

Abstract
G protein‐coupled receptors (GPCRs) modulate cell function over short‐ and long‐term timescales. GPCR signaling depends on biochemical parameters that define the what, when, and where of receptor function: what proteins mediate and regulate receptor signaling, where within the cell these interactions occur, and how long these interactions persist. These parameters can vary significantly depending on the activating ligand. Collectivity, differential agonist activity at a GPCR is called bias or functional selectivity. Here we review agonist bias at GPCRs with a focus on ligands that show dramatically different cellular responses from their unbiased counterparts.
Keywords: agonist bias, desensitization, GPCR, signaling
1. INTRODUCTION
G protein‐coupled receptors (GPCRs) are a superfamily of membrane‐bound signaling proteins that upon activation change the cellular program. Agonist binding to a GPCR activates signaling and regulatory molecules to produce a stereotypical response.1, 2 Many of these proteins have been identified in the decades long work of molecular pharmacology, including: heterotrimeric G proteins, GPCR kinases (GRKs), β‐arrestins, adenylyl cyclases, ion channels, kinases, and trafficking proteins. This abbreviated list only scratches the surface, and it is clear that new proteins await discovery.3, 4, 5, 6 In addition to “what” proteins function with GPCRs, it is increasingly clear that the cellular response to an agonist is shaped by “where” and “when” protein interactions occur (Figure 1).7, 8, 9, 10 Yet, these biochemical parameters are not inviolable. Structurally different agonists acting on the same receptor can alter these parameters and, as a consequence, change the downstream signaling and regulatory processes of the cell.11, 12 This phenomenon is called functional selectivity or biased agonism. Agonists exist on a continuum of bias and more strongly biased compounds are being developed.13, 14 Thus, it is important to determine the possible cellular responses to a biased agonist. For cases of extreme bias, how different are the cellular pathways of GPCR signaling and regulation?
FIGURE 1.
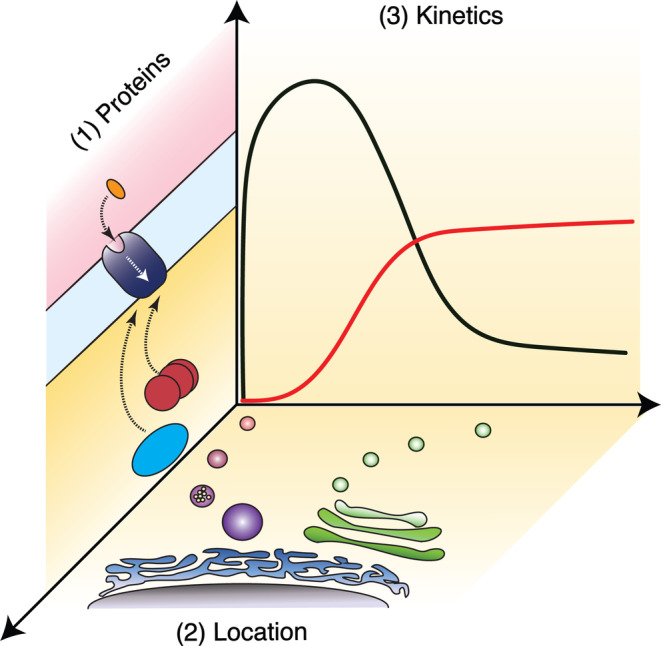
Cellular dimensions of GPCR function. Different ligands acting at the same receptor can alter the “what,” “where,” and “when” parameters of GPCR function. Panel 1 shows an extracellular agonist (orange) activating a GPCR (purple) and subsequent coupling to intracellular proteins (red and blue). Panel 2 highlights intracellular locations including endosomes (magenta) and Golgi (green) where GPCRs may function. Panel 3 provides a hypothetical example comparing activity of two ligands (black and red traces). Observed efficacy (y‐axis) can be dependent on when (x‐axis) an assay is performed. Ligands which show agonist bias, location bias, or kinetic bias can change these parameters compared to their unbiased counterparts. Panel 2 was adapted from Lobingier and von Zastrow 2019. 10
2. CLASSICAL PHARMACOLOGY AND AN INTRODUCTION TO BIAS
2.1. Full, partial, and biased agonism
An important step from the early concept of a “receptive substance” to modern receptor theory was the recognition of partial agonism.15, 16, 17 The differing capability of agonists to cause a response is called efficacy. Agonists can be classified based on relative efficacy with a full agonist causing a maximum response while a partial agonist, despite saturating concentrations, only stimulating a submaximal response. The above definition of efficacy is monodynamic. The cellular response to GPCR activation, however, is composed of the simultaneous, competitive, and sequential action of different proteins. The biased agonism hypothesis has two key tenets: (a) biased agonists stabilize receptor conformations distinct from full agonists and the result is differential coupling to receptor interacting proteins; (b) the response to GPCR activation arises from many cellular pathways and differential coupling to G proteins, GPCR kinases (GRKs), and β‐arrestins can preferentially activate one pathway over another.12, 18, 19 Recent biophysical, structural, and cell biological studies have provided important support to this model.20, 21, 22, 23, 24, 25 Thus, a G protein biased agonist will have greater relative efficacy in activation of the heterotrimeric G protein and subsequent signaling cascades. 26 In contrast, an arrestin biased agonist will have greater relative efficacy for the GRK/β‐arrestin pathway, which includes: phosphorylation of the receptor by GRKs, formation of a GPCR/β‐arrestin complex, and β‐arrestin terminating G protein signaling, acting as an adaptor for endocytosis, and scaffolding additional signaling events.1, 11, 27
2.2. Receptor reserve and system bias
Another important development in receptor theory was the recognition that full agonists need not occupy all receptors to drive a maximum response.15, 16 These unoccupied receptors are called spare receptors or receptor reserve. Receptor reserve represents a technical challenge when studying partial or biased agonists because of the inherent amplification in GPCR signaling combined with the fact that each tissue/cell type has a limited maximal response. With a large receptor reserve, lower efficacy agonists can appear as full agonists for G protein activity.28, 29 However, not all GPCR pathways are signal amplifying: binding of β‐arrestin to the phosphorylated receptor and subsequent endocytosis requires a one‐to‐one interaction. 27 Thus, receptor reserve can differ between G protein and GRK/β‐arrestin pathways and care must be taken in investigation of bias. 30 Receptor reserve is not the only variable between cell types. System bias describes the concept that the expression of any component in the GPCR pathway can vary between tissues. 19 Thus, an agonist/receptor pair can produce distinct activities in different cell types. In this review, we will note if agonists are considered full, partial, or biased with the recognition that these are system‐dependent terms.
2.3. Other forms of bias: Location bias and kinetic bias
Biased agonists can change the duration and location of receptor signaling by altering GPCR coupling to the G protein or GRK/β‐arrestin pathways. However, these changes do not encapsulate the extent to which ligands can bias the location and timing of GPCR activity. Two recently described types of bias involve these where and when variables: location bias and kinetic bias. Location bias is a two part phenomenon: (a) GPCRs can activate signaling events from intracellular compartments as well as the plasma membrane; (b) certain ligands can preferentially drive GPCR activity from intracellular compartments or restrict activity to the plasma membrane and, as consequence, change signaling.9, 10 One type of intracellular signaling occurs at compartments such as the Golgi or nucleus. GPCRs at these compartments are activated by agonists which are actively transported into the cell or are inherently capable of passive diffusion through the cell membrane.31, 32, 33, 34 A second type of intracellular signaling occurs at endosomes following agonist‐induced endocytosis.9, 10 Importantly, intracellular GPCR activation can drive distinct signaling events compared to agonists which restrict receptor activity to the cell surface.34, 35, 36, 37, 38, 39, 40 Kinetic bias (also called temporal bias or kinetic context) highlights the interplay of ligand binding rates and the duration of GPCR signaling and regulatory pathways.7, 8, 41 For example, when ligands with slow dissociation rates are examined for efficacy in transient signaling events, the degree and even the direction of bias can change depending on when the assay takes place after agonist stimulation. Examples include serotonin and dopamine receptors while opioid receptors provide a counterexample.42, 43, 44 Together, agonist bias, location bias, and kinetic bias can all shape the cellular response to GPCR activation.
3. A “CELLULAR PERSPECTIVE” OF GPCR SIGNALING: PHOSPHOPROTEOMICS AND TRANSCRIPTOMICS
3.1. Phosphoproteomics
GPCR activation stimulates cellular kinases. Activity of these kinases can be probed with phospho‐specific antibodies or genetically encoded reporters. 45 What these approaches miss is the breadth of cellular remodeling evoked by GPCR activation. Phosphoproteomics is an alternative approach to understand how GPCR activation changes kinase and phosphatase activity in the cell. One advantage to phosphoproteomics is that it can be used to detect and identify thousands of phosphosites; thus, examining GPCR activity from the “cellular perspective.” For example, GPCR activation is estimated to change 1–10% of all phosphorylation sites in the cell.46, 47, 48, 49 As a comparison, an estimated 16–20% of phosphosites can be regulated upon receptor tyrosine kinase (RTK) stimulation.50, 51
Before discussing examples in which biased agonists change the targets of GPCR signaling when compared to their unbiased counterparts, it is important to highlight an example demonstrating that this is not always the case. Tsvetanova et al. compared the cellular phosphoproteome of HEK293 cells endogenously expressing β2‐adrenergic receptor (β2AR) when stimulated by the full agonist isoproterenol compared to a partial agonist with G protein bias, salmeterol.48, 52, 53 The authors found that salmeterol induced changes in the same phosphorylation sites as isoproterenol and the only difference was that the changes were smaller in magnitude with salmeterol. 48 In contrast, a pair of highly complementary studies examined the cellular phosphoproteome downstream of the angiotensin II type 1 receptor (AT1R) when activated by the endogenous agonist angiotensin II (AngII) or a biased agonist, SII (Sar1, Ile4, Ile8‐angiotensin).46, 47 AngII is a full agonist that activates Gαq and GRK/β‐arrestin pathways while SII is a partial agonist for GRK/β‐arrestin and shows minimal activation of Gαq.54, 55, 56, 57 Both studies employed HEK293 cells stably overexpressing AT1R. Together, these papers demonstrated that one‐third of phosphosites responding to AT1R activation were linked to GRK/β‐arrestin activity (observed with both SII and AngII), while two‐thirds were linked to Gαq (observed with only AngII).46, 47 Thus, the majority of the cellular phosphorylation sites remodeled by AngII were untouched by SII‐mediated AT1R signaling. Additionally, the authors noted it was unlikely that SII caused AT1R to gain to new signaling targets. 47
A more recent study examined how the cellular phosphoproteome was remodeled by activation of the protease‐activated receptor‐1 (PAR1) with thrombin or anticoagulant protease (APC). 49 Thrombin cleavage of PAR1 causes coupling to Gα12/13, Gαq, and β‐arrestin while APC causes PAR1 to couple to β‐arrestin.58, 59, 60, 61 Phosphoproteomic comparison of PAR1 signaling in endothelial cell derived EA.hy926 cells revealed broadly different targets of thrombin and APC‐induced activity. 49 Targets of thrombin activated PAR1 signaling were enriched in proteins linked to endothelial barrier function and adherens junctions while APC‐induced signaling modulated phosphorylation of proteins linked to gene transcription. 49 Additionally, APC modulated a much smaller number of phosphosites than the full agonist thrombin.47, 49 Thus, certain biased agonist/GPCR pairs can greatly change the cellular phosphoproteome—and consequently cellular function—compared to their unbiased counterparts.
3.2. Transcriptomics
GPCR signaling can alter gene transcription and cause persistent changes to the proteome. Multiple methods, including RT‐qPCR and transcriptional reporters, allow study of transcription factor activity or levels of specific mRNAs. Yet analogous to what was discussed above, these transcriptional assays cannot capture the breadth of changes induced by GPCR activation. Microarrays and RNAseq provide an alternative approach to more holistically capture how GPCR activation changes the cell. Such approaches show that GPCR signaling can activate the transcription of hundreds of genes.39, 62, 63, 64 As a point of comparison, activation of RTKs can induce changes to the transcription of ~1,000 genes.65, 66, 67 Christensen et al. compared how the full agonist AngII and the biased agonist SII changed transcription downstream of AT1R. 62 They observed that SII regulated only 12% of the 212 transcripts that were regulated by AngII, and had no additional transcriptional targets. They also observed kinetic bias: AngII‐induced transcription returned to basal after 48 hr of stimulation while SII induced transcripts remained upregulated at 48 hr. 62 In an investigation of location bias, endogenous β2AR‐induced transcription in HEK293 cells was compared under normal conditions or when signaling was restricted to the cell surface by inhibition of endocytosis. 39 Fifty‐five genes were found to be induced by activation of β2AR, and transcription of over half of these targets was suppressed by inhibition of endocytosis. 39 In these two studies, agonist bias or location bias caused a strong reduction in the number of transcribed genes.
3.3. Bias in GPCR signaling
As more holistic methods, transcriptomics and proteomics provide a cellular perspective of agonist bias at GPCRs. 39, 47, 49, 62 These studies provide insight about whether biased agonists cause GPCRs to gain new signaling targets or simply change the number/magnitude of full agonist targets. In several cases, biased agonists strongly reduced the number of targets while in one case bias caused GPCR signaling to target new phosphosites.47, 48, 49, 62 Why this occurs is not currently understood and these phenomena deserves further study. Together, these data raise the question: if the cellular state induced by GPCR signaling can differ significantly between full agonists and biased agonists, how then can GPCR regulation be effected by bias?
4. REGULATION OF GPCR SIGNALING
4.1. GPCR kinases: Partial agonism and changes in GRK/GPCR pairing
Acute termination of GPCR signaling—called desensitization—requires GRKs to phosphorylate the agonist‐activated receptor. Genetic or chemical inhibition of GRK function, or mutation of GPCR Ser/Thr phosphorylation sites, inhibits desensitization.68, 69 Nonvisual GRKs are grouped into two subfamilies by sequence homology: GRK2/3 and GRK4/5/6. 68 Two important questions remain incompletely resolved: (a) why GRKs pair with certain GPCRs; (b) what are the consequences of these pairings? A critical difference between GRKs is the mechanism by which they are recruited to activated GPCRs. GRK2/3 proteins contain a pleckstrin homology (PH) domain which directly binds free Gβ/Gγ while GRK4/5/6 proteins lack this PH domain and are instead membrane associated through palmitoylation or direct lipid binding. 27 Thus, a component of GRK2/3 recruitment requires GPCR signaling to liberate free Gβ/Gγ. 25 Additionally, it has been shown that GRKs can phosphorylate nonreceptor proteins and the targets of this “extramural” activity appear to be GRK‐specific.68, 70 Here we review studies in cultured cells showing how partial and biased agonists can change the GPCR/GRK pairing.
Studies of agonist‐driven phosphorylation of β2AR in model human cells demonstrated that both GRK2 and GRK6 are required to obtain full phosphorylation of β2AR.71, 72, 73 RNAi‐mediated knockdown of GRK6 reduced agonist‐driven phosphorylation of two sites in β2AR (Ser355 and Ser356) while GRK2 knockdown reduced phosphorylation on the other six (Thr360, Ser364, Ser396, Ser401, Ser407, and Ser411). 73 Intriguing, the ligand carvedilol—an inverse agonist for the G protein pathway and partial agonist for the GRK/β‐arrestin pathway—was shown to only stimulate phosphorylation of the GRK6‐sensitive sites, Ser355/Ser356.73, 74 One possible consequence of ligand‐specific GPCR phosphorylation was defined in the “bar‐code” hypothesis: differential patterns of GPCR phosphorylation cause distinct activities of β‐arrestin and/or preferential coupling to different signaling pathways.73, 75, 76, 77, 78 Thus, in cultured cells, a full agonist at β2AR can activate both GRK2 and GRK6 while carvedilol drives a partial version of the same response and only activates GRK6. 73
The mu opioid receptor (MOR) provides a contrasting example of the cellular response to partial/biased agonism. In cultured cell models, the full agonist DAMGO was shown to cause GRK2/3‐mediated phosphorylation of MOR phosphorylation sites: S356, T357, T370, S375, T376, and T379.79, 80, 81, 82, 83, 84 This result was compared with the lower efficacy agonist morphine, which is typically considered a partial agonist in vitro although with systems‐dependent efficacy.44, 85, 86, 87, 88 Morphine caused phosphorylation of S375 in MOR, produced minimal phosphorylation of T370, T376, and T379, and thus drove 5‐ to 15‐fold‐less multiphosphorylation of MOR.79, 80, 81, 82, 83, 84 Unexpectedly, and in contrast to β2AR, morphine caused a switch to GRK5 (part of the GRK4/5/6 subfamily) as the kinase primarily required for phosphorylation of MOR in cultured cells.80, 82 Importantly, agonist‐dependent and GRK‐specific phosphorylation of MOR is largely conserved in animal models. GRK3 knockout mice, but not GRK5 knockout mice, showed reduction in MOR phosphorylation stimulated by higher efficacy agonists.80, 89 In comparison, MOR phosphorylation driven by morphine was reduced in both GRK5 and GRK3 knockout mice.80, 89 These data suggest that the lower efficacy agonist morphine can cause a fundamental re‐wiring of GRK/GPCR pairing compared to higher efficacy opioids.
4.2. Atypical regulation of opioid receptors via PKC and JNK
The canonical mechanism for homologous desensitization of GPCRs involves GRKs and β‐arrestin. However, not all agonists efficiently recruit these proteins. How do GPCRs desensitize under these conditions? A series of observations demonstrated that lower efficacy opioid ligands can fundamentally re‐write the mechanisms of cellular desensitization and physiological tolerance. 90 Here we refer to this process as atypical regulation to differentiate it from canonical homologous regulation involving GRK/β‐arrestin.
In cultured cells, desensitization of MOR caused by the full agonist DAMGO was sensitive to dominant negative mutants of GRK2 but not PKC inhibitors.83, 91 In contrast, MOR desensitization driven by the partial agonist morphine was sensitive to PKC but not GRK inhibition.83, 91 These findings were extended to electrophysiological recordings in the locus coeruleus (LC) from acute brain slice.92, 93 Cell‐type differences play a role in atypical desensitization of MOR: the PKC dependence of morphine induced desensitization could be seen in naive HEK293 or AtT20 cells while neurons assayed in brain slice required co‐activation of PKC or pretreatment of the animal with morphine.83, 91, 92, 93, 94, 95, 96 Thus, morphine opens the door to an atypical mode of GPCR regulation but cell‐type specific parameters determine if it occurs. One model suggests that the mechanism of MOR desensitization exist on a continuum informed by ligand efficacy and shaped by cell‐type specific parameters. 90 In this model, morphine and DAMGO occupy ends of the continuum, and primarily use either PKC or GRK, while other opioids may be able to use both mechanisms.90, 92, 97, 98
In vitro studies have provided insight into how PKC may be mediating atypical regulation. Efficient β‐arrestin binding to a GPCR is thought to require three or more correctly spaced phosphorylations in the receptor. 12 In cultured cell models, PKC activity can drive phosphorylation of two residues in MOR, S363 and T370.81, 99, 100 Thus, it is possible that the desensitizing activity of PKC may not occur exclusively at the receptor but at other points in the pathway. 90 In support of this model, a mutant MOR lacking all phosphorylation sites in its carboxy‐terminal tail was shown to still be able to desensitize in response to morphine. 83 Additionally, it has been demonstrated that Gαi is directly phosphorylated by PKC downstream of morphine‐activated MOR and this phosphorylation contributed to atypical regulation. 101
Development of opioid tolerance in vivo can also occur through opioid‐specific mechanisms involving canonical pathways (GRK) or atypical pathways (PKC or JNK).102, 103 Here we will discuss behavioral assays comparing opioid analgesic activity when combined with genetic or chemical inhibition of GRKs, PKC, or JNK. Such experimental paradigms are powerful but have potential caveats: (a) opioids have different pharmacokinetic/pharmacodynamic properties such that dosing can vary significantly between compounds; (b) tissue‐specific expression of proteins in the opioid receptor pathway means opioids can behave differently between physiological assays; (c) kinases phosphorylate many proteins and thus the kinase inhibitors will be pleiotropic in their effects as well as having potential off‐target activities. With these caveats in mind, data from multiple groups suggest the mechanisms for development of opioid tolerance differ between the lower efficacy agonist morphine and higher efficacy opioids.
In a GRK3 KO mouse, tolerance to the high efficacy agonist fentanyl but not the lower efficacy agonist morphine was attenuated in the hot plate assay for analgesia.97, 104 In a separate study, GRK/β‐arrestin function was disrupted by mutation of S375 in MOR. 105 S375 is the initiating site for MOR multiphosphorylation and mutation of this site to alanine reduced overall MOR phosphorylation, β‐arrestin recruitment, and agonist‐dependent MOR endocytosis.79, 82, 84 In a S375A‐MOR knock‐in mouse, development of tolerance to high efficacy opioids was reduced while development of tolerance to morphine was retained in the electrical tail root assay for analgesia. 105 In conjunction with these genetic studies, kinase inhibitors have provided insight into the pathway(s) by which tolerance to morphine can develop in the animal. Inhibitors to PKC were shown to inhibit the development of tolerance to morphine in both the tail‐flick and hot plate assays for analgesia.98, 106, 107, 108 Importantly, PKC inhibitors did not inhibit the development of tolerance to the high efficacy agonist DAMGO in assays for opioid‐induced analgesia. 98 In addition to PKC, inhibition of JNK has been broadly reported to inhibit the development of morphine tolerance but not affect tolerance to higher efficacy opioids.104, 109, 110 Consistent with the model that JNK activity is involved in the development of morphine tolerance, a JNK2 knockout mouse was shown to have reduced tolerance to morphine in tail‐flick and hotplate assays of analgesia.104, 111 However, it is important to underscore pathway/tissue specific differences in opioid efficacy and mechanisms of desensitization. For example, in opioid‐induced respiratory depression, PKC inhibitors—but not JNK inhibitors—reduced the development of morphine tolerance. 112 Together these in vitro data examining MOR desensitization and in vivo data studying development of tolerance suggest that the cell contains the ability to muster different homeostatic responses to opioid receptor activation: a “canonical” response utilizing GRK/β‐arrestin and an “atypical” response which utilizes kinases such as PKC or JNK.
Is atypical regulation a broader phenomenon observed with other GPCRs? While not conclusively resolved, evidence suggests that D2 dopamine receptor, CB1 cannabinoid receptor, and 7A serotonin receptor may undergo analogous types of regulation.113, 114, 115, 116 Another intriguing example involves kappa opioid receptor (KOR). Nor‐binaltorphimine (nor‐BNI) was originally identified as a KOR antagonist. 117 It was subsequently recognized that nor‐BNI, as well as the KOR antagonists JDTic and 5′‐GNTI, had properties of biased agonists in that they inhibited G protein signaling but activated JNK signaling through KOR. 118 Additionally, several atypical properties were noted about these KOR ligands: (a) they evoked long‐lasting (~3 weeks) antagonism of the G protein pathway in vivo without any sign of covalently modifying the receptor; (b) inhibition of JNK blocked long‐term inactivation of KOR; (c) inhibition of PKC blocked phosphorylation of JNK, implying that PKC was part of atypical KOR regulation.104, 115, 118, 119 A recent study identified an increased association between KOR and Gαi after nor‐BNI treatment, and proposed a mechanism for nor‐BNI mediated inactivation of KOR proceeding through loss of G protein palmitoylation via a JNK‐mediated increase in local reactive oxygen species production. 115
5. THE BOUNDARIES OF LIGAND BIAS AT GPCRS
Much is still unknown about how agonist bias, location bias, and kinetic bias shape GPCR activity. In this review, we have highlighted examples in which biased agonists can substantially change the targets of GPCR signaling or the mechanisms by which GPCRs are regulated. While it is clear that not all biased ligands evoke such distinct cellular responses compared to their unbiased counterparts, the examples reviewed here help to draw boundaries around what is possible in a cellular response to GPCR activation. It is noteworthy that many of the biased agonists discussed in this review are, in fact, partial agonists for the pathway(s) they activate. This raises the intriguing question of how to disentangle partial agonism from differential pathway efficacy (e.g., bias)? Toward that point, a recent paper investigating bias at opioid receptors developed an approach to minimize the effects of system bias and receptor reserve and found that many opioids previously described as biased agonists are in fact low efficacy partial agonists without significant bias. 88 In this same paper, they show that many of these low efficacy partial agonists have an improved therapeutic window for analgesia compared to higher efficacy opioids. 88 Thus, further study of partial agonism and bias in all its forms will be necessary to define the boundaries of what is possible and harness those understandings for improved GPCR‐targeting therapeutics.
AUTHOR CONTRIBUTIONS
Thomas Fernandez: Conceptualization; writing‐original draft; writing‐review and editing. Monica De Maria: Conceptualization; writing‐original draft; writing‐review and editing. Braden Lobingier: Conceptualization; funding acquisition; supervision; writing‐original draft; writing‐review and editing.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
The authors thank John Williams, Roshanak Irannejad, Nina Tsvetanova, and Brandon Novy for critical discussion and suggestions. We also thank the members of the Lobingier lab and other colleagues for additional advice. B. T. L. acknowledges NIH/NIDA DA043607.
Fernandez TJ, De Maria M, Lobingier BT. A cellular perspective of bias at G protein‐coupled receptors. Protein Science. 2020;29:1345–1354. 10.1002/pro.3872
Funding information National Institutes of Health, Grant/Award Number: DA043607
REFERENCES
- 1. Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol. 2018;25:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weis WI, Kobilka BK. The molecular basis of G protein‐coupled receptor activation. Annu Rev Biochem. 2018;87:897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ritter SL, Hall RA. Fine‐tuning of GPCR activity by receptor‐interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hay DL, Pioszak AA. Receptor activity‐modifying proteins (RAMPs): New insights and roles. Annu Rev Pharmacol Toxicol. 2016;56:469–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lobingier BT, Hüttenhain R, Eichel K, et al. An approach to spatiotemporally resolve protein interaction networks in living cells. Cell. 2017;169:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paek J, Kalocsay M, Staus DP, et al. Multidimensional tracking of GPCR signaling via peroxidase‐catalyzed proximity labeling. Cell. 2017;169:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lane JR, May LT, Parton RG, Sexton PM, Christopoulos A. A kinetic view of GPCR allostery and biased agonism. Nat Chem Biol. 2017;13:929–937. [DOI] [PubMed] [Google Scholar]
- 8. Grundmann M, Kostenis E. Temporal bias: Time‐encoded dynamic GPCR signaling. Trends Pharmacol Sci. 2017;38:1110–1124. [DOI] [PubMed] [Google Scholar]
- 9. Thomsen ARB, Jensen DD, Hicks GA, Bunnett NW. Therapeutic targeting of endosomal G‐protein‐coupled receptors. Trends Pharmacol Sci. 2018;39:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lobingier BT, von Zastrow M. When trafficking and signaling mix: How subcellular location shapes G protein‐coupled receptor activation of heterotrimeric G proteins. Traffic. 2019;20:130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wootten D, Christopoulos A, Marti‐Solano M, Madan Babu M, Sexton PM. Mechanisms of signalling and biased agonism in G protein‐coupled receptors. Nat Rev Mol Cell Biol. 2018;19:638–653. [DOI] [PubMed] [Google Scholar]
- 12. Gurevich VV, Gurevich EV. Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol Ther. 2020;107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kenakin T, Watson C, Muniz‐Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Nerosci. 2012;3:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stahl EL, Zhou L, Ehlert FJ, Bohn LM. A novel method for analyzing extremely biased agonism at G protein‐coupled receptors. Mol Pharmacol. 2015;87:866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stephenson RP. A modification of receptor theory. Br J Pharmacol Chemother. 1956;11:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nickerson M. Receptor occupancy and tissue response. Nature. 1956;178:697–698. [DOI] [PubMed] [Google Scholar]
- 17. Furchgott RF, Bursztyn P. Comparison of dissociation constants and of relative efficacies of selected agonists acting on parasympathetic receptors. Ann NY Acad Sci. 1967;144:882–899. [Google Scholar]
- 18. Luttrell LM. Minireview: More than just a hammer: Ligand “bias” and pharmaceutical discovery. Mol Endocrinol. 2014;28:281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: From simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17:243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi M, Staus DP, Wingler LM, et al. G protein‐coupled receptor kinases (GRKs) orchestrate biased agonism at the β2‐adrenergic receptor. Sci Signal. 2018;11:eaar7084. [DOI] [PubMed] [Google Scholar]
- 21. Wingler LM, McMahon C, Staus DP, Lefkowitz RJ, Kruse AC. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell. 2019;176:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wingler LM, Elgeti M, Hilger D, et al. Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell. 2019;176:468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wingler LM, Skiba MA, McMahon C, et al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science. 2020;367:888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suomivuori C‐M, Latorraca NR, Wingler LM, et al. Molecular mechanism of biased signaling in a prototypical G protein‐coupled receptor. Science. 2020;367:881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stoeber M, Jullié D, Li J, et al. Agonist‐selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. Elife. 2020;9:e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luttrell LM, Maudsley S, Bohn LM. Fulfilling the promise of “biased” G protein‐coupled receptor agonism. Mol Pharmacol. 2015;88:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gurevich VV, Gurevich EV. GPCR signaling regulation: The role of GRKs and arrestins. Front Pharmacol. 2019;10:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adham N, Ellerbrock B, Hartig P, Weinshank RL, Branchek T. Receptor reserve masks partial agonist activity of drugs in a cloned rat 5‐hydroxytryptamine1B receptor expression system. Mol Pharmacol. 1993;43:427–433. [PubMed] [Google Scholar]
- 29. Kenakin T. Receptor theory. Curr Protoc Pharmacol 2008;Chapter 1:Unit 1.2. [DOI] [PubMed]
- 30. Kelly E. Efficacy and ligand bias at the μ‐opioid receptor. Br J Pharmacol. 2013;169:1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Audet N, Dabouz R, Allen BG, Hébert TE. Nucleoligands‐repurposing G protein‐coupled receptor ligands to modulate nuclear‐localized G protein‐coupled receptors in the cardiovascular system. J Cardiovasc Pharmacol. 2018;71:193–204. [DOI] [PubMed] [Google Scholar]
- 32. Jong Y‐JI, Harmon SK, O'Malley KL. GPCR signalling from within the cell. Br J Pharmacol. 2018;175:4026–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Irannejad R, Pessino V, Mika D, et al. Functional selectivity of GPCR‐directed drug action through location bias. Nat Chem Biol. 2017;13:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stoeber M, Jullié D, Lobingier BT, et al. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron. 2018;98:963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Calebiro D, Nikolaev VO, Gagliani MC, et al. Persistent cAMP‐signals triggered by internalized G‐protein‐coupled receptors. PLoS Biol. 2009;7:e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferrandon S, Feinstein TN, Castro M, et al. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jensen DD, Lieu T, Halls ML, et al. Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief. Sci Transl Med. 2017;9:eaal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Godbole A, Lyga S, Lohse MJ, Calebiro D. Publisher correction: Internalized TSH receptors en route to the TGN induce local Gs‐protein signaling and gene transcription. Nat Commun. 2018;9:5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol. 2014;10:1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nash CA, Wei W, Irannejad R, Smrcka AV. Golgi localized β1‐adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife. 2019;8:e48167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Costa‐Neto CM, Parreiras‐E‐Silva LT, Bouvier M. A pluridimensional view of biased agonism. Mol Pharmacol. 2016;90:587–595. [DOI] [PubMed] [Google Scholar]
- 42. Unett DJ, Gatlin J, Anthony TL, et al. Kinetics of 5‐HT2B receptor signaling: Profound agonist‐dependent effects on signaling onset and duration. J Pharmacol Exp Ther. 2013;347:645–659. [DOI] [PubMed] [Google Scholar]
- 43. Klein Herenbrink C, Sykes DA, Donthamsetti P, et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. 2016;7:10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pedersen MF, Wróbel TM, Märcher‐Rørsted E, et al. Biased agonism of clinically approved μ‐opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology. 2020;166:107718. [DOI] [PubMed] [Google Scholar]
- 45. Lee HN, Mehta S, Zhang J. Recent advances in the use of genetically encodable optical tools to elicit and monitor signaling events. Curr Opin Cell Biol. 2020;63:114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiao K, Sun J, Kim J, et al. Global phosphorylation analysis of beta‐arrestin‐mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc Natl Acad Sci U S A. 2010;107:15299–15304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christensen GL, Kelstrup CD, Lyngsø C, et al. Quantitative phosphoproteomics dissection of seven‐transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics. 2010;9:1540–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsvetanova NG, Trester‐Zedlitz M, Newton BW, et al. G protein‐coupled receptor endocytosis confers uniformity in responses to chemically distinct ligands. Mol Pharmacol. 2017;91:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin Y, Wozniak JM, Grimsey NJ, et al. Phosphoproteomic analysis of protease‐activated receptor‐1 biased signaling reveals unique modulators of endothelial barrier function. Proc Natl Acad Sci U S A. 2020;117:5039–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Batth TS, Papetti M, Pfeiffer A, Tollenaere MAX, Francavilla C, Olsen JV. Large‐scale phosphoproteomics reveals Shp‐2 phosphatase‐dependent regulators of Pdgf receptor signaling. Cell Rep. 2018;22:2784–2796. [DOI] [PubMed] [Google Scholar]
- 51. Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site‐specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. [DOI] [PubMed] [Google Scholar]
- 52. van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. Quantification of ligand bias for clinically relevant β2‐adrenergic receptor ligands: Implications for drug taxonomy. Mol Pharmacol. 2014;85:492–509. [DOI] [PubMed] [Google Scholar]
- 53. Gimenez LE, Baameur F, Vayttaden SJ, Clark RB. Salmeterol efficacy and bias in the activation and kinase‐mediated desensitization of β2‐adrenergic receptors. Mol Pharmacol. 2015;87:954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wei H, Ahn S, Shenoy SK, et al. Independent beta‐arrestin 2 and G protein‐mediated pathways for angiotensin II activation of extracellular signal‐regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003;100:10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Holloway AC, Qian H, Pipolo L, et al. Side‐chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. [DOI] [PubMed] [Google Scholar]
- 56. Saulière A, Bellot M, Paris H, et al. Deciphering biased‐agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol. 2012;8:622–630. [DOI] [PubMed] [Google Scholar]
- 57. Hoare SRJ, Tewson PH, Quinn AM, Hughes TE. A kinetic method for measuring agonist efficacy and ligand bias using high resolution biosensors and a kinetic data analysis framework. Sci Rep. 2020;10:1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McLaughlin JN, Shen L, Holinstat M, Brooks JD, DiBenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease‐activated receptor‐1. J Biol Chem. 2005;280:25048–25059. [DOI] [PubMed] [Google Scholar]
- 59. Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. β‐Arrestins regulate protease‐activated receptor‐1 desensitization but not internalization or down‐regulation. J Biol Chem. 2002;277:1292–1300. [DOI] [PubMed] [Google Scholar]
- 60. Soh UJK, Trejo J. Activated protein C promotes protease‐activated receptor‐1 cytoprotective signaling through‐arrestin and dishevelled‐2 scaffolds. Proc Natl Acad Sci U S A. 2011;108:E1372–E1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease‐activated receptors. Front Endocrinol. 2014;5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Christensen GL, Knudsen S, Schneider M, et al. AT1 receptor Gαq protein‐independent signalling transcriptionally activates only a few genes directly, but robustly potentiates gene regulation from the β2‐adrenergic receptor. Mol Cell Endocrinol. 2011;331:49–56. [DOI] [PubMed] [Google Scholar]
- 63. Maudsley S, Martin B, Gesty‐Palmer D, et al. Delineation of a conserved arrestin‐biased signaling repertoire in vivo. Mol Pharmacol. 2015;87:706–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kamato D, Bhaskarala VV, Mantri N, et al. RNA sequencing to determine the contribution of kinase receptor transactivation to G protein coupled receptor signalling in vascular smooth muscle cells. PLoS One. 2017;12:e0180842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nava M, Dutta P, Zemke NR, Farias‐Eisner R, Vadgama JV, Wu Y. Transcriptomic and ChIP‐sequence interrogation of EGFR signaling in HER2+ breast cancer cells reveals a dynamic chromatin landscape and S100 genes as targets. BMC Med Genomics. 2019;12:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Blumenberg M. Profiling and metaanalysis of epidermal keratinocytes responses to epidermal growth factor. BMC Genomics. 2013;14:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Köstler WJ, Zeisel A, Körner C, et al. Epidermal growth‐factor‐induced transcript isoform variation drives mammary cell migration. PLoS One. 2013;8:e80566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gurevich EV, Tesmer JJG, Mushegian A, Gurevich VV. G protein‐coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rajagopal S, Shenoy SK. GPCR desensitization: Acute and prolonged phases. Cell Signal. 2018;41:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Anon . G protein‐coupled receptor kinases: Specific phosphorylation of 7TM receptors and beyond. Drug Discov Today Technol. 2010;7:e1–e94. [DOI] [PubMed] [Google Scholar]
- 71. Trester‐Zedlitz M, Burlingame A, Kobilka B, von Zastrow M. Mass spectrometric analysis of agonist effects on posttranslational modifications of the beta‐2 adrenoceptor in mammalian cells. Biochemistry. 2005;44:6133–6143. [DOI] [PubMed] [Google Scholar]
- 72. Fredericks ZL, Pitcher JA, Lefkowitz RJ. Identification of the G protein‐coupled receptor kinase phosphorylation sites in the human β2‐adrenergic receptor. J Biol Chem. 1996;271:13796–13803. [DOI] [PubMed] [Google Scholar]
- 73. Nobles KN, Xiao K, Ahn S, et al. Distinct phosphorylation sites on the β(2)‐adrenergic receptor establish a barcode that encodes differential functions of β‐arrestin. Sci Signal. 2011;4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wisler JW, DeWire SM, Whalen EJ, et al. A unique mechanism of beta‐blocker action: Carvedilol stimulates beta‐arrestin signaling. Proc Natl Acad Sci U S A. 2007;104:16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tobin AB, Butcher AJ, Kong KC. Location, location, location…site‐specific GPCR phosphorylation offers a mechanism for cell‐type‐specific signalling. Trends Pharmacol Sci. 2008;29:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Butcher AJ, Prihandoko R, Kong KC, et al. Differential G‐protein‐coupled receptor phosphorylation provides evidence for a signaling bar code. J Biol Chem. 2011;286:11506–11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yang F, Yu X, Liu C, et al. Phospho‐selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and 19F‐NMR. Nat Commun. 2015;6:8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yang Z, Yang F, Zhang D, et al. Phosphorylation of G protein‐coupled receptors: From the barcode hypothesis to the flute model. Mol Pharmacol. 2017;92:201–210. [DOI] [PubMed] [Google Scholar]
- 79. Lau EK, Trester‐Zedlitz M, Trinidad JC, et al. Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci Signal. 2011;4:ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Doll C, Pöll F, Peuker K, Loktev A, Glück L, Schulz S. Deciphering μ‐opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br J Pharmacol. 2012;167:1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen YJ, Oldfield S, Butcher AJ. Identification of phosphorylation sites in the COOH‐terminal tail of the μ‐opioid receptor. J Neurochem. 2013;124:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Just S, Illing S, Trester‐Zedlitz M, et al. Differentiation of opioid drug effects by hierarchical multi‐site phosphorylation. Mol Pharmacol. 2013;83:633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yousuf A, Miess E, Sianati S, Du Y‐P, Schulz S, Christie MJ. Role of phosphorylation sites in desensitization of μ‐opioid receptor. Mol Pharmacol. 2015;88:825–835. [DOI] [PubMed] [Google Scholar]
- 84. Miess E, Gondin AB, Yousuf A, et al. Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid μ‐opioid receptor desensitization. Sci Signal. 2018;11:eaas9609. [DOI] [PubMed] [Google Scholar]
- 85. Ishii K, Yamamoto S, Muraki T, Kato R. Partial agonistic action of morphine in the rat vas deferens. Jpn J Pharmacol. 1981;31:905–909. [DOI] [PubMed] [Google Scholar]
- 86. Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of Mu‐opioid receptors. J Biol Chem. 2003;278:18776–18784. [DOI] [PubMed] [Google Scholar]
- 87. McPherson J, Rivero G, Baptist M, et al. μ‐Opioid receptors: Correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010;78:756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gillis A, Gondin AB, Kliewer A, et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal. 2020;13:eaaz3140. [DOI] [PubMed] [Google Scholar]
- 89. Glück L, Loktev A, Moulédous L, Mollereau C, Law P‐Y, Schulz S. Loss of morphine reward and dependence in mice lacking G protein‐coupled receptor kinase 5. Biol Psychiatry. 2014;76:767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kelly E, Bailey CP, Henderson G. Agonist‐selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153:S379–S388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Johnson EA, Oldfield S, Braksator E, et al. Agonist‐selective mechanisms of μ‐opioid receptor desensitization in human embryonic kidney 293 cells. Mol Pharmacol. 2006;70:676–685. [DOI] [PubMed] [Google Scholar]
- 92. Bailey CP, Kelly E, Henderson G. Protein kinase C activation enhances morphine‐induced rapid desensitization of μ‐opioid receptors in mature rat locus ceruleus neurons. Mol Pharmacol. 2004;66:1592–1598. [DOI] [PubMed] [Google Scholar]
- 93. Bailey CP, Oldfield S, Llorente J, et al. Involvement of PKCα and G‐protein‐coupled receptor kinase 2 in agonist‐selective desensitization of μ‐opioid receptors in mature brain neurons. Br J Pharmacol. 2009;158:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bailey CP, Llorente J, Gabra BH, et al. Role of protein kinase C and mu‐opioid receptor (MOPr) desensitization in tolerance to morphine in rat locus coeruleus neurons. Eur J Neurosci. 2009;29:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Levitt ES, Williams JT. Morphine desensitization and cellular tolerance are distinguished in rat locus ceruleus neurons. Mol Pharmacol. 2012;82:983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lowe JD, Bailey CP. Functional selectivity and time‐dependence of μ‐opioid receptor desensitization at nerve terminals in the mouse ventral tegmental area. Br J Pharmacol. 2015;172:469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Terman GW, Jin W, Cheong Y‐P, et al. G‐protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol. 2004;141:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hull LC, Llorente J, Gabra BH, et al. The effect of protein kinase C and G protein‐coupled receptor kinase inhibition on tolerance induced by μ‐opioid agonists of different efficacy. J Pharmacol Exp Ther. 2010;332:1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Feng B, Li Z, Wang JB. Protein kinase C‐mediated phosphorylation of the μ‐opioid receptor and its effects on receptor signaling. Mol Pharmacol. 2011;79:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Illing S, Mann A, Schulz S. Heterologous regulation of agonist‐independent μ‐opioid receptor phosphorylation by protein kinase C. Br J Pharmacol. 2014;171:1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chu J, Zheng H, Zhang Y, Loh HH, Law P‐Y. Agonist‐dependent mu‐opioid receptor signaling can lead to heterologous desensitization. Cell Signal. 2010;22:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Al‐Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor‐dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Williams JT, Ingram SL, Henderson G, et al. Regulation of μ‐opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev. 2013;65:223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand‐directed c‐Jun N‐terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci U S A. 2010;107:11608–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Grecksch G, Just S, Pierstorff C, et al. Analgesic tolerance to high‐efficacy agonists but not to morphine is diminished in phosphorylation‐deficient S375A μ‐opioid receptor knock‐in mice. J Neurosci. 2011;31:13890–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Smith FL, Lohmann AB, Dewey WL. Involvement of phospholipid signal transduction pathways in morphine tolerance in mice. Br J Pharmacol. 1999;128:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Granados‐Soto V, Kalcheva I, Hua X‐Y, Newton A, Yaksh TL. Spinal PKC activity and expression: Role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. [DOI] [PubMed] [Google Scholar]
- 108. Smith FL, Gabra BH, Smith PA, Redwood MC, Dewey WL. Determination of the role of conventional, novel and atypical PKC isoforms in the expression of morphine tolerance in mice. Pain. 2007;127:129–139. [DOI] [PubMed] [Google Scholar]
- 109. Morgan MM, Reid RA, Saville KA. Functionally selective signaling for morphine and fentanyl antinociception and tolerance mediated by the rat periaqueductal gray. PLoS One. 2014;9:e114269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Marcus DJ, Zee M, Hughes A, et al. Tolerance to the antinociceptive effects of chronic morphine requires C‐Jun N‐terminal kinase. Mol Pain. 2015;11:s12990–s12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kuhar JR, Bedini A, Melief EJ, Chiu Y‐C, Striegel HN, Chavkin C. Mu opioid receptor stimulation activates c‐Jun N‐terminal kinase 2 by distinct arrestin‐dependent and independent mechanisms. Cell Signal. 2015;27:1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Withey SL, Hill R, Lyndon A, Dewey WL, Kelly E, Henderson G. Effect of tamoxifen and brain‐penetrant protein kinase C and c‐Jun N‐terminal kinase inhibitors on tolerance to opioid‐induced respiratory depression in mice. J Pharmacol Exp Ther. 2017;361:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick‐Davis K, Teitler M. Risperidone irreversibly binds to and inactivates the h5‐HT7 serotonin receptor. Mol Pharmacol. 2006;70:1264–1270. [DOI] [PubMed] [Google Scholar]
- 114. Knight JA, Smith C, Toohey N, Klein MT, Teitler M. Pharmacological analysis of the novel, rapid, and potent inactivation of the human 5‐Hydroxytryptamine7 receptor by risperidone, 9‐OH‐Risperidone, and other inactivating antagonists. Mol Pharmacol. 2009;75:374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Schattauer SS, Land BB, Reichard KL, et al. Peroxiredoxin 6 mediates Gαi protein‐coupled receptor inactivation by cJun kinase. Nat Commun. 2017;8:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Henderson‐Redmond AN, Nealon CM, Davis BJ, et al. c‐Jun N terminal kinase signaling pathways mediate cannabinoid tolerance in an agonist‐specific manner. Neuropharmacology. 2020;164:107847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor‐binaltorphimine, potent and selective kappa‐opioid receptor antagonists. Life Sci. 1987;40:1287–1292. [DOI] [PubMed] [Google Scholar]
- 118. Bruchas MR, Yang T, Schreiber S, et al. Long‐acting κ opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating C‐Jun N‐terminal kinase. J Biol Chem. 2007;282:29803–29811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long‐lasting antagonistic actions of nor‐binaltorphimine (nor‐BNI) in the mouse tail‐flick test. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]