

Abstract
Exocyst is an evolutionarily conserved hetero‐octameric tethering complex that plays a variety of roles in membrane trafficking, including exocytosis, endocytosis, autophagy, cell polarization, cytokinesis, pathogen invasion, and metastasis. Exocyst serves as a platform for interactions between the Rab, Rho, and Ral small GTPases, SNARE proteins, and Sec1/Munc18 regulators that coordinate spatial and temporal fidelity of membrane fusion. However, its mechanism is poorly described at the molecular level. Here, we determine the molecular architecture of the yeast exocyst complex by an integrative approach, based on a 3D density map from negative‐stain electron microscopy (EM) at ~16 Å resolution, 434 disuccinimidyl suberate and 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride cross‐links from chemical‐crosslinking mass spectrometry, and partial atomic models of the eight subunits. The integrative structure is validated by a previously determined cryo‐EM structure, cross‐links, and distances from in vivo fluorescence microscopy. Our subunit configuration is consistent with the cryo‐EM structure, except for Sec5. While not observed in the cryo‐EM map, the integrative model localizes the N‐terminal half of Sec3 near the Sec6 subunit. Limited proteolysis experiments suggest that the conformation of Exo70 is dynamic, which may have functional implications for SNARE and membrane interactions. This study illustrates how integrative modeling based on varied low‐resolution structural data can inform biologically relevant hypotheses, even in the absence of high‐resolution data.
Keywords: chemical cross‐linking mass spectrometry, EM, exocytosis, integrative modeling, membrane fusion, protein cross‐linking, SNAREs, structural models, yeast exocyst complex
Abbreviations
- CATCHR
complexes associated with tethering containing helical rods
- CXMS
chemical‐crosslinking mass spectrometry
- DSS
disuccinimidyl suberate
- EDC
1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride
- EM
electron microscopy
- GMM
Gaussian mixture model
- IMP
integrative modeling platform
- LC/MS
liquid chromatography and mass spectrometry
- MTC
multisubunit tethering complex
- PH domain
Pleckstrin homology
- PMI
python modeling interface
- RMSD
root‐mean‐square deviation
- RMSF
root‐mean‐square fluctuation
- SNARE
soluble NSF attachment protein receptor
1. INTRODUCTION
Vesicle trafficking and fusion in eukaryotic cells are accurately regulated to ensure that protein and lipid cargos are delivered to the correct destinations. At the plasma membrane, regulation is carried out by a multisubunit tethering complex (MTC) called exocyst, which is required for quality control of fusion of secretory vesicles at appropriate exocytosis sites.1, 2, 3, 4, 5, 6, 7 Exocyst and other MTCs function to recognize the correct vesicles and regulate SNARE complex assembly for membrane fusion. These functions are required for many fundamental cellular processes, such as secretion, cell growth, cell–cell communication, and cell division.1, 6, 8
The exocyst complex is a hetero‐octameric MTC composed of eight subunits, including Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70, and Exo84, ranging in size from ~650 to 1,300 amino acid residues (Figure 1a). Most of these subunits were discovered in a yeast secretory pathway screen.2, 9, 10 The phenotype of the yeast mutants at the nonpermissive temperature is the accumulation of secretory vesicles that do not fuse, resulting in failed cargo delivery. High‐resolution crystal structures of Exo70, Exo84, Sec3, Sec15, Sec10, and Sec6 domains indicate that the subunits are mainly elongated, consisting of helical bundles stacked on top of each other. Subunits without available crystal structures were predicted to contain similar helical bundles.11, 12, 13, 14, 15 These helical bundles indicate that the exocyst belongs to the complexes associated with tethering containing helical rods (CATCHRs) family of MTCs, 16 along with Dsl1, Golgi‐associated retrograde protein, endosome‐associated retrograde protein, and conserved oligomeric Golgi complexes.17, 18, 19, 20 In addition to the helical bundles, the exocyst subunits Sec3 and Exo84 (and Sec5 in mammals) have domains that bind small GTPases and/or PI(4,5)P2.21, 22, 23
FIGURE 1.
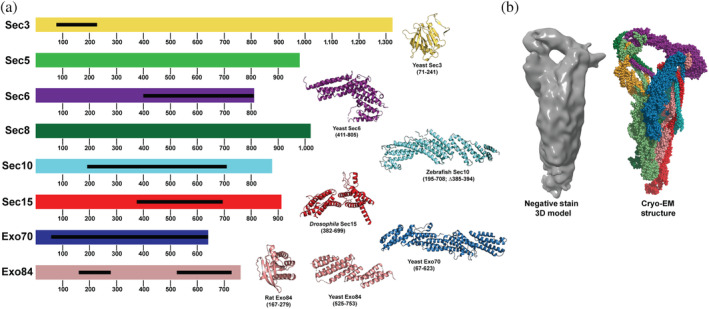
Diagram of the exocyst subunit domain arrangements and high‐resolution structures. (a) Saccharomyces cerevisiae exocyst complex consists of eight subunits ranging from ~650 to 1,300 residues. Black bars show crystallized regions of each subunit, which are shown on the right. Structures were determined of exocyst domains from yeast,11, 12, 22, 23 rat, 21 zebrafish, 14 and Drosophila. 13 (b) Comparison between the 16 Å negative stain electron microscopy (EM) density and the cryo‐EM model of exocyst, showing similar architectures and overall structures
We previously obtained negative‐stain EM 2D class averages of the yeast exocyst and used an auxin‐inducible degron system to selectively degrade exocyst subunits, 24 which showed that the yeast exocyst is stably assembled from two 4‐subunit modules or half‐complexes. Full molecular architecture of the yeast exocyst was revealed by the cryo‐EM structure determined at 4.4 Å resolution. 25 Each 4‐subunit module is stabilized by a four‐helix bundle, called CorEx. Each CorEx bundle consists of one helix from the N‐terminus of each of the subunits, except for Sec3 that contributes a helix from the middle of the sequence. Recently, we also determined a 3D negative‐stain EM map of the entire octameric exocyst 26 (Figure 1b). As seen by both negative‐stain EM24, 26 and cryo‐EM, 25 the structure appears compact and “closed.” This form is in stark contrast with structures of other CATCHR MTCs, whose subunits protrude from the complex in extended conformations,18, 20 suggesting direct roles in tethering vesicles to the appropriate target membranes. Many protein–protein interactions between exocyst subunits and its partners have been determined over the years, but most have not been characterized in relation to the fully assembled complex. Our recent studies indicate that the closed conformation of exocyst needs to be activated by one or more of its partners to open into a functional conformation. 26
Here, we determined the molecular architecture of the yeast exocyst complex using an integrative approach that depends on multiple types of experimental data to maximize the accuracy, precision, completeness, and efficiency of structure characterization. By bringing together varied low‐resolution structural information, we aimed to illustrate the power of the integrative approach in the absence of high‐resolution data. We also aimed to annotate the exocyst function by comparing our integrative structure with a previously determined cryo‐EM structure in light of new biochemical data.
2. RESULTS
2.1. Chemical cross‐linking of exocyst
To obtain structural information about the exocyst, purified complexes were subjected to extensive chemical cross‐linking with both EDC (0 Å linker length) and DSS (11.4 Å linker length), followed by mass spectrometry (CXMS) identification of the cross‐linked peptides (Section 4). This approach identified 434 unique intra‐ and intersubunit cross‐linked pairs of residues (Figure 2 and Table S1).
FIGURE 2.
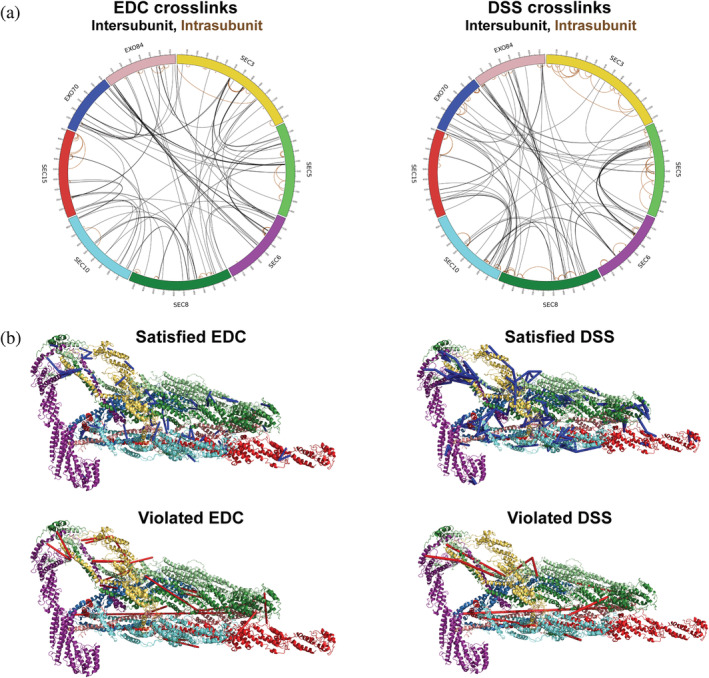
Chemical cross‐linking and mass spectrometry reveal connectivity within the exocyst. (a) Both EDC (left) and DSS (right) crosslinks are shown with intermolecular (black) and intramolecular (brown) crosslinks indicated. The map was generated using the Circos‐CX software (http://cx-circos.net/). (b) The EDC and DSS crosslinks are mapped onto the previously published cryo‐EM structure of exocyst, 25 showing those that satisfy the distance restraint (<35 Å for DSS and <25 Å for EDC; blue) or violate it (>35 Å for DSS and >25 Å for EDC; red)
2.2. Integrative structure determination of the exocyst complex
To model the octameric yeast exocyst structure, we followed the previously described four‐stage workflow (Figure 3, Supplementary Methods, Table S2).27, 28, 29, 30, 31, 32 Input information included the 16 Å negative‐stain EM density map, 26 the 434 chemical cross‐links (Section 2.1), as well as the X‐ray structures,11, 12, 13, 14, 22 and comparative models for 51% of the residues in the 8 exocyst subunits. Stereochemically feasible exocyst structures were sampled to compute an ensemble containing 9,741 models that satisfy the experimental restraints sufficiently well (Figure S1, Section 4, and Supplementary Methods). The ensemble corresponds to a single cluster of models with the root‐mean‐square fluctuation (RMSF) of 38 Å (defining the precision of the integrative structure; Figure 4a,b). The centroid structure (the structure with the minimal sum of root‐mean‐square deviations (RMSD) from all the other structures in the ensemble) is used as the representative model.
FIGURE 3.
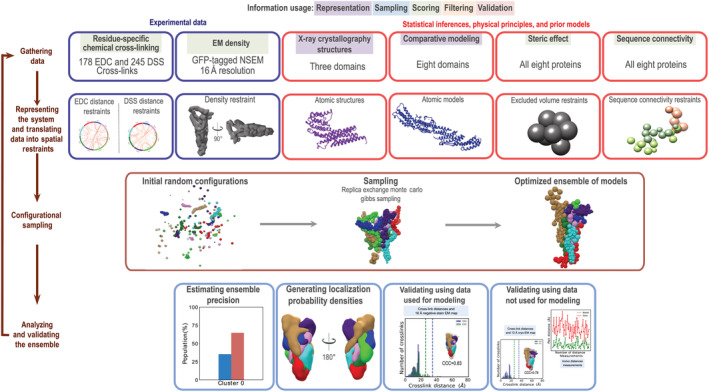
Integrative structure determination of the yeast exocyst complex. Integrative structure determination of the architecture of exocyst complex proceeded through four stages: (1) Gathering data, (2) representing subunits and translating data into spatial restraints, (3) configurational sampling to produce an ensemble of structures that satisfies the restraints, and (4) analyzing and validating the ensemble structures and data. The modeling protocol (i.e., Stages 2, 3, and 4) was scripted using the python modeling interface (PMI) package, version 4d97507, a library for modeling macromolecular complexes based on our open‐source integrative modeling platform (IMP) package, version 2.8 (https://integrativemodeling.org). Details are provided in Supplementary Methods; files containing the input data, scripts, and output results are available at http://salilab.org/exocyst
FIGURE 4.
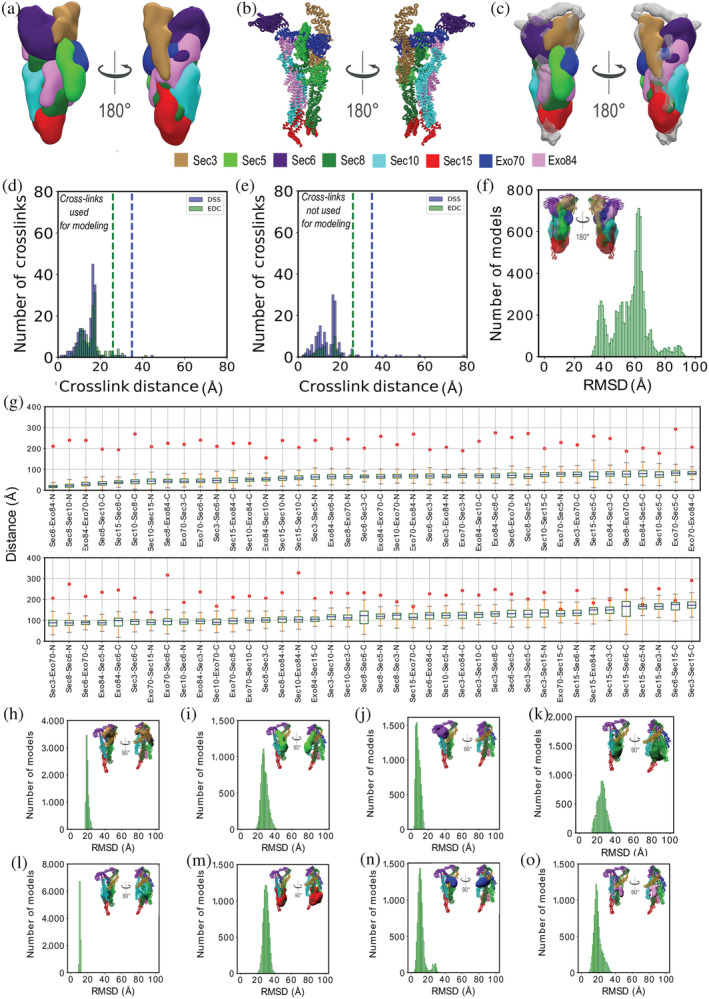
Validation of the integrative exocyst structure. (a) Localization probability densities of the exocyst subunits from the solution cluster. Left and right images represent two views at 180° rotation. (b) Atomic representation of the centroid structure of the solution cluster. Left and right images represent two distinct views at 180° rotation. (c) Fit of the localization probability density of the exocyst subunits from the solution cluster to input EM map shown in mesh representation. (d) Euclidean Cα–Cα distance distributions of our measured cross‐links in the ensemble of solutions for the cluster (Table S1). The y‐axis details the normalized number of cross‐links that were mapped onto the model, x‐axis details the distances. Dashed blue and green lines denotes the expected maximum reach of a DSS and EDC cross‐link respectively. Satisfied DSS and EDC cross‐links, with Cα–Cα distances that fall within the distance threshold of 35 and 25 Å in at least one exocyst structure in the solution ensemble, are shown in blue and green, respectively. (e) Euclidean Cα–Cα distance distributions of all previously reported cross‐links 25 in the solution ensemble. Satisfied DSS and EDC cross‐links, with Cα–Cα distances that fall within the distance threshold of 35 and 25 Å in at least one exocyst structure in the solution ensemble, are shown in blue and green, respectively. (f) RMSD distance distribution between regions represented at atomic resolution (51% of the structure) in the solution ensemble and PDB ID http://firstglance.jmol.org/fg.htm?mol=5YFP, after a global least‐squares superposition (Section 2). The structure on the inset shows the fit of the localization probability densities of the exocyst subunits in the solution cluster to the published cryo‐EM map (EMDB ID 6827). (g) Euclidean Cα–Cα distance measurements of previously published in vivo distance measurements 33 in the solution ensemble. Box plot for each measurement across the solution ensemble is compared with the reported mean distance (red circles). (h–o) Comparison of subunits in the integrative structure with the cryo‐EM structure (PDB: http://firstglance.jmol.org/fg.htm?mol=5YFP) upon a global least‐squares superposition. Figures h–o represent RMSD distributions of subunits Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70, and Exo84, respectively
2.3. Validation of the integrative structure
We begin by assessing how well the model ensemble satisfies the input cross‐links and negative‐stain EM density map. The ensemble satisfies the data well. First, 98% (255 DSS and 170 EDC) of cross‐links are satisfied by at least one model in the cluster (Figure 4a,d). Most of the violations are small (<5%), and can be rationalized by experimental uncertainty, local structural fluctuations, coarse‐grained representation of the model, and/or finite structural sampling. Second, the shape of the negative‐stain EM density map also overlaps well with that of the ensemble; the correlation coefficient between the map and the ensemble is 0.83 (Figure 4c).
Next, we assessed the model by cross‐validation against the previously published cryo‐EM density map, 25 cross‐links, 25 and distances from in vivo fluorescence microscopy in live yeast cells. 33
First, as for the negative‐stain EM map, the shape of the cryo‐EM density map also overlaps well with that of the ensemble; the correlation coefficient between the cryo‐EM map and the ensemble is 0.78 (Figure 4f). However, the Sec15 and Sec6 subunits do not fit the map as well as other subunits, resulting in an unoccupied volume in the EM map (shaded gray region in Figure 4c,f). Both these subunits are also poorly resolved in the negative‐stain EM 2D averages 24 and the cryo‐EM structure. 25 This could be due to three reasons, (a) sample heterogeneity or the presence of multiple states in the sample, (b) dynamics or fluctuations in Sec15 and Sec6, or (c) lack of sufficient data.
Second, the integrative model satisfied 284 (211 DSS and 73 EDC, or 98%) out of the 290 previously published inter‐ and intrasubunit DSS and EDC cross‐links 25 (Figure 4e), even though these cross‐links were not used to compute the model; four of the six violated cross‐links are violated by less than 10 Å. Out of these 290 cross‐links, 139 overlap between the datasets (119 DSS and 20 EDC cross‐links).
Finally, we also validated our model based on 80 intersubunit distance measurements obtained by in vivo fluorescence imaging experiments. 33 The authors converted these distance measurements into a unique threshold for each of the distances between the two labeled subunits 33 (Figure 4g). All 80 distance thresholds were satisfied by at least one model in the ensemble (Figure 4g). Each model in the ensemble satisfies at least 70 of the 80 distance thresholds (Figure 4g).
In conclusion, our confidence in the integrative model is increased by its consistency with the data used to compute it (our negative‐stain EM map and cross‐links) and the data not used to compute it (previously published cryo‐EM map, cross‐links, and in vivo fluorescence images).
2.4. Comparison of integrative and cryo‐EM structures
We now compare the representative integrative structure (Supplementary Methods) and the cryo‐EM structure (PDB: http://firstglance.jmol.org/fg.htm?mol=5YFP). 25 Upon a global least‐squares superposition, the average Cα RMSD value for the regions represented at atomic resolution in both structures is 57 Å (Figure 4f). Using these global superpositions, we also quantified the similarity in individual subunit positions, orientations, and conformations. The subunit Cα RMSD values range from 10 to 32 Å. The subunit with one of the highest atomic coverages (74% of the atoms are represented explicitly), Sec10, has the highest degree of similarity in position, orientation, and conformation in the two structures (Cα RMSD of 10 Å). In contrast, the position, orientation, and conformation of the subunit with the lowest atomic coverage (33%), Sec15, is most different with an RMSD of 30 Å (Table S2).
We now interpret the observed differences between the two structures. In general, differences between two models can arise from a combination of two factors. First, a difference can appear because of the uncertainty of both models. Second, a difference can also appear because of the actual variation between the two structures, resulting from the different samples and/or conditions used to obtain the data for the two structure determinations. We attempt to deconvolute the two sources of difference as follows. The uncertainty of the integrative model (model precision is 38 Å) is most likely substantially higher than that of the cryo‐EM structure (based on a cryo‐EM map at resolution of 4.4 Å). Therefore, to visualize the significance of the structural differences between the integrative and cryo‐EM structures, we plot the distribution of the RMSD values between the cryo‐EM structure and each of the ensemble models (Figure 4h–o). The cryo‐EM structure is within the integrative ensemble density except for Sec5, indicating there is no significant difference between the two structures other than for Sec5. In other words, at the precision of the integrative model, the actual structural difference between the two structures is limited to the position, orientation, and conformation of Sec5. Most of the difference is due to changes in position and orientation, as the conformations of the Sec5 models are similar (Cα RMSD of 5 Å).
2.5. Comparison of cross‐links and negative‐stain EM map with the cryo‐EM structure
We now assess how well the cryo‐EM structure satisfies our cross‐links and negative‐stain EM density map. The cryo‐EM structure satisfies 78% of the mapped EDC cross‐links (83 of 106) and 95% of the mapped DSS cross‐links (189 of 199) (Figure 2b). While these cross‐link satisfactions are less than that for the integrative structure (96 and 99%, respectively), our cross‐links were not used in computing the cryo‐EM structure and the degree of satisfaction is still high. The cryo‐EM structure also fits the negative‐stain EM density map well, with a cross correlation coefficient of 0.86, which is comparable to that for the integrative model (0.83). In conclusion, our cross‐links and negative‐stain EM density map are consistent with the cryo‐EM structure.
2.6. Structural interpretations for biochemical data
While the exocyst complex has been studied for many decades, its structure and function are only beginning to be understood. There is considerable published and unpublished biochemical data on the yeast complex as well as exocyst complexes from other species. Here, we use the structures to help interpret several types of biochemical data.
2.6.1. Protein–protein interactions
Exocyst subunits interact with each other as well as with a number of different protein and lipid factors.1, 3, 6, 7, 12, 15, 34, 35, 36, 37 Many of these interactions are critical for the stability of exocyst, the recognition of secretory vesicles (via Rab GTPases and the type V myosin, Myo2) at specific sites on the plasma membrane (Rho GTPases, PI(4,5)P2, and Ral GTPase in mammals), and the regulation of membrane fusion through interactions with the SNARE proteins and Sec1/Munc18. The regions of the exocyst that interact with many of these partners are poorly resolved in our integrative model, the negative‐stain EM map, 24 and the cryo‐EM structure. 25 Examples include Sec6 interacting with SNARE and Sec1 binding partners,38, 39, 40, 41 the Sec3 N‐terminal region interacting with Rho GTPases, PIP2 and the Sso1/2 SNARE,22, 23, 42 and Sec15 interacting with the Rab GTPase Sec4 and Myo2.43, 44 This observation suggests that these regions are flexible in the absence of their binding partners.
2.6.2. Limited proteolysis
To reveal exposed surfaces of exocyst, the assembled complex was incubated with trypsin in solution overnight, and the digested peptides were analyzed by liquid chromatography and mass spectrometry (LC/MS). Generally, the digested regions mapped to flexible loops and short helices in the cryo‐EM structure, consistent with the solvent accessibility of those regions (Figure 5a). Exo70 was the second‐most cleaved subunit after Sec3 (discussed in the next section). Not surprisingly, an exposed helix that links two of the Exo70 helical bundles was digested. However, several buried regions of Exo70, including its CorEx helix, were also cleaved. This result suggests that Exo70 is sufficiently dynamic to make its CorEx bundle residues accessible. We showed that gain‐of‐function mutants of Exo70 led to a dynamic conformational change in which Exo70 shifted away from the rest of the complex,26, 45 potentially exposing regions that were cleaved in our limited proteolysis experiment. These dynamics of Exo70 were not observed in the cryo‐EM structure. However, the cryo‐EM data were obtained using glutaraldehyde‐crosslinked complexes, 25 which likely stabilized the complex on grids at the cost of limiting configurational and conformational variability. In contrast, the integrative model reflects a substantial configurational and conformational uncertainty of Exo70 (Figure 4n), although it is not clear to what degree it originates from the lack of data or actual flexibility/heterogeneity (above). Our limited proteolysis data support a model in which exocyst is autoinhibited and undergoes structural rearrangements as part of its tethering and SNARE regulation function.7, 26, 46
FIGURE 5.
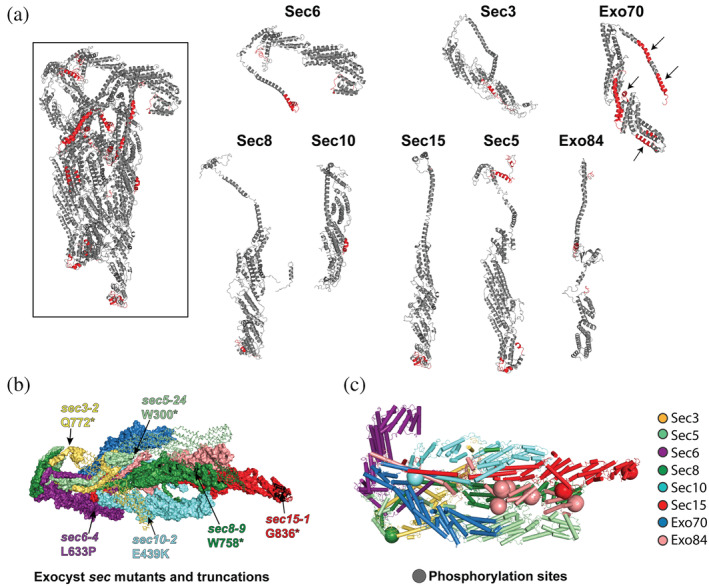
Limited trypsin proteolysis, mutations, and phosphorylation of the assembled exocyst. (a) Limited proteolysis using trypsin identified flexible and exposed regions within the intact exocyst complex. Regions colored in red indicate peptides that were cleaved after an overnight trypsin digest in solution. Arrows point to regions of Exo70 that are buried in the cryo‐EM structure. (b) Regions of exocyst that are truncated or mutated in the original sec mutant screen: 9 sec6‐4 (L633P);11, 50 sec3‐2 is Q772*; sec5‐24 is W300*; sec8‐9 is W758*; sec10‐2 is E439K; sec15‐1 is G836*. Truncated regions are shown as lines. (c) Phospho‐sites mapped to the cryo‐EM structure (Table S3)
2.6.3. Deletions and mutations
Deletion of most exocyst subunits are lethal in eukaryotes. 6 In yeast, the secretory (sec) mutants 9 as well as auxin‐induced degradation studies provided insights into the function of the complex.2, 24, 47, 48, 49 The yeast sec6‐4 mutant (L633P) had previously been sequenced.11, 50 We sequenced several others (sec3‐2 is Q772*; sec5‐24 is W300*; sec8‐9 is W758*; sec10‐2 is E439K; sec15‐1 is G836*). Surprisingly, many mutations result in premature stop codons that truncate exocyst subunits (Figure 5b). These truncated proteins appear to function reasonably well at permissive temperatures; for example, while the yeast cells grow at 20°C, the deletions are destabilizing at higher temperatures and the cells die. 2 In each case, the truncation occurs in the sequence after the CorEx helical bundles, indicating that an essential function of most of the subunits is to stabilize the complex through their CorEx bundles.
2.6.4. Phosphorylation
To investigate possible structural and functional consequences of phosphorylation, we mapped phosphorylation sites (Table S3) from high throughput studies51, 52, 53, 54, 55, 56 and our own MS studies, onto the current cryo‐EM model (Figure 5c). Phosphorylation sites often occur in intrinsically disordered protein regions, and may control disorder–order transitions. 57 Of the total of 49 phosphorylation sites, 40 are in regions absent from the cryo‐EM structure. The remaining nine phosphorylated sites all map on interhelical loops. All 24 of the phosphorylated sites on Sec3 are in the Sec3N region, mostly clustered around the PH domain (Figure 6b). Seventeen sites are in Exo84. In particular, five of these sites lie near interaction surfaces with other subunits (including Sec5, Sec8, and Sec15). These phospho‐sites have previously been implicated in the assembly of the complex, as their mutation leads to defects in secretion and cell growth.58, 59 Other phospho‐sites on Sec5, Sec8, and Sec10 are in the vicinity of regions critical for conformational changes that activate Sec6 for SNARE binding or near SNARE binding sites.26, 39, 40 A phosphorylated serine in the C‐terminus of Sec15 is located in a region near putative Rab and Myo2 binding sites,13, 43 although this region of the cryo‐EM structure is dynamic and poorly resolved. These phosphorylated regions may be critical for regulation of exocyst assembly, vesicle binding, and/or SNARE function.
FIGURE 6.
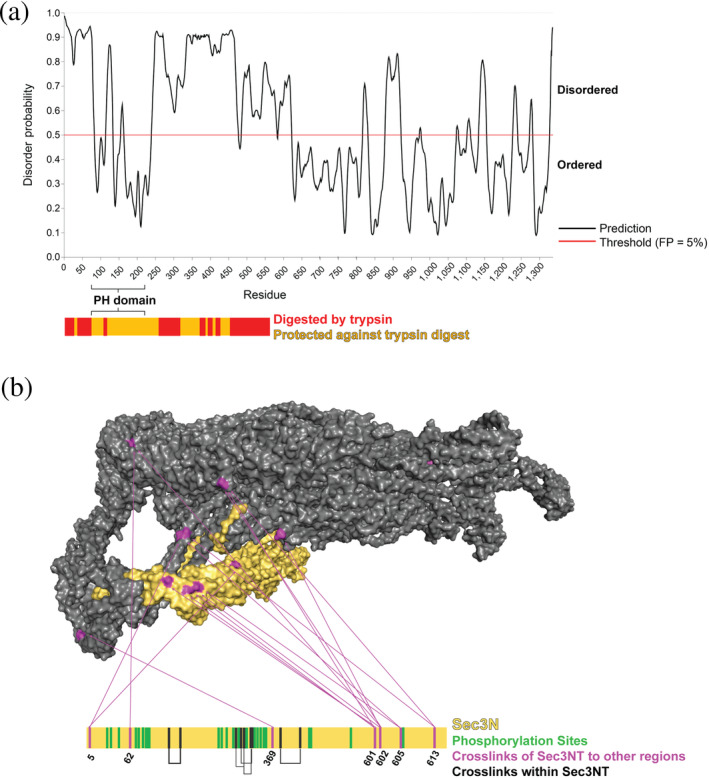
The N‐terminal domain of Sec3 (Sec3N) is disordered and localizes near the C‐terminal region of Sec6. (a) Sec3N (1–611 residues) is predicted to be disordered using PrDOS, 73 with the exception of the PH domain (71–241). The y‐axis gives the probability of each residue to be disordered, and the threshold is the false positive rate. The graph below shows the results from the limited trypsin digestion; the digestion showed that most of Sec3N is susceptible to proteolysis (red), consistent with the predicted disorder in most of this region. (b) CXMS revealed a large cluster of contacts between Sec3N and the region near the C‐terminal end of Sec6 (purple), suggesting that Sec3N can occupy this area in various positions, presumably transiently. Intramolecular Sec3N crosslinks and phosphorylation sites in this domain are shown in black and green, respectively (Table S3)
2.7. The Sec3 N‐terminal region is flexible and proximal to the SNARE‐binding region of Sec6
We now focus on Sec3—the entire Sec3 protein is included in the integrative model, while the N‐terminal half of Sec3 is missing in the cryo‐EM structure. Sec3 is unique among the budding yeast exocyst subunits, as it appears to be substantially longer (1,336 residues) than its homologs in other species, ranging from fission yeast to humans (~800 residues). Most of this difference in length lies in the N‐terminal region (1–611 residues), which is considerably longer in Saccharomyces cerevisiae.60, 61, 62, 63 The length difference may explain the structural differences among the exocyst complexes in different organisms; biochemical studies of the S. cerevisiae exocyst indicate that it functions as a single octameric complex, 24 while smaller subcomplexes appear to exist in other species.6, 64, 65, 66, 67, 68, 69, 70 For example, Sec3 appears to dissociate from the mammalian complex after tethering. 65 In contrast, the longer budding yeast Sec3 protein may extend the physical reach of the complex to promote interactions with other factors during tethering, thus avoiding post‐tethering dissociation by maintaining the complex as a stable octamer and bypassing the need for subcomplexes.
Residues 71–241 of Sec3 fold into a Pleckstrin homology (PH) domain (Figure 1a), which is a key functional region in the N‐terminal half of Sec3 (Sec3N).22, 23, 42 Association of the polybasic region in the PH domain with PI(4,5)P2 was suggested to play an important role in localization of the complex and in tethering. 23 Moreover, this domain binds Sso2 and catalyzes assembly of SNARE complexes, likely by interacting with the inhibitory N‐terminal Habc domain of Sso2 and relieving autoinhibition.42, 71 Interestingly, deletion of Sec3N is not lethal unless combined with phosphoinositide‐binding defective mutants of Exo70, suggesting that functional redundancy exists between the two subunits. 48 This functional redundancy also extends to binding of Rho GTPases, including Cdc42, by both Sec3 and Exo70, 72 suggesting that binding of exocyst to both PI(4,5)P2 and Rho GTPases are essential for cell growth and secretion.
Apart from the PH domain, the rest of Sec3N is predicted by the PrDOS and PONDR algorithms73, 74 to be disordered (Figure 6a). In contrast, the disordered regions in the C‐terminal half of Sec3 were predicted to be only in short loop regions. The N‐terminal flexible regions were confirmed by limited proteolysis experiments of exocyst in solution (Figure 6a). In contrast, in the C‐terminal half of Sec3, only a single loop was cleaved (Figure 6a). The predicted flexibility of the N‐terminal half of Sec3 is consistent with the lack of density for Sec3N in the cryo‐EM model. Heterogeneity in Sec3N phosphorylation could also lead to structural heterogeneity (Table S3 and Figure 6b). Differential phosphorylation of Sec3N is likely to provide regulation; Pkc1 phosphorylation of Sec3 upon wounding of the plasma membrane was shown to lead to Sec3 degradation, exocyst relocalization, and inhibition of exocytosis. 75
Although the position of Sec3N was unresolved in the cryo‐EM model, our CXMS experiments identified the region of exocyst that Sec3N interacts with, albeit transiently (Figure 6b). It had been suggested that Sec3N is located near Sec15, based on a single crosslink between Sec3 and Sec15. 25 In contrast, our more extensive EDC and DSS CXMS revealed that Sec3N forms multiple crosslinks in a region at the other end of the complex, near Sec6 and its SNARE interacting domains (Figure 6b). This crosslinking data informed the integrative model, which localizes the N‐terminal domain of Sec3 near Sec6 (Figure 4a), in agreement with two independent considerations as follows. First, both Exo70 and Sec3 interact with PI(4,5)P2. Therefore, we expect them to be positioned on the same face of exocyst for optimal membrane binding (Figure 4a). Second, as Sec3 and Sec6 are the SNARE‐binding exocyst subunits, they are likely to be close to each other to coordinate SNARE complex assembly.
3. DISCUSSION
3.1. Utility of integrative structure modeling
No experimental method for structure determination is universally applicable. For example, crystals suitable for X‐ray crystallography cannot always be produced, especially for insufficiently homogenous assemblies of multiple components. While cryo‐EM can be used to study large assemblies, it often produces structural models at worse than atomic resolution due to sample heterogeneity and/or a limited number of single particle images. Finally, molecular biology, biochemistry, and proteomics techniques, such as yeast two‐hybrid, affinity purification, and mass spectrometry, yield information about the interactions between proteins, but not the positions of these proteins within the assembly or the atomic structures of the proteins themselves.
One approach to overcome the limitations of individual methods is integrative structure modeling.27, 28, 29, 30, 31, 32, 76, 77, 78, 79 Integrative modeling aims to simultaneously use all available information from any method, including from varied experiments (e.g., EM and chemical cross‐linking), physical theories (e.g., molecular mechanics force field), statistical analyses (e.g., statistical potentials), and prior models (e.g., atomic structures of complex subunits). Therefore, integrative modeling maximizes the accuracy, precision, completeness, and efficiency of structure determination. Numerous structures refractive to traditional methods have already been solved using this approach. 28
This study illustrates how integrative modeling based on low‐resolution structural data can be useful even in the absence of high‐resolution data. In particular, we determined the molecular architecture of the yeast exocyst complex at 38 Å precision (Supplementary Methods) by an integrative approach as implemented in our open source IMP package. 32 Modeling relied on a negative‐stain EM density map at 16 Å resolution, 434 intra‐ and intermolecular cross‐links, partial atomic models of the 8 exocyst subunits, sequence connectivity, and excluded volume. The model was largely consistent with the previously published cryo‐EM structure, 25 chemical cross‐links, and intersubunit distances from in vivo fluorescence measurements in yeast. Importantly, the integrative model reproduces locations of critical functional regions, such as the C‐terminal regions of Sec15 (for vesicle binding) and Sec6 (for SNARE interactions) at opposite ends of the complex, as found in the cryo‐EM structure.
3.2. Biochemical experiments indicate regulatory regions in exocyst
We examined the yeast exocyst structure in relation to previously published as well as new biochemical and genetic data regarding flexible/disordered regions, mutations, and phosphorylation sites (Figures 5 and 6). The data suggests regions that are likely involved in (a) the regulation of the conformation and function of exocyst, (b) the assembly and stability of the complex, and (c) the function of exocyst controlled by phosphorylation. Our cross‐linking data also places the N‐terminal region of Sec3, which interacts with PI(4,5)P2, Rho GTPases, and the plasma membrane SNARE Sso1/2, nearby the region of Sec6 that has been implicated in SNARE and Sec1 binding. This information is critical for beginning to tease apart exocyst's mechanism of action, although further structural and biochemical information will be necessary for a molecular understanding of this fundamental process.
In conclusion, our integrative exocyst model is similar to the previous structure based on a cryo‐EM density map at 4.4 Å resolution. Therefore, it allows us to reach many of the same conclusions about the function of exocyst. Because chemical cross‐links and negative‐stain EM density map are often much easier to determine than a higher resolution cryo‐EM density map, this study illustrates the efficacy of integrative structure determination. Thus, the integrative approach can facilitate rationalizing existing data and suggesting hypotheses for future experiments, even in the absence of higher resolution cryo‐EM data.
4. MATERIALS AND METHODS
4.1. Cross‐linking and mass spectrometry analyses
4.1.1. EDC and DSS cross‐linking of the exocyst complex
Exocyst was isolated from cells based on the previously described protocol. 24 Briefly, 8 L of either GFP‐Exo84/Sec15‐PrA (for EDC crosslinking) or Sec8‐GFP (for DSS crosslinking) were grown to an approximate OD600 of 1.5, and lysed using a ball mill grinder. 80 The frozen powder was resuspended in 40 ml of 40 mM Tris pH 8.0 and 200 mM sodium citrate lysis buffer with a Roche protease inhibitor tablet and spun at 13,000 rpm to remove cell debris. The soluble lysate was then incubated with either IgG‐conjugated Dynabeads (EDC) or anti‐GFP nanobody Dynabeads (DSS) for 1 hr at 4°C. The beads were washed with 600 μl of lysis buffer three times, then washed with 600 μl of 20 mM MES pH 6.5, 250 mM NaCl, 2 mM MgCl2, 0.1% CHAPs, and 1 mM DTT, 3×. The slurry for EDC was resuspended in 100 μl of 20 mM MES pH 6.5, 250 mM NaCl, 2 mM MgCl2, 0.1% CHAPs, and 1 mM DTT, plus PreScission protease, for 1.5 hr at 4°C. The eluted exocyst was then treated with either 2.5 mM EDC and 5 mM NHS for 30 min at 25°C. The reaction was quenched by adding Tris pH 8.0 and β‐mercaptoethanol to a final concentration of 50 and 20 mM, respectively. For DSS, either 5 or 10 g of frozen yeast powder was used, the cross‐linking was carried out on beads, and the peptides eluted by trypsin digest. 81
4.1.2. Proteolytic digestion of EDC crosslinked exocyst complex
After EDC cross‐linking, the complex was reduced by 10 mM tris‐(2‐carboxyethyl)‐phosphine (Invitrogen, MA) at 80°C for 15–20 min, cooled to room temperature and alkylated by 50 mM iodoacetamide for 20 min in the dark to block the formation of disulfide bonds. After reduction and alkylation, the cross‐linked complexes were separated by 3–12% SDS‐PAGE (NuPAGE Bis‐Tris, Invitrogen) to reduce the complexity of the sample. For in‐gel digestion, the high‐molecular‐weight‐region gel bands (>220 kDa, estimated by the high‐molecular‐weight protein markers, Invitrogen) corresponding to the cross‐linked exocyst proteins were sliced and proteolysed by trypsin as previously described. 81 In brief, gel plugs were crushed into small pieces, 0.5 μg of sequencing‐grade trypsin (Promega, Madison, WI) per ~10 μg protein was added with subsequent 6–8 hr incubation. Peptides were extracted by formic acid and acetonitrile, desalted on C18 cartridges (Sep‐Pak, Waters, MA) and snap‐frozen before fractionation.
4.1.3. Fractionation of cross‐linked peptides by high pH chromatography
For the high pH chromatography fractionation, the tryptic peptides were separated in a home‐made reverse‐phase C18 column in a pipet tip. Peptides were eluted into seven fractions using a stepwise gradient of increasing acetonitrile (6, 9, 12, 15, 18, 22, and 35%) at pH 10 adjusted with ammonium hydroxide, and the 6 and 35% fractions were combined to one fraction. All of the six fractions were lyophilized in a vacuum concentrator (Thermo Fisher Scientific, MA), and analyzed by LC/MS.
4.1.4. Mass spectrometric analysis of cross‐linked peptides
For cross‐link identification, the fractionated peptides were dissolved in the sample loading buffer (2% ACN and 0.2% formic acid) and loaded onto a Picochip® column with an integrated electrospray ionization emitter tip (360 outer diameter, 75 inner diameter with 15 μm tip, New Objective, Woburn, MA). The column was packed with 25 cm of reverse‐phase C18 material (3 μm porous silica, 120‐Å pore size, Dr. Maisch GmbH, Entringen, Germany). Mobile phase A consisted of 2% acetonitrile with 0.1% acetic acid, and mobile phase B consisted of 80% acetonitrile with 0.1% acetic acid. The peptides were eluted in a 90‐min LC gradient (5% B to 12% B, 0–10 min; 12% B to 32% B, 10–75 min; 32% B to 100% B, 75–80 min, and followed by 100% B until 90 min) using an easy nLC‐1200 system (Thermo Fisher) and analyzed with an Q Exactive HFX or QE Plus mass spectrometer (Thermo Fisher). The flow rate was 350 nl/min. The capillary temperature was 250°C, and Funnel RF level was set at 55%. The instrument was operated in the data‐dependent mode, where the top 10 or top 6 most abundant ions were fragmented by HCD (HCD energy 30) and analyzed in the Orbitrap mass analyzer. The target resolution for MS1 was 60,000, and for MS2 it was 7,500. Peptide ions with a charge state of >3 were selected for fragmentation. An auto dynamic exclusion of was used. Other instrumental parameters include the following: Isolation window of 1.8 Th, and minimal threshold of 50,000 to trigger an MS/MS event. The AGC targets were set as 1 × 106 for full mass and 2 × 105 for MS2. The maximal ion injection times for MS1 and MS2 were set as 100 and 200 ms, respectively.
4.1.5. Identification of cross‐linked peptides using pLink
The raw data were transformed to Mascot generic format by pXtract 2.0 and searched by pLink3 using a target decoy search strategy with a concatenated FASTA protein sequence database containing the eight subunits of the exocyst complex. We also included the BSA sequence for the target‐decoy database search to quickly prefilter the false positive identifications containing the BSA sequence. Other search parameters included cysteine carbamidomethylation as a fixed modification, methionine oxidation, and protein N‐terminal methionine cleavage as a variable modification. A maximum of two trypsin missed‐cleavage sites were allowed. The data were filtered using a mass accuracy of MS1 ≤ 10 ppm (parts per million) and MS2 ≤ 20 ppm as specified in the software. We then manually applied additional filters to remove potential false positive identifications from our dataset as previously described. 81 The cross‐linking data were analyzed and plotted by an online software tool, CX‐Circos (http://cx-circos.net).
4.2. Integrative structure modeling of the exocyst complex
The architecture of the yeast exocyst complex was determined using the integrative structure modeling approach described previously.27, 28, 29, 30, 31, 32, 76, 77, 78, 79 All available structural information on the exocyst complex was used for computational analyses (Table S2, Figure 3, and Section 4). Representations of individual subunits relied on (a) atomic structures of three yeast exocyst subunit domains, determined by X‐ray crystallography (PDB codes http://firstglance.jmol.org/fg.htm?mol=2FJI, http://firstglance.jmol.org/fg.htm?mol=2D2S, and http://firstglance.jmol.org/fg.htm?mol=2B1E)11, 12, 22 and (b) eight comparative models built with MODELLER 9.13 82 based on known template structures detected by HHPred83, 84 (Table S2 and Section 4). The atomic regions of the exocyst subunits were represented by 12 rigid bodies, corresponding to different domains of the eight constituent proteins (Table S2). Regions with unknown structures were modeled as flexible strings of beads. The EM density map was approximated by a Gaussian mixture model (GMM) containing 200 components and every 50–100 residues of each subunit was approximated by a GMM component. Finally, the proximity between specific residue pairs was determined by DSS and EDC XL‐MS experiments, which identified a total of 256 DSS and 178 EDC (Table S2 and Figure 4) inter‐ and intrasubunit interactions. The maximum Cα–Cα distance between any two residues cross‐linked by DSS was estimated to be ∼35 Å, and by EDC was estimated to be ~25 Å, based on spacer length, flexibility of side chains, and backbone dynamics.
Next, 2,000,000 exocyst models were computed by optimizing spatial proximities using a Monte Carlo method, as informed by cross‐linking data, EM map, excluded volume, and sequence connectivity from 200 random initial models. The sampling yielded an ensemble of 9,741 models that sufficiently satisfied the input restraints; this entire ensemble is the model of the exocyst, with its uncertainty reflecting both the lack of input data and the actual heterogeneity of the samples used to collect the data. The clustering of these models revealed a single cluster containing the majority (99% or 9,669 models) of the models (Figure S1 and Supplementary Methods). The precision of the cluster defines the overall precision of the structural model (Figure S1). It is quantified by the average root‐mean‐RMSF with respect to the centroid model and is 38 Å for the modeled exocyst structure.
In general, an ensemble can be visualized as a localization probability density map. The map specifies the probability of any volume element being occupied by a given bead in superposed models. The probability localization density for the subunits of exocyst is sufficiently precise to define their position, but not their orientation (Figure 4a).
4.3. In solution limited trypsin proteolysis of native complexes
4.3.1. In‐solution digestion
Ten μg of eluted exocyst (purified as above) was added to 20 μl of 0.1% ProteaseMAX Surfactant (Promega) in 50 mM ammonium bicarbonate, an additional aliquot of 20 μl of 50 mM ammonium bicarbonate was then added. For reduction, a 2 μl aliquot of 45 mM DTT was added and the samples were incubated at 50°C for 30 min. For alkylation of cysteines, a 2 μl aliquot of 100 mM iodoacetamide was added and the samples were incubated at room temperature for 30 min. Tryptic digestion was initiated with a 1 μl aliquot of trypsin (0.2 μg/μl Sigma Proteomics grade). Samples were incubated at 37°C for 18 hr. Tryptic digests were acidified with 4 μl of 5% trifluoroacetic acid (TFA) and lyophilized in Speed Vacc.
4.3.2. LC–MS/MS analysis
A 3 μl injection was loaded by a Thermo Easy‐nLC II in 5% acetonitrile (0.1% formic acid) at 4.0 μl/min for 4.0 min onto a 100 μm I.D. fused‐silica precolumn packed with 2 cm of 5 μm (200 Å) Magic C18AQ (Bruker‐Michrom). Peptides were eluted at 300 nl/min from a 75 μm I.D. gravity‐pulled analytical column packed with 25 cm of 3 μm (100 Å) Magic C18AQ particles using a linear gradient from 5 to 35% of mobile phase B (acetonitrile + 0.1% formic acid) in mobile phase A (water + 0.1% formic acid) over 60 min. Ions were introduced by positive electrospray ionization via liquid junction at 1.4 kV into a Thermo Scientific Velos Pro mass spectrometer. Mass spectra were acquired over m/z 350–2,000 at 60,000 resolution (m/z 200) with an AGC target of 1e5, and data‐dependent acquisition selected the top 10 most abundant precursor ions for tandem mass spectrometry by CID fragmentation using an isolation width of 2 Da, max fill time of 110 ms, and AGC target of 1e5. Peptides were fragmented by a normalized collisional energy of 35, and fragment spectra acquired at a resolution of 17,500 (m/z 200).
4.3.3. Data analysis
Data was analyzed by Mascot Server (version 2.4, Matrix Science) against the S. cerevisiae (Swissprot) FASTA file (downloaded 06/2013). Search parameters included Trypsin/P specificity, up to two missed cleavages, a fixed modification of carbamidomethyl cysteine, and variable modifications of oxidized methionine, pyroglutamic acid for Q, and N‐terminal acetylation. Assignments were made using a 15 ppm mass tolerance for the precursor and 0.05 Da mass tolerance for the fragments. All nonfiltered search results were processed by Scaffold (version 4.0.5, Proteome Software, Inc.) utilizing the Trans‐Proteomic Pipeline (Institute for Systems Biology) with threshold values set at 1% FDR for peptides and 1% FDR for proteins (two peptide minimum) and quantitative comparisons made using the total unique peptide count method with all samples normalized by total ion current for the run.
AUTHOR CONTRIBUTIONS
Sai Ganesan: Conceptualization; methodology; validation; visualization; writing‐original draft; writing‐review and editing. Michael Feyder: Conceptualization; formal analysis; investigation; visualization; writing‐original draft; writing‐review and editing. Ilan Chemmama: Conceptualization; validation; visualization; writing‐review and editing. Fei Fang: Data curation; formal analysis; methodology; validation; visualization; writing‐review and editing. Michael Rout: Conceptualization; resources; writing‐review and editing. Brian Chait: Conceptualization; formal analysis; funding acquisition; project administration; validation; writing‐review and editing. Yi Shi: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; supervision; validation; visualization; writing‐review and editing. Mary Munson: Conceptualization; funding acquisition; project administration; writing‐original draft; writing‐review and editing. Andrej Sali: Conceptualization; funding acquisition; project administration; supervision; writing‐original draft; writing‐review and editing.
CODE AVAILABILITY
The integrative structure modeling scripts are available at https://salilab.org/exocyst.
MODEL AVAILABILITY
The integrative exocyst structure has been deposited in the nascent public PDB repository, PDB‐dev (https://pdb-dev.rcsb.rutgers.edu/), accession number PDBDEV‐00000041. Raw data for Figure 4 and Figure S1 are available at https://salilab.org/exocyst.
Supporting information
Appendix S1. Supporting Information.
Figure S1. Thoroughness of configurational sampling and model precision for yeast exocyst complex. (a) Convergence of the model score, for the 9,741 good‐scoring models; the scores do not continue to improve as more models are computed essentially independently. The error bars represent the standard deviations of the best scores, estimated by repeating sampling of models 50 times. (b) Distribution of model scores for model Samples 1 (scarlet) and 2 (blue), comprising the 9,741 good‐scoring structures. The Kolmogorov–Smirnov two‐sample test indicates that the difference between the two score distributions is insignificant (p‐value is .1), thus, the two score distributions are effectively equal. (c) Three criteria for determining the sampling precision (y‐axis), evaluated as a function of the RMSD clustering threshold (x‐axis). First, the p‐value is computed using the χ 2‐test for homogeneity of proportions (red dots). Second, an effect size for the χ 2‐test is quantified by the Cramer's V value (blue triangles). Third, the population of models in sufficiently large clusters (containing at least 10 models from each sample) is shown as green stars. The vertical dotted black line indicates the RMSD clustering threshold at which three conditions are satisfied (p‐value > .05, Cramer's V < 0.10, and the population of clustered models >0.80), thus defining the sampling precision of 51 Å. (d) Populations of Sample 1 and 2 models in the two clusters obtained by threshold‐based clustering using the sampling precision value as threshold. The resulting cluster (Cluster 1) contains 99% of the models and has a precision of 38 Å. The precision of this cluster defines the model or cluster precision. (e) Comparison of localization probability densities of models from Sample 1 (left), Sample 2 (right), and the complete ensemble (center); the cross‐correlation coefficient between the localization densities from Samples 1 and 2 is 0.9.
Table S1. Four hundred and thirty‐four unique disuccinimidyl suberate and 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride cross‐linked pairs of residues.
Table S2. Summary of integrative structure determination of yeast exocyst complex (PDBDEV_00000041).
Table S3. Phosphorylation sites mapped on exocyst from this study, and from high throughput studies (51–56), as well as (58,59).
ACKNOWLEDGMENTS
Thanks to members of the Sali and Munson labs for advice and critical feedback on the manuscript, and the UMass Medical School Proteomics facility for the trypsin limited proteolysis MS data. Many thanks for the technical assistance of Dante Lepore, Billie Reneker, Alexander Czuchra, and Anne Mirza. Work in our laboratories is supported by National Institutes of Health grants GM068803 (M. M), R01 GM112108 (M. P. R.), P41 GM109824 (M. P. R., B. T. C., and A. S.), GM103314 (B. T. C.), R01GM083960 (A. S.), and National Science Foundation graduate research Fellowship 1650113 (to I. E. C.).
Ganesan SJ, Feyder MJ, Chemmama IE, et al. Integrative structure and function of the yeast exocyst complex. Protein Science. 2020;29:1486–1501. 10.1002/pro.3863
Sai J. Ganesan and Michael J. Feyder authors contributed equally to this work.
Funding information National Science Foundation, Grant/Award Number: 1650113; National Institutes of Health, Grant/Award Numbers: GM068803, R01GM083960, GM103314, P41 GM109824, R01 GM112108
Contributor Information
Mary Munson, Email: mary.munson@umassmed.edu.
Andrej Sali, Email: sali@salilab.org.
DATA AVAILABILITY STATEMENT
The EM density map has been deposited in the EMDB, accession code 21226.
REFERENCES
- 1. Heider MR, Munson M. Exorcising the exocyst complex. Traffic. 2012;13:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. TerBush DR, Maurice T, Roth D, Novick P. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae . EMBO J. 1996;15:6483–6494. [PMC free article] [PubMed] [Google Scholar]
- 3. Munson M, Novick P. The exocyst defrocked, a framework of rods revealed. Nat Struct Mol Biol. 2006;13:577–581. [DOI] [PubMed] [Google Scholar]
- 4. Mei K, Guo W. The exocyst complex. Curr Biol. 2018;28:R922–R925. [DOI] [PubMed] [Google Scholar]
- 5. Novick PJ. A Rab effector called the exocyst and related vesicle tether complexes. Ras Superfamily Small G Proteins: Biol Mech. 2014;2:67–79. [Google Scholar]
- 6. Martin‐Urdiroz M, Deeks MJ, Horton CG, Dawe HR, Jourdain I. The exocyst complex in health and disease. Front Cell Dev Biol. 2016;4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lepore DM, Martinez‐Nunez L, Munson M. Exposing the elusive exocyst structure. Trends Biochem Sci. 2018;43:714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu B, Guo W. The exocyst at a glance. J Cell Sci. 2015;128:2957–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post‐translational events in the yeast secretory pathway. Cell. 1980;21:205–215. [DOI] [PubMed] [Google Scholar]
- 10. Guo W, Grant A, Novick P. Exo84p is an exocyst protein essential for secretion. J Biol Chem. 1999;274:23558–23564. [DOI] [PubMed] [Google Scholar]
- 11. Sivaram MV, Furgason ML, Brewer DN, Munson M. The structure of the exocyst subunit Sec6p defines a conserved architecture with diverse roles. Nat Struct Mol Biol. 2006;13:555–556. [DOI] [PubMed] [Google Scholar]
- 12. Dong G, Hutagalung AH, Fu C, Novick P, Reinisch KM. The structures of exocyst subunit Exo70p and the Exo84p C‐terminal domains reveal a common motif. Nat Struct Mol Biol. 2005;12:1094–1100. [DOI] [PubMed] [Google Scholar]
- 13. Wu S, Mehta SQ, Pichaud F, Bellen HJ, Quiocho FA. Sec15 interacts with Rab11 via a novel domain and affects Rab11 localization in vivo. Nat Struct Mol Biol. 2005;12:879–885. [DOI] [PubMed] [Google Scholar]
- 14. Chen J, Yamagata A, Kubota K, Sato Y, Goto‐Ito S, Fukai S. Crystal structure of Sec10, a subunit of the exocyst complex. Sci Rep. 2017;7:40909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Croteau NJ, Furgason ML, Devos D, Munson M. Conservation of helical bundle structure between the exocyst subunits. PLoS One. 2009;4:e4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu IM, Hughson FM. Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol. 2010;26:137–156. [DOI] [PubMed] [Google Scholar]
- 17. Ha JY, Chou HT, Ungar D, Yip CK, Walz T, Hughson FM. Molecular architecture of the complete COG tethering complex. Nat Struct Mol Biol. 2016;23:758–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ren Y, Yip CK, Tripathi A, et al. A structure‐based mechanism for vesicle capture by the multisubunit tethering complex Dsl1. Cell. 2009;139:1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schindler C, Chen Y, Pu J, Guo X, Bonifacino JS. EARP is a multisubunit tethering complex involved in endocytic recycling. Nat Cell Biol. 2015;17:639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chou HT, Dukovski D, Chambers MG, Reinisch KM, Walz T. CATCHR, HOPS and CORVET tethering complexes share a similar architecture. Nat Struct Mol Biol. 2016;23:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin R, Junutula JR, Matern HT, Ervin KE, Scheller RH, Brunger AT. Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J. 2005;24:2064–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamashita M, Kurokawa K, Sato Y, et al. Structural basis for the Rho‐ and phosphoinositide‐dependent localization of the exocyst subunit Sec3. Nat Struct Mol Biol. 2010;17:180–186. [DOI] [PubMed] [Google Scholar]
- 23. Baek K, Knodler A, Lee SH, et al. Structure‐function study of the N‐terminal domain of exocyst subunit Sec3. J Biol Chem. 2010;285:10424–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heider MR, Gu M, Duffy CM, et al. Subunit connectivity, assembly determinants and architecture of the yeast exocyst complex. Nat Struct Mol Biol. 2016;23:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mei K, Li Y, Wang S, et al. Cryo‐EM structure of the exocyst complex. Nat Struct Mol Biol. 2018;25:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rossi G, Lepore D, Kenner L, et al. Exocyst structural changes associated with activation of tethering downstream of Rho/Cdc42 GTPases. J Cell Biol. 2020;219:31904797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alber F, Dokudovskaya S, Veenhoff LM, et al. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. [DOI] [PubMed] [Google Scholar]
- 28. Rout MP, Sali A. Principles for integrative structural biology studies. Cell. 2019;177:1384–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schneidman‐Duhovny D, Pellarin R, Sali A. Uncertainty in integrative structural modeling. Curr Opin Struct Biol. 2014;28:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim SJ, Fernandez‐Martinez J, Nudelman I, et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature. 2018;555:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lasker K, Forster F, Bohn S, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A. 2012;109:1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Russel D, Lasker K, Webb B, et al. Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012;10:e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Picco A, Irastorza‐Azcarate I, Specht T, et al. The in vivo architecture of the exocyst provides structural basis for exocytosis. Cell. 2017;168:400–412. [DOI] [PubMed] [Google Scholar]
- 34. Katoh Y, Nozaki S, Hartanto D, Miyano R, Nakayama K. Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J Cell Sci. 2015;128:2351–2362. [DOI] [PubMed] [Google Scholar]
- 35. Zarsky V, Kulich I, Fendrych M, Pecenkova T. Exocyst complexes multiple functions in plant cells secretory pathways. Curr Opin Plant Biol. 2013;16:726–733. [DOI] [PubMed] [Google Scholar]
- 36. Zeng J, Feng S, Wu B, Guo W. Polarized exocytosis. Cold Spring Harb Perspect Biol. 2017;9:a027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tarassov K, Messier V, Landry CR, et al. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. [DOI] [PubMed] [Google Scholar]
- 38. Sivaram MV, Saporita JA, Furgason MLM, Boettcher AJ, Munson M. Dimerization of the exocyst protein Sec6p and its interaction with the t‐SNARE Sec9p. Biochemistry. 2005;44:6302–6311. [DOI] [PubMed] [Google Scholar]
- 39. Dubuke ML, Maniatis S, Shaffer SA, Munson M. The exocyst subunit Sec6 interacts with assembled exocytic SNARE complexes. J Biol Chem. 2015;290:28245–28256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen D, Yuan H, Hutagalung A, et al. The synaptobrevin homologue Snc2p recruits the exocyst to secretory vesicles by binding to Sec6p. J Cell Biol. 2013;202:509–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morgera F, Sallah MR, Dubuke ML, et al. Regulation of exocytosis by the exocyst subunit Sec6 and the SM protein Sec1. Mol Biol Cell. 2012;23:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yue P, Zhang Y, Mei K, et al. Sec3 promotes the initial binary t‐SNARE complex assembly and membrane fusion. Nat Commun. 2017;8:14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jin Y, Sultana A, Gandhi P, et al. Myosin V transports secretory vesicles via a Rab GTPase cascade and interaction with the exocyst complex. Dev Cell. 2011;21:1156–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo W, Roth D, Walch‐Solimena C, Novick P. The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 1999;18:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu H, Turner C, Gardner J, Temple B, Brennwald P. The Exo70 subunit of the exocyst is an effector for both Cdc42 and Rho3 function in polarized exocytosis. Mol Biol Cell. 2010;21:430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Irastorza‐Azcarate I, Castano‐Diez D, Devos DP, Gallego O. Live‐cell structural biology to solve biological mechanisms: The case of the exocyst. Structure. 2019;27:886–892. [DOI] [PubMed] [Google Scholar]
- 47. Songer JA, Munson M. Sec6p anchors the assembled exocyst complex at sites of secretion. Mol Biol Cell. 2009;20:973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He B, Xi F, Zhang X, Zhang J, Guo W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 2007;26:4053–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang X, Zajac A, Zhang J, et al. The critical role of Exo84p in the organization and polarized localization of the exocyst complex. J Biol Chem. 2005;280:20356–20364. [DOI] [PubMed] [Google Scholar]
- 50. Lamping E, Tanabe K, Niimi M, Uehara Y, Monk BC, Cannon RD. Characterization of the Saccharomyces cerevisiae sec6‐4 mutation and tools to create S. cerevisiae strains containing the sec6‐4 allele. Gene. 2005;361:57–66. [DOI] [PubMed] [Google Scholar]
- 51. Gruhler A, Olsen JV, Mohammed S, et al. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol Cell Proteomics. 2005;4:310–327. [DOI] [PubMed] [Google Scholar]
- 52. Swaney DL, Beltrao P, Starita L, et al. Global analysis of phosphorylation and ubiquitylation cross‐talk in protein degradation. Nat Methods. 2013;10:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in‐depth phosphoproteome analysis. Mol Cell Proteomics. 2008;7:1389–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ficarro SB, McCleland ML, Stukenberg PT, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae . Nat Biotechnol. 2002;20:301–305. [DOI] [PubMed] [Google Scholar]
- 56. Soulard A, Cremonesi A, Moes S, Schutz F, Jeno P, Hall MN. The rapamycin‐sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol Biol Cell. 2010;21:3475–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nishi H, Shaytan A, Panchenko AR. Physicochemical mechanisms of protein regulation by phosphorylation. Front Genet. 2014;5:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Duan Y, Guo Q, Zhang T, et al. Cyclin‐dependent kinase‐mediated phosphorylation of the exocyst subunit Exo84 in late G1 phase suppresses exocytic secretion and cell growth in yeast. J Biol Chem. 2019;294:11323–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Luo G, Zhang J, Luca FC, Guo W. Mitotic phosphorylation of Exo84 disrupts exocyst assembly and arrests cell growth. J Cell Biol. 2013;202:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Haarer BK, Corbett A, Kweon Y, Petzold AS, Silver P, Brown SS. SEC3 mutations are synthetically lethal with profilin mutations and cause defects in diploid‐specific bud‐site selection. Genetics. 1996;144:495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matern HT, Yeaman C, Nelson WJ, Scheller RH. The Sec6/8 complex in mammalian cells: Characterization of mammalian Sec3, subunit interactions, and expression of subunits in polarized cells. Proc Natl Acad Sci U S A. 2001;98:9648–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jourdain I, Dooley HC, Toda T. Fission yeast sec3 bridges the exocyst complex to the actin cytoskeleton. Traffic. 2012;13:1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bendezu FO, Vincenzetti V, Martin SG. Fission yeast Sec3 and Exo70 are transported on actin cables and localize the exocyst complex to cell poles. PLoS One. 2012;7:e40248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Moskalenko S, Tong C, Rosse C, et al. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. 2003;278:51743–51748. [DOI] [PubMed] [Google Scholar]
- 65. Ahmed SM, Nishida‐Fukuda H, Li Y, McDonald WH, Gradinaru CC, Macara IG. Exocyst dynamics during vesicle tethering and fusion. Nat Commun. 2018;9:5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bodemann BO, Orvedahl A, Cheng T, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murthy M, Ranjan R, Denef N, Higashi ME, Schupbach T, Schwarz TL. Sec6 mutations and the Drosophila exocyst complex. J Cell Sci. 2005;118:1139–1150. [DOI] [PubMed] [Google Scholar]
- 68. Kulich I, Pecenkova T, Sekeres J, et al. Arabidopsis exocyst subcomplex containing subunit EXO70B1 is involved in autophagy‐related transport to the vacuole. Traffic. 2013;14:1155–1165. [DOI] [PubMed] [Google Scholar]
- 69. Riquelme M, Bredeweg EL, Callejas‐Negrete O, et al. The Neurospora crassa exocyst complex tethers Spitzenkorper vesicles to the apical plasma membrane during polarized growth. Mol Biol Cell. 2014;25:1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Inamdar SM, Hsu SC, Yeaman C. Probing functional changes in exocyst configuration with monoclonal antibodies. Front Cell Dev Biol. 2016;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Munson M, Chen X, Cocina AE, Schultz SM, Hughson FM. Interactions within the yeast t‐SNARE Sso1p that control SNARE complex assembly. Nat Struct Biol. 2000;7:894–902. [DOI] [PubMed] [Google Scholar]
- 72. Zhang X, Orlando K, He B, et al. Membrane association and functional regulation of Sec3 by phospholipids and Cdc42. J Cell Biol. 2008;180:145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ishida T, Kinoshita K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007;35:W460–W464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR‐FIT: A meta‐predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kono K, Saeki Y, Yoshida S, Tanaka K, Pellman D. Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell. 2012;150:151–164. [DOI] [PubMed] [Google Scholar]
- 76. Sali A, Berman HM, Schwede T, et al. Outcome of the first wwPDB hybrid/integrative methods task force workshop. Structure. 2015;23:1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Berman HM, Adams PD, Bonvin AA, et al. Federating structural models and data: Outcomes from a workshop on archiving integrative structures. Structure. 2019;27:1745–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gutierrez C, Chemmama IE, Mao H, et al. Structural dynamics of the human COP9 signalosome revealed by cross‐linking mass spectrometry and integrative modeling. Proc Natl Acad Sci U S A. 2020;117:4088–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jishage M, Yu X, Shi Y, et al. Architecture of Pol II(G) and molecular mechanism of transcription regulation by Gdown1. Nat Struct Mol Biol. 2018;25:859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. LaCava J, Fernandez‐Martinez J, Hakhverdyan Z, Rout MP. Optimized Affinity capture of yeast protein complexes. Cold Spring Harb Protoc. 2016;2016:27371596. [DOI] [PubMed] [Google Scholar]
- 81. Shi Y, Pellarin R, Fridy PC, et al. A strategy for dissecting the architectures of native macromolecular assemblies. Nat Methods. 2015;12:1135–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 83. Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Soding J. Protein homology detection by HMM‐HMM comparison. Bioinformatics. 2005;21:951–960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Figure S1. Thoroughness of configurational sampling and model precision for yeast exocyst complex. (a) Convergence of the model score, for the 9,741 good‐scoring models; the scores do not continue to improve as more models are computed essentially independently. The error bars represent the standard deviations of the best scores, estimated by repeating sampling of models 50 times. (b) Distribution of model scores for model Samples 1 (scarlet) and 2 (blue), comprising the 9,741 good‐scoring structures. The Kolmogorov–Smirnov two‐sample test indicates that the difference between the two score distributions is insignificant (p‐value is .1), thus, the two score distributions are effectively equal. (c) Three criteria for determining the sampling precision (y‐axis), evaluated as a function of the RMSD clustering threshold (x‐axis). First, the p‐value is computed using the χ 2‐test for homogeneity of proportions (red dots). Second, an effect size for the χ 2‐test is quantified by the Cramer's V value (blue triangles). Third, the population of models in sufficiently large clusters (containing at least 10 models from each sample) is shown as green stars. The vertical dotted black line indicates the RMSD clustering threshold at which three conditions are satisfied (p‐value > .05, Cramer's V < 0.10, and the population of clustered models >0.80), thus defining the sampling precision of 51 Å. (d) Populations of Sample 1 and 2 models in the two clusters obtained by threshold‐based clustering using the sampling precision value as threshold. The resulting cluster (Cluster 1) contains 99% of the models and has a precision of 38 Å. The precision of this cluster defines the model or cluster precision. (e) Comparison of localization probability densities of models from Sample 1 (left), Sample 2 (right), and the complete ensemble (center); the cross‐correlation coefficient between the localization densities from Samples 1 and 2 is 0.9.
Table S1. Four hundred and thirty‐four unique disuccinimidyl suberate and 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride cross‐linked pairs of residues.
Table S2. Summary of integrative structure determination of yeast exocyst complex (PDBDEV_00000041).
Table S3. Phosphorylation sites mapped on exocyst from this study, and from high throughput studies (51–56), as well as (58,59).
Data Availability Statement
The EM density map has been deposited in the EMDB, accession code 21226.