

Abstract
Epidermolysis bullosa simplex (EBS) is an inherited skin disorder characterized by increased skin and mucous membrane fragility. The majority of cases are caused by mutations in keratin 5 (KRT5) and keratin 14 (KRT14). Mutations of these genes result in cytoskeletal disruption of the basal keratinocytes. Gross and histopathologic findings of two clinically affected homozygous rhesus macaques with an insertion variant mutation in KRT5 are described and compared with six deceased phenotypically normal animals that were heterozygous for the KRT5 insertion variant. Animals that were homozygous for the KRT5 insertion variant were stillborn and had widespread loss of the epidermis. Microscopic examination confirmed severe ulceration and basal cell vacuolation, with basilar vesicle formation in the remaining intact epidermis. Immunohistochemistry for cytokeratin 5 demonstrated lack of epidermal immunoreactivity in homozygotes. DNA sequencing identified a 34 base pair insertion variant in exon 5 of the KRT5 gene. To our knowledge, this is the first report of epidermolysis bullosa in rhesus macaques.
Keywords: Epidermolysis bullosa simplex, genetic skin diseases, rhesus macaque, primate, animal model
Two stillborn macaques were initially recognized in 2012 based on their phenotype at necropsy. It was later identified through retrospective DNA sequence analysis that they were homozygous for a KRT5 insertion variant. These were compared to four living and six previously euthanized rhesus macaques that were heterozygous for the KRT5 insertion variant. The animals were of Indian- origin ancestry and born at the Oregon National Primate Research Center (ONPRC).
Animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the ONPRC. They were conducted to ensure compliance with the U.S. Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the 2011 edition of the National Research Council’s Guide for the Care and Use of Laboratory Animals. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Of the two stillborn fetuses that were homozygous for the KRT5 insertion variant: one was a full term female (case 1) and one was a third trimester male (case 2), both were in fair to good postmortem condition. Both fetuses were born to apparently healthy, outdoor-housed, female rhesus macaques that were still alive at the time of publication. At necropsy, gross findings in both fetuses ranged from multifocal cutaneous vesicles that were frequently ruptured, resulting in multifocal to coalescing ulcers, to severe generalized epidermal loss involving the head, trunk, and extremities (Fig. 1–2). The lips and gingiva were also affected, but to a much lesser extent. Case 2 was extremely small (198.8 g) for the apparent gestational age, with a 16.6 cm crown-rump length, 3.4 cm right hand length, an enlarged, domed cranium, and inferior brachygnathia. The cerebrum bulged from the dura, the sulci were flattened, the occipital lobes were moderately enlarged, and the occipital fontanelle was enlarged and widened.
Figures 1–2.
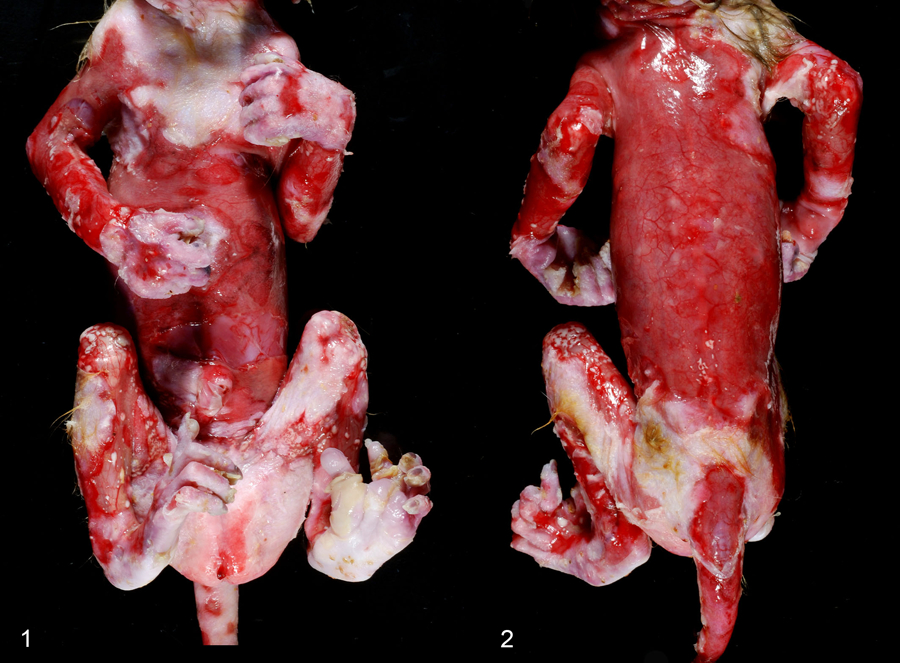
Epidermolysis bullosa simplex, integument, stillborn rhesus macaque, case 1. Widespread epidermal loss affecting 75% of the body including the head, back, and extremities.
Microscopic findings in both KRT5 insertion variant homozygous animals included multifocal to coalescing, variably sized intraepidermal clefts (Figs. 3–5). Clefting sites were characterized by prominent basal cell vacuolation and fragmentation (Fig. 5, inset). Within regions of clefting, there were multiple areas where low numbers of basilar keratinocytes remained attached to the subjacent basement membrane zone (Fig. 4–5). Multifocally, the intraepidermal vesicles contained extravasated erythrocytes and/or eosinophilic proteinaceous fluid (Suppl. Fig. 1). In regions of complete epidermal loss, there were variable amounts of superficial dermal edema and hemorrhage, with small amounts of fibrin. In sections stained by periodic acid-Schiff (PAS) reaction, the cleavage was above the magenta-stained basement membrane zone (Fig. 6). Immunohistochemistry (IHC) for cytokeratin 5 (see Supplemental Materials) demonstrated an absence of cytoplasmic immunoreactivity throughout all layers of epidermal keratinocytes in KRT5 insertion variant homozygotes (Fig. 7), whereas there was robust cytoplasmic immunoreactivity in epidermal keratinocytes in the skin from the KRT5 insertion variant heterozygotes and from a normal rhesus macaque (Fig. 7 inset). Additional microscopic findings in case 1included low numbers of intra-alveolar and intra-bronchiolar squamous epithelial cells admixed with small amounts of mucus and low numbers of neutrophils. Case 1 also had segmental renal dysplasia, characterized by corticomedullary interstitial fibrosis which entrapped primitive and irregular tubules, as well as underdeveloped fetal glomeruli, with multifocal compensatory hypertrophy and glomerulocystic change. Case 2 had marked thyroid hyperplasia, with collapsed follicles that contained little to no colloid and were lined by hypertrophic and hyperplastic follicular epithelium. Pulmonary alveoli contained high numbers of sloughed squamous epithelial cells and scant meconium. Despite the gross abnormalities of the cerebrum, it was microscopically unremarkable.
Figures 3–7.
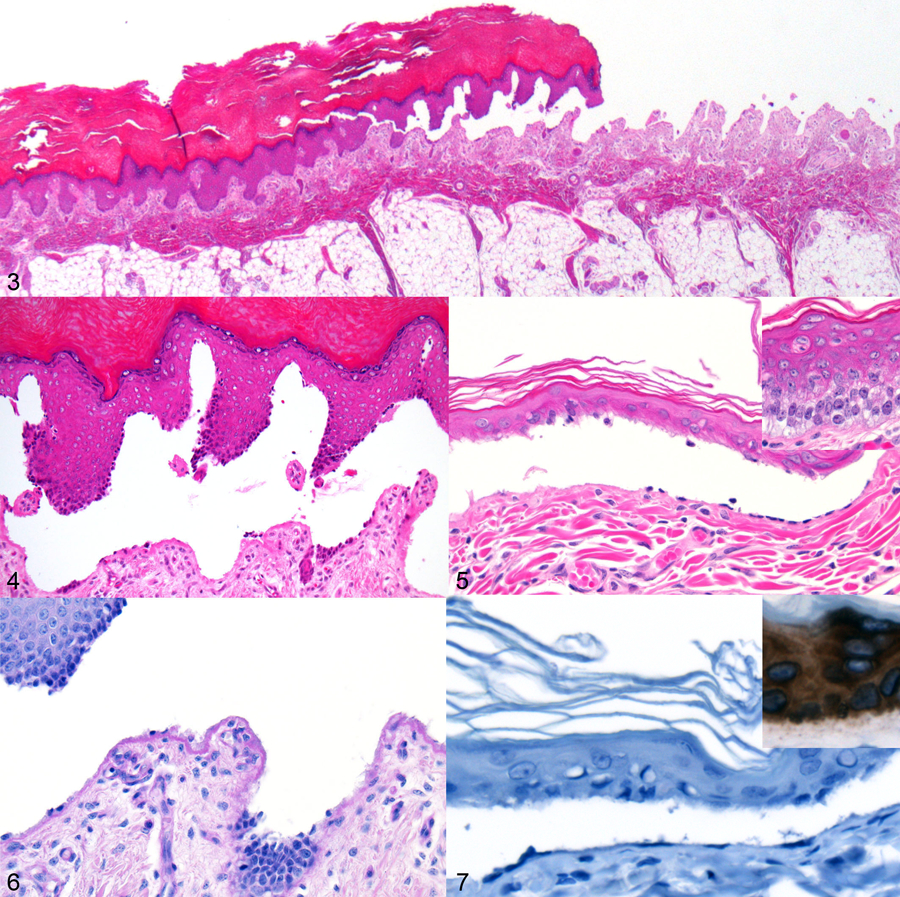
Epidermolysis bullosa simplex, integument, stillborn rhesus macaque, case 1. Figure 3. Intraepidermal clefting that transitions to complete loss of epithelium. Hematoxylin and eosin (HE). Figure 4. Cleavage sites with low numbers of basilar keratinocytes attached to the subjacent basement membrane zone. HE. Figure 5. Intraepidermal clefts with basilar keratinocytes adherent to the base of the cleft. Inset: Basilar keratinocytes adjacent to regions of cleavage have cytoplasmic vacuolation. HE. Figure 6. The intraepidermal cleft is present above the basement membrane, which stains magenta with periodic acid-Schiff reaction. Figure 7. Immunohistochemistry for cytokeratin 5. In a KRT5 insertion variant homozygous macaque (case 2), keratinocytes lack cytoplasmic immunoreactivity. Inset: Positive control (macaque lacking KRT5 mutation). Basilar keratinocytes exhibit strong, diffuse cytoplasmic immunoreactivity.
Genetic variants identified from whole-genome sequencing of both stillborn animals were evaluated using the Macaque Genotype and Phenotype Resource (mGAP; https://mgap.ohsu.edu). A 34 base-pair (bp) insertion variant in exon 5 of the KRT5 gene (KRT5:c.1087_1088insGTCTTGGTACCAGCTTGGTCTTGGTACCAGACCA; p.Lys363fs) was identified as a possible causal mutation (Suppl. Fig. 2). The frameshift variant was predicted to cause an altered protein starting at amino acid 363 and truncating the protein after the 13th altered amino acid, which is predicted to disrupt the fourth coil domain (Suppl. Fig. 3). The mGAP database had genotype information for 278 non-symptomatic ONPRC macaques in which the observed allele frequency was 0.0054 with no observed KRT5 insertion variant homozygotes. The insertional allele was validated with targeted PCR and Sanger sequencing. Expanded pedigree genotyping demonstrated an autosomal recessive pattern of inheritance and identified a total of 11 KRT5 insertion variant heterozygotes with no additional KRT5 insertion variant homozygous monkeys (Suppl. Fig. 4).
Here we report two cases of EBS in rhesus macaques homozygous for a KRT5 34 bp insertion mutation, as well as six asymptomatic KRT5 insertion variant heterozygous rhesus macaques. All cases were evenly distributed between males and females. Diagnosis of EBS was established by identification of characteristic gross and microscopic features, specifically intra-epidermal clefting and absent anti-CK5 immunoreactivity in all layers of keratinocytes. Based on the presence of skin lesions identified at birth and the widespread distribution of lesions, these cases are further classified as EBS generalized severe. Within the rhesus macaque population at the ONPRC, the mutant KRT5 allele frequency was considered low and only asymptomatic KRT5 insertion variant heterozygotes are currently present in the colony.
In humans, epidermolysis bullosa (EB) is broadly classified into four groups: epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB), dystrophic epidermolysis bullosa (DEB), and Kindler syndrome.3 EBS is attributed to keratinocyte disruption resulting in intraepidermal clefts and is further categorized as suprabasilar or basilar depending on the location of the cleft within the epidermis.3 In contrast, JEB is classified by clefting within the lamina lucida resulting from disruption of anchoring filament-hemidesmosome complexes.3 In contrast, DEB is characterized by superficial dermal clefting within the sub-lamina densa region of anchoring filaments.3 Lastly, Kindler syndrome is recognized as a mixed pattern with clefting within or beneath the basement membrane zone accompanied by clinical phenotypic features of photosensitization and blistering.3
The four most common forms of EBS are those in which the clefting occurs within the basal epidermis and include localized (Weber-Cockayne), generalized intermediate (Koebner), EBS with mottled pigmentation, and generalized severe EBS (EBS-gen sev)(Dowling-Meara ) forms.3,9 Mutations in KRT5 and KRT14 account for ~75% of EBS cases, and heterozygous missense mutations are the most common.1 Mutations in KRT5 and KRT14 have been associated with increased mortality with generalized severe forms of EBS.9,10 Cases of EBS-gen sev typically present at birth with severe widespread blistering of the skin and mucosal surfaces, transitioning to progressive hyperkeratosis during childhood.9 Despite the severity, EBS-gen sev is not typically fatal; however, mortality due to sepsis or unspecified respiratory failure occurs in a small percentage of these patients.4
Few animal models of EBS are reported; even fewer models are specific to KRT5 gene mutations and are restricted to cattle and the Krt5 knockout mouse.2,5–8 Phenotypic features of the mouse model include a high degree of neonatal deaths in homozygotes, and loss of the keratin cytoskeleton and widespread cytolysis within the basal layer of the epidermis.8 In contrast, heterozygous mice lacked any overt phenotypic changes.8 These findings are consistent with those described in this report of KRT5 insertion variant homozygous and heterozygous rhesus macaques with the KRT5 34 bp insertion mutation. The phenotypic and genotypic changes described in the rhesus macaque make it a viable alternative to the currently available animal models of KRT5 EBS. Both the KRT5 knockout mouse and macaque models of EBS face similar drawbacks, including increased mortality experienced by the KRT5 insertion variant homozygotes. However, the potential advantages to the macaque model include a spontaneous model of a naturally occurring disease in a species more closely related to humans, the opportunity for delivery by cesarean section to avoid birthing trauma, and the possibility of more intensive peri-parturient management to reduce neonatal loss. To our knowledge, no cases of EB or EBS have been reported in the rhesus macaque or any other species of nonhuman primates.
Supplementary Material
Acknowledgements
The authors thank Wendy Price and Allie Meristem for their technical contributions. We also thank Erinn Stefanich for his expertise with histology, special stains and immunohistochemistry. Lastly, we thank the staff and personnel of the Pathology Services Unit and Integrated Pathology Core for the handling and processing of archival materials.
Funding
This work was supported in part by grant P51OD011092 from the National Institutes of Health and R24 OD021234.
References
- 1.Bolling MC, Lemmink HH, Jansen GH, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol 2011;164:637–44. [DOI] [PubMed] [Google Scholar]
- 2.Bruckner-Tuderman L, McGrath JA, Robinson EC, Uitto J. Animal models of epidermolysis bullosa: update 2010. J Invest Dermatol 2010;130:1485–8. [DOI] [PubMed] [Google Scholar]
- 3.Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014. June;70(6):1103–26. [DOI] [PubMed] [Google Scholar]
- 4.Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr 2008. February;152(2):276–80. [DOI] [PubMed] [Google Scholar]
- 5.Ford CA, Stanfield AM, Spelman RJ, et al. A mutation in bovine keratin 5 causing epidermolysis bullosa simplex, transmitted by a mosaic sire. J Invest Dermatol 2005;124:1170–6. [DOI] [PubMed] [Google Scholar]
- 6.Jiang QJ, Uitto J. Animal models of epidermolysis bullosa--targets for gene therapy. J Invest Dermatol 2005;124:xi–xiii. [DOI] [PubMed] [Google Scholar]
- 7.Mauldin EA, Wang P, Olivry T, Henthorn PS, Casal ML. Epidermolysis bullosa simplex in sibling Eurasier dogs is caused by a PLEC non-sense variant. Vet Dermatol 2017. February;28(1):10–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters B, Kirfel J, Büssow H, Vidal M, Magin TM. Complete cytolysis and neonatal lethality in keratin 5 knockout mice reveal its fundamental role in skin integrity and in epidermolysis bullosa simplex. Mol Biol Cell. 2001;12:1775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfendner EG, Bruckner AL. Epidermolysis Bullosa Simplex In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2019. 1998. October 7 [updated 2016 Oct 13]. [Google Scholar]
- 10.Sathishkumar D, Orrin E, Terron-Kwiatkowski A, et al. The p.Glu477Lys Mutation in Keratin 5 Is Strongly Associated with Mortality in Generalized Severe Epidermolysis Bullosa Simplex. J Invest Dermatol 2016;136:719–721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.