

Abstract
Follicular helper T cells provide signals that promote B cell development, proliferation, and production of affinity matured and appropriately isotype switched antibodies. In addition to their classical locations within B cell follicles and germinal centers therein, B cell helper T cells are also be found in extrafollicular spaces – either in secondary lymphoid or non-lymphoid tissues. Both follicular and extrafollicular T helper cells drive autoantibody-mediated autoimmunity. Interfering with B cell help provided by T cells can ameliorate autoimmune disease in animal models and human patients. The next frontier in Tfh cell biology will be identification of Tfh-cell specific pathogenic changes in autoimmunity and exploiting them for therapeutic purposes.
Classical Model of Tfh Cell Function
Adaptive immunity allows the host to respond specifically and defend against a diverse array of insults, from pathogens to tumors, that threaten homeostasis. The cellular and soluble mediators of the immune system detect pathogens, producing a coordinated response that is tailored to salient features of the pathogen such as intracellular vs extracellular, mucosal vs intravascular, and cutaneous vs visceral, and in proportion to the danger posed. Humoral immunity, a key part of this defense strategy, is a biological high-throughput mechanism of generating high-affinity ligands, antibodies, against targets of interest, antigens. Depending on antibody isotype, different effector functions can be engaged upon ligand recognition. Thereby, the immune system can, in relatively short order, neutralize, opsonize, sensitize or destroy the inciting pathogen specifically and methodically.
Optimal antibody responses are the result of T and B cell collaboration. Much of our understanding of the details of an antibody response comes from studies of experimental viral infection or immunization. In these settings, antibody production is regulated within secondary lymphoid organs (SLOs) with an early induction of short-lived plasmablasts in extrafollicular regions followed by a later, durable, follicular response initiated in germinal centers (GCs) within B cell follicles [1]. Upon antigen engagement, activated T cells in the T cell zone of SLOs and B cells from the follicle proliferate and migrate to interfollicular regions of lymph nodes or the T-B cell border of the spleen, sites of their initial interaction and where T cells initiate steps for B cell maturation. B cells can then migrate to extrafollicular regions where they may undergo isotype switch and even somatic hypermutation leading to the production of low to moderate affinity antibodies that aid in pathogen elimination early following challenge. Alternatively, nascent follicular helper T (Tfh) cells, expressing their canonical transcription factor Bcl-6, and B cells migrate into the follicle, as both express the chemokine receptor CXCR5 (C-X-C motif chemokine receptor 5) enabling migration toward its ligand CXCL13 (chemokine C-X-C motif ligand 13) expressed in the follicle, acting together to develop the GC. Tfh cells in the follicle, and subsequently the GC, guide the antibody response using cell-surface proteins including PD-1 (programmed cell death protein 1), CD40 ligand (CD40L, CD154) and ICOS (inducible co-stimulator), as well as secreted factors such as IL-21, the signature Tfh-cell cytokine. In the absence of functional Tfh cell help, GC B cells exhibit impaired isotype switching and reduced survival, proliferation and affinity maturation [2–4]. As GC B cells undergo somatic hypermutation, Tfh cells provide repetitive selection to ensure immunoglobulin (Ig) affinity maturation. Tfh cells also help skew isotype choice in order to ensure the humoral response is appropriately coordinated within the larger immune response to the inciting pathogen. For example, intracellular pathogens, such as viruses, elicit a Th1 cell response driven by IFN-γ. During such a response, Tfh cells will also produce IFN-γ which helps GC B cells switch to inflammatory IgG2a/c (IgG1 in humans) [5,6]. By contrast, allergens or helminths will drive Th2 responses via IL-4 and other type 2 cytokines. Tfh cells generated during a Th2 response express IL-4 which helps induce IgG1 (IgG4 in humans) and IgE antibodies. These findings give rise to the paradigm that the basic Tfh-cell developmental program can be modified by the cytokine milieu towards Tfh1 or Tfh2 fates. IL-17 and IL-21 co-secreting Tfh cells, thought to arise directly from Th17 or Treg cells, have also been described in small intestine Peyer’s patches and may be important for supporting IgA production again gut microbes [7–9]. Thus, Tfh cells can adopt fates similar to how T effector cells can become Th1, Th2 or Th17 cells (Fig. 1), thereby allowing the humoral response to match the overall immune response to a particular pathogen. After completing their maturation, B cells exit the GC as memory B cells or long-lived plasma cells that together provide sustained humoral immunity for the host. The detailed migration patterns, molecular basis for Tfh cell differentiation and function, and mechanisms of Tfh cell skewing have recently been reviewed [5].
Figure 1. Functional heterogeneity of T cells that help B cells.
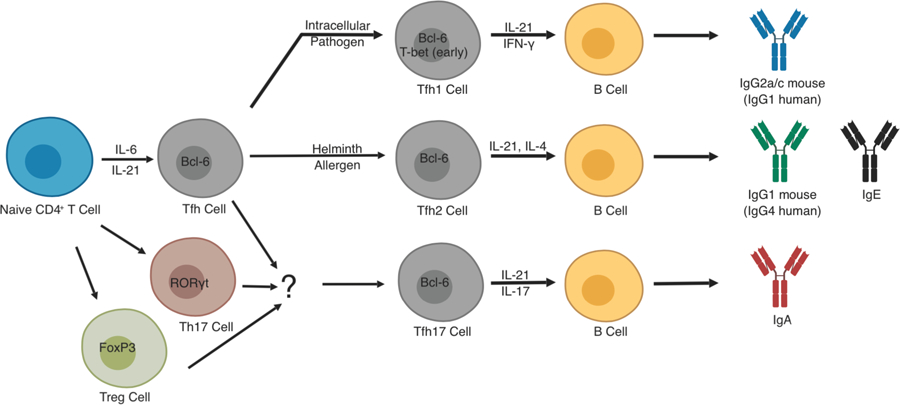
Tfh cells can adopt functional subtypes that correlate with the fate decisions of their T effector counterparts. This is best exemplified for type 1 and 2 immune responses where Tfh cells can adopt Tfh1 or Tfh2 subtypes, respectively. IFN-γ expressed early during a type I immune to virus, for example, will lead to early T-bet expression in Bcl6+ Tfh cells. This leads to IL-21 and IFN-γ co-expression by Tfh1 cells and skews GC B cells to switch to pro-inflammatory IgG2a/c isotypes for aid in clearance of intracellular pathogens. In the case of type 2 infection, Tfh cells adopt a Tfh2 subtype characterized by sequential expression of IL-21 and IL-4 leading to GC B cells isotype switching to IgG1 or IgE for pathogen neutralization and activation of mast cells, respectively. The situation for Tfh17 development is more complex. Current thinking suggests that Tfh17 subsets exist, particularly in Peyer’s patches or other lymphoid tissue associated with mucosal surfaces. These Tfh17 cells are thought to arise primarily from Th17 cells, or perhaps, Treg cells, which can then induce IgA production by GC B cells. All of these subtypes of Tfh cells, Tfh1, Tfh2, and Tfh17 have been implicated in various autoimmune conditions as described in the text.
This model of Tfh cell function is largely based on results in mice. Such detailed experiments are generally not feasible in humans where direct access to lymphoid tissue is limited. However, experiments using tonsils have been largely consistent with the model above [10]. CXCR5+CD4+ T cells are also found in human circulation. The majority of such cells are central memory cells, carrying markers of this phenotype, and thus have the potential to recirculate to follicles of secondary lymphoid organs to promote B cell responses upon immune re-5 challenge. Growing evidence, primarily from vaccination studies that show induction and temporal correlation of activated CXCR5+ cells with antibody responses, suggests a portion of such cells are related to effector Tfh cells, and thus have been termed circulating Tfh (cTfh) cells [11]. Despite the correlation with antibody responses, cTfh cells are phenotypically distinct from their SLO Tfh cell counterparts. Most strikingly, cTfh cells lack Bcl-6, the Tfh-defining transcription factor. They also have less expression of the canonical Tfh cell markers CXCR5, PD-1 and ICOS [6]. cTfh cells have been phenotypically and functionally subsetted using chemokine receptors as surrogates for subset-defining transcription factors and cytokines, primarily CXCR3 and CCR6: CXCR3hiCCR6lo express T-bet and IFN-γ and are termed cTfh1 cells, CXCR3loCCR6lo cells express Gata3, IL-4, IL-5 and IL-13 and are termed cTfh2 cells, and CXCR3loCCR6hi cells express RORγt and IL-17 as cTfh17 cells [11,12]. Those cells expressing CXCR3 and CCR6 express both IFN-γ and IL-17. Within these subtypes, activated cells are ICOShiPD1hi while ICOSloPD1lo cells are considered quiescent, central memory cells that also express the memory marker CCR7 [13]. The different subtypes exhibit varying degrees of fitness to provide B cell help - as measured by proliferation, differentiation and antibody secretion - in vitro. Direct evidence that cTfh cells promote Ig affinity maturation is also lacking, although, admittedly, this would be challenging to demonstrate in vitro. In particular, cTfh1 cells, which are not particularly active in vitro, have nevertheless been found to correlate in vivo with generation of high-avidity antibodies following influenza vaccination [12]. The detailed function and regulation of cTfh cells in vivo remains to be established.
Tfh Cells in Systemic Autoimmunity
Systemic autoimmunity is the result of an immune response to host cells or tissues, leading to organ damage and dysfunction. Many autoimmune diseases are associated with the production of specific autoantibodies, including, but not limited to, rheumatoid arthritis (RA, antibodies to citrullinated peptides), granulomatosis with polyangitis (GPA, anti-neutrophil cytoplasmic antibodies), myasthenia gravis (MG, anti-acetylcholine receptor antibodies) and type 1 diabetes mellitus (T1DM, anti-B islet cell antibodies) [14]. Systemic lupus erythematosus (lupus, SLE) is the prototypic, systemic autoimmune disease characterized by anti-nucleoprotein antibodies, with those directed against chromatin, including dsDNA, the earliest identified [15] and best characterized [16]. Data from murine lupus models indicate that anti-dsDNA antibodies undergo somatic hypermutation and affinity maturation, with similar findings analyzing these antibodies in lupus patients [17,18], indicating that autoreactive 6 B cell maturation is T-, and thus Tfh-, cell dependent. Although multiple immune mechanisms are dysregulated in systemic autoimmunity and likely contribute to pathogenesis, autoantibodies can be pathogenic, develop before symptoms of disease, and may correlate with disease activity [14]. Further, the frequency of activated cTfh cells is increased or correlated with disease activity in patients with lupus, as well as in RA, juvenile dermatomyositis, MG, multiple sclerosis, psoriasis vulgaris and Sjögren syndrome [11,13,19–25]. While correlating therapeutic responses has been difficult, recent analysis of patients with relapsing-remitting multiple sclerosis found that efficacy of dimethyl fumarate treatment correlated with reduction in cTfh percentages [26]. Together, these results indicate that understanding the mechanisms that lead to autoantibody development are likely crucial for understanding the pathogenesis of autoimmune diseases.
Lupus is a heterogenous disease. While autoantibodies are universal features required for clinical diagnosis, disease manifestations can vary from mild arthralgias and skin rashes to life-threatening glomerulonephritis. Perhaps reflecting this variability, mouse models of lupus exhibit heterogenous immunological features. For example, murine lupus models like C57BL/6J (B6) Sle1.Yaa [27–30], BXSB-Yaa [31–33], B6.Sle1.Sle2.Sle3 (triple congenic, or TC, mice) [34], or sanroque [35] are characterized by early, exuberant germinal center expansion, while others such as MRL/MpJ-Faslpr (MRLlpr) [36–39] distinctly demonstrate reduced GC responses but increased extrafollicular responses in SLOs, with NZB/NZW F1 [39] mice having GCs and extrafollicular responses [40,41]. For the former “GC-amplified” lupus-prone mice, the canonical model of Tfh cell function in GCs, described above, is a useful starting point to dissect pathogenic mechanisms. Recent studies have demonstrated a direct link between excessive IFN-γ signaling and Tfh cell expansion and pathogenic function in sanroque mice [42], suggesting that lupus Tfh cells may skew towards a Tfh1 cell phenotype. This would also be consistent with the observed production of IFN-γ in lupus Tfh cells [42–44] and the accumulation of IgG2a antibodies that mediate complement fixation and glomerulonephritis in lupus prone mice [2]. In murine lymphocytic choriomenigitis virus (LCMV) infection, the transcription factors T-bet and STAT4 are required for production of IFN-γ by Tfh1 cells [45–47]. Interestingly, STAT4 has been found in GWAS studies to be associated with both human rheumatoid arthritis and lupus [48]. The detailed roles of T-bet and STAT4 in lupus Tfh cells have yet to be defined, but T-bet deficiency does not rescue sanroque mice [42], while activated STAT4 was found to be elevated in cTfh1 cells in patients with SLE [49]. Other mechanisms may also play a role in Tfh expansion in autoimmunity. Mice deficient in RORγt, the lineage defining transcription factor of 7 Th17 cells, spontaneously develop germinal center expansion and autoantibody production [50]. Surprisingly, autoimmunity in this model is driven by IL-17 producing Tfh17 cells. Thus, animal models may yield significant insight into the diverse cellular and molecular pathways that can drive Tfh-cell mediated autoimmunity.
In lupus nephritis, the classical autoantibodies that can deposit in the kidney and activate downstream immune mechanisms to cause disease are IgG1 anti-dsDNA [14]. Recent work in Lyn-deficient mice demonstrated these animals develop spontaneous Th2 skewing with development of IgE anti-dsDNA antibodies that mediate nephritis in a basophil-dependent manner [51]. A follow-up study in human lupus patients, demonstrated a strong correlation between IgE anti-dsDNA antibodies and active disease [52]. Whether these antibodies are high-avidity, and thus Tfh-cell dependent, has not been addressed, but assuming so, as for conventional IgE responses, the prediction would be that they are regulated by Tfh2 cells. The finding that human patients can have IgG anti-DNA (presumably IgG1 given the presence of lupus nephritis in these patients) and IgE anti-DNA antibodies concurrently [52] would suggest that Tfh1 and Tfh2 cells may be activated independently during an immune response, a phenomenon that has been described in mice infected with Leishmania [53]. Further, IgE autoantibodies have now been described in a variety of autoimmune diseases [54], so determining their Tfh-cell dependence and the role of Tfh2 cells in autoimmunity become important clinical questions. These studies demonstrate how using animal models and human samples allow novel insights into biological and disease processes and identification of new areas for investigation.
Tfh Cells as Therapeutic Targets
Successful therapeutic targeting of Tfh cells in murine lupus highlights the importance of these cells in autoimmune disease pathogenesis. CD40L signaling is critical for Tfh cell-dependent B cell maturation to memory and plasma cell formation. Genetic disruption of CD40L or inhibition of CD40L signaling with blocking antibodies was the first Tfh-targeted therapy in murine lupus almost two decades ago (although Tfh cells had not been formally identified as such at that time) [55–57]. Anti-CD40L therapy even demonstrated promise in patients with SLE [58]; however, this strategy was eventually halted because of thrombotic complications that developed presumably due to interaction of the antibody with CD40L expressed on the surface of platelets [59]. The hallmark cytokine expressed by Tfh cells is IL-21. Blockade of IL-21 [32,43,60], its receptor [61], or genetic deletion of the receptor [33] all have been shown to ameliorate disease in a variety of murine lupus models. Similarly, using a monoclonal antibody to signal through death receptor 6 (DR6), a negative regulator of NFAT signaling on the surface of Tfh cells, reduces Tfh cell accumulation and delays disease progression in NZB/W F1 mice [62]. One of the most exciting recent developments in this area is the identification of characteristics of lupus Tfh cells that are not found in their physiological counterparts. For example, virus-induced Tfh cells are relatively less glycolytic and more reliant on oxidative phosphorylation than Th1 cells [63]. Lupus Tfh cells, by contrast, have high rates of glycolysis [64,65]. Inhibiting glucose metabolism in lupus prone mice reduced lupus Tfh cells and autoantibody titers in these mice but left humoral responses to immunization or infection by influenza unchanged [66]. Identifying lupus Tfh specific aberrations and exploiting these for therapeutic purposes has the promise of delivering safe, yet specific and potent treatments for autoimmunity.
The therapeutic benefits of targeting Tfh cells in autoimmunity likely go beyond simply reducing autoantibody titers. Indeed, the observation that autoantibodies can precede the onset of clinical disease by years [67], suggests that autoantibodies are not sufficient to cause disease. Autoantibodies are also not simply epiphenomena of overactive immune responses, as has been suggested by some [3]. Rather, multiple levels of immune dysfunction, in both innate and adaptive immune systems, are likely required to initiate clinical autoimmunity [68]. Different immune abnormalities likely drive each other to produce a feed-forward mechanism of amplifying inflammation and immunopathology [69,70]. One manifestation of this is the observation that in addition to the GC expansion characteristic of many lupus-prone mouse strains, these animals exhibit expansion of multiple immune cells including B, T, and myeloid cells [29,32,71]. When IL-21 signaling is disrupted in these mice, Tfh and GC responses are clearly reduced, but other immune cell populations, not directly affected by IL-21 blockade, are also reduced [33,43]. These results suggest that blocking Tfh cell function has a ripple effect that extends to other dysfunctional parts of the immune system, confirming the interdependence of immunopathology in these animal models. Thus, the benefits of Tfh-targeted therapies may extend beyond autoantibody production to settings where autoantibodies have not yet been demonstrated to be pathogenic.
Extrafollicular B Helper T Cells
While the discussion this far has focused on the canonical model of Tfh cell regulation of GC-derived high-affinity autoantibodies, several emerging lines of evidence suggest that extrafollicular responses may also be important in humoral autoimmunity and, in some cases, may dominate the pathogenic response (Fig. 2). Early models 9 posited that extrafollicular responses occurred early after immunization or acute viral infection to supply low to moderate affinity antibodies that could temper the burgeoning infection until the production of Tfh-dependent, high-affinity, GC-derived antibodies [1]. The idea that sustained, high-affinity antibodies could be produced by extrafollicular foci was thought to happen exclusively in the setting of autoreactivity as first identified in the lupus prone MRLlpr mouse strain [40], and expanded upon in transgenic lupus-prone mice [40,72]. These autoreactive extrafollicular responses were found to be controlled by extrafollicular helper T cells (Tefh) expressing markers such as ICOS, CD40L, as well as IL-21, and identified as P-selection glycoprotein ligandlo (PSGL1lo, later found to be a selective marker for murine and human follicular and GC Tfh cells [73,74]) but with CXCR4 expression rather than CXCR5 to drive extrafollicular rather than follicular localization [40]. Subsequently, the extrafollicular response was observed in mouse models of immunization and infection and reported to depend on PD-1loCD4+ T cells expressing Bcl6 (as opposed to Tfh cells which are PD-1hi) [75]. Qualitatively, extrafollicular foci tend to be loosely organized groups of cells rather than the precise anatomical structures seen in germinal centers. Functional differences between Tfh cells and Tefh cells have not been carefully studied, nor is it known whether Tfh-driven antibodies differ qualitatively or quantitatively from Tefh-driven antibodies. It is now appreciated that for some pathogens such as Ehrlichia muris, Borrelia burgdorferi and Salmonella enterica the extrafollicular response is the dominant pathway of high-affinity antibody response as these pathogens actively interfere with GC responses [76–78]. Tefh cells were recently reported in human tonsillar tissue [74]. These cells were found to express PD-1, CXCR5, CD40L, Bcl-6 and IL-21 and were confirmed to localize in an extrafollicular niche via microscopy. Thus, it appears that Tefh responses can occur in humans and can be a component of physiologic immune responses.
Figure 2. Spatial heterogeneity of T cells that help B cells.
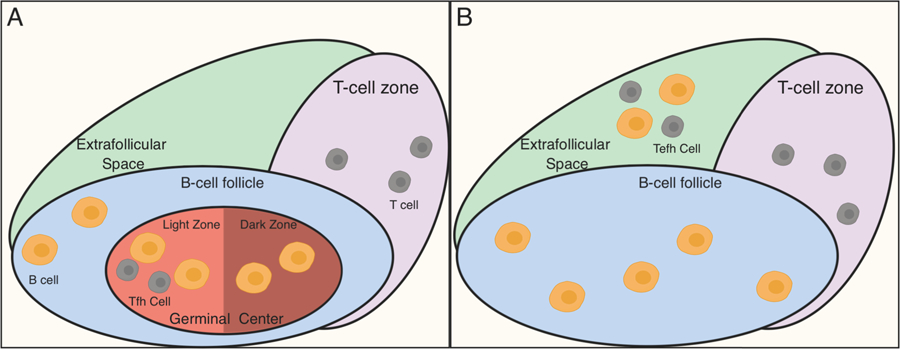
A) The classical model of Tfh function is shown. In this model, T:B cell collaboration occurs mainly in germinal centers (GCs), highly anatomically organized structures within B follicles of lymphoid tissue. Both Tfh cells and GC B cells contribute to GC organization. GCs can be further subdivided into light zones and dark zones. GC B cells cycle between the dark and light zones, proliferating in the former and receiving selective and instructive cues from Tfh cells in the latter where they undergo selection. Upon maturation, GC B cells exit the GCs as long-lived plasmablasts or memory B cells. B) An alternate mode of T:B cell collaboration can occur in extrafollicular spaces. Here, Tefh provide soluble and cell-contact dependent cues for B cell maturation with affinity maturation and isotype switch. This mode of B cell help has been demonstrated both during physiologic immune responses to pathogens as well as in settings of autoimmunity. Further, extrafollicular responses need not be confined to lymphoid tissue (as shown), but may also occur within local tissues, such as the kidneys, lungs or brain as discussed in the text.
Given the growing recognition of physiologic Tefh cell driven extrafollicular antibody responses, recent studies have focused on identifying evidence of extrafollicular antibody responses in mouse models of autoimmunity as well as human patients with autoimmune disease. Considering different lupus-prone mouse strains have disparate proportions of follicular vs. extrafollicular responses, it is unknown whether lupus patients exhibit heterogeneity in terms of follicular vs. extrafollicular aberrant autoantibody responses and whether this has any bearing on the observed clinical heterogeneity between lupus patients. A recent study in MRLlpr mice found Tfh-like cells expressing ICOS, PD-1, CXCR5, Bcl-6, IL-21, and IFN-γ in the choroid plexus suggesting a role for these cells in establishing the neuropsychiatric manifestations of human lupus that are replicated in this mouse model [79]. Another recent study using an airway inflammation model reported GC B cell-like cells in inflamed lung tissues along with CD4+ T cells expressing PD-1, ICOS, CD40L and IL-21 but not Bcl-6 [80]. These cells were able to stimulate B cell proliferation in vitro suggesting functional ability to help B cells. CD4+ T cells bearing classic Tfh cell markers including Bcl-6, ICOS, PD-1 and IL-21 reside in close proximity with and in contact with renal-infiltrating B-cells, as assessed by computational microscopy on human lupus renal biopsy samples, raising the possibility that local antibody production may contribute to renal disease in human lupus [81]. Another population of Tefh cells was reported in patients with rheumatoid arthritis [82], with mass cytometry used to analyze activated T cells from inflamed joint tissue. A subpopulation of cells expressing PD-1, ICOS and IL-21, but not the Tfh-cell transcription factor Bcl-6, termed peripheral helper (Tph) cells, could provide B-cell help in vitro. Chronic graft versus host disease (cGVHD) is another autoimmune disease often used to model autoantibody mediated disease. Mouse models of cGVHD reveal a role for PSGL-1loCD4+ T cells that express ICOS and Bcl-6 in the pathogenesis of this disease, similar to the phenotype of Tefh cells seen in lupus mice [83]. In a recent study in human patients with cGVHD, circulating Tefh-like (c-Tefh-like) cells were isoloated from blood samples based on phenotypic markers of murine Tefh cells [84]. The frequency and numbers of c-Tefh-like cells correlated with disease activity and these cells exhibited the ability to provide B cell help in vitro. Recent work identified in SLE patients a population of circulating IL-10 producing CXCR3+PD1hi CD4+ T cells (Th10 cells), activated by plasmacytoid dendritic cells stimulated with oxidized mitochondrial DNA, that promotes B cell maturation via succinate delivery. Th10 cells lack the canonical Tfh cell markers CXCR5, Bcl-6 or IL-21, unlike Tfh cells. In addition to the circulation, these cells also found in the lupus nephritic kidney in proximity to B cells, suggesting an extrafollicular B cell help. Taken together, these studies confirm the existence of Tefh cells in various settings of autoimmunity. But questions remain. Will any T cell outside the follicle/GC that has the capacity to help B cells be considered a Tefh cell? What are the rules governing extrafollicular tissue-specific B helper cells in terms of B cell selection, if any? Are Tefh cells drivers of B memory cell reactivation, given the extrafollicular location of the latter cells? The in vivo functional significance and the role of targeting these different populations of Tefh cells remains to be established.
Conclusions
While Tfh cell biology is based on the simple premise of a T helper cell that can provide supportive and instructive cues to developing B cells, the actual scope of Tfh cell function in physiologic and pathologic immune responses is quite complex. It is clear that Tfh cells do not represent a homogenous developmental program. Rather, the host immune response shapes developing Tfh cells to acquire phenotypic and functional properties that will allow them to orchestrate a humoral immune response that is best-suited to the inciting pathogen or immunologic insult. Bcl-6 is the lineage defining transcriptional regulator associated with the classical Tfh program, but the lack of Bcl-6 expression in certain cTfh or Tefh populations suggests either plasticity of Bcl-6 expression or that the ability to provide B cell help can be achieved through different developmental pathways. Currently, our knowledge of specific signaling pathways and developmental requirements of Bcl6-expressing Tfh cells is quite robust and has enabled selective therapeutic targeting of these cells in animal models of autoimmunity and human patients. As we continue to parse B helper T cell development and function, we hope to identify those population(s) of Tfh cells that are truly pathogenic whether they be in GCs, extrafollicular sites, and/or in the circulation and then target those cells to prevent the aberrant T-B collaboration that enables autoimmunity.
Highlights.
Tfh cells drive isotype switch and affinity maturation of pathogenic autoantibodies.
Tfh cell subsets acquire unique functional properties based on the cytokine milieu.
T cells that help B cells localize to germinal centers or extrafollicular spaces.
Tfh cells are therapeutic targets in autoimmune disease.
Acknowledgements
We thank Wenzhi Song for critical reading of the manuscript and helpful discussions. This work was supported by grants from the National Institutes of Health (R37 AR40072 and R01 AR074545), and the Lupus Research Alliance.
Footnotes
Declaration of Interests: None
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.DeFranco AL: Germinal centers and autoimmune disease in humans and mice. Immunol Cell Biol 2016, 94:918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craft JE: Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol 2012, 8:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crotty S: T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinuesa CG, Linterman MA, Goodnow CC, Randall KL: T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol Rev 2010, 237:72–89. [DOI] [PubMed] [Google Scholar]
- 5.Song W, Craft J: T follicular helper cell heterogeneity: Time, space, and function. Immunol Rev 2019, 288:85–96.•• This recent thorough review describes our current understanding of physiologic Tfh cell development and function.
- 6.Ueno H: Human Circulating T Follicular Helper Cell Subsets in Health and Disease. J Clin Immunol 2016, 36 Suppl 1:34–39. [DOI] [PubMed] [Google Scholar]
- 7.Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, Stockinger B: Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol 2013, 14:372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S: Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 2009, 323:1488–1492. [DOI] [PubMed] [Google Scholar]
- 9.Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, Kuchroo VK: The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol 2009, 10:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueno H, Banchereau J, Vinuesa CG: Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol 2015, 16:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, Foucat E, Dullaers M, Oh S, Sabzghabaei N, et al. : Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentebibel SE, Khurana S, Schmitt N, Kurup P, Mueller C, Obermoser G, Palucka AK, Albrecht RA, Garcia-Sastre A, Golding H, et al. : ICOS(+)PD −1(+)CXCR3(+) T follicular helper cells contribute to the generation of high-avidity antibodies following influenza vaccination. Sci Rep 2016, 6:26494.• cTfh1 cell expansion correlates with an in vivo humoral response to influenza vaccination.
- 13.He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, Sun X, Vandenberg K, Rockman S, Ding Y, et al. : Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39:770–781. [DOI] [PubMed] [Google Scholar]
- 14.Suurmond J, Diamond B: Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 2015, 125:2194–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robbins WC, Holman HR, Deicher H, Kunkel HG: Complement fixation with cell nuclei and DNA in lupus erythematosus. Proc Soc Exp Biol Med 1957, 96:575–579. [DOI] [PubMed] [Google Scholar]
- 16.Pisetsky DS: Anti-DNA antibodies--quintessential biomarkers of SLE. Nat Rev Rheumatol 2016, 12:102–110. [DOI] [PubMed] [Google Scholar]
- 17.Radic MZ, Weigert M: Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol 1994, 12:487–520. [DOI] [PubMed] [Google Scholar]
- 18.Sakakibara S, Arimori T, Yamashita K, Jinzai H, Motooka D, Nakamura S, Li S, Takeda K, Katayama J, El Hussien MA, et al. : Clonal evolution and antigen recognition of anti-nuclear antibodies in acute systemic lupus erythematosus. Sci Rep 2017, 7:16428.•• Affinity maturation in human ANA and anti-dsDNA antibodies demonstrates the requirement for T cell help for autoantibody production.
- 19.Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, Manku H, Vyse TJ, Roncador G, Huttley GA, et al. : Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum 2010, 62:234–244. [DOI] [PubMed] [Google Scholar]
- 20.Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, Borba EF, Goncalves CR, Costa PR, Kallas EG, et al. : Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol 2015, 67:988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu R, Wu Q, Su D, Che N, Chen H, Geng L, Chen J, Chen W, Li X, Sun L: A regulatory effect of IL-21 on T follicular helper-like cell and B cell in rheumatoid arthritis. Arthritis Res Ther 2012, 14:R255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, Jiang Y: High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol 2013, 174:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romme Christensen J, Bornsen L, Ratzer R, Piehl F, Khademi M, Olsson T, Sorensen PS, Sellebjerg F: Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One 2013, 8:e57820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo C, Li Y, Liu W, Feng H, Wang H, Huang X, Qiu L, Ouyang J: Expansion of circulating counterparts of follicular helper T cells in patients with myasthenia gravis. J Neuroimmunol 2013, 256:55–61. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wang L, Shi Y, Wang F, Yang H, Han S, Bai Y: Altered circulating T follicular helper cell subsets in patients with psoriasis vulgaris. Immunol Lett 2017, 181:101–108. [DOI] [PubMed] [Google Scholar]
- 26.Cunill V, Massot M, Clemente A, Calles C, Andreu V, Nunez V, Lopez-Gomez A, Diaz RM, Jimenez MLR, Pons J, et al. : Relapsing-Remitting Multiple Sclerosis Is Characterized by a T Follicular Cell Pro-Inflammatory Shift, Reverted by Dimethyl Fumarate Treatment. Front Immunol 2018, 9:1097.•• Successful treatment of MS correlates with reduction in cTfh cells.
- 27.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S: Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 2006, 312:1669–1672. [DOI] [PubMed] [Google Scholar]
- 28.Murphy ED, Roths JB: A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum 1979, 22:1188–1194. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, et al. : A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A 2006, 103:9970–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S: Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, Hwu P, Shaffer DJ, Akilesh S, Roopenian DC, et al. : Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol 2004, 173:5361–5371. [DOI] [PubMed] [Google Scholar]
- 32.Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC 3rd, Leonard WJ, Roopenian DC: A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A 2009, 106:1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McPhee CG, Bubier JA, Sproule TJ, Park G, Steinbuck MP, Schott WH, Christianson GJ, Morse HC 3rd, Roopenian DC: IL-21 is a double-edged sword in the systemic lupus erythematosus-like disease of BXSB.Yaa mice. J Immunol 2013, 191:4581–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK: Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A 2000, 97:6670–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, et al. : A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435:452–458. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson BA, Panka DJ, Nguyen KA, Erikson J, Abbas AK, Marshak-Rothstein A: Anatomy of autoantibody production: dominant localization of antibody-producing cells to T cell zones in Fas-deficient mice. Immunity 1995, 3:509–519. [DOI] [PubMed] [Google Scholar]
- 37.William J, Euler C, Christensen S, Shlomchik MJ: Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 2002, 297:2066–2070. [DOI] [PubMed] [Google Scholar]
- 38.William J, Euler C, Leadbetter E, Marshak-Rothstein A, Shlomchik MJ: Visualizing the onset and evolution of an autoantibody response in systemic autoimmunity. J Immunol 2005, 174:6872–6878. [DOI] [PubMed] [Google Scholar]
- 39.Luzina IG, Atamas SP, Storrer CE, daSilva LC, Kelsoe G, Papadimitriou JC, Handwerger BS: Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol 2001, 70:578–584. [PubMed] [Google Scholar]
- 40.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J: ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med 2008, 205:2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, Manz RA: Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med 2004, 199:1577–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee SK, Silva DG, Martin JL, Pratama A, Hu X, Chang PP, Walters G, Vinuesa CG: Interferon-gamma excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity 2012, 37:880–892. [DOI] [PubMed] [Google Scholar]
- 43.Choi JY, Seth A, Kashgarian M, Terrillon S, Fung E, Huang L, Wang LC, Craft J: Disruption of Pathogenic Cellular Networks by IL-21 Blockade Leads to Disease Amelioration in Murine Lupus. J Immunol 2017, 198:2578–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coquery CM, Loo WM, Wade NS, Bederman AG, Tung KS, Lewis JE, Hess H, Erickson LD: BAFF regulates follicular helper t cells and affects their accumulation and interferon-gamma production in autoimmunity. Arthritis Rheumatol 2015, 67:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, Nutt SL: The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol 2012, 13:491–498. [DOI] [PubMed] [Google Scholar]
- 46.Carpio VH, Opata MM, Montanez ME, Banerjee PP, Dent AL, Stephens R: IFN-gamma and IL-21 Double Producing T Cells Are Bcl6-Independent and Survive into the Memory Phase in Plasmodium chabaudi Infection. PLoS One 2015, 10:e0144654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinstein JS, Laidlaw BJ, Lu Y, Wang JK, Schulz VP, Li N, Herman EI, Kaech SM, Gallagher PG, Craft J: STAT4 and T-bet control follicular helper T cell development in viral infections. J Exp Med 2018, 215:337–355.• This study elucidates transcriptional regulation of Tfh1 subtype development.
- 48.Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, et al. : STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 2007, 357:977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma X, Nakayamada S, Kubo S, Sakata K, Yamagata K, Miyazaki Y, Yoshikawa M, Kitanaga Y, Zhang M, Tanaka Y: Expansion of T follicular helper-T helper 1 like cells through epigenetic regulation by signal transducer and activator of transcription factors. Ann Rheum Dis 2018, 77:1354–1361.•• Stat1 and Stat4 are implicated in the differentiation of cTfh1 cell expansion in SLE patients.
- 50.Wichner K, Stauss D, Kampfrath B, Kruger K, Muller G, Rehm A, Lipp M, Hopken UE: Dysregulated development of IL-17- and IL-21-expressing follicular helper T cells and increased germinal center formation in the absence of RORgammat. FASEB J 2016, 30:761–774. [DOI] [PubMed] [Google Scholar]
- 51.Charles N, Hardwick D, Daugas E, Illei GG, Rivera J: Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat Med 2010, 16:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dema B, Charles N, Pellefigues C, Ricks TK, Suzuki R, Jiang C, Scheffel J, Hasni S, Hoffman V, Jablonski M, et al. : Immunoglobulin E plays an immunoregulatory role in lupus. J Exp Med 2014, 211:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reinhardt RL, Liang HE, Locksley RM: Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 2009, 10:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maurer M, Altrichter S, Schmetzer O, Scheffel J, Church MK, Metz M: Immunoglobulin E-Mediated Autoimmunity. Front Immunol 2018, 9:689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohan C, Shi Y, Laman JD, Datta SK: Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol 1995, 154:1470–1480. [PubMed] [Google Scholar]
- 56.Ma J, Xu J, Madaio MP, Peng Q, Zhang J, Grewal IS, Flavell RA, Craft J: Autoimmune lpr/lpr mice deficient in CD40 ligand: spontaneous Ig class switching with dichotomy of autoantibody responses. J Immunol 1996, 157:417–426. [PubMed] [Google Scholar]
- 57.Daikh DI, Finck BK, Linsley PS, Hollenbaugh D, Wofsy D: Long-term inhibition of murine lupus by brief simultaneous blockade of the B7/CD28 and CD40/gp39 costimulation pathways. J Immunol 1997, 159:3104–3108. [PubMed] [Google Scholar]
- 58.Grammer AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, Illei GG, Lipsky PE: Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154-CD40 interactions. J Clin Invest 2003, 112:1506–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peters AL, Stunz LL, Bishop GA: CD40 and autoimmunity: the dark side of a great activator. Semin Immunol 2009, 21:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K: IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol 2007, 178:3822–3830. [DOI] [PubMed] [Google Scholar]
- 61.Rankin AL, Guay H, Herber D, Bertino SA, Duzanski TA, Carrier Y, Keegan S, Senices M, Stedman N, Ryan M, et al. : IL-21 receptor is required for the systemic accumulation of activated B and T lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J mice. J Immunol 2012, 188:1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fujikura D, Ikesue M, Endo T, Chiba S, Higashi H, Uede T: Death receptor 6 contributes to autoimmunity in lupus-prone mice. Nat Commun 2017, 8:13957.• This study highlights a novel pathway of T:B cell communication within the germinal center required for Tfh cell development.
- 63.Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, Laidlaw BJ, Araki K, Ahmed R, Kaech SM, et al. : The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity 2015, 43:690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morel L: Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol 2017, 13:280–290. [DOI] [PubMed] [Google Scholar]
- 65.Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, Morel L: Glucose Oxidation Is Critical for CD4+ T Cell Activation in a Mouse Model of Systemic Lupus Erythematosus. J Immunol 2016, 196:80–90.•• Metabolic dysregulation is central to the phenotype of murine lupus.
- 66.Choi SC, Titov AA, Abboud G, Seay HR, Brusko TM, Roopenian DC, Salek-Ardakani S, Morel L: Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat Commun 2018, 9:4369.•• Autoreactive Tfh cells can be selectively targeted over physiologic Tfh cells to regulate autoimmunity while preserving normal immune function.
- 67.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB: Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 2003, 349:1526–1533. [DOI] [PubMed] [Google Scholar]
- 68.Liu Z, Davidson A: Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med 2012, 18:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003, 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, Li QZ, Lian Y, Wu T, Reimold AM, et al. : SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS One 2013, 8:e67003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fairhurst AM, Hwang SH, Wang A, Tian XH, Boudreaux C, Zhou XJ, Casco J, Li QZ, Connolly JE, Wakeland EK: Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol 2008, 38:1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sweet RA, Christensen SR, Harris ML, Shupe J, Sutherland JL, Shlomchik MJ: A new site-directed transgenic rheumatoid factor mouse model demonstrates extrafollicular class switch and plasmablast formation. Autoimmunity 2010, 43:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poholek AC, Hansen K, Hernandez SG, Eto D, Chandele A, Weinstein JS, Dong X, Odegard JM, Kaech SM, Dent AL, et al. : In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol 2010, 185:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim ST, Choi JY, Lainez B, Schulz VP, Karas DE, Baum ED, Setlur J, Gallagher PG, Craft J: Human Extrafollicular CD4(+) Th Cells Help Memory B Cells Produce Igs. J Immunol 2018, 201:1359–1372.• This study characterizes important features of extrafollicular T helper cells in human lymphoid tissue.
- 75.Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, et al. : B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med 2011, 208:1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Racine R, Jones DD, Chatterjee M, McLaughlin M, Macnamara KC, Winslow GM: Impaired germinal center responses and suppression of local IgG production during intracellular bacterial infection. J Immunol 2010, 184:5085–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hastey CJ, Elsner RA, Barthold SW, Baumgarth N: Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J Immunol 2012, 188:5612–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Niro R, Lee SJ, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, Gupta NT, Kleinstein SH, Vigneault F, Gilbert TJ, et al. : Salmonella Infection Drives Promiscuous B Cell Activation Followed by Extrafollicular Affinity Maturation. Immunity 2015, 43:120–131.•• Extrafollicular T:B colloboration can be the primary mode of humoral immunity in a physiologic immune reaction.
- 79.Jain S, Stock A, Macian F, Putterman C: A Distinct T Follicular Helper Cell Subset Infiltrates the Brain in Murine Neuropsychiatric Lupus. Front Immunol 2018, 9:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vu Van D, Beier KC, Pietzke LJ, Al Baz MS, Feist RK, Gurka S, Hamelmann E, Kroczek RA, Hutloff A: Local T/B cooperation in inflamed tissues is supported by T follicular helper-like cells. Nat Commun 2016, 7:10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liarski VM, Kaverina N, Chang A, Brandt D, Yanez D, Talasnik L, Carlesso G, Herbst R, Utset TO, Labno C, et al. : Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med 2014, 6:230ra246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, et al. : Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017, 542:110–114.•• Identification and characterization of extrafollicular T helper cells in rheumatoid arthritis patients.
- 83.Deng R, Hurtz C, Song Q, Yue C, Xiao G, Yu H, Wu X, Muschen M, Forman S, Martin PJ, et al. : Extrafollicular CD4(+) T-B interactions are sufficient for inducing autoimmune-like chronic graft-versus-host disease. Nat Commun 2017, 8:978.• Extrafollicular helper T cells drive autoimmunity in mouse model of cGVHD.
- 84.Jin H, Yang K, Zhang H, Chen Y, Qi H, Fan Z, Huang F, Xuan L, Lin R, Zhao K, et al. : Expansion of circulating extrafollicular helper T-like cells in patients with chronic graft-versus-host disease. J Autoimmun 2019, 100:95–104.•• This study identifies characteristics of circulating extrafollicular T helper cells in patients with cGVHD.
- 85.Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, Banchereau R, Xu Z, Chandra M, Chung CH, et al. : A CD4(+) T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med 2019, 25:75–81.•• Extrafollicular Th10 cells in SLE patients promote B cell IgG responses in vitro through IL-10 and the metabolite, succinate, engaging succinate receptors on B cells.