annotations to UTRs, exons, or introns. Each variant received the highest severity annotation (as reported in Ensembl) such that each variant had only one annotation. The clinical consequence of variants in the gene body of AP‐4 subunits were obtained from ClinVar on 12/2019.
4.2. Calculating nonsynonymous and synonymous substitution rates
We downloaded unaligned AP4B1, AP4E1, AP4M1, and AP4S1 nucleotide and amino acid ortholog sequences from Ensembl GRCh37. The ortholog sequences were filtered to retain only species present in the Ensembl vertebrate phylogeny. Vertebrate species with outlier nucleotide sequence lengths were further removed. Outliers were defined as 1.5 times the interquartile range above and below the third and first quartiles, respectively. After applying aforementioned filters, AP4B1, AP4E1, AP4M1, and AP4S1 had 174, 65, 126, and 107 remaining vertebrate species sequences, respectively. We used MAFFT v7.407_1 implemented at http://ngphylogeny.fr to align the amino acid orthologs.46, 47 We obtained a codon alignment using PAL2NAL v14 based on the aligned amino acid and unaligned nucleotide orthologs. 48 The codon alignment was used for input to calculate nonsynonymous and synonymous substitution rates using Single Likelihood Ancestor Counting (SLAC) implemented at http://datamonkey.org.49, 50, 51 Default settings were used for all software applications unless otherwise specified. SLAC tests for statistically significant difference between dN and dS using an extended binomial distribution to test that the number of inferred synonymous sites is no less or greater than the expected proportion of synonymous sites. 49 All coordinates are in hg19/GRCh37.
4.3. Detecting evolutionary signatures at coding variants in AP‐4 subunits
To evaluate the population and evolutionary history of coding variants in AP‐4 subunits, we tested variants in exons occurring in any transcript for signatures of sequence conservation, acceleration, population differentiation, and recent positive selection. 52 Exon ranges for AP‐4 subunits were obtained from the UCSC genome browser. 53 Since these variants can overlap an exon from any transcript, some of variants may be classified as intronic by other databases. All analyses were conducted in the European population from 1,000 Genomes unless otherwise specified. Each coding variant was matched to 5,000 control variants on minor allele frequency, linkage disequilibrium structure, distance to nearest gene, and gene density using default settings in SNPSNAP.30, 54 Coding variants and the corresponding control variants were annotated with different evolutionary measures. FST and XP‐EHH between 1,000 Genomes superpopulations was computed using VCFTools v0.1.14 and the R package rehh 2.0, respectively.30, 55, 56 The remaining coding variants present in the SNPSNAP database and with nonmissing values for FST and XP‐EHH are reported in Figure 3. Each evolutionary annotation value for a coding variant was compared to a background distribution comprised of the control SNPs. A z‐score quantified the deviation from the mean. An empirical p‐value was also derived based the background distribution defined as the proportion of control variants with a value equal to or greater than the coding variant.
4.4. AP‐4 structural model generation
The primary sequences of each AP‐4 subunit (AP4E1: NP_001239056.1, AP4B1: NP_001240781.1, AP4M1: NP_001350600.1, AP4S1: NP_001121598.1) were used as inputs for one‐to‐one threading in PHYRE2. 20 The AP4E1, AP4B1, AP4M1, and AP4S1 subunits found in the core were threaded onto the open core structure of AP‐2 core domains AP2A1, AP2B1, AP2M1, and AP2S1 (PDB: 2XA7). The appendage domain of AP4E1 was threaded onto the AP2A1 appendage domain structure (PDB: 1B9K). The appendage domain of AP4B1 was threaded onto the AP2B1 appendage domain structure (PDB: 1E42). Each resulting AP‐4 core subunit model was superimposed over its respective AP‐2 counterpart in the AP‐2 open core structure in Chimera. 57 Intrinsically disordered linkers were then modeled in Chimera to attach the AP4E1 and AP4B1 appendage domains to the complete AP‐4 heterotetrameric core.
4.5. Pathogenic proximity (PathProx) calculation
We quantified the spatial proximity of each residue in the AP‐4 model to annotated pathogenic variants and genetic variants observed in individuals without severe disease using the previously described PathProx score.34, 35 The data set of reported pathogenic variants was derived from clinical cases and variants annotated from ClinVar as either pathogenic or likely pathogenic. 32 The likely benign variants were taken from gnomAD. The PathProx metric computes the average distance of a position of interest to all known pathogenic variants minus the average distance to all known benign variants. Thus, positions that are closer on average to pathogenic variants receive positive scores, and those that are closer to benign receive negative scores.
AUTHOR CONTRIBUTIONS
John Gadbery: Data curation; formal analysis; supervision; writing‐original draft; writing‐review and editing. Abin Abraham: Data curation; formal analysis; writing‐original draft; writing‐review and editing. Carli Needle: Formal analysis; visualization. Chris Moth: Methodology; validation. Jonathan Sheehan: Methodology; supervision; validation. John Capra: Conceptualization; resources; supervision; writing‐original draft; writing‐review and editing. Lauren Jackson: Conceptualization; funding acquisition; supervision; writing‐original draft; writing‐review and editing.
Supporting information
Figure S1 AP‐4 closed core and electrostatic surface representations of AP‐4 homology model, Related to Figure 1. (A, B) AP‐4 in its closed conformation. (C, D) Side views of the electrostatic surface of AP‐4 in its open conformation. Blue residues are more basic while red residues are more acidic. Views are equivalent to Figure 1A. (E) Top‐down view of AP‐4 core in its open conformation, as viewed down through the membrane. (F) Bottom‐up view of AP‐4 core. Overall, AP‐4 core appears to be a very acidic protein.
Figure S2. AP‐4 core surface conservation, Related to Figure 2. Residues are colored from cyan to dark magenta based on conservation scores calculated using ConSurf. AP‐4 core with conservation scores mapped to residues are shown for ε on the left and β4 on the right; C‐μ4 is shown as gray ribbons. Many surface‐exposed residues in both large subunits are conserved across vertebrates. Views are rotated 90° relative to Figure 2a,b.
Figure S3. Confidence measures of the PathProx calculation, Related to Figure 4. Leave‐one‐out cross‐validation was performed with 11 pathogenic and 699 benign variants to evaluate the constraint values of the spatial distributions across the modeled structure. The ROC (Receiver Operating Curve) on the left shows that the disease‐associated (Pathogenic) variants display more significant 3D clustering than the putative benign (Neutral) variants. The PR (Precision‐Recall) curve on the right also shows that the model has partial predictive power, although the low AUC of 0.18 for the Pathogenic Constraint is due to the small number of pathogenic variants available to map to the structure.
Figure S4. AP‐4 subunits have strong evidence of purifying selection. (a‐d) Top four figures quantify the difference between nonsynonymous (dN) and synonymous (dS) substitution rates (dN‐dS, y‐axis) at each amino acid position (x‐axis) for the AP‐4 subunits. Sites that differ significantly from dN‐dS = 0 (p < .05, Methods) are colored in red. The bottom four figures (e‐h) display the distribution of dN‐dS values for the AP‐4 subunits. A majority of amino acid sites across AP‐4 subunits have evidence of purifying selection (dN‐dS < 0). Two sites in AP4E1 (204 and 207) have dN estimates significantly greater than dS, which suggests positive selection.
Figure S5. Yxxφ and YKFFE binding sites mapped on AP‐4 open core. (A, B) Two views of AP‐4 core in its open conformation, with putative binding sites for Yxxφ (orange) and YKFFE (yellow) motifs (C, D) Close‐up ribbon (C) and electrostatic surface (D) views of the β4/C‐μ4 interface with YKFFE peptide in yellow (PDB: 3L81). The binding pocket for the YKFFE peptide (yellow) is occluded by residues from β4 in the open conformation.
Table S1 Supplement information
ACKNOWLEDGMENTS
J. G., A. A., C. N., C. M., and J. S. built structural homology models and ran computational analyses. The manuscript was written by J. G., A. A., J. A. C., and L. P. J. with input from all authors. J. A. C. and L. P. J. conceived the project. J. G., C. N., and L. P. J. are supported by NIH R35GM119525. L. P. J. is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts. J. A. C. was supported by NIH R35GM127087. A. A. was supported by NIH T32GM007347. This work was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University. We thank Dr. Abigail L. LaBella for sharing her calculations of XP‐EHH selection measures. We also thank Dr. Jennifer Hirst for sharing her expertise in identifying and compiling patient mutations. The authors declare no competing conflicts of interest.
Gadbery JE, Abraham A, Needle CD, et al. Integrating structural and evolutionary data to interpret variation and pathogenicity in adapter protein complex 4. Protein Science. 2020;29:1535–1549. 10.1002/pro.3870
John E. Gadbery and Abin Abraham contributed equally to this work.
Funding information National Institute of General Medical Sciences, Grant/Award Numbers: R35GM119525, R35GM127087, T32GM007347; Pew Charitable Trusts, Grant/Award Number: Pew Scholars Award
REFERENCES
- 1. Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. [DOI] [PubMed] [Google Scholar]
- 2. Hirst J, Irving C, Borner GH. Adaptor protein complexes AP‐4 and AP‐5: New players in endosomal trafficking and progressive spastic paraplegia. Traffic. 2013;14:153–164. [DOI] [PubMed] [Google Scholar]
- 3. Robinson MS, Bonifacino JS. Adaptor‐related proteins. Curr Opin Cell Biol. 2001;13:444–453. [DOI] [PubMed] [Google Scholar]
- 4. Robinson MS. Forty years of clathrin‐coated vesicles. Traffic. 2015;16:1210–1238. [DOI] [PubMed] [Google Scholar]
- 5. Boehm M, Aguilar RC, Bonifacino JS. Functional and physical interactions of the adaptor protein complex AP‐4 with ADP‐ribosylation factors (ARFs). EMBO J. 2001;20:6265–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barlow LD, Dacks JB, Wideman JG. From all to (nearly) none: Tracing adaptin evolution in fungi. Cell Logist. 2014;4:e28114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davies AK, Itzhak DN, Edgar JR, et al. AP‐4 vesicles contribute to spatial control of autophagy via RUSC‐dependent peripheral delivery of ATG9A. Nat Commun. 2018;9:3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mattera R, Park SY, De Pace R, Guardia CM, Bonifacino JS. AP‐4 mediates export of ATG9A from the trans‐Golgi network to promote autophagosome formation. Proc Natl Acad Sci U S A. 2017;114:E10697–E10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abou Jamra R, Philippe O, Raas‐Rothschild A, et al. Adaptor protein complex 4 deficiency causes severe autosomal‐recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 2011;88:788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdollahpour H, Alawi M, Kortum F, et al. An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP‐4 deficiency syndrome. Eur J Hum Genet. 2015;23:256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hardies K, May P, Djemie T, et al. Recessive loss‐of‐function mutations in AP4S1 cause mild fever‐sensitive seizures, developmental delay and spastic paraplegia through loss of AP‐4 complex assembly. Hum Mol Genet. 2015;24:2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tuysuz B, Bilguvar K, Kocer N, et al. Autosomal recessive spastic tetraplegia caused by AP4M1 and AP4B1 gene mutation: Expansion of the facial and neuroimaging features. Am J Med Genet A. 2014;164:1677–1685. [DOI] [PubMed] [Google Scholar]
- 13. Lamichhane DM. New AP4B1 mutation in an African–American child associated with intellectual disability. J Pediatr Genet. 2013;2:191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ebrahimi‐Fakhari D, Cheng C, Dies K, et al. Clinical and genetic characterization of AP4B1‐associated SPG47. Am J Med Genet A. 2018;176:311–318. [DOI] [PubMed] [Google Scholar]
- 15. Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. [DOI] [PubMed] [Google Scholar]
- 16. Bettencourt C, Salpietro V, Efthymiou S, et al. Genotype‐phenotype correlations and expansion of the molecular spectrum of AP4M1‐related hereditary spastic paraplegia. Orphanet J Rare Dis. 2017;12:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ebrahimi‐Fakhari D, Behne R, Davies AK, Hirst J. AP‐4‐associated hereditary spastic paraplegia In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle, Washington: University of Washington, 2018. [PubMed] [Google Scholar]
- 18. Teinert J, Behne R, Wimmer M, Ebrahimi‐Fakhari D. Novel insights into the clinical and molecular spectrum of congenital disorders of autophagy. J Inherit Metab Dis. 2019;43:51–62. [DOI] [PubMed] [Google Scholar]
- 19. Burgos PV, Mardones GA, Rojas AL, et al. Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP‐4 complex. Dev Cell. 2010;18:425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ashkenazy H, Abadi S, Martz E, et al. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Borner GH, Antrobus R, Hirst J, et al. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol. 2012;197:141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paolini L, Radeghieri A, Civini S, Caimi L, Ricotta D. The epsilon hinge‐ear region regulates membrane localization of the AP‐4 complex. Traffic. 2011;12:1604–1619. [DOI] [PubMed] [Google Scholar]
- 24. Frazier MN, Davies AK, Voehler M, et al. Molecular basis for the interaction between AP4 beta4 and its accessory protein, tepsin. Traffic. 2016;17:400–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mattera R, Guardia CM, Sidhu SS, Bonifacino JS. Bivalent motif‐ear interactions mediate the association of the accessory protein tepsin with the AP‐4 adaptor complex. J Biol Chem. 2015;290:30736–30749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olesen LE, Ford MG, Schmid EM, et al. Solitary and repetitive binding motifs for the AP2 complex alpha‐appendage in amphiphysin and other accessory proteins. J Biol Chem. 2008;283:5099–5109. [DOI] [PubMed] [Google Scholar]
- 27. Ross BH, Lin Y, Corales EA, Burgos PV, Mardones GA. Structural and functional characterization of cargo‐binding sites on the mu4‐subunit of adaptor protein complex 4. PLoS One. 2014;9:e88147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 2002;109:523–535. [DOI] [PubMed] [Google Scholar]
- 29. Kelly BT, McCoy AJ, Spate K, et al. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature. 2008;456:976–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Genomes Project C , Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer‐Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce‐Hoffman E, Zappala Z, O'Donnell‐Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano‐Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv, 531210.
- 32. Landrum MJ, Lee JM, Benson M, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutat. 2018;39:1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sivley RM, Dou X, Meiler J, Bush WS, Capra JA. Comprehensive analysis of constraint on the spatial distribution of missense variants in human protein structures. Am J Hum Genet. 2018;102:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sivley RM, Sheehan JH, Kropski JA, et al. Three‐dimensional spatial analysis of missense variants in RTEL1 identifies pathogenic variants in patients with familial interstitial pneumonia. BMC Bioinf. 2018;19:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Behne R, Teinert J, Wimmer M, et al. Adaptor protein complex 4 deficiency: A paradigm of childhood‐onset hereditary spastic paraplegia caused by defective protein trafficking. Hum Mol Genet. 2020;29:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bustamante CD, Fledel‐Alon A, Williamson S, et al. Natural selection on protein‐coding genes in the human genome. Nature. 2005;437:1153–1157. [DOI] [PubMed] [Google Scholar]
- 38. Robinson MS. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. [DOI] [PubMed] [Google Scholar]
- 39. Traub LM, Bonifacino JS. Cargo recognition in clathrin‐mediated endocytosis. Cold Spring Harb Perspect Biol. 2013;5:a016790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pryor PR, Mullock BM, Bright NA, et al. Combinatorial SNARE complexes with VAMP7 or VAMP8 define different late endocytic fusion events. EMBO Rep. 2004;5:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jackson LP, Kelly BT, McCoy AJ, et al. A large‐scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell. 2010;141:1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aguilar RC, Boehm M, Gorshkova I, et al. Signal‐binding specificity of the mu4 subunit of the adaptor protein complex AP‐4. J Biol Chem. 2001;276:13145–13152. [DOI] [PubMed] [Google Scholar]
- 43. Canagarajah BJ, Ren X, Bonifacino JS, Hurley JH. The clathrin adaptor complexes as a paradigm for membrane‐associated allostery. Protein Sci. 2013;22:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu X, Breitman M, Goldberg J. A structure‐based mechanism for Arf1‐dependent recruitment of coatomer to membranes. Cell. 2012;148:530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fumagalli M, Camus SM, Diekmann Y, et al. Genetic diversity of CHC22 clathrin impacts its function in glucose metabolism. Elife. 2019;8:e41517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lemoine F, Correia D, Lefort V, et al. NGPhylogeny.fr: New generation phylogenetic services for non‐specialists. Nucleic Acids Res. 2019;47:W260–W265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suyama M, Torrents D, Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kosakovsky Pond SL, Frost SD. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–1222. [DOI] [PubMed] [Google Scholar]
- 50. Pond SL, Frost SD, Muse SV. HyPhy: Hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. [DOI] [PubMed] [Google Scholar]
- 51. Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 2018;35:773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abigail L, LaBella AA, Pichkar Y, Fong SL, Zhang G, Muglia LJ, Abbot P, Rokas A, Capra JA (2019) Accounting for diverse evolutionary forces reveals the mosaic nature of selection on genomic regions associated with human preterm birth. bioRxiv. doi: 10.1101/816827 [DOI] [PMC free article] [PubMed]
- 53. Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pers TH, Timshel P, Hirschhorn JN. SNPsnap: A web‐based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gautier M, Klassmann A, Vitalis R. Rehh 2.0: A reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol Ecol Resour. 2017;17:78–90. [DOI] [PubMed] [Google Scholar]
- 57. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 AP‐4 closed core and electrostatic surface representations of AP‐4 homology model, Related to Figure 1. (A, B) AP‐4 in its closed conformation. (C, D) Side views of the electrostatic surface of AP‐4 in its open conformation. Blue residues are more basic while red residues are more acidic. Views are equivalent to Figure 1A. (E) Top‐down view of AP‐4 core in its open conformation, as viewed down through the membrane. (F) Bottom‐up view of AP‐4 core. Overall, AP‐4 core appears to be a very acidic protein.
Figure S2. AP‐4 core surface conservation, Related to Figure 2. Residues are colored from cyan to dark magenta based on conservation scores calculated using ConSurf. AP‐4 core with conservation scores mapped to residues are shown for ε on the left and β4 on the right; C‐μ4 is shown as gray ribbons. Many surface‐exposed residues in both large subunits are conserved across vertebrates. Views are rotated 90° relative to Figure 2a,b.
Figure S3. Confidence measures of the PathProx calculation, Related to Figure 4. Leave‐one‐out cross‐validation was performed with 11 pathogenic and 699 benign variants to evaluate the constraint values of the spatial distributions across the modeled structure. The ROC (Receiver Operating Curve) on the left shows that the disease‐associated (Pathogenic) variants display more significant 3D clustering than the putative benign (Neutral) variants. The PR (Precision‐Recall) curve on the right also shows that the model has partial predictive power, although the low AUC of 0.18 for the Pathogenic Constraint is due to the small number of pathogenic variants available to map to the structure.
Figure S4. AP‐4 subunits have strong evidence of purifying selection. (a‐d) Top four figures quantify the difference between nonsynonymous (dN) and synonymous (dS) substitution rates (dN‐dS, y‐axis) at each amino acid position (x‐axis) for the AP‐4 subunits. Sites that differ significantly from dN‐dS = 0 (p < .05, Methods) are colored in red. The bottom four figures (e‐h) display the distribution of dN‐dS values for the AP‐4 subunits. A majority of amino acid sites across AP‐4 subunits have evidence of purifying selection (dN‐dS < 0). Two sites in AP4E1 (204 and 207) have dN estimates significantly greater than dS, which suggests positive selection.
Figure S5. Yxxφ and YKFFE binding sites mapped on AP‐4 open core. (A, B) Two views of AP‐4 core in its open conformation, with putative binding sites for Yxxφ (orange) and YKFFE (yellow) motifs (C, D) Close‐up ribbon (C) and electrostatic surface (D) views of the β4/C‐μ4 interface with YKFFE peptide in yellow (PDB: 3L81). The binding pocket for the YKFFE peptide (yellow) is occluded by residues from β4 in the open conformation.
Table S1 Supplement information