

Abstract
The orchestration of mitochondria within the cell represents a critical aspect of cell biology. At the center of this process is the outer mitochondrial membrane protein, Miro. Miro coordinates diverse cellular processes by regulating connections between organelles and the cytoskeleton that range from mediating contacts between the endoplasmic reticulum and mitochondria to the regulation of both actin and microtubule motor proteins. Recently, a number of cell biological, biochemical, and protein structure studies have helped to characterize the myriad roles played by Miro. In addition to answering questions regarding Miro's function, these studies have opened the door to new avenues in the study of Miro in the cell. This review will focus on summarizing recent findings for Miro's structure, function, and activity while highlighting key questions that remain unanswered.
Keywords: GTPase, microtubule transport, Miro, mitochondria, neurodegeneration, organelle contact sites
Abbreviations
- cGTPase
C‐terminal GTPase domain of Miro
- ER
endoplasmic reticulum
- ERMES
ER‐mitochondria encounter structure
- ERMCS
ER‐mitochondria contact site
- GAP
GTPase activating protein
- GEF
GTPase exchange factor
- HDAC6
histone deacetylase 6
- iPSCs
induced pluripotent stem cells
- LRRK2
leucine‐rich repeat kinase 2
- MICOS
mitochondrial cristae organizing structure
- MCU
mitochondria calcium uniporter
- Miro
mitochondrial Rho GTPases
- nGTPase
N‐terminal GTPase domain of Miro
- OMM
outer‐mitochondrial membrane
1. INTRODUCTION
Mitochondria represent a key component of the cell, sitting at the interface of cellular metabolism, energy production, and cell death. These diverse functions of mitochondria require a host of proteins that function within mitochondria as well as on the outer mitochondrial membrane. Continued proteomic efforts have mapped 1,000+ components to the mitochondria,1, 2 despite the fact that the mitochondrial genome only encodes for 13 genes. This indicates that a significant percentage of cellular factors participate in mitochondrial biology.
Over the past twenty years, a growing body of evidence has implicated the mitochondrial Rho GTPase (Miro) family of proteins as regulators of mitochondrial cellular function. Originally classified as a member of the Rho family, human Miro1 and Miro23 (RHOT1 and RHOT2, respectively) are now considered to be founding members of its own family. The Miro family are atypical GTPases within the Ras superfamily, composed of two GTPase domains flanking two EF‐hand motifs (Figure 1a). These domains indicate that the Miro family likely exploits allostery in order to regulate cellular processes. Indeed, over the course of 20 years, numerous studies have implicated Miro activity as playing important roles through mutagenesis of these domains.
Figure 1.
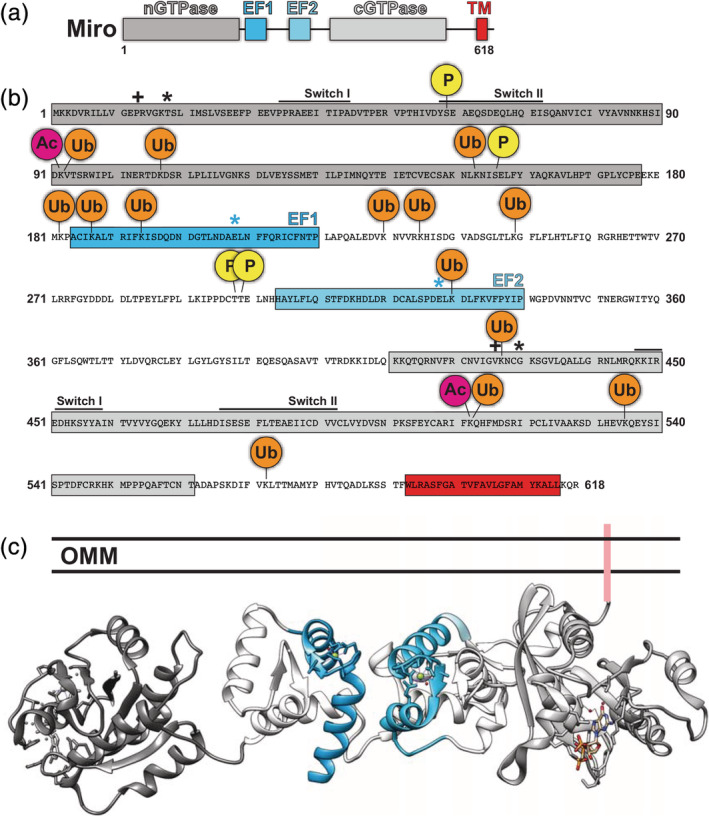
Structural features of Miro. (a) Domain organization. (b) Annotated primary sequence of Miro. Black “+” indicates constitutively active GTP mutant whereas “*” indicates constitutively inactive GTP mutant. Blue “*” indicate mutations in EF‐hands that block calcium binding. (c) Model of Miro structure based on published coordinates of Miro's nGTPase (PDB: 6D71) and the C‐terminus of Miro (PDB: 5KSZ)
In this review, we will highlight recent publications that connect the Miro family to diverse cellular processes. Considering that the majority of studies on the Miro family have focused on Miro1, the term “Miro” will refer to Miro1 unless otherwise noted. We will begin with a discussion about the role that Miro family members play with inter‐organelle contacts as they stitch together a number of cell biology processes. Then, we will narrow our focus onto how Miro family members facilitate cytoskeletal trafficking with a particular emphasis on recent biochemical and molecular characterizations.
2. MIRO: AN ATYPICAL GTPASE
Members of the Miro family are single‐pass, Type IV transmembrane proteins, consisting of two GTPase domains flanking two calcium‐binding domains (EF‐hands) with a C‐terminal tail‐anchored in the mitochondrial outer membrane3, 4, 5 (Figure 1a). Although the sequences of the GTPases and EF‐hand motifs vary, the overall domain architecture of Miro is conserved throughout single‐celled eukaryotes to plants and animals.
A hallmark feature of Miro is the presence of two GTPase domains (Figure 1a). Typically, small GTPases like Miro act as molecular switches and cycle between two states: an active, GTP‐bound state and an inactive, GDP‐bound state.6 Like other Ras family G‐proteins, G1, G4, and G5 loop sequence motifs are conserved in Miro.7 However, Miro GTPases vary from other members of the Ras superfamily in the Switch I/II regions. In general, Switch I regions are involved in the stabilization of nucleotide, whereas Switch II is involved in the regulation of nucleotide hydrolysis. The Switch regions for both the C‐terminal (cGTPase) and N‐terminal (nGTPase) domains of Miro1 are shown in Figure 1b. For Miro2, these regions vary slightly in the C‐terminal GTPase domain, with Switch I corresponding to residues 446–455 and Switch II comprising residues 472–489.7 Typically, in the GTP‐bound state, the surface loops are known as Switch I (G2) and Switch II (G3) and exist in close proximity to one another.8, 9 However, the Rho‐conserved DxxG Switch II motif is absent from Miro1.10 Other differences in these regions are variations in the key catalytic residues of Ras (Y32, T35, G60, and Q61).7 Together, current research demonstrates that Miro is a distinct protein, with unique GTPase domains.
As a small G‐protein with two GTPases, both GTPase domains may act as potential regulators of Miro activity. In order to dissect the role of the GTP state of Miro, characteristic mutations are typically introduced into Miro's nGTPase or cGTPase in order to stabilize Miro in the GTP or GDP form. For the nGTPase, GTP is stabilized via the mutation P13V and whereas the GDP form is promoted by T18N3, 11, 12 (Figure 1b). For the cGTPase, the GTP and GDP‐locked forms correspond to V425/V427 and N430/N432 for Miro1/2, respectively3 (Figure 1b0. While some authors3 have described the N432 mutant in Miro2 to be GDP‐locked, others have described this mutant to behave similarly to wild‐type Miro.13
While a full‐length structure of Miro has yet to be determined, there are two separate structural studies on Miro comprising a C‐terminal fragment (MiroS: EF1‐EF2‐cGTPase)5, 8 and a recent structure of the nGTPase9 (Figure 1c). Previous work on MiroS indicated (a) the EF‐hands form a tightly packed interaction with the cGTPase and (b) Ca2+ binding to the EF‐hands did not cause dramatic structural changes. Moreover, in the structure of MiroS, Miro's cGTPase was found to be bound to GDP, GDP‐Pi, or GMPPCP in conjunction with either Ca2+ or Mg2+,5, 8 indicating that both EF‐hands and the cGTPase can engage ligands simultaneously.
Recently, the structure of the isolated nGTPase was determined using X‐ray crystallography.9 Surprisingly, despite the absence of nucleotide during protein purification and crystallization, the nGTPase crystallized with GTP bound to the active site.9 This suggests that the nGTPase alone may not be sufficient for GTP hydrolysis. Interestingly, both nGTPase and cGTPase structures both showed the presence of dimers within the asymmetric unit of the structure, which suggests that Miro may be able to exist in higher‐order structures. However, size‐exclusion chromatography and small‐angle X‐ray scattering indicated that these constructs were monomers in solution, allowing the authors to conclude that Miro does not dimerize in solution. Finally, recent small‐angle X‐ray scattering studies suggest that the nGTPase may be loosely tethered to the MiroS.9 This suggests that the nGTPase may exist in multiple conformations or that the nGTPase is capable of interacting with MiroS.
Throughout the review, the structure of Miro will serve as a framework with which to understand the diverse cellular activities. This will involve mutations that alter the GTP state, phosphorylation sites, and calcium binding. To help the reader, we have constructed an in silico model of Miro based on superimposing Miro's nGTPase9 with the EF1‐EF2‐cGTPase structure5 (Figure 1c).
3. MIRO FACILITATES INTER‐ORGANELLE INTERACTIONS WITH CYTOPLASMIC MACHINERY
Before discussing Miro's well‐known role in cytoskeletal transport of mitochondria, we will first discuss the role that Miro plays in facilitating mitochondrial interactions with other cellular compartments. To date, Miro has been implicated in mediating contact and interaction with the endoplasmic reticulum (ER), peroxisomal movement, and mitochondrial shape transition necessary for mitophagy and mitochondrial quality control. In this section, we will summarize recent literature exploring Miro's role as a facilitator of mitochondrial contact with other organelles.
3.1. Miro at ER–mitochondria contact sites
Contact between the mitochondria and the ER was discovered over half a century ago.14 The interaction of these organelles is of such frequency that contact sites can be physically isolated as mitochondria‐associated membranes.15 A robust body of research16, 17, 18, 19, 20 has developed around the structure and function of this contact where both calcium and phospholipids are exchanged. Recent evidence indicates that Miro plays a critical role in facilitating and regulating this interaction.
Before discussing the regulation of ER–mitochondrial contact sites by Miro, we will first discuss the composition of this interaction. Many proteins localize to the area of contact between ER and mitochondria, forming a physical bridge (Figure 2). This interaction is known as the ER–mitochondria encounter structure (ERMES) in yeast and as the ER–mitochondria contact site (ERMCS) in mammals.21, 22 The partners involved in the connection differ depending on the process being engaged. The less complex organism is perhaps a better‐suited place to examine the basic functionality of the complex.
Figure 2.
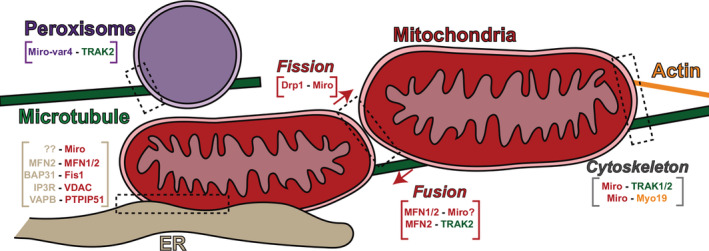
Miro mediates organelle–cytoskeleton and organelle–organelle contact sites. Miro contributes to diverse cellular activities involved in organelle–cytoskeleton contacts (peroxisome–microtubule, mitochondria–microtubule, & mitochondria–actin) as well as organelle–organelle contacts (ER–mitochondria & mitochondria–mitochondria)
In yeast, Gem1p is an OMM protein within the Miro family that is responsible for mitochondrial inheritance and is required for regulation of the ERMES complex. Gem1p was first identified by its homology to human Miro and characterized as a factor recognizing cell‐signaling events to affect mitochondrial dynamics.23 Deletion strains of gem1 had difficulty growing on non‐fermentable carbon source media and suffered from anomalous mitochondrial morphology and inheritance but maintained cristae organization. Its functional correlation to Miro was further demonstrated by mutations to disrupt GTPase activity, demonstrating their necessity for mitochondrial inheritance.24 This was also the first study to demonstrate the GTPase activity of a Miro homolog in vitro and determined the impact of active site mutations on hydrolase activity.
Subsequent studies determined that Gem1p regulates the formation of the ERMES complex. This was verified by colocalization of a recombinant fluorescent Gem1p with another ERMES component at the ER‐mitochondria interface in addition to mass‐spectrometry‐based approaches.25, 26 Additionally, in gem1 deletion strains, there is reduced formation of the ERMES complex, indicating that Gem1p facilities ERMES formation. Further, mutations affecting GTP binding and hydrolysis decreased localization of exogenous fluorescently‐labeled Gem1p to ERMES foci.25 Mutation of the first EF‐hand to impede calcium‐binding mimicked the distribution of the hydrolysis mutant. Similar mutations in the cGTPase domain did not affect localization but failed to rescue the synthetic lethality of a deletion mutant deficient in phospholipid exchange. These experiments incorporated mutations in the second EF‐ hand as well, but the phenotypes were indistinguishable from wild type and respective mutant backgrounds. This work establishes that the nGTPase and EF‐hand 1 play important roles in ERMES complex formation, suggesting that allostery from these domains contributes to Gem1p function.
Gem1p's role in mitochondrial dynamics was expanded with tomography.27 ERMES components and resident ER and mitochondria components were modified with fluorescent fusion proteins. Gem1p localized at contact sites where mitochondria were undergoing ER‐associated mitochondrial division, in which the ER tubules form and constrict mitochondrial membranes, contributing to the creation of “daughter” mitochondria.28 Once divided, the Gem1p proteins were found to be clustered at one tip after segregation. Taken together, the yeast version of Miro appears to be involved in directing the function of ERMES, instructing ER components to share membrane components, initiate fission, or perform other roles in ER–mitochondria interaction.
Mammalian Miro retains this role but has a broader array of potential regulatory targets in the ERMCS. ER–outer mitochondrial membrane (OMM) partners include PS2‐MFN2, BAP31‐Fis1, IP3R‐VDAC, VAPB‐PTPIP51, and ORP5/8‐PTPIP5129 (Figure 2). Some clearly function in biochemical regulation. For instance, the high concentration of calcium in the ER lumen is shuttled out through the inositol 1,4,5‐trisphosphate receptor (IP3R)30 and accepted by the OMM voltage‐dependent anion‐selective channel.31 ORP5/8 facilitates the metabolism of phospholipids by exchanging lipid transfer proteins.32, 33 BAP31‐Fis134 and PACS‐235 have been implicated in pro‐apoptotic pathways36 while others have currently unelucidated functions.
Many studies have examined the components and functional linkage of ERMCS to regulatory pathways, but how and according to what signal they form has only recently come under scrutiny. The earliest observation of ERMCS disruption in a multicellular eukaryote came from an examination of neural stem cells from Drosophila bearing a protein‐null mutant allele of Miro.4 Homozygous crosses of dMiro B682 suffered greatly reduced brain size and lacked the Deadpan marker that accompanies brain development.22 The authors manipulated protein levels using Gal4 induction to suppress or overexpress dMiro, manipulate other genes involved in calcium homeostasis for complementation experiments, and employed fluorescent probes for calcium ions to investigate the cause of the phenotype. They found that defects arising from alterations to dMiro expression could be partially or fully rescued with complements of ER‐resident calcium channels and receptors. They further demonstrated that dMiro was specifically phosphorylated at conserved residue S66 (S59 in human) by Polo kinase and that this modification resulted in the accumulation of dMiro at the ER surface. Co‐immunoprecipitation revealed that phospho‐S66 dMiro associated with VDAC/Porin and Mitofusin/Marf, components of the ERMCS. Further, S66 phosphorylation enhanced the activity of the N‐terminal GTPase domain, implying that the GTP hydrolysis activity of Miro plays an active role in recruiting Miro to the ERMCS. Thus, these results indicate that Miro plays a critical role in supporting the formation of the ERMCS.
Lee et al. followed this study by investigating the effect of the Miro‐ERMCS interaction on calcium flux in a Parkinson's disease model.37 They examined dopaminergic neurons bearing mutations in PINK1, the kinase responsible for priming Miro for degradation or bearing transgenes for overexpression or knockdown of Miro. When Miro was upregulated, either by removal of PINK1 or Gal4 overexpression, the calcium concentration in mitochondria increased. This correlated with increased mitochondria size and eventual cell death. Removing Miro by knockdown attenuated the effect in the PINK1 mutant, bringing mitochondrial calcium levels close to wild type. Further, knockdown of genes involved in calcium transfer within the ERMCS attenuated the calcium elevation from Miro overexpression, as did inhibition of IP3R and the mitochondrial calcium uniporter with small molecules. Likewise, knockdown of Polo rescued neuron loss from Miro overexpression, while overexpression of Polo exacerbated neuron loss except when Miro was knocked down or mutated to a phospho‐null state (dMiro S66A). These results heavily support a role for Miro in directing calcium influx through the ERMCS into mitochondria.
Recently, a pair of heterozygous mutations in Miro were identified in a cohort of Parkinson's patients.38 Patient fibroblasts bearing R272Q and R450C mutations were treated with thapsigargin, which depletes ER calcium into the cytosol. Control cells were able to re‐establish normal cytosolic calcium levels, but calcium levels remained high in the mutant cells. Simultaneous blockade of the mitochondria calcium uniporter (MCU) resulted in elevated cytosolic calcium in both mutant and wild‐type cells, implying a defect in mitochondrial calcium uptake or recycling to the ER. Imaging with tracking dyes revealed an overall decrease in ERMCS, which in turn induced a reduction in the ER area and an increase in mitochondrial clearance by LC3‐independent autophagy.
Miro's role in ERMCS was further characterized using super‐resolution microscopy techniques.39 Double knockout of Miro1 and Miro2 in mouse fibroblasts caused a decrease in ER–mitochondria overlap and distinct changes to mitochondrial morphology. They found that the absence of Miro causes a shift toward short, round mitochondria that partially lacked normal cristae architecture, while reintroduction returned morphology to longer, thinner mitochondria with normal, contiguous cristae. Immunoprecipitation of Miro identified interactions with components of the mitochondrial cristae organizing structure (MICOS). Microscopic analysis confirmed a positive correlation for co‐localization of outer‐membrane Miro and inner‐membrane MICOS, with Miro forming clusters containing both Miro‐1 and Miro‐2. In contrast, double knockout of Miro caused the dispersal of MICOS components throughout the inner membrane, with occasional areas completely devoid of the subject species.
Miro's presence at the ERMCS and its apparent regulatory partners have been identified, but how Miro's recognition of its environment alters its function to regulate the activities involved remains unclear. Miro's calcium‐binding EF‐hands are almost certainly involved in environmental sensing, but how does the molecule react to this information to translate it into action? Are the GTPase domains involved at all? How does phosphorylation by Polo initiate the process? The molecular details may hold the keys to decipher the regulatory timing and decode its role in disease states.
3.2. Miro as a microtubule motor protein receptor for peroxisomes
Peroxisomes are “multipurpose organelles” whose contents are diverse, varying between organisms but also between tissues or cells of single organisms.40 Their contents are frequently toxic to the cell at large, consisting of metabolic enzymes required for redox equilibrium, reactive oxygen species neutralization, and bile acid synthesis, as well as signaling proteins modulating inflammation and innate immune signaling, among others.41 Like mitochondria, peroxisomes carry critical functions that must be distributed throughout the cell. Indeed, peroxisomes can assist in regulating the redox environment around mitochondria, so the two organelles often need to be in proximity for proper function.
It is, therefore, not surprising that both peroxisomes and mitochondria utilize a similar long‐range Miro‐based transport mechanism. Miro has recently been identified as a component of peroxisomal trafficking.42 Specifically, a splicing variant of Miro1 (named Miro1‐var4) contains an insert between the C‐terminal GTPase domain and membrane anchor that is recognized by the cytosolic receptor Pex19p for post‐translational targeting to the peroxisomal membrane.43 When the C‐terminal membrane anchor was replaced with a peroxisome‐targeted sequence, the resulting recombinant expression in COS‐7 cells demonstrated differential peroxisome migration.44 Recombinant peroxisome‐targeted Miro bearing a constitutively active N‐terminal GTPase domain caused accumulation of peroxisomes at the cell periphery, implying the activated GTPase adopts a preference for kinesin‐driven movement. On the other hand, a peroxisome‐targeted dominant‐negative N‐terminal GTPase mutant scattered the peroxisomes throughout the cytoplasm. Expression of the wild‐type peroxisome‐targeted construct in healthy and diseased patient‐derived fibroblasts caused proliferation rather than the accumulation of peroxisomes. This points to a potential role in which Miro employs the force contributed by bound microtubule motors to exert pull forces that are likely to be at least partially responsible for organelle movement or dynamics.
The role of Miro in peroxisome transport has been supported by another recent study,45 however, in this work, Miro was implicated in peroxisome fission. In fibroblasts bearing knockout mutations of Miro1, Miro2, and double knockouts, they observed the same number of long‐range peroxisomal transport events as wild‐type cells. However, in the Miro knockout cells, the authors observed a decreased peroxisomal size. From this, they conclude that Miro's primary role in the context of the peroxisome is to negatively regulate Drp1 recruitment, inhibiting fission. This indicates that Miro may play multiple roles on peroxisomal surfaces, facilitating transport or regulating fission, or both roles simultaneously. Further work will be needed to arrive at a coherent model of Miro's role in peroxisome dynamics.
The mechanism of microtubule attachment through Miro occurs via Pex1 and KIFC346 (Figure 1), which is in contrast to axonal mitochondria that utilizes TRAK1 and KIF5B.47 Interestingly, the retrograde transport machinery for Miro1‐var4 is shared with mitochondria given that TRAK1, TRAK2, and dynein/dynactin are required for both peroxisome and mitochondrial transport.48 Importantly, this process appears to be distinct from a hitch‐hiking model of peroxisome transport, whereby peroxisomes are tethered to endosomes via Pex1 to facilitate transport.49 Peroxisomes can also bind directly to microtubules via Pex14,50 an essential component of peroxisome import machinery. Thus, these data indicate that peroxisomes utilize a similar mode of microtubule‐based transport as mitochondria.
While it would be reasonable to assume that the mode of transport of the two organelles is similar, the molecular details have not been determined. The Miro variant study did not address whether the peroxisomal movement is arrested by calcium influx, like mitochondrial movement.51 Also, there are several components in mitochondria implicated in motor adapter attachment, including mitofusin52 and MICOS,39 which are absent from peroxisomes. How similar these processes are and the extent to which Miro is involved remain open for study.
4. MIRO COORDINATES MITOCHONDRIAL TRANSPORT ON BOTH MICROTUBULE AND ACTIN CYTOSKELETON
In order to maintain the health of neurons, mitochondria must be transported and positioned throughout neuronal bodies. As the required distances can reach up to 1 m in length, the creation, transport, and positioning of mitochondria is a formidable task for the neuron.
The bi‐directional transport of mitochondria along microtubules is well established, considering that the first evidence of mitochondrial transport occurred nearly 30 years ago.53 Since its discovery, the key players and pieces of the regulatory mechanism have been characterized. As a C‐terminally anchored OMM protein, Miro plays a key role as the receptor onto which microtubule motor proteins assemble4, 11 (Figure 3a). Miro cooperates with the adaptor protein Milton (Drosophila), also known as TRAK1 and TRAK2 in mammals, to recruit kinesin and dynein to drive anterograde and retrograde mitochondrial transport3, 11, 47, 48, 54, 55, 56 (Figures 3b and 2c). Based on mouse knockout studies, Miro1 plays a dominant role in both axonal and dendritic transport, as loss of Miro1 leads to a dramatic reduction in mitochondria motility.57 As discussed below, even though Miro2 does play a role in connecting mitochondria to microtubules,52 there are multiple pathways that connect mitochondria to microtubules considering that Miro1‐Miro2 double knockout neurons still have axonal mitochondria (albeit with reduced velocity).52
Figure 3.
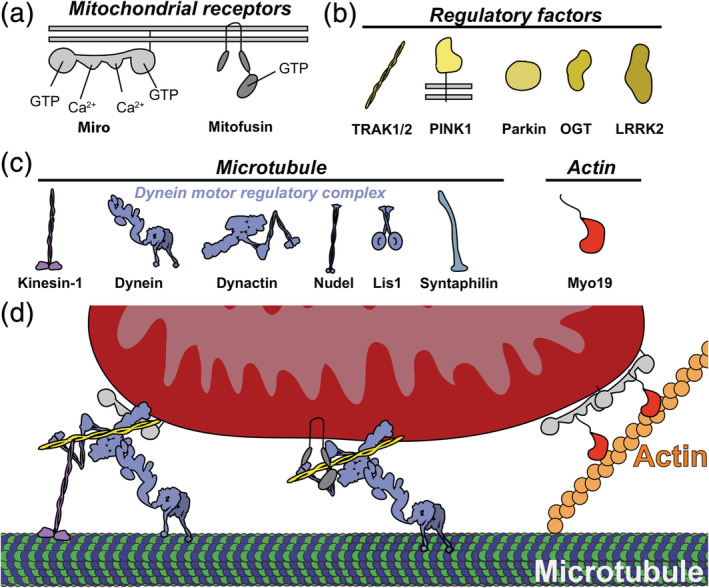
Miro‐directed transport of mitochondria. (a) Receptors localized on outer mitochondrial membrane. (b) Regulatory factors that influence transport on microtubules and actin. (c) Motor protein machinery required for transport. (d) Example receptor‐motor complexes docked onto mitochondria & cytoskeletal elements
Considering that modes of microtubule motor regulation are reviewed elsewhere,56, 58 here we will review some of the latest findings regarding mitochondrial movement patterns, actin‐based transport, and Miro‐independent mitochondrial movement.
4.1. Actin anchors stationary axonal mitochondria
The dynamic of the axonal mitochondrial transport changes as the neuron matures. In primary neuronal cultures, visualization of axonal mitochondrial transport indicates that roughly one‐third of mitochondria are motile, whereas two‐thirds are stationary.53, 59, 60, 61 However, examining the axonal mitochondrial transport in vivo revealed that motility of mitochondria is gradually decreased with neuronal maturation, which coincides with increased localization to presynaptic sites and axonal boutons.62, 63, 64 While previous work indicated that regulatory factors such as syntaphilin could anchor mitochondria in the presence of calcium,65, 66 there were very few tools available with which to probe this phenomenon.
In order to study this process, a new photocleavable small molecule was developed that could be used to artificially dimerize and then cleave two proteins in living cells.67 This molecule—“Zapalog”—induces a physical interaction between two target proteins by rapidly dimerizing recombinant fusions bearing FKBP and DHFR domains. Then, exposure to 405 nm wavelength radiation instantaneously photocleaves zapalog, causing rapid dissociation of the two proteins. Importantly, replenishing zapalog replaces the cleaved molecule to re‐establish dimerization.
By exploiting these properties, the authors utilized zapalog to recruit a highly processive kinesin motor, KIF1a, to the mitochondrial surface in order to manipulate positioning. Consistent with the previous literature,56 ~30% of mitochondria were motile and ~70% were stationary before adding zapalog. Adding zapalog caused the majority (60.3%) of mitochondria to rapidly switch to anterograde movement. After the photocleavage of zapalog, the behavior of the mitochondrial population returned to normal, indicating the endogenous mitochondrial motor complex retained its activity during the zapalog treatment.
A surprising result from this study was that 36% of axonal mitochondria remained stationary after inducing KIF1a recruitment via zapalog.67 This work shows that immovable mitochondria are mostly found at presynaptic sites. Depolymerizing actin filaments reduces the number of stationary mitochondria, indicating actin‐based anchoring plays a role in tethering mitochondria at presynaptic sites.
This work expands the classification of mitochondrial movement status as either “motile” or “stationary” to three categories: motile, immotile but moveable, and immoveable. Immobilization of mitochondria at presynaptic sites ensures the stable production of ATP and calcium buffering capacity68 to meet the local demands of synapses.
4.2. Myo19—A new link between Miro and Actin cytoskeleton
In addition to microtubule‐based motility, actin‐based mechanisms also play a role in the distribution of mitochondria inside cells. In budding yeast, the inheritance of mitochondria by the bud is mediated by the actin‐based motor myosin‐V.69 In human neurons, actin‐based motors may transport mitochondria to regions where microtubules are interrupted or may facilitate mitochondrial anchoring.70
Over ten years ago, Myo19 was implicated in actin‐mediated mitochondrial movement and positioning in human cell lines.71 Human Myo19 is a 970 amino acid length protein consisting of a myosin motor domain, a neck region, and a short tail that is widely expressed in multiple tissues and cell lines.71 Immunostaining and truncation assays demonstrated that Myo19 localizes to mitochondria through its tail domain. Given that overexpression of full‐length Myo19 leads to dramatic increases in the velocity of motile mitochondria, Myo19 acts as an actin‐based motor that facilitates mitochondrial transport.71
After the discovery of Myo19, biochemical studies have started to shed light on its kinetic properties. Myo19 is a plus‐end‐directed,72 actin‐activated ATPase.73 It is able to translocate actin filaments in gliding assays.72, 73 Myo19 motor activity is necessary for mitochondrial motility, as Myo19 containing a mutation impairing ATPase activity did not alter mitochondrial network dynamics.73
The physiological function of Myo19 has been elusive since its discovery. Initial studies revealed that Myo19 connects mitochondria to actin and plays a role in controlling the distribution of mitochondria during cell division, thus ensuring their fair inheritance into the daughter cells.74 Following this observation, additional work found that Myo19 facilitated the formation of starvation‐induced filopodia by positioning mitochondria into the filopodia.75, 76
How is Myo19 tethered to the mitochondria? Originally, Myo19 was thought to act as a peripheral membrane protein,75 interacting with lipids directly. However, recent work discovered that Miro1/2 are receptors for recruiting and stabilizing Myo19 to the mitochondria based on knockout studies of Miro1 and Miro2, in addition to proximity labeling approaches52, 77, 78 (Figure 3c,d). Biochemical studies demonstrated that the Myo19 tail contains both Miro1/2‐independent and Miro1/2‐dependent mitochondrial targeting sites.77, 78 Interestingly, Miro1/2‐mediated recruitment is regulated by the nucleotide state of Miro1/2 N‐terminal GTPase domain.77 Miro bearing a point mutation (T18N) in the N‐terminal GTPase domain failed to recruit Myo19 tail to the mitochondria. These results suggest Miro1/2 plays a critical role in coordinating microtubule‐ and actin‐based mitochondrial movements.
To summarize, Myo19 is a novel myosin that transports mitochondria along the actin filament. How it coordinates (or competes) with microtubule‐based motors remains unclear. Given that Miro functions as a receptor for both TRAK1/2 and Myo19, further studies are needed to understand how Miro preferentially interacts with actin vs. microtubule cytoskeleton.
4.3. Miro‐independent transport of mitochondria along microtubules via Mitofusin‐TRAK1/2
Recently, Miro‐independent transport of mitochondria along microtubules was found in Miro knockout cells for Miro1 and Miro2.52 In this study, the authors explored the ability of mitochondria to undergo directed cytoskeletal transport in the absence of Miro. Considering that knocking out Miro1 in mice is lethal,57 the authors derived mouse embryonic fibroblast cells in order to study mitochondrial transport. When monitoring mitochondrial transport in Miro1 and Miro2 knockout cells, the authors noticed that motor proteins remained associated with mitochondria and that mitochondria underwent short‐range transport. Specifically, kinesin‐1 and dynein/dynactin motor proteins in addition to their TRAK1/2 adaptors remained associated with mitochondria in Miro knockout cells.52 Interestingly, even though long‐range fast axonal transport was inhibited, mitochondria were distributed within axons, indicating that additional mechanisms can connect mitochondria to microtubules.57 Furthermore, co‐expression of TRAK1 or TRAK2 with kinesin‐1 (KIF5C) still induced a dramatic redistribution of mitochondria to the cell periphery,52 suggesting the existence of other TRAK1/2 receptors on the mitochondrial membrane.
How do motor proteins remain attached to mitochondria in Miro knock‐out cells? Previous work indicated that mitofusins (Mfn1 and Mfn2) could be potential TRAK1/2 receptors given that they interact with Miro1/2 and TRAK1/2 in co‐immunoprecipitation assays79 and 3D‐SIM super‐resolution imaging analysis.80 Mitofusins are dynamin family GTPases that mediate mitochondrial fusion by tethering opposing membranes81, 82 (Figure 3a).
Mfn2 can regulate mitochondrial transport directly in neurons. Mitochondria exhibit decreased velocity and increased pausing time in Mfn2 knockout neurons or neurons expressing mutated Mfn2,79 suggesting that Mfn2 is required for mitochondrial transport. On the other side, TRAK1 has been shown to cooperate with Mitofusins to regulate mitochondrial fusion. The depletion of TRAK1 resulted in the fragmentation of mitochondria into small tubules and spheres, with decreased mitochondrial fusion rate.80 Overexpression of TRAK1 caused the appearance of elongated and enlarged mitochondria, a sign of mitochondrial hyperfusion.80 TRAK1 was hypothesized to promote mitochondrial fusion by tethering mitochondria together. In mitofusin knockout cells, mitochondria appear to be fragmented with small globular structures.80, 83 Overexpression of TRAK1 results in extensive clustering of mitochondria in these cells.80 Finally, electron microscopic analysis of mitochondrial structure revealed that the distance between the adjacent mitochondrial outer membrane is about 12 nm,80 which is similar to the reported distance for Mfn1‐mediated mitochondrial tethering.82
4.4. Mitochondrial transport requires acetylation of Miro
Recent work has highlighted a new dimension of Miro regulation by acetylation. In this study, the authors initially noticed that inhibition of HDAC6 (histone deacetylase 6) promoted axonal growth.84 Upon closer examination, the authors determined that when HDAC6 deacetylated Miro, mitochondrial transport was inhibited. This indicated that Miro‐directed mitochondrial transport requires acetylation. Proteomic studies of lysine acetylation previously indicated that sites K92, K512, and K616 are acetylated.85 Expanding on this, Kalinski et al. demonstrated that lysine 105 is critical for transport, although the mode of transport regulation remains to be determined.
4.5. Mitochondrial motor protein “shedding” during mitosis
During mitosis, mitochondria must be inherited equally between mother and daughter cells. To achieve this, mitochondria detach from microtubule motors in order to facilitate mitochondrial inheritance by the daughter cells.86 Motor protein detachment occurs via phosphorylation of motor proteins by the kinases CDK1 and Aurora A. This detachment is critical considering that inappropriate tethering of mitochondria to microtubules during early stages of mitosis leads to binucleate cells and asymmetric mitochondrial inheritance.86 Interestingly, during the later stages of mitosis, mitochondria regain microtubule attachment via Miro to Cenp‐F in order to track the growing ends of microtubules for proper distribution into daughter cells.13, 87, 88 Therefore, the regulation of microtubule attachment by Miro serves as a critical step in the proper inheritance of mitochondria to daughter cells.
5. MIRO: ALLOSTERIC REGULATION WITH WIDE‐SPREAD CELLULAR CONSEQUENCES
Cell biology‐based investigation of Miro's GTP state indicates that specific nucleotide states are required for Miro's cellular activities. For example, when the nGTPase is GDP‐bound, Miro facilitates Drp1‐dependent mitochondrial fission.89 However, when a similar mutation is studied within the context of microtubule‐based transport, nGTPase in the GDP‐bound form leads to a block in both microtubule and actin‐based transport.77, 90 This effect on transport appears to be limited to the nGTPase as GDP‐stabilizing or GTP‐stabilizing mutations to the cGTPase have little to no effect on transport.90 These results indicate that further work is needed to understand how Miro's GTP state connects to its diverse cellular activities.
Miro's GTPase activity has been best characterized in Gem1p, the yeast homolog of human Miro. In Gem1p, both of the GTPase domains were shown to hydrolyze GTP.24 Drosophila Miro was also shown to have GTPase activity,22 however, a full enzymatic analysis was not performed on Drosophila Miro, making it difficult to compare with Gem1p. Recently, both the nGTPase and the cGTPase of human Miro1/Miro2 were shown to be catalytically active,7 although this study did not investigate intact Miro enzymes nor did they characterize enzyme mechanisms. More detailed investigations into the catalytic activity of Miro by comparing the effect of calcium and binding to other cellular factors (e.g., TRAK1/2) will be necessary to draw a conclusion about the GTPase within the context of Miro activity.
Due to the putative GTPase activity of Miro, the role of GTPase‐activating proteins (GAPs) and guanine‐nucleotide exchange factors (GEFs) in regulating Miro activity remains an active area of research. A few potential GAPs/GEFs have been discovered recently, although much more research needs to be done to further characterize their role in Miro regulation. Interestingly, the only published GAP for Miro is VopE, a Vibrio cholera Type III secretion system effector.91 During infection, VopE stimulated mitochondrial redistribution, whereby instead of mitochondria clustering around the nucleus they were transported to the cell periphery via kinesin. The authors proposed that VopE acted as a GAP for Miro based on the observed interaction of VopE with Miro and the increased Miro GTPase activity when VopE was added. It remains to be determined whether there is a native GAP for Miro.
To date, two potential GEFs for Miro have been identified, GBF1 and Vimar.89, 92 For GBF1, a physical interaction between GBF1/GTP‐bound Arf1 and Miro was demonstrated.93, 94, 95 The combination of mitochondrial phenotype changes and the physical interaction between GBF1 and Miro demonstrates that GBF1 could act as a GEF for Miro.92 In Drosophila, Vimar was identified to partially localize to the mitochondria and physically interact with/act as a GEF for Miro.89 For characterizing the GTPase activity of Miro, a GAP or GEF may be the missing piece that assists Miro with the hydrolysis of GTP and subsequent replacement with unhydrolyzed GTP.
5.1. Miro: Calcium regulation
Beyond its GTPase activity, Miro possesses tandem EF‐hands allowing Miro to bind to Ca2+. EF‐hands consist of a helix–loop–helix motif that binds to calcium96 (Figure 1c). There is extensive evidence to suggest that the EF‐hands of Miro act as calcium‐dependent molecular switches to regulate mitochondrial transport in axons and dendrites in mammalian cell lines. This is supported by live‐cell imaging studies indicating that the influx of cytoplasmic Ca2+ paused mitochondrial transport.12, 51 Moreover, when the EF‐hands of Miro were mutated to E208K and E328K to block Ca2+ binding, mitochondrial transport was no longer inhibited by Ca2+.51
While there is agreement on the inhibition of mitochondrial transport via Ca2+, there are competing models for the molecular mechanism. Three distinct models have emerged related to kinesin‐mediated inhibition: (a) dissociation of kinesin (KIF5B) from Miro‐TRAK complexes;12 (b) allosteric control of motor protein activity;51 (c) recruitment of an anchoring factor (syntaphilin).66 Using in vitro translated proteins, Macaskill et al. saw that the addition of Ca2+ led kinesin to separate from Miro‐TRAK complexes.12 In contrast, cell‐based transfection studies by Wang and Schwarz observed that Miro‐TRAK‐kinesin complexes remained intact, but instead lost microtubule‐binding abilities due to the binding of kinesin to Miro directly.51 Finally, using a combination of cell‐based transfection pull‐downs and reconstituted recombinant proteins, Chen and Sheng demonstrated that kinesin binds to syntaphilin directly.66 These competing models indicate that further work is needed in order to provide decisive insight into how Ca2+ inhibits transport.
Finally, beyond the transport of mitochondria, the shape of mitochondria is also regulated by Ca2+. A recent study links Miro's calcium‐binding capacity to mitochondrial shape transition that occurs to differentiate mitochondria between long networked organelles that are more efficient at energy production and shorter, round organelles that are frequently associated with pathological states or apoptotic signals.97 Live‐cell imaging revealed that mitochondrial shape change occurred prior to the opening of the permeability transition pore, mitochondrial swelling, and calcium uptake through the mitochondrial calcium uniporter. Likewise, shape transition was independent of Drp1/dynamin‐dependent fission machinery. Mutations made to Miro‐1 and Miro‐2's EF‐hands confirmed that EF‐hand 1 of Miro1 was the calcium‐sensing determinant for shape transition, and the process did not require Miro's GTPase activity.
In addition to directly binding Ca2+, Miro interacts with the mitochondrial calcium uniporter, MCU, which has been shown to regulate Ca2+ influx into the mitochondria.98 While the N‐terminus of MCU is not required for mitochondrial targeting, it is essential for binding to Miro‐1.99 Calcium elevation triggers proteolytic cleavage of MCU, which disrupts the interactions between MCU and Miro1, thereby reducing calcium influx and mitochondrial movement.98, 99 This coupling between MCU and Miro1 suggests that there may be an interplay between Ca2+ influx and mitochondrial motility.
Considering that mitochondria buffer cytosolic Ca2+ levels and Miro's ability to interact with MCU, Miro may be able to sustain or limit the influence of Ca2+ on mitochondrial processes. This line of thinking suggests that if Miro alters MCU activity, Miro may promote longer periods of high cytosolic Ca2+ levels, thus altering mitochondrial biology (inhibiting transport, mitochondrial shape). Further work will be needed to determine how Miro may be able to regulate Ca2+ levels directly through its interaction with MCU.
6. MIRO REGULATION IN NEURODEGENERATIVE DISEASE
As a critical component of axonal mitochondrial transport, Miro occupies a central role in regulating mitochondrial position and turnover, a process critical to the health of neurons.100 In this section, we will discuss the link between Miro and mitochondrial biology with neurodegeneration.
Accumulating evidence is providing a link between proper mitochondrial function and dendritic tree development, a distinct process from axonal transport. These observations come from studying Miro1 knockout mouse animal models, where the deletion of Miro1 resulted in reduced mitochondria within dendrites.101 This, in turn, reduced the size and complexity of dendrites, indicating that Miro1 is critical for supporting dendritic health.101 The importance of Miro1 for dendrites was further underscored by conditional knockouts, where Miro1 was deleted from mature neurons. In these cells, loss of Miro1 leads to loss of dendritic mitochondria and dendritic complexity, ultimately leading to neurodegeneration.101 Considering that axonal mitochondrial distribution was unaffected in Miro1 knockout cells, the dramatic loss of dendritic complexity in Miro1 knockout cells mirrors evidence from other forms of neurodegeneration where dendritic complexity is reduced.102 Indeed, in a mouse model for Alzheimer's disease, loss of dendritic complexity was linked to changes in the electrical properties of the neurons.103 This all indicates that, since mitochondria are critical for supporting dendritic complexity, Miro may occupy a central role in many forms of neurodegeneration.
While proper mitochondrial function may be critical in many forms of neurodegenerative disease, we will focus on one such disease—Parkinson's disease—as there is a growing body of work linking Miro and mitochondrial clearance to Parkinson's disease.
6.1. Parkinson's disease
Parkinson's disease is a neurodegenerative disorder that causes the progressive loss of dopaminergic neurons, leading to impaired motor function associated with α‐synuclein aggregates (Lewy bodies).104 Affecting nearly 680,000 individuals in North America as of 2010,105 Parkinson's disease represents a growing malady with an estimated impact of 10 million individuals worldwide.106 While there are many different aspects of cell biology that are impaired in Parkinson's disease, we will focus this review on the role of mitochondrial turnover and the impact that Miro has on this process.
Over the past 20 years, studies of human patient mutations in combination with cell biological studies have produced a model of mitochondrial surveillance via PINK1 (kinase: PTEN‐induced kinase 1) and Parkin (E3 ubiquitin ligase) that is critical to support healthy neurons.107 In healthy mitochondria, PINK1 is translated into the OMM, at which point it is translocated into mitochondria for eventual proteolytic degradation. This indicates that PINK1 levels are typically low in healthy mitochondria. During mitochondrial damage such as membrane depolarization, PINK1 is no longer degraded and instead resides as a membrane‐anchored component in the OMM. In this new localization, PINK1‐mediated phosphorylation of Parkin leads to Parkin activation. Once activated, Parkin‐mediated ubiquitination signals the initiation of mitophagy, which is the selective degradation of mitochondria by the autophagosome.
While human genetics, cell biology, and biochemical studies have implicated PINK1 and Parkin as critical components necessary for stimulating mitochondrial turnover, the mechanism connecting these proteins to mitophagy remained poorly understood until recently. A big breakthrough occurred nearly 10 years ago with the identification of PINK1 as a subunit of a multi‐subunit complex comprising Miro and TRAK1.108 Subsequent work identified Miro as a target of both PINK1 phosphorylation and Parkin ubiquitination in order to degrade damaged mitochondria.109 Moreover, recent studies suggest that Parkin directly associates with Miro prior to activation by PINK1,110 suggesting that Miro has direct physical connections to both Parkin and PINK1. These initial results suggested that dissociating mitochondria from the microtubule network is a requirement for the successful turnover of mitophagy.
6.2. Phosphorylation & ubiquitination of Miro is required for mitophagy
Miro phosphorylation regulates mitophagy, serving to promote or inhibit mitochondrial turnover. PINK1 phosphorylates Miro1 residues S156, T298, and T299109, 111 (Figure 1a). Probing the role of these phosphorylation sites through phosphomimetic mutants, S156E effectively recruited Parkin although this was insufficient to drive mitophagy. However, mimicking phosphorylation of Miro at T298/T299 inhibited the ubiquitination of Miro, the recruitment of Parkin, and the Parkin‐dependent arrest of mitochondria.111 This indicates that the specific phosphorylation status of Miro may serve as a cellular signal regulating mitophagy.
After phosphorylation by PINK1, Miro is ubiquitinated by Parkin.112 In mouse embryonic fibroblasts, mitochondrial damage resulted in Miro ubiquitination on multiple lysine residues (Figure 1a).112 Preventing the ubiquitination of Miro stabilized the level of Miro after mitochondrial damage and slowed mitophagy.112 Interestingly, the ubiquitination kinetics between mutant K572R and a Miro mutant where all lysines were mutated to arginines were not significantly different.112 These results suggest that K572 plays a critical role in the ubiquitination of Miro, however, other residues may still play a role. While K572 seems to be fundamental to the ubiquitination of Miro1, more work is needed to determine the necessity of the other ubiquitination sites for the regulation of Miro and subsequent effects on mitophagy.
Recently, these results connecting targeted Miro degradation in order to promote mitophagy have been further supported through studies with Parkinson's disease‐derived patient samples. First, by comparing induced pluripotent stem cells (iPSCs) from both healthy and Parkinson's disease patients, Wang and coworkers extended the initial work of PINK1 and Parkin to include leucine‐rich repeat kinase 2 (LRRK2).113 Specifically, the authors compared mitochondrial motility after mitochondrial stress in LRRK2‐mutation backgrounds, sporadic Parkinson's disease, and healthy individuals to demonstrate that while healthy iPSC‐derived neurons showed slowing and degradation of mitochondria following mitochondrial stress, both LRRK2 and sporadic Parkinsons' disease neurons did not have any change in mitochondrial motility or turnover. Beyond the role of LRRK2, parallel investigations of Parkinson's disease patient samples demonstrated that increased α‐synuclein levels are associated with increased Miro expression and mitochondrial motility where α‐synuclein may be directly interacting with Miro.114
Based on these recent findings, a more detailed model of PINK1 and Parkin‐mediated mitophagy is emerging, placing Miro at the center of the process (Figure 4). Under normal conditions, prior to mitochondrial damage, Miro directs microtubule‐based transport via kinesin and dynein. In this condition, Parkin is associated with Miro but is not activated. Upon damage, PINK1 accumulates on the OMM but is unable to phosphorylate Miro until LRRK2 phosphorylates Miro. After Miro is phosphorylated by LRRK2, PINK1 phosphorylates both Miro and Parkin to activate Parkin's activity as an E3‐ubiquitin ligase. Once activated, Parkin ubiquitinates Miro, thus targeting Miro for proteasomal degradation and subsequent mitochondrial clearance via mitophagy.
Figure 4.
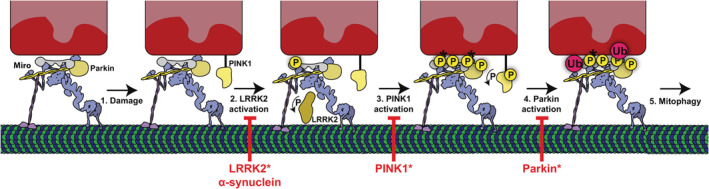
Potential model of mitochondrial clearance via mitophagy. Under normal cellular conditions, Miro directs microtubule‐based transport via kinesin and dynein. Note that Miro is also pre‐bound to Parkin. (1) Mitochondrial damage (e.g., membrane depolarization) occurs. (2) Following damage, LRRK2 is activated and phosphorylates Miro. (3) Once phosphorylated by LRRK2, PINK1 phosphorylates Miro in addition to Parkin. (4) Parkin ubiquitinates Miro. (5) Mitophagy clears damaged mitochondria. Note that mutations to LRRK2, PINK1, and Parkin (denoted “*”) in addition to α‐synuclein accumulation likely inhibits this process to prevent mitochondrial clearance
Given that this process is modified by changes to the regulatory factors LRRK2, PINK1, and Parkin (Figure 4), there may be therapeutic strategies to increase the amount of mitophagy. In Parkinson's disease patient‐derived neurons, impairment of flux through this pathway resulted in decreased mitochondrial clearance via mitophagy. Indeed, there are increased levels of mitochondria in addition to higher levels of Miro expression.115 Based on these observations, an in silico small molecule inhibitor of Miro was developed with the goal of reducing Miro levels in order to promote mitophagy.115 Promisingly, “Miro reducer” decreased Miro expression levels in Parkinson's disease neurons and increased mitophagy, potentially opening the door to future therapeutic strategies to promote the clearance of damaged mitochondria from Parkinson's disease patients.
7. FUTURE DIRECTIONS AND QUESTIONS
Over the past 20 years, there has been significant progress in our understanding of Miro's role in the regulation of mitochondrial cell biology. We believe this review has demonstrated that Miro sits at the center of a number of mitochondrial biology processes: organelle contact sites, mitochondrial transport, and mitophagy. By sitting at this nexus, Miro may act a lynchpin to allow cellular processes to have multiple rippling effects; through changes in Miro, the cell can simultaneously redistribute mitochondria (via microtubules, shape transitions), degrade old mitochondria, or alter other organelle processes. This aspect of Miro is both the most exciting and the most daunting to study. Future work will require a systems‐level understanding of cellular physiological in order to unpack the pleiotropic nature of Miro within mitochondrial biology.
We are excited for the next 20 years as we believe this field will be ushered into an era of mechanistic understanding of mitochondrial biology. During this time, we hope that the following questions will help to guide future cell biological, biochemical, and structural understandings of Miro with the goal of developing a deep understanding that can be translated into the clinical setting:
How does Miro interact with myriad binding partners and how is binding preference determined?
Does the GTP state of Miro regulate the context through which Miro acts on cellular processes?
How does Ca2+ inhibit mitochondrial motility?
What is the stoichiometry of Miro's interaction with binding partners?
How does PINK1/LRRK2/Parkin uncouple Miro from microtubules to promote mitophagy?
We believe that continued advances in our cell biological and biochemical understanding of Miro will help to characterize how a single protein is able to sit at the center of many critical cellular processes.
ACKNOWLEDGMENTS
E.L.E. is supported by NIH T‐32‐GM007315; Z.T. is supported by NSF OAC 1925476.
Eberhardt EL, Ludlam AV, Tan Z, Cianfrocco MA. Miro: A molecular switch at the center of mitochondrial regulation. Protein Science. 2020;29:1269–1284. 10.1002/pro.3839
Emily L. Eberhardt, Anthony V. Ludlam, and Zhenyu Tan contributed equally to this study.
Funding information National Institutes of Health, Grant/Award Number: T‐32‐GM007315; National Science Foundation, Grant/Award Number: OAC 1925476
REFERENCES
- 1. Hung V, Lam SS, Udeshi ND, et al. Proteomic mapping of cytosol‐facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife. 2017;6:e24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fransson A, Ruusala A, Aspenström P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. [DOI] [PubMed] [Google Scholar]
- 4. Guo X, Macleod GT, Wellington A, et al. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. [DOI] [PubMed] [Google Scholar]
- 5. Klosowiak JL, Park S, Smith KP, et al. Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Sci Rep. 2016;6:33019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Syrovatkina V, Alegre KO, Dey R, Huang X‐Y. Regulation, signaling, and physiological functions of G‐proteins. J Mol Biol. 2016;428:3850–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peters DT, Kay L, Eswaran J, Lakey JH, Soundararajan M. Human Miro proteins act as NTP hydrolases through a novel, non‐canonical catalytic mechanism. Int J Mol Sci. 2018;19:3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klosowiak JL, Focia PJ, Chakravarthy S, Landahl EC, Freymann DM, Rice SE. Structural coupling of the EF hand and C‐terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 2013;14:968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith KP, Focia PJ, Chakravarthy S, et al. Structural assembly of the human Miro1/2 GTPases based on the crystal structure of the N‐terminal GTPase domain. bioRxiv. 2019;729251 https://www.biorxiv.org/content/10.1101/729251v1. [Google Scholar]
- 10. Reis K, Fransson A, Aspenström P. The Miro GTPases: At the heart of the mitochondrial transport machinery. FEBS Lett. 2009;583:1391–1398. [DOI] [PubMed] [Google Scholar]
- 11. Fransson S, Ruusala A, Aspenström P. The atypical Rho GTPases Miro‐1 and Miro‐2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. [DOI] [PubMed] [Google Scholar]
- 12. Macaskill AF, Rinholm JE, Twelvetrees AE, et al. Miro1 is a calcium sensor for glutamate receptor‐dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanfer G, Courthéoux T, Peterka M, et al. Mitotic redistribution of the mitochondrial network by Miro and Cenp‐F. Nat Commun. 2015;6:8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bernhard W, Rouiller C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J Biophys Biochem Cytol. 1956;2:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009;4:1582–1590. [DOI] [PubMed] [Google Scholar]
- 16. Michel AH, Kornmann B. The ERMES complex and ER‐mitochondria connections. Biochem Soc Trans. 2012;40:445–450. [DOI] [PubMed] [Google Scholar]
- 17. Eisenberg‐Bord M, Shai N, Schuldiner M, Bohnert M. A tether is a tether is a tether: Tethering at membrane contact sites. Dev Cell. 2016;39:395–409. [DOI] [PubMed] [Google Scholar]
- 18. Ellenrieder L, Rampelt H, Becker T. Connection of protein transport and organelle contact sites in mitochondria. J Mol Biol. 2017;429:2148–2160. [DOI] [PubMed] [Google Scholar]
- 19. Tamura Y, Endo T. Role of intra‐ and inter‐mitochondrial membrane contact sites in yeast phospholipid biogenesis. Adv Exp Med Biol. 2017;997:121–133. [DOI] [PubMed] [Google Scholar]
- 20. Zecchini E, Siviero R, Giorgi C, Rizzuto R, Pinton P. Mitochondrial calcium signalling: Message of life and death. Ital J Biochem. 2007;56:235–242. [PubMed] [Google Scholar]
- 21. Kornmann B, Currie E, Collins SR, et al. An ER‐mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee S, Lee K‐S, Huh S, et al. Polo kinase phosphorylates Miro to control ER‐mitochondria contact sites and mitochondrial Ca(2+) homeostasis in neural stem cell development. Dev Cell. 2016;37:174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koshiba T, Holman HA, Kubara K, et al. Structure‐function analysis of the yeast mitochondrial rho GTPase, Gem1p: Implications for mitochondrial inheritance. J Biol Chem. 2011;286:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kornmann B, Osman C, Walter P. The conserved GTPase Gem1 regulates endoplasmic reticulum‐mitochondria connections. Proc Natl Acad Sci USA. 2011;108:14151–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stroud DA, Oeljeklaus S, Wiese S, et al. Composition and topology of the endoplasmic reticulum‐mitochondria encounter structure. J Mol Biol. 2011;413:743–750. [DOI] [PubMed] [Google Scholar]
- 27. Murley A, Lackner LL, Osman C, et al. ER‐associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife. 2013;2:e00422.23682313 [Google Scholar]
- 28. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee S, Min K‐T. The interface between ER and mitochondria: Molecular compositions and functions. Mol Cells. 2018;41:1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. [DOI] [PubMed] [Google Scholar]
- 31. Rapizzi E, Pinton P, Szabadkai G, et al. Recombinant expression of the voltage‐dependent anion channel enhances the transfer of Ca 2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. D'Angelo G, Vicinanza M, De Matteis MA. Lipid‐transfer proteins in biosynthetic pathways. Curr Opin Cell Biol. 2008;20:360–370. [DOI] [PubMed] [Google Scholar]
- 33. Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F. ORP5/ORP8 localize to endoplasmic reticulum‐mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016;17:800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iwasawa R, Mahul‐Mellier A‐L, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria‐ER interface to establish a platform for apoptosis induction. EMBO J. 2011;30:556–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simmen T, Aslan JE, Blagoveshchenskaya AD, et al. PACS‐2 controls endoplasmic reticulum‐mitochondria communication and Bid‐mediated apoptosis. EMBO J. 2005;24:717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: Fact and fiction. J Bioenerg Biomembr. 2005;37:129–142. [DOI] [PubMed] [Google Scholar]
- 37. Lee K‐S, Huh S, Lee S, et al. Altered ER‐mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc Natl Acad Sci USA. 2018;115:E8844–E8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grossmann D, Berenguer‐Escuder C, Bellet ME, et al. Mutations in RHOT1 disrupt endoplasmic reticulum‐mitochondria contact sites interfering with calcium homeostasis and mitochondrial dynamics in Parkinson's disease. Antioxid Redox Signal. 2019;31:1213–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Modi S, López‐Doménech G, Halff EF, et al. Miro clusters regulate ER‐mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat Commun. 2019;10:4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Islinger M, Cardoso MJR, Schrader M. Be different––The diversity of peroxisomes in the animal kingdom. Biochim Biophys Acta. 2010;1803:881–897. [DOI] [PubMed] [Google Scholar]
- 41. Fransen M, Lismont C, Walton P. The peroxisome‐mitochondria connection: How and why? Int J Mol Sci. 2017;18:1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Costello JL, Castro IG, Camões F, et al. Predicting the targeting of tail‐anchored proteins to subcellular compartments in mammalian cells. J Cell Sci. 2017;130:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okumoto K, Ono T, Toyama R, Shimomura A, Nagata A, Fujiki Y. New splicing variants of mitochondrial Rho GTPase‐1 (Miro1) transport peroxisomes. J Cell Biol. 2018;217:619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Castro IG, Richards DM, Metz J, et al. A role for mitochondrial Rho GTPase 1 (MIRO1) in motility and membrane dynamics of peroxisomes. Traffic. 2018;19:229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Covill‐Cooke C, Toncheva VS, Drew J, Birsa N, López‐Doménech G, Kittler JT. Peroxisomal fission is modulated by the mitochondrial Rho‐GTPases, Miro1 and Miro2. EMBO Rep. 2020;21:e49865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dietrich D, Seiler F, Essmann F, Dodt G. Identification of the kinesin KifC3 as a new player for positioning of peroxisomes and other organelles in mammalian cells. Biochim Biophys Acta. 2013;1833:3013–3024. [DOI] [PubMed] [Google Scholar]
- 47. Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires Milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Spronsen M, Mikhaylova M, Lipka J, et al. TRAK/Milton motor‐adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. [DOI] [PubMed] [Google Scholar]
- 49. Salogiannis J, Egan MJ, Reck‐Peterson SL. Peroxisomes move by hitchhiking on early endosomes using the novel linker protein PxdA. J Cell Biol. 2016;212:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bharti P, Schliebs W, Schievelbusch T, et al. PEX14 is required for microtubule‐based peroxisome motility in human cells. J Cell Sci. 2011;124:1759–1768. [DOI] [PubMed] [Google Scholar]
- 51. Wang X, Schwarz TL. The mechanism of Ca2+ −dependent regulation of kinesin‐mediated mitochondrial motility. Cell. 2009;136:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. López‐Doménech G, Covill‐Cooke C, Ivankovic D, et al. Miro proteins coordinate microtubule‐ and actin‐dependent mitochondrial transport and distribution. EMBO J. 2018;37:321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. [DOI] [PubMed] [Google Scholar]
- 54. Brickley K, Stephenson FA. Trafficking kinesin protein (TRAK)‐mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem. 2011;286:18079–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stowers RS, Megeath LJ, Górska‐Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on Milton, a novel drosophila protein. Neuron. 2002;36:1063–1077. [DOI] [PubMed] [Google Scholar]
- 56. Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5:a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. López‐Doménech G, Higgs NF, Vaccaro V, et al. Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 2016;17:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin M‐Y, Sheng Z‐H. Regulation of mitochondrial transport in neurons. Exp Cell Res. 2015;334:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ligon LA, Steward O. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol. 2000;427:340–350. [DOI] [PubMed] [Google Scholar]
- 60. Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. [DOI] [PubMed] [Google Scholar]
- 61. Russo GJ, Louie K, Wellington A, et al. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29:5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smit‐Rigter L, Rajendran R, Silva CAP, et al. Mitochondrial dynamics in visual cortex are limited in vivo and not affected by axonal structural plasticity. Curr Biol. 2016;26:2609–2616. [DOI] [PubMed] [Google Scholar]
- 63. Moutaux E, Christaller W, Scaramuzzino C, et al. Neuronal network maturation differently affects secretory vesicles and mitochondria transport in axons. Sci Rep. 2018;8:13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lewis TL Jr, Turi GF, Kwon S‐K, Losonczy A, Polleux F. Progressive decrease of mitochondrial motility during maturation of cortical axons in vitro and in vivo. Curr Biol. 2016;26:2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kang J‐S, Tian J‐H, Pan P‐Y, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short‐term facilitation. Cell. 2008;132:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen Y, Sheng Z‐H. Kinesin‐1‐syntaphilin coupling mediates activity‐dependent regulation of axonal mitochondrial transport. J Cell Biol. 2013;202:351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gutnick A, Banghart MR, West ER, Schwarz TL. The light‐sensitive dimerizer zapalog reveals distinct modes of immobilization for axonal mitochondria. Nat Cell Biol. 2019;21:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yi M, Weaver D, Hajnóczky G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J Cell Biol. 2004;167:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frederick RL, Shaw JM. Moving mitochondria: Establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pathak D, Sepp KJ, Hollenbeck PJ. Evidence that myosin activity opposes microtubule‐based axonal transport of mitochondria. J Neurosci. 2010;30:8984–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Quintero OA, DiVito MM, Adikes RC, et al. Human Myo19 is a novel myosin that associates with mitochondria. Curr Biol. 2009;19:2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu Z, Ma X‐N, Zhang H‐M, et al. Mouse myosin‐19 is a plus‐end‐directed, high‐duty ratio molecular motor. J Biol Chem. 2014;289:18535–18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Adikes RC, Unrath WC, Yengo CM, Quintero OA. Biochemical and bioinformatic analysis of the myosin‐XIX motor domain. Cytoskeleton. 2013;70:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rohn JL, Patel JV, Neumann B, et al. Myo19 ensures symmetric partitioning of mitochondria and coupling of mitochondrial segregation to cell division. Curr Biol. 2014;24:2598–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shneyer BI, Ušaj M, Henn A. Myo19 is an outer mitochondrial membrane motor and effector of starvation‐induced filopodia. J Cell Sci. 2016;129:543–556. [DOI] [PubMed] [Google Scholar]
- 76. Ušaj M, Henn A. Kinetic adaptation of human Myo19 for active mitochondrial transport to growing filopodia tips. Sci Rep. 2017;7:11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Oeding SJ, Majstrowicz K, Hu X‐P, et al. Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. J Cell Sci. 2018;131: jcs219469. https://jcs.biologists.org/content/131/17/jcs219469.long. [DOI] [PubMed] [Google Scholar]
- 78. Bocanegra JL, Fujita BM, Melton NR, et al. The MyMOMA domain of MYO19 encodes for distinct Miro‐dependent and Miro‐independent mechanisms of interaction with mitochondrial membranes. Cytoskeleton. 2019;109:2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee CA, Chin L‐S, Li L. Hypertonia‐linked protein Trak1 functions with mitofusins to promote mitochondrial tethering and fusion. Protein Cell. 2018;9:693–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pernas L, Scorrano L. Mito‐morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–531. [DOI] [PubMed] [Google Scholar]
- 82. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. [DOI] [PubMed] [Google Scholar]
- 83. Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003;134:333–344. [DOI] [PubMed] [Google Scholar]
- 84. Kalinski AL, Kar AN, Craver J, et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol. 2019;218:1871–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science. 2009;325:834–840. [DOI] [PubMed] [Google Scholar]
- 86. Chung JY‐M, Steen JA, Schwarz TL. Phosphorylation‐induced motor shedding is required at mitosis for proper distribution and passive inheritance of mitochondria. Cell Rep. 2016;16:2142–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Peterka M, Kornmann B. Miro‐dependent mitochondrial pool of CENP‐F and its farnesylated C‐terminal domain are dispensable for normal development in mice. PLoS Genet. 2019;15:e1008050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kanfer G, Peterka M, Arzhanik VK, et al. CENP‐F couples cargo to growing and shortening microtubule ends. Mol Biol Cell. 2017;28:2343–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ding L, Lei Y, Han Y, Li Y, Ji X, Liu L. Vimar is a novel regulator of mitochondrial fission through Miro. PLoS Genet. 2016;12:e1006359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Babic M, Russo GJ, Wellington AJ, Sangston RM, Gonzalez M, Zinsmaier KE. Miro's N‐terminal GTPase domain is required for transport of mitochondria into axons and dendrites. J Neurosci. 2015;35:5754–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Suzuki M, Danilchanka O, Mekalanos JJ. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe. 2014;16:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Walch L, Pellier E, Leng W, et al. GBF1 and Arf1 interact with Miro and regulate mitochondrial positioning within cells. Sci Rep. 2018;8:17121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gillingham AK, Munro S. The small G proteins of the Arf family and their regulators. Annu Rev Cell Dev Biol. 2007;23:579–611. [DOI] [PubMed] [Google Scholar]
- 94. Ye K, Hurt KJ, Wu FY, et al. Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell. 2000;103:919–930. [DOI] [PubMed] [Google Scholar]
- 95. Ackema KB, Hench J, Böckler S, et al. The small GTPase Arf1 modulates mitochondrial morphology and function. EMBO J. 2014;33:2659–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chazin WJ. Relating form and function of EF‐hand calcium binding proteins. Acc Chem Res. 2011;44:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nemani N, Carvalho E, Tomar D, et al. MIRO‐1 determines mitochondrial shape transition upon GPCR activation and Ca2+ stress. Cell Rep. 2018;23:1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Raffaello A, De Stefani D, Sabbadin D, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant‐negative pore‐forming subunit. EMBO J. 2013;32:2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Niescier RF, Hong K, Park D, Min K‐T. MCU interacts with Miro1 to modulate mitochondrial functions in neurons. J Neurosci. 2018;38:4666–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Franco‐Iborra S, Vila M, Perier C. Mitochondrial quality control in neurodegenerative diseases: Focus on Parkinson's disease and Huntington's disease. Front Neurosci. 2018;12:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. López‐Doménech G, Covill‐Cooke C, Ivankovic D, et al. Miro proteins coordinate microtubule‐and actin‐dependent mitochondrial transport and distribution. EMBO J. 2018;37:321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kulkarni VA, Firestein BL. The dendritic tree and brain disorders. Mol Cell Neurosci. 2012;50:10–20. [DOI] [PubMed] [Google Scholar]
- 103. Šišková Z, Justus D, Kaneko H, et al. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer's disease. Neuron. 2014;84:1023–1033. [DOI] [PubMed] [Google Scholar]
- 104. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. [DOI] [PubMed] [Google Scholar]
- 105. Marras C, Beck JC, Bower JH, et al. Prevalence of Parkinson's disease across North America. NPJ Parkinsons Dis. 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Anon Parkinson's Disease Statistics––Parkinson's News Today. Parkinson's News Today. Available from: https://parkinsonsnewstoday.com/parkinsons-disease-statistics/
- 107. Jin SM, Youle RJ. PINK1‐ and Parkin‐mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. 2009;48:2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang X, Winter D, Ashrafi G, et al. PINK1 and parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Safiulina D, Kuum M, Choubey V, et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019;38:e99384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci USA. 2016;113:E6097–E6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. López‐Doménech G, Covill‐Cooke C, Howden JH, et al. Miro ubiquitination is critical for efficient damage‐induced PINK1/parkin‐mediated mitophagy. BioRXiv. 10.1101/414664. [DOI] [Google Scholar]
- 113. Hsieh C‐H, Shaltouki A, Gonzalez AE, et al. Functional impairment in Miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell. 2016;19:709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shaltouki A, Hsieh C‐H, Kim MJ, Wang X. Alpha‐synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson's models. Acta Neuropathol. 2018;136:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hsieh C‐H, Li L, Vanhauwaert R, et al. Miro1 marks Parkinson's disease subset and Miro1 reducer rescues neuron loss in Parkinson's models. Cell Metab. 2019;30:1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]